

Abstract
Background
The number of tumor suppressor genes for which germline mutations have been linked to cancer risk is steadily increasing. However, while recent reports have linked constitutional normal tissue promoter methylation of BRCA1 and MLH1 to ovarian and colon cancer risk, the role of epigenetic alterations as cancer risk factors remains largely unknown, presenting an important area for future research. Currently, we lack fast and sensitive methods for assessment of promoter methylation status across known tumor suppressor genes.
Results
In this paper, we present a novel NGS-based approach assessing promoter methylation status across a large panel of defined tumor suppressor genes to base-pair resolution. The method omits the limitations related to commonly used array-approaches. Our panel includes 565 target regions covering the promoters of 283 defined tumor suppressors, selected by pre-specified criteria, and was applied for rapid targeted methylation-specific NGS. The feasibility of the method was assessed by analyzing normal tissue DNA (white blood cells, WBC) samples from 34 healthy postmenopausal women and by performing preliminary assessment of the methylation landscape of tumor suppressors in these individuals. The mean target coverage was 189.6x providing a sensitivity of 0.53%, sufficient for promoter methylation assessment of low-level methylated genes like BRCA1. Within this limited test-set, we detected 206 regions located in the promoters of 149 genes to be differentially methylated (hyper- or hypo-) at > 99% confidence level. Seven target regions in gene promoters (CIITA, RASSF1, CHN1, PDCD1LG2, GSTP1, XPA, and ZNF668) were found to be hyper-methylated in a minority of individuals, with a > 20 percent point difference in mean methylation across the region between individuals. In an exploratory hierarchical clustering analysis, we found that the individuals analyzed may be grouped into two main groups based on their WBC methylation profile across the 283 tumor suppressor gene promoters.
Conclusions
Methylation-specific NGS of our tumor suppressor panel, with detailed assessment of differential methylation in healthy individuals, presents a feasible method for identification of novel epigenetic risk factors for cancer.
Keywords: Methylation, Epimutations, Cancer risk, Promoter, Massive parallel sequencing
Introduction
The number of tumor suppressor genes for which germline mutations are linked to elevated cancer risk is steadily increasing [1–3]. Mutations across different genes present a continuum of penetrance, ranging from moderately to massively elevated risk of different cancer forms. Further, while mutations in some genes (so far) are restricted to increased risk of a single, or a few cancer forms, mutations in other genes may increase the risk of multiple different types of cancer [4, 5].
Some of the best described “classical” high penetrance genes include BRCA1/2, for which germline mutations are associated with an elevated risk of ovarian and breast cancer [6], MLH1/MSH2 linked to colorectal cancer [7], CDKN2A and RB1, associated with melanoma and retinoblastoma, respectively [8–10], as well as TP53, associated with the Li-Fraumeni syndrome with an elevated risk for multiple cancer forms [11]. However, the list of genes for which germline mutations are ascertained to confer cancer risk is continuously increasing due to application of massive parallel sequencing [12, 13]. Still, for many families with multiple cases of a specific tumor form (like breast, ovary, or melanomas), no pathogenic germline gene variant has been identified.
Epigenetic gene inactivation may occur through different mechanisms [14, 15]. So far, promoter methylation is the best studied of all the epigenetic modifications, and such methylation is well established as a mechanism of inactivation of tumor suppressor genes. While many germline mutations affecting tumor suppressor genes are well studied as cancer risk factors, knowledge regarding constitutional epigenetic inactivation [16] as a potential cancer risk factor remains limited. Somatic promotor methylation in tumor suppressor genes is a common event in cancer [17], but the role of aberrant epigenetic events, or constitutional promoter methylation of tumor suppressor genes in normal cells as potential cancer risk factors, remains largely unexplored. While mosaic methylation of the MLH1 gene in normal leukocytes has been observed in colorectal cancer patients [18, 19] and a haplotype leading to secondary constitutional methylation in the MGM2 promoter [20] has been found in a cancer-prone family [21], in general, data on normal tissue methylation patterns and cancer risk are scarce [22].
Recently, in a large study, we reported low-grade mosaic (< 10% of alleles) normal tissue BRCA1 promoter methylation to confer a significantly increased risk of high-grade serous ovarian cancer (HGSOC) [23]. In our study, we found > 4% of healthy adult females in a Caucasian population to harbor mosaic BRCA1 promoter methylation in their normal white blood cells (WBC). Individuals carrying such methylation had a 2-3 fold increased risk of HGSOC. Importantly, WBC BRCA1 promoter methylation was strongly associated with corresponding methylation in other normal tissues, and, in HGSOC patients, also associated with methylation in the tumor. Taken together, this indicated that methylated normal cells in the ovary may act as tumor precursors.
Based on these results and the findings of others [19, 24–29], we hypothesized that additional tumor suppressors could be hyper-methylated in normal cells, thereby causing an elevated risk for certain cancer forms within subgroups of healthy individuals in the general population [30].
To explore such a hypothesis, there is a need for improved methodologies. Although methylation status may be analyzed by conventional arrays, such assessments are limited to the selection of CpGs covered by the array probes. These selected CpGs may not necessarily represent all the CpGs crucial for gene silencing [23]. An alternative is methylation-specific whole genome sequencing, but this remains prohibitively costly. In the present study, we aimed to establish, and provide proof-of-concept for, a novel strategy assessing the full CpG spectrum across promoter areas of tumor suppressor genes. The assay applies methylation-specific massive parallel sequencing of the promoter areas of a panel of 283 tumor suppressor genes. We show the feasibility of the method by depicting promoter methylation variation across the promoter panel in a set of white blood cell (WBC) DNA obtained from 34 healthy individuals. Further, by performing an exploratory hierarchical clustering, our findings indicate that the profiles of normal cell promoter methylation of tumor suppressor genes fall into two main clusters defined by differences in genes regulating key biological pathways.
Results
Methylation specific sequencing
We analyzed WBC DNA from 34 healthy individuals. After bisulfite conversion of the DNA, we performed methylation-specific sequencing of 565 capture regions representing 356 target regions from 283 tumor suppressor gene promoters (the full list of genes and regions is presented as Supplementary Table S1). Sequencing was performed on an Illumina MiSeq, running 8 samples per run. Regarding average values per sample, we obtained 4.95 × 106 reads (range 3.36-7.85 × 106) (Fig. 1a; for details per sample see Table 1). Subsequent to quality filtering, 88% of the reads, were retained. Thus, after filtering, 4.30 × 106 reads were attempted mapped to the genome, yielding 4.08 × 106 mapped single reads. Out of these, 3.6 × 106 reads mapped with properly paired reads for each sample (average values; Fig. 1a). These reads led to a mean primary target coverage of 189.6x (114.8x-269.5x) and a mean capture target coverage of 199.4x (120.7x-283.4x). Every sample had almost equal percentage of reads mapped to capture targets and primary targets (Fig. 1b).
Fig. 1.
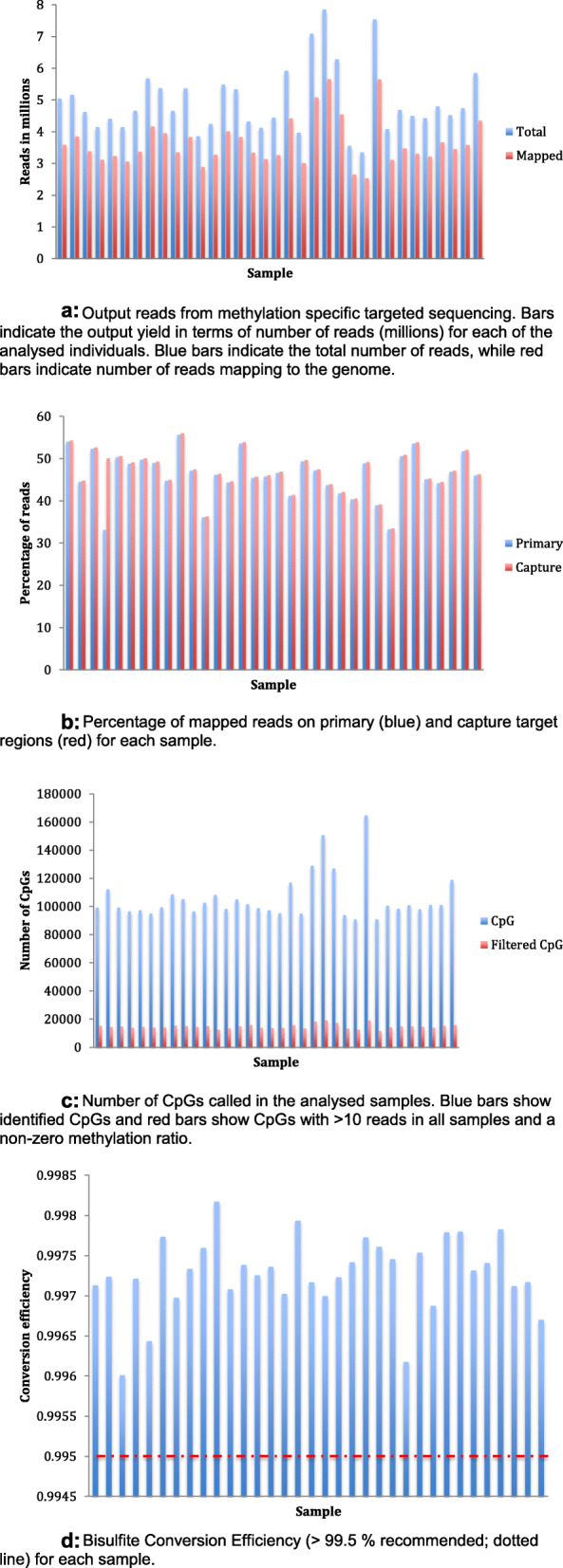
a Output reads from methylation specific targeted sequencing. Bars indicate the output yield in terms of number of reads (millions) for each of the analyzed individuals. Blue bars indicate the total number of reads, while red bars indicate number of reads mapping to the genome. b Percentage of mapped reads on primary (blue) and capture target regions (red) for each sample. c Number of CpGs called in the analyzed samples. Blue bars show identified CpGs and red bars show CpGs with > 10 reads in all samples and a non-zero methylation ratio. d Bisulfite conversion efficiency (> 99.5% recommended; dotted line) for each sample
Table 1.
