

Abstract
Engineering protein and peptide-based materials for drug delivery applications has gained momentum due to their biochemical and biophysical properties over synthetic materials, including biocompatibility, ease of synthesis and purification, tunability, scalability, and lack of toxicity. These biomolecules have been used to develop a host of drug delivery platforms, such as peptide- and protein-drug conjugates, injectable particles, and drug depots to deliver small molecule drugs, therapeutic proteins, and nucleic acids. In this review, we discuss progress in engineering the architecture and biological functions of peptide-based biomaterials —naturally derived, chemically synthesized and recombinant— with a focus on the molecular features that modulate their structure-function relationships for drug delivery.
Keywords: Drug delivery, Polypeptides, Recombinant proteins, Bioinspired materials, Hierarchical self-assembly
1. Introduction
Research in the past several decades has made it evident that the overall therapeutic benefit of a drug is not proportional to its in vitro potency. Under physiological conditions, drugs encounter biological barriers, such as insolubility, aggregation, degradation, the impermeability of vascular endothelial cell layers, clearance by the kidney and the reticulo-endothelial system that contribute to a drug's short in vivo half-life, non-specific tissue distribution and poor tissue penetration, inefficient cellular internalization, undesired immunogenicity, and off-target toxicities [1,2]. In addition, a drug's potency can be severely compromised due to environmental changes such as pressure, temperature, humidity, and pH, which can occur during storage, administration, or systemic circulation. These factors create a narrow therapeutic window and can result in dismal in vivo performance, thus making the clinical translation of an otherwise potent drug an uphill task. To overcome these challenges, controlled drug delivery systems have been developed to improve the stability, efficacy, and tolerability of existing drugs while mitigating their off-target toxicity and promoting patient compliance. An ideal drug delivery system should be non-toxic, non-immunogenic, and biodegradable, and the architecture, chemical functionality, biological interactions, and mode of administration should all be tailored to optimize the drug's pharmacokinetic (PK) and pharmacodynamic (PD) properties [1,2].
Three major drug delivery strategies have been widely exploited to favorably alter the PK and PD properties of a drug: (i) using a prodrug in which a small moiety is covalently conjugated to the drug, masking its bioactivity until it is activated by a disease-specific stimulus at the desired site; (ii) encapsulating the drug in a delivery vehicle that dictates the PK and PD through its physicochemical properties; and (iii) using an implantable drug-eluting depot or device [3]. It is important to note that various hybrid approaches —combining more than one delivery strategy discussed above— are used to create a bespoke delivery system. Researchers have explored many synthetic and natural carrier molecules to create drug delivery systems [4]. Of these, proteins and peptides have garnered significant attention due to their structural diversity, biocompatibility, ability to form hierarchical self-assembly ranging from the nano- to meso- scale, exquisite tunability, non-immunogenicity, ease of synthesis, and scalability [[5], [6], [7], [8]]. Because of these diverse traits, peptide materials have been the focus of many innovative drug delivery systems in the past several decades. In this review, we will focus on drug delivery systems that have been engineered from protein materials, including silk, albumin, keratin, collagen, gelatin, elastin, and resilin ( Fig. 1 ). We will discuss the guiding principles for the design of peptide- and protein-based delivery systems and customization of their physicochemical properties for specific applications in drug delivery.
Fig. 1.

List of protein-based materials discussed in this review. Created with Biorender.com
2. Early advances in protein biomaterials
Controlled drug delivery was first conceptualized at the turn of the 20th century by Paul Ehrlich, a Nobel laureate who envisioned a “magic bullet” therapy that could deliver drugs to a specific target while avoiding off-target toxicity [9]. At that time, drugs were typically administered in pill formulations that instantly released the drug upon contact with water. There was no control over the release kinetics, which led to unsteady drug concentrations [10]. Small molecule drugs were rapidly cleared from circulation, thus necessitating multiple doses to maintain the drug's minimum effective concentration. Not only are such dosing regimens inconvenient to the patient, they also increase the occurrence of dose-dependent side effects. The lack of target specificity made it impossible to deliver a therapeutic to the organ of interest without impacting other cells, which caused off-target toxicity.
In the 1950s, scientists began to focus on developing drug delivery systems (Fig. 2 ). In the next thirty years, they established the principles of drug diffusion, dissolution, and pharmacokinetics [11,12]. Research then shifted toward prolonging drug release and increasing the drug's retention time in circulation [11]. Scientists also focused on how to specifically deliver drugs to a disease site by engineering systems for local drug release, passive drug accumulation in the diseased tissue, and active targeting [12]. This research led to the first FDA approval of a drug delivery system —liposomal amphotericin B— in 1990 for treatment of fungal infections [13]. Since then, controlled delivery has been leveraged to improve the bioavailability and efficacy of numerous therapeutics [14].
Fig. 2.

A brief timeline of advances made in engineering protein-based biomaterials for controlled drug delivery.
Biomaterials have been critical to the success of drug delivery systems. Many drug formulations employ biomaterials to extend the therapeutic window by both sustaining its release from the formulation and slowing its elimination from the body. Beginning in the late 1960s, scientists used synthetic materials, especially polymers, as drug carriers [11,12]. These systems successfully extended the drug's circulation time by increasing its molecular weight to slow renal excretion. Biodegradable systems were also engineered to prolong drug release, thus reducing the frequency of administration [15]. Despite these accomplishments, synthetic materials can have high immunogenicity or toxic degradation products [16]. Furthermore, there is limited control over the stereochemistry, structure, and molecular weight of synthetic polymers, which impacts the drug's biodistribution and pharmacokinetics [17,18]. The production of synthetic polymer drug carriers can be difficult and expensive to scale up [19].
These challenges associated with synthetic polymers led to an interest in the use of natural materials, such as polysaccharides, lipids, nucleic acids, and proteins, as drug carriers. Herein, we will focus on the progress made in the design of protein-based drug delivery systems [20]. Using protein-based materials in a biomedical setting is not a new concept, as such materials had been used to treat injury or illness for centuries [21]. The renewed interest in protein-based materials for drug delivery was driven, in part, by the development of better methods to extract proteins from their natural sources, and techniques to characterize them [6]. Protein-based materials are relatively biocompatible, and their degradation products —amino acids— are nontoxic [5].
The next major advance that transformed this field was the advent of recombinant DNA technology, which has made it possible to design polypeptides de novo as drug delivery carriers and to customize native proteins for drug delivery applications by manipulating their amino acid sequences. The first class of polypeptides used for biomedical applications are biopolymers based on consensus sequences from naturally derived proteins [10]. These polypeptides likewise benefit from sequence-level control over their structures and bioactivities, low monodispersity, and lack of toxicity. A second class are de novo designed polypeptides [22]. For both classes of polypeptide carriers, these methods provide near-absolute control over the carrier's sequence, self-assembly, stimuli-responsiveness, and dispersity that cannot be matched by synthetic polymers [23]. Further, active functional groups can be readily introduced into the sequence for chemical modification and drug conjugation [24]. Recombinant techniques are also used to optimize native proteins for a given application by creating fusions of protein and peptide drugs with carriers or targeting proteins. Because these fusions are genetically encoded, this is accomplished with greater precision than is possible synthetic carriers by site-specific introduction of new functional groups in recombinant proteins by unnatural amino acids or post-translational modification [5,25,26]. Furthermore, molecular simulations have enabled de novo design of proteins for a given application [27].
As the field shifts its focus toward “smart” drug delivery systems, the unique properties and exquisite tunability of protein-based materials continue to be attractive. They have been designed to respond to a variety of stimuli, including temperature, pH, oxidative conditions, or the presence of specific biomolecules [6,28]. Furthermore, they can be engineered to self-assemble into a variety of architectures ranging from nanomaterials to hydrogels to porous scaffolds [29]. These architectures provide numerous opportunities to create precisely engineered drug delivery systems ( Fig. 3 ).
Fig. 3.

Recent advances in the engineering of the peptide-based biomaterials as delivery vehicles. Created with Biorender.com.
3. Pathophysiological and translational challenges in drug delivery
Drug delivery systems are designed by optimizing pharmacokinetic–pharmacodynamic behavior, which involves modifying components of the delivery system to overcome the pathophysiological and translational challenges discussed below.
Solubility: Drugs must be soluble in blood to achieve prolonged circulation following systemic administration. Hydrophobic drugs with low aqueous solubility can be administered systemically by combining them with surfactants; however, these surfaces often pose health risks [30]. An alternative is to sequester hydrophobic drugs in peptide-based or polymeric delivery systems (which are surfactant-free) to improve drug safety and efficacy.
Degradation and clearance: Enzymes in blood can degrade or deactivate drugs, shortening their half-lives. Drug carriers shield drugs from enzymes and other degradative factors. The size, charge, and hydrophobicity of a drug dictate its clearance rate. If a drug is below the glomerular filtration cutoff of ~60 kDa or 6 nm diameter, it will be cleared rapidly from systemic circulation via renal filtration. Drug carriers can increase the half-life of small-molecule drugs in plasma by increasing their effective size to greater than the renal filtration cutoff [31]. Small, positively-charged drugs are cleared preferentially from circulation via negatively-charged capillary walls in the kidney's glomerulus [32]. A drug's charge and hydrophobicity also affect its clearance via opsonization, a process in which blood proteins adsorb to a drug and trigger degradation by the mononuclear phagocytic system [33]. Shielding the solvent-accessible interface of the drug with a “stealth” carrier can prevent protein adsorption and minimize opsonin-mediated uptake by macrophages.
Accumulation: Drug accumulation in the target tissue remains the major challenge for optimizing therapeutic efficacy. Drug carriers have been developed to improve accumulation in diseased tissue by passive targeting (e.g. in tumor tissue via the enhanced permeation and retention (EPR) effect [34] and by active targeting, wherein drug carriers are decorated with a ligand that binds a receptor that is overexpressed in the diseased tissue [35,36].
Tissue penetration: Once a drug extravasates to the target tissue, it often faces an environment rich in endothelial cells and extracellular matrix that prohibits further transport deep into the tissue [[37], [38], [39]]. This reduced drug transport reduces drug efficacy and can lead to drug resistance. Drug delivery systems can be loaded with penetrating moieties or modified to tune their physicochemical properties to enhance delivery into the target tissue [38].
Cell uptake and subcellular trafficking: After reaching the desired tissue, the drug must traverse cell membranes to reach its intracellular target. Cell membranes are permeable to small hydrophobic drugs, but for large and/or hydrophilic drugs, the cell membrane is an impermeable barrier [[39], [40], [41]]. Once a drug is internalized by a cell, additional barriers within the cell may separate the drug from its therapeutic target. Directing a drug to its subcellular site of molecular action is the penultimate challenge in drug delivery [42].
Release kinetics: Once a drug carrier permeates the cell membrane, its drug cargo can be released by passive mechanisms such as diffusion or by active mechanisms such as stimuli-responsive release [43]. Drug release can be triggered by the acidic and enzyme-rich environment of the endosome and lysosome, or by the reducing environment of the cytosol. Most drug carriers are internalized by endocytosis and thus must be designed to escape the endosome to prevent degradation of the drug before it can reach its intracellular therapeutic target.
The delivery system must be stable while also allowing spatiotemporal control of drug release. The simplest method to load a drug into a carrier is by physical entrapment, in which the physicochemical properties of the drug are matched to the properties of the carrier to drive encapsulation. Alternatively, drugs can be covalently conjugated to a functionalized carrier. This approach requires reactive groups that do not interfere with the therapeutic function of the drug or the self-assembly of the carrier [43].
Translational challenges: Once a drug formulation is optimized, its potency is evaluated in preclinical in vitro and in vivo experiments. Preclinical testing is required by regulatory agencies prior to clinical studies. The low correlation of preclinical results with clinical efficacy and the limited relevance of in vitro and non-human in vivo models used in preclinical evaluation are challenges in translating novel protein-based drug carriers to the clinic. The high cost of drug development—up to $10 million for Phase I, $20 million for Phase II, and $50–100 million for Phase III clinical trials [44]—creates a prerequisite that intellectual property cover a new drug delivery system to ensure exclusivity so as to recover the cost of drug delivery development.
4. Design of peptide-based drug delivery systems to overcome pathophysiological and translational challenges
The physicochemical properties of peptide-based delivery systems can be engineered with near absolute precision, which is impossible with synthetic polymers. The vast repertoire of natural, recombinant, and artificial proteins and the ability to customize their amino acid sequences allow construction of on-demand delivery systems. In this section we discuss intrinsic design modules (Fig. 4 ) that can be systematically engineered to create a drug delivery system with tunable physicochemical properties, to overcome the physiological and translational challenges discussed above. The relationship between the structure of these modules and their function as a therapeutic delivery system is described below for each major component of the modules.
Fig. 4.

Intrinsic design modules of a peptide amenable to precision engineering for drug delivery application. Created with Biorender.com.
Targeting ligand and linker: Like traditional drug formulations, peptide-based drug carriers accumulate in diseased tissue either by passive targeting, which includes diffusion and, in the case of tumor tissue, the EPR effect, or by active targeting via receptor-ligand interactions [[34], [35], [36]]. Tissue penetration can be enhanced by using cell-penetrating peptides [45]. The valency and surface density of targeting ligands impacts the affinity of the delivery system for its target receptors and impacts tissue accumulation [46]. Suboptimal display of targeting peptides on the vehicle surface may inhibit targeting. To address this issue, Wang et al. used a heuristic approach to determine the optimal presentation of a targeting peptide on a delivery vehicle [47]. By using 98 combinations of 15 tumor-homing peptides presented via 8 peptide linkers (differing in length and charge) on the surface of a delivery vehicle, they showed that two factors—nanoparticle charge and surface hydrophilicity—are critical in determining peptide presentation, and that an intervening peptide linker consisting of hydrophilic and charged residues (e.g. lysine and aspartic acid) prevents undesirable insertion of hydrophobic ligands into the micelle corona or micelle core. These charged linker residues can also be used to counteract extra charged groups within the micelle to create a neutral nanoparticle surface that minimally interferes with electrostatic interactions between ligand and receptor.
Peptide backbone and self-assembling block: The peptide backbone and self-assembling block are the heart of the peptide-based drug delivery system. The choice of peptide backbone and self-assembling block dictates the physicochemical properties of the system, which include its (soluble vs. gel), stability, size (nanoscale to mesoscale), morphology (spherical vs. elongated), charge, solubility, payload encapsulation and release, and response to external stimuli. Collagen-based peptides form a stable gel with a slow rate of degradation; gelatin is used when more rapid degradation is required [48]. A crosslinkable peptide can be used to increase the in vivo stability of a gel [49]. Albumin is widely used as a soluble drug carrier [50]. Gliadin (from gluten) facilitates penetration of drug carriers into the gastric mucosa and is used to deliver drugs to the stomach [51].
The choice of peptide building block can also impact the encapsulation and release of a drug. Due to their negative charge, keratin [52] and silks [53] are used to entrap positively-charged molecules and are hence rarely used to deliver negatively charged nucleic acid-based therapeutics. A hydrophilic peptide is desirable to deliver a hydrophobic drug. For example, a paclitaxel-loaded nanoparticle composed of hydrophilic zwitterionic polypeptide showed a wider therapeutic window —the range of drug dose that could treat solid tumors without toxic side-effects— than a neutral elastin-like polypeptide with the same molecular weight [54].
The stimuli-responsiveness of the delivery system is also governed by the choice of peptide building blocks. Elastin and collagen are widely used to synthesize temperature-responsive drug delivery vehicles, whereas silk is often used as a pH-responsive building block.
The hydrophobic / self-assembling block impacts the molecular architecture of the self-assembled peptide-based delivery system. For example, increased resilin content in the self-assembling block in an elastin-resilin fusion peptide shifts the self-assembled morphology from spherical micelles to elongated “worm-like” micelles, and can increase the avidity of a micelle with exposed peptide ligands that bind the αvβ3 integrin receptor by 1000-fold compared to a monomeric ligand [55]. Lipids and hydrophobic peptides like resilin are used not only to drive self-assembly but also to entrap hydrophobic drugs. Similarly, incorporation of a β-sheet-forming peptide into an elastin-lipid fusion drives self-assembly of morphologies ranging from worm-like micelles to bundled fibers [56].
Drug-binding domain: Lysine and cysteine are the two most widely used amino acids for covalent conjugation of a drug to a peptide carrier; a drug can be functionalized with N-hydroxy succinimide to target the amino group of a lysine, or with a maleimide to target the thiol group of a cysteine. Selective release of a drug from a drug-binding domain at the target tissue can be achieved by using pH-, redox-, or enzyme-labile bonds between the drug and carrier [57]. However, chemical conjugation of a therapeutic payload should not interfere with the activity of the drug and disrupt the self-assembly of the carrier in an obstructive manner. For this reason, recombinant strategies are emerging for site specific conjugation of small-molecule drugs to therapeutic or targeting proteins. One such approach exploits sortase mediated ligation that relies on the specificity of the transpeptidase Sortase A (SrtA) for short peptide sequences. SrtA retains its specificity while accepting a wide range of potential substrates [[58], [59], [60], [61]].
5. Applications in drug delivery
Protein-based biomaterials have revolutionized drug delivery by providing many unique structural and physico-chemical properties to delivery systems. Numerous techniques have been implemented to engineer protein materials with exceptional release profiles, pharmacokinetics, targeting capacity, and safety. In this section, we will discuss how proteins have been modified and leveraged to improve the delivery of a wide range of therapeutic agents.
5.1. Silk
Silk is a water insoluble, fibrous protein produced by silkworms like Bombyx mori and spiders such as Araneus diadematus and Nephila clavipes [62]. Silk is composed of two main proteins - sericin and silk fibroin (SF). Sericin is a hydrophilic, amorphous protein composed of 18 nonrepetitive amino acids and forms approximately 25% of the total weight of raw silk. It acts as an adhesive to join fibroin filaments. Sericin may cause immunogenic reactions and is thus separated from the SF for biomedical applications [63]. SF offers a repertoire of materials systems for biomedical applications, such as injectable particles, bioadhesives, hydrogels, implantable scaffolds, and recombinant or chemical conjugates.
5.1.1. Structure and properties of silk fibroin
SF is a high MW protein complex composed of a light chain (MW ~26 kDa) and a heavy chain (MW ~390 kDa) covalently held together with a single disulfide bond while non-covalently encapsulating a 25 kDa glycoprotein, P25. The fibroin heavy chain is an amphiphilic block copolymer consisting of alternating hydrophobic and hydrophilic blocks. The hydrophobic, crystallizable blocks, responsible for forming the β-sheet structure, are composed of a highly repetitive dipeptide motif of Gly-X, where X can be alanine, serine, tyrosine, or valine in decreasing frequency. The shorter, hydrophilic, amorphous blocks are composed of nonrepetitive sequences [64,65]. Its unique structure endows SF with highly adaptable properties: (i) its high thermal stability and mechanical malleability are suitable for further processing, such as chemical modification, material fabrication, and sterilization [4,64,[66], [67], [68], [69]]; (ii) in response to external stimuli, SF can self-assemble into various structures ranging from nanoparticles to hydrogels by modulating its β-sheet content [64,67]; (iii) the side chains of SF contain an abundance of active functional groups that enable chemical modification, and new functional groups can be incorporated to tune self-assembly, biodegradation, and payload release [69]; (iv) its anionic charge can be exploited to deliver a positively charged payload [53]; (v) recombinant DNA technology provides a modular platform to further engineer SF and create fusions with bioactive peptides [[70], [71], [72], [73]]; and (iv) SF is completely biodegradable and biocompatible and has high immunogenic tolerance [67,68].
5.1.2. Silk nanoparticles
Silk fibroin nanoparticles (SFNPs) have been extensively studied as injectable drug carriers to control the release of bioactive substances both in vivo and in vitro [74]. Their wide therapeutic application stems from the fact that their properties, including size, shape, zeta potential, and secondary structure, can be modified during self-assembly by external stimuli, such as pH [53], salt concentration [53], or the amount of co-solvent [75,76]. Lammel and coworkers prepared SFNPs in which the pH of the solution could control the secondary structures and zeta potential of the nanoparticles by salting out a silk fibroin aqueous solution with potassium phosphate [53]. At pH 6, the SFNPs predominantly resembled a silk II (crystalline) structure, whereas at pH 9, particles were composed of silk I (less crystalline). The authors also proposed a model to predict the effect of pH and kosmotropic salts on particle formation. Model small molecule drugs, such as alcian blue, rhodamine B, and crystal violet, were loaded into SFNPs by absorption, and their release was governed by SF crystallinity; more crystalline structures demonstrated a greater release rate. Shi et al. synthesized a SFNP for loading and release of hydrophobic small molecules and protein therapeutics [77]. Over 50 days, 23% FITC-BSA and 34% rhodamine B were released from the SFNPs and internalized by cells, as seen by microscopy and flow cytometry. Crivelli et al. synthesized a SFNP using a desolvation technique to encapsulate the anti-inflammatory drugs celecoxib (CXB) or curcumin for osteoarthritis (OA) treatment [78]. The release of the drug was controlled by varying the drug loading into the SFNP. In vitro release experiments indicated that the release reached equilibrium after 24 h, which was much faster than the release of the same drugs from silk based hydrogel systems.
Covalent functionalization can be used to tune self-assembly and interactions with therapeutics and the biological environment. The active amino acid residues on SF, such as serine, threonine, aspartic acid, glutamic acid, and tyrosine, make it amenable to chemical functionalization to modulate its properties for a given application. For example, SFNPs 40–120 nm in diameter were synthesized by an acetone extraction method and conjugated to insulin with glutaraldehyde as a crosslinker [79]. Insulin-conjugated SFNPs were resistant to trypsin digestion and had a half-life 2.5 times higher in human serum compared to bare insulin, which demonstrates the potential of SF nanoconjugates for peptide or enzyme delivery.
Recombinant silk-like peptides (SLPs) have also been used to create nanoparticles with reproducible sizes for drug and gene delivery. The negatively charged SF cannot complex with nucleic acids through electrostatic interaction, thus limiting its applications in gene therapy. To address this limitation, Numata et al. recombinantly synthesized silk-based block copolymers with poly(l-lysine) domains for gene delivery. The pDNA complexes of silk-polylysine prepared at a polymer:nucleotide ratio of 10:1 showed the highest transfection efficiency. The pDNA complexes were also immobilized on silk films and could directly transfect cells from these surfaces [80]. Recombinant SLP sequences derived from the native sequence of the dragline protein MaSp1 sequence from the spider Nephila clavipes were combined with a polylysine domain to produce hybrid systems that formed nanocomplexes of varying sizes based on the polymer to pDNA ratio or the molecular weight of the polylysine domain [80,81]. Transfection efficiency was also significantly enhanced by introducing cell-specific targeting groups like the arginine-glycine-aspartic acid (RGD) tripeptide [82]. Fusion of a cell-penetrating peptide with an Sp1-based SLP produced a delivery vehicle that was 45-fold more efficient at transfection than poly(ethyleneimine) at low pDNA concentrations.
Hybrid SFNPs have also been reported for injectable drug delivery. Yang et al. synthesized a multifunctional SF@MnO2 nanoparticle-based platform using SF as a reductant and template via a one-step biomineralization-inspired crystallization process (Fig. 5 ) [83]. The authors took advantage of the mesoporous structure and carboxyl residues of the SF@MnO2 nanoparticles to conjugate the photodynamic agent indocyanine green (ICG) and the chemotherapeutic drug doxorubicin (DOX) to form a SF@MnO2/ICG/DOX nanocomplex (SMID). TEM images suggested that the SMID nanocomplex possesses a well-defined spheroid structure and an average diameter of 60 nm. The presence of MnO2 made it highly reactive with endogenous hydrogen peroxide, which decomposed into O2 to enhance tumor-specific photodynamic therapy (PDT). In addition, the SMID nanocomplex demonstrated a stable photothermal effect upon near-infrared (NIR) irradiation for photothermal therapy (PTT) due to the photothermal response of SF@MnO2 and conjugated ICG. In vivo NIR fluorescence and magnetic resonance (MR) imaging indicated significant accumulation of the SMID nanocomplex in the tumor, which consequently improved tumor regression efficacy after a combination of PTT, PDT, and DOX chemotherapy [83]. Mao et al. conjugated the cyclic pentapeptide cRGDfk that targets the αvβ3 integrin and the photodynamic agent Chlorin e6 (Ce6) to SF polypeptides using a simple acid-amine coupling reaction and genipin peptide as a crosslinker to formulate a 5-fluorouracil (5-FU) -doped SFNP (Fig. 6 ) [84]. The authors investigated the active targeting properties and photodynamic effects of the nanoparticles in vitro. Results revealed that treatment with multifunctional SFNP and infrared radiation produced high levels of reactive oxygen species (ROS) and induced cell death in MGC-803 gastric cancer cells. The multifunctional SFNP, which combined targeted 5-FU chemotherapy with PDT, induced a remarkable antitumor effect in a xenograft mouse model for gastric cancer. However, the low colloidal stability of SFNPs under physiological conditions restricts widespread use of these system in vivo. Shao and colleagues addressed this issue by fabricating SFNP composite materials with a core–shell structure (CS-SFNPs). The authors electrostatically coated the negatively charged SFNPs with four different cationic polymers —glycol chitosan, N,N,N-trimethyl chitosan, polyethyleneimine, and PEGylated polyethyleneimine. Dynamic light scattering and nanoparticle tracking analysis revealed that CS-SFNPs had much greater colloidal stability than bare SFNPs in biological media.
Fig. 5.

Synthesis procedure of SF@MnO2/ICG/DOX (SMID) nanoparticles. SMID nanoparticle acts as a multifunctional drug delivery platform for in vivo MR/fluorescence imaging-assisted tri-modal therapy of cancer. Adapted with permission from [83].
Fig. 6.

Conjugation of the cyclic pentapeptide cRGDfk and the photodynamic agent Chlorin e6 (Ce6) to SF was achieved using a simple acid-amine coupling reaction and the resulting conjugate was doped with 5-fluorouracil (5-FU) using genipin peptide as a crosslinker. Adapted with permission from [84].
5.1.3. Silk hydrogels and depots
The adaptable mechanical properties and thermal stability of SF make it an ideal candidate material for forming hydrogels and scaffolds. Its degradation can be tuned by controlling the self-assembly of SF which can stably encapsulate the therapeutics and release them on demand. Kaplan and colleagues have extensively studied the abilities of SF and its hybrids to form hydrogels. They developed a method to synthesize a thixotropic silk nanofiber hydrogel from aqueous solution, which otherwise requires an organic co-solvent [85,86]. The injectable nanofiber hydrogel stably entraps DOX, solidifies in situ, and demonstrates pH-triggered sustained release of DOX [86]. Kaplan et al. also used an injectable SF-hydrogel system to sustain the delivery of anti-vascular endothelial growth factor (anti-VEGF) therapeutics [87], and small molecule drugs [88]. However, the prolong gelation time (weeks-months) of SF hydrogel acts as a major roadblock for their practical application. To decrease the gelation time Gong et al. synthesized another class of thixotropic hydrogels by blending regenerated SF and hydroxyl propyl cellulose (HPC), a natural cellulose ether approved by FDA [89]. HPC has lower critical solution temperature (LCST) of about 40°C above which it precipitates in water. The HPC-SF blend gelled at 37°C within 1 h. Results from confocal laser scanning microscopy (CLSM), Raman spectroscopy, and 13C NMR spectroscopy suggested that the conformational transition of SF from random coil to β-sheet during phase separation resulted in gel formation through β-sheet crosslinking and immobilization of the molecules of SF and HPC in the dispersed phase. The blended hydrogel encapsulated mice fibroblasts and protected them against high shear force during injection, which suggests that the hydrogel can be used for cell delivery [89]. Germershaus et al. developed heuristics to decipher protein interactions with SF using protamine and polylysine as model proteins [90]. The author concluded that the interaction between protein and SF arises primarily from entropy-driven complex coacervation, which depends on the ionic strength of the solution and presence of kosmotropic and chaotropic salts.
SF-hydrogels have also been fabricated with various nanoassemblies including SFNPs, carbon nanotubes and hybrid nanoparticles of different materials. Mao and colleagues encapsulated curcumin-loaded cationic nanoparticles of RRR-α-tocopheryl succinate-grafted-ε-polylysine conjugate into a SF-hydrogel, which promoted the penetration of curcumin into the thickening corneum of psoriatic mice and thus inhibited skin inflammation. Compared to 49% of curcumin released from the nanoparticle, only 30% was released from the mixed hydrogel-nanoparticle system. The author concluded that this slow release of curcumin might be due to adherence of the nanoparticles into the SF hydrogel, making it difficult for the embedded curcumin to diffuse out of the gel. This delayed release profile resulted into significant accumulation of curcumin in the stratum corneum at the 48 h [91]. Wu et al. incorporated salinomycin and paclitaxel-loaded SF-nanoparticles into a SF hydrogel to inhibit cancer stem cell and tumor growth. The dual drug-loaded hydrogel had homogeneous drug distribution and exhibited increased tumor inhibition compared to the single drug-loaded hydrogel, as evidenced by fewer CD44 + CD133+ tumor cells in vivo. Because paclitaxel and salinomycin interacted differently with SF, their release profiles were different, with paclitaxel showing sustained release and salinomycin an initial burst release [92]. Gangrade et al. introduced carbon nanotubes into SF hydrogels to construct an on-demand, tumor-targeting system [93]. They synthesized folic acid functionalized, DOX-loaded, single-walled carbon nanotubes (SWCNT) and incorporated them into an SF hydrogel matrix. Only 7% of DOX was released from the composite material over a period of five days under physiological conditions. However, intermittent exposure to near-infrared light stimulated on-demand DOX release (~15%) due to the photothermal property of SWCNT. He et al. developed an injectable silk fibroin nanofiber hydrogel system complexed with upconversion nanoparticles and nano-graphene oxide (SF/UCNP@NGO) for upconversion luminescence imaging and photothermal therapy [94]. The NaLuF4:Er3+,Yb3+ upconversion nanoparticles were complexed with the nano-graphene oxide and then doped into an aqueous SF solution to form a hybrid hydrogel system. SF/UCNP@NGO hydrogels efficiently ablated 4T1 breast cancer cells via the photothermal effect both in vitro and in vivo.
Recombinant techniques can also be utilized to modulate the properties of silk-based scaffolds and hydrogels. Anderson et al. recombinantly synthesized SLP segments conjugated to a human fibronectin segment and cell attachment domain [95]. Crystal structure characterization of (GAGAGS)-based SLPs revealed the hydrophobic domains of silk collapsed to form anti-parallel β-sheets that assembled into crystallite whiskers at the nanometer scale. This self-assembled material is mechanically tough, can undergo processing in the presence of bioactive molecules, and can be processed for into thin films, hydrogels, and three-dimensional scaffolds. Schacht et al. fabricated highly porous foams made from recombinant spider silk protein eADF4(C16) and a variant containing an RGD motif using a salt-leaching technique [96]. In contrast to other salt-leached silk scaffolds, the swelling behaviors of these scaffolds as measured by Discovery V20 stereomicroscope were low, and the mechanical properties were suitable for soft tissue engineering. The compressive moduli of the foam in a hydrated state was 3.24 ± 1.03 kP at a protein concentration of 8% (w/v). The pore size and porosity of the foams were optimized by altering the salt crystal size, thus rendering them suitable to adhere and culture fibroblasts.
As silk fibroin can self-assemble into hydrogels from an aqueous solution, it has been widely used for ocular delivery of various drugs ranging from small molecules to antibodies and other therapeutic proteins. In one such example, silk hydrogel formulations of bevacizumab, a clinically used antibody as angiogenesis inhibitor showed sustained release of the antibody over a three months' period in an intravitreal injection model in Dutch-belted rabbits [87]. The bevacizumab concentration in the vitreous humor on day 90 using hydrogel formulation at both standard (1.25 mg /50 μl injection) and high dose (5.0 mg /50 μl injection) was equivalent to the levels achieved with positive control on day 30 (1.25 mg bevacizumab/50 μl injection). [87]. This concentration is estimated to be the therapeutic threshold based on the current dosage of 1 injection/month. These gels also got degraded after 3 months, indicating a repetitive dosing may be possible. Additionally, the propensity of SF to bind positively charged molecules through electrostatic interaction can be exploited for topical drug delivery on the eye surface. Dong et al. electrostatically coated an ibuprofen-encapsulated liposome of cationic lipids with SF for ocular drug delivery [97]. The SF being a potential mucoadhesive biopolymer aided in retention of the drug on eye surface and facilitated its sustained release. Recently a phase II clinical trial (NCT03889886) has been undertaken to evaluate the ocular and systemic safety and efficacy of SDP-4, a naturally occurring silk-based ophthalmic solution in subjects with moderate to severe dry eye disease over a 12-week treatment period.
5.2. Keratin
Keratin is a fibrous structural protein abundant in epithelial cells of human and animal skin, hair, nails, scales, feathers, and other epidermal appendages. It forms the body's protective barrier by facilitating cell-to inter-cellular adhesions. Keratins are highly tunable and responsive, which makes them ideal materials for drug delivery applications. Like other protein carriers, keratins benefit from a high MW and avoid rapid renal clearance, thus increasing the circulation half-life of their cargo. They are very durable and stable due to their high level of intramolecular bond formation. Furthermore, the unique amino acid composition of keratins allows them to stably interact with a variety of therapeutics and respond to numerous biological stimuli [98].
5.2.1. Structure and properties of keratin
Unlike other proteins reviewed herein, keratins have a high cysteine content that provides the protein with mechanical, chemical, and thermal stability. Typically, a greater cysteine content, which results in more disulfide bonds within the protein, creates harder keratins such as those found in hair and nails. Soft keratin, which is found in skin, has fewer disulfide bonds. In addition to being biocompatible, biodegradable, non-toxic and tunable, keratins can also contain cell adhesion motifs, a useful characteristic for facilitating drug delivery [99].
Keratins can be subdivided into three molecular configurations. The first, α-keratin, has a relatively low sulfur content and an α-helical structure with four intertwined, right-handed helices [100]. These α-helices form the fibers that are abundantly found in soft tissues. The second, β-keratin, is rich in glycine, lysine, histidine, alanine, serine, and tryptophan. It forms a β-sheet structure stabilized by hydrogen bonds and provides rigidity to skin, scales, and nails. The third configuration, γ-keratin, has high levels of cysteine, glycine, and tyrosine. It has an amorphous structure that forms many intermolecular and intramolecular disulfide bonds. It is a key matrix protein that primarily holds α-keratin fibers together and provides mechanical strength to hair.
Keratins are negatively charged; thus, positively charged therapeutics can easily adhere to the surface of keratin hydrogels or nanoparticles. Carboxyl, amine, and carbonyl groups in the protein also provide useful drug attachment sites, either through hydrogen bond formation or chemical conjugation. Keratins have cell-targeting capacity due to the presence of cell attachment sites, including RGD sequences and leucine-aspartic acid-valine (LDV) sequences, within the protein. Additionally, disulfide bonding, electrostatic interactions, and hydrogen bonding provide keratin with mucoadhesive properties, which make it a useful material for delivery of drugs to the gastrointestinal tract [101].
Furthermore, keratins have been employed as “smart” materials for stimulus responsive drug delivery. The large number of carboxyl groups within their sequence makes them pH-sensitive so that in response to an increase in pH, release their cargo in a controlled manner as these groups become deprotonated [102]. Additionally, their rich cysteine content renders them redox-responsive. Tuning the number or level of crosslinking of disulfide bonds within the protein alters the duration of drug release from a keratin-based material. Similarly, keratin is responsive to changes in glutathione (GSH) concentration. This is particularly useful for delivery of chemotherapeutics to metastatic cancer cells, which have significantly higher concentrations of GSH than healthy cells. Moreover, the high lysine and arginine content in some keratins enables their cleavage by high concentrations of trypsin [103], which is frequently overexpressed in inflamed tissue [104]. Thus, keratin could be a useful material when targeting injured or tumorous tissues.
5.2.2. Keratin nanoparticles
Due to its unique material properties, keratin has been used to create nanoparticles that encapsulate and sequester a therapeutic cargo before releasing it in response to biological stimuli. These nanoparticles can stably carry therapeutics via electrostatic interactions, hydrogen bonding, disulfide bond formation, or chemical conjugation. Keratins are durable and remain stable in the bloodstream, which increases the half-life of its cargo [100].
Positively charged drugs electrostatically adsorb to the surface of negatively charged keratin nanoparticles; this interaction provides long-term, sustained release of the drug from its carrier. Zhi et al. first explored this strategy by complexing the model drug chlorhexidine (CHX) with keratin nanoparticles generated by ionic gelation [102]. Carboxylate groups on the nanoparticle surface stabilized the polyanion complex with CHX. This interaction provided a remarkable CHX encapsulation efficiency of 91.2% and a loading content of 9.2%. Zhi et al. demonstrated that for 140 h, the drug was released in a pH-dependent manner with greater release observed at neutral and slightly acidic pH, which indicates the utility of this system to deliver chemotherapeutics to the acidic tumor microenvironment [102].
Similarly, Li et al. demonstrated that keratin-based drug-loaded nanoparticles (KDNPs) can be loaded with DOX via electrostatic interactions [105]. These nanoparticles were designed to exploit keratin's responsiveness to pH and glutathione concentration to release the drug, which is a useful strategy to treat solid tumors, which have a pH of 6.2–6.9 and GSH concentration of 0.5–10 mM, which is approximately 10-fold greater than the GSH concentration of healthy tissue [105]. KDNPs were created with a desolvation method that destabilizes keratin with ethanol and causes it to aggregate into nanoparticles that are crosslinked with glutaraldehyde. In an environment with a low pH or high concentrations of glutathione, KDNPs experienced a stark increase in zeta potential and underwent a negative-to-positive charge conversion. The positive charge facilitated internalization by cells and electrostatically repelled the drug, thus accelerating its release (Fig. 7 ). Moreover, the charge conversion destabilized the nanoparticles, causing them to aggregate and accumulate at the tumor due to the enhanced permeation and retention (EPR) effect, which further enhanced their anti-cancer potency [105].The stimulus-responsive properties of KDNPs were expanded by Li et al., whoe engineered triple stimuli-responsive KDNPs via drug-induced ionic gelation. In addition to releasing DOX in response to changes in pH and glutathione concentration, these nanoparticles broke down in the presence of high concentrations of trypsin, which digested the peptide bonds within the keratin [106]. Similarly, keratin graft poly(ethylene glycol) nanoparticles have also been explored as glutathione-responsive vehicles; DOX-HCl entrapped in the disulfide-crosslinked keratin core was released in response to increased intracellular glutathione concentrations [107].
Fig. 7.