Summary of samples and analyses
| Sample | Input reads | % reads post QC | % reads mapped | Reads (paired and clipped) | % reads on target (primary) | % reads on target (capture) | Coverage on target (primary) | Coverage on target (capture) |
|---|---|---|---|---|---|---|---|---|
| 10046_S2 | 5043578 | 90.39 | 78.71 | 3588074 | 54.01 | 54.31 | 209.99 | 220.84 |
| 10071_S7 | 5167738 | 89.08 | 83.62 | 3849392 | 44.54 | 44.81 | 186.40 | 196.12 |
| 10077_S3 | 4622790 | 90.13 | 81.18 | 3382228 | 52.35 | 52.66 | 198.17 | 208.34 |
| 10078_S8 | 4148304 | 89.91 | 83.59 | 3117970 | 33.13 | 50.13 | 175.26 | 184.27 |
| 10081_S4 | 4408742 | 89.53 | 81.99 | 3236368 | 50.31 | 50.60 | 186.37 | 195.79 |
| 10082_S5 | 4146244 | 88.90 | 82.83 | 3053058 | 48.82 | 49.10 | 171.00 | 179.62 |
| 10086_S6 | 4665150 | 89.39 | 80.91 | 3373894 | 49.76 | 50.06 | 186.09 | 195.68 |
| 10088_S2 | 5683572 | 88.12 | 83.26 | 4169990 | 49.01 | 49.29 | 219.04 | 230.48 |
| 10097_S3 | 5372752 | 87.46 | 84.09 | 3951544 | 44.74 | 45.00 | 195.13 | 205.17 |
| 10107_S1 | 4659718 | 89.67 | 80.24 | 3352898 | 55.68 | 56.00 | 213.95 | 224.78 |
| 10110_S6 | 5369964 | 86.87 | 82.21 | 3835038 | 47.17 | 47.45 | 199.76 | 210.11 |
| 10113_S7 | 3862862 | 89.44 | 83.54 | 2886124 | 36.11 | 36.31 | 120.48 | 126.52 |
| 10117_S5 | 4243470 | 89.93 | 85.80 | 3274194 | 46.15 | 46.42 | 169.10 | 177.78 |
| 10126_S7 | 5490894 | 87.68 | 83.36 | 4013294 | 44.39 | 44.65 | 198.79 | 209.00 |
| 10131_S1 | 5338282 | 87.00 | 82.62 | 3836988 | 53.58 | 53.88 | 232.54 | 244.35 |
| 10146_S6 | 4326882 | 90.13 | 85.57 | 3337118 | 45.47 | 45.73 | 168.34 | 177.00 |
| 10149_S7 | 4129130 | 90.01 | 84.46 | 3139126 | 45.78 | 46.04 | 159.55 | 167.77 |
| 10155_S5 | 4450890 | 86.31 | 84.99 | 3264796 | 46.65 | 46.91 | 170.60 | 179.37 |
| 20011_S4 | 5923516 | 87.27 | 85.49 | 4419628 | 41.19 | 41.42 | 203.82 | 214.20 |
| 20019_S1 | 3972578 | 89.46 | 84.62 | 3007020 | 49.39 | 49.68 | 163.80 | 172.27 |
| 20022_S4 | 7091616 | 88.48 | 81.02 | 5083584 | 47.19 | 47.47 | 257.88 | 271.25 |
| 20023_S1 | 7854298 | 88.16 | 81.78 | 5662728 | 43.72 | 43.98 | 269.47 | 283.35 |
| 20024_S5 | 6284900 | 88.04 | 82.23 | 4549698 | 41.85 | 42.09 | 215.05 | 225.92 |
| 20062_S2 | 3554848 | 88.54 | 84.13 | 2648098 | 40.36 | 40.54 | 127.76 | 133.78 |
| 20068_S3 | 3355298 | 89.51 | 84.38 | 2534292 | 48.89 | 49.18 | 138.43 | 145.54 |
| 20078_S2 | 7541786 | 88.33 | 84.85 | 5651984 | 38.94 | 39.16 | 246.67 | 259.20 |
| 20088_S4 | 4083996 | 90.28 | 84.33 | 3109332 | 33.27 | 33.46 | 114.79 | 120.70 |
| 20092_S3 | 4686060 | 89.01 | 83.38 | 3477852 | 50.58 | 50.88 | 196.38 | 206.39 |
| 20098_S1 | 4501632 | 88.21 | 83.23 | 3305210 | 53.58 | 53.89 | 197.69 | 207.78 |
| 20106_S2 | 4430250 | 86.96 | 83.43 | 3214408 | 45.12 | 45.32 | 173.69 | 181.84 |
| 20117_S4 | 4802994 | 88.70 | 86.01 | 3664364 | 44.22 | 44.48 | 178.18 | 187.37 |
| 20119_S5 | 4525080 | 89.19 | 85.57 | 3453670 | 46.90 | 47.16 | 180.35 | 189.59 |
| 20122_S6 | 4741150 | 88.79 | 85.18 | 3585870 | 51.76 | 52.07 | 204.62 | 215.16 |
| 20160_S3 | 5848296 | 88.30 | 84.30 | 4353508 | 46.02 | 46.28 | 220.04 | 231.29 |
The overall number of informative CpGs identified for each sample were on average 1.0 × 105 (range 0.9 × 105 to 1.6 × 105). Restricting the CpGs to those with a methylation ratio > 0, and more than 10 reads in coverage, the number was reduced to 1.5 × 104 (range 1.1 × 104-1.9 × 104; Fig. 1c).
We defined the sensitivity of our strategy as 1/x, where x = sequencing depth at any given CpG. With the average primary target depth being 189.6x, the sensitivity was 0.53%. In theory, the fragility of this sensitivity estimate lies in that, for some samples, the results may depend on a single read, rendering them more sensitive to artifacts such as inadequate bisulfite conversion. However, assessing the bisulfite conversion rate (C to T) of the internal Lambda DNA control (see the “Methods” section), we found the conversion efficiency to be on average 99.7% (range 99.6-99.8%) across the analyzed samples (Fig. 1d). This indicates a rate of technical artifacts (falsely retained C’s instead of T’s) to be lower than 0.2-0.4%, thus approaching the error rate in the sequencing per se (Q30 threshold).
Reproducibility was assessed in a separate standard sample (pooled DNA from 5 healthy donors) that was run in 6 parallels per run over 2 independent runs. In a selection of 12 out of the 565 regions, we found the mean coefficient of variation to be 7.1% (median 4.4%; Supplementary Table S2). As such, the technical variability in this standard sample was considerably lower than the detected biological variation (see below) in our study set of 34. Variability was considerably lower when assessing all CpGs in a region than when limiting analyses to randomized selections of CpGs within the regions (e.g., for PRDM2, the coefficient of variation was 1.5% when considering all CpGs while it was on average 4.7% when assessing randomized selections of 5 CpGs within the region).
Methylation landscape of tumor suppressors
For each sample, we calculated the mean methylation for each of the 565 capture regions based on individual CpG methylation ratios within each actual region (see the “Methods” section for details). We observed large inter-region variation in the methylation levels of the regions within the 283 tumor suppressor gene promoters analyzed (Fig. 2). Some regions were completely methylated (e.g., regions within the promoters of AIP, PRDM2, ATR, DICER1, SFPQ), while others in general were non-methylated in most individuals (e.g., regions within ARID2, TRIM33, SETD2, IKZF1, and ARID1B; Supplementary Figure 1).
Fig. 2.
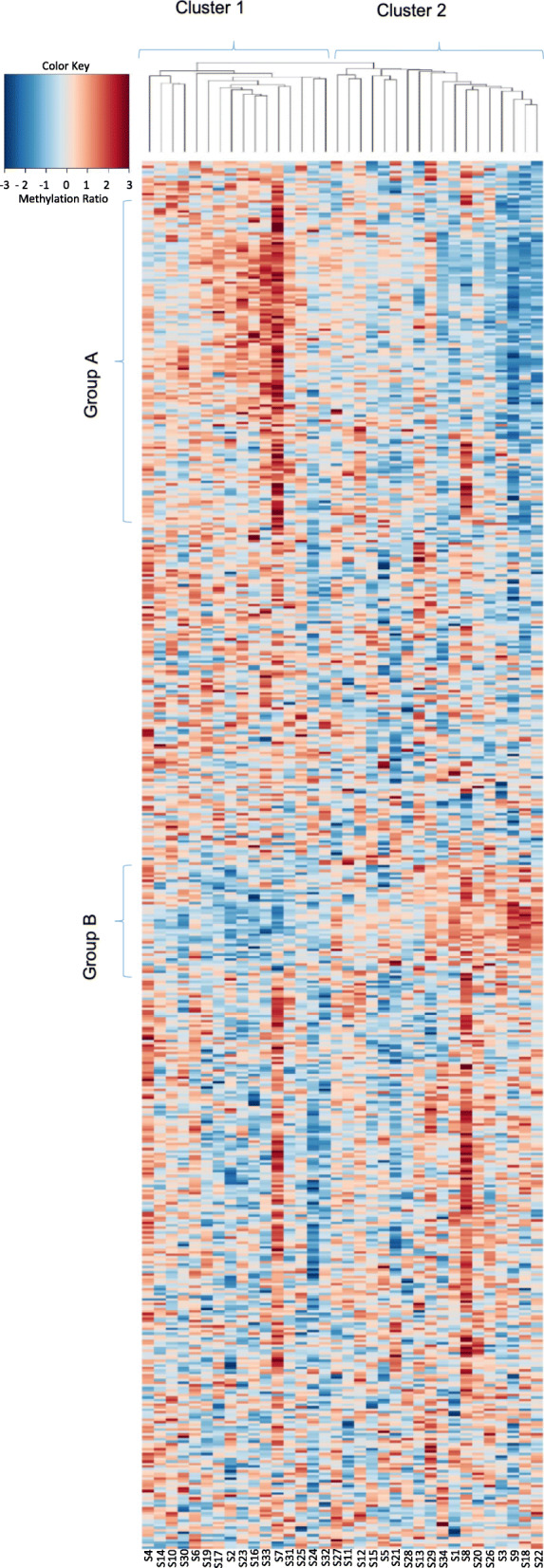
Heatmap showing average methylation ratio for all samples and genes. Scale: Red indicates high methylation and blue indicate low methylation
In some regions, there was a large variability between CpGs within the promoter region, indicating that some CpGs may be constitutively methylated, while others (perhaps more crucial for transcriptional regulation) had a lower methylation level and may be more dynamically methylated (Supplementary Figure 2).
Constitutional promoter hyper-methylation has been classified either as secondary due to a rare genetic/SNP variant [16], typically resulting in high methylation levels [31, 32] or primary, in which case, methylation may occur at a low mosaic level (VAF of < 10%) [23]. As for both cases, we may not expect identifying several affected individuals in a small dataset like the present; thus, lack of differential methylation here may not exclude a gene as a potential epigenetic pathogenic factor. Still, to validate the feasibility of our method, we aimed at exploring potential differential methylation between individuals across our data set. To do so, we took three approaches: first, we assessed differential methylation across the dataset in general. Second, we specifically assessed for individual hyper-methylation, assuming this to be the most relevant alteration regarding inactivating tumor suppressors. Third, we specifically assessed those tumor suppressors where previous data have linked promoter methylation to cancer risk.
Differential methylation
Subsequent to methylation calling, we identified promoter regions differentially methylated across our sample set. Although low levels of methylation (allele methylation frequency of < 5%) have been shown to affect cancer risk [23], in the present sample set we focused on identifying those genes presenting the largest inter individual methylation variation as a proof-of-concept for our methodological approach. We defined methylation variation in a region according to the difference in absolute but also relative methylation level. First, we assessed the difference in absolute methylation as the difference in percentage of alleles methylated between individual (i.e., difference presented as percent points). Second, we assessed the relative difference between individuals, i.e., the ratio between the highest and lowest methylated individual with respect to percentage of methylated alleles.
Based on a Z-score assessment of a methylation matrix consisting of averaged methylation ratios for each of the 565 capture regions across all 34 samples (see the “Methods” section for details), we identified 206 regions (within the promoters of 149 genes) where a minority (one-third or less) of the samples analyzed were significantly differentially methylated as compared to the majority of samples at a ≥ 99% confidence level (i.e., outside the 99% confidence interval; Supplementary Table S3). Assessing the difference between the samples with the highest and the lowest level of methylation within these 206 regions, about half of the regions (n = 101) displayed less than 5 percent point difference. However, several of the tumor suppressor regions displayed a large variation in methylation, with 72 regions displaying > 10 percent point difference and 22 regions displaying > 20 percent points difference between the highest and the lowest methylated samples (Table 2). The largest difference was observed for GAS7, where the difference between the highest and the lowest methylated sample was 66.6 percent points.
Table 2.
Differentially methylated genes. Gene regions with > 20 percent points difference in methylation ratio, between least methylated sample to most methylated sample along with fold change differences are listed. Hyper-methylated target region of those genes are shown in bold
| Gene name | Gene capture region | Min. methylation ratio | Max. methylation ratio | Difference in methylation ratio | Fold change |
|---|---|---|---|---|---|
| GAS7 | chr17: 10199716 - 10200316 | 0.2670 | 0.9332 | 0.6662 | 3.4951 |
| ELAC2 | chr17: 13019069 - 13019845 | 0.4581 | 0.8332 | 0.3751 | 1.8188 |
| GSTM1 | chr1: 109686327 - 109687046 | 0.6438 | 1.0000 | 0.3562 | 1.5533 |
| THBS1 | chr15: 39579298 - 39579871 | 0.4671 | 0.7885 | 0.3214 | 1.6881 |
| CIITA | chr16: 10874982 - 10875928 | 0.2511 | 0.5577 | 0.3066 | 2.221 |
| RASSF1 | chr3: 50339388 - 50340021 | 0.1786 | 0.4720 | 0.2934 | 2.6428 |
| CHN1 | chr2: 174846842 - 174848034 | 0.2141 | 0.5074 | 0.2933 | 2.3699 |
| MSH2 | chr2: 47401613 - 47402319 | 0.5897 | 0.8734 | 0.2838 | 1.4811 |
| PALB2 | chr16: 23642511 - 23643136 | 0.6333 | 0.9134 | 0.2801 | 1.4423 |
| RUNX3 | chr1: 24964233 - 24965550 | 0.3920 | 0.6479 | 0.2559 | 1.6528 |
| TP63 | chr3: 189789769 - 189790448 | 0.6612 | 0.9059 | 0.2446 | 1.3701 |
| PDCD1LG2 | chr9: 5510022 - 5511326 | 0.3182 | 0.5511 | 0.2330 | 1.7319 |
| AIP | chr11: 67481632 - 67482276 | 0.6716 | 0.9002 | 0.2286 | 1.3404 |
| GPC3 | chrX: 133986729 - 133987434 | 0.5842 | 0.8036 | 0.2194 | 1.3756 |
| AIP | chr11: 67482202 - 67482880 | 0.1035 | 0.3214 | 0.2180 | 3.1053 |
| GSTP1 | chr11: 67581895 - 67582976 | 0.2673 | 0.4834 | 0.2162 | 1.8085 |
| AIP | chr11: 67481257 - 67481869 | 0.7857 | 1.0000 | 0.2143 | 1.2728 |
| XPA | chr9: 97698585 - 97699193 | 0.6991 | 0.9130 | 0.2139 | 1.306 |
| APC | chr5: 112736082 - 112736959 | 0.6361 | 0.8479 | 0.2118 | 1.333 |
| CTCFL | chr20: 57524096 - 57527440 | 0.6554 | 0.8663 | 0.2109 | 1.3218 |
| CASP8 | chr2: 201259179 - 201260169 | 0.3698 | 0.5799 | 0.2102 | 1.5681 |
| ZNF668 | chr16: 31064314 - 31065859 | 0.4513 | 0.6584 | 0.2070 | 1.4589 |
| -- | -- | -- | -- | -- | -- |
| RABEP1 | chr17: 5281240 - 5283045 | 0.0415 | 0.1033 | 0.0618 | 2.4902 |
| AIP | chr11: 67482382 - 67483805 | 0.0517 | 0.1229 | 0.0712 | 2.3783 |
| RASSF1 | chr3: 50338258 - 50339618 | 0.0976 | 0.2178 | 0.1202 | 2.2322 |
| FOXO4 | chrX: 71094692 - 71096928 | 0.1256 | 0.2592 | 0.1335 | 2.0629 |
| ZRSR2 | chrX: 15789350 - 15791219 | 0.0559 | 0.1021 | 0.0462 | 1.8252 |
| RUNX1T1 | chr8: 92102449 - 92105016 | 0.0558 | 0.1008 | 0.0450 | 1.8068 |
| RHOH | chr4: 40196452 - 40197679 | 0.1059 | 0.1914 | 0.0855 | 1.8067 |
Assessing the relative difference (ratio between the highest and lowest methylated sample), again GAS7 was the top-ranking promoter, showing a relative difference of 3.5 fold between the highest and the lowest methylated sample. As expected, in addition to GAS7, we found a substantial overlap between top-ranking regions based on absolute differences and the top-ranking regions based on relative differences (ratio) in methylation levels (Table 2). Especially the AIP gene also had a region that was highly differentially methylated both in terms of percentage difference (> 20%) and fold difference (> 3 fold). The only regions with less than 20 percent point difference but a high fold difference (> 2 fold), were regions in RABEP1, RASSF1, AIP, and FOXO4 (Table 2, lower section).