Dual stimulus responsiveness of keratin-based drug loaded nanoparticles (KDNPs). (A) Schematic of nanoparticle fabrication and GSH- or pH- stimulated drug release (B) An acidic environment shifts the size and zeta potential of KDNPs and accelerates the release of entrapped DOX (C) The presence of GSH shifts the size and zeta potential of KDNPs and accelerates the release of entrapped DOX. Adapted with permission from [105].
Due to its exquisite and versatile loading capacity, keratin has also been harnessed to create bimodal nanoformulations that combine chemotherapeutics and photodynamic therapy. Martella et al. functionalized high molecular weight keratin with the photosensitizer Chlorin-e6 (Ce6) and induced spontaneous nanoparticle formation by mixing the protein with paclitaxel, a chemotherapeutic that aggregates with the hydrophobic residues in keratin [108]. This is an attractive bottom-up nanoparticle fabrication strategy, as it did not require any toxic crosslinkers or downstream purification steps for nanoparticle fabrication. When administered to an osteosarcoma cell line, the nanoparticles localized to the cell lysosome. Although Ce6 typically experiences a drop in fluorescence in acidic conditions, the keratin protected the photosensitizer and no decrease in fluorescence was observed. Further, the nanoparticle formulation transported the paclitaxel in a three-dimensional tumor model system without reducing the drug's potency [108]. Keratin nanoparticles have also been engineered for mucoadhesive drug delivery. Kerateine (KTN) and keratose (KOS) are two forms of keratin that have been extracted and processed in its reduced and oxidized forms, respectively. Cheng et al. demonstrated that, by altering the KTN:KOS ratio, the mucoadhesive properties and thus drug release, gastric retention time, and bioavailability of keratin nanoparticles could be tuned [109]. An evaluation of the hydrophobicity, surface charge, and terminal groups of the nanoparticles revealed that the mucoadhesive properties of KTN were dominated by electrostatic interactions, whereas those of KOS were primarily due to hydrogen bonding with gastric mucin. Moreover, Cheng et al. discovered that gastric retention time decreased with an increase in KOS, and release of the model drug, amoxicillin, increased with a larger proportion of KTN due to its pH responsiveness [109].
5.2.3. Keratin films
Keratin-based films have been widely explored for biomedical applications due to the presence of cell attachment sites, their biocompatibility, and their large surface area. These mechanically and chemically stable materials have been useful for delivering a variety of drugs and peptides.
In an early study of keratin films, Fuji et al. extracted keratin from human hair in the absence of surfactant, thus creating a water-soluble film comprised primarily of α-keratins [110]. Alkaline phosphatase, a model enzyme, was incorporated into the film by mixing it with the keratin prior to gelation. The biochemical properties and bioactivity of alkaline phosphatase were maintained for two weeks after loading [110].
While keratin films have excellent biocompatibility, they lack mechanical strength. Thus, many studies combine keratin with other materials or treat the protein with a crosslinking agent to improve its rigidity, stiffness, and stability. In one example, keratin was blended with SF to create a composite film for the delivery of Bowman-Birk inhibitors (BBI), synthetic peptides designed to inhibit elastase in wound healing applications [111]. SF provided structural stability to the film while keratin governed the degradation and BBI release rates. Using FITC -tagged bovine serum albumin (BSA) as a model protein, Vasconcelos et al. demonstrated that, although the SF was compact and rigid, increasing the percentage of hydrolytic keratin could increase the rate of FITC-BSA release by film degradation and diffusion [111]. Another study used transglutaminase (TGase) to crosslink keratin films to improve the mechanical strength and chemical stability of the material [112]. Treatment with TGase resulted in more compact network formation and increased the mechanical strength from 5.18 MPa to 6.22 MPa. This reduced the solubility of the film and thus delayed the release of the model drug, diclofenac [112]. These studies illustrate how keratin materials can be modified to improve drug elution for wound healing and tissue engineering applications.
5.2.4. Keratin hydrogels
Keratin hydrogels have been widely studied for biomedical applications, due to their stability, durability, and large number and variety of attachment sites for drugs or cells. The latter makes keratin hydrogels attractive for tissue engineering and regenerative medicine applications [100,101,113]. Additionally, keratin hydrogels have been explored for local, controlled delivery of small molecule drugs and macromolecules. Drug release is mediated by keratin degradation, the rate of which can be systematically tuned by controlling the number and type of bonds within the hydrogel [111].
The simplest keratin hydrogels rely on electrostatic interactions to load the drug of interest, which is typically positively charged, onto a keratose-based hydrogel. This strategy was used by Saul et al. to locally deliver and sustain the release of ciprofloxacin, a broad-range antibiotic [114]. Ciprofloxacin was loaded into the hydrogel by mixing it with the keratose solution before gelation. Because keratose is processed with sulfonic acid, it lacks disulfide bonds, and its gelation relies on hydrophobic interactions and physical chain entanglement. Saul et al. observed that ciprofloxacin release strongly correlated with keratose degradation. Approximately 40% of the drug was released within the first 24 h, followed by a period of linear release for the next six days, with release detectable for up to three weeks. Drug bioactivity was not impacted by its incorporation into the hydrogel [114]. Halofuginone, a type I collagen synthesis inhibitor, was loaded into a keratose hydrogel by the same technique [115]. One day after administration, the drug was released at a steady rate from the hydrogel for four days. After seven days, 60% of the halofuginone had been released, consistent with the degradation profile of the keratose hydrogel. As with the ciprofloxacin study, halofuginone remained bioactive [115]. These studies highlight the applications for keratin hydrogels in local drug release.
The rate of drug release from keratin hydrogels can be modified by altering the crosslinking density through alkylation. Han et al. alkylated KTN by treating keratin with iodoacetamide to “cap” the cysteine thiol groups and thus modulate the number of disulfide bonds within the protein [116]. Three therapeutics – ciprofloxacin, recombinant human insulin-like growth factor 1 (rhIGF-I), and recombinant human bone morphogenic protein (rhBMP-2) – were loaded into the hydrogel. Han et al. found that the increased rate of hydrogel degradation correlated with the amount of iodoacetamide used, though it was not directly proportional due to the binding affinity of the model drug to the keratin [116]. A disulfide shuffling strategy was used by Cao et al. to explore how disulfide bond formation impacts drug release. They cleaved intramolecular disulfide bonds with a reductive agent (e.g., cysteine) to free thiol groups that could then form intermolecular disulfide bonds ( Fig. 8A) [117]. This tactic increased the mechanical strength of the hydrogel, reduced its gelation time, and required a lower amount of keratin for gelation. The release rates of ciprofloxacin and DOX were inversely proportional to the level of cysteine in the hydrogels. Hydrogels that entrapped ciprofloxacin also demonstrated zero-order release kinetics in PBS (Fig. 8B & C). Moreover, when these hydrogels were exposed to increasing concentrations of glutathione, the degradation rate increased in response to the more rapid degradation of disulfide bonds (Fig. 8D & E) [117]. These studies indicate that the degradation rate of keratin hydrogels can be altered for specific disease states and drugs.
Fig. 8.

Disulfide shuffling to modulate drug release from keratin hydrogels. (A) Schematic of gelation by the disulfide shuffling strategy. Crosslinking density and thus microstructure, mechanical strength, and degradation can be tuned with this method (B,C) Hydrogel degradation and ciprofloxacin release in PBS demonstrate that greater crosslinking, represented by a greater number of Cys residues, slows degradation and release. (D,E) Hydrogel degradation and DOX release in the presence of GSH demonstrates the redox responsiveness of these hydrogels. Adapted with permission from [117].
pH-responsive keratin hydrogels have also been designed to reversibly swell in response to their environment to modulate the release of their cargo. In many of these examples, keratin has been combined with itaconic acid, N-isopropyl acrylamide, or methacrylic acid to achieve pH responsiveness [[118], [119], [120]]. More recently, Peralta Ramos et al. reported a stimulus-responsive hydrogel that did not depend on chemical grafting to achieve its mechanical properties and pH-responsivity [121]. This was credited to a novel synthesis strategy during which disulfide bridges were broken and the α-keratin reorganized. At an acidic pH, the carboxyl groups were protonated and formed hydrogen bonds, which created a higher fraction of β-sheet conformations and kept the gel in a collapsed state. At a basic pH, hydrogen bonds broke to create more sites for water adsorption, thus forcing chain reorganization and allowing the material to swell [121]. Villanueva et al. expanded upon this work by incorporating antimicrobial ZnO nanoplates into the pH-responsive hydrogel [122]. Upon administration to a chronic wound, the hydrogel swelled in response to the basic environment and locally released the biocidal agent. As the wound healed and the presence of microbes decreased, the gel collapsed and inhibited the release of ZnO [122]. This study demonstrates how keratin hydrogels can be used to adjust the delivery of a therapeutic agent in response to alterations in pH.
5.3. Albumin
Albumin is the most abundant protein in human plasma and has a set of properties that make it a unique molecular carrier for drugs: (i) it is a natural physiological carrier of native ligands and nutrients; (ii) it bypasses systemic clearance and degradation by the body's own innate mechanisms, so that it has an exceptionally long half-life of 19 days in humans, and similarly long half-lives in most animal species [[123], [124], [125], [126]]; (iii) it preferentially accumulates at sites of vascular leakiness; (iv) it is highly internalized and metabolized by rapidly growing, nutrient-starved cancer cells; and (v) it is biodegradable and has no known systemic toxicity. Because of these properties especially its extraordinarily long plasma circulation, albumin has attracted a lot interest as a carrier for diverse drugs.
5.3.1. Types of albumin
Three naturally derived albumin molecules have been used to address the delivery challenges associated with small molecule drugs. Ovalbumin (OVA), a highly functional food protein with a molecular weight of 47 kDa and isoelectric point (pI) of 4.8, is a monomeric phospho-glycoprotein consisting of 386 amino acid residues [127]. Each OVA molecule has one internal disulfide bond and four free sulfhydryl groups. OVA was originally chosen as a carrier for drug delivery because of its availability, low cost, ability to form gel networks and stabilize emulsions and foams, and pH- and temperature-sensitive properties [128]. Bovine serum albumin (BSA), which has a molecular weight of 69 kDa and a pI of 4.7 in water (at 25°C), has also been widely used for drug delivery because of its abundance, low cost, ease of purification, unusual ligand-binding properties, and wide acceptance in the pharmaceutical industry [[129], [130], [131], [132], [133], [134]].
However, due to possible human immunologic response to OVA and BSA in vivo, human serum albumin (HSA) is now exclusively used for drug delivery [50]. HSA, which has a MW of 66.5 kDa, is the most abundant serum protein with a concentration of 30–50 g/l in human serum. It is long-circulating with an exceptionally long in vivo half-life of ~19 days [[123], [124], [125],135]. About 10–15 g of albumin is synthesized by liver hepatocytes daily and released into circulation [136]. When albumin extravasates into tissue, it is naturally recycled and returned to the vascular space via the lymphatic system. The same approximate mass of 10–15 g of albumin entering the intravascular space is also catabolized daily. HSA is a carrier of a wide variety of endogenous and exogenous compounds and facilitates the colloidal solubilization and transport of hydrophobic molecules, such as long chain fatty acids, and variety of other ligands, including bilirubin, hormones, amino acids, metal ions, and drugs [136,137].
5.3.2. Structure and properties of HSA
HSA is a soluble, globular, monomeric protein consisting of 585 amino acid residues. It contains 35 cysteinyl residues that form one sulfhydryl group and 17 disulfide bridges [123,135,138]. Fig. 9A shows the crystal structure of albumin and the sites where ligands can bind [139]. Three distinct features of albumin make it ideal for use in drug delivery applications: binding, trafficking, and recycling.
Fig. 9.

Ribbon diagram of the three-dimensional structure of human serum albumin (A). Schematic of the in vivo thiol-maleimide conjugation reaction (B). Chemical structure of DOX-EMCH/Aldoxorubicin (C). Adapted with permission from [140] (A) and [144] (B).
Binding: Each class of drugs has a distinct binding affinity to different binding sites of albumin. Dicarboxylic acids and sterically demanding anionic heterocyclic molecules (e.g., Warfarin) bind to Sudlow's site I (Fig. 9A), whereas aromatic carboxylic acids with a single negatively charged acid group separated by a hydrophobic center (e.g., diazepam, ibuprofen) bind to Sudlow's site II [140]. HSA has seven long-chain fatty acid binding sites (FA1–7 in Fig. 9A, asymmetrically distributed throughout its three domains) that promote binding of up to two moles of fatty acid per mole of HSA under normal physiological condition [[141], [142], [143]]. Compounds with p-isothiocyanate (p-SCN) or NHS ester (N-hydroxysuccinimide) functional moieties covalently react with albumin's lysine, with Lys199 residue being the most reactive [144,145]. However, due to multiple other (at least ten) surface accessible lysine residues in HSA, reaction with lysine residues leads to poorly defined conjugates with a range of stoichiometries [146,147]. Alternatively, the solvent accessible cysteine34 in HSA can be selectively modified with a prodrug containing a maleimide functionality, as (i) 70% of circulating serum albumin possess one free cysteine; (ii) other major serum proteins lack a solvent-accessible free cysteine; and (iii) the 34th cysteine residue of albumin has the most reactive thiol group (pKa ~ 7) in human plasma [144,148].
Trafficking: Albumin naturally transcytoses across the vascular endothelium, which normally creates an impermeable barrier to most plasma proteins. This process is attributed to the 60 kDa vascular endothelium receptor sialoglycoprotein (gp60). Albumin binds to gp60 and forms a cluster in association with Cav-1, the main protein critical to caveolae formation. Clustered albumin-gp60 receptors and compounds bound to albumin are then internalized and transported to the basolateral membrane to complete transcytosis [149]. Interestingly, modified albumins show preferential binding to gp18 and gp30 [150]. Preferential internalization of albumin in cancer cells has also been correlated with albumin binding to SPARC (secreted protein acidic and rich in cysteine). For example, immunohistochemical staining detected stromal SPARC in ∼70% of non-small cell lung cancer (NSCLC) cases, and increased chemotherapeutic efficacy of albumin-bound paclitaxel was correlated with high stromal SPARC reactivity of NSCLC cells [151].
Recycling: The long half-life of albumin is attributed to the neonatal Fc receptor (FcRn), a widely distributed intracellular receptor responsible for salvaging albumin from cellular catabolism [143]. FcRn binds to albumin in the acidic endosome, diverts it from the lysosomal degradation pathway, and the complex is exocytosed. Albumin is released from FcRn at extracellular pH and enters circulation through the lymphatics, thus prolonging its half-life. Albumin also avoids renal clearance by reabsorption through megalin and cubilin receptor-mediated endocytosis in the renal proximal tubule.
5.3.3. Endogenous albumin for drug delivery
In situ albumin-binding drugs have been designed to covalently react with or noncovalently bind to endogenous albumin, which capitalizes on the long-circulating property of the protein to enhance the PK and hence PD of the drug. Indeed, exploitation of endogenous serum albumin has several advantages over the use of exogenous albumin: first, although commercial exogenous albumin can be isolated with high yield and purity, it is often contaminated with pathogens. Second, the cost, effort, and time required for manufacturing exogenous albumin is altogether avoided. Third, as no external macromolecular carrier is involved, the quality control associated with endogenous albumin is comparable to small molecule drug candidates.
Irreversible covalent bond formation between endogenous albumin molecules and a therapeutic payload can be utilized to enhance the pharmacokinetics of the latter. Kratz et al. pioneered this strategy by synthesizing a prodrug that selectively reacts with the 34th cysteine residue of circulating serum albumin after systemic administration, which improves the drug's plasma half-life and protects it from premature degradation. The therapeutic cargo (e.g., DOX) was anchored with a thiol reactive maleimide group via a pH-sensitive hydrazone bond and a carefully optimized alkyl spacer. Upon intravenous injection, the highly reactive maleimide group formed an irreversible thioether bond with the thiol group of the albumin. The pH-sensitive hydrazone bond facilitated the liberation of the covalently attached DOX from albumin after en endocytosis by cells. Aldoxorubicin, also known as DOX-EMCH (Fig. 9B & C) is currently under various phases of clinical trial for the treatment of soft tissue sarcomas (NCT01673438 and [152]) and small cell lung cancer (NCT02235688). Inspired by DOX-EMCH, many other albumin-binding prodrugs have been developed. These prodrugs often consist of an anticancer drug, a maleimide group as the thiol-binding moiety, and a cleavable linker [144].
Native albumin-binding ligands such as fatty acids can be directly conjugated to therapeutics, which results in docking of the drug to endogenous albumin upon systemic administration. A notable example of this class is Semaglutide, an FDA approved drug that is a glucagon-like peptide-1 (GLP-1) analog with an octadecyl fatty diacid conjugated to the epsilon amine of a lysine residue in the peptide via a glutamyl ethylene glycol spacer [153]. The plasma half-life (t1/2, seven days in humans) and GLP-1 receptor (GLP-1R) binding affinity (Kd ~ 0.38 nM) of Semaglutide make it suitable for once-weekly administration for treatment of type 2 diabetes [153,154]. This was an important advancement, as its predecessor Liraglutide —which contains a hexadecyl monoacid conjugated to the same lysine residue of GLP-1 via a γ-Glutamic acid spacer— has a t1/2 of only 0.5 days in humans, and is hence only approved for once-daily treatment of type 2 diabetes, despite having a three-fold higher affinity for GLP-1R (Kd ~ 0.11 nM) than Semaglutide. This dramatic improvement was attributed to both the spacer and ligand chemistry of the GLP-1 analog in Semaglutide, which influenced in vivo albumin binding [153]. Other therapeutic payloads are bound to endogenous HSA by conjugation of the drug to fatty acids through various cleavable linkers [125,144].
Synthetic small molecules can also bind endogenous albumin [144]. Most drugs and small molecules that interact with HSA are anionic, although a few cationic drugs have detectable affinity [142]. Evans blue (EB), an aromatic dye with four anionic charges, reversibly binds to serum albumin with a micromolar affinity. The affinity of EB for albumin allows 100% of the injected dose to be retained in the blood; thus, it can be used to quantify total plasma volume of a test subject [155]. Albumin-bound EB can also be used to assess blood brain barrier (BBB) permeability, as the BBB is impermeable to the complex in healthy, non-pathological conditions [156]. EB derivatives with various affinities to albumin have been used to deliver therapeutic cargo [144,157]. Yamamoto et al. developed an o-toludine EB analogue that chelates gadolinium (III), as a T1-weighted MRI contrast agent for imaging of blood vessels [158]. Following this seminal work, the Chen group developed the o-toludine EB analogue as a positron emission tomography (PET) tracer for in vivo labeling of serum albumin. They conjugated o-toludine EB to 1,4,7- triazacyclononane-N,N′,N″-triacetic acid (NOTA) to prepare a new analog named NEB. The NOTA chelator provides a simple radiolabeling route that allows labeling with different PET isotopes (e.g., 68Ga, 64Cu) [[159], [160], [161]]. Chen et al. evaluated labeled NEB in mice as a blood pool imaging agent under various pathological conditions, including myocardial infarction (MI) and serum leakage from permeable or abnormal blood vessels. Sentinel lymph nodes (SLNs) were visualized in all 24 pathologically diagnosed breast cancer patients within 4.0–10.0 (5.6 ± 1.4) min [161]. by analyzing 68Ga-NEB PET/CT images. To identify molecular features required for albumin binding, the Neri group screened a DNA-encoded chemical library comprising >600 oligonucleotide-conjugates of potential albumin-binding molecules coded with unique six-base-pair sequences for identification [162]. The HSA-binding tags were chosen through Systematic Evolution of Ligands by Exponential Enrichment (SELEX). The selection, amplification, and microarray analysis of the pool suggested that following structural features are important for albumin binding: (i) the presence of a 4-phenylbutanoic acid moiety is essential; (ii) a butanoyl acid moiety increases albumin affinity as propanoyl and pentanoyl acid residue was not enriched in the pool; and (iii) substitution at the para position of the phenyl ring with a hydrophobic functionality increases the affinity toward albumin.
Albumin-binding domains (ABDs) are small protein domains that non-covalently associate with serum albumin. ABDs are structurally robust and stable, as evidenced by their ability to withstand denaturation at extremely low pH (2.4) and high temperatures. and have also been explored for drug delivery. ABDs are used for drug delivery by either fusion with a therapeutic protein at the gene level if the protein is recombinantly produced or are chemically synthesized to conjugate small molecule drugs for systemic administration. Sjöbring and colleagues isolated a 14-kDa albumin binding protein fragment from Streptococcal protein G. This 46-amino acid native ABD (ABDN) is a 3-helix bundle and has nanomolar affinity for HSA [163,164]. Using ABDN as a template, Jonsson et al. identified an engineered version of ABDN from a phage library. Specifically, they targeted 15 residues in a combinatorial protein engineering strategy to identify an albumin -binding sequence with improved HSA affinity. The high -affinity ABD (ABDH) has superior thermal stability and binds HSA with femtomolar affinity.
Chilkoti and colleagues recombinantly fused ABDN and ABDH to the N-terminus of a hydrophilic, thermally responsive chimeric polypeptide (CP) that contained multiple C-terminal cysteine residues [126]. The recombinant polypeptides formed highly monodisperse micellar nanoparticles upon covalent conjugation of multiple copies of hydrophobic DOX molecules to the.
cysteine residues through the pH-sensitive hydrazone bond (using maleimide-thiol chemistry) (Fig. 10A). The DOX-loaded ABDN/ABDH-decorated polypeptide micelles —termed ABD-CP-DOX— bind both human and mouse serum albumin with high affinity, as seen by native PAGE and isothermal titration calorimetry (Fig. 10B & D). Hence, upon systemic injection, they instantaneously bind endogenous albumin with high affinity and are coated with an albumin corona. The albumin decorated ABD-CP-DOX nanoparticles had significantly superior PK in both murine and canine models compared to a negative control, CP-DOX nanoparticles that do not present ABD domains on the surface of the nanoparticle (Fig. 10E & F) The long plasma circulation of the ABD-CP-DOX nanoparticles also resulted in higher accumulation in tumors than the CP-DOX nanoparticles, and these nanoparticles also showed better tumor regression, and a wider therapeutic window than the CP-DOX nanoparticles. These results clearly show the enhancement in PK and PD conferred by decorating a drug-loaded nanoparticle with endogenous albumin.
Fig. 10.

Design of doxorubicin(DOX)-conjugated albumin-binding nanoparticles. DOX was conjugated via a pH-sensitive hydrazone linker. (B) Albumin-binding properties of ABDN-CP-DOX and ABDH-CP-DOX were qualitatively demonstrated using native PAGE. (C) Cryo-TEM images of ABDN-CP-DOX (I) and CP-DOX (II) micelles. (D) Isothermal titration calorimetry of ABDN-CP-DOX and ABDH-CP-DOX micelles with MSA. The solid red line represents the best fit of the binding isotherm. (E, F) Pharmacokinetics of ABD-decorated nanoparticles in murine (E) and canine (F) models. Adapted with permission from [126]. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Furthermore, the same group took advantage of the native sortase reaction to directly install DOX onto the ABD (ABD-DOX) through a pH-sensitive linker, without forming nanoparticles [59]. ABD-DOX bound to both human and mouse serum albumin with nanomolar affinity, had a terminal t1/2 of 29.4 h in mice, and increased tumor accumulation by ~120 fold compared to free DOX (Fig. 11A). In multiple mouse xenograft models, ABD-DOX resulted in greater tumor regression than Aldoxorubicin, DOX-prodrug under clinical development that was designed to covalently bind endogenous serum albumin. Other notable examples of albumin-based delivery systems involve the genetic fusion of ABD to various therapeutic proteins including affibodies [165,166], human soluble complement receptor type 1 [167], single chain antibody-drug conjugates [168], insulin-like growth factor II [169], immunotoxins [170], and respiratory syncytial virus subgroup A (RSV-A) G protein (G2Na) [171].
Fig. 11.

(A) Synthetic scheme of ABD–DOX. Elastin-like polypeptide (ELP) was used as a purification tag and removed following drug conjugation using sortase A. Inclusion of the KEKE peptide at the N-terminus disrupted micellar self-assembly upon DOX conjugation and enabled the subsequent sortase A cleavage of ELP from the ABD–DOX conjugate. (B) Synthetic scheme of albumin−polymer−drug conjugates. Drug-containing monomer (drug: panobinostat, dark blue) was copolymerized with HPMA using RAFT agents, which allow one-step conjugation of albumin. Adapted with permission from [59] (A) and [172] (B). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
5.3.4. Exogenous albumin formulations for drug delivery
Albumin-bound therapeutics can also be formulated ex vivo with purified albumin molecules prior to administration. In contrast to other physiological proteins, albumin tolerates a wide range of pH (stable in the range of 4–9), temperatures (can be heated at 60°C for up to 10 h), and organic solvents [50]. These properties have motivated researchers to covalently and non-covalently append various payloads, including radioisotopes (e.g., 18F, 68Ga, 111In) and anticancer drugs (e.g., DOX, curcumin, methotrexate), to albumin for imaging and delivery purposes [125,144]. However, modification of exogenous albumin with drugs is often associated with suboptimal drug loading, as denaturation and albumin molecule crosslinking potentially compromise its physiological behavior. Smith et al. addressed these challenges by synthesizing an albumin-polymer-drug conjugate with high drug loading efficiency [172]. Using a convergent, reversible addition−fragmentation chain transfer (RAFT) polymerization reaction, they synthesized a polymeric prodrug of panobinostat, an FDA-approved anticancer agent, with HPMA (N-2-hydroxypropylacrylamide). Multiple copies of panobinostat were attached to the polymeric prodrug, which was then conjugated to albumin using NHS chemistry under physiological conditions, thereby minimally affecting the albumin molecule (Fig. 11B). The first albumin-drug conjugate that entered phase I/II trial was a methotrexate-albumin (MTX-HSA) conjugate synthesized using covalent coupling between carboxylate of the MTX and lysine residues of HSA. Seventeen patients not amenable to standard care were treated with up to eight injections given in weekly intervals. Tumor responses were seen in three with no sign of toxicity and drug accumulation. The MTD for weekly administration was found to be 4 × 50 mg/m2 and a injection of 50 mg/m2 every 2 weeks was recommended for a further study [173]. However, in a subsequent phase II study no objective responses were seen, although eight patients did not have disease progression (stable disease) for up to 8 months (median 121 days) [174]. A phase II study showed combination of MTX-HSA with cisplatin as an effective treatment modality against urothelial carcinomas with an acceptable toxicity profile [175]. Patients were treated with a loading dose of 110 mg/m2 of MTX-HSA followed by a weekly dose of 40 mg/m2 starting on day 8. Tumor response was observed in 7 patients. Complete response (CR) and partial response (PR) was observed in 1 patient each (overall response rate: 29%) but no follow up clinical investigation was undertaken.
Albumin nanoparticle formulations represent the most widespread utilization of exogenous albumin as a carrier. Fabrication techniques such as desolvation, emulsification, thermal gelation, nano-spray drying, and self-assembly have been used to formulate albumin nanoparticles [125]. The most notable albumin nanoparticle is Abraxane, also known as nab-paclitaxel, an FDA approved paclitaxel formulation for the treatment of multiple cancer types [176,177]. Abraxane was developed by American Bioscience using the so-called nab-technology in which paclitaxel and human serum albumin are passed through a jet under high pressure to form nanoparticles with a mean diameter of 130 nm. Several albumin nanoparticle of small molecule drugs has been developed using nab technology and are under various phases of clinical trials [178]. Nab-rapamycin is currently under phase I clinical trial to treat non-muscle invasive bladder cancer (NCT02009332) and under phase II clinical trial to treat progressive high-grade glioma (NCT03463265). Nab-docetaxel proved to be effective against hormone refractory prostate cancer (NCT00477529) and metastatic breast tumors (NCT00531271). Nab-5404, a novel albumin formulation of thiocolchicine dimer exhibits dual inhibition of tubulin polymerization and topoisomerase I activities. It showed antiangiogenic and vascular targeting activities and was found to be effective against solid tumors and lymphomas (NCT01163071). Lin et al. reported a method to simultaneously encapsulate paclitaxel by denaturing albumin with sodium borohydride at high urea concentration, followed by addition of hydrophobic drugs to the denatured protein, and diluting the mixture to spontaneously refold albumin and form nanoparticles [179]. This method avoids chemical crosslinkers and high-pressure emulsification techniques. By adding a cell-penetrating peptide to the dual drug-loaded albumin nanoparticles, they achieved BBB penetration through interaction with SPARC, which resulted in greater survivability in a U87 orthotopic glioma model [179]. Albumin has also been derivatized into a diblock format in which a hydrophilic albumin block gets exposed at the surface of the hybrid nanoparticle and a synthetic hydrophobic polymer, such as polycaprolactone, forms the core. To achieve this, Jiang et al. synthesized two different polymers based on ring-opening polymerization: poly(oligo-(ethylene glycol) methyl ether acrylate)–poly(ε-caprolactone) (POEGMA-PCL) and maleimide–functionalized polycaprolactone (MI-PCL). MI-PCL was conjugated to the free cysteine on bovine serum albumin (BSA-PCL) [180]. The authors formulated hybrid nanoparticles by co-assembling POEGMA-PCL, BSA-PCL, and curcumin with increasing albumin content. Cellular uptake of the nanoparticles in cancer cell lines increased with albumin content [120]. Additionally, albumin nanoparticles can be decorated with targeting ligands, such as mannose [181], folic acid [182], antibodies [183,184], and aptamers [185], to provide greater receptor specificity.
Recombinant albumin is another alternative to animal-derived albumin and is increasingly being used in exogenous formulations. Direct genetic fusion to recombinant albumin enables one-step fabrication of therapeutic proteins. For instance, rlX-FP, a genetically encoded fusion protein linking recombinant human albumin with human coagulation factor IX, has a 5-fold longer plasma half-life than unmodified coagulation factor IX (FIX) products. An advanced phase III clinical trial (PROLONG-9FP) demonstrated the long-term safety, tolerability, and efficacy of rIX-FP for prophylactic and on-demand treatment of bleeding episodes in children [186]. Li et al. synthesized an albumin-lidamycin conjugate by recombinant DNA technology. Lidamycin, a cyclic enediyne antibiotic is ~1000 fold more potent than DOX against multiple cultured cancer cell line and it consists of an apoprotein (LDP) and an enediyne chromophore (AE) that can be separated and reassembled in vitro [187]. A DNA fragment encoding HSA-LDP was constructed and the fusion protein was purified from Pichia pastoris using IMAC. The HSA-LDP conjugate was then reconstituted with the AE to form the albumin-lidamycin conjugate that had a sub nanomolar IC50 value in various cancer cell lines, significant tumor retention, and potent in vivo tumor regression efficacy against mouse hepatoma H22 model [188]. Other recombinant albumin proteins include genetic fusions with interleukin-2 [189] and interferons [190,191]. Albuferon® aka Albinterferon-α-2b was developed as a fusion protein of albumin and interferon-α-2b (INFα-2b) and is under phase III for the treatment of hepatitis C infection (NCT00724776). Several other phase III trials are under progress aimed at evaluating the efficacy and safety of Albinterferon-α-2b [177]. However, high-yield synthesis of pure recombinant albumin and albumin fusions requires a complex procedure and specialized yeast expression system that are not readily available to most researchers. Nguyen et al. made significant progress by purifying recombinant HSA from a bacterial expression system using maltose-binding protein and protein disulfide isomerase [192]. But an optimized bacterial expression system to purify recombinant albumin is still needed.
5.4. Collagen
Collagen is the most abundant protein in mammals, making up nearly one-third of all proteins in the body. A diverse family of structural proteins, collagen is a major component of the extracellular matrix (ECM) and connective tissue. It has a broad spectrum of functions, including cell adhesion, cell migration, tissue scaffolding, and repair [193]. To date, thirty different classes of collagen have been identified and characterized [194], though not all are relevant for this discussion. The most abundant class of collagen —fibrillar collagen— represents more than 90% of human collagen and includes types I, II, III, V, and XI. This class is a major component of skin, hair, ligament, tendon, cartilage, bone, and placenta. Other common types of collagens, like IV and VIII, construct the network structure of basement membranes [193,194]. Before discussing their molecular architecture and biomedical applications, it is important to define the term “collagens.” Gelatin, a thermally degraded, collagen-derived product, is also termed “collagen” in the literature despite losing the characteristics of real collagen, and will hence be discussed separately [195].
5.4.1. Principles of collagen self-assembly
Although the molecular architectures and functions of collagens vary greatly, they all share the same tertiary structure —the collagen triple helix [196]. Under the electron microscope, native collagen shows a thread-like structure —fibrils— consisting of collagen molecules organized in three interlocking polytripeptide chains that form a triple helix, with each chain having the general sequence GIy-X-Y, where X and Y are occupied by Proline (Pro) and (4R)-hydroxyproline with the highest statistical frequency. Each collagen polypeptide chain is twisted around a threefold screw axis and exists in a secondary structure analogous to the left-handed polyproline II-helix. The three polyproline II-helices are held together by hydrogen bonding, which repeats periodically between the glycine amide in one helix and the carbonyl group of the amino acid residue of the neighboring helix. The three polyproline II-helices form a right-handed triple helix stabilized by hydrogen bonds [194,196,197]. With tunable self-assembly, gel-forming ability, biodegradability, and responsiveness to external stimuli, collagens have been increasingly used for drug delivery and tissue regeneration [195,[198], [199], [200]]. Current research is focused on developing short, bioinspired model collagens, termed as collagen-mimetic peptide (CMPs) or collagen-like peptides (CLPs) [201,202]. CLPs are short, synthetic peptides arranged in the same triple-helical conformation as native collagens. CLPs have been used: (i) to elucidate the triple helix structure and the molecular forces responsible for this architecture; (ii) to target pathological collagen in vivo through triple helix hybridization; and (iii) as a bioactive domain to construct smart biomaterials through hierarchical self-assembly. Each of these applications will be discussed separately as they are relevant for drug delivery.
5.4.2. Elucidation of the triple helix structure
To exploit CLPs to fabricate biomaterials, it is critical to elucidate the intrinsic parameters affecting the sequence-based thermal stability of the triple helix. In contrast to native collagen, CLPs exhibit reversible phase transition behavior. Their slow folding rate upon denaturation and small size allow researchers to thermodynamically characterize the folding and melting processes of CLPs by X-ray crystallography, atomic force microscopy (AFM), light scattering, and circular dichroism (CD) and nuclear magnetic resonance (NMR) spectroscopy. Many researchers have synthesized and studied polypeptides with Gly-X-Y sequences that fold into a triple-helical structure. Persikov et al. investigated the role of guest amino acid residues at the X and Y positions on the thermal stability of the triple helix [203]. They documented that the guest triplets Gly-X-Hyp and Gly-Pro-Y can be used to quantitate the conformational propensities of all 20 amino acids to form a triple helix at the X and Y positions in a (Gly-Pro-Hyp)8 host peptide. Persikov proposed that the triple-helical structure of CLPs is the direct result of the propensity of amino acids to adopt a polyproline II-like conformation, which is driven by the high propensity of ionizable residues in the X position for interchain hydrogen bonding and a low propensity of bulky residues in the Y position for effective solvation. Building upon this concept, many studies have deciphered the structure-function relationship of the CLP phase transition [201,202,204].
To improve the stability of the triple-helical structure of CLPs, C-terminal covalent crosslinking was performed using an interchain cystine knot derived from collagen type III [[205], [206], [207]]. Such covalent modification also includes the crosslinking of three α-chains at the C-terminus using various templates such as cis,cis-1,3,5-trimethylcyclohexane-1,3,5-tricarboxylic acid (KTA) and tris(2-aminoethyl)amine-(succinate-OH)3 (TREN) [208,209]. Comparative analysis using CD and NMR spectroscopy revealed that the flexibility of the TREN scaffold is superior to that of the KTA scaffold for the induction of triple helicity [208]. However, to avoid cumbersome covalent crosslinking, simple noncovalent crosslinking strategies have been developed based on: (i) interchain metal bridging by functionalization of the terminus of CLPs with a chelating ligand [210,211] and (ii) hydrophobic interactions using a single saturated hydrocarbon or lipid tail [[212], [213], [214]]. The thermal stability of the triple helix of lipid-modified CLPs increased as the monoalkyl chain length increased from C6 to C16 [214]. Furthermore, introducing amino acids with guest residues that have a Cγ substitution with a highly electronegative atom, such as fluorine and chlorine, in place of hydroxyproline also increased triple helix stability [[215], [216], [217], [218]]. It is important to note that the triple-helical structure restricts the sequence space available for synthetic CLPs, and until recently, was thought to be intolerant to substitution of glycine in the Gly-X-Y triplet. Multiple recent reports have shown that modification of glycine with a thioamide, nitrogen atom, or azaglycine (azGly) yielded comparable or better stability of CLPs compared to their non-substituted counterparts. Another recent approach incorporated metal-binding sites at the ends of CLPs to drive thermodynamically stable, nano-micro scale self-assemblies of various shapes [219,220]. Zheng et al. elucidated the role of surface electrostatics and hydrogen bonding on the stability of the triple helix and provided computational tools for de novo design of CLPs beyond the Gly-X-Y triplet [221]. Taken together, these structure-function studies indicate that nature may not have optimized the stability of the collagen triple helix, and that there is plenty of room for further improvement through precise design of CLPs beyond tandem arrays of the Gly-X-Y triplet sequence.
5.4.3. Single-stranded CLPs in targeting native collagens
Although the major impediment in CLP research is deciphering the triple helix conformation and supramolecular assembly formation (discussed in the next section), biomedical applications of monomeric CLPs have gained attention in recent years using denatured collagen. Degradation of the ECM is a crucial element governing the progression of tissue remodeling in many life-threatening diseases, including cancer, cardiovascular diseases, and organ fibrosis, as well as prevalent debilitating conditions, such as arthritis and intervertebral disc degeneration. During tissue remodeling, collagen molecules within the collagen fibers and networks are degraded by proteases, such as MMPs or cathepsin, and denature at body temperature. In a seminal work, Yu and coworkers demonstrated that unfolded, single-stranded CLPs with a sequence (GPO)n (where n=6–10) have a high propensity for binding denatured collagen through a “strand hybridization process”, which is similar to the binding of complementary DNA strands [222,223]. Due to the high serum stability of such collagen -hybridizing peptides [224] and their affinity for native collagen [222], they have been used to selectively stain native collagen both in vitro and in vivo by fluorescently labeled CLPs [[225], [226], [227], [228], [229]]. Fluorescently labeled CLPs can also target solid tumors through the triple helix hybridization process, as high MMP-9 activity in tumor tissue exposes denatured collagen [230]. Because homotrimeric CLPs have little driving force for collagen hybridization, CLPs must be thermally dissociated by heating at 80°C to the monomeric state before binding to a collagen substrate. To avoid the preheating step, the Yu group developed a sequence (GfO)9 by replacing the hydroxyproline with a fluoroproline (f) [231]. This new sequence cannot self-trimerize at body temperature but maintains the ability to hybridize with natural collagen chains. By appending an octa-glutamic acid residue at the N-terminus of the (GPO)9, the authors attracted vascular endothelial growth factors (VEGFs) to the collagen-binding site of the endothelial cells through charge-charge interactions, which resulted in tubulogenesis [232]. Triple helix hybridization has also been used to impart collagen-targeting properties to nanoparticles [233,234] and for sustained release of nucleic acids from collagen films and depots [235,236]. Recently, the Hubbell lab recombinantly fused a collagen-binding domain (CBD) to interleukin-12 (Fig. 12 ), a potent cytokine that stimulates the innate and adaptive immune system (CBD–IL-12). Intravenously administered CBD–IL-12 preferably accumulated in the tumor stroma and provided a sustained intratumoral level of interferon-γ, thereby eliciting superior anti-tumor effects and fewer off-target toxicities when compared to naked IL-12 [237].
Fig. 12.