Hyper-methylated tumor suppressors
Regarding tumor suppressor genes, we hypothesized that in case constitutional methylation is associated with a significantly elevated cancer risk, we may expect a minor sub-fraction of healthy individuals to have hyper-methylated promoters. We therefore performed additional sub-analyses restricting 206 genes identified above, to the genes/region with positive Z-scores with > 99% confidence level, i.e., genes/regions that were significantly hyper-methylated in a minority of individuals as compared to the majority of individuals (see the “Methods” section). Among the 206 differentially methylated regions, 115 revealed positive Z-scores. Out of these 115, 25 displayed > 10 percent points difference from the highest to the lowest methylated sample. The corresponding number of regions revealing > 20 percent points difference was 7. These 7 regions were within the promoters of CIITA, RASSF1, CHN1, PDCD1LG2, GSTP1, XPA, and ZNF668, with the three former genes revealing a difference of more than 30 percent points (Table 2). Re-assessing these data based on fold difference instead of percent points, we identified three regions (in AIP, RABEP1, and RASSF1) with a lower than 20 percent point absolute difference but a relative ratio > 2. Since another region of RASSF1 was already identified as having a difference > 20 percent points, this left us with 9 different genes with substantial differences in methylation levels.
Further, we reasoned that if methylation of any of these genes may act as a cancer risk factor, then somatic methylation of the same genes should be present in a fraction of human cancers. We therefore mined the COSMIC data base [33] for reported somatic methylation of the 9 genes. Six of these genes (CHN1, PDCD1LG2, XPA, ZNF668, RABEP1, AIP) were not reported to be aberrantly somatically methylated in tumors, while one gene (CIITA) was reported to be hypo-methylated in a very small fraction (0.19-1.53%) of various solid tumors. In contrast, somatic hyper-methylation of RASSF1 was reported in > 4% of endometrial cancers and > 1% of breast cancers. Further, somatic hyper-methylation of GSTP1 was reported in > 7% of prostate cancers and > 1% of breast cancers. Thus, this finding indicates that some genes found hyper-methylated in tumor tissue are also differentially methylated in normal tissue of healthy individuals. Although these data do not provide any conclusive evidence per se, the findings warrant further investigations exploring constitutional methylation as a potential cause of cancer risk.
Methylation in established cancer risk genes
Among some of the best-characterized cancer risk genes in terms of mutations (BRCA1, TP53, and RB1), we found the mean methylation level to be 0.7% in the known regulatory region of the BRCA1 promoter, in line with our previous findings [23]. For TP53, the mean methylation level was 7.9%, while the corresponding number for RB1 was 24.9%. For some additional genes where methylation has been found as a cancer risk factor, MLH1 and MGMT, these revealed mean methylation levels of 6.4% and 18.6%, respectively. Among these established cancer risk genes (BRCA1, TP53, RB1, MLH1, and MGMT), we found no significant differences between the individuals in the present data set.
Co-methylated tumor suppressors
The cause of differential DNA methylation, and, in particular, tumor suppressor promoter methylation, remains poorly understood. Thus, in an exploratory analysis, we assessed potential covariation between promoter methylation on an individual basis. For this purpose, we performed hierarchical clustering of the samples by applying the Z-scores from average methylation ratio across the 565 capture regions. Doing so, all samples could be classified into two distinct major clusters, each harboring distinguishable sub-clusters (Fig. 2). Interestingly, the two major clusters (1 and 2) were characterized by different promoter methylation in two groups of genes (A and B), where cluster 1 had high methylation in genes in group A and low methylation in genes in group B, while the opposite methylation pattern was seen for samples in cluster 2 (Fig. 2).
We identified genes falling into these two groups (A and B), and analyzed their involvement in functional pathways by KEGG pathway analysis and GO enrichment analysis via Gather. Many of the genes involved in group A were important in development and regulation of cellular processes like Wnt signaling and TGF-beta signaling pathways. In contrast, genes from group B showed involvement in apoptotic pathways and leukocyte differentiation (Supplementary Table S4).
Notably, some individuals were characterized by having a majority of genes either hyper- or hypo-methylated as compared to the rest of individuals. Applying a 95% confidence interval across samples with respect to the overall methylation level of the regions analyzed, one sample (S24) fell below the lower limit of the CI, while three fell above the upper limit of the CI (Supplementary Figure 3). However, these individuals were distributed across the two main clusters with no preference for one group over the other. Assessing the available general clinical data for these individuals, no notable associations were observed between methylation and factors such as age or BMI (data not shown).
Validations in external data sets
Although our data are unique since they are generated by targeted massive parallel sequencing analyses, we sought to validate our biological findings by mining available data sets generated by application of methylation arrays.
A technical concern is that methylation could potentially vary between subfractions of leukocytes and differential methylation between individuals could then potentially be a result of individuals having different compositions of leukocyte subfractions in their blood. Assessing the 7 most differentially methylated regions in our data set, in the leukocyte subfractions published in the Bioconductor Experiment Data Package FlowSorted.Blood.450K revealed no major difference in any of the 7 regions (Supplementary Table S5, with figures). In GSTP1, 6 out of 19 CpGs revealed lower methylation in CD14+ T cells and/or CD56+ NK cells than other subfractions, but the impact of this on the average levels in total WBC was negligible. Very similar observations were made in another data set of cord blood (R package FlowSorted. CordBlood Norway.450 K in Bioconductor [34]; Supplementary Table S6, with figures). This confirmed potentially varying composition of leukocyte subfractions not to be a likely cause of the observed methylation differences.
Further, we sought to validate the biological differences observed for the 7 most differentially methylated regions in our sample set, by assessing their methylation in a sample of blood DNA from 845 individuals (GSE51032). In this sample set, data was available for CHN1, PDCD1LG2, GSTP1, and ZNF668. In addition, we here included the two top-ranking genes with high differential methylation calculated as ratio, but where percent point difference was below 20 (see above; RABEP1 and AIP; Table 2). In general, the methylation levels were called as slightly higher in the GSE51032 set than by our own sequencing. However, the differences between individuals were confirmed for all genes and the difference in percent points between the highest and lowest methylated individual was similar (Supplementary Table S7). The exception was ZNF668, where our maximum observation was 66% methylation, while in the GSE51032 set, some individuals were scored as 100% methylated. This difference probably relates to a substantially higher number of individuals analyzed in the validation set increasing the chance of observing outliers.
Discussion
While to this end constitutional epimutations of tumor suppressors have been linked to cancer risk for a few genes only [23, 27, 31, 35–37], one may postulate that constitutional epimutations affect other tumor suppressors as well. This may have implications to our understanding of cancer risk. A substantial number of cancer-prone families in which no underlying germline mutation have been identified, and it is tempting to postulate that some of these individuals may be at increased cancer risk due to constitutional epimutations in tumor suppressor genes [30]. In addition, germline mutations in several tumor suppressor genes have been associated with other conditions such as skin and limb development deficiencies, Cowden syndrome, and Fanconi anemia [38–40]. Thus, exploring constitutional promoter methylation across tumor suppressor genes may be of importance to other medical conditions as well.
To this end, the vast majority of epigenetic data reported in respect to different health conditions are based on global methylation-array analyses or single gene promoter analyses by methods like MSP or MLPA. While the array-based approaches do provide data for single CpGs, a large number of (potentially important) CpGs are lacking from the arrays, limiting the possibilities to identify methylation pattern across all regions of interest (e.g., as seen for BRCA1 [23]). As for MSP and MLPA, such methods are fast and cheap but they are sensitive only to a general methylation presence in the CpGs covered by the primers and probes, precluding assessment at a single CpG resolution level.
Here, we established a massive parallel sequencing-based approach, enabling base-pair resolution analyses of methylation status in gene promoters. The method provides several advantages as compared to previous methods. First, as compared to conventional methods like MSP and MLPA, our method allows for detailed single-CpG resolution analyses of multiple promoter regions in concert. Second, our method limits both workload and costs compared to application whole-genome methylation sequencing for promoter methylation analysis. Third, the benefit of determining exact methylation levels, instead of binary assessments, has been confirmed in clinical studies [23], underlining the importance of high sensitivity required to detect low-grade mosaic methylation [30]. Fourth, as compared to available array-based approaches, our NGS-assay allows for methylation assessment of all CpGs in the region of interest, not only those covered by array probes. As mentioned above, this proved to be crucial in analyses of the cancer risk associated with mosaic BRCA1 methylation [23].
In principle, the sequencing of the DNA-libraries we prepared could be run on any Illumina instrument. As such, the method is flexible and scalable. Here, we used the MiSeq instrument due to the rapid run time. In our set-up, we chose to run 8 samples in one run, yielding an average coverage of 189.6x, corresponding to a mean sensitivity limit of 0.53%. Although indicating a very sensitive method, this is an average value, and some regions reveal lower coverage. If needed, however, coverage could be increased in order to improve the sensitivity of the method [23]. Notably, the reproducibility of the assay may vary between the different covered regions. However, we show that the reproducibility is very good even in regions with low levels of methylation. Importantly, the observed technical variation was consistently negligible compared to the biological variations described. Further, we found that technical variations were lower when assessing all CpGs across a given region than when assessing randomized selections of CpGs as “representative” for a region. This emphasizes the value of applying assays where all CpGs in a given region are covered, instead of relying on scattered, selected CpGs.
While constitutional methylation is considered an early life event affecting different germinal layers, methylation status is also prone to environmental influences and other factors and has been found to change during lifetime [41], causing differential methylation of many genes across different tissues [42]. One potentially important caveat when analyzing WBCs as surrogate markers for constitutional methylation is the fact that different leukocyte fractions may harbor different methylation patterns [43]. While such differences, so far, have been linked to global methylation patterns, it remains unclear whether this may represent a problem with respect to specific tumor suppressor methylation. Notably, differential methylation across WBC subfractions was found not be an issue regarding BRCA1 promoter methylation [23], and in the present study, it was not found to be an issue in the most differentially methylated promoter regions either.