CBD–IL-12 binds to collagen with high affinity without compromising functionality. (A) Schematic of the fusion sites of the von Willebrand factor A3 CBD to the mouse p35 and p40 subunits via a (GGGS)2 linker. (B) Dose–response of phosphorylated STAT4 to IL-12 and CBD–IL-12 in preactivated primary mouse CD8+ T cells. EC50, half-maximum effective concentration; MFI, mean fluorescence intensity. (C, D) Binding of CBD–IL-12 to collagen I (C) and collagen III (D) as measured by SPR. The curves represent the specific responses (in resonance units (RU)) to CBD–IL-12. (E, F) Affinity of bare IL-12 (E) or CBD–IL-12 (F) to human melanoma cryosections was imaged using fluorescence microscopy. Scale bars, 100 μm. Adapted with permission from [237].
5.4.4. Hierarchical self-assembly of CLPs and their hybrids: biomedical implications
In the past few decades, the wealth of knowledge accumulated about the structure of collagen has driven a surge in research focused on the supramolecular assembly of collagen-like peptides. In the previous section, we described how various molecular interactions can stabilize the formation of a triple helix. In this section, we delve deeper into the molecular determinants of higher-order self-assembly of CLP–based biomaterials —beyond the triple helix— and how they can be used in drug delivery. We have organized this section by the specific molecular stimuli that trigger self-assembly.
Cysteine knot in CLP self-assembly: In an early example, Raines and coworkers used two cysteine knots to covalently link three CLPs, one of which protruded out to produce a sticky end during triple helix assembly [238]. The sticky ends forced the trimers to configure themselves in a head-to-tail fashion, which resulted in a long, single collagen triple helix. Such collagen-like fibrils were much longer (400 nm) than the native type I collagen (300 nm). In a similar approach, the Koide group developed a synthetic CLP system in which three CLPs were preorganized in a staggered configuration that was locked in place by two cysteine knots [239]. CD, ultrafiltration, and laser diffraction analysis indicated that the staggered trimers formed large supramolecular architectures through intermolecular triple helix formation. However, hydrogels made of these collagen fibers did not successfully form due to concentration-dependent aggregation. To address this limitation, Yamazaki et al. appended a hydrophilic arginine residue at the cysteine knotted end of the synthetic CLPs [240]. The disulfide-linked trimers of the Gly-X-Y triplet repeats formed hydrogels by spontaneous intermolecular triple helix formation upon cooling. The thermal sol-gel transition is reversible, and the design of the peptides can tune the transition temperature. Koide's group also incorporated an integrin-binding sequence GFOGER into one CLP strand of the same knotted trimeric base unit. The supramolecular structure exhibited adhesion to human dermal fibroblasts that was comparable to natural collagens [241]. To further tune the gel properties, Ichise et al. appended multiple end-to-end disulfides through crosslinking of chemically synthesized triple-helical CLP. Rheology showed that gel stiffness is controlled by the number of cysteine residues up to three, at which point it plateaus. With the incorporation of an integrin α2β1-binding sequence, the peptide polymer showed receptor-specific cell binding in vitro. Moreover, cell signaling activity and biodegradability of such a system can be tuned by altering the content of integrin binding ligand in the peptide polymer and by varying the weight percentage of CLP [242].
π–π interaction in CLP self-assembly: The Maryanoff group introduced an aromatic-aromatic recognition motif at either the N- or C-terminus of each peptide chain of the CLP triple helix to drive the supramolecular assembly of CLPs [243,244]. The aromatic π–π interaction facilitated head-to-tail stacking of CLPs into a micrometer-sized fibrillar structure, as evidenced by CD, 1H NMR, dynamic light scattering (DLS), TEM, AFM, and computational energy dynamics. The fibrillar CLPs induced the aggregation of human blood platelets, an ability lacking in less organized CLPs, with nearly the same potency as native type I collagen [245]. Kar et al. investigated how the presence of aromatic residues on neither end, one end, or both ends of a collagen model peptide affected the kinetics of self-association. CLPs with aromatic residues at both termini self-assembled and aggregated too quickly to be monitored by turbidimetry at a concentration of 7 mg/ml [246]. Similar interactions, such as CH-π interaction between amino acids (Proline and Hydroxyproline) and aromatic residues (Phenylalanine and Tyrosine) and cation-π interaction between a positively charged N-terminal Arginine and a C-terminal Phenylalanine, were also introduced to fabricate collagen-like fibrillar structures in a head-to-tail manner [246,247].
Hydrophobic interaction in CLP self-assembly: Hydrophobic interactions also contribute to CLP-hybrid self-assembly and collagen-mimicking properties. The Field and Tirrell research groups independently synthesized CLP-hybrids by conjugating collagen peptides with single or double hydrocarbon tails. Lipidation of CLPs increased the triple helix stability, triggered self-assembly of the CLP-hybrid in spherical micelles, induced the formation of lipid film, and promoted cell adhesion [248,249]. To induce even higher-order assembly, the Tong research group designed a series of CLPs consisting of four segments: an N-terminal palmitoyl segment, a β-sheet-forming domain, a polylysine spacer, and a bioactive CLP domain [250]. A CLP with the sequence (GPO)3GFOGER(GPO)3 and a C-terminal C16 hydrocarbon tail self-assembled into micrometer-long fibers with a 16 nm diameter in aqueous solution. Self-assembled peptide nanofibers made of triple-helical CLP constructs with bioactive GFOGER sequences recognized and adhered to several integrin receptors and promoted cell spreading, thereby mimicking native collagen [250].
Electrostatic interaction in CLP self-assembly: The Conticello and Chaikof research groups first presented evidence of a self-assembled, fibrous, collagen-like structure with a well-defined D-periodicity using lateral electrostatic interaction through a sticky-ended nucleation strategy, reminiscent of that commonly used in the assembly of DNA [251]. They used a synthetic peptide sequence (PRG)4(POG)4(EOG)4 consisting of three distinct segments: a central tetrameric POG sequence flanked by an arginine-containing positively charged domain at the N-terminus and a glutamic acid-containing negatively charged domain at the C-terminus (Fig. 13 ). TEM suggested that in the absence of thermal annealing, non-banded fibers are formed. In contrast, fiber growth was observed within several hours after thermal annealing. A nucleation-growth mechanism was followed by the self-association of multiple triple helices, which was driven by electrostatic interactions between oppositely charged terminal residues and hydrophobic interactions between central domains from neighboring triple helices. Building upon this concept, O'Leary et al. proposed a new sequence (PKG)4(POG)4(DOG)4 by replacing arginine and glutamic acid with lysine and aspartic acid, respectively [252]. They argued that arginine has a higher propensity to hydrogen bond with a carbonyl group of a neighboring strand compared to lysine, which inhibits its ability to form a salt bridge with the carboxylate group of the glutamic acid and thus forms homogenous collagen mimetic nanofibers. The modified peptide sequence formed both homogeneous fibers and hydrogels, and the hydrogel was degraded by collagenase at a rate similar to that for natural collagen. Raines's group adapted this approach to generate extended fibers a few microns in length with a highly symmetrical and uniform “tesselated” geometry that was dictated by the precise placement of lysine-aspartate salt bridges in the staggered triple helix building blocks. The extended geometry of this construct maximizes the surface area available to support the growth and maturation of cells [253]. Notably, the banding periodicity of these peptide fibers (18 nm) is significantly longer than the length of the peptide itself (10 nm) [251]. This elongation suggests that the units driving self-assembly are not the individual triple-helical peptides but oligomers of multiple triple helices that are preorganized such that their assembly into mature fibers leads to periodic packing and charge distribution, as visualized by TEM. This contrasts with the native type I collagen fiber, in which staggered assembly of individual collagen molecules is responsible for the periodic banding pattern and suggests that true mimicry of the natural collagen assembly process has yet to be achieved.
Fig. 13.

(A) Amino acid sequence of synthetic collagen-like peptide CPII. Distinct domain structure of collagen triads is shown. (B) Proposed triple-helical wheel diagram of CPII. (C) Side view of higher-order assembly of a CPII homotrimer illustrating the proposed interhelical electrostatic interactions to yield triple-helical protomers. (D) Schematic representation of CPII fibrillogenesis. Adapted with permission from [251].
pH-responsive domains and metal ions have also been used to induce electrostatically driven self-assembly of CLPs. Chmielewski et al. developed a new CLP sequence by replacing the hydroxyproline of the parent (POG)7 with an anionic carboxylate containing a derivative of hydroxyproline (O-propionate hydroxyproline, PE) that shows pH-sensitive folding [254]. Melting and folding experiments using CD indicated that introducing 1–3 units of the unnatural amino acid PE into the backbone had little effect on triple helix stability at acidic and neutral pH, although the rate of folding into the triple helical structures were different. They also observed that a peptide in which all seven Hydroxyproline residues were substituted with PE underwent pH-triggered folding with an even lower transition temperature than the parent CLPs [254]. Research group led by Chmielewski and Hsu also placed metal-binding ligands at various points along the CLP backbone for chelation-controlled higher-order assembly. They appended histidine or iminodiacetic acid to the termini of the (POG)n backbone and upon adding divalent metal ions, such as Ni2+, Zn2+, and Cu2+, the CLPs co-assembled into petal-like microstructures with a periodic banding at the nanometer scale [255]. Subsequent work from the Chmielewski group demonstrated that a diverse range of distinct structures, including microflorettes, stacked sheet microsaddles, and fiber-like meshes, could be made by varying the length of the (POG)n core of the peptides and the metal ion concentration. [256]. As demonstrated by Hsu and coworkers, a similar metal-promoted supramolecular assembly can also be obtained by introducing histidine residues at both termini as well as into a CLP backbone lacking hydroxyproline. A variety of higher-order structures were obtained, ranging from the nano- to micro-scale [219]. Metal-induced self-assembly is advantageous over lateral electrostatic interaction because diverse supramolecular structures can be obtained under mild physiological conditions. A repertoire of unsatisfied metal-ligand interactions are also available in these self-assembled structures that enable incorporation of functional peptide sequences [[257], [258], [259], [260]] for biomedical applications, including 3D culture of human endothelial cells, protein purification, and fluorescence tagging.
Temperature -triggered self-assembly of CLPs: Kiick and co-workers recently reported the temperature -triggered self-assembly of CLP-hybrid materials [[261], [262], [263], [264]]. They appended a synthetic polymer, poly(diethylene glycol methyl ether methacrylate) (PDEGMEMA) or human tropoelastin -derived elastin-like polypeptide (ELP, to be discussed in a later section) with CLPs. Both PDEGMEMA and ELPs are thermally responsive and exhibit lower LCST-phase behavior. Above a transition temperature (Tt), they undergo reversible phase transition from a soluble phase to an insoluble coacervate, which drives the self-assembly of the CLP-hybrid into vesicular- or platelet -like structures. Conversely, triple helix formation of the CLP chains drives the formation of a bilayer of the supramolecular assembly (Fig. 14 ) [265,266]. It is important to note that the Tt of the short ELP sequence is drastically reduced in the CLP-ELP hybrid due to formation of a CLP-triple helix that increases the local concentration of the ELP. Above the melting temperature of the CLPs, these nanostructures disassemble into unimers. Moreover, the CLP-ELP vesicle can target type II collagen, which is overexpressed in various diseases, including cancer, through triple helix hybridization. These vesicles exhibited sustained release of an encapsulated fluorophore over a period of three weeks, and complete release was triggered by destroying the CLP -helical structure with heat [267].
Fig. 14.

Chemical conjugation of ELP and CLP using a Cu-mediated click reaction. (B) Proposed assembly and disassembly behavior of the CLP-ELP vesicle. (C) Conjugation and two-step assembly mechanism of the CLP-PDEGMEMA conjugate. Adapted with permission from [265,266].
5.5. Gelatin
Gelatin is a well-known, biodegradable, biocompatible, and nonimmunogenic biomaterial obtained either by partial acid or alkaline hydrolysis or by thermal or enzymatic degradation of type 1 collagen [268]. Gelatin is composed primarily (85–92%) of proteins, with minerals and water constituting the rest. Gelatin is used in a wide range of food, cosmetic, and pharmaceutical applications [268,269]. The triple helix conformation of gelatin endows it with high thermal stability in solution [270]. The use of gelatin has gained popularity for the following reasons: (i) gelatin is readily available and inexpensive; (ii) gelatin is highly soluble and easy to use; (iii) gelatin's structure is highly similar to that of collagen and contains peptide binding motifs for cell attachment; (iv) gelatin is biocompatible, biodegradable, and does not induce antigenicity or toxicity in cells; (v) the physicochemical properties of gelatin can be modulated by external stimuli for different applications; and (vi) gelatin can be co-fabricated with various other natural and synthetic polymers to further modulate its properties.
5.5.1. Structure and function of gelatin
At the sequence level, gelatin molecules contain repeating (Gly-X-Pro)n triplets, where X represents the amino acid (usually lysine, arginine, methionine, or valine), that is responsible for the triple helical structure of gelatin. Gelatin is a polyampholyte that is ~13% positively charged (lysine and arginine), ~12% negatively charged (glutamic and aspartic acid), and ~11% hydrophobic (leucine, isoleucine, methionine and valine) [271]. Another important trait shared by collagen and gelatin is the low frequency of aromatic amino acids —tryptophan, tyrosine, and phenylalanine— in their primary sequence. The low frequency of these amino acids is the primary reason for the low antigenicity and toxicity of both gelatin and native collagen [272]. Commercially, two different types of natural gelatin —cationic type A & anionic type B— are available and differ based on the details of the manufacturing process —collagen hydrolysis — used to obtain them [273]. Gelatin A is obtained from porcine skin following acid pre-treatment, which minimally hydrolyses the amide groups of glutamine and asparagine and results in a macromolecule with a pI ranging from 7 to 9. Conversely, gelatin B is extracted from ossein —cut bovine hide— and is pre-treated at alkaline conditions, which leads to hydrolysis of asparagine and glutamine to aspartate and glutamate, respectively, thus resulting in a lower pI of 4–6. Recombinant gelatin, which can serve as a substitute for the natural protein, is also commercially available [274,275]. However, unlike collagen, gelatin suffers from mechanical instability and has high rate of degradation. It is also highly susceptible to several proteases. Gelatin nanoparticles (GNPs) prepared without crosslinking were unstable and formed aggregates [271,273]. For these reasons, crosslinking of gelatin-based materials is required to give it mechanical stability, shape, and enhanced circulation time in vivo [8,271,276]. Crosslinking of gelatin can improve its thermal and mechanical stability, as well as lower its degradation in vivo. Crosslinking, which modifies cleavage sites of gelatin molecules, inhibits enzyme-substrate interactions and hence increases the resistance to degradation [277]. Various crosslinking agents, including aldehydes, genipin, carbodiimide/N-hydroxysuccinimide, and enzymes, have been explored to covalently crosslink gelatin [268,271]. The crosslinking density, defined as bloom number, of the gelatin biomaterials can be altered by the crosslinking reaction conditions and/or increasing the concentration of crosslinker, thus regulating its physicochemical and biomedical properties. In addition, the mechanical properties of gelatin can also be tuned by temperature and pH. At a sufficiently high concentration, gelatin forms a semisolid hydrogel at lower temperatures. As the gelatin solution cools to below 30°C, a random coil to triple helix transition, promoted by stable hydrogen bond formation, occurs [278]. Changes in pH affect the melting temperature of gelatin and hence the propensity for gel formation. Moreover, changes in pH modify ionic interactions between gelatin monomers and consequently affect hydrogen bond-mediated crosslinking [279]. Physical crosslinking is also possible and can reversibly modulate gel strength and has the advantage that it does not need enzymes or chemical reagents to effect crosslinking [280]. Various methods such as desolvation, coacervation, emulsification, nanoprecipitation, and layer-by-layer coating have also been used to fabricate gelatin-based systems for drug and gene delivery [268,271].
5.5.2. Gelatin nanoparticles
Gelatin nanoparticles (GNPs) range from 200 to 400 nm in size [281]. Colloidal stability, drug release, drug encapsulation, tissue distribution, and cellular uptake are strongly dictated by the charge and size of GNPs. The effects of various parameters, such as temperature, pH, cosolvents, degree of crosslinking, and type of gelatin, on the physicochemical properties of GNPs have been investigated by several groups [[282], [283], [284]]. A temperature of 40°C was found to be optimal to attain a narrow size distribution of GNPs because at q high temperature (50°C), gelatin forms a random coil, which destroys the self-assembly and the nanoparticle while at room temperature, gelatin forms a highly viscous semi-solid. In contrast, at 40°C, a controlled uncoiling of the triple-helical structure results in monodisperse GNPs. The size and dispersity of GNPs is highly dependent on solution pH, and pH 3 and pH 11 were found to be optimal for nanoparticle formation of cationic type A and anionic type B gelatins, respectively. Increasing the degree of crosslinking results in greater polydispersity and a reduction in particle size.
GNPs and their hybrids have been explored to encapsulate both hydrophobic and hydrophilic bioactive molecules including antimalarial drugs [285], such as chloroquine phosphate; anti-cancer drugs [271,286], such as paclitaxel, DOX, cisplatin, curcumin, and methotrexate; growth factors [287], such as osteogenic bone morphogenetic protein-2 (BMP-2), and angiogenic basic fibroblast growth factor (bFGF); anti-inflammatory drugs [288], such as ibuprofen; photo-responsive dyes [289,290]; anti-HIV drugs [291] such as didanosine; and therapeutic nucleic acids [271,292]. In all cases, an on-demand release profile was achieved by controlling the degree of crosslinking, pH, temperature, and fabrication method and thus the physicochemical properties of the GNPs. For example, Amjadi et al. designed a pH-responsive nanocarrier by decorating the GNPs with (methoxy poly (ethylene glycol)-poly ((2-dimethylamino) ethyl methacrylate-co-itaconic acid) (PGNPs) (Fig. 15A) [286]. The nanoparticles (162 nm diameter) were encapsulated with DOX and betanin with high efficiency (loading capacities of 20.5% and 16.25%, respectively), and demonstrated pH- and temperature -responsive drug release, and synergistically inhibited the growth of MCF-7 breast cancer cell lines. In another example, Rose Bengal -conjugated gelatin nanoparticles (RB-GNPs) were developed for antimicrobial photodynamic therapy (Fig. 15B) [290]. RB-GNPs generated singlet oxygen that damaged the microbial cell membrane, which demonstrated their potential for treating multi-drug-resistant microbial infections.
Fig. 15.

(A) Three-step preparation of gelatin nanoparticles coated with pH-sensitive polymer. (B) Schematic presentation of the synthesis of RB-GNP. Adapted with permission from [286] (A) and [290] (B).
5.5.3. Gelatin microparticles
Gelatin microparticles (GMPs) have been extensively investigated for many biomedical applications. Solorio et al. engineered self-assembled, gelatin microsphere-incorporated human mesenchymal stem cell (hMSC) sheets [293]. The microspheres were loaded with growth factor TGF-β1, distributed within the hMSC sheets, and released chondrogenic growth factor to the interior of the sheets, thus enabling spatially homogenous differentiation of the hMSCs. The differentiation rate could be tailored by adjusting the microsphere crosslinking levels, which controls the release kinetics of the growth factor. This system not only produces hMSC sheets with superior mechanical properties but also decreases the pre-implantation culture time. In addition, the hMSCs are continuously exposed to growth factor by microparticles, which prevents loss of the chondrogenic phenotype in vivo. Similar controlled release of growth factor and proteins from GMPs to stimulate tissue regeneration have been achieved for vascular endothelial growth factor (VEGF) [294], bone morphogenetic protein-2 (BMP-2) [295], basic fibroblast growth factor (bFGF) [296], and matrix metalloproteinases (MMPs) [297]. In all cases, the release kinetics were dependent on the degree of crosslinking of GMPs.
It is well known that the use of most hydrophobic drugs is limited by their poor aqueous solubility, short circulation time, low bioavailability, and narrow therapeutic window. GMPs have been used to address some of these challenges. For instance, the antibacterial activity of curcumin —a hydrophobic polyphenol derived from turmeric with a wide range of biological properties— is negligible at concentrations up to 100 mg/ml. However, curcumin encapsulated in GMPs at 4 mg/ml reduced the microbial population by 2.08, 1.67, 2.70, and 2.18 log counts (CFU/ml) for L. monocytogenes, S. enterica, S. aureus, and E. coli, respectively [298]. GMPs encapsulated with noscapine, an anticancer drug that targets human NSCLC, demonstrated first order release kinetics and increased the plasma half-life of the drug by ~10-fold with an accumulation of ~50% drug in the lungs [299]. Kim et al. used a microfluidic approach to fabricate monodisperse GMPs with a microshell structure for embolization, a minimally invasive, nonsurgical procedure that deliberately blocks a blood vessel [300]. Uniform gelatin emulsion precursors were fabricated using the microfluidic technique and consecutively crosslinked by inbound diffusion of glutaraldehyde from the oil phase to the suspending droplets of gelatin precursor. The authors also performed a model micromechanics study in an artificial blood vessel, which revealed that the microshell structure results intorapid degradation of the GMPs within a narrow time period leading to burst release of the drug under chemoembolic conditions.
5.5.4. Gelatin fibers
Gelatin fibers are used for various biomedical applications such as tissue-engineering and wound-healing, due to their morphological resemblance to the structure of the native ECM. Gelatin fibers can be fabricated by several methods, such as melt spinning, wet spinning, gel spinning, electrospinning, centrifugal spinning, and solution/melt blow spinning. Multiple processing parameters, including solution properties, humidity, and temperature, can influence the morphology of nanofibers and release rate of the entrapped drug. For instance, nanofibers fabricated by electrospinning of poly (D, l-lactide-co-glycolide) (PLGA) and gelatin were loaded with fenbufen. The hydrophilicity of the fibers was enhanced by increasing the gelatin content, which in turn increased the release rate of fenbufen. In contrast, the addition of a crosslinker (glutaraldehyde vapor) reduced the drug release rate [41]. Vitamins A and E incorporated in gelatin nanofibers via electrospinning promoted the proliferation of fibroblasts, increased the expression of collagen-specific genes, and supported wound healing [301]. Another research group demonstrated that genipin-crosslinked gelatin fibers loaded with human VEGF sustained viability, promoted endothelial differentiation, attracted human mesenchymal stem cells (hMSCs) and stimulated early angiogenesis [302].
5.5.5. Gelatin hydrogel and adhesives
Chemically modified gelatin has been extensively used as hydrogel because of its tunable mechanical and biochemical properties. Gelatin-methacryloyl (GelMA), a synthetic, photosensitive crosslinkable derivative of gelatin, is widely used as a precursor to fabricate gelatin hydrogels and is commercially available as a bio-ink for 3D printing. Various crosslinking methods, such as ultraviolet stereolithography, gamma irradiation, and two-photon polymerization, have been used to fabricate gelatin-based hydrogel and scaffolds [303]. Light-activated crosslinking of methacrylated gelatin in air or in an aqueous solution resulted in an injectable hydrogel that supported hMSC growth and TGF-b3-induced chondrogenesis [304]. Mazaki et al. developed furfurylamine-conjugated gelatin (gelatin-FA) that was rapidly cross-linked by visible light with Rose Bengal [305]. Hybrid gelatin hydrogels can also be made from various synthetic polymers, including PLGA, poly (hydroxyethyl methacrylate), and polyvinyl alcohol, or natural polymers, such as chitosan and alginate. Composite hydrogels consisting of gelatin, polyvinyl alcohol, and the anticancer drug cisplatin were synthesized as a controlled-release drug delivery system. In vivo, inhibition of tumor growth was similar for hydrogels containing a low dose of cisplatin and conventional intraperitoneal administration of high doses of free cisplatin [306]. Kinsella and colleague fabricated various alginate-gelatin bio-printable composite hydrogels that resemble the microscopic architecture of native tumor stroma [[307], [308], [309]]. 3D-printed hydrogels embedded with breast cancer cells, and fibroblasts promoted the self-assembly of breast cancer cells into multicellular tumor spheroids (MCTS), which were viable for more than 30 days [307].
Gelatin -based tissue bioadhesives, fabricated alone or with other synthetic and natural polymers, have also been used in drug delivery. Composite tissue adhesives, developed by carbodiimide -mediated crosslinking of gelatin and alginate, were used to entrap the anti-inflammatory drugs bupivacaine and ibuprofen, and the resulting adhesive was used for local pain management [310,311]. Hydrophilicity of the adhesive and its swelling properties control the release of drugs from this matrix. The incorporation of a drug —bupivacaine— improved the bonding strength of the adhesive, whereas incorporation of ibuprofen adversely affected the bonding strength. This is due to the inert nature of bupivacaine and the reinforcing effect of its fibrous crystals. In contrast, ibuprofen reacts with the crosslinking reagent —carbodiimide— and decreases the crosslinking density and hence bonding strength of the adhesive networks [311]. To address this issue, N-hydroxysuccinimide can be added during crosslinking of gelatin and alginate to decrease the carbodiimide content without compromising the bonding strength. The resulting patch is less cytotoxic (due to low carbodiimide content) and can be used for controlled release of various antibiotics such as clindamycin, vancomycin, and ofloxacin [310].
5.6. Elastin
Like collagen, elastin is a key protein found in the ECM and connective tissues. It is abundant in organs that require elasticity and resilience for their function, including skin, ligaments, lungs, and blood vessels [312]. Its inherently disordered structure provides elastin with unique properties that impact its recombinant production and purification, drug-loading capacity, and in vivo behavior.
5.6.1. Structure and properties of elastin
The soluble precursor to elastin, tropoelastin, is composed of alternating hydrophobic and hydrophilic domains that undergo enzymatic crosslinking via their lysine residues in the extracellular space. The resulting elastin fibrils are durable and insoluble. Although these characteristics are critical for its function in biological tissue, they were a hindrance during early efforts to manipulate elastin as a biomaterial [313]. Consequently, soluble and recombinant forms of elastin were explored. The most widely studied elastin-based materials used for drug delivery are elastin-like polypeptides (ELPs) [314]. ELPs are biopolymers with the Val-Pro-Gly-X-Gly pentapeptide motif, in which X represents any amino acid besides proline [315].
Perhaps the most interesting property of ELPs is their thermal responsiveness, as they demonstrate LCST phase behavior. In an aqueous solution, they are soluble below a characteristic transition temperature (Tt). Above their Tt, ELPs reversibly phase transition and forms an insoluble, polymer-rich phase. This behavior is thermodynamically driven by the favorable entropy of demixing above the Tt (Fig. 16A & B) [316]. Spontaneous mixing occurs when the Gibbs free energy change is negative. For an ELP to be solubilized, water molecules must be ordered along the polypeptide chain. This interaction results in a negative entropy term. A small temperature term compensates for this hit in entropy, which keeps the Gibbs free energy change negative and allows the ELP to stay in solution. However, as the temperature term increases, ELP-water mixing becomes more energetically unfavorable. The entropy term becomes dominant, causing the ELP to aggregate into a “coacervate” phase. The temperature at which this phenomenon occurs is dependent on multiple features of the ELP and the solution environment [317,318] The ELP's molecular weight and concentration in the aqueous solution are inversely related to its Tt, which is also impacted by the guest residue. High concentrations of hydrophobic residues, such as lysine or valine, depress the Tt and high concentrations of hydrophilic residues, such as alanine or glycine, increase the Tt [319]. ELPs retain their LCST behavior when recombinantly fused to a drug, targeting moiety, or other molecule, but the Tt can be similarly affected by the surface hydrophobicity and charge of its fusion partner [320]. The Tt of an ELP solution can be easily quantified by optical turbidity measurements of an ELP solution at 350 nm (OD350) as a function of solution temperature. As the temperature is increased, the ELP will transition from an optically transparent solution to an opaque mixture within a narrow, 1–2°C temperature window. The Tt can be graphically determined at the inflection point of an OD350 vs temperature plot, which is typically a sigmoidal curve (Fig. 16C) [321]. Because the Tt is concentration dependent, it should be analyzed over a range of application -relevant concentrations.
Fig. 16.

Lower critical solution temperature behavior of elastin-like polypeptides (ELPs). (A, B) At temperatures below the transition temperature (Tt), water molecules order themselves along the ELP chain such that the ELP remains soluble and appears optically clear. At temperatures above the transition temperature, water is expelled from the polymer, and the polypeptide chain aggregates, leading to a turbid suspension. C) The Tt can be determined from the temperature dependent optical turbidity and increases with guest residue hydrophobicity, molecular weight, and concentration. Adapted with permission from [316] (A & B) and [321].
The LCST behavior of ELPs can also be used in a chromatography-free purification process termed inverse transition cycling (ITC). Initially developed by Meyer and Chilkoti, this procedure cycles cell lysate through a series of hot and cold centrifugation steps [322]. After insoluble cell debris is removed from the lysate via centrifugation, the ELP-containing supernatant is heated above the Tt of the ELP. Kosmotropic salts, which interact more favorably with the water along the ELP chain, may be added to facilitate the phase transition and promote aggregation. The solution is centrifuged to pellet the ELP while soluble contaminants remain in the supernatant. The ELP pellet is then dissolved in cold buffer and centrifuged below the Tt, which causes insoluble contaminants to precipitate while the ELP remains in solution. This process is rapid and inexpensive, avoids the use of harsh chemicals, and can be scaled up [322]. ELPs have hence been used as a purification tag for other proteins [323]. After ITC, the ELP can be cleaved from the protein of interest using a proteolytic cleavage site or self-cleaving intein that is incorporated between the ELP and protein [324,325].
5.6.2. ELP unimers
Though small molecule drugs and peptides demonstrate potency and —in the case of peptides— specificity, their efficacy is frequently hindered by their rapid clearance from circulation or poor solubility (Kalepu & Nekkanti, 2015), necessitating frequent dosing [326,327]. [328] Scientists have addressed these limitations by conjugating macromolecular carriers to these therapeutics to improve their circulation time, target-site accumulation, and solubility [327,329]. Soluble ELP unimers have been recombinantly fused to therapeutic peptides and chemically conjugated to small molecule drugs for these purposes [[330], [331], [332]]. ELP fusions benefit from the large molecular weight of the ELP, which extends the circulation time of the fusion partner, and enhance the solubility of the drug, and improve their biodistribution, and pharmacokinetic profiles. Compared to synthetic drug carriers, ELPs benefit from monodispersity and a lack of toxicity [315].
Despite the advantages of employing ELPs as soluble macromolecular carriers, they lack the stealth properties provided by some hydrophilic polymers, such as poly(ethylene glycol) and zwitterionic polymers [333]. These “stealth” polymers create a hydration sphere around the payload and polymers, which improves the pharmacokinetic profile of the conjugate by protecting it from opsonization and premature clearance from circulation [334]. Recently, Banskota and coworkers modified the canonical ELP sequence to impart stealth behavior to biopolymers [335]. They designed zwitterionic polypeptides (ZIPPs), which are composed of repeat units of the Val-Pro-X1-X2-Gly motif, where X1 and X2 are positively and negatively charged amino acids, respectively (Fig. 17A). Like ELPs, ZIPPs can be recombinantly expressed in E. coli, and exhibit the favorable properties of ELPs including monodispersity, ability to recombinantly be fused to a therapeutic peptide, and sequence-level control of their properties. Additionally, ZIPPs can be purified without chromatography by inverse transition cycling, similar to ELPs. ZIPPs provide greater solubility to their payload than ELPs as a result of their charged residues [335]. Most importantly, ZIPPs had a 3-fold longer half-life and 2–3-fold greater bioavailability compared to an uncharged ELP control. Consequently, when GLP-1 was fused to either a ZIPP or a MW-matched uncharged ELP control, the ZIPP achieved 1.5-fold longer blood glucose control, which demonstrates that the stealth behavior of ZIPPs can increase the efficacy of their therapeutic cargo (Fig. 17B) [335].
Fig. 17.