The methylation level of the genes found to confer cancer risk, so far, is highly variable. Regarding MLH1, normal cell methylation affecting ~ 50% of the alleles has been reported in a limited number of probands with familial colorectal cancer (for original references, see [30]). Recently, two families with a high breast and ovarian cancer incidence were found to harbor secondary constitutional BRCA1 methylation, also with a methylation level of ~ 50% [31]. In contrast, about 4% of females in a Caucasian population was found to carry low-level mosaic constitutional BRCA1 methylation (4-10% of alleles). Among these low-level methylated individuals, the incidence of high-grade serous ovarian cancer was significantly elevated with an odds ratio between 2 and 3 across two large cohorts [23]. As for the method presented here, this has the sensitivity required for exploring both scenarios.
While the limited number of samples analyzed precludes formal assessments of methylation frequency and/or potential correlations to health outcome, importantly, our findings confirm differential constitutional promoter methylation across a panel of tumor suppressor genes in healthy individuals. Interestingly, among those promoter regions found to be hyper-methylated in the normal tissue of some of the analyzed individuals, we found promoters in genes previously reported to be hyper-methylated in tumors (such as RASSF1 and GSTP1). The presence of epigenetic deregulation of a distinct tumor suppressor at the somatic (tumor) level provides no evidence for constitutional methylation of the same gene. However, the examples related to MLH1 and BRCA1 suggest that potential relationships may occur for other genes as well. Thus, it is tempting to speculate that, at least some of the genes detected here (e.g., RASSF1 and GSTP1) could be constitutionally methylated and, in such cases, methylated tumor cells may have originated from the constitutionally methylated normal cells [30]. Notably, although not directly comparable to our data, due to a restricted selection of CpGs covered, mining of a large external data set revealed similar interindividual differences largely confirming our findings.
Interestingly the methylation patterns revealed across our gene panel indicated that the individuals analyzed could be classified into two different methylation clusters. These findings should be interpreted with caution due to the limited number of individuals analyzed. However, the fact that the clusters were separated by differential methylation across important biological pathways involving Wnt- and TGF-beta signaling pathways as well as genes involved in apoptotic pathways and leukocyte differentiation indicate potential underlying biological differences to be explored in future studies.
Conclusions
We provide a relatively fast and affordable strategy for detailed assessments of differential methylation of tumor suppressors. This strategy is attractive in the warranted search for additional tumor suppressors that may be cancer risk factors when methylated in normal tissues.
Methods
Samples
The samples analyzed in the present study were from 34 individuals, selected from a set of 114 healthy postmenopausal women previously described [44]. Subsequent to providing informed consent, each individual donated anonymized blood samples in accordance with Norwegian regulations. All women were recruited during routine mammographic screening at Haukeland University Hospital, Bergen, Norway. Individuals with diabetes or other types of endocrine diseases as well as individuals using hormone replacement therapy were excluded. All samples were drawn > 2 years after the last menstrual period. Within the selection of 34 individuals analyzed in the present study, the mean age was 64 years (range 56-71 years) and the mean BMI was 24.8 (range 19.4-39.6) at the time of sample collection.
DNA isolation
Genomic DNA was extracted from EDTA-whole blood, using QIAamp DNA Mini kit (Qiagen). The procedure was performed according to the manufacturer’s instructions with the exception that 400 μl of whole blood was used as input.
Selection of tumor suppressor promoter regions
Regions of interest were defined as 356 regions from the promoters of 283 tumor suppressor genes. The selection of genes was based on the cancer gene panel previously described as “CGPv2/3” [45, 46], Roche’s “Comprehensive Cancer Design” as well as a manual literature review, in order to cover all well-established tumor suppressor genes, independent of cancer type. As such, the selection was independent of previous knowledge about methylation status. For each transcription start site (TSS), we designed probes covering a region spanning from −1500 to +500 relative to TSS. Positions of TSS were determined by NCBI and Ensembl-curated transcripts, literature search, and use of the FANTOM5 RNA expression resource (fantom.gsc.riken.jp/5/). Probes for hybridization to the included regions were manufactured by Roche and designed to bind the target DNA of all possible methylation configurations (fully methylated, partially methylated, and completely unmethylated). Importantly, both strands were targeted, in order to enable correction for potential overlap between CpGs and SNPs. By probe design, the 356 target regions were split into 565 capture regions. Full lists of included tumor suppressor genes and target regions are given in Supplementary Table S1.
Library preparation and methylation sequencing
Processing of the sample libraries was performed using the solution-based bead capture method for enrichment of bisulfite-converted DNA, SeqCap Epi Enrichment System (Roche) according to the user guide (version 1.2).
For each sample, 1 μg DNA isolated from blood was mixed with bisulfite-conversion control (Lambda DNA, negative for methylation). DNA was fragmented to the range of 180-220 bp using Covaris M220 followed by end repair, A-tailing, ligation of index/adapters, and dual size selection. Using the Zymo Research EZ DNA Methylation-Lightning kit, the DNA was bisulfite-converted according to manufactures protocol, and the resulting sample was amplified prior to nanodrop quantification. Based on these measurements, 1 μg bisulfite-converted DNA was put into the hybridization with custom-made probes for 68 h prior to capture by streptavidin-coated beads, extensive washing, and a final library amplification step.
The protocol was combined with the use of a custom-made probe design enabling analysis of only regions of interest (consisting of 356 promoter regions from 283 tumor suppressor genes, described above and in Supplementary Table S1). In addition, the probe set included probes targeting (Lambda DNA for conversion control). The targeted regions were enriched by a bead capturing method that captures both strands of DNA. Purified libraries were pooled, spiked with 10% PhiX, and sequenced on an Illumina MiSeq sequencer, using v2 chemistry and 2 × 100 (200 cycles) paired-end reads. RTA v1.18.54 and MCS v2.5.0.5 software was used to generate data. Eight samples were multiplexed per run, and resulting data were de-multiplexed based on sample-specific indexes attached to the sequencing adaptors. De-multiplexing was run automatically by the MiSeq Reporter software before further processing.
Methylation calling
Raw sequencing data was analyzed using an in-house workflow designed in collaboration with Roche, comprised of publicly available tools, implemented using shell script (Fig. 3; for a detailed description see Supplementary information). In brief, the first analytic steps involved quality checking of fastq files by FASTQC. Paired-end reads were filtered based on quality and clipped using Trimmomatic [47]. Trimmed sequences were aligned to the human genome (GRCh38) from NCBI as well as Enterobacteria phage lambda (NC_001416.1) complete genome, added for bisulfite conversion efficiency control using the bisulfite mapping algorithm BSMAP [48]. The aligned read statistics and format conversions were carried out using SAMtools [49]. After bisulfite conversion, the DNA strands are no longer complementary. To achieve methylation information from both strands, aligned reads were split into the top and bottom strand [50]. Subsequently, the sequences were sorted, and duplicates were removed and merged back using Picard tools. In the next step, the analysis was further restricted to those read pairs where both mates in the pair could be mapped in the correct orientation and at given distance consistent with the library insert size (properly paired reads) using BamTools [51]. To avoid bias, overlapping reads were clipped using BamUtils. Various statistics for reads, alignment, and coverage were calculated using SamTools.
Fig. 3.
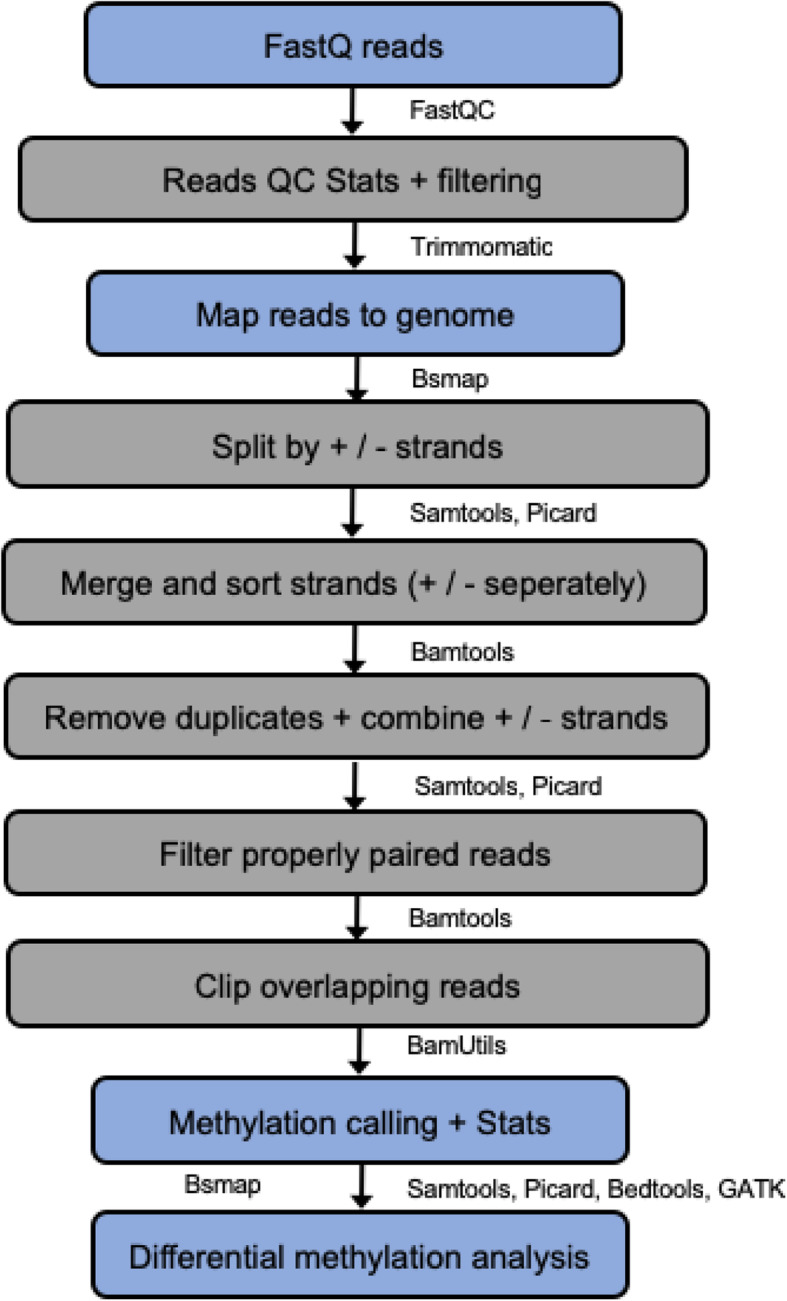
Workflow of the methylation analysis. Flow chart of the steps taken within the informatics analysis pipeline from raw FastQ files to processed data used for biological interpretations. Main steps are indicated by blue background; smaller steps are indicated by gray background (figure adapted from original design by Roche)
For each sample, methylation analysis was carried out using methratio.py package in BSMAP by calculating methylation percentage. An additional step involves SNP calling for the targeted regions with BisSNP [52] from aligned reads.
DNA conversion rate was calculated based on all original Cs in the Lambda DNA sequence. For all Cs in the untreated sequence the following formula was used on sequencing data post bisulfite treatment:
Assay reproducibility
To assess reproducibility of the assay, we performed 2 independent experiments with 6 parallels of a standard sample in each experiment. The standard sample consisted of pooled DNA of equal amounts from WBC of 5 healthy donors. Reproducibility was assessed across 12 regions, selected based on three separate criteria: First, we selected 4 regions found to have high biological variance in our original sample set of 34 healthy women (GAS7, ELAC2, AIP, ZRSR2). Further, we selected 6 regions in genes known to be high penetrance genes when either mutated or hypermethylated (BRCA1, TP53 (2 regions), RB1, MLH1, MGMT). Finally, we selected 2 regions at random (PRDM2, TMEFF2). Based on the 12 replicate analyses, we calculated mean methylation, standard deviation and coefficient of variation for all the regions (Supplementary Table S2a). Further, within the 2 randomly selected genes (PRDM2, TMEFF2), we performed a randomized selection of 5 CpGs per region, using the mean methylation in these 5 as “representative” for the region. Then, we calculated mean methylation, standard deviation and coefficient of variation across the 12 replicate analyses of these 5 CpGs. This randomization was repeated 5 times, yielding a general overview of the variability when applying limited numbers of CpGs as “representative” for a region (Supplementary Table S2b).
Differential methylation assessment
Among all CpGs in the 565 capture regions as well as 250 flanking bps at each end, the analysis was restricted only to include CpGs with minimum of 10 reads in coverage in all of the 34 samples. For each sample, we then calculated the mean methylation per region, based on individual CpG methylation ratios within the region. Based on these data, we generated a methylation matrix for all the common regions across all the samples (n = 34 in the present study), and calculated Z-scores for that matrix. Then we assessed the Z-scores and identified all the regions where a minority of individuals were differentially methylated as compared to the majority. Differential methylation was here defined as Z-scores that were outside of the 99% confidence interval. We used an arbitrary definition of minority, set to one-third, or less, of the total number of samples, i.e., minimum 1 individual and maximum 12 individuals (this definition may need adjustment according to the size of subsequent studies). Regions that had confidence level more than 99% were then categorized into negatively and positively methylated regions based on the Z-score value and whether the minority of individuals had higher or lower methylation levels than the majority.
To find the differentially methylated regions, we calculated the mean methylation for these regions across CpGs within individual samples and measured the difference in methylation between individuals with the lowest and highest methylation mean. Although relatively small differences in methylation levels have been shown to modulate cancer risk [23], we here sought to identify the regions with larger differences, applying arbitrary thresholds of 5, 10, and 20 percent point difference in methylation. Further, we performed additional analyses assessing ratios (fold difference) between individuals, taking into account that biological important differences may have high ratios, not necessarily reaching a certain threshold set by percent point difference (e.g., a difference between 1% and 10% may be important, even if the percent point difference is only 9).
Hierarchical clustering
We created a matrix of methylation ratios for all genes across patients. We then calculated a variance for each gene across patients to identify differential methylation. Heatmap was produced with heatplot function from made 4 package [53], with mean linkage cluster analysis and a correlation metric distance. For the purpose of clustering, missing values for regions in individual patients were filled in using the impute R package [54, 55]. (Impute-knn function from impute R package, finds k-nearest neighbors using a Euclidean metric and uses their mean to substitute the missing value). Missing values affected one region of GSTM1 in 16 samples, another region of GSTM1 in 7 samples, and a region of AIP in 3 samples.
Pathway analysis
We identified groups of genes from cluster analysis and explored their functional roles by pathway analyses with GATHER. GATHER is an online platform that predicts functional molecular patterns and biological context by incorporation of several biological databases [56]. In GATHER, we analyzed KEGG pathways and gene ontology enrichment analyses [57].
External data sets
We performed data mining and extracted detailed methylation status for all available CpGs for a given region (defined by our NGS-panel) from the Bioconductor Experiment Data Package FlowSorted.Blood.450K (https://bioconductor.org/packages/release/data/experiment). This data set was generated by methylation array analyses across 6 independent samples from adult individuals and contains information on 10 different categories of leukocytes. The categories include the major groups of granulocytes and lymphocytes.
We obtained similar data for umbilical cord blood from newborns [34]. These data were available as the R package FlowSorted.CordBloodNorway.450K in Bioconductor. This data set was also based on methylation array and holds information about 7 categories of leukocytes, including the major groups of granulocytes and lymphocytes, across 11 independent cord blood samples from newborns.
For validation of methylation differences in blood DNA from healthy individuals, we mined data from GSE51032, available through Gene Expression Omnibus (GEO). This data set was generated by methylation array and consists of 845 samples from the EPIC-Italy cohort (out of which 188 were males and 657 were females).
Supplementary information
Additional file 1: Supplementary Figure 1. Fraction of methylated alleles in promoter region of selected tumour suppressor genes. (A) Regions with high methylation levels across samples from all 34 healthy individuals. (B) Regions with low methylation levels across the same samples. Note the different scale on the Y-axis for panel A and B. Data for AIP were lacking for samples 32, 33, 34 due to low coverage (see details in Materials and methods). Supplementary Figure 2. Plot examplifying consistent high and low methylated CpGs in the same promoter, across patients. Fraction of methylated alleles across CpGs in the promoter region of RB1 in the two samples S7 and S24 are displayed. These two samples were selected because they were the one with highest and lowest overall methylation across the 283 investigated tumour suppressor genes, respectively (ref. Supplementary figure 3), and as such should represent the extremes. Still within the RB1 promoter, they reveal a very similar pattern of some CpGs being highly methylated, while others are hardly methylated at all. Supplementary Figure 3. Distribution of overall average methylation across 283 tumour suppressor gene promoters in 34 healthy individuals. (A) Bars indicate the average fraction of methylated alleles for all CpGs covered per patient. Dotted red lines indicate the upper and lower border of the 95% confidence interval for the average values per patient (CI for individual observations). Sample S24 falls below the lower border of the CI, indicating general hypo-methylation. Samples S4, S8 and S7 fall above the upper border of the CI, indicating general hyper-methylation. (B) Q-Q plot based on the same data as displayed in (A). S24 is encircled in green, while S4, S8 and S7 are encircled in red.
Additional file 2: Supplementary information – workflow
Additional file 3: Supplementary Table S1. Pan-cancer panel of 283 tumor suppressor genes for which promoters are included in methylation analyses. The panel was generated based on CGPv2/3-panels [1], Roche’s Comprehensive Cancer Design along with manual literature search.
Additional file 4: Supplementary Table S2a. Reproducibility test. Supplementary Table S2b. Reproducibility test restricted to randomised CpGs.
Additional file 5: Supplementary Table S3. Genes with >99 confidence level difference in methylation ratio between a minority (one third or less) of samples versus the majority.
Additional file 6: Supplementary Table S4. groupAB_GE
Additional file 7: Supplementary Table S5. WBC fractions
Additional file 8: Supplementary Table S6. Coord blood
Additional file 9: Supplementary Table S7. EPIC
Acknowledgements
We thank Beryl Leirvaag and Christine Eriksen for technical assistance.
Abbreviations
- BMI
Body mass index
- CpG
Cytosine-phosphate-guanine
- HGSOC
High grade serous ovarian cancer
- MSP
Methylation-specific polymerase chain reaction
- MLPA
Multiplex ligation-dependent probe amplification
- NGS
Next-generation sequencing
- SNP
Single nucleotide polymorphism
- WBC
White blood cells
Authors’ contributions
Study design: DBP, EO, PEL, SK. Generation of data: DBP, EO, ZS, EV, GTI, LM. Interpretation of data: DBP, EO, ZS, PEL, SK. Funding/grants: PEL, SK. Writing of manuscript: DBP, PEL, SK. Approval of final manuscript: All authors
Funding
This work was performed in the Mohn Cancer Research Laboratory and was funded by grants from the Bergen Research Foundation, the Norwegian Cancer Society, the Norwegian Research Council, and the Norwegian Health Region West.