Design and efficacy of zwitterionic polypeptides (ZIPPs). (A) ZIPPs are homopolymers comprised of repeating monomers of the pentapeptide Val-Pro-X1-X2-Gly, where X1 and X2 are cationic and anionic residues, respectively. (B) ZIPPs have a higher plasma concentration than length- and MW-matched ELPs after intravenous injection (C, D) A ZIPP fusion to the antidiabetic peptide GLP1 resulted in bettr blood-glucose control than a MW matched ELP. Adapted with permission from [335].
5.6.3. ELP nanoparticles
Owing to their ease of production and thermal phase behavior, ELPs are useful materials for design of self-assembling nanostructures and nanoparticles to deliver a variety of therapeutic agents. ELP nanoparticle self-assembly is driven by hydrophobic interactions. Depending on the design, the hydrophobic component might be either an ELP segment or its conjugated hydrophobic cargo. ELP amphiphiles, which contain two connected ELP blocks with different hydrophobicities and hence Tt, form micelles at temperatures between the Tt of each block. At an intermediate temperature, the hydrophilic block is hydrated and favorably interacts with water, allowing it to form the corona and interact with the environment. Meanwhile, the hydrophobic block dehydrates and aggregates to form the core. The temperature at which this occurs is termed the critical micellization temperature (CMT) and can be tuned by changing the guest residue in the ELP sequence. Similarly, the micelle diameter and aggregation number can be tuned by adjusting the molecular weight of the ELP and the ratio of the length of the hydrophobic to hydrophilic ELP blocks. Hydrophobic drugs can be sequestered in the micelle core, while hydrophilic targeting moieties or ligands can be displayed on the corona.
The first examples of diblock ELP nanoparticles were developed by Conticello and coworkers. The hydrophilic component of the diblock that they synthesized consisted of 14 repeats of [VPGEG-(IPGAG)4], wherein glutamic acid and alanine were the guest residues. The hydrophobic component of the diblock consisted of 16 repeats of [VPGFG-(IPGVG)4], wherein phenylalanine and valine were the guest residues. Due to the presence of ionizable residues in the sequence, the size and shape of the self-assembled nanoparticles were pH-sensitive. Above the CMT, these ELP fusions self-assembled into spherical and cylindrical morphologies [336,337]. Conticello et al. later designed triblock ELPs, which were made of a hydrophilic block between hydrophobic end blocks [338]. The hydrophobic blocks phase transitioned and aggregated to act as “crosslinks” in an elastomeric network.
Subsequently Dreher and coworkers systematically explored the design parameters that controlled ELP nanoparticle self-assembly by creating a library of ten diblock ELPs [339]. They discovered that the temperature at which unimers assemble into micelles depends upon the length of the hydrophobic domain. In contrast, micelle diameter is controlled by the length of the copolymer and the ratio of hydrophobic to hydrophilic blocks. Dreher et al. also incorporated a functional ligand – the tumor-homing NGR peptide sequence at the terminus of the hydrophilic segment of a diblock ELP. These micelles were readily internalized by cells that recognized the displayed ligand, which demonstrates that the ELP diblock design can be engineered for targeted drug delivery [339].
Several models have contributed to our understanding of diblock ELP self-assembly. Janib and coworkers developed a mathematical model to predict the phase behavior of diblock ELPs based on the Tt values of each ELP domain. They found that although the CMT was dependent on the hydrophobic domain, the bulk transition temperature was dependent on the hydrophilic block [340]. A second, theoretical model developed by Hassouneh et al. used polymer physics to study micelle assembly. Upon examination of six diblock ELPs, they found that these copolymers form “weak” micelles with dense cores and unstretched coronas, which is unique for diblock polymer micelles [341].
The initial studies on diblock ELP micelle targeting were expanded by the development of dynamic affinity modulation, a strategy in which local hyperthermia induces micelle formation to increase the avidity of a low-affinity ligand that is fused to the N-terminal end of the hydrophilic segment of diblock ELP for site-specific accumulation and targeting. Simnick and coworkers first used this strategy in a proof -of -concept study in which a low-affinity ligand specific to αvβ3 integrin was recombinantly fused to the N-terminus of an ELP [342]. Cysteine residues were incorporated at the C-terminus for drug conjugation. At temperatures below the CMT of the ELP, the fusion protein existed as a soluble unimer and the low-affinity ligand interacted weakly with the αvβ3 integrin. In the presence of local hyperthermia, the ELP underwent temperature -triggered self-assembly into spherical micelles, thus increasing the valency and avidity of the ligand such that it could efficiently bind to and be taken up by cells [342]. This strategy was then modulated to achieve more specific cell internalization of cell-penetrating peptides (CPPs) [343]. CPPs are arginine -rich sequences that promote uptake by electrostatically interacting with the cell membrane of multiple cell types. CPPs have been conjugated to nanoparticle surfaces to improve their internalization but lack specificity. MacEwan and Chilkoti addressed this issue by incorporating five arginine residues onto a diblock ELP. They found that below the CMT, the number of arginine residues was below the threshold of six necessary to induce cell uptake [344]. Local hyperthermia drove the formation of micelles that displayed a high valency of arginine on their surfaces, resulting in 8-fold increase in cell internalization [343]. The design of these nanoparticles and their thermal “switch” allow for control over the site of action of ELP nanoparticles, which can reduce off-target drug delivery and mitigate systemic toxicity. Furthermore, adding these targeting moieties did not disturb micelle formation. Hassouneh and coworkers showed that when proteins ~10 kDa in size are incorporated at the end of the hydrophilic block, the diblocks still self-assembled [345]. Diblock ELPs have also been designed for applications beyond cancer, including specific targeting to liver cells [346] and lacrimal gland cells [347].
To improve tumor-specific drug delivery of diblock ELP micelles, Callahan and coworkers engineered a diblock ELP that disassembled in the acidic tumor microenvironment [348]. Histidine, which has a pKa near physiological pH, was incorporated into the hydrophobic block. These micelles assembled at 37°C, but upon exposure to the low PH conditions mimicking the tumor microenvironment (pH ~6), the histidine became ionized. Consequently, the Tt of the hydrophobic block increased, and it was no longer energetically favorable for ELPs to self-assemble. The micelles broke down into ELP unimers, resulting in better penetration and accumulation of the ELP in the tumor compared to pH insensitive ELP micelles. This study demonstrated that diblock ELPs can be engineered to overcome the diffusion barrier many nanocarriers face when delivering drugs to tumors [348].
MacKay and coworkers have adapted diblock ELPs nanoparticles for two-phase rapamycin release by nonspecifically entrapping the drug in the nanoparticle core as well as specifically displaying the drug on the nanoparticle corona [349,350]. This type of display was achieved by appending FK506 binding protein12 (FKBP12), the cognate protein target of rapamycin, to the hydrophilic block of the ELP fusion. The entrapped rapamycin was rapidly released upon administration, while the release of FKBP12-bound rapamycin was prolonged. The two-phase release increased the terminal t1/2 of rapamycin to 57.8 h compared to 2.2 h for nanoparticles without FKBP12-bound rapamycin [350]. The nanoparticle formulation also reduced off-target toxicity compared to the free drug. In the aggressive breast cancer model MDA-MB-468, treatment with the nanoparticle resulted in more potent reduction in tumor volume and extended survival time [350]. In a model for the chronic autoimmune disease Sjögren's syndrome, the treatment downregulated inflammation in the endocrine gland and reduced lymphocytic infiltration into the lacrimal gland [349].
Other ELP nanoparticle designs fuse an ELP segment with a peptide or protein to drive self-assembly. In an early example of this method, McDaniel and coworkers incorporated repeats of the sequence (XGy)z onto the ELP C-terminus, where X is a hydrophobic residue [351]. Relative to the ELP, the (XGy)z domain is only a few percent by mass in the diblock. Despite the low mass fraction of the second segment, it was surprising and notable that these highly asymmetric amphiphiles self-assembled into cylindrical micelles with lengths that could be controlled by altering the identity of X or the value of y— the number of glycine spacers [351]. MacKay et al. further explored how small proteins and peptides impact nanoparticle assembly. They discovered that fusing an scFv –a single chain antibody—to a hydrophilic ELP block drives the fusion to self-assembly into a worm-like micelle, with the ELP forming the corona [352]. Despite being entrapped in the core, the scFv remained active due to the low packing density of the corona. These nanoworms outperformed their antibody equivalent in an in vivo xenograft model of lymphoma [352]. Another study by Mckay and coworkers was driven by the amphipathic nature of L4F, whose four phenylalanine residues orient face-to-face to create vesicles with 8 nm thick lamellae and a 49 nm radius [353]. L4F maintained its anti-inflammatory properties despite being in the vesicle lamellae. This study demonstrates how amphipathic peptides may be used to drive ELP vesicle formation, which can be leveraged to encapsulate soluble therapeutics into the aqueous vesicle interior.
Hydrophobic drug conjugation can also trigger ELP self-assembly into nanoparticles, masking the drug inside the core, thus increasing the aqueous solubility of the drug and protecting it from premature release and degradation. This results in improved drug half-life, biodistribution, tumor accumulation, and efficacy while reducing off-target toxicity. An early example of this strategy was developed by Chilkoti and coworkers. In their bottom-up nanofabrication method, termed attachment directed assembly of micelles (ADAM), a hydrophobic (CGG)8 domain was conjugated to the C terminus of a hydrophobic ELP [354] to create a “chimeric polypeptide” (CP). The cysteine-rich domain of the CP enabled the attachment of up to eight maleimide -functionalized small molecules (Fig. 18 ). When the hydrophobicity of the small molecule exceeds a threshold (log ≥ ~1.5), it provides the conjugate sufficient amphiphilicity to drive self-assembly [354]. This platform was adapted for drug delivery by chemical conjugation with DOX to create CP-DOX nanoparticles, which self-assemble at physiological pH at concentrations above 3 μM. DOX is rapidly released upon exposure to an acidic pH (5), which mimics the conditions of late endosomes and lysosomes. A single injection of CP-DOX had 4-fold higher maximum tolerated dose and reduced tumor size nearly 13-fold compared to free drug, indicating that this nanoparticle formulation can be harnessed to enhance potency while reducing off-target effects such as cardiotoxicity. ADAM has also been leveraged to deliver the hydrophobic chemotherapeutics paclitaxel [355] and niclosamide [356].
Fig. 18.

Design and efficacy of CP-DOX nanoparticles. (A) ELPs are conjugated to DOX at Cys residues via a heterobifunctional linker. (B) CP nanoparticles self-assemble to entrap hydrophobic DOX in the core and display the hydrophilic polypeptide on the corona. (C) DOX release from CP-DOX is pH-dependent. (D, E) CP-DOX formulation promotes DOX accumulation in the tumor while reducing its presence in heart tissue. (F) A single injection of CP-DOX outperforms free DOX in reducing tumor volume. Adapted with permission from [354].
While the original ADAM strategy was successful at enhancing the efficacy and safety of hydrophobic chemotherapeutics, many cancer drugs are hydrophilic and cannot be encapsulated by this strategy. To address this, the Chilkoti and coworkers developed a new strategy by the design of an asymmetric triblock polypeptides [357]. ATBPs consist of three segments—a hydrophilic ELP segment that is fused to a hydrophobic ELPs segment that in turn is fused to a drug attachment (GGC)8 segment. The ATBP is deigned to retain its temperature triggered amphiphilicity even upon conjugation of hydrophilic small molecules with a logD ≤1.0 that the drug-conjugated ATBP self-assembles into rod-shaped micelles above its CMT, which is designed to be below body temperature. They showed that conjugation of hydrophilic drugs, such as gemcitabine, to the cysteine residues of the ATBP did not abrogate the formation of micelles, and these drug-loaded ATBP micelles had greater efficacy in an aggressive in vivo model of colon cancer compared to free drug [357].
While cysteine-maleimide covalent conjugation can incorporate a structurally diverse set of drugs, molecules, and imaging agents into the nanoparticle's core, this strategy relies on functionalization of a naturally occurring amino acid. These residues can be promiscuously distributed across other peptides that may be appended to the ELP such as targeting peptides or proteins, and, consequently, site-specific drug attachment for these constructs is challenging. To address this concern, Costa and coworkers inserted p-acetylphenylalanine, an unnatural amino acid, onto an ELP micelles that displayed a functional nanobody [358]. The nanobody contains a disulfide bond that is essential for its stability and functionality, so that site specific covalent conjugation of a drug by introduction of cysteine residues in a drug attachment was not possible. The p-acetylphenylalanine residue provided a biorthogonal ketone moiety to site-specifically conjugate DOX via a pH-sensitive bond without affecting the disulfide bond of the nanobody. As a result, this platform enabled site-specific drug conjugation while enabling the display of a functional nanobody on an ELP nanoparticle [358].
Fatty acids have also been incorporated by a post-translation modification into ELPs to drive nanoparticle assembly. Luginbuhl and coworkers incorporated a myristoyl group at the N-terminus of an ELP that presented a peptide substrate for N-myristoyltransferase. The enzyme was co-expressed with the ELP from a bicostronic plasmid in E. coli that grown with exogenous myristic acid added to the culture medium [359]. This led to close to 100% myristoylation of the ELP in E. coli, and the myristoylated ELP (M-ELP) formed spherical or rod-like micelles depending on the ELP chain length. Chemotherapeutic drugs could be physically loaded into the micelles via hydrophobic interactions with the fatty acid, which resulted in a greater encapsulation efficiency than that possible with diblock ELPs [359]. This strategy thus provides a useful mechanism for entrapping drugs without covalent conjugation, making it possible to deliver drugs that do not contain an active functional group for conjugation, or drugs that lose potency upon conjugation.
ELPs have been similarly combined with silk to form silk-elastin like protein (SELP) nanoparticles [71,360]. Nanoparticle self-assembly is driven by both hydrophobic interactions and hydrogen bonding between the silk blocks. The radius of the nanoparticle is controlled by altering the ELP guest residue as well as the ratio of silk blocks to ELP blocks [361]. Xia and coworkers demonstrated that these nanoparticles can be used to deliver hydrophobic small molecule drugs by loading DOX into the nanoparticles [362]. DOX was entrapped in the core by hydrophobic interactions with the silk blocks. The SELP nanoparticles were endocytosed into the cell, thus allowing drug to accumulate in the cytoplasm. The cytotoxicity of the SELP-DOX nanoparticles was 1.8-fold greater than that of free DOX [362].
5.6.4. ELP depots
ELPs provide a useful platform for sustained drug release due to their LCST behavior. Their Tt can be precisely tuned by modulating the molecular weight and guest residue of the sequence, making it possible to engineer the ELP with a Tt below physiological temperature. The ELP thus remains soluble at room temperature, enabling injection through narrow-bore syringe, but rapidly coacervates upon exposure to body temperature, forming a subcutaneous depot. Because the Tt is concentration -dependent and the boundary layer of the coacervate is diluted by interstitial flow, the depot slowly undergoes surface-to-core dissolution. As it dissolves, the therapeutic cargo is released into circulation. This sustained -release platform, coupled with the ability of ELPs to impede renal filtration, extends the half-life of small molecules and peptides to which the ELP is fused.
Prolonged -release systems are advantageous for diseases that require long-term treatment, such as cancer or chronic illnesses. Liu et al. systematically altered ELP parameters to adapt the molecule for in situ radiotherapy [363,364]. They recombinantly fused a hydrophobic ELP, comprised of 60 repeats of a Val-Pro-Gly-Val-Gly pentapeptide, to a tyrosine-rich domain that could be radiolabeled with 131I. An intratumoral injection of the 131I-ELP was retained in the tumor for more than one week. The ELP shielded the radionuclide from dehalogenase degradation and increased its tumor retention time [363]. Liu and coworkers found that doubling the molecular weight of the ELP increased tumor retention time by 14-fold; however, the effect plateaued with the Tt once the ELP surpassed 120 repeats. Similarly, increasing the injection concentration of 131I-ELP 16-fold resulted in 5-fold greater tumor retention [364]. Furthermore, 131I-ELPs with seven repeats of the hydrophobic, tyrosine-rich peptide domain self-assembled into rod-shaped micelles and formed long-lasting “seeds”. The seed-like particles exhibited potent anti-tumor efficacy, remarkable intratumoral retention, and minimal toxicity [364]. The coacervate was further stabilized by radiation-induced crosslinking of the tyrosine residues in the hydrogel, which resulted in retention of 52% and 70% of its radioactivity over 60 days in a prostate and pancreatic tumor model, respectively [365].
More recently, Wang and coworkers engineered an ELP depot for the sustained release of interferon-α, an anti-cancer protein that has been clinically hindered due to its short half-life and dose-dependent toxicity [366]. Fusion of the protein to 90 repeats of a Val-Pro-Gly-Val-Gly pentapeptide resulted in near zero-order release of IFN-α over the course of a month. Compared to free IFN-α and IFN-α fused to a soluble ELP, the depot-forming IFN-α-ELP exhibited greater antitumor efficacy in a melanoma model. The fusion was well tolerated and demonstrated little renal toxicity, key advantages over free IFN-α [366].
ELP depots have also been extensively engineered for the treatment of diabetes. Antidiabetic peptides such as insulin and glucagon-like peptide-1 (GLP-1) have short half-lives that necessitate frequent and repeated injections to achieve long-term blood glucose control. There is hence a need for sustained -release systems that prolong the efficacy of these peptides, and which reduce the need for multiple injections and improve patient compliance. Amiram et al. developed the first example of an injectable depot -forming GLP-1 by fusion of GLP-1 to and ELP that was designed to coacervate at body temperature. The depots were retained in the subcutaneous space and lowered blood glucose levels for up to 5 days [367]. Luginbuhl and coworkers expanded upon this work and optimized the ELP parameters for depot retention and GLP-1 half-life [368]. They discovered that ELPs with Tt values of approximately 30°C and molecular weights greater than 36 kDa achieved near zero-order release kinetics for GLP-1. The optimized GLP1-ELP fusion formed a depot in the subcutaneous space upon injection as a solution and provided glycemic control in three different diabetic mouse models for up to 10 days after a single injection (Fig. 19 ). In a cynomolgus monkey model, circulating levels of the ELP fusion were detected for 17–21 days post-injection. GLP-1 delivery has also been achieved by introducing protease cleavage sites between oligomeric repeats of GLP-1 on an ELP. Once these so-called “protease-operated depots” are injected subcutaneously, proteases under the skin free GLP-1 from the ELP in the depot. This system extended blood glucose control for 5 days [367].
Fig. 19.

Design and efficacy of an optimized subcutaneous depot of GLP1-ELP. A single injection of the ELP fusion maintained blood glucose control in (A) diet-induced obese mice, (B) ob/ob mice, and (C) db/db mice for up to 10 days. (D) Circulating levels of the GLP1-ELP fusion were detectable for up to 21 days in cynomolgus monkeys. (E) The enhanced pharmacokinetics of GLP1 are attributed to the increased residence time of depot-forming GLP1-ELP compared to the soluble control or GLP1 alone. Adapted with permission from [368].
The antidiabetic protein fibroblast growth factor 21 (FGF21) has also been fused to an ELP by Gilroy et al. to improve its release, half-life, and production [369]. Fusion to the ELP enhances FGF21 solubility, thus allowing it to be expressed in the bacterial cytosol rather than inclusion bodies and consequently avoiding costly, complex refolding steps during purification. The ELP-FGF21 depot was retained in the subcutaneous space for up to 4 days and achieved glycemic control in an ob/ob mouse model for up to 5 days. In a long-term study, treatment with an ELP-FGF21 fusion resulted in 40% reduction in mean serum insulin and triglyceride levels when mice received subcutaneous injections every 5 days for up to 60 days [369]. Other ELP depots have been engineered for the treatment of dry eye disease [370], infectious illnesses [371], and inflammation [372].
5.6.5. Clinical trials
Several ELP fusions have entered clinical trials with the goal of extending the half-life of their therapeutic cargo. These formulations have primarily been developed by PhaseBio, a biotechnology company that has commercialized ELP fusions for the treatment of diabetes and cardiovascular disease. One of their products, Glyerma, or PB1023, is an ELP fusion of GLP-1 for the treatment of Type 2 diabetes. It entered a Phase 2b clinical trial in 2013 (NCT01658501) to evaluate its ability to control HbA1c levels with once-weekly dosing. The rights to Glymera were acquired by ImmunoForge [373] in 2019 for further development. A second anti-diabetic formulation, PE0139, is an ELP fusion of insulin. After completing a Phase I clinical trial in 2014 (NCT01835730), it began a Phase 2a clinical trial in 2015 (NCT02581657) to further assess its safety, tolerability, and pharmacokinetics. The results of these studies have not yet been disclosed. PhaseBio also has several clinical trials ongoing for the treatment of cardiovascular disease. The company's lead candidate formulation, PB1046, is comprised of a vasoactive intestinal peptide (VIP) fused to an ELP. In a recent Phase 1b/2a clinical trial [374] (NCT03315507), three patients received weekly subcutaneous injections of PB1046 over the course of 8 weeks. This resulted in a decreased pulmonary aterial pressure, decreased pulmonary resistance, increase in stroke volume and increase in cardiac output. Phase 2 clinical trials of PB1046 are ongoing to evaluate its efficacy in patients with pulmonary hypertension (NCT03556020) and in COVID-19 patients with acute reparatory distress syndrome (NCT04433546).
5.7. Resilin
Resilin is another elastomeric protein that has been extensively engineered for biomedical applications. Natural resilin is a rubbery protein with remarkable elasticity that is derived from the exoskeletons of insects and arthropods, where it is a critical component of locomotive systems such as jumping and flight [375]. It possesses high resilience and extensibility, low stiffness, and the ability to efficiently store energy [376]. These unique mechanical properties, coupled with its biodegradability, make resilin an attractive protein for regenerative medicine and drug delivery [377].
5.7.1. Structure and properties of resilin
After Ardell and Anderson identified tentative resilin gene sequences in Drosophila melanogaster [378], Elvin and coworkers engineered the first example of a recombinantly expressed resilin-like polypeptide (RLP) in 2005 [379]. The resulting polypeptide, termed Rec1-resilin, is composed of the first exon of the D. melanogaster CG15920 gene, which contains seventeen copies of the GGRPSDSYGAPGGGN motif [379]. Rec1-resilin possesses the favorable mechanical properties of native resilin with the added benefit of customizability provided by its recombinant synthesis. Upon photochemical crosslinking of the tyrosine residues, Rec1-resilin demonstrated up to 92% resilience that is comparable to that of many synthetic rubbers [379].
Lyons and coworkers expanded the RLP library with the development of An16 and Dros16. An16 is composed of sixteen repeats of an eleven amino acid motif found in Anopheles gambiae while Dros16 is composed of sixteen copies of a fifteen amino acid motif in the first exon of the CG15920 gene from which Rec1-resilin is derived [380]. Similar to Rec1-resilin, these RLPs were highly resilient, elastic, and capable of photo-crosslinking [380].
RLPs are intrinsically disordered polypeptides that contain high concentrations of glycine, which provides them with high flexibility and makes secondary structure formation entropically unfavorable. The high proline content of RLPs also prevents secondary structure formation, as the bulky residue reduces hydrogen bonding [381]. Further, unlike other elastin-mimetic proteins, RLPs contain high frequencies of polar and charged residues and are thus hydrophilic. RLPs are responsive to a variety of stimuli, including temperature [382,383]. RLPs demonstrate dual phase behavior (DPB), an unusual property characterized by both a LCST and upper critical solution temperature (UCST), the temperature above which the RLP is fully miscible in solution. The DPB of RLPs is fully reversible and largely dependent on the molecular composition and architecture of the RLP. Their UCST behavior is also strongly impacted by pH [384]. At pH values greater than the pI, the charged residues enhance RLP solubility and the UCST increases. At low pH values, carboxylic acid residues on the RLP become protonated, which reduces the molecule's solubility and drives up the UCST [383,385]. The tunable DPB of resilin is useful to fabricate modular drug delivery systems, described in the next section.
5.7.2. Resilin nanoparticles
The unique, temperature-driven phase behavior of RLPs provides numerous opportunities for designing nanoparticles, whose size, shape, and aggregation number can be tuned to create drug delivery vehicles for a variety of applications.
Li et al. initially described the factors that impact RLP nanoparticle formation using a novel RLP based on twelve repeats of a fifteen amino acid consensus motif derived from D. melanogaster [385]. They replaced the tyrosine residues with phenylalanine and methionine, which can be leveraged for subsequent chemical modification. To form nanoparticles, the polypeptide was heated to its LCST of 65°C, at which point it irreversibly aggregated [385]. Li and coworkers found that the LCST was concentration- and pH-dependent; transition temperature decreased with increasing concentration or decreasing pH (Fig. 20 ). Interestingly, the presence of salt improved RLP solubility and increased the transition temperature, which is the opposite of ELPs (Fig. 20). This phenomenon is attributed to the ionizable residues in the RLP. The resulting nanoparticles can be tuned to have diameters ranging from 20 nm below the transition temperature up to nearly 400 nm when heated beyond their transition temperature [385]. This study demonstrates how the temperature-triggered phase behavior of RLPs can be exploited to design nanoparticles with precision.
Fig. 20.

RLP nanoparticle formation in response to various stimuli. (A-D) Representative TEM images of RLP nanoparticle formations at 25°C (A), 50°C (B), 65°C (C), and 85°C (D). The hydrodynamic radius of RLP nanoparticles is dependent on (E) RLP concentration, (F) salt concentration, (G) salt identity, and (H) pH. Adapted with permission from [385].
Block copolypeptides that incorporate RLPs have also been used to create nanoparticles. The self-assembly of diblock copolymers containing both UCST and LCST blocks is strongly impacted by temperature. At low temperatures, the UCST block is relatively hydrophobic and aggregates at the core while the LCST block remains soluble. As the temperature increases, the UCST block becomes hydrophilic and relocates to the nanoparticle corona as the LCST block aggregates [[386], [387], [388]]. This phenomenon motivated Weitzhandler et al. to combine RLPs with ELPs to better characterize temperature-triggered phase behavior of UCST-LCST block co-polypeptides with the goal of nanoparticle formation [389]. Using an RLP composed of repeat units of the Gln-Tyr-Pro-Ser-Asp-Gly-Arg-Gly octapeptide and an ELP, they identified two principles that affect nanoparticle formation – the RLP:ELP mass ratio and ELP hydrophilicity. With an increase in the RLP:ELP ratio, they observed an increase in the nanoparticle size. In contrast, the hydrophobicity of the ELP impacted morphology; a hydrophobic ELP resulted in spherical micelles, while a hydrophilic ELP promoted the formation of worm-like structures [389].
Dzuricky and coworkers expanded upon this work to observe how nanoparticle morphology impacted cell uptake [55]. A fibronectin type-III (Fn3) -binding domain that binds the αvβ3 integrin was fused to the C-terminus of an RLP-ELP diblock copolymer. They found that the Fn3 domain did not perturb self-assembly of the RLP-ELP diblock copolymer, so that it was displayed on the nanoparticle surface upon self-assembly (Fig. 21 ). The tunable morphology and high avidity of the RLP-ELP-Fn3 nanoparticles significantly improved the binding affinity to the αvβ3 integrin via multivalency. They found that shape played an important role in promoting multivalency though the avidity effect. While a spherical RLP-ELP micelle that presented a Fn3 domain showed a respectable 10-fold increase in affinity for the αvβ3 integrin compared to the monovalent RLPELP-Fn3 unimer, the worm-like RLP-ELP-Fn3 nanoparticles showed a remarkable 1000-fold increase in affinity for the integrin (Fig. 21) [55]. The combination of morphology and high avidity yielded a picomolar KD, a remarkable achievement considering that a therapeutic antibody against the same integrin exhibits a KD of only 20 nM. Consequently, the worm-like RLP-ELP-Fn3 was more readily taken up by cells than the antibody [55]. These studies demonstrate that RLP-ELP nanoparticles can be harnessed for robust, specific cellular targeting and uptake of therapeutic agents.
Fig. 21.

Cell binding and uptake by RLP-ELP diblock nanomaterials. (A) Cellular uptake of RLP-ELP unimers, spherical micelles, and worm-shaped micelles with an integrin-targeting Fn3 protein domain. The increased avidity and worm-like structure of the micelle promoted its ability to enter the cell. (B) Shape-dependent avidity of RLP-ELP diblocks. Multivalency increased the affinity of the nanoparticles for its target by decreasing the off-rate. Adapted from [55].
5.7.3. Resilin hydrogels
Because of their exquisite stimulus responsiveness and mechanical properties, RLPs have been engineered as hydrogels for many biomedical applications. Although many of the hydrogels were designed for use in tissue engineering applications, the principles derived from these studies can are also useful for the design of RLP-based controlled drug delivery systems.
Charati and coworkers reported an early example of a RLP hydrogel [390]. To test its utility for biomedical applications, they incorporated several biologically active domains into the sequence to control cell adhesion, hydrogel degradation, and biocompatibility. Upon purification from E. coli, the protein was crosslinked with [tris(hydroxymethyl) phosphine] propionic acid (THPP), which linked the lysine residues in the RLP sequence. The resulting hydrogel was mechanically stable and capable of supporting hMSCs and mouse NIH-3T3 fibroblasts [390]. Li and coworkers later evaluated the mechanical properties of RLP hydrogels more precisely and showed that the extent of THPP crosslinking impacts factors such as swelling ratio, elastic shear modulus, and resilience [391].
Su and coworkers adapted RLPs for cell adhesion by combining cell binding domains from fibronectin with a consensus sequence derived from A. gambiae. After it was crosslinked with tris(hydroxylmethyl)phosphine, the novel RLP scaffold exhibited mechanical properties consistent with those of Rec1-resilin and a compression modulus comparable to that of cartilage [392]. The material supported MSCs at 95% viability for up to 3 days, and the cells exhibited extensive spreading on the material surface [392]. While this hydrogel was primarily engineered for tissue engineering applications, it provides an example of how peptides can be appended to the RLP sequence for other biomedical applications such as drug delivery. Other moieties, such as heparin [390,391], VEGF-mimicking peptides [393], and chitin -binding domains [394], have similarly been appended onto RLPs.
Taking advantage of the recombinant nature of RLPs, Su et al. engineered their novel RZ10-RGD hydrogel to respond to changes in redox [395]. They used 3,3′-dithiobis(sulfosuccinimidyl propionate) (DTSSP), a redox-responsive crosslinker, to create RLP hydrogels that degrade in a reducing environment (Fig. 22A). Various molecular weights of FITC-labeled dextran were loaded into the crosslinked RZ10-RGD hydrogel. In reducing conditions, there was no observed difference in the release rates of dextrans with different molecular weights. In non-reducing conditions, release rate decreased with an increase in dextran MW (Fig. 22B) [395]. The redox-responsiveness of this novel hydrogel makes it a promising candidate for tumor-specific drug delivery.
Fig. 22.

The fabrication and release profiles of redox-sensitive RZ10-RGD hydrogels. (A) Schematic of the crosslinking and degradation reactions of RZ10-RGD and DTSSP. Images of the crosslinked hydrogel in a non-reducing (left) or reducing environment (right). In a reducing environment (B), dextran release was not MW-dependent. In a non-reducing, PBS environment (C), dextran release was dependent on MW. Adapted from [395].
Other “smart” RLP hydrogels have been designed to degrade in response to elevated MMP concentrations [396]. When an MMP-sensitive domain was incorporated into a RLP sequence, the hydrogel underwent 90% degradation in the presence of physiological levels of MMPs over the course of 48 RLP hydrogels without the MMP-sensitive domain did not degrade [396]. Because the concentration of MMP-sensitive domains within the RLP hydrogel may be modulated to control degradation, this design can be adapted for drug delivery to tissues with elevated MMP concentrations, such as tumors or wound sites.
Resilin has also been combined with other proteins, including elastin, collagen, silk, and peptide amphiphiles (PAs), to enhance the properties of RLPs. Bracalello and coworkers designed a chimeric polypeptide comprised of an RLP, ELP, and CLP in tandem, which they termed the REC polypeptide [397]. The purified material formed highly aligned fibrils that, over time, self-assembled into large fibers and fiber bundles. The fibers and bundles possessed a Young's modulus of 0.1–3 MPa, which demonstrated that the material retained the elastic properties of resilin and elastin [397]. More recently, Okesola and coworkers developed a highly tunable, multicomponent hydrogel using a covalent co-assembly strategy to combine an RLP with a peptide amphiphile (PA). This approach enabled control over the hierarchical self-assembly of the RLP hydrogel and enhanced its mechanical properties. The hydrogels were synthetized using a thiol-ene photoclick reaction and, depending on the PA concentration, assembled into a variety of nanostructures. In the absence of PA, the RLP hydrogels formed nanospheres upon photo-crosslinking, a low concentration of PA resulted in co-assembled beaded strings, and a high concentration of PA created co-assembled nanofibers. The photoclicked hydrogels maintained the remarkable elastic and energy-storing properties of RLP hydrogels, thus demonstrating that the co-assembly strategy can improve the mechanical properties of more brittle proteins such as PAs. These results further support that RLP materials can be precisely tuned to achieve desirable properties for a drug release system.
5.8. Silk-Elastin-Like polypeptides
Silk-Elastin-Like Polypeptides, or SELPs, are block co-polymers consisting of tandem repeats of silk-like and elastin-like blocks [360]. These hybrid polypeptides combine the mechanical strength and chemical stability of silk with the unique thermal responsivity of ELPs, resulting in a material that can assemble into a variety of architectures ranging from nanomaterials to hydrogels [350]. Like many other biopolymers, these materials are recombinantly produced, offering precise control over their length, the ratio of SLP to ELP blocks, and the ability to introduce biologically active molecules into the sequence [398]. An interesting characteristic of SELPs is their ability to undergo an irreversible sol-gel transition under physiological conditions, creating a matrix in situ that is useful for local delivery [360].
5.8.1. Nanoparticles
SELPs have been engineered to self-assemble into micellar structures that can be used as nanocarriers for drugs. The core of the micelle is formed by the SLP blocks which form intermolecular hydrogen bonds to stabilize the micelle while the relatively hydrophilic ELP forms the corona [361]. The hydrodynamic radius of the micelle can be tuned by altering the SLP to ELP block ratio or by altering the choice of guest residue in the ELP block [361].
The self-assembly of SELP nanoparticles may also be triggered by encapsulation of a hydrophobic drug. Xia et al. first explored this concept by mixing DOX with SELPs with a SLP to ELP block ratio of 1:8, 1:4 or 1:2 [362]. The DOX hydrophobically interacted with the silk domains, driving the formation of nanoparticles with hydrodynamic radii between 50 and 142 nm (Fig. 23A). These nanoparticles are in the appropriate size range to be passively targeted to solid tumors via the EPR effect [399]. This approach is attractive as it avoids the need for harsh solvents and provides a respectable 6.5% drug loading efficiency [362]. DOX-loaded SELP nanoparticles enter HeLa cells via endocytosis rather than diffusion like free DOX (Fig. 23B). This allowed for potent accumulation of the drug in the cytoplasm and nearly 2-fold greater cytotoxicity than the free drug [362].
Fig. 23.

(A) Doxorubicin-triggered self-assembly of SELP nanoparticles (B) Delivery of SELP-encapsulated DOX to HeLa cells at 40 min and 4 h. SELP encapsulation improves cell uptake and drug accumulation in the cytoplasm. Adapted from [362].
More recently, Parker et al. developed mucoadhesive SELP nanoparticles for transmucosal drug delivery [400]. To identify potential mucoadhesive sequences, they generated a series of SELPs with a 1:4 SLP to ELP block ratio with four different guest residues – Cys, Arg, Lys, and Glu. These amino acids were chosen due to the potential of charged residues and thiol groups to interact with mucus. ANS, a hydrophobic, solvatochromatic dye, was loaded into the nanoparticles via simple mixing to serve as both a model compound and reporter. In an in vitro biosimilar mucus model, the SELP with Cys guest residues demonstrated the strongest affinity for mucus, followed by SELPs containing Lys, Arg, or Glu as the guest residue, respectively. Interestingly, when incubated with mucus-secreting HT29-MTX cells, the Cys-containing SELP demonstrated little cell retention, suggesting it may have mucolytic properties. Meanwhile, SELPs with charged guest residues were well retained by the cells, indicating their potential for transmucosal delivery.
5.8.2. Hydrogels
Perhaps the most interesting property of SELPs is their ability to undergo a sol-gel transition under physiological conditions. These materials can be engineered to be soluble at room temperature, at which point drugs and other therapeutic molecules can be loaded by simple mixing without the use of toxic or denaturing solvents. Upon injection, the material coacervates to form a polymeric matrix in situ. This property, along with the biocompatibility and sequence control of SELPs, makes them an attractive material for engineering local, controlled drug delivery systems.
This approach was first explored by Cappello et al. in 1998. In this study, fluorescently labeled macromolecules of varying molecular weights were loaded into SELP hydrogels and their release rates were monitored [401]. For all compounds, release from the hydrogels was near first-order; however, there was no correlation between the release kinetics and the molecular weight of the molecule. This was attributed to the large size of the hydrogel pores, which are not small enough to impede diffusion [401]. The release rate decreased with an increase in SELP concentration. The hydrogels were biocompatible when implanted in vivo with no sign of immunological activity or inflammation [401]. Dinerman et al. further evaluated the diffusion behavior of molecules out of SELP hydrogels by using cytochrome c, theophylline, and vitamin B12 as model compounds [402]. They observed Fickian, diffusion-controlled release behavior wherein diffusion slowed with an increase in molecular weight and polymer volume fraction [402].
Due to the simplicity of drug loading and their gelation behavior, SELPs are also attractive materials for gene delivery. Nucleic acids can be incorporated into the SELP hydrogel by physical mixing, and upon injection, the SELP hydrogel protects its cargo from rapid degradation while controlling the rate and location or release. This concept was first explored by Megeed et al. in 2002 using an SELP containing positively charged residues. The negatively charged DNA molecules electrostatically adsorbed onto the SELP before injection [403]. They discovered that the release of DNA was inversely related to the SELP concentration and gelation time and that the ionic strength of the buffer directly correlated with DNA release rate; as ionic strength increases, the greater concentration of counter ions disrupt the electrostatic interaction between the SELP and DNA, facilitating the release of the DNA [403]. In a later study, Megeed et al. discovered that the release rate of plasmids from the SELP hydrogel was inversely proportional to plasmid MW. Furthermore, plasmid conformation impacted release rate, with linearized plasmids diffusing from the hydrogel fastest followed by supercoiled and circular plasmids, respectively [404]. The hydrogel maintained the stability and bioactivity of plasmid DNA for 28 days and adenoviruses for 22 days. In an in vivo model with athymic nude mice bearing subcutaneous MDA-MD-435 tumors, mice treated with SELPs loaded with a plasmid for luciferase exhibited greater gene expression than mice treated with naked DNA [404].
These studies aid the groundwork for SELP design for DNA delivery to treat cancer. Hatefi et al. loaded SELP-47K, an SELP comprised of four SLP and 7 ELP blocks with one ELP block containing a Lys substitution, with the adenoviruses Ad.GFP or Ad.CMV.LacZ that encode green fluorescent protein (GFP) or β-galactosidase, respectively [405]. In an in vitro study, the SELP hydrogel maintained the release of the adenoviruses over a 28 day period with only a 30–40% loss in virus activity (Fig. 24A) [405]. The Ad.GFP-loaded SELP was intratumorally administered in two mouse models – one for breast cancer (MDA-MB-435) and one for head and neck cancer (FaDu). A dose of 50 μl of SELP with 4 × 109 plaque forming units of virus resulted in high GFP expression in the solid tumors, with better expression observed in the breast cancer model compared to the head and neck cancer model, due to the higher level of Coxsackie and Adenovirus Receptor on the MDA-MB-435 cell surface. Tumors treated with Ad.GFP alone showed significantly lower GFP expression than tumors receiving the SELP formulation (Fig. 24B) [405]. This study demonstrated the potential of SELPs for adenoviral delivery to solid tumors.
Fig. 24.

Adenovirus delivery by SELP hydrogels. (A) Ad.GFP was released from a 4% SELP hydrogel over the course of 28 days. (B) GFP expression in tumor cells receiving Ad.GFP or Ad.GFP mixed with SELP. Panels a-d show GFP expression 3, 7, 11 and 15 days after injection. Adapted from [405].
In a subsequent study, the same research group evaluated factors impacting adenovirus delivery in a head and neck cancer model. Using three different SELPs, they administered Ad.LacZ, the β-galactosidase gene, to nude mice bearing JHU-022 head and neck squamous cell carcinoma (HNSCC) tumors. The SELPs were selected to evaluate the effect of SELP structure on adenovirus biodistribution. They found that SELP-815K, the SELP with a high number of silk blocks and the lowest degradation rate, demonstrated the most prolonged gene expression and highest transfection efficiency. Compared to intratumoral injections of free virus, the virus-loaded SELPs all exhibited higher and more localized transfection. These observations indicate the utility of SELP hydrogels for delivering adenoviruses to tumors while avoiding off-target toxicity.
The ability of SELP-815K to deliver adenoviruses was further tested in a head-to-head comparison with poloxamer 407, a commercially available synthetic copolymer used for gene delivery. Both platforms were used to deliver a genetically encoded prodrug with an antiviral vector to a xenogenic model of HNSCC. Compared to poloxamer 407 and free virus, SELP-815 exhibited the highest initial gene expression in tumors followed by prolonged expression for up to three weeks [406]. This high and sustained level of gene expression resulted in the greatest tumor size reduction and survivability across all groups. Furthermore, SELP-815K showed superior safety than poloxamer 407, as adipocytic necrosis was observed in animals treated with the latter [406]. A subsequent study demonstrated that, compared to free virus, virus-loaded SELPs also had reduced systemic toxicity [407].
More recent studies have provided new insights into the stability of viral vectors in SELP hydrogels. Two different SELPs were loaded with the oncolytic adenovirus Ad-ΔB7-KOX and evaluated with AFM imaging to visualize the interaction between the adenovirus and SELP nanofibers. AFM showed that as adenovirus concentration increased, the density of the SELP nanofibers also increased despite their fixed concentration [408]. This is likely attributed to the Brownian motion of the adenovirus during assembly, as it acts as a stimulus for SELP network formation. Although most adenoviruses exhibited a round morphology for the first half-hour of being attached to SELP, they began to deform after approximately 1 h. This is likely due to multiple strong interactions forming between adenovirus and different SELP fibers [408]. It was also discovered that SELP analogues with shorter elastin units exhibited lower temperature responsivity and more rapid elastase degradation [408]. With this fundamental understanding of SELP degradation and adenovirus complexation, better gene delivery systems can be engineered from this material.
SELP hydrogels have also been used for sustained release of polysaccharides. The Ghandehari group synthesized positively charged SELP hydrogels that could electrostatically interact with an anionic polysaccharide —semisynthetic glycosaminoglycan ether (SAGE)— that is an anti-inflammatory drug for mucosa restoration. Unfortunately when SAGE is administered, it does not achieve a high enough therapeutic concentration due to its insufficiently long residence time [409,410]. To solve this problem for treatment of radiation-induced proctitis (RIP), the Ghandehari group designed an in situ gelling enema (Fig. 25A) [410]. In a murine model of RIP, SAGE was present in the rectal tissue of mice 12 h after receiving a SAGE-loaded SELP enema, while it was undetectable in mice receiving SAGE with saline (Fig. 25B) [410]. The Ghanedhari group further adapted the SELP platform for the sustained release of intravesically delivered SAGE for treatment of interstitial cystitis (IC) model. They found that employing a softer SELP with a slower gelation time could provide an analgesic effect for 24 h without exacerbating the discomfort associated with IC, whereas stiffer gels could reduce the capacity of or obstruct the bladder [409].
Fig. 25.

SAGE delivery with SELP hydrogels for the treatment of radiation-induced proctitis (RIP). (A) A schematic of the treatment platform. The glycosaminoglycan (GM-0111) was mixed with SELP and rectally delivered to a murine model for RIP. The injectable SELP solution forms a solid hydrogel upon administration to slowly release GM-0111. (B) Representative fluorescent micrograph images of rectal tissue 3 and 12 h after treatment with GM-0111 loaded SELP or GM-0111 alone. GM-0111 accumulation is enhanced and prolonged when delivered with the SELP hydrogel. Adapted from [410].
5.9. De Novo designed polypeptides
More recently, researchers have focused on the design of de novo artificial polypeptides as drug carriers. These genetically engineered polypeptides are conformationally disordered, hydrophilic, and inert. Like other protein polymers, they are recombinantly expressed and purified in bacterial systems, providing exquisite control over their length and sequence. They can also be genetically fused to therapeutic peptides or proteins to increase the half-life, stability, and biodistribution of their therapeutic partner. In addition to their stability and non-immunogenicity, these hydrophilic biopolymers have long plasma circulation, making them an intriguing alternative to PEGylation. In the last decade, two polypeptide sequences in particular– XTEN and PAS – have gained traction as biopolymeric drug carriers.
5.9.1. XTENylation
The first XTEN molecule was designed and synthesized by Schellenberger et al. in 2009 [411]. To create a stable, soluble and unstructured stealth polymer, they eliminated hydrophobic amino acids known to contribute to aggregation or secondary structure formation, positively charged molecules that could bind cell membranes, and amide-containing residues that reduce stability. These criteria limited their pool of potential amino acids to six – alanine, glutamate, glycine, proline, serine and tyrosine. Schellenberger et al. then screened a library of non-repetitive, randomized sequences to obtain an 864-residue biopolymer with optimal expression levels, yield, solubility, and stability [411]. This original XTEN molecule was genetically fused to exenatide, an antidiabetic peptide that has an in vivo half-life of 2.4 h. Fusion to XTEN increased the peptide's half-life to 60 h in cynomolgous monkeys, with a predicted half-life of 139 h in a 75 kg human (Fig. 26 ) [411]. XTEN similarly enhanced the terminal half-life of GFP, human growth hormone, glucagon and factor VII. Since this initial study, different MW variants of XTEN have been produced [412] and different functional groups for drug conjugation – including azide, maleimide, and alkyne groups - have been introduced into the polypeptide [413].
Fig. 26.
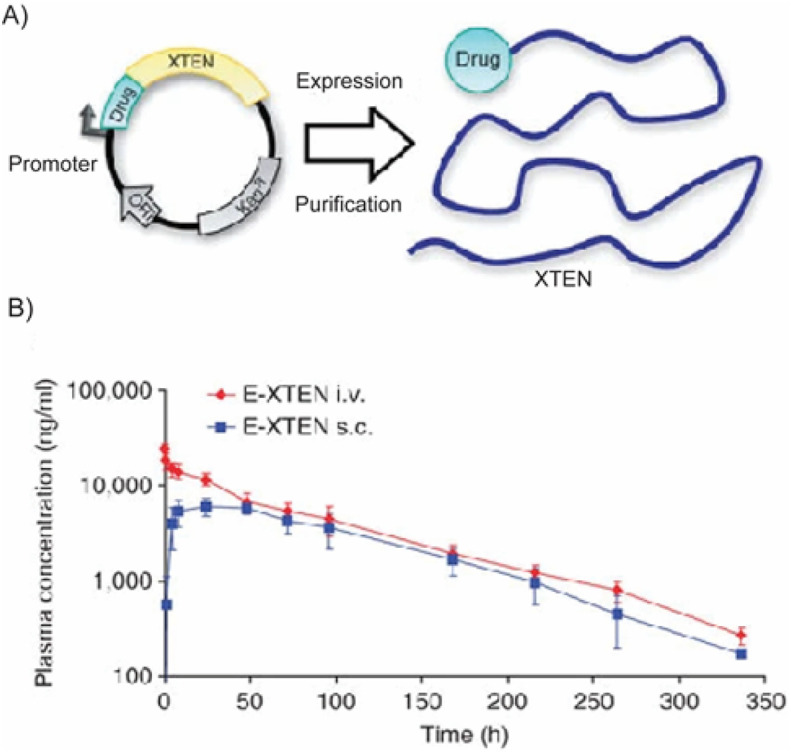
XTENylation of therapeutics for improved pharmacokinetic properties. (A) Design and production of XTENylated protein drugs. A plasmid is designed such that the gene encoding the drug is fused to XTEN for recombinant expression (B) Pharmacokinetic profile of XTENylated exenatide (E-XTEN) delivered intravenously or subcutaneously to cynomolgous monkeys (C) A single injection of E-XTEN protected mice from a glucose challenge 48 h post administration. Adapted from [411].
The biopolymer's large size and stealth behavior has made XTENylation an attractive strategy to improve the pharmacokinetic profile of many therapeutic peptides. It has primarily been used to create long-lasting formulations of therapeutics that would otherwise require frequent dosing or have dose-dependent side effects [413]. One of the first examples of this was VRS-859, an exenatide-XTEN fusion developed by Diartis [414]. In a Phase I clinical trial reported by Diartis, VRS-859 achieved a half-life of 128 h, suggesting the formula could be administered once-monthly. The gastrointestinal side effects associated with exenatide dosing were mild and dissipated within 24 h, further demonstrating the clinical utility of XTENylated therapeutics [414].
XTEN has been similarly employed to counteract nocturnal hypoglycemia in diabetic patients. Geething et al. fused the biopolymer to Gcg, a peptide hormone that converts glucagon stores into glucose but suffers from a minutes-long half-life and poor stability in liquid formulations [415]. In addition to improving the stability of Gcg 60-fold, fusion to XTEN prolonged the efficacy of Gcg in a fasted beagle dog model. Gcg-XTEN provided strong resistance to a glucose challenge 6 h after prophylactic administration without affecting baseline blood glucose level [415]. The effect tapered off at 12 h post-administration, whereas free Gcg was ineffective 2 h post-administration. XTEN also remarkably enhanced the pharmacokinetic profile of teduglutide, a glucagon-like peptide 2 analog used to treat an inflammatory malabsorption disorder in the small intestine with a half-life of ~3 h in humans [416]. The teduglutide-XTEN fusion achieved a 120 h half-life in cynomolgous monkeys, with a predicted half-life of 240 h in humans, further indicating how XTEN can be employed for once-monthly administration. The formulation also reduced the occurrence of ulcerations and presence of inflammatory cytokines in the small intestine [416]. Additionally, this study was the first instance where the payload was chemically conjugated to XTEN; in this case, teduglutide was chemically conjugated via a C-terminal cysteine residue. Since then, XTEN has been chemically conjugated to other therapeutics including antivirals [412] and imaging agents such as annexin 5 [417] to improve their circulation time.
XTENylation has been used to improve th P to human growth hormone (hGH), which typically suffers from an hours-long half-life and requires daily administration. Somavaratan (VRS-317), an XTENylated formulation of hGH, achieved a half-life of 131 h, a 30–60 fold increase compared to recombinant hGH in a Phase I clinical trial (NCT01718041) [418]. Interestingly, fusion to XTEN reduced the potency of hGH 12-fold, yet the prolonged tissue exposure XTEN provided a 3 to 5-fold increase in efficacy compared to recombinant hGH [418]. In a Phase II clinical trial VISTA study (NCT02068521), patients treated with somavaratan demonstrated increasing height velocity with increasing doses of the drug. and dose-dependent adverse events were minimal [419]. Unfortunately, Somavaratan failed to achieve the primary endpoint of non-inferiority versus Genotropin, a recombinant formulation of hGH, in a Phase III VELOCITY clinical trial (NCT02339090) for pediatric patients with growth hormone deficiency. A phase II clinical trial (NCT02526420) has also been completed for growth hormone deficiency in adults. These studies demonstrate the promise of XTEN in improving drug half-life, bioavailability, and efficacy.
Due to its ability to prolong the therapeutic effects of its fusion partner, XTEN has also been used as a carrier for clotting factors for treating hemophilia. Infusions of factor VIII (FVIII) or factor IX (FIX) greatly improve the clotting activity in hemophiliacs, but their short half-life necessitates multiple injections per week. Recombinant FVII and FIX, which is used when a patient develops neutralizing antibodies against the natural FVII and FIX, suffers from a shorter half-life, is less effective at controlling bleeding, and requires infusions every 2 h to promote clotting. Thus, prolonging the half-life of clotting factors via XTENylation is an attractive strategy to reduce the dosing frequency. In a collaboration between Biogen and Amunix, XTEN was fused to recombinant FIX and a FIX-Fc fusion [420]. A 288 residue XTEN molecule improved the area under the curve of recombinant FIX 40-fold and increased the maximum concentration in circulation 30-fold. Further, XTENylated FIX exhibited a 3-fold greater bioavailability compared to unmodified recombinant FIX. A 72 residue XTEN molecule fused to FIX-Fc fusion improved the AUC 6-fold relative to the non-XTENylated Fc fusion, leading to a 3-fold increase in Cmax [420]. More recently, a von-Willebrand factor independent FVIII-XTEN fusion, termed BIVV001, has demonstrated good safety and efficacy in early stage clinical trials (NCT03205163) and is currently recruiting for Phase 3 (NCT04161495) [421,422]. XTENylation of FVIII increases its plasma half-life three to four-fold relative to recombinant FVIII, achieving a half-life of up to 34 h in cynomolgous monkeys [423]. This resulted in four-fold longer hemostatic control, suggesting that this formulation could provide clotting protection for patients for up to one week [423].
XTEN's large size has also been harnessed to take advantage of the EPR effect to passively deliver anticancer therapeutics to tumors. To demonstrate its utility, Haeckel et al. designed a multicomponent fusion protein to improve the delivery of Killin, a cytostatic and cytotoxic protein [424]. Despite the promising antitumor effects of Killin, its expression in bacterial systems was impossible due to its toxicity. C-terminal fusion to XTEN deactivated the protein so it could be stably expressed and purified. Upon administration, XTEN would further serve to enhance the circulation time of Killin and exploit the EPR effect for tumor-specific targeting. A matrix metalloproteinase cleavage site was incorporated between the XTEN and Killin, allowing the Killin to be released and activated upon tumor penetration. To improve cell membrane penetration and DNA binding, a cell penetrating peptide (CPP) was also added onto the C-terminus of Killin [424]. The fusion protein was preferentially taken up into cancer cells with high MMP-2 or MMP-9 expression. This XTEN-Killin-CPP fusion also arrested growth and induced concentration-dependent apoptosis in Killin-sensitive cell lines, with the effect peaking at 48 h after treatment [424]. This study demonstrates the potential of XTEN as a carrier for anticancer therapeutics.
This concept has recently been adapted by Amunix to develop XTENylated Protease-Activated T-Cell Engagers, or XPAT. In this technology, the bispecific T-cell engager Blincyto, which engaged CD3 and and CD19, is XTENylated to reduce its immunogenicity. Tandem protease recognition sites are incorporated between the XTEN chain and tandem scFvs of the bispecific T-cell engager to promote its release upon protease exposure. This technology has been adapted to target HER2+ (AMX-818) and EGFR+ cells [425]. In recent pre-clinical studies, these fusions achieved picomolar affinity EC50s against cell lines expressing their target proteins while reducing T-cell toxicity 15,000-fold. They also induced significant protease-dependent tumor regression in xenograft models. HER2- and EGFR-XPAT were also well tolerated by cynomolgous monkeys, with a 1000-fold greater Cmax for the HER 2-XTEN fusion and a 100-fold greater Cmax for the EGFR-XPAT than their non-XTENylated counterparts [425].
5.9.2. PASylation
A second recombinant polypeptide, termed PAS, was engineered by XL-Protein GmbH Company in 2013 and is conceptually similar to XTEN [426]. Both are intrinsically disordered, hydrophilic polypeptides that are designed to enhance the plasm half-life of fused proteins and peptides [426]. PAS are reported to be non-immunogenic, biodegradable, and are produced in bacterial systems [426]. Their recombinant synthesis enables precise control over their sequence and length enable gene-level fusion to a therapeutic peptide or protein of interest in order to enhance the pharmacokinetic profile of the fusion partner [427].
Whereas XTEN incorporates negatively charged residues and consists of Pro, Tyr, Glu, Gly, Ala and Ser, PAS do not contain charged amino acids and their sequence is limited to Pro, Ala and Ser [427]. In the initial study published by XL-Protein GmbH Company, PAS was recombinantly fused to three proteins —a recombinant Fab region of a humanized anti-HER2 antibody, human interferon a2b, and human growth hormone— to evaluate its utility as a drug delivery carrier. Fusion to PAS increased the hydrodynamic volume of the cargo up to 26-fold and extended the blood circulation of the fusions [426]. The plasma half-life and AUC of PASylated IFN was 32-fold and 100-fold greater than the non-PASylated IFN and the protein's binding affinity for its receptor was not affected by fusion to PAS (Fig. 27A). Similarly, PASylated hGH exhibited a 94-fold greater plasma half-life than control hGH and exhibited a 3-fold greater growth promoting effect than native hGH on an equimolar basis (Fig. 27B) [426].
Fig. 27.