Availability of data and materials
All data generated or analyzed during this study are included in this published article (and its supplementary information files). Unprocessed raw files are available from the corresponding author on reasonable request.
Ethics approval and consent to participate
Subsequent to providing informed consent, each individual included in the present work donated anonymized blood samples for research purposes. This was done in accordance with Norwegian legislation at the time of sample collection (late 1990s).
Consent for publication
Not applicable
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s13148-020-00920-7.
References
- 1.Clark DF, Maxwell KN, Powers J, Lieberman DB, Ebrahimzadeh J, Long JM, et al. Identification and confirmation of potentially actionable germline mutations in tumor-only genomic sequencing. JCO Precis Oncol. 2019;3 Epub 2019/09/13. doi: 10.1200/PO.19.00076. PubMed PMID: 31511844; PubMed Central PMCID: PMCPMC6738953. [DOI] [PMC free article] [PubMed]
- 2.Hata C, Nakaoka H, Xiang Y, Wang D, Yang A, Liu D, et al. Germline mutations of multiple breast cancer-related genes are differentially associated with triple-negative breast cancers and prognostic factors. J Hum Genet. 2020. 10.1038/s10038-020-0729-7 Epub 2020/02/08PubMed PMID: 32029870. [DOI] [PubMed]
- 3.Jansen AML, Ghosh P, Dakal TC, Slavin TP, Boland CR, Goel A. Novel candidates in early-onset familial colorectal cancer. Familial Cancer. 2020;19(1):1–10. doi: 10.1007/s10689-019-00145-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li FP, Fraumeni JF., Jr Soft-tissue sarcomas, breast cancer, and other neoplasms. A familial syndrome? Ann Intern Med. 1969;71(4):747–752. doi: 10.7326/0003-4819-71-4-747. [DOI] [PubMed] [Google Scholar]
- 5.Nichols KE, Malkin D, Garber JE, Fraumeni JF, Jr, Li FP. Germ-line p53 mutations predispose to a wide spectrum of early-onset cancers. Cancer Epidemiol Biomark Prev. 2001;10(2):83–87. [PubMed] [Google Scholar]
- 6.Donaldson A, Murray A, Antoniou AC, Brewer C, Houghton C, Evans DG, et al. Cancer risks for BRCA1 and BRCA2 mutation carriers: results from prospective analysis of EMBRACE. J Natl Cancer Inst. 2013;105(11):812–822. doi: 10.1093/jnci/djt095. [DOI] [PubMed] [Google Scholar]
- 7.Bonadona V, Bonaiti B, Olschwang S, Grandjouan S, Huiart L, Longy M, et al. Cancer risks associated with germline mutations in MLH1, MSH2, and MSH6 genes in Lynch syndrome. JAMA. 2011;305(22):2304–2310. doi: 10.1001/jama.2011.743. [DOI] [PubMed] [Google Scholar]
- 8.Hussussian CJ, Struewing JP, Goldstein AM, Higgins PA, Ally DS, Sheahan MD, et al. Germline p16 mutations in familial melanoma. Nat Genet. 1994;8(1):15–21. doi: 10.1038/ng0994-15. [DOI] [PubMed] [Google Scholar]
- 9.Borg A, Sandberg T, Nilsson K, Johannsson O, Klinker M, Masback A, et al. High frequency of multiple melanomas and breast and pancreas carcinomas in CDKN2A mutation-positive melanoma families. J Natl Cancer Inst. 2000;92(15):1260–1266. doi: 10.1093/jnci/92.15.1260. [DOI] [PubMed] [Google Scholar]
- 10.Knudson AG., Jr Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A. 1971;68(4):820–823. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li FP, Fraumeni JF., Jr Rhabdomyosarcoma in children: epidemiologic study and identification of a familial cancer syndrome. J Natl Cancer Inst. 1969;43(6):1365–1373. [PubMed] [Google Scholar]
- 12.Tung N, Domchek SM, Stadler Z, Nathanson KL, Couch F, Garber JE, et al. Counselling framework for moderate-penetrance cancer-susceptibility mutations. Nat Rev Clin Oncol. 2016;13(9):581–588. doi: 10.1038/nrclinonc.2016.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Plichta JK, Griffin M, Thakuria J, Hughes KS. What’s new in genetic testing for cancer susceptibility? Oncology (Williston Park, NY) 2016;30(9):787–799. [PubMed] [Google Scholar]
- 14.Berdasco M, Esteller M. Clinical epigenetics: seizing opportunities for translation. Nat Rev Genet. 2019;20(2):109–127. doi: 10.1038/s41576-018-0074-2. [DOI] [PubMed] [Google Scholar]
- 15.Llinas-Arias P, Esteller M. Epigenetic inactivation of tumour suppressor coding and non-coding genes in human cancer: an update. Open Biol. 2017;7(9) Epub 2017/09/22. doi: 10.1098/rsob.170152. PubMed PMID: 28931650; PubMed Central PMCID: PMCPMC5627056. [DOI] [PMC free article] [PubMed]
- 16.Sloane MA, Ward RL, Hesson LB. Defining the criteria for identifying constitutional epimutations. Clin Epigenetics. 2016;8:39. doi: 10.1186/s13148-016-0207-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Esteller M. CpG island hypermethylation and tumor suppressor genes: a booming present, a brighter future. Oncogene. 2002;21(35):5427–5440. doi: 10.1038/sj.onc.1205600. [DOI] [PubMed] [Google Scholar]
- 18.Damaso E, Canet-Hermida J, Vargas-Parra G, Velasco A, Marin F, Darder E, et al. Highly sensitive MLH1 methylation analysis in blood identifies a cancer patient with low-level mosaic MLH1 epimutation. Clin Epigenetics. 2019;11(1):171. doi: 10.1186/s13148-019-0762-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gazzoli I, Loda M, Garber J, Syngal S, Kolodner RD. A hereditary nonpolyposis colorectal carcinoma case associated with hypermethylation of the MLH1 gene in normal tissue and loss of heterozygosity of the unmethylated allele in the resulting microsatellite instability-high tumor. Cancer Res. 2002;62(14):3925–3928. [PubMed] [Google Scholar]
- 20.Welin S, Sorbye H, Sebjornsen S, Knappskog S, Busch C, Oberg K. Clinical effect of temozolomide-based chemotherapy in poorly differentiated endocrine carcinoma after progression on first-line chemotherapy. Cancer. 2011;117(20):4617–4622. doi: 10.1002/cncr.26124. [DOI] [PubMed] [Google Scholar]
- 21.Hitchins MP. Constitutional epimutation as a mechanism for cancer causality and heritability? Nat Rev Cancer. 2015;15(10):625–634. doi: 10.1038/nrc4001. [DOI] [PubMed] [Google Scholar]
- 22.Verma M, Rogers S, Divi RL, Schully SD, Nelson S, Su LJ, et al. Epigenetic research in cancer epidemiology: trends, opportunities, and challenges. Cancer Epidemiol Biomark Prev. 2014;23(2):223–233. doi: 10.1158/1055-9965.epi-13-0573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lonning PE, Berge EO, Bjornslett M, Minsaas L, Chrisanthar R, Hoberg-Vetti H, et al. White blood cell BRCA1 promoter methylation status and ovarian cancer risk. Ann Intern Med. 2018;168(5):326–334. doi: 10.7326/M17-0101. [DOI] [PubMed] [Google Scholar]
- 24.Chan TL, Yuen ST, Kong CK, Chan YW, Chan ASY, Ng WF, et al. Heritable germline epimutation of MSH2 in a family with hereditary nonpolyposis colorectal cancer. Nat Genet. 2006;38(10):1178–1183. doi: 10.1038/ng1866. [DOI] [PubMed] [Google Scholar]
- 25.Hesson LB, Hitchins MP, Ward RL. Epimutations and cancer predisposition: importance and mechanisms. Curr Opin Genet Dev. 2010;20(3):290–298. doi: 10.1016/j.gde.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 26.Hitchins MP. The role of epigenetics in Lynch syndrome. Familial Cancer. 2013;12(2):189–205. doi: 10.1007/s10689-013-9613-3. [DOI] [PubMed] [Google Scholar]
- 27.Ligtenberg MJ, Kuiper RP, Chan TL, Goossens M, Hebeda KM, Voorendt M, et al. Heritable somatic methylation and inactivation of MSH2 in families with Lynch syndrome due to deletion of the 3' exons of TACSTD1. Nat Genet. 2009;41(1):112–117. doi: 10.1038/ng.283. [DOI] [PubMed] [Google Scholar]
- 28.Miyakura Y, Sugano K, Akasu T, Yoshida T, Maekawa M, Saitoh S, et al. Extensive but hemiallelic methylation of the hMLH1 promoter region in early-onset sporadic colon cancers with microsatellite instability. Clin Gastroenterol Hepatol. 2004;2(2):147–156. doi: 10.1016/S1542-3565(03)00314-8. [DOI] [PubMed] [Google Scholar]
- 29.Shen L, Kondo Y, Rosner GL, Xiao L, Hernandez NS, Vilaythong J, et al. MGMT promoter methylation and field defect in sporadic colorectal cancer. J Natl Cancer Inst. 2005;97(18):1330–1338. doi: 10.1093/jnci/dji275. [DOI] [PubMed] [Google Scholar]
- 30.Lonning PE, Eikesdal HP, Loes IM, Knappskog S. Constitutional mosaic epimutations - a hidden cause of cancer? Cell Stress. 2019;3(4):118–135. doi: 10.15698/cst2019.04.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Evans DGR, van Veen EM, Byers HJ, Wallace AJ, Ellingford JM, Beaman G, et al. A dominantly inherited 5′ UTR variant causing methylation-associated silencing of BRCA1 as a cause of breast and ovarian cancer. Am J Hum Genet. 2018;103(2):213–220. doi: 10.1016/j.ajhg.2018.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morak M, Schackert HK, Rahner N, Betz B, Ebert M, Walldorf C, et al. Further evidence for heritability of an epimutation in one of 12 cases with MLH1 promoter methylation in blood cells clinically displaying HNPCC. Eur J Hum Genet. 2008;16(7):804–811. doi: 10.1038/ejhg.2008.25. [DOI] [PubMed] [Google Scholar]
- 33.Forbes SA, Beare D, Boutselakis H, Bamford S, Bindal N, Tate J, et al. COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res. 2017;45(D1):D777–DD83. doi: 10.1093/nar/gkw1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gervin K, Page CM, Aass HC, Jansen MA, Fjeldstad HE, Andreassen BK, et al. Cell type specific DNA methylation in cord blood: a 450 K-reference data set and cell count-based validation of estimated cell type composition. Epigenetics. 2016;11(9):690–698. doi: 10.1080/15592294.2016.1214782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hansmann T, Pliushch G, Leubner M, Kroll P, Endt D, Gehrig A, et al. Constitutive promoter methylation of BRCA1 and RAD51C in patients with familial ovarian cancer and early-onset sporadic breast cancer. Hum Mol Genet. 2012;21(21):4669–4679. doi: 10.1093/hmg/dds308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hitchins MP, Wong JJ, Suthers G, Suter CM, Martin DI, Hawkins NJ, et al. Inheritance of a cancer-associated MLH1 germ-line epimutation. N Engl J Med. 2007;356(7):697–705. doi: 10.1056/NEJMoa064522. [DOI] [PubMed] [Google Scholar]
- 37.Prajzendanc K, Domagala P, Hybiak J, Rys J, Huzarski T, Szwiec M, et al. BRCA1 promoter methylation in peripheral blood is associated with the risk of triple-negative breast cancer. Int J Cancer. 2020;146(5):1293–1298. doi: 10.1002/ijc.32655. [DOI] [PubMed] [Google Scholar]
- 38.Blumenthal GM, Dennis PA. PTEN hamartoma tumor syndromes. Eur J Hum Genet. 2008;16(11):1289–1300. doi: 10.1038/ejhg.2008.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Celli J, Duijf P, Hamel BC, Bamshad M, Kramer B, Smits AP, et al. Heterozygous germline mutations in the p53 homolog p63 are the cause of EEC syndrome. Cell. 1999;99(2):143–153. doi: 10.1016/s0092-8674(00)81646-3. [DOI] [PubMed] [Google Scholar]
- 40.Fiesco-Roa MO, Giri N, McReynolds LJ, Best AF, Alter BP. Genotype-phenotype associations in Fanconi anemia: a literature review. Blood Rev. 2019;37:100589. doi: 10.1016/j.blre.2019.100589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci U S A. 2005;102(30):10604–10609. doi: 10.1073/pnas.0500398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cavalli G, Heard E. Advances in epigenetics link genetics to the environment and disease. Nature. 2019;571(7766):489–499. doi: 10.1038/s41586-019-1411-0. [DOI] [PubMed] [Google Scholar]
- 43.Houseman EA, Molitor J, Marsit CJ. Reference-free cell mixture adjustments in analysis of DNA methylation data. Bioinformatics. 2014;30(10):1431–1439. doi: 10.1093/bioinformatics/btu029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Helle SI, Ekse D, Holly JMP, Lønning PE. The IGF-system in healthy pre- and postmenopausal women: relations to demographic variables and sex-steroids. J Steroid Biochem Mol Biol. 2002;81(1):95-102. doi: http://dx.doi.org/10.1016/S0960-0760(02)00052-3. [DOI] [PubMed]
- 45.Yates LR, Gerstung M, Knappskog S, Desmedt C, Gundem G, Van Loo P, et al. Subclonal diversification of primary breast cancer revealed by multiregion sequencing. Nat Med. 2015;21(7):751–759. doi: 10.1038/nm.3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yates LR, Knappskog S, Wedge D, Farmery JHR, Gonzalez S, Martincorena I, et al. Genomic evolution of breast cancer metastasis and relapse. Cancer Cell. 2017;32(2):169-184.e7. doi: 10.1016/j.ccell.2017.07.005. PubMed PMID: 28810143; PubMed Central PMCID: PMCPMC5559645. [DOI] [PMC free article] [PubMed]
- 47.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xi Y, Li W. BSMAP: whole genome bisulfite sequence MAPping program. BMC Bioinformatics. 2009;10:232. doi: 10.1186/1471-2105-10-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27(21):2987–2993. doi: 10.1093/bioinformatics/btr509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Law JA, Jacobsen SE. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat Rev Genet. 2010;11(3):204–220. doi: 10.1038/nrg2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Barnett DW, Garrison EK, Quinlan AR, Stromberg MP, Marth GT. BamTools: a C++ API and toolkit for analyzing and managing BAM files. Bioinformatics. 2011;27(12):1691–1692. doi: 10.1093/bioinformatics/btr174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu Y, Siegmund KD, Laird PW, Berman BP. Bis-SNP: Combined DNA methylation and SNP calling for Bisulfite-seq data. Genome Biol. 2012;13(7):1–14. doi: 10.1186/gb-2012-13-7-r61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Culhane AC, Thioulouse J, Perriere G, Higgins DG. MADE4: an R package for multivariate analysis of gene expression data. Bioinformatics. 2005;21(11):2789-2790. doi: 10.1093/bioinformatics/bti394. Epub 2005/03/31. PubMed PMID: 15797915. [DOI] [PubMed]
- 54.Hastie T, Tibshirani R, Sherlock G, Eisen M, Brown P, Botstein D. Imputing missing data for gene expression arrays. Stanford University Statistics Department Technical report. 1999. [Google Scholar]
- 55.Troyanskaya O, Cantor M, Sherlock G, Brown P, Hastie T, Tibshirani R, et al. Missing value estimation methods for DNA microarrays. Bioinformatics. 2001;17(6):520–525. doi: 10.1093/bioinformatics/17.6.520. [DOI] [PubMed] [Google Scholar]
- 56.Chang JT, Nevins JR. GATHER: a systems approach to interpreting genomic signatures. Bioinformatics. 2006;22(23):2926–2933. doi: 10.1093/bioinformatics/btl483. [DOI] [PubMed] [Google Scholar]
- 57.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Supplementary Figure 1. Fraction of methylated alleles in promoter region of selected tumour suppressor genes. (A) Regions with high methylation levels across samples from all 34 healthy individuals. (B) Regions with low methylation levels across the same samples. Note the different scale on the Y-axis for panel A and B. Data for AIP were lacking for samples 32, 33, 34 due to low coverage (see details in Materials and methods). Supplementary Figure 2. Plot examplifying consistent high and low methylated CpGs in the same promoter, across patients. Fraction of methylated alleles across CpGs in the promoter region of RB1 in the two samples S7 and S24 are displayed. These two samples were selected because they were the one with highest and lowest overall methylation across the 283 investigated tumour suppressor genes, respectively (ref. Supplementary figure 3), and as such should represent the extremes. Still within the RB1 promoter, they reveal a very similar pattern of some CpGs being highly methylated, while others are hardly methylated at all. Supplementary Figure 3. Distribution of overall average methylation across 283 tumour suppressor gene promoters in 34 healthy individuals. (A) Bars indicate the average fraction of methylated alleles for all CpGs covered per patient. Dotted red lines indicate the upper and lower border of the 95% confidence interval for the average values per patient (CI for individual observations). Sample S24 falls below the lower border of the CI, indicating general hypo-methylation. Samples S4, S8 and S7 fall above the upper border of the CI, indicating general hyper-methylation. (B) Q-Q plot based on the same data as displayed in (A). S24 is encircled in green, while S4, S8 and S7 are encircled in red.
Additional file 2: Supplementary information – workflow
Additional file 3: Supplementary Table S1. Pan-cancer panel of 283 tumor suppressor genes for which promoters are included in methylation analyses. The panel was generated based on CGPv2/3-panels [1], Roche’s Comprehensive Cancer Design along with manual literature search.
Additional file 4: Supplementary Table S2a. Reproducibility test. Supplementary Table S2b. Reproducibility test restricted to randomised CpGs.
Additional file 5: Supplementary Table S3. Genes with >99 confidence level difference in methylation ratio between a minority (one third or less) of samples versus the majority.
Additional file 6: Supplementary Table S4. groupAB_GE
Additional file 7: Supplementary Table S5. WBC fractions
Additional file 8: Supplementary Table S6. Coord blood
Additional file 9: Supplementary Table S7. EPIC
Data Availability Statement
All data generated or analyzed during this study are included in this published article (and its supplementary information files). Unprocessed raw files are available from the corresponding author on reasonable request.