PASylation of therapeutics for improved pharmacokinetic properties. (A) Pharmacokinetic profile of recombinant and PASylated IFN in BALB/c mice. The number within the parentheses in the PASylated versions indicates the length of the PAS peptide. (B) Pharmacokinetic profile of recombinant and PASylated hGH injected intravenously or subcutaneously. Adapted from [426].
Since this initial study, PASylation has been employed to enhance the half-life and pharmacokinetics of numerous therapeutics. Like other biopolymers, many applications that harness PASylation require long-lasting therapeutics to reduce dosing frequency and side effects. As such, XL-Protein GmbH Company has developed several formulations to improve the therapeutic efficacy of several peptides for the treatment of many diseases, including exendin for type II diabetes, hGH for pediatric growth hormone deficiency, and anti-inflammatory agents for rheumatoid arthritis [428]. Many of these fusions are currently in preclinical trials.
Other groups have also explored PASylation as a strategy to improve the circulation and efficacy of interferons for the treatment of multiple sclerosis. In a study conducted by Harari et al., a superagonistic analog of IFNβ, YNSα8, was fused to the C terminus of a 600 amino acid PAS polypeptide chain [429]. When administered to a transgenic mouse model harboring humanized IFNAR receptors (HyBNAR model), PAS-YNSα8 exhibited a 10-fold increase in half-life compared to YNSα8 alone without impacting the biological activity of the superagonist. Despite being administered at an overall 16-fold lower dosage than IFNβ alone, PAS-YNSα8 outperformed the control group in an experimental autoimmune encephalomyelitis model, as indicated by a decrease in infiltrating macrophages in the central nervous system and an increase in regulatory T cells [429]. In a separate study by Zvonona et al., PAS was fused to the C-terminus of IFN-β1b and resulted in a 4-fold increase in hydrodynamic radius compared to free IFN-β1b [430]. The fusion protein also notably increased IC50 2-fold compared to free IFN-β1b, an accomplishment considering PEGylation of the same interferon decreases its activity. Furthermore, this formulation was stable when stored for 22 months at −20°C, indicating how PASylation can contribute to the maintenance of biological stability [430]. Similarly, PASylation has contributed to the improved half-life and efficacy of numerous proteins including leptin [431,432], erythropoietin [433], Conversin [434] and clotting factor VIII [435].
PASylation has also been harnessed to improve the delivery of nanocarriers, most notably for the treatment of cancer. In one study, PAS 40 or 75 residues long were recombinantly fused to ferritin, an iron storage protein that self-assembles into a hollow sphere and binds internalizing transferrin receptor I, which is upregulated on many cancer cells [436]. PASylation stabilized the ferritin during purification and drug loading, resulting a greater yield of nanocarriers and 3-fold higher DOX loading. The half-life of the PASylated nanocarrier was 5-fold greater than that of the control nanocarrier and 55-fold greater than that of free DOX [436]. The same research group further developed this platform by incorporating an MMP-cleavable linker between PAS and ferritin. This design resulted in a 5–6 fold decrease in affinity between ferritin and its receptor, transferrin receptor I. Thus, healthy cells with low expression of transferrin receptor I were not affected by the encapsulated DOX [437]. Upon exposure to high MMP concentrations in the tumor microenvironment, the PAS shield was cleaved from the ferritin, increasing the affinity of the nanocarrier for its target. In mice bearing xenogeneic PaCa-44 pancreatic tumors, treatment with the PASylated formulation led to 4-fold greater tumor regression than a non-PASylated nanocarriers and 8-fold greater regression than seen with free DOX, respectively [437]. These studies indicate how PASylated nanocarriers can be used to improve drug potency while reducing off-target effects.
PASylation is also useful for nucleic acid delivery by providing stealth behavior to the delivery system. Morys et al. conjugated short PAS sequences only four repeats long to polyethylenimine, a cationic polymer frequently employed in nucleic acid delivery systems [438]. The PAS provided enough hydrophilicity to prevent aggregation and dissociation of the PEI/DNA polycomplexes. Furthermore, PASylation of the PEI-DNA complex prevented interaction with blood serum proteins, preventing polyplex opsonization and clearance in vivo [438]. Although this system needs further optimization for in vivo gene transfer, this study indicates that PASylation is potentially a useful strategy for improving the stability and delivery of nucleic acids.
5.10. Other proteins
5.10.1. Gliadin
Gliadin is a gluten protein extracted from wheat. Its monomeric form, which has a MW of 25–100 kDa, is up to 70% soluble in alcohol, and its polymeric form, which has a MW of 106 kDa, contains a high level of disulfide bonds that renders it largely insoluble [439]. As such, gliadin materials show excellent stability without the need for toxic crosslinking agents. Gliadin is rich in neutral residues that provide it with mucoadhesive properties and make it an ideal platform for oral drug delivery [440]. Gliadin also contains many lipophilic residues that allow it to protect its cargo for prolonged release and hydrophobic interaction with tissues, including skin. Due to its apolarity, gliadin has been most useful as a carrier for hydrophobic or amphiphilic drugs [441].
The solubility, hydrophobicity, and bioadhesive properties of gliadin contribute to its ability to sustain the release of therapeutics in drug delivery applications. Polymeric gliadin has been widely explored as a material for nanoparticulate systems targeting the gastrointestinal mucosa. Arangoa et al. demonstrated that upon oral administration of the lipophilic molecule carbazole in a gliadin nanoparticle to mice, the gliadin delivery vehicle improved drug adsorption through the stomach mucosa [442]. This phenomenon resulted in a sustained carbazole concentration in circulation and improved bioavailability. Further research evaluated gliadin as a nanoparticulate carrier for the anti-cancer drug cyclophosphamide [443]. After loading cyclophosphamide onto the nanoparticles with a remarkable 72% loading efficiency, Gulfam et al. demonstrated that the drug was released in a two-step manner. After a brief period of rapid release from the nanoparticles, cyclophosphamide was steadily released over the course of 48 h and induced apoptosis in breast cancer cells, which highlighted the utility of this system utility as a carrier for anti-cancer therapeutics [443].
Because gliadin nanoparticles improve drug penetration into the gastric mucosa, it has been extensively engineered to protect and deliver antibiotics to the stomach to treat Helicobacter pylori infections [51]. Ramteke et al. loaded gliadin nanoparticles with clarithromycin and omeprazole via a desolvation technique [51]. The nanoparticles showed sustained release of the drugs at an acidic pH that mimicked conditions in the stomach and were more effective in eradicating H. pylori in vitro than free drug. In a similar study, Umamaheshwari et al. demonstrated that gliadin nanoparticles increased the gastrointestinal residence time of amoxicillin, thus necessitating a lower dose to eradicate H. pylori infection than the free drug in a Mongolian gerbil model [444]. These studies indicate that the bioadhesive properties of gliadin can be leveraged to improve gastric absorption and drug availability.
5.10.2. Soy protein isolate
Another abundant source of plant protein is the soybean, from which soy protein isolate (SPI) is extracted. SPI contains glycinin and β-conglycinin, which compose nearly 60% and 40% of the protein, respectively [445]. These proteins exist as globular structures in an aqueous environment and aggregate upon the addition of a crosslinking agent or solvent [446]. This can drive the formation of hydrogels, polymer blends, or microspheres. Moreover, because SPIs are enriched in glutamine, aspartic acid, and leucine that offer a variety of secondary bonding interactions, many types of drugs can be incorporated onto the proteins for delivery purposes. [447].
SPI nanoparticles have been designed to carry a variety of molecules. Teng et al. engineered soy nanoparticles to sequester and relase curcumin [448]. They prepared the nanoparticles using a desolvation technique and achieved an encapsulation efficiency of 97%. Curcumin release from the nanoparticles followed a biphasic trend. Initial burst release was observed as the nanoparticles swelled, followed by sustained release [448]. The pharmacokinetics, gastrointestinal uptake, and encapsulation efficiency of soy nanoparticles have also been improved by combining the protein with other materials such as folic acid [449], zein [450,451], or alginate [452].
SPI films have also been engineered to release a variety of drugs to treat bacterial infections and promote wound healing. In one example, Peles et al. incorporated the antibiotic gentamicin into the matrix of an SPI film prepared by solvent-casting. After an initial period of burst release, gentamicin exhibited prolonged release over the course of four weeks [453]. Other studies have explored various crosslinking agents to improve the structural integrity and degradation kinetics of SPI films. To circumvent the toxicity associated with chemical crosslinkers, Song et al. employed genipin, a natural crosslinking agent, to control the swelling ratio and enhance the strength of the film [454]. Films crosslinked with genipin exhibited slowed release rates of BSA, the model protein drug, compared to SPI films without genipin. Furthermore, the film exhibited pH-sensitivity that could be employed for delivery to the gastrointestinal tract [454]. Chen et al. also designed SPI films to release drug in the gastrointestinal tract by using formaldehyde as a crosslinking agent [455]. Formaldehyde crosslinking increased the tensile strength of the film. Methylene blue —a model drug— and rifampicin—and antibiotic —interacted strongly with the SPI such that they were not released until bulk erosion occurred, which was accelerated by gastric enzymes [455]. Another pH-sensitive SPI film was engineered by Vaz et al. using glyoxal as a crosslinking agent for SPIs produced via a melt extrusion technique [456]. Because the pI of soy protein isolate is between 4.3 and 4.8, rapid release of the model drug theophylline was observed at a pH of 5, as there was little interaction between the drug and soy. At physiological pH, however, soy protein isolate was negatively charged and more strongly interacted with the drug than in acidic conditions, thereby slowing its release [456].
SPI fibers have also been investigated as a drug delivery scaffold. Xu et al. studied the sorption, kinetics and thermodynamics of drug release from SPI fibers for three different drugs [457]. As the temperature was increased, the diffusion coefficient of drug sorption onto the fibers increased. For metformin and diclofenac, sorption and release relied on the ionic interactions between the drug and SPI scaffold. Conversely, these parameters were governed more strongly by van der Waals forces for drugs such as 5-fluorouracil. For all model drugs, burst release was controlled by altering the drug-loading concentration. Because release relies on the affinity of the drug for the SPI, a lower loading concentration resulted in less burst release [457]. Collectively, these studies demonstrate that SPI vehicles can be designed and tuned to deliver a variety of therapeutics.
5.10.3. Corn zein
Zein, the major storage protein in corn, is a low-molecular weight protein in maize endosperm [458]. Depending on its molecular weight and extraction process, zein is classified asα-zein, β-zein, γ-zein, or δ-zein. The most abundant form, α-zein, is a 22- to 24-kDa protein that forms a triple super-helix [459]. Though the structure is not fully elucidated, small-angle X-ray scattering data suggests that nine or ten homologous, helical repeating units arrange into an anti-parallel helix that is stabilized by hydrogen bonding [460,461]. All classes of zein are rich in alanine, leucine, proline, and glutamine residues, but α-zein is the most frequently used class in drug delivery applications due to its abundance. Though zein contains both hydrophilic and hydrophobic regions, its large fraction of non-polar residues makes it insoluble in water without the addition of alcohol, urea, or anionic or alkaline surfactants [459]. The hydrophobicity of zein also renders it mucoadhesive [462].
Zein has been incorporated into “smart” hydrogels to control the release of drugs in response to a biological or environmental stimulus. Liu et al. generated complex hydrogel beads composed of zein and pectin [463]. The hydrophobic zein suppressed bead swelling in physiological conditions, while pectin protected the zein from protease degradation in the gastrointestinal tract, thus promoting colon-specific drug delivery [463]. Zein has also been engineered into an injectable, biodegradable gel that forms in situ. Gao et al. combined zein with sucrose acetate isobutyrate (SAIB) to reduce burst release of the model drug, pingyangmycin hydrochloride [464]. When administered to venous malformations as a sclerosant, the zein-SAIB hydrogel provided four days of prolonged drug release [464]. Zein hydrogels have also been designed for sustained release of chemotherapeutics and reduce their off-target effects. Cao et al. demonstrated that an intratumoral injection of an in situ zein hydrogel extended the release of entrapped DOX, resulting in increased anti-tumor efficacy [465]. They also showed that DOX release could be modulated by adjusting the zein concentration [465].
Most zein-based materials for drug delivery have been microspheres or nanoparticles. Zein is rich in nonpolar residues, which allows it to stably complex with drugs and entrap them in the nanoparticle core. The first reported zein microparticle was designed by Matsuda et al. to improve the therapeutic index of polysaccharide-Kureha (PS-K), a protein polysaccharide that stimulates anti-cancer immune activity [466]. Monodisperse microspheres were generated by sonication and produced 1-μm microspheres that could be phagocytosed by macrophages [466]. Subsequent studies by this group involved combinations of zein with three other anti-cancer drugs to improve their pharmacokinetics, which demonstrates the versatility of the platform [467]. Since then, zein microparticles have been engineered to deliver antibiotics such as ciproflaxin [468], anti-parasitic drugs such as ivermectin [458], and corticosteroids such as prednisolone [469].
Muthuselvi et al. investigated how the solvent used for microsphere fabrication impacts drug release [470]. Using the cardiotonic glycoside gitoxin as a model drug, they fabricated zein microspheres with ethanol, methanol, and isopropanol mixtures. While microspheres made with methanol and isopropanol rapidly released the drug over four days, microspheres prepared with ethanol exhibited biphasic release. Following an initial phase of burst release, drug release was sustained for 80 h before tapering off into a slower, final stage of release [470].
Their ability to harness the EPR effect provides another reason for the use of zein nanoparticles in chemotherapeutics. In one example, Lai et al. loaded zein nanoparticles with 5-fluorouracil for delivery to the liver [471]. When labeled with rhodamine-B intravenously administered, the zein nanoparticles increased liver-targeting efficiency 31% vs. free rhodamine B and 5-fluorouracil uptake in the liver was ~3-fold higher than that of free drug [471]. Dong et al. loaded zein nanoparticles with DOX and achieved 90% loading efficiency [472]. DOX release was pH-dependent, with more rapid release occurring in acidic conditions. Confocal laser scanning microscopy in HeLa cells showed that the zein nanoparticles were effectively endocytosed into the cytoplasm and that smaller nanoparticles could enter the nucleus [472]. More recent work conducted by Alhakamy et al. demonstrated that zein nanoparticles can improve the anti-cancer potency of statins using lovastatin as the model drug [473]. Lovastatin-loaded zein nanoparticles promoted anti-proliferative activity in HepG2 cells by accumulating in the cytoplasm during the G2/M and pre-G phases of mitosis [473]. Further, zein nanoparticles have improved the pharmacokinetics, solubility, and potency of anti-tuberculosis drugs—including rifampicin, pyrazinamide, and isoniazid [474]- and vitamin D3 [475].
5.10.4. Casein
Caseins are milk proteins that evolved from a family of secreted calcium (phosphate)-binding phosphoproteins [476]. Accounting for nearly 80% of all bovine milk proteins, caseins exist in four different forms – αs1, αs2, β-, and κ [477,478]. Though they are all amphiphilic and proline-rich, the four caseins differ in their overall amino acid, phosphorous, and carbohydrate content [479]. Caseins are naturally unstructured, which endows them with the flexibility to exhibit intermolecular interactions. Consequently, they assemble into micelles 50–500 nm in diameter with negatively charged surfaces [479]. The size distribution of these micelles is readily impacted by environmental factors, including pH, ionic strength, and hydrostatic pressure, due to their amphiphilicity and lack of tertiary structure [480,481]. Although they withstand high temperatures of up to ~71°C, they are quickly destabilized by acidic environments, which makes them a good option for oral delivery [482].
Because casein naturally takes on a micellar conformation, it has been readily adapted as a drug nanocarrier. An early example of this idea was demonstrated by Semo et al. They encapsulated the fat-soluble vitamin D2 in the core of casein micelles and demonstrated that the casein barrier protected vitamin D2 from UV degradation [483]. Soon after this finding was published, casein micelles were used to entrap poorly soluble chemotherapeutics, such as curcumin. In this study, Sahu and coworkers used steady state fluorescence spectroscopy to show that curcumin shows hydrophobic interactions with casein. The complexes self-assembled into micelles ~166 nm in diameter, which were efficiently internalized by HeLa cells [484]. The pH-sensitivity of casein nanoparticles also makes them useful for gastrointestinal delivery. The Livney group loaded β-casein nanoparticles with the chemotherapeutic drug mitoxantrone to create stable nanoparticles 100–300 nm in diameter that clustered to trap the drug between micelles as well as within their cores [485]. These micelles rapidly disintegrated in an acidic microenvironment, suggesting their potential for targeting gastric cancer [486]. Casein micelles have similarly been adapted to improve the bioavailability and targeting of other drugs, such as DOX [487], folic acid [488] and flutamide [489].
Casein films have also been applied to tablets to slow drug release. These films, which are typically prepared with transglutaminase as the crosslinker, exhibit remarkable tensile strength, and are gradually hydrolyzed by chymotrypsin [490]. Abu Diak and coworkers coated diltiazem-HCl tablets with casein using four different plasticizing agents and ultimately derived a continuous tablet coat with oleic acid as the olasticizer [491]. At pH 1.2, casein-coated tablets slowed the release of diltiazem-HCl compared to uncoated tablets, which released 80% of drug within 2 h. The rate at which the coated tablets released the drug decreased with increasing post-coating heat treatment temperature. Similarly, at pH 6.8, only coated tablets that underwent post-coating heat treatment at 100°C successfully showed sustained drug release, which was likely due to casein crosslinking under these conditions [491].
The favorable hydrophilicity and biocompatibility of casein, coupled with the availability of reactive sites for chemical modification within its sequence, make it an ideal material for forming hydrogels. In an early example, Song and coworkers created genipin-crosslinked casein hydrogels to encapsulate BSA [492]. Hydrogel swelling and subsequent BSA release was governed by the environmental pH. At pH 1.2, there was minimal hydrogel swelling and the BSA remained entrapped in the gel. When the pH was increased to 7.4, the hydrogel swelled and released greater amounts of BSA, making this hydrogel useful for intestinal delivery. The swelling behavior was similarly influenced by the degree of crosslinking by genipin [492]. In a later study, the same group employed microbial transglutaminase to crosslink casein hydrogels into a fractal network structure compared to uncrosslinked casein hydrogels [493]. Vitamin B12, was loaded into the hydrogel under mild conditions and was slowly released from the hydrogel at pH 7.4. The rate of release slowed with increased crosslinking [493].
Casein-drug composites have also been designed to improve the solubility and dissolution of various drugs. Millar and Corrigan first demonstrated that freeze-dried compacts made from sodium caseinate and acidic casein enhanced the dissolution rates of chlorothiazide and hydrochlorothiazide 35-fold and 1.5-fold, respectively. The improvement in dissolution was related to the complexation with casein and processing of the drug [494]. Millar et al. further characterized the dissolution rate of the compacts over a range of compositions and found that the dissolution rate of acidic casein was half that of sodium caseinate [495]. Using ibuprofen as the model drug, they showed that acidic casein compacts significantly slowed drug dissolution compared to sodium caseinate compacts. They attributed the difference to the changing form of casein that took place due to microenvironmental pH differences in the boundary layer. Acidic casein was also more viscous and rigid than sodium caseinate, which decreased its solubility and reduced drug diffusivity [495]. A subsequent research group demonstrated that acidic casein and sodium caseinate could be mixed with other materials, such as hydroxypropylmethocellulose matrices, to slow drug dissolution [496].
5.10.5. Iron binding proteins
Iron binding proteins – most notably transferrin and ferritin – have been widely explored as drug delivery vehicles due to the increased need for iron in many diseases, including cancer. In these applications, iron binding proteins serve to both improve the pharmacokinetics and stability of their cargo, but also to target the drug by taking advantage of the body's natural iron transport mechanisms.
Transferrin is an abundant glycoprotein that plays a critical role in the systemic transport of iron. As a natural iron-chelating agent, it reversibly binds one or two ferric ions to maintain their solubility and prevent toxic free-radical generation [497]. It also promotes iron endocytosis by binding the transferrin receptor 1, which is upregulated on malignant cells to meet their increased iron needs [498]. Transferrin receptor 1 is also expressed on brain endothelial cells, making transferrin an attractive option for delivering therapeutic cargo across the blood brain barrier [499]. It should be noted that, due to its unique targeting capacity, transferrin has been used as a targeting moiety to deliver both small molecule chemotherapeutics and peptides/proteins.
Transferrin-drug conjugates have been employed to target chemotherapeutics to tumors delivery and reduce the off-target toxicity of the drug. Szwed et al. coupled DOX to transferrin and showed that transferrin could overcome anthracycline resistance by impairing the P-glycoprotein activity in several anthracycline-resistant cell lines such as DOX resistant K562 cells [500]. In a subsequent study, they demonstrated that a DOX-transferrin conjugate successfully induces apoptosis in leukemia cells while avoiding toxicity to non-malignant cells [501]. Transferrin was also used to target cytochrome C to lung tumors by conjugating it to transferrin using Sulfo-LC SPDP as a crosslinker [502]. This strategy resulted in nanocarriers ~10 nm in diameter, allowing them to avoid rapid kidney filtration. Upon endocytosis by cells, the redox-sensitive bond in the linker was cleaved, releasing cytochrome c from transferrin and initiating apoptosis [502].
Transferrin based nanoparticles have also been engineered to deliver photothermal and photodynamic agents for cancer therapy. Wang et al. treated transferrin with DTT, leading to exposure of hydrophobic residues that could interact with the near-infrared dye IR780 through hydrophobic interactions [503]. These dye-loaded nanoparticles had a half-life of 20 h and were readily endocytosed by malignant cells within 2 h. They accumulated in which tumor with a high tumor-to-background ratio and, when pulsed with an 808 nm laser, caused a temperature increase to 49.5°C that caused tumor regression, whereas tumors treated with PBS only showed a temperature increase to 36.3°C, which had no effect on tumor growth [503]. This study demonstrates how transferrin could be used to enhance the pharmacokinetics and delivery of hydrophobic agents.
Transferrin has similarly been fused to GLP-1 to improve the with improved pharmacokinetics of this peptide drug for treatment of type 2 diabetes [504]. This technology, developed by BioRexis and later acquired by Pfizer, consists of non-glycosylated transferrin genetically fused to GLP-1, creating a fusion protein that is expressed in yeast cells. Fusion with transferrin did not affect the insulinotrophic properties of GLP-1 and yielded a half-life of 1–2 days, compared to a half-life of minutes for native GLP-1 [504]. Furthermore, the GLP-1-transferrin fusion promoted greater β-cell proliferation and β-cell area compared to transferrin alone. Two days after receiving an intraperitoneal injection of GLP-1-transferrin, treated mice demonstrated a significant increase in β-cell proliferation with nearly 10% of total islet cell possessing BrdU+ nuclei. Meanwhile, β-cells in animals treated with transferrin alone did not show signs of proliferation after two days [504]. As such, GLP-1-transferrin may provide a novel strategy for restoring β-cell function in diabetics while maintaining blood glucose control.
Transferrin has also been exploited to treat autoimmune diseases such as myasthenia gravis (MG). In patients with MG, autoantibodies recognize and block nicotinic acetylcholine receptors (AChR). By fusing the α-subunit of AChR to transferrin, Keefe et al. created SHG2210, a fusion protein that is recognized and bound by anti-AChR autoantibodies and is internalized by cells via the transferrin receptor [505]. Results from in vitro binding and internalization studies show that anti-AChR autoantibodies bind SHG2210 with an IC50 of 0.37 μM, while no binding to transferrin alone was observed [505]. When the SHG2210:antibody complex was added to HeLa cells in culture, the complex was readily internalized via the transferrin receptor, as seen by the negligible uptake by the cells in the presence of saturating levels of a soluble AChR inhibitor. After internalization, the complex was trafficked to the lysosome and was degraded [505].
Transferrin delivery platforms were further optimized by Chen et al., who evaluated the effect of various linkers on the binding affinity and pharmacokinetics of transferrin fusion proteins. In this work, growth hormone (GH) or granulocyte colony-stimulating factor (G-CSF) were genetically linked to transferrin via a dipeptide linker, a helical peptide linker, or a thrombin-cleavable, disulfide cyclopeptide linker [506]. For both GH-Tf and G-CSF-Tf, the longer –helical or cyclopeptide linkers– linkers resulted in less steric hindrance and thus provided greater binding affinity between GH or G-CSF and their receptors as well as transferrin and its receptor. Fusion proteins with the highest affinity for their targets also had the greatest plasma half-life [506]. The fusion containing the cyclopeptide linker had the greatest affinity for the transferrin receptor (IC50 of 0.9 nM) and achieved a half-life of 5.7 h. In contrast, the fusion with the dipeptide linker had an 8-fold lower affinity for the transferrin receptor and exhibited a shorter half-life of 4.2 h [506]. This study demonstrates how delivery of a protein drug via a transferrin carrier can be modulated by tuning the length and type of linker between the transferrin and its cargo.
Like transferrin, ferritin has also been exploited to target and carry drugs. Ferritin is an iron-storage protein composed of 24 subunits that assemble into a spherical nanocage [507]. While this nanocage naturally houses up to 4500 iron atoms, it can also be engineered to house a variety of therapeutics [508,509]. Its loading capacity depends on several factors, including MW of the drug, the encapsulation strategy, and choice of co-solvent for entrapping drugs. Metal salts achieve the highest encapsulation efficiency with nearly 5000 particles of AgNO3 fitting in a single cage, whereas small, hydrophobic molecules – including many chemotherapeutics, exhibit lower loading efficiency [510,511].
Taking advantage of ferritin's natural ability to traverse the blood brain barrier, Fan et al. loaded a ferritin nanocage with an infrared dye and administered it to mice bearing U87MB orthotopic gliomas [512]. Accumulation of the ferritin nanocage in the glioma-bearing mice was 10-fold greater than in the control group that received the dye alone, with maximum accumulation 4–6 h after administration. Interestingly, nanocage accumulation was limited to the tumor and no significant accumulation was observed in healthy brain tissue [512]. Fan et al. then loaded the nanocages with DOX. and administered them intravenously. The DOX-loaded nanocages nearly doubled survival time compared to treatment with free DOX and significantly reduced tumor volume. Additionally, mice treated with the DOX-loaded nanocages exhibited slowed weight loss, indicating that the formulation reduced toxicity compared to free DOX [512]. In a separate study, DOX loaded ferritin nanocages increased the half-life of DOX 12-fold and the AUC by over 100-fold [513]. Ferritin nanocages have also been used to deliver the poly ADP ribose polymerase inhibitor Olaparib to treat multiple forms of breast cancer. A study conducted by Mazzucchelli et al. demonstrated that the encapsulation of the drug in a ferritin nanocage led to 1000-fold greater uptake of the drug by triple negative breast cancer cells compared to the free drug [514]. The nanocage enhanced the cytotoxicity of the drug in cancer cells and reduced off-target toxicity in healthy cells. These studies demonstrate that ferritin nanocages can be harnessed to improve the delivery of chemotherapeutics in a variety of cancers.
In another application, Jiang et al. synthesized a cobalt nanozyme - a nanomaterial with intrinsic enzyme-like activity - within a ferritin nanocage [515]. Synthesis of the nanozyme within the confines of the nanocage provided better control over the size and structure of the nanozyme compared to nanozymes synthesized outside of the nanocage. The hepatocellular carcinoma targeting peptide SP94 was recombinantly fused to ferritin such that it was displayed on the nanocage exterior for optimal targeting [515]. Though this design was used for diagnostic purposes, the remarkable targeting specificity it achieved demonstrates how it can be readily adapted as a drug delivery vehicle.
Ferritin nanocages can also be employed to deliver nucleic acids. Li et al. successfully encapsulated siRNA into the nanocage, protecting it from rapid in vivo degradation [516]. This system exhibited highly efficient transfection into primary human tumorigenic cell lines, PMBCs, and mesenchymal stem cells. Notably, up to 85% gene knockdown was achieved by the nanocage, while Lipofectamine —the gold standard— only achieved 40% gene knockdown [516]. At the low siRNA concentration of 10 nM used in this study, Lipofectamine is relatively inefficient at transfection as it requires a higher siRNA concentration for optimal activity, whereas the nanocage is more effective at gene knockdown at this low siRNA concentration. Because the biophysical properties and encapsulation of siRNA are sequence-independent, this approach could be adapted for siRNA delivery for numerous applications [516].
6. Conclusion, challenges, and future prospects
The research discussed in this review highlights the versatility and multifunctionality of protein-based materials for drug delivery. These carriers benefit from exceptional biocompatibility and biodegradability and minimal toxicity. Most importantly, protein-based materials are highly customizable. Their chemical, physical, and biological properties can be tailored for a specific application with sequence-level precision, thus providing the researcher with full control over the formulation's pharmacokinetics, pharmacodynamics, biodistribution, and in vivo interactions. Much of this work has been made possible by continuing advances in genetic engineering and synthetic biology, which have made it possible to incorporate new chemistries and structural motifs into the polypeptide sequence to improve drug conjugation, targeting, stimulus responsiveness, and self-assembly.
Researchers continue to explore new ways to engineer proteins to possess desirable chemical and mechanical properties. One such strategy is to create hybrid materials by combining a protein with other proteins or synthetic components to generate a material with the favorable physicochemical properties of its building blocks. Another method focuses on conferring new functionalities onto proteins through chemical processing, post-translational modification, or the incorporation of unnatural amino acids into the protein sequence. This approach expands the range of drugs that can be recombinantly fused to, chemically conjugated to, or physically encapsulated by the material. New protein-based materials also continue to be developed from existing proteins and by de novo design of peptide sequences that can introduce new attributes to protein materials to improve drug delivery.
As researchers continue to make progress in adapting existing and developing new protein materials, there is also great opportunity for biological and materials science research that will maximize the clinical success of protein drug carriers. Despite the progress made in this field, formulations using protein-based materials rarely reach or survive clinical trials. They are hindered by poor in vivo stability and performance, which results from an insufficient understanding of how to create materials that remain stable and functional in the harsh environments present during storage, administration, and circulation. This is further highlighted by the fact that, even though the half-life of a drug can be significantly increased by its protein carrier, the half-life of the formulation still falls short of that of the native protein. Alterations to the chemical and physical structure can change how a material and drug are endocytosed by their target, degraded and cleared from circulation, or recognized by immune cells. As new chemistries and hierarchical structures are explored, this knowledge becomes more critical. Through increased understanding of these fundamental principles, we can better design and predict the in vivo behavior of protein-based drug delivery vehicles.
As new materials are developed, enhanced biological and materials science research can improve tools used to predict their behavior and improve their design. For example, experimental results will boost theoretical models and molecular simulations, which will in turn enhance de novo design of protein architectures. This will inform our understanding of structure-function relationships such that new morphologies can be engineered to control the delivery of therapeutics. It is also necessary to continue developing tools to improve the production of protein materials. Current methods suffer from low yields, limited options for post-translational modification, or high endotoxin levels. By exploring alternate systems – including bacterial, yeast, mammalian, and plant cell-based – researchers can address these challenges.
Acknowledgements
A.V. and S.S. contributed equally to this work. A.V. acknowledges the support of the NSF through a graduate research fellowship. A.C. acknowledges support from the National Institutes of Health (NIH) through an MIRA grant R35GM127042 and R21CA237705.
References
- 1.Petrak K. Essential properties of drug-targeting delivery systems. Drug Discov. Today. 2005;10:1667–1673. doi: 10.1016/S1359-6446(05)03698-6. [DOI] [PubMed] [Google Scholar]
- 2.Fenton O.S., Olafson K.N., Pillai P.S., Mitchell M.J., Langer R. Advances in biomaterials for drug delivery. Adv. Mater. 2018;30:1705328. doi: 10.1002/adma.201705328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li C., Wang J., Wang Y., Gao H., Wei G., Huang Y., Yu H., Gan Y., Wang Y., Mei L., Chen H., Hu H., Zhang Z., Jin Y. Recent progress in drug delivery. Acta Pharm. Sin. B. 2019;9:1145–1162. doi: 10.1016/j.apsb.2019.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tong X., Pan W., Su T., Zhang M., Dong W., Qi X. Recent advances in natural polymer-based drug delivery systems. React. Funct. Polym. 2020;148:104501. doi: 10.1016/j.reactfunctpolym.2020.104501. [DOI] [Google Scholar]
- 5.Jao D., Xue Y., Medina J., Hu X. Protein-based drug-delivery materials. Materials (Basel) 2017;10 doi: 10.3390/ma10050517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abascal N.C., Regan L. The past, present and future of protein-based materials. Open Biol. 2018;8 doi: 10.1098/rsob.180113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saha S., Banskota S., Roberts S., Kirmani N., Chilkoti A. Engineering the architecture of elastin-like polypeptides: from Unimers to hierarchical self-assembly. Adv. Ther. 2020;3:1900164. doi: 10.1002/adtp.201900164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Elzoghby A.O., Samy W.M., Elgindy N.A. Protein-based nanocarriers as promising drug and gene delivery systems. J. Control. Release. 2012;161:38–49. doi: 10.1016/j.jconrel.2012.04.036. [DOI] [PubMed] [Google Scholar]
- 9.Strebhardt K., Ullrich A. Paul Ehrlich's magic bullet concept: 100 years of progress. Nat. Rev. Cancer. 2008;8:473–480. doi: 10.1038/nrc2394. [DOI] [PubMed] [Google Scholar]
- 10.Yun Y.H., Lee B.K., Park K. Controlled drug delivery: historical perspective for the next generation. J. Control. Release. 2015;219:2–7. doi: 10.1016/j.jconrel.2015.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hoffman A.S. The origins and evolution of “controlled” drug delivery systems. J. Control. Release. 2008;132:153–163. doi: 10.1016/j.jconrel.2008.08.012. [DOI] [PubMed] [Google Scholar]
- 12.Park K. Controlled drug delivery systems: past forward and future back. J. Control. Release. 2014;190:3–8. doi: 10.1016/j.jconrel.2014.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang Y., Chan H.F., Leong K.W. Advanced materials and processing for drug delivery: the past and the future. Adv. Drug Deliv. Rev. 2013;65:104–120. doi: 10.1016/j.addr.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rosen H., Abribat T. The rise and rise of drug delivery. Nat. Rev. Drug Discov. 2005;4:381–385. doi: 10.1038/nrd1721. [DOI] [PubMed] [Google Scholar]
- 15.Uhrich K.E., Cannizzaro S.M., Langer R.S., Shakesheff K.M. Polymeric Systems for Controlled Drug Release. Chem. Rev. 1999;99:3181–3198. doi: 10.1021/cr940351u. [DOI] [PubMed] [Google Scholar]
- 16.Ekladious I., Colson Y.L., Grinstaff M.W. Polymer–drug conjugate therapeutics: advances, insights and prospects. Nat. Rev. Drug Discov. 2019;18:273–294. doi: 10.1038/s41573-018-0005-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Simone E.A., Dziubla T.D., Muzykantov V.R. Polymeric carriers: role of geometry in drug delivery. Expert Opin. Drug Deliv. 2008;5:1283–1300. doi: 10.1517/17425240802567846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hickey J.W., Santos J.L., Williford J.-M., Mao H.-Q. Control of polymeric nanoparticle size to improve therapeutic delivery. J. Control. Release. 2015;219:536–547. doi: 10.1016/j.jconrel.2015.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hamid Akash M.S., Rehman K., Chen S. Natural and synthetic polymers as drug carriers for delivery of therapeutic proteins. Polym. Rev. 2015;55:371–406. doi: 10.1080/15583724.2014.995806. [DOI] [Google Scholar]
- 20.Kobsa S., Saltzman W.M. Bioengineering approaches to controlled protein delivery. Pediatr. Res. 2008;63:513–519. doi: 10.1203/PDR.0b013e318165f14d. [DOI] [PubMed] [Google Scholar]
- 21.Choi S.M., Chaudhry P., Zo S.M., Han S.S. In: Advances in Protein-Based Materials: From Origin to Novel Biomaterials BT - Cutting-Edge Enabling Technologies for Regenerative Medicine. Chun H.J., Park C.H., Kwon I.K., Khang G., editors. Springer Singapore; Singapore: 2018. pp. 161–210. [DOI] [PubMed] [Google Scholar]
- 22.Boyle A.L. Pept Appl. Biomed. Biotechnol. Bioeng. Elsevier Inc.; 2018. Applications of de novo designed peptides; pp. 51–86. [DOI] [Google Scholar]
- 23.Song R., Murphy M., Li C., Ting K., Soo C., Zheng Z. Current development of biodegradable polymeric materials for biomedical applications. Drug Des. Devel. Ther. 2018;12:3117–3145. doi: 10.2147/DDDT.S165440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rodríguez Cabello J.C., González De Torre I., Santos M., Testera A.M., Alonso M. Chemical Modification of Biomaterials from Nature. Biomater. from Nat. Adv. Devices Ther. 2016:444–474. doi: 10.1002/9781119126218.ch24. [DOI] [Google Scholar]
- 25.Sengupta D., Heilshorn S.C. Protein-engineered biomaterials: highly tunable tissue engineering scaffolds. Tissue Eng. Part B. Rev. 2010;16:285–293. doi: 10.1089/ten.teb.2009.0591. [DOI] [PubMed] [Google Scholar]
- 26.Wals K., Ovaa H. Unnatural amino acid incorporation in E. coli: current and future applications in the design of therapeutic proteins. Front. Chem. 2014;2:15. doi: 10.3389/fchem.2014.00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang P.-S., Boyken S.E., Baker D. The coming of age of de novo protein design. Nature. 2016;537:320–327. doi: 10.1038/nature19946. [DOI] [PubMed] [Google Scholar]
- 28.Hollingshead S., Lin C.-Y., Liu J.C. Designing Smart Materials with Recombinant Proteins. Macromol. Biosci. 2017;17 doi: 10.1002/mabi.201600554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sun H., Luo Q., Hou C., Liu J. Nanostructures based on protein self-assembly: from hierarchical construction to bioinspired materials. Nano Today. 2017;14:16–41. doi: 10.1016/j.nantod.2017.04.006. [DOI] [Google Scholar]
- 30.Gelderblom H., Verweij J., Nooter K., Sparreboom A. Cremophor EL: the drawbacks and advantages of vehicle selection for drug formulation. Eur. J. Cancer. 2001;37:1590–1598. doi: 10.1016/S0959-8049(01)00171-X. [DOI] [PubMed] [Google Scholar]
- 31.Du B., Yu M., Zheng J. Transport and interactions of nanoparticles in the kidneys. Nat. Rev. Mater. 2018;3:358–374. doi: 10.1038/s41578-018-0038-3. [DOI] [Google Scholar]
- 32.Brenner B.M., Hostetter T.H., Humes H.D. Glomerular permselectivity: Barrier function based on discrimination of molecular size and charge. Am. J. Physiol. - Ren. Fluid Electrolyte Physiol. 1978;3 doi: 10.1152/ajprenal.1978.234.6.f455. [DOI] [PubMed] [Google Scholar]
- 33.Zahr A.S., Davis C.A., Pishko M.V. Macrophage uptake of core-shell nanoparticles surface modified with poly(ethylene glycol) Langmuir. 2006;22:8178–8185. doi: 10.1021/la060951b. [DOI] [PubMed] [Google Scholar]
- 34.Park J., Choi Y., Chang H., Um W., Ryu J.H., Kwon I.C. Alliance with EPR effect: combined strategies to improve the EPR effect in the tumor microenvironment. Theranostics. 2019;9:8073–8090. doi: 10.7150/thno.37198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Salahpour Anarjan F. Active targeting drug delivery nanocarriers: ligands. Nano Struct. Nano-Objects. 2019;19:100370. doi: 10.1016/j.nanoso.2019.100370. [DOI] [Google Scholar]
- 36.Attia M.F., Anton N., Wallyn J., Omran Z., Vandamme T.F. An overview of active and passive targeting strategies to improve the nanocarriers efficiency to tumour sites. J. Pharm. Pharmacol. 2019;71:1185–1198. doi: 10.1111/jphp.13098. [DOI] [PubMed] [Google Scholar]
- 37.Azzi S., Hebda J.K., Gavard J. Vascular permeability and drug delivery in cancers. Front. Oncol. 2013;3:211. doi: 10.3389/fonc.2013.00211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barua S., Mitragotri S. Challenges associated with penetration of nanoparticles across cell and tissue barriers: a review of current status and future prospects. Nano Today. 2014;9:223–243. doi: 10.1016/j.nantod.2014.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tee J.K., Yip L.X., Tan E.S., Santitewagun S., Prasath A., Ke P.C., Ho H.K., Leong D.T. Nanoparticles' interactions with vasculature in diseases. Chem. Soc. Rev. 2019;48:5381–5407. doi: 10.1039/c9cs00309f. [DOI] [PubMed] [Google Scholar]
- 40.Nelemans L.C., Gurevich L. Drug delivery with polymeric nanocarriers-cellular uptake mechanisms. Materials (Basel) 2020;13 doi: 10.3390/ma13020366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang R., Qin X., Kong F., Chen P., Pan G. Improving cellular uptake of therapeutic entities through interaction with components of cell membrane. Drug Deliv. 2019;26:328–342. doi: 10.1080/10717544.2019.1582730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rajendran L., Knölker H.J., Simons K. Subcellular targeting strategies for drug design and delivery. Nat. Rev. Drug Discov. 2010;9:29–42. doi: 10.1038/nrd2897. [DOI] [PubMed] [Google Scholar]
- 43.Son G.H., Lee B.J., Cho C.W. Mechanisms of drug release from advanced drug formulations such as polymeric-based drug-delivery systems and lipid nanoparticles. J. Pharm. Investig. 2017;47:287–296. doi: 10.1007/s40005-017-0320-1. [DOI] [Google Scholar]
- 44.Sertkaya A., Wong H.H., Jessup A., Beleche T. Key cost drivers of pharmaceutical clinical trials in the United States. Clin. Trials. 2016;13:117–126. doi: 10.1177/1740774515625964. [DOI] [PubMed] [Google Scholar]
- 45.Patel S.G., Sayers E.J., He L., Narayan R., Williams T.L., Mills E.M., Allemann R.K., Luk L.Y.P., Jones A.T., Tsai Y.H. Cell-penetrating peptide sequence and modification dependent uptake and subcellular distribution of green florescent protein in different cell lines. Sci. Rep. 2019;9:1–9. doi: 10.1038/s41598-019-42456-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cao J., Zhang Y., Wu Y., Wu J., Wang W., Wu Q., Yuan Z. The effects of ligand valency and density on the targeting ability of multivalent nanoparticles based on negatively charged chitosan nanoparticles. Colloids Surfaces B Biointerfaces. 2018;161:508–518. doi: 10.1016/j.colsurfb.2017.11.015. [DOI] [PubMed] [Google Scholar]
- 47.Wang J., Saha S., Schaal J.L., Yousefpour P., Li X., Chilkoti A. Heuristics for the optimal presentation of bioactive peptides on polypeptide micelles. Nano Lett. 2019;19:7977–7987. doi: 10.1021/acs.nanolett.9b03141. [DOI] [PubMed] [Google Scholar]
- 48.Antoine E.E., Vlachos P.P., Rylander M.N. Review of collagen i hydrogels for bioengineered tissue microenvironments: characterization of mechanics, structure, and transport. Tissue Eng. - Part B Rev. 2014;20:683–696. doi: 10.1089/ten.teb.2014.0086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xing Q., Yates K., Vogt C., Qian Z., Frost M.C., Zhao F. Increasing mechanical strength of gelatin hydrogels by divalent metal ion removal. Sci. Rep. 2014;4:1–10. doi: 10.1038/srep04706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Elzoghby A.O., Samy W.M., Elgindy N.A. Albumin-based nanoparticles as potential controlled release drug delivery systems. J. Control. Release. 2012;157:168–182. doi: 10.1016/j.jconrel.2011.07.031. [DOI] [PubMed] [Google Scholar]
- 51.Ramteke S., Jain N. Clarithromycin- and omeprazole-containing gliadin nanoparticles for the treatment of Helicobacter pylori. J. Drug Target. 2008;16:65–72. doi: 10.1080/10611860701733278. [DOI] [PubMed] [Google Scholar]
- 52.Vasconcelos A., Cavaco-Paulo A. The use of keratin in biomedical applications. Curr. Drug Targets. 2013;14:612–619. doi: 10.2174/1389450111314050010. [DOI] [PubMed] [Google Scholar]
- 53.Lammel A.S., Hu X., Park S.H., Kaplan D.L., Scheibel T.R. Controlling silk fibroin particle features for drug delivery. Biomaterials. 2010;31:4583–4591. doi: 10.1016/j.biomaterials.2010.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Banskota S., Saha S., Bhattacharya J., Kirmani N., Yousefpour P., Dzuricky M., Zakharov N., Li X., Spasojevic I., Young K., Chilkoti A. Genetically encoded stealth nanoparticles of a Zwitterionic polypeptide-paclitaxel conjugate have a wider therapeutic window than Abraxane in multiple tumor models. Nano Lett. 2020;20:2396–2409. doi: 10.1021/acs.nanolett.9b05094. [DOI] [PubMed] [Google Scholar]
- 55.Dzuricky M., Xiong S., Weber P., Chilkoti A. Avidity and cell uptake of integrin-targeting polypeptide micelles is strongly shape-dependent. Nano Lett. 2019;19:6124–6132. doi: 10.1021/acs.nanolett.9b02095. [DOI] [PubMed] [Google Scholar]
- 56.Mozhdehi D., Luginbuhl K.M., Simon J.R., Dzuricky M., Berger R., Varol H.S., Huang F.C., Buehne K.L., Mayne N.R., Weitzhandler I., Bonn M., Parekh S.H., Chilkoti A. Genetically encoded lipid-polypeptide hybrid biomaterials that exhibit temperature-triggered hierarchical self-assembly. Nat. Chem. 2018;10:496–505. doi: 10.1038/s41557-018-0005-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee J.H., Yeo Y. Controlled drug release from pharmaceutical nanocarriers. Chem. Eng. Sci. 2015;125:75–84. doi: 10.1016/j.ces.2014.08.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bellucci J.J., Bhattacharyya J., Chilkoti A. A noncanonical function of sortase enables site-specific conjugation of small molecules to lysine residues in proteins. Angew. Chemie - Int. Ed. 2015;54:441–445. doi: 10.1002/anie.201408126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yousefpour P., Ahn L., Tewksbury J., Saha S., Costa S.A., Bellucci J.J., Li X., Chilkoti A. Conjugate of doxorubicin to albumin-binding peptide outperforms Aldoxorubicin. Small. 2019;15:1804452. doi: 10.1002/smll.201804452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Qi Y., Amiram M., Gao W., McCafferty D.G., Chilkoti A. Sortase-catalyzed initiator attachment enables high yield growth of a stealth polymer from the C terminus of a protein. Macromol. Rapid Commun. 2013;34:1256–1260. doi: 10.1002/marc.201300460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen L., Cohen J., Song X., Zhao A., Ye Z., Feulner C.J., Doonan P., Somers W., Lin L., Chen P.R. Improved variants of SrtA for site-specific conjugation on antibodies and proteins with high efficiency. Sci. Rep. 2016;6:1–12. doi: 10.1038/srep31899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jastrzebska K., Kucharczyk K., Florczak A., Dondajewska E., Mackiewicz A., Dams-Kozlowska H. Silk as an innovative biomaterial for cancer therapy. Reports Pract. Oncol. Radiother. 2015;20:87–98. doi: 10.1016/j.rpor.2014.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pandey V., Haider T., Jain P., Gupta P.N., Soni V. Silk as a leading-edge biological macromolecule for improved drug delivery. J. Drug Deliv. Sci. Technol. 2020;55:101294. doi: 10.1016/j.jddst.2019.101294. [DOI] [Google Scholar]
- 64.Mottaghitalab F., Farokhi M., Shokrgozar M.A., Atyabi F., Hosseinkhani H. Silk fibroin nanoparticle as a novel drug delivery system. J. Control. Release. 2015;206:161–176. doi: 10.1016/j.jconrel.2015.03.020. [DOI] [PubMed] [Google Scholar]
- 65.Altman G.H., Diaz F., Jakuba C., Calabro T., Horan R.L., Chen J., Lu H., Richmond J., Kaplan D.L. Silk-based biomaterials. Biomaterials. 2003;24:401–416. doi: 10.1016/S0142-9612(02)00353-8. [DOI] [PubMed] [Google Scholar]
- 66.Gil E.S., Park S.H., Hu X., Cebe P., Kaplan D.L. Impact of sterilization on the enzymatic degradation and mechanical properties of silk biomaterials. Macromol. Biosci. 2014;14:257–269. doi: 10.1002/mabi.201300321. [DOI] [PubMed] [Google Scholar]
- 67.Yucel T., Lovett M.L., Kaplan D.L. Silk-based biomaterials for sustained drug delivery. J. Control. Release. 2014;190:381–397. doi: 10.1016/j.jconrel.2014.05.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang Y., Guo J., Zhou L., Ye C., Omenetto F.G., Kaplan D.L., Ling S. Design, fabrication, and function of silk-based nanomaterials. Adv. Funct. Mater. 2018;28:1805305. doi: 10.1002/adfm.201805305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Murphy A.R., Kaplan D.L. Biomedical applications of chemically-modified silk fibroin. J. Mater. Chem. 2009;19:6443–6450. doi: 10.1039/b905802h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Numata K., Kaplan D.L. Silk-based delivery systems of bioactive molecules. Adv. Drug Deliv. Rev. 2010;62:1497–1508. doi: 10.1016/j.addr.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Megeed Z., Cappello J., Ghandehari H. Genetically engineered silk-elastinlike protein polymers for controlled drug delivery. Adv. Drug Deliv. Rev. 2002;54:1075–1091. doi: 10.1016/S0169-409X(02)00063-7. [DOI] [PubMed] [Google Scholar]
- 72.Frandsen J.L., Ghandehari H. Recombinant protein-based polymers for advanced drug delivery. Chem. Soc. Rev. 2012;41:2696–2706. doi: 10.1039/c2cs15303c. [DOI] [PubMed] [Google Scholar]
- 73.Aigner T.B., DeSimone E., Scheibel T. Biomedical applications of recombinant silk-based materials. Adv. Mater. 2018;30:1704636. doi: 10.1002/adma.201704636. [DOI] [PubMed] [Google Scholar]
- 74.Mathur A.B., Gupta V. Silk fibroin-derived nanoparticles for biomedical applications. Nanomedicine. 2010;5:807–820. doi: 10.2217/nnm.10.51. [DOI] [PubMed] [Google Scholar]
- 75.Wang X., Yucel T., Lu Q., Hu X., Kaplan D.L. Silk nanospheres and microspheres from silk/pva blend films for drug delivery. Biomaterials. 2010;31:1025–1035. doi: 10.1016/j.biomaterials.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shi P., Goh J.C.H. Self-assembled silk fibroin particles: tunable size and appearance. Powder Technol. 2012;215–216:85–90. doi: 10.1016/j.powtec.2011.09.012. [DOI] [Google Scholar]
- 77.Shi P., Goh J.C.H. Release and cellular acceptance of multiple drugs loaded silk fibroin particles. Int. J. Pharm. 2011;420:282–289. doi: 10.1016/j.ijpharm.2011.08.051. [DOI] [PubMed] [Google Scholar]
- 78.Crivelli B., Bari E., Perteghella S., Catenacci L., Sorrenti M., Mocchi M., Faragò S., Tripodo G., Prina-Mello A., Torre M.L. Silk fibroin nanoparticles for celecoxib and curcumin delivery: ROS-scavenging and anti-inflammatory activities in an in vitro model of osteoarthritis. Eur. J. Pharm. Biopharm. 2019;137:37–45. doi: 10.1016/j.ejpb.2019.02.008. [DOI] [PubMed] [Google Scholar]
- 79.Yan H.B., Zhang Y.Q., Ma Y.L., Zhou L.X. Biosynthesis of insulin-silk fibroin nanoparticles conjugates and in vitro evaluation of a drug delivery system. J. Nanopart. Res. 2009;11:1937–1946. doi: 10.1007/s11051-008-9549-y. [DOI] [Google Scholar]
- 80.Numata K., Subramanian B., Currie H.A., Kaplan D.L. Bioengineered silk protein-based gene delivery systems. Biomaterials. 2009;30:5775–5784. doi: 10.1016/j.biomaterials.2009.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Numata K., Hamasaki J., Subramanian B., Kaplan D.L. Gene delivery mediated by recombinant silk proteins containing cationic and cell binding motifs. J. Control. Release. 2010;146:136–143. doi: 10.1016/j.jconrel.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Numata K., Kaplan D.L. Silk-based gene carriers with cell membrane destabilizing peptides. Biomacromolecules. 2010;11:3189–3195. doi: 10.1021/bm101055m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yang R., Hou M., Gao Y., Lu S., Zhang L., Xu Z., Li C.M., Kang Y., Xue P. Biomineralization-inspired crystallization of manganese oxide on silk fibroin nanoparticles for in vivo MR/fluorescence imaging-assisted tri-modal therapy of cancer. Theranostics. 2019;9:6314–6333. doi: 10.7150/thno.36252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mao B., Liu C., Zheng W., Li X., Ge R., Shen H., Guo X., Lian Q., Shen X., Li C. Cyclic cRGDfk peptide and Chlorin e6 functionalized silk fibroin nanoparticles for targeted drug delivery and photodynamic therapy. Biomaterials. 2018;161:306–320. doi: 10.1016/j.biomaterials.2018.01.045. [DOI] [PubMed] [Google Scholar]
- 85.Yang Y.J., Kwon Y., Choi B.H., Jung D., Seo J.H., Lee K.H., Cha H.J. Multifunctional adhesive silk fibroin with blending of RGD-bioconjugated mussel adhesive protein. Biomacromolecules. 2014;15:1390–1398. doi: 10.1021/bm500001n. [DOI] [PubMed] [Google Scholar]
- 86.Wu H., Liu S., Xiao L., Dong X., Lu Q., Kaplan D.L. Injectable and pH-responsive silk Nanofiber hydrogels for sustained anticancer drug delivery. ACS Appl. Mater. Interfaces. 2016;8:17118–17126. doi: 10.1021/acsami.6b04424. [DOI] [PubMed] [Google Scholar]
- 87.Lovett M.L., Wang X., Yucel T., York L., Keirstead M., Haggerty L., Kaplan D.L. Silk hydrogels for sustained ocular delivery of anti-vascular endothelial growth factor (anti-VEGF) therapeutics. Eur. J. Pharm. Biopharm. 2015;95:271–278. doi: 10.1016/j.ejpb.2014.12.029. [DOI] [PubMed] [Google Scholar]
- 88.Ding Z., Zhou M., Zhou Z., Zhang W., Jiang X., Lu X., Zuo B., Lu Q., Kaplan D.L. Injectable silk Nanofiber hydrogels for sustained release of Small-molecule drugs and vascularization. ACS Biomater. Sci. Eng. 2019;5:4077–4088. doi: 10.1021/acsbiomaterials.9b00621. [DOI] [PubMed] [Google Scholar]
- 89.Gong Z., Yang Y., Ren Q., Chen X., Shao Z. Injectable thixotropic hydrogel comprising regenerated silk fibroin and hydroxypropylcellulose. Soft Matter. 2012;8:2875–2883. doi: 10.1039/c2sm06984a. [DOI] [Google Scholar]
- 90.Germershaus O., Werner V., Kutscher M., Meinel L. Deciphering the mechanism of protein interaction with silk fibroin for drug delivery systems. Biomaterials. 2014;35:3427–3434. doi: 10.1016/j.biomaterials.2013.12.083. [DOI] [PubMed] [Google Scholar]
- 91.Mao K.L., Fan Z.L., Yuan J.D., Chen P.P., Yang J.J., Xu J., ZhuGe D.L., Jin B.H., Zhu Q.Y., Shen B.X., Sohawon Y., Zhao Y.Z., Xu H.L. Skin-penetrating polymeric nanoparticles incorporated in silk fibroin hydrogel for topical delivery of curcumin to improve its therapeutic effect on psoriasis mouse model. Colloids Surfaces B Biointerfaces. 2017;160:704–714. doi: 10.1016/j.colsurfb.2017.10.029. [DOI] [PubMed] [Google Scholar]
- 92.Wu P., Liu Q., Wang Q., Qian H., Yu L., Liu B., Li R. Novel silk fibroin nanoparticles incorporated silk fibroin hydrogel for inhibition of cancer stem cells and tumor growth. Int. J. Nanomed. 2018;13:5405–5418. doi: 10.2147/IJN.S166104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gangrade A., Mandal B.B. Injectable carbon nanotube impregnated silk based multifunctional hydrogel for localized targeted and on-demand anticancer drug delivery. ACS Biomater. Sci. Eng. 2019;5:2365–2381. doi: 10.1021/acsbiomaterials.9b00416. [DOI] [PubMed] [Google Scholar]
- 94.He W., Li P., Zhu Y., Liu M., Huang X., Qi H. An injectable silk fibroin nanofiber hydrogel hybrid system for tumor upconversion luminescence imaging and photothermal therapy. New J. Chem. 2019;43:2213–2219. doi: 10.1039/c8nj05766d. [DOI] [Google Scholar]
- 95.Anderson J.P., Cappello J., Martin D.C. Morphology and primary crystal structure of a silk-like protein polymer synthesized by genetically engineered Escherichia coli bacteria. Biopolymers. 1994;34:1049–1058. doi: 10.1002/bip.360340808. [DOI] [PubMed] [Google Scholar]
- 96.Schacht K., Vogt J., Scheibel T. Foams made of engineered recombinant spider silk proteins as 3D scaffolds for cell growth. ACS Biomater. Sci. Eng. 2016;2:517–525. doi: 10.1021/acsbiomaterials.5b00483. [DOI] [PubMed] [Google Scholar]
- 97.Dong Y., Dong P., Huang D., Mei L., Xia Y., Wang Z., Pan X., Li G., Wu C. Fabrication and characterization of silk fibroin-coated liposomes for ocular drug delivery. Eur. J. Pharm. Biopharm. 2015;91:82–90. doi: 10.1016/j.ejpb.2015.01.018. [DOI] [PubMed] [Google Scholar]
- 98.Costa F., Silva R., Boccaccini A.R. Pept. Proteins as Biomater. Tissue Regen. Repair. Elsevier; 2018. Fibrous protein-based biomaterials (silk, keratin, elastin, and resilin proteins) for tissue regeneration and repair; pp. 175–204. [DOI] [Google Scholar]
- 99.Numata K., Kaplan D.L. Adv. Wound Repair Ther. Elsevier Inc.; 2011. Biologically derived scaffolds; pp. 524–551. [DOI] [Google Scholar]
- 100.Rouse J.G., Van Dyke M.E. A review of keratin-based biomaterials for biomedical applications. Materials (Basel). 2010;3:999–1014. doi: 10.3390/ma3020999. [DOI] [Google Scholar]
- 101.Vasconcelos A., Cavaco-Paulo A. The use of keratin in biomedical applications. Curr. Drug Targets. 2013;14:612–619. doi: 10.2174/1389450111314050010. [DOI] [PubMed] [Google Scholar]
- 102.Zhi X., Wang Y., Li P., Yuan J., Shen J. Preparation of keratin/chlorhexidine complex nanoparticles for long-term and dual stimuli-responsive release. RSC Adv. 2015;5:82334–82341. doi: 10.1039/c5ra16253j. [DOI] [Google Scholar]
- 103.Ghaffari R., Eslahi N., Tamjid E., Simchi A. Dual-sensitive hydrogel nanoparticles based on conjugated thermoresponsive copolymers and protein filaments for triggerable drug delivery. ACS Appl. Mater. Interfaces. 2018;10:19336–19346. doi: 10.1021/acsami.8b01154. [DOI] [PubMed] [Google Scholar]
- 104.Shah D., Mital K. The role of trypsin:chymotrypsin in tissue repair. Adv. Ther. 2018;35:31–42. doi: 10.1007/s12325-017-0648-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Li Y., Zhi X., Lin J., You X., Yuan J. Preparation and characterization of DOX loaded keratin nanoparticles for pH/GSH dual responsive release. Mater. Sci. Eng. C. 2017;73:189–197. doi: 10.1016/j.msec.2016.12.067. [DOI] [PubMed] [Google Scholar]
- 106.Li Y., Lin J., Zhi X., Li P., Jiang X., Yuan J. Triple stimuli-responsive keratin nanoparticles as carriers for drug and potential nitric oxide release. Mater. Sci. Eng. C. 2018;91:606–614. doi: 10.1016/j.msec.2018.05.073. [DOI] [PubMed] [Google Scholar]
- 107.Li Q., Zhu L., Liu R., Huang D., Jin X., Che N., Li Z., Qu X., Kang H., Huang Y. Biological stimuli responsive drug carriers based on keratin for triggerable drug delivery. J. Mater. Chem. 2012;22:19964–19973. doi: 10.1039/c2jm34136k. [DOI] [Google Scholar]
- 108.Martella E., Ferroni C., Guerrini A., Ballestri M., Columbaro M., Santi S., Sotgiu G., Serra M., Donati D.M., Lucarelli E., Varchi G., Duchi S. Functionalized keratin as nanotechnology-based drug delivery system for the pharmacological treatment of osteosarcoma. Int. J. Mol. Sci. 2018;19 doi: 10.3390/ijms19113670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Cheng Z., Chen X., Zhai D., Gao F., Guo T., Li W., Hao S., Ji J., Wang B. Development of keratin nanoparticles for controlled gastric mucoadhesion and drug release. J. Nanobiotechnol. 2018;16:1–13. doi: 10.1186/s12951-018-0353-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Fujii T., Ogiwara D., Arimoto M. Convenient procedures for human hair protein films and properties of alkaline phosphatase incorporated in the film. Biol. Pharm. Bull. 2004;27:89–93. doi: 10.1248/bpb.27.89. [DOI] [PubMed] [Google Scholar]
- 111.Vasconcelos A., Pêgo A.P., Henriques L., Lamghari M., Cavaco-Paulo A. Protein matrices for improved wound healing: Elastase inhibition by a synthetic peptide model. Biomacromolecules. 2010;11:2213–2220. doi: 10.1021/bm100537b. [DOI] [PubMed] [Google Scholar]
- 112.Cui L., Gong J., Fan X., Wang P., Wang Q., Qiu Y. Transglutaminase-modified wool keratin film and its potential application in tissue engineering. Eng. Life Sci. 2013;13:149–155. doi: 10.1002/elsc.201100206. [DOI] [Google Scholar]
- 113.Shavandi A., Silva T.H., Bekhit A.A., Bekhit A.E.D.A. Keratin: dissolution, extraction and biomedical application. Biomater. Sci. 2017;5:1699–1735. doi: 10.1039/c7bm00411g. [DOI] [PubMed] [Google Scholar]
- 114.Saul J.M., Ellenburg M.D., De Guzman R.C., Van Dyke M. Keratin hydrogels support the sustained release of bioactive ciprofloxacin. J. Biomed. Mater. Res. - Part A. 2011;98(A):544–553. doi: 10.1002/jbm.a.33147. [DOI] [PubMed] [Google Scholar]
- 115.Peyton C.C., Keys T., Tomblyn S., Burmeister D., Beumer J.H., Holleran J.L., Sirintrapun J., Washburn S., Hodges S.J. Halofuginone infused keratin hydrogel attenuates adhesions in a rodent cecal abrasion model. J. Surg. Res. 2012;178:545–552. doi: 10.1016/j.jss.2012.07.053. [DOI] [PubMed] [Google Scholar]
- 116.Han S., Ham T.R., Haque S., Sparks J.L., Saul J.M. Alkylation of human hair keratin for tunable hydrogel erosion and drug delivery in tissue engineering applications. Acta Biomater. 2015;23:201–213. doi: 10.1016/j.actbio.2015.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Cao Y., Yao Y., Li Y., Yang X., Cao Z., Yang G. Tunable keratin hydrogel based on disulfide shuffling strategy for drug delivery and tissue engineering. J. Colloid Interface Sci. 2019;544:121–129. doi: 10.1016/j.jcis.2019.02.049. [DOI] [PubMed] [Google Scholar]
- 118.Guo J., Pan S., Yin X., He Y.F., Li T., Wang R.M. PH-sensitive keratin-based polymer hydrogel and its controllable drug-release behavior. J. Appl. Polym. Sci. 2015;132:1–8. doi: 10.1002/app.41572. [DOI] [Google Scholar]
- 119.Sun K., Guo J., He Y., Song P., Xiong Y., Wang R.M. Fabrication of dual-sensitive keratin-based polymer hydrogels and their controllable release behaviors. J. Biomater. Sci. Polym. Ed. 2016;27:1926–1940. doi: 10.1080/09205063.2016.1239955. [DOI] [PubMed] [Google Scholar]
- 120.Li T., Yin X., Zhai W., He Y.F., Wang R.M. Enzymatic digestion of keratin for preparing a pH-sensitive biopolymer hydrogel. Aust. J. Chem. 2016;69:191–197. doi: 10.1071/CH15224. [DOI] [Google Scholar]
- 121.Peralta Ramos M.L., González J.A., Fabian L., Pérez C.J., Villanueva M.E. Sustainable and smart keratin hydrogel with pH-sensitive swelling and enhanced mechanical properties. Mater. Sci. Eng. C. 2017;78:619–626. doi: 10.1016/j.msec.2017.04.120. [DOI] [PubMed] [Google Scholar]
- 122.Villanueva M.E., Cuestas M.L., Pérez C.J., V. Campo Dall’ Orto, G.J. Copello Smart release of antimicrobial ZnO nanoplates from a pH-responsive keratin hydrogel. J. Colloid Interface Sci. 2019;536:372–380. doi: 10.1016/j.jcis.2018.10.067. [DOI] [PubMed] [Google Scholar]
- 123.Carter D.C., He X.M., Munson S.H., Twigg P.D., Gernert K.M., Broom M.B., Miller T.Y. Three-dimensional structure of human serum albumin. Science (80-. ) 1989;244:1195–1198. doi: 10.1126/science.2727704. [DOI] [PubMed] [Google Scholar]
- 124.Sleep D., Cameron J., Evans L.R. Albumin as a versatile platform for drug half-life extension. Biochim. Biophys. Acta - Gen. Subj. 2013;1830:5526–5534. doi: 10.1016/j.bbagen.2013.04.023. [DOI] [PubMed] [Google Scholar]
- 125.Hoogenboezem E.N., Duvall C.L. Harnessing albumin as a carrier for cancer therapies. Adv. Drug Deliv. Rev. 2018;130:73–89. doi: 10.1016/j.addr.2018.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Yousefpour P., McDaniel J.R., Prasad V., Ahn L., Li X., Subrahmanyan R., Weitzhandler I., Suter S., Chilkoti A. Genetically encoding albumin binding into chemotherapeutic-loaded polypeptide nanoparticles enhances their antitumor efficacy. Nano Lett. 2018;18:7784–7793. doi: 10.1021/acs.nanolett.8b03558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Huntington J.A., Stein P.E. Structure and properties of ovalbumin. J. Chromatogr. B Biomed. Sci. Appl. 2001;756:189–198. doi: 10.1016/S0378-4347(01)00108-6. [DOI] [PubMed] [Google Scholar]
- 128.Wongsasulak S., Patapeejumruswong M., Weiss J., Supaphol P., Yoovidhya T. Electrospinning of food-grade nanofibers from cellulose acetate and egg albumen blends. J. Food Eng. 2010;98:370–376. doi: 10.1016/j.jfoodeng.2010.01.014. [DOI] [Google Scholar]
- 129.Liu W., Griffith M., Li F. Alginate microsphere-collagen composite hydrogel for ocular drug delivery and implantation. J. Mater. Sci. Mater. Med. 2008;19:3365–3371. doi: 10.1007/s10856-008-3486-2. [DOI] [PubMed] [Google Scholar]
- 130.Tantra R., Tompkins J., Quincey P. Characterisation of the de-agglomeration effects of bovine serum albumin on nanoparticles in aqueous suspension. Colloids Surfaces B Biointerfaces. 2010;75:275–281. doi: 10.1016/j.colsurfb.2009.08.049. [DOI] [PubMed] [Google Scholar]
- 131.Hu Y.J., Liu Y., Sun T.Q., Bai A.M., Lü J.Q., Pi Z.B. Binding of anti-inflammatory drug cromolyn sodium to bovine serum albumin. Int. J. Biol. Macromol. 2006;39:280–285. doi: 10.1016/j.ijbiomac.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 132.Yu Z., Yu M., Zhang Z., Hong G., Xiong Q. Bovine serum albumin nanoparticles as controlled release carrier for local drug delivery to the inner ear. Nanoscale Res. Lett. 2014;9:1–7. doi: 10.1186/1556-276X-9-343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Lam P.L., Kok S.H.L., Gambari R., Kok T.W., Leung H.Y., Choi K.L., Wong C.S., Hau D.K.P., Wong W.Y., Lam K.H., Bian Z.X., Lee K.K.H., Chui C.H. Evaluation of berberine/bovine serum albumin nanoparticles for liver fibrosis therapy. Green Chem. 2015;17:1640–1646. doi: 10.1039/c4gc01815j. [DOI] [Google Scholar]
- 134.Li J., Yao P. Self-assembly of ibuprofen and bovine serum albumin-dextran conjugates leading to effective loading of the drug. Langmuir. 2009;25:6385–6391. doi: 10.1021/la804288u. [DOI] [PubMed] [Google Scholar]
- 135.Carter D.C., Ho J.X. Structure of serum albumin. Adv. Protein Chem. 1994;45:153–176. doi: 10.1016/S0065-3233(08)60640-3. [DOI] [PubMed] [Google Scholar]
- 136.Peters J.T. Elsevier; 1995. All About Albumin. [DOI] [Google Scholar]
- 137.Curry S., Mandelkow H., Brick P., Franks N. Crystal structure of human serum albumin complexed with fatty acid reveals an asymmetric distribution of binding sites. Nat. Struct. Biol. 1998;5:827–835. doi: 10.1038/1869. [DOI] [PubMed] [Google Scholar]
- 138.Sugio S., Kashima A., Mochizuki S., Noda M., Kobayashi K. Crystal structure of human serum albumin at 2.5 Å resolution. Protein Eng. Des. Sel. 1999;12:439–446. doi: 10.1093/protein/12.6.439. [DOI] [PubMed] [Google Scholar]
- 139.Arroyo V., García-Martinez R., Salvatella X. Human serum albumin, systemic inflammation, and cirrhosis. J. Hepatol. 2014;61:396–407. doi: 10.1016/j.jhep.2014.04.012. [DOI] [PubMed] [Google Scholar]
- 140.Kragh-Hansen U., Chuang V.T.G., Otagiri M. Practical aspects of the ligand-binding and enzymatic properties of human serum albumin. Biol. Pharm. Bull. 2002;25:695–704. doi: 10.1248/bpb.25.695. [DOI] [PubMed] [Google Scholar]
- 141.Cistola D.P., Small D.M. Fatty acid distribution in systems modeling the normal and diabetic human circulation: a 13C nuclear magnetic resonance study. J. Clin. Invest. 1991;87:1431–1441. doi: 10.1172/JCI115149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Lee P., Wu X. Review: modifications of human serum albumin and their binding effect. Curr. Pharm. Des. 2015;21:1862–1865. doi: 10.2174/1381612821666150302115025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Knudsen Sand K.M., Bern M., Nilsen J., Noordzij H.T., Sandlie I., Andersen J.T. Unraveling the interaction between FcRn and albumin: opportunities for design of albumin-based therapeutics. Front. Immunol. 2015;5:682. doi: 10.3389/fimmu.2014.00682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Liu Z., Chen X. Simple bioconjugate chemistry serves great clinical advances: albumin as a versatile platform for diagnosis and precision therapy. Chem. Soc. Rev. 2016;45:1432–1456. doi: 10.1039/C5CS00158G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Wang S., Liu S., He J., David H. Human serum albumin (HSA) and its applications as a drug delivery vehicle abstract. Heal. Sci. J. 2020;14:1–8. doi: 10.36648/1791-809X.14.2.698. [DOI] [Google Scholar]
- 146.Liu Q., Simpson D.C., Gronert S. The reactivity of human serum albumin toward trans-4-hydroxy-2-nonenal. J. Mass Spectrom. 2012;47:411–424. doi: 10.1002/jms.2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Walker J.E. Lysine residue 199 of human serum albumin is modified by acetylsalicylic acid. FEBS Lett. 1976;66:173–175. doi: 10.1016/0014-5793(76)80496-6. [DOI] [PubMed] [Google Scholar]
- 148.Turell L., Radi R., Alvarez B. The thiol pool in human plasma: the central contribution of albumin to redox processes. Free Radic. Biol. Med. 2013;65:244–253. doi: 10.1016/j.freeradbiomed.2013.05.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Schnitzer J.E. gp60 is an albumin-binding glycoprotein expressed by continuous endothelium involved in albumin transcytosis. Am. J. Physiol. - Hear. Circ. Physiol. 1992;262 doi: 10.1152/ajpheart.1992.262.1.h246. [DOI] [PubMed] [Google Scholar]
- 150.Schnitzer J.E., Oh P. Albondin-mediated capillary permeability to albumin. Differential role of receptors in endothelial transcytosis and endocytosis of native and modified albumins. J. Biol. Chem. 1994;269:6072–6082. [PubMed] [Google Scholar]
- 151.Komiya K., Nakamura T., Nakashima C., Takahashi K., Umeguchi H., Watanabe N., Sato A., Takeda Y., Kimura S., Aragane N.S. SPARC is a possible predictive marker for albumin-bound paclitaxel in non-small-cell lung cancer. Onco. Targets. Ther. 2016;9:6663–6668. doi: 10.2147/OTT.S114492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Gong J., Yan J., Forscher C., Hendifar A. Aldoxorubicin: a tumor-targeted doxorubicin conjugate for relapsed or refractory soft tissue sarcomas. Drug Des. Devel. Ther. 2018;12:777–786. doi: 10.2147/DDDT.S140638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Lau J., Bloch P., Schäffer L., Pettersson I., Spetzler J., Kofoed J., Madsen K., Knudsen L.B., McGuire J., Steensgaard D.B., Strauss H.M., Gram D.X., Knudsen S.M., Nielsen F.S., Thygesen P., Reedtz-Runge S., Kruse T. Discovery of the once-weekly glucagon-like Peptide-1 (GLP-1) analogue Semaglutide. J. Med. Chem. 2015;58:7370–7380. doi: 10.1021/acs.jmedchem.5b00726. [DOI] [PubMed] [Google Scholar]
- 154.Hall S., Isaacs D., Clements J.N. Pharmacokinetics and clinical implications of Semaglutide: a new glucagon-like peptide (GLP)-1 receptor agonist. Clin. Pharmacokinet. 2018;57:1529–1538. doi: 10.1007/s40262-018-0668-z. [DOI] [PubMed] [Google Scholar]
- 155.Keith N.M., Rowntree L.G., Geraghty J.T. A method for the determination of plasma and blood volume. Arch. Intern. Med. XVI. 1915:547–576. doi: 10.1001/archinte.1915.00080040043004. [DOI] [Google Scholar]
- 156.de Goldim M.P.S., Giustina A. Della, Petronilho F. Using Evans Blue Dye to Determine Blood-Brain Barrier Integrity in Rodents. Curr. Protoc. Immunol. 2019;126 doi: 10.1002/cpim.83. [DOI] [PubMed] [Google Scholar]
- 157.Jacobson O., Kiesewetter D.O., Chen X. Albumin-binding Evans blue derivatives for diagnostic imaging and production of long-acting therapeutics. Bioconjug. Chem. 2016;27:2239–2247. doi: 10.1021/acs.bioconjchem.6b00487. [DOI] [PubMed] [Google Scholar]
- 158.Yamamoto T., Ikuta K., Oi K., Abe K., Uwatoku T., Murata M., Shigetani N., Yoshimitsu K., Shimokawa H., Katayama Y. First functionalized MRI contrast agent recognizing vascular lesions. Anal. Sci. 2004;20:5–7. doi: 10.2116/analsci.20.5. [DOI] [PubMed] [Google Scholar]
- 159.Niu G., Lang L., Kiesewetter D.O., Ma Y., Sun Z., Guo N., Guo J., Wu C., Chen X. In vivo labeling of serum albumin for PET. J. Nucl. Med. 2014;55:1150–1156. doi: 10.2967/jnumed.114.139642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Wang Y., Lang L., Huang P., Wang Z., Jacobson O., Kiesewetter D.O., Ali I.U., Teng G., Niu G., Chen X. In vivo albumin labeling and lymphatic imaging. Proc. Natl. Acad. Sci. U. S. A. 2015;112:208–213. doi: 10.1073/pnas.1414821112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Zang J., Liu Q., Sui H., Guo H., Peng L., Li F., Lang L., Jacobson O., Zhu Z., Mao F., Chen X. Combined 68Ga-NOTA-Evans blue Lymphoscintigraphy and 68Ga-NOTA-RM26 PET/CT evaluation of sentinel lymph node metastasis in breast Cancer patients. Bioconjug. Chem. 2020;31:396–403. doi: 10.1021/acs.bioconjchem.9b00789. [DOI] [PubMed] [Google Scholar]
- 162.Dumelin C.E., Trüssel S., Buller F., Trachsel E., Bootz F., Zhang Y., Mannocci L., Beck S.C., Drumea-Mirancea M., Seeliger M.W., Baltes C., Müggler T., Kranz F., Rudin M., Melkko S., Scheuermann J., Neri D. A portable albumin binder from a DNA-encoded chemical library. Angew. Chemie - Int. Ed. 2008;47:3196–3201. doi: 10.1002/anie.200704936. [DOI] [PubMed] [Google Scholar]
- 163.Sjobring U., Falkenberg C., Nielsen E., Akerstrom B., Bjorck L. Isolation and characterization of a 14-kDa albumin-binding fragment of streptococcal protein G. J. Immunol. 1988;140:1595–1599. [PubMed] [Google Scholar]
- 164.Kraulisa P.J., Jonasson P., Nygren P.Å., Uhlén M., Jendeberga L., Nilsson B., Kördel J. The serum albumin-binding domain of streptococcal protein G is a three-helical bundle: a heteronuclear NMR study. FEBS Lett. 1996;378:190–194. doi: 10.1016/0014-5793(95)01452-7. [DOI] [PubMed] [Google Scholar]
- 165.Malm M., Bass T., Gudmundsdotter L., Lord M., Frejd F.Y., Ståhl S., Löfblom J. Engineering of a bispecific affibody molecule towards HER2 and HER3 by addition of an albumin-binding domain allows for affinity purification and in vivo half-life extension. Biotechnol. J. 2014;9:1215–1222. doi: 10.1002/biot.201400009. [DOI] [PubMed] [Google Scholar]
- 166.Löfblom J., Feldwisch J., Tolmachev V., Carlsson J., Ståhl S., Frejd F.Y. Affibody molecules: engineered proteins for therapeutic, diagnostic and biotechnological applications. FEBS Lett. 2010;584:2670–2680. doi: 10.1016/j.febslet.2010.04.014. [DOI] [PubMed] [Google Scholar]
- 167.Makrides S.C., Nygren P.A., Andrews B., Ford P.J., Evans K.S., Hayman E.G., Adari H., Uhlén M., Toth C.A. Extended in vivo half-life of human soluble complement receptor type 1 fused to a serum albumin-binding receptor. J. Pharmacol. Exp. Ther. 1996;277 [PubMed] [Google Scholar]
- 168.Nessler I., Khera E., Vance S., Kopp A., Qiu Q., Keating T.A., Abu-Yousif A.O., Sandal T., Legg J., Thompson L., Goodwin N., Thurber G.M. Increased tumor penetration of single-domain antibody-drug conjugates improves in vivo efficacy in prostate cancer models. Cancer Res. 2020;80:1268–1278. doi: 10.1158/0008-5472.CAN-19-2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Hammarberg B., Nygren P.A., Holmgren E., Elmblad A., Tally M., Hellman U., Moks T., Uhlen M. Dual affinity fusion approach and its use to express recombinant human insulin-like growth factor II. Proc. Natl. Acad. Sci. U. S. A. 1989;86:4367–4371. doi: 10.1073/pnas.86.12.4367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Guo R., Guo W., Cao L., Liu H., Liu J., Xu H., Huang W., Wang F., Hong Z. Fusion of an albumin-binding domain extends the half-life of immunotoxins. Int. J. Pharm. 2016;511:538–549. doi: 10.1016/j.ijpharm.2016.07.046. [DOI] [PubMed] [Google Scholar]
- 171.Libon C., Corvaïa N., Haeuw J.F., Nguyen T.N., Ståhl S., Bonnefoy J.Y., Andreoni C. The serum albumin-binding region of streptococcal protein G (BB) potentiates the immunogenicity of the G130-230 RSV-A protein. Vaccine. 1999;17:406–414. doi: 10.1016/S0264-410X(98)00198-4. [DOI] [PubMed] [Google Scholar]
- 172.Smith A.A.A., Zuwala K., Pilgram O., Johansen K.S., Tolstrup M., Dagnæs-Hansen F., Zelikin A.N. Albumin-polymer-drug conjugates: long circulating, high payload drug delivery vehicles. ACS Macro Lett. 2016;5:1089–1094. doi: 10.1021/acsmacrolett.6b00544. [DOI] [PubMed] [Google Scholar]
- 173.Hartung G., Stehle G., Sinn H., Wunder A., Schrenk H.H., Heeger S., Kränzle M., Edler L., Frei E., Fiebig H.H., Heene D.L., Maier-Borst W., Queisser W. Phase I trial of methotrexate-albumin in a weekly intravenous bolus regimen in cancer patients. Phase I Study Group of the Association for Medical Oncology of the German Cancer Society. Clin. Cancer Res. 1999;5:753–759. http://www.ncbi.nlm.nih.gov/pubmed/10213209 [PubMed] [Google Scholar]
- 174.Vis A., Van Der Gaast A., Van Rhijn B., Catsburg T., Schmidt C., Mickisch G. A phase II trial of methotrexate-human serum albumin (MTX-HSA) in patients with metastatic renal cell carcinoma who progressed under immunotherapy. Cancer Chemother. Pharmacol. 2002;49:342–345. doi: 10.1007/s00280-001-0417-z. [DOI] [PubMed] [Google Scholar]
- 175.Bolling C., Graefe T., Lübbing C., Jankevicius F., Uktveris S., Cesas A., Meyer-Moldenhauer W.H., Starkmann H., Weigel M., Burk K., Hanauske A.R. Phase II study of MTX-HSA in combination with Cisplatin as first line treatment in patients with advanced or metastatic transitional cell carcinoma. Investig. New Drugs. 2006;24:521–527. doi: 10.1007/s10637-006-8221-6. [DOI] [PubMed] [Google Scholar]
- 176.Desai N., Trieu V., Yao Z., Louie L., Ci S., Yang A., Tao C., De T., Beals B., Dykes D., Noker P., Yao R., Labao E., Hawkins M., Soon-Shiong P. Increased antitumor activity, intratumor paclitaxel concentrations, and endothelial cell transport of cremophor-free, albumin-bound paclitaxel, ABI-007, compared with cremophor-based paclitaxel. Clin. Cancer Res. 2006;12:1317–1324. doi: 10.1158/1078-0432.CCR-05-1634. [DOI] [PubMed] [Google Scholar]
- 177.Elsadek B., Kratz F. Impact of albumin on drug delivery - new applications on the horizon. J. Control. Release. 2012;157:4–28. doi: 10.1016/j.jconrel.2011.09.069. [DOI] [PubMed] [Google Scholar]
- 178.Van de Sande L., Cosyns S., Willaert W., Ceelen W. Albumin-based cancer therapeutics for intraperitoneal drug delivery: a review. Drug Deliv. 2020;27:40–53. doi: 10.1080/10717544.2019.1704945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Lin T., Zhao P., Jiang Y., Tang Y., Jin H., Pan Z., He H., Yang V.C., Huang Y. Blood-brain-barrier-penetrating albumin nanoparticles for biomimetic drug delivery via albumin-binding protein pathways for Antiglioma therapy. ACS Nano. 2016;10:9999–10012. doi: 10.1021/acsnano.6b04268. [DOI] [PubMed] [Google Scholar]
- 180.Jiang Y., Wong S., Chen F., Chang T., Lu H., Stenzel M.H. Influencing selectivity to Cancer cells with mixed nanoparticles prepared from albumin-polymer conjugates and block copolymers. Bioconjug. Chem. 2017;28:979–985. doi: 10.1021/acs.bioconjchem.6b00698. [DOI] [PubMed] [Google Scholar]
- 181.Li F.-Q., Su H., Chen X., Qin X.-J., Liu J.-Y., Zhu Q.-G., Hu J.-H. Mannose 6-phosphate-modified bovine serum albumin nanoparticles for controlled and targeted delivery of sodium ferulate for treatment of hepatic fibrosis. J. Pharm. Pharmacol. 2009;61:1155–1161. doi: 10.1211/jpp.61.09.0004. [DOI] [PubMed] [Google Scholar]
- 182.Sun Y., Zhao Y., Teng S., Hao F., Zhang H., Meng F., Zhao X., Zheng X., Bi Y., Yao Y., Lee R.J., Teng L. Folic acid receptor-targeted human serum albumin nanoparticle formulation of cabazitaxel for tumor therapy. Int. J. Nanomedicine. 2019;14:135–148. doi: 10.2147/IJN.S181296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Fadaeian G., Shojaosadati S.A., Kouchakzadeh H., Shokri F., Soleimani M. Targeted delivery of 5-fluorouracil with monoclonal antibody modified bovine serum albumin nanoparticles. Iran. J. Pharm. Res. 2015;14:395–405. doi: 10.22037/ijpr.2015.1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Xiong J., Han S., Ding S., He J., Zhang H. Antibody-nanoparticle conjugate constructed with trastuzumab and nanoparticle albumin-bound paclitaxel for targeted therapy of human epidermal growth factor receptor 2-positive gastric cancer. Oncol. Rep. 2018;39:1396–1404. doi: 10.3892/or.2018.6201. [DOI] [PubMed] [Google Scholar]
- 185.Saleh T., Soudi T., Shojaosadati S.A. Aptamer functionalized curcumin-loaded human serum albumin (HSA) nanoparticles for targeted delivery to HER-2 positive breast cancer cells. Int. J. Biol. Macromol. 2019;130:109–116. doi: 10.1016/j.ijbiomac.2019.02.129. [DOI] [PubMed] [Google Scholar]
- 186.Kenet G., Brainsky A., Powell J.S., Li Y., Seifert W. Recombinant fusion protein linking coagulation Factor IX with albumin (RIX-FP) in previously treated children < 12 years of age with Hemophilia B: long-term efficacy and safety from an ongoing phase 3b extension clinical trial. Blood. 2017;130:2370. doi: 10.1182/blood.V130.Suppl_1.2370.2370. [DOI] [Google Scholar]
- 187.Shao R., Zhen Y. Enediyne anticancer antibiotic Lidamycin: chemistry, biology and pharmacology. Anti Cancer Agents Med. Chem. 2008;8:123–131. doi: 10.2174/187152008783497055. [DOI] [PubMed] [Google Scholar]
- 188.Li L., Hu L., Zhao C.Y., Zhang S.H., Wang R., Li Y., Shao R.G., Zhen Y.S. The recombinant and reconstituted novel albumin-Lidamycin conjugate shows lasting tumor imaging and intensively enhanced therapeutic efficacy. Bioconjug. Chem. 2018;29:3104–3112. doi: 10.1021/acs.bioconjchem.8b00456. [DOI] [PubMed] [Google Scholar]
- 189.Lei J., Guan B., Li B., Duan Z., Chen Y., Li H., Jin J. Expression, purification and characterization of recombinant human interleukin-2-serum albumin (rhIL-2-HSA) fusion protein in Pichia pastoris. Protein Expr. Purif. 2012;84:154–160. doi: 10.1016/j.pep.2012.05.003. [DOI] [PubMed] [Google Scholar]
- 190.Melder R.J., Osborn B.L., Riccobene T., Kanakaraj P., Wei P., Chen G., Stolow D., Halpern W.G., Migone T.S., Wang Q., Grzegorzewski K.J., Gallant G. Pharmacokinetics and in vitro and in vivo anti-tumor response of an interleukin-2-human serum albumin fusion protein in mice. Cancer Immunol. Immunother. 2005;54:535–5475. doi: 10.1007/s00262-004-0624-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Sung C., Shah D., Moody G., Gentz S., Beebe L., Moore P.A., Nardelli B., Lafleur D.W., Blatter E., Corcoran M., Olsen H.S., Birse C.E., Pickeral O.K., Zhang J. An IFN-β-albumin fusion protein that displays improved pharmacokinetic and pharmacodynamic properties in nonhuman primates. J. Interf. Cytokine Res. 2003;23:25–36. doi: 10.1089/10799900360520423. [DOI] [PubMed] [Google Scholar]
- 192.Nguyen M.T., Heo Y., Do B.H., Baek S., Kim C.J., Jang Y.J., Lee W., Choe H. Bacterial overexpression and purification of soluble recombinant human serum albumin using maltose-binding protein and protein disulphide isomerase. Protein Expr. Purif. 2020;167:105530. doi: 10.1016/j.pep.2019.105530. [DOI] [PubMed] [Google Scholar]
- 193.Gelse K., Pöschl E., Aigner T. Collagens - structure, function, and biosynthesis. Adv. Drug Deliv. Rev. 2003;55:1531–1546. doi: 10.1016/j.addr.2003.08.002. [DOI] [PubMed] [Google Scholar]
- 194.Ricard-Blum S. The collagen family. Cold Spring Harb. Perspect. Biol. 2011;3:1–19. doi: 10.1101/cshperspect.a004978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Ruszczak Z., Friess W. Collagen as a carrier for on-site delivery of antibacterial drugs. Adv. Drug Deliv. Rev. 2003;55:1679–1698. doi: 10.1016/j.addr.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 196.Ramachandran G.N., Kartha G. Structure of collagen. Nature. 1955;176:593–595. doi: 10.1038/176593a0. [DOI] [PubMed] [Google Scholar]
- 197.Heidemann E., Roth W. Synthesis and investigation of collagen model peptides. Adv. Polym. Sci. 1982:143–203. doi: 10.1007/3-540-11048-8_3. [DOI] [Google Scholar]
- 198.Chak V., Kumar D., Visht S., Chak* Vikash, Kumar Dharmendra, Visht Sharad. 2013. A Review on Collagen Based Drug Delivery Systems. [Google Scholar]
- 199.Mandal A., Panigrahi S., Zhang C. Collagen as biomaterial for medical application-drug delivery and scaffolds for tissue regeneration: a review. Biol. Eng. 1934;2:63–88. [Google Scholar]
- 200.M. Violeta GHICA, M. Georgiana ALBU, L. Popa, M. Leca, L. Brăzdaru, C. Cotruţ, V. Trandafir . 2009. Drug Delivery Systems Based on Collagen-Tannic Acid Matrices. [Google Scholar]
- 201.Luo T., Kiick K.L. Collagen-like peptides and peptide-polymer conjugates in the design of assembled materials. Eur. Polym. J. 2013;49:2998–3009. doi: 10.1016/j.eurpolymj.2013.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Luo T., Kiick K.L. Collagen-Like Peptide Bioconjugates. Bioconjug. Chem. 2017;28:816–827. doi: 10.1021/acs.bioconjchem.6b00673. [DOI] [PubMed] [Google Scholar]
- 203.Persikov A.V., Ramshaw J.A.M., Kirkpatrick A., Brodsky B. Amino acid propensities for the collagen triple-helix. Biochemistry. 2000;39:14960–14967. doi: 10.1021/bi001560d. [DOI] [PubMed] [Google Scholar]
- 204.Yu S.M., Li Y., Kim D. Collagen mimetic peptides: Progress towards functional applications. Soft Matter. 2011;7:7927–7938. doi: 10.1039/c1sm05329a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Chung E., Keele E.M., Miller E.J. Isolation and characterization of the cyanogen bromide peptides from the α1(III)chain of human collagen. Biochemistry. 1974;13:3459–3464. doi: 10.1021/bi00714a006. [DOI] [PubMed] [Google Scholar]
- 206.Barth D., Kyrieleis O., Frank S., Renner C., Moroder L. The role of cystine knots in collagen folding and stability, part II. Conformational properties of (Pro-Hyp-Gly)n model trimers with N- and C-terminal collagen type III cystine knots. Chem. - A Eur. J. 2003;9:3703–3714. doi: 10.1002/chem.200304918. [DOI] [PubMed] [Google Scholar]
- 207.BÄCHINGER H.P., BRUCKNER P., TIMPL R., PROCKOP D.J., ENGEL J. Folding mechanism of the triple Helix in type-III collagen and type-III pN–collagen: role of disulfide bridges and peptide bond isomerization. Eur. J. Biochem. 1980;106:619–632. doi: 10.1111/j.1432-1033.1980.tb04610.x. [DOI] [PubMed] [Google Scholar]
- 208.Kwak J., De Capua A., Locardi E., Goodman M. TREN (tris(2-aminoethyl)amine): An effective scaffold for the assembly of triple helical collagen mimetic structures. J. Am. Chem. Soc. 2002;124:14085–14091. doi: 10.1021/ja0209621. [DOI] [PubMed] [Google Scholar]
- 209.Feng Y., Melacini G., Taulane J.P., Goodman M. Acetyl-terminated and template-assembled collagen-based polypeptides composed of Gly-Pro-Hyp sequences. 2. Synthesis and conformational analysis by circular dichroism, ultraviolet absorbance, and optical rotation. J. Am. Chem. Soc. 1996;118:10351–10358. doi: 10.1021/ja961260c. [DOI] [Google Scholar]
- 210.Koide T., Yuguchi M., Kawakita M., Konno H. Metal-assisted stabilization and probing of collagenous triple helices. J. Am. Chem. Soc. 2002;124:9388–9389. doi: 10.1021/ja026182+. [DOI] [PubMed] [Google Scholar]
- 211.Parmar A.S., Xu F., Pike D.H., Belure S.V., Hasan N.F., Drzewiecki K.E., Shreiber D.I., Nanda V. Metal stabilization of collagen and de novo designed mimetic peptides. Biochemistry. 2015;54:4987–4997. doi: 10.1021/acs.biochem.5b00502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Yu Y.C., Berndt P., Tirrell M., Fields G.B. Self-assembling amphiphiles for construction of protein molecular architecture. J. Am. Chem. Soc. 1996;118:12515–12520. doi: 10.1021/ja9627656. [DOI] [Google Scholar]
- 213.Gore T., Dori Y., Talmon Y., Tirrell M., Bianco-Peled H. Self-assembly of model collagen peptide amphiphiles. Langmuir. 2001;17:5352–5360. doi: 10.1021/la010223i. [DOI] [Google Scholar]
- 214.Yu Y.C., Tirrell M., Fields G.B. Minimal lipidation stabilizes protein-like molecular architecture. J. Am. Chem. Soc. 1998;120:9979–9987. doi: 10.1021/ja981654z. [DOI] [Google Scholar]
- 215.Inouy K., Sakakibara S., Prockop D.J. Effects of the stereo-configuration of the hydroxyl group in 4-hydroxyproline on the triple-helical structures formed by homogeneous peptides resembling collagen. BBA - Protein Struct. 1976;420:133–141. doi: 10.1016/0005-2795(76)90352-4. [DOI] [PubMed] [Google Scholar]
- 216.Eberhardt E.S., Panasik N., Raines R.T. Inductive effects on the energetics of prolyl peptide bond isomerization: implications for collagen folding and stability. J. Am. Chem. Soc. 1996;118:12261–12266. doi: 10.1021/ja9623119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Steven R.T.R., Holmgren K., Taylor Kimberly M., Bretscher Lynn E. Code for collagen's stability deciphered. Nature. 1998;392:666–667. doi: 10.1038/33573. [DOI] [PubMed] [Google Scholar]
- 218.Holmgren S.K., Bretscher L.E., Taylor K.M., Raines R.T. A hyperstable collagen mimic. Chem. Biol. 1999;6:63–70. doi: 10.1016/S1074-5521(99)80003-9. [DOI] [PubMed] [Google Scholar]
- 219.Hsu W., Chen Y.-L., Horng J.-C. Promoting self-assembly of collagen-related peptides into various higher-order structures by metal–Histidine coordination. Langmuir. 2012;28:3194–3199. doi: 10.1021/la204351w. [DOI] [PubMed] [Google Scholar]
- 220.Pires M.M., Chmielewski J. Self-assembly of collagen peptides into microflorettes via metal coordination. J. Am. Chem. Soc. 2009;131:2706–2712. doi: 10.1021/ja8088845. [DOI] [PubMed] [Google Scholar]
- 221.Zheng H., Lu C., Lan J., Fan S., Nanda V., Xu F. How electrostatic networks modulate specificity and stability of collagen. Proc. Natl. Acad. Sci. U. S. A. 2018;115:6207–6212. doi: 10.1073/pnas.1802171115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Wang A.Y., Mo X., Chen C.S., Yu S.M. Facile modification of collagen directed by collagen mimetic peptides. J. Am. Chem. Soc. 2005;127:4130–4131. doi: 10.1021/ja0431915. [DOI] [PubMed] [Google Scholar]
- 223.Li Y., Yu S.M. Targeting and mimicking collagens via triple helical peptide assembly. Curr. Opin. Chem. Biol. 2013;17:968–975. doi: 10.1016/j.cbpa.2013.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Bennink L.L., Smith D.J., Foss C.A., Pomper M.G., Li Y., Yu S.M. High serum stability of collagen hybridizing peptides and their Fluorophore conjugates. Mol. Pharm. 2017;14:1906–1915. doi: 10.1021/acs.molpharmaceut.7b00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Converse M.I., Walther R.G., Ingram J.T., Li Y., Yu S.M., Monson K.L. Detection and characterization of molecular-level collagen damage in overstretched cerebral arteries. Acta Biomater. 2018;67:307–318. doi: 10.1016/j.actbio.2017.11.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.San B.H., Hwang J., Sampath S., Li Y., Bennink L.L., Yu S.M. Self-assembled water-soluble nanofibers displaying collagen hybridizing peptides. J. Am. Chem. Soc. 2017;139:16640–16649. doi: 10.1021/jacs.7b07900. [DOI] [PubMed] [Google Scholar]
- 227.Hwang J., San B.H., Turner N.J., White L.J., Faulk D.M., Badylak S.F., Li Y., Yu S.M. Molecular assessment of collagen denaturation in decellularized tissues using a collagen hybridizing peptide. Acta Biomater. 2017;53:268–278. doi: 10.1016/j.actbio.2017.01.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Wang A.Y., Foss C.A., Leong S., Mo X., Pomper M.G., Yu S.M. Spatio-temporal modification of collagen scaffolds mediated by triple helical propensity. Biomacromolecules. 2008;9:1755–1763. doi: 10.1021/bm701378k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Li Y., Ho D., Meng H., Chan T.R., An B., Yu H., Brodsky B., Jun A.S., Michael Yu S. Direct detection of collagenous proteins by fluorescently labeled collagen mimetic peptides. Bioconjug. Chem. 2013;24:9–16. doi: 10.1021/bc3005842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Li Y., Foss C.A., Summerfield D.D., Doyle J.J., Torok C.M., Dietz H.C., Pomper M.G., Yu S.M. Targeting collagen strands by photo-triggered triple-helix hybridization. Proc. Natl. Acad. Sci. U. S. A. 2012;109:14767–14772. doi: 10.1073/pnas.1209721109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Bennink L.L., Li Y., Kim B., Shin I.J., San B.H., Zangari M., Yoon D., Yu S.M. Visualizing collagen proteolysis by peptide hybridization: from 3D cell culture to in vivo imaging. Biomaterials. 2018;183:67–76. doi: 10.1016/j.biomaterials.2018.08.039. [DOI] [PubMed] [Google Scholar]
- 232.Wang A.Y., Leong S., Liang Y.C., Huang R.C.C., Chen C.S., Yu S.M. Immobilization of growth factors on collagen scaffolds mediated by polyanionic collagen mimetic peptides and its effect on endothelial cell morphogenesis. Biomacromolecules. 2008;9:2929–2936. doi: 10.1021/bm800727z. [DOI] [PubMed] [Google Scholar]
- 233.Mo X., An Y., Yun C.-S., Yu S.M. Nanoparticle-assisted visualization of binding interactions between collagen mimetic peptide and collagen fibers. Angew. Chemie. 2006;118:2325–2328. doi: 10.1002/ange.200504529. [DOI] [PubMed] [Google Scholar]
- 234.Santos J.L., Li Y., Culver H.R., Yu M.S., Herrera-Alonso M. Conducting polymer nanoparticles decorated with collagen mimetic peptides for collagen targeting. Chem. Commun. 2014;50:15045–15048. doi: 10.1039/c4cc06056c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Urello M.A., Kiick K.L., Sullivan M.O. A CMP-based method for tunable, cell-mediated gene delivery from collagen scaffolds. J. Mater. Chem. B. 2014;2:8174–8185. doi: 10.1039/c4tb01435a. [DOI] [PubMed] [Google Scholar]
- 236.Urello M.A., Kiick K.L., Sullivan M.O. Integration of growth factor gene delivery with collagen-triggered wound repair cascades using collagen-mimetic peptides. Bioeng. Transl. Med. 2016;1:207–219. doi: 10.1002/btm2.10037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Mansurov A., Ishihara J., Hosseinchi P., Potin L., Marchell T.M., Ishihara A., Williford J.M., Alpar A.T., Raczy M.M., Gray L.T., Swartz M.A., Hubbell J.A. Collagen-binding IL-12 enhances tumour inflammation and drives the complete remission of established immunologically cold mouse tumours. Nat. Biomed. Eng. 2020;4:531–543. doi: 10.1038/s41551-020-0549-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Kotch F.W., Raines R.T. Self-assembly of synthetic collagen triple helices. Proc. Natl. Acad. Sci. U. S. A. 2006;103:3028–3033. doi: 10.1073/pnas.0508783103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Koide T., Homma D.L., Asada S., Kitagawa K. Self-complementary peptides for the formation of collagen-like triple helical supramolecules. Bioorganic Med. Chem. Lett. 2005;15:5230–5233. doi: 10.1016/j.bmcl.2005.08.041. [DOI] [PubMed] [Google Scholar]
- 240.Yamazaki C.M., Asada S., Kitagawa K., Koide T. Artificial collagen gels via self-assembly of de novo designed peptides. Biopolymers. 2008;90:816–823. doi: 10.1002/bip.21100. [DOI] [PubMed] [Google Scholar]
- 241.Yamazaki C.M., Kadoya Y., Hozumi K., Okano-Kosugi H., Asada S., Kitagawa K., Nomizu M., Koide T. A collagen-mimetic triple helical supramolecule that evokes integrin-dependent cell responses. Biomaterials. 2010;31:1925–1934. doi: 10.1016/j.biomaterials.2009.10.014. [DOI] [PubMed] [Google Scholar]
- 242.Ichise S.F., Takeuchi S., Aoki S., Kuroda K.C., Nose H., Masuda R., Koide T. Development of a collagen-like peptide polymer via end-to-end disulfide cross-linking and its application as a biomaterial. Acta Biomater. 2019;94:361–371. doi: 10.1016/j.actbio.2019.06.010. [DOI] [PubMed] [Google Scholar]
- 243.Cejas M.A., Kinney W.A., Chen C., Leo G.C., Tounge B.A., Vinter J.G., Joshi P.P., Maryanoff B.E. Collagen-related peptides: self-assembly of short, single strands into a functional biomaterial of micrometer scale. J. Am. Chem. Soc. 2007;129:2202–2203. doi: 10.1021/ja066986f. [DOI] [PubMed] [Google Scholar]
- 244.Cejas M.A., Kinney W.A., Chen C., Vinter J.G., Almond H.R., Balss K.M., Maryanoff C.A., Schmidt U., Breslav M., Mahan A., Lacy E., Maryanoff B.E. Thrombogenic collagen-mimetic peptides: self-assembly of triple helix-based fibrils driven by hydrophobic interactions. Proc. Natl. Acad. Sci. 2008;105:8513–8518. doi: 10.1073/PNAS.0800291105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Rao G.H., Fields C.G., White J.G., Fields G.B. Promotion of human platelet adhesion and aggregation by a synthetic, triple-helical “mini-collagen”. J. Biol. Chem. 1994;269:13899–13903. http://www.ncbi.nlm.nih.gov/pubmed/8188668 accessed February 17, 2020. [PubMed] [Google Scholar]
- 246.Kar K., Ibrar S., Nanda V., Getz T.M., Kunapuli S.P., Brodsky B. Aromatic interactions promote self-association of collagen triple-helical peptides to higher-order structures. Biochemistry. 2009;48:7959–7968. doi: 10.1021/bi900496m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Chen C.C., Hsu W., Kao T.C., Horng J.C. Self-assembly of short collagen-related peptides into fibrils via cation-π interactions. Biochemistry. 2011;50:2381–2383. doi: 10.1021/bi1018573. [DOI] [PubMed] [Google Scholar]
- 248.Yu Y.C., Pakalns T., Dori Y., Mccarthy J.B., Tirrell M., Fields G.B. Construction of biologically active protein molecular architecture using self-assembling peptide-amphiphiles. Methods Enzymol. 1997;289:571–582. doi: 10.1016/S0076-6879(97)89065-9. [DOI] [PubMed] [Google Scholar]
- 249.Yu Y.C., Roontga V., Daragan V.A., Mayo K.H., Tirrell M., Fields G.B. Structure and dynamics of peptide-amphiphiles incorporating triple- helical proteinlike molecular architecture. Biochemistry. 1999;38:1659–1668. doi: 10.1021/bi982315l. [DOI] [PubMed] [Google Scholar]
- 250.Luo J., Tong Y.W. Self-assembly of collagen-mimetic peptide amphiphiles into biofunctional nanofiber. ACS Nano. 2011;5:7739–7747. doi: 10.1021/nn202822f. [DOI] [PubMed] [Google Scholar]
- 251.Rele S., Song Y., Apkarian R.P., Qu Z., Conticello V.P., Chaikof E.L. D-periodic collagen-mimetic microfibers. J. Am. Chem. Soc. 2007;129:14780–14787. doi: 10.1021/ja0758990. [DOI] [PubMed] [Google Scholar]
- 252.O'Leary L.E.R., Fallas J.A., Bakota E.L., Kang M.K., Hartgerink J.D. Multi-hierarchical self-assembly of a collagen mimetic peptide from triple helix to nanofibre and hydrogel. Nat. Chem. 2011;3:821–828. doi: 10.1038/nchem.1123. [DOI] [PubMed] [Google Scholar]
- 253.Tanrikulu I.C., Forticaux A., Jin S., Raines R.T. Peptide tessellation yields micrometre-scale collagen triple helices. Nat. Chem. 2016;8:1008–1014. doi: 10.1038/nchem.2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Lee S.-G., Lee J.Y., Chmielewski J. Investigation of pH-dependent collagen triple-Helix formation. Angew. Chemie Int. Ed. 2008;47:8429–8432. doi: 10.1002/anie.200802224. [DOI] [PubMed] [Google Scholar]
- 255.Pires M.M., Przybyla D.E., Rubert Pérez C.M., Chmielewski J. Metal-mediated tandem Coassembly of collagen peptides into banded microstructures. J. Am. Chem. Soc. 2011;133:14469–14471. doi: 10.1021/ja2042645. [DOI] [PubMed] [Google Scholar]
- 256.Pires M.M., Lee J., Ernenwein D., Chmielewski J. Controlling the morphology of metal-promoted higher ordered assemblies of collagen peptides with varied Core lengths. Langmuir. 2012;28:1993–1997. doi: 10.1021/la203848r. [DOI] [PubMed] [Google Scholar]
- 257.Pires M.M., Przybyla D.E., Chmielewski J. A metal-collagen peptide framework for three-dimensional cell culture. Angew. Chemie Int. Ed. 2009;48:7813–7817. doi: 10.1002/anie.200902375. [DOI] [PubMed] [Google Scholar]
- 258.Pires M.M., Ernenwein D., Chmielewski J. Selective decoration and release of his-tagged proteins from metal-assembled collagen peptide Microflorettes. Biomacromolecules. 2011;12:2429–2433. doi: 10.1021/bm2004934. [DOI] [PubMed] [Google Scholar]
- 259.Nepal M., Sheedlo M.J., Das C., Chmielewski J. Accessing three-dimensional crystals with incorporated guests through metal-directed coiled-coil peptide assembly. J. Am. Chem. Soc. 2016;138:11051–11057. doi: 10.1021/jacs.6b06708. [DOI] [PubMed] [Google Scholar]
- 260.Strauss K., Chmielewski J. Advances in the design and higher-order assembly of collagen mimetic peptides for regenerative medicine. Curr. Opin. Biotechnol. 2017;46:34–41. doi: 10.1016/j.copbio.2016.10.013. [DOI] [PubMed] [Google Scholar]
- 261.Luo T., David M.A., Dunshee L.C., Scott R.A., Urello M.A., Price C., Kiick K.L. Thermoresponsive elastin-b-collagen-like peptide bioconjugate nanovesicles for targeted drug delivery to collagen-containing matrices. Biomacromolecules. 2017;18:2539–2551. doi: 10.1021/acs.biomac.7b00686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Luo T., Kiick K.L. Noncovalent modulation of the inverse temperature transition and self-assembly of elastin-b-collagen-like peptide bioconjugates. J. Am. Chem. Soc. 2015;137:15362–15365. doi: 10.1021/jacs.5b09941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Qin J., Luo T., Kiick K.L. Self-Assembly of Stable Nanoscale Platelets from Designed Elastin-like Peptide-Collagen-like Peptide Bioconjugates. Biomacromolecules. 2019 doi: 10.1021/acs.biomac.8b01681. [DOI] [PubMed] [Google Scholar]
- 264.Prhashanna A., Taylor P.A., Qin J., Kiick K.L., Jayaraman A. Effect of peptide sequence on the LCST-like transition of elastin-like peptides and elastin-like peptide-collagen-like peptide conjugates: simulations and experiments. Biomacromolecules. 2019;20:1178–1189. doi: 10.1021/acs.biomac.8b01503. [DOI] [PubMed] [Google Scholar]
- 265.Luo T., Kiick K.L. Noncovalent modulation of the inverse temperature transition and self-assembly of elastin-b-collagen-like peptide bioconjugates. J. Am. Chem. Soc. 2015;137:15362–15365. doi: 10.1021/jacs.5b09941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Luo T., He L., Theato P., Kiick K.L. Thermoresponsive self-assembly of nanostructures from a collagen-like peptide-containing Diblock copolymer. Macromol. Biosci. 2015;15:111–123. doi: 10.1002/mabi.201400358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Luo T., David M.A., Dunshee L.C., Scott R.A., Urello M.A., Price C., Kiick K.L. Thermoresponsive elastin-b-collagen-like peptide bioconjugate Nanovesicles for targeted drug delivery to collagen-containing matrices. Biomacromolecules. 2017;18:2539–2551. doi: 10.1021/acs.biomac.7b00686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Bello A.B., Kim D., Kim D., Park H., Lee S.H. Engineering and functionalization of gelatin biomaterials: from cell culture to medical applications. Tissue Eng. - Part B Rev. 2020;26:164–180. doi: 10.1089/ten.teb.2019.0256. [DOI] [PubMed] [Google Scholar]
- 269.Djagny K.B., Wang Z., Xu S. Gelatin: a valuable protein for food and pharmaceutical industries: review. Crit. Rev. Food Sci. Nutr. 2001;41:481–492. doi: 10.1080/20014091091904. [DOI] [PubMed] [Google Scholar]
- 270.Flory P.J., Weaver E.S. Helix ⇆ Coil Transitions in Dilute Aqueous Collagen Solutions. J. Am. Chem. Soc. 1960;82:4518–4525. doi: 10.1021/ja01502a018. [DOI] [Google Scholar]
- 271.Elzoghby A.O. Gelatin-based nanoparticles as drug and gene delivery systems: reviewing three decades of research. J. Control. Release. 2013;172:1075–1091. doi: 10.1016/j.jconrel.2013.09.019. [DOI] [PubMed] [Google Scholar]
- 272.Gorgieva S., Kokol V. Biomater. Appl. Nanomedicine. InTech; 2011. Collagen- vs. Gelatine-Based Biomaterials and Their Biocompatibility: Review and Perspectives. [DOI] [Google Scholar]
- 273.Sahoo N., Sahoo R.K., Biswas N., Guha A., Kuotsu K. Recent advancement of gelatin nanoparticles in drug and vaccine delivery. Int. J. Biol. Macromol. 2015;81:317–331. doi: 10.1016/j.ijbiomac.2015.08.006. [DOI] [PubMed] [Google Scholar]
- 274.Su K., Wang C. Recent advances in the use of gelatin in biomedical research. Biotechnol. Lett. 2015;37:2139–2145. doi: 10.1007/s10529-015-1907-0. [DOI] [PubMed] [Google Scholar]
- 275.Olsen D., Yang C., Bodo M., Chang R., Leigh S., Baez J., Carmichael D., Perälä M., Hämäläinen E.R., Jarvinen M., Polarek J. Recombinant collagen and gelatin for drug delivery. Adv. Drug Deliv. Rev. 2003;55:1547–1567. doi: 10.1016/j.addr.2003.08.008. [DOI] [PubMed] [Google Scholar]
- 276.Kommareddy S., Shenoy D.B., Amiji M.M. Wiley-VCH Verlag GmbH & Co. KGaA; Weinheim, Germany: 2007. Gelatin Nanoparticles and their Biofunctionalization, in: Nanotechnologies Life Sci. [DOI] [Google Scholar]
- 277.Liang H.C., Chang W.H., Liang H.F., Lee M.H., Sung H.W. Crosslinking structures of gelatin hydrogels crosslinked with genipin or a water-soluble carbodiimide. J. Appl. Polym. Sci. 2004;91:4017–4026. doi: 10.1002/app.13563. [DOI] [Google Scholar]
- 278.Harrington W.F., Rao N.V. Collagen Structure in Solution. I. Kinetics of Helix Regeneration in Single-Chain Gelatins. Biochemistry. 1970;9:3714–3724. doi: 10.1021/bi00821a010. [DOI] [PubMed] [Google Scholar]
- 279.Bello J., Riese H.C.A., Vinograd J.R. Mechanism of gelation of gelatin. Influence of certain electrolytes on the melting points of gels of gelatin and chemically modified gelatins. J. Phys. Chem. 1956;60:1299–1306. doi: 10.1021/j150543a035. [DOI] [Google Scholar]
- 280.Djabourov M., Leblond J., Papon P. Gelation of aqueous gelatin solutions. I. Structural investigation. J. Phys. 1988;49:319–332. doi: 10.1051/jphys:01988004902031900. [DOI] [Google Scholar]
- 281.Lee E.J., Khan S.A., Lim K.H. Gelatin nanoparticle preparation by nanoprecipitation. J. Biomater. Sci. Polym. Ed. 2011;22:753–771. doi: 10.1163/092050610X492093. [DOI] [PubMed] [Google Scholar]
- 282.Li Z., Gu L. Effects of mass ratio, pH, temperature, and reaction time on fabrication of partially purified pomegranate ellagitannin-gelatin nanoparticles. J. Agric. Food Chem. 2011;59:4225–4231. doi: 10.1021/jf200024d. [DOI] [PubMed] [Google Scholar]
- 283.FARRUGIA C.A., GROVES M.J. Gelatin behaviour in dilute aqueous solution: designing a Nanoparticulate formulation. J. Pharm. Pharmacol. 1999;51:643–649. doi: 10.1211/0022357991772925. [DOI] [PubMed] [Google Scholar]
- 284.Saxena A., Sachin K., Bohidar H.B., Verma A.K. Effect of molecular weight heterogeneity on drug encapsulation efficiency of gelatin nano-particles. Colloids Surfaces B Biointerfaces. 2005;45:42–48. doi: 10.1016/j.colsurfb.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 285.Bajpai A.K., Choubey J. Design of gelatin nanoparticles as swelling controlled delivery system for chloroquine phosphate. J. Mater. Sci. Mater. Med. 2006;17:345–358. doi: 10.1007/s10856-006-8235-9. [DOI] [PubMed] [Google Scholar]
- 286.Amjadi S., Hamishehkar H., Ghorbani M. A novel smart PEGylated gelatin nanoparticle for co-delivery of doxorubicin and betanin: a strategy for enhancing the therapeutic efficacy of chemotherapy. Mater. Sci. Eng. C. 2019;97:833–841. doi: 10.1016/j.msec.2018.12.104. [DOI] [PubMed] [Google Scholar]
- 287.Wang H., Zou Q., Boerman O.C., Nijhuis A.W.G., Jansen J.A., Li Y., Leeuwenburgh S.C.G. Combined delivery of BMP-2 and bFGF from nanostructured colloidal gelatin gels and its effect on bone regeneration in vivo. J. Control. Release. 2013;166:172–181. doi: 10.1016/j.jconrel.2012.12.015. [DOI] [PubMed] [Google Scholar]
- 288.Narayanan D., Geena M.G., Lakshmi H., Koyakutty M., Nair S., Menon D. Poly-(ethylene glycol) modified gelatin nanoparticles for sustained delivery of the anti-inflammatory drug ibuprofen-sodium: An in vitro and in vivo analysis. Nanomed. Nanotechnol. Biol. Med. 2013;9:818–828. doi: 10.1016/j.nano.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 289.Carvalho J.A., da Silva Abreu A., Tedesco A.C., Junior M.B., Simioni A.R. Functionalized photosensitive gelatin nanoparticles for drug delivery application. J. Biomater. Sci. Polym. Ed. 2019;30:508–525. doi: 10.1080/09205063.2019.1580664. [DOI] [PubMed] [Google Scholar]
- 290.Kirar S., Thakur N.S., Laha J.K., Bhaumik J., Banerjee U.C. Development of gelatin nanoparticle-based biodegradable Phototheranostic agents: advanced system to treat infectious diseases. ACS Biomater. Sci. Eng. 2018;4:473–482. doi: 10.1021/acsbiomaterials.7b00751. [DOI] [PubMed] [Google Scholar]
- 291.Yarchoan R., Mitsuya H., Thomas R.V., Pluda J.M., Hartman N.R., Perno C.F., Marczyk K.S., Allain J.P., Johns D.G., Broder S. In vivo activity against HIV and favorable toxicity profile of 2′,3′-dideoxyinosine. Science (80-. ) 1989;245:412–415. doi: 10.1126/science.2502840. [DOI] [PubMed] [Google Scholar]
- 292.Zhang W., Han B., Lai X., Xiao C., Xu S., Meng X., Li Z., Meng J., Wen T., Yang X., Liu J., Xu H. Stiffness of cationized gelatin nanoparticles is a key factor determining RNAi efficiency in myeloid leukemia cells. Chem. Commun. 2020;56:1255–1258. doi: 10.1039/c9cc09068a. [DOI] [PubMed] [Google Scholar]
- 293.Solorio L.D., Vieregge E.L., Dhami C.D., Dang P.N., Alsberg E. Engineered cartilage via self-assembled hMSC sheets with incorporated biodegradable gelatin microspheres releasing transforming growth factor-β1. J. Control. Release. 2012;158:224–232. doi: 10.1016/j.jconrel.2011.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Patel Z.S., Ueda H., Yamamoto M., Tabata Y., Mikos A.G. In vitro and in vivo release of vascular endothelial growth factor from gelatin microparticles and biodegradable composite scaffolds. Pharm. Res. 2008;25:2370–2378. doi: 10.1007/s11095-008-9685-1. [DOI] [PubMed] [Google Scholar]
- 295.Patel Z.S., Yamamoto M., Ueda H., Tabata Y., Mikos A.G. Biodegradable gelatin microparticles as delivery systems for the controlled release of bone morphogenetic protein-2. Acta Biomater. 2008;4:1126–1138. doi: 10.1016/j.actbio.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Huang S., Wang Y., Liang T., Jin F., Liu S., Jin Y. Fabrication and characterization of a novel microparticle with gyrus-patterned surface and growth factor delivery for cartilage tissue engineering. Mater. Sci. Eng. C. 2009;29:1351–1356. doi: 10.1016/j.msec.2008.10.036. [DOI] [Google Scholar]
- 297.Nguyen A.H., Wang Y., White D.E., Platt M.O., McDevitt T.C. MMP-mediated mesenchymal morphogenesis of pluripotent stem cell aggregates stimulated by gelatin methacrylate microparticle incorporation. Biomaterials. 2016;76:66–75. doi: 10.1016/j.biomaterials.2015.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Gómez-Estaca J., Balaguer M.P., López-Carballo G., Gavara R., Hernández-Muñoz P. Improving antioxidant and antimicrobial properties of curcumin by means of encapsulation in gelatin through electrohydrodynamic atomization. Food Hydrocoll. 2017;70:313–320. doi: 10.1016/j.foodhyd.2017.04.019. [DOI] [Google Scholar]
- 299.Madan J., Pandey R.S., Jain U.K., Katare O.P., Aneja R., Katyal A. Sterically stabilized gelatin microassemblies of noscapine enhance cytotoxicity, apoptosis and drug delivery in lung cancer cells. Colloids Surfaces B Biointerfaces. 2013;107:235–244. doi: 10.1016/j.colsurfb.2013.02.010. [DOI] [PubMed] [Google Scholar]
- 300.Kim B., Han S.W., Choi S.E., Yim D., Kim J.H., Wyss H.M., Kim J.W. Monodisperse microshell structured gelatin microparticles for temporary chemoembolization. Biomacromolecules. 2018;19:386–391. doi: 10.1021/acs.biomac.7b01479. [DOI] [PubMed] [Google Scholar]
- 301.Li H., Wang M., Williams G.R., Wu J., Sun X., Lv Y., Zhu L.M. Electrospun gelatin nanofibers loaded with vitamins a and e as antibacterial wound dressing materials. RSC Adv. 2016;6:50267–50277. doi: 10.1039/c6ra05092a. [DOI] [Google Scholar]
- 302.Del Gaudio C., Baiguera S., Boieri M., Mazzanti B., Ribatti D., Bianco A., Macchiarini P. Induction of angiogenesis using VEGF releasing genipin-crosslinked electrospun gelatin mats. Biomaterials. 2013;34:7754–7765. doi: 10.1016/j.biomaterials.2013.06.040. [DOI] [PubMed] [Google Scholar]
- 303.Jaipan P., Nguyen A., Narayan R.J. Gelatin-based hydrogels for biomedical applications. MRS Commun. 2017;7:416–426. doi: 10.1557/mrc.2017.92. [DOI] [Google Scholar]
- 304.Lin H., Cheng A.W.M., Alexander P.G., Beck A.M., Tuan R.S. Cartilage tissue engineering application of injectable gelatin hydrogel with in situ visible-light-activated gelation capability in both air and aqueous solution. Tissue Eng. - Part A. 2014;20:2402–2411. doi: 10.1089/ten.tea.2013.0642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Mazaki T., Shiozaki Y., Yamane K., Yoshida A., Nakamura M., Yoshida Y., Zhou D., Kitajima T., Tanaka M., Ito Y., Ozaki T., Matsukawa A. A novel, visible light-induced, rapidly cross-linkable gelatin scaffold for osteochondral tissue engineering. Sci. Rep. 2014;4:1–10. doi: 10.1038/srep04457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Oun R., Plumb J.A., Wheate N.J. A cisplatin slow-release hydrogel drug delivery system based on a formulation of the macrocycle cucurbit[7]uril, gelatin and polyvinyl alcohol. J. Inorg. Biochem. 2014;134:100–105. doi: 10.1016/j.jinorgbio.2014.02.004. [DOI] [PubMed] [Google Scholar]
- 307.Jiang T., Munguia-Lopez J., Flores-Torres S., Grant J., Vijayakumar S., de Leon-Rodriguez A., Kinsella J.M. Bioprintable alginate/gelatin hydrogel 3D in vitro model systems induce cell spheroid formation. J. Vis. Exp. 2018;2018 doi: 10.3791/57826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Jiang T., Munguia-Lopez J.G., Gu K., Bavoux M.M., Flores-Torres S., Kort-Mascort J., Grant J., Vijayakumar S., De Leon-Rodriguez A., Ehrlicher A.J., Kinsella J.M. Engineering bioprintable alginate/gelatin composite hydrogels with tunable mechanical and cell adhesive properties to modulate tumor spheroid growth kinetics. Biofabrication. 2020;12 doi: 10.1088/1758-5090/ab3a5c. [DOI] [PubMed] [Google Scholar]
- 309.Jiang T., Munguia-Lopez J.G., Flores-Torres S., Grant J., Vijayakumar S., De Leon-Rodriguez A., Kinsella J.M. Directing the Self-Assembly of Tumour Spheroids by Bioprinting Cellular Heterogeneous Models within Alginate/Gelatin Hydrogels. Sci. Rep. 2017;7 doi: 10.1038/s41598-017-04691-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Foox M., Raz-Pasteur A., Berdicevsky I., Krivoy N., Zilberman M. In vitro microbial inhibition, bonding strength, and cellular response to novel gelatin-alginate antibiotic-releasing soft tissue adhesives. Polym. Adv. Technol. 2014;25:516–524. doi: 10.1002/pat.3278. [DOI] [Google Scholar]
- 311.Cohen B., Shefy-Peleg A., Zilberman M. Novel gelatin/alginate soft tissue adhesives loaded with drugs for pain management: structure and properties. J. Biomater. Sci. Polym. Ed. 2014;25:224–240. doi: 10.1080/09205063.2013.849904. [DOI] [PubMed] [Google Scholar]
- 312.Mithieux S.M., A.S.B.T.-A. in P.C. Weiss . Fibrous Proteins: Coiled-Coils, Collagen and Elastomers. Academic Press; 2005. Elastin; pp. 437–461. [DOI] [Google Scholar]
- 313.Almine J.F., Bax D.V., Mithieux S.M., Nivison-Smith L., Rnjak J., Waterhouse A., Wise S.G., Weiss A.S. Elastin-based materials. Chem. Soc. Rev. 2010;39:3371–3379. doi: 10.1039/B919452P. [DOI] [PubMed] [Google Scholar]
- 314.MacEwan S.R., Chilkoti A. Applications of elastin-like polypeptides in drug delivery. J. Control. Release. 2014;190:314–330. doi: 10.1016/j.jconrel.2014.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.MacEwan S.R., Chilkoti A. Elastin-like polypeptides: biomedical applications of tunable biopolymers. Pept. Sci. 2010;94:60–77. doi: 10.1002/bip.21327. [DOI] [PubMed] [Google Scholar]
- 316.Li N.K., Quiroz F.G., Hall C.K., Chilkoti A., Yingling Y.G. Molecular description of the LCST behavior of an elastin-like polypeptide. Biomacromolecules. 2014;15:3522–3530. doi: 10.1021/bm500658w. [DOI] [PubMed] [Google Scholar]
- 317.Ougizawa T., Inoue T., Kammer H.W. UCST and LCST behavior in polymer blends. Macromolecules. 1985;18:2089–2092. doi: 10.1021/ma00152a052. [DOI] [Google Scholar]
- 318.McDaniel J.R., Radford D.C., Chilkoti A. A unified model for de novo design of elastin-like polypeptides with tunable inverse transition temperatures. Biomacromolecules. 2013;14:2866–2872. doi: 10.1021/bm4007166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Urry D.W., Haynes B., Harris R.D. Temperature dependence of length of elastin and its polypentapeptide. Biochem. Biophys. Res. Commun. 1986;141:749–755. doi: 10.1016/S0006-291X(86)80236-4. [DOI] [PubMed] [Google Scholar]
- 320.Trabbic-Carlson K., Meyer D.E., Liu L., Piervincenzi R., Nath N., LaBean T., Chilkoti A. Effect of protein fusion on the transition temperature of an environmentally responsive elastin-like polypeptide: a role for surface hydrophobicity? Protein Eng. Des. Sel. 2004;17:57–66. doi: 10.1093/protein/gzh006. [DOI] [PubMed] [Google Scholar]
- 321.Christensen T., Hassouneh W., Trabbic-Carlson K., Chilkoti A. Predicting transition temperatures of elastin-like polypeptide fusion proteins. Biomacromolecules. 2013;14:1514–1519. doi: 10.1021/bm400167h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Meyer D.E., Chilkoti A. Purification of recombinant proteins by fusion with thermally-responsive polypeptides. Nat. Biotechnol. 1999;17:1112–1115. doi: 10.1038/15100. [DOI] [PubMed] [Google Scholar]
- 323.Trabbic-Carlson K., Liu L., Kim B., Chilkoti A. Expression and purification of recombinant proteins from Escherichia coli: comparison of an elastin-like polypeptide fusion with an oligohistidine fusion. Protein Sci. 2004;13:3274–3284. doi: 10.1110/ps.04931604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Banki M.R., Feng L., Wood D.W. Simple bioseparations using self-cleaving elastin-like polypeptide tags. Nat. Methods. 2005;2:659–662. doi: 10.1038/nmeth787. [DOI] [PubMed] [Google Scholar]
- 325.Shen Y., Ai H.-X., Song R., Liang Z.-N., Li J.-F., Zhang S.-Q. Expression and purification of moricin CM4 and human β-defensins 4 in Escherichia coli using a new technology. Microbiol. Res. 2010;165:713–718. doi: 10.1016/j.micres.2010.01.002. [DOI] [PubMed] [Google Scholar]
- 326.Bruno B.J., Miller G.D., Lim C.S. Basics and recent advances in peptide and protein drug delivery. Ther. Deliv. 2013;4:1443–1467. doi: 10.4155/tde.13.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Takakura Y., Hashida M. Macromolecular carrier Systems for Targeted Drug Delivery: pharmacokinetic considerations on biodistribution. Pharm. Res. 1996;13:820–831. doi: 10.1023/A:1016084508097. [DOI] [PubMed] [Google Scholar]
- 328.Kalepu S., Nekkanti V. Insoluble drug delivery strategies: review of recent advances and business prospects. Acta Pharm. Sin. B. 2015;5:442–453. doi: 10.1016/j.apsb.2015.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Tiwari G., Tiwari R., Sriwastawa B., Bhati L., Pandey S., Pandey P., Bannerjee S.K. Drug delivery systems: An updated review. Int. J. Pharm. Investig. 2012;2:2–11. doi: 10.4103/2230-973X.96920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Conley A.J., Joensuu J.J., Jevnikar A.M., Menassa R., Brandle J.E. Optimization of elastin-like polypeptide fusions for expression and purification of recombinant proteins in plants. Biotechnol. Bioeng. 2009;103:562–573. doi: 10.1002/bit.22278. [DOI] [PubMed] [Google Scholar]
- 331.Conrad U., Plagmann I., Malchow S., Sack M., Floss D.M., Kruglov A.A., Nedospasov S.A., Rose-John S., Scheller J. ELPylated anti-human TNF therapeutic single-domain antibodies for prevention of lethal septic shock. Plant Biotechnol. J. 2011;9:22–31. doi: 10.1111/j.1467-7652.2010.00523.x. [DOI] [PubMed] [Google Scholar]
- 332.Phan H.T., Hause B., Hause G., Arcalis E., Stoger E., Maresch D., Altmann F., Joensuu J., Conrad U. Influence of elastin-like polypeptide and hydrophobin on recombinant hemagglutinin accumulations in transgenic tobacco plants. PLoS One. 2014;9(6):e99347. doi: 10.1371/journal.pone.0099347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Holloway J.L. Drug delivery in stealth mode. Sci. Transl. Med. 2018;10 doi: 10.1126/scitranslmed.aaw0523. eaaw0523. [DOI] [Google Scholar]
- 334.Salmaso S., Caliceti P. Stealth properties to improve therapeutic efficacy of drug nanocarriers. J. Drug Deliv. 2013 doi: 10.1155/2013/374252. (2013) 374252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Banskota S., Yousefpour P., Kirmani N., Li X., Chilkoti A. Long circulating genetically encoded intrinsically disordered zwitterionic polypeptides for drug delivery. Biomaterials. 2019;192:475–485. doi: 10.1016/j.biomaterials.2018.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Lee T.A.T., Cooper A., Apkarian R.P., Conticello V.P. Thermo-reversible self-assembly of nanoparticles derived from elastin-mimetic polypeptides. Adv. Mater. 2000;12:1105–1110. doi: 10.1002/1521-4095(200008)12:15<1105::AID-ADMA1105>3.0.CO;2-1. [DOI] [Google Scholar]
- 337.Wright E.R., Conticello V.P. Self-assembly of block copolymers derived from elastin-mimetic polypeptide sequences. Adv. Drug Deliv. Rev. 2002;54:1057–1073. doi: 10.1016/S0169-409X(02)00059-5. [DOI] [PubMed] [Google Scholar]
- 338.Wright E.R., McMillan R.A., Cooper A., Apkarian R.P., Conticello V.P. Thermoplastic elastomer hydrogels via self-assembly of an elastin-mimetic Triblock polypeptide. Adv. Funct. Mater. 2002;12:149–154. doi: 10.1002/1616-3028(20020201)12:2<149::AID-ADFM149>3.0.CO;2-N. [DOI] [Google Scholar]
- 339.Dreher M.R., Simnick A.J., Fischer K., Smith R.J., Patel A., Schmidt M., Chilkoti A. Temperature triggered self-assembly of polypeptides into multivalent spherical micelles. J. Am. Chem. Soc. 2008;130:687–694. doi: 10.1021/ja0764862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Janib S.M., Pastuszka M.F., Aluri S., Folchman-Wagner Z., Hsueh P.Y., Shi P., Lin Y.A., Cui H., MacKay J.A. A quantitative recipe for engineering protein polymer nanoparticles. Polym. Chem. 2014;5:1614–1625. doi: 10.1039/C3PY00537B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Hassouneh W., Zhulina E.B., Chilkoti A., Rubinstein M. Elastin-like polypeptide Diblock copolymers self-assemble into weak micelles. Macromolecules. 2015;48:4183–4195. doi: 10.1021/acs.macromol.5b00431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Simnick A.J., Valencia C.A., Liu R., Chilkoti A. Morphing low-affinity ligands into high-avidity nanoparticles by thermally triggered self-assembly of a genetically encoded polymer. ACS Nano. 2010;4:2217–2227. doi: 10.1021/nn901732h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Macewan S.R., Chilkoti A. Digital switching of local arginine density in a genetically encoded self-assembled polypeptide nanoparticle controls cellular uptake. Nano Lett. 2012;12:3322–3328. doi: 10.1021/nl301529p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Wender P.A., Mitchell D.J., Pattabiraman K., Pelkey E.T., Steinman L., Rothbard J.B. The design, synthesis, and evaluation of molecules that enable or enhance cellular uptake: peptoid molecular transporters. Proc. Natl. Acad. Sci. U. S. A. 2000;97:13003–13008. doi: 10.1073/pnas.97.24.13003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Hassouneh W., Fischer K., MacEwan S.R., Branscheid R., Fu C.L., Liu R., Schmidt M., Chilkoti A. Unexpected multivalent display of proteins by temperature triggered self-assembly of elastin-like polypeptide block copolymers. Biomacromolecules. 2012;13:1598–1605. doi: 10.1021/bm300321n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Sun G., Hsueh P.-Y., Janib S.M., Hamm-Alvarez S., MacKay J.A. Design and cellular internalization of genetically engineered polypeptide nanoparticles displaying adenovirus knob domain. J. Control. Release. 2011;155:218–226. doi: 10.1016/j.jconrel.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Hsueh P.-Y., Edman M.C., Sun G., Shi P., Xu S., Lin Y.-A., Cui H., Hamm-Alvarez S.F., MacKay J.A. Tear-mediated delivery of nanoparticles through transcytosis of the lacrimal gland. J. Control. Release. 2015;208:2–13. doi: 10.1016/j.jconrel.2014.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Callahan D.J., Liu W., Li X., Dreher M.R., Hassouneh W., Kim M., Marszalek P., Chilkoti A. Triple stimulus-responsive polypeptide nanoparticles that enhance intratumoral spatial distribution. Nano Lett. 2012;12:2165–2170. doi: 10.1021/nl300630c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Shah M., Edman M.C., Janga S.R., Shi P., Dhandhukia J., Liu S., Louie S.G., Rodgers K., Mackay J.A., Hamm-Alvarez S.F. A rapamycin-binding protein polymer nanoparticle shows potent therapeutic activity in suppressing autoimmune dacryoadenitis in a mouse model of Sjögren's syndrome. J. Control. Release. 2013;171:269–279. doi: 10.1016/j.jconrel.2013.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Shi P., Aluri S., Lin Y.-A., Shah M., Edman M., Dhandhukia J., Cui H., MacKay J.A. Elastin-based protein polymer nanoparticles carrying drug at both corona and core suppress tumor growth in vivo. J. Control. Release. 2013;171:330–338. doi: 10.1016/j.jconrel.2013.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.McDaniel J.R., Weitzhandler I., Prevost S., Vargo K.B., Appavou M.-S., Hammer D.A., Gradzielski M., Chilkoti A. Noncanonical self-assembly of highly asymmetric genetically encoded polypeptide amphiphiles into cylindrical micelles. Nano Lett. 2014;14:6590–6598. doi: 10.1021/nl503221p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Aluri S.R., Shi P., Gustafson J.A., Wang W., Lin Y.-A., Cui H., Liu S., Conti P.S., Li Z., Hu P., Epstein A.L., MacKay J.A. A hybrid protein-polymer nanoworm potentiates apoptosis better than a monoclonal antibody. ACS Nano. 2014;8:2064–2076. doi: 10.1021/nn403973g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Pastuszka M.K., Wang X., Lock L.L., Janib S.M., Cui H., DeLeve L.D., MacKay J.A. An amphipathic alpha-helical peptide from apolipoprotein A1 stabilizes protein polymer vesicles. J. Control. Release. 2014;191:15–23. doi: 10.1016/j.jconrel.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.McDaniel J.R., Bhattacharyya J., Vargo K.B., Hassouneh W., Hammer D.A., Chilkoti A. Self-assembly of thermally responsive nanoparticles of a genetically encoded peptide polymer by drug conjugation. Angew. Chem. Int. Ed. Engl. 2013;52:1683–1687. doi: 10.1002/anie.201200899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Bhattacharyya J., Bellucci J.J., Weitzhandler I., McDaniel J.R., Spasojevic I., Li X., Lin C.-C., Chi J.-T.A., Chilkoti A. A paclitaxel-loaded recombinant polypeptide nanoparticle outperforms Abraxane in multiple murine cancer models. Nat. Commun. 2015;6:7939. doi: 10.1038/ncomms8939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Bhattacharyya J., Ren X.-R., Mook R.A., Wang J., Spasojevic I., Premont R.T., Li X., Chilkoti A., Chen W. Niclosamide-conjugated polypeptide nanoparticles inhibit Wnt signaling and colon cancer growth. Nanoscale. 2017;9:12709–12717. doi: 10.1039/c7nr01973d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Bhattacharyya J., Weitzhandler I., Ho S.B., McDaniel J.R., Li X., Tang L., Liu J., Dewhirst M., Chilkoti A. Encapsulating a hydrophilic chemotherapeutic into rod-like nanoparticles of a genetically encoded asymmetric Triblock polypeptide improves its efficacy. Adv. Funct. Mater. 2017;27:1605421. doi: 10.1002/adfm.201605421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Costa S.A., Mozhdehi D., Dzuricky M.J., Isaacs F.J., Brustad E.M., Chilkoti A. Active targeting of cancer cells by Nanobody decorated polypeptide micelle with bio-orthogonally conjugated drug. Nano Lett. 2019;19:247–254. doi: 10.1021/acs.nanolett.8b03837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359.Luginbuhl K.M., Mozhdehi D., Dzuricky M., Yousefpour P., Huang F.C., Mayne N.R., Buehne K.L., Chilkoti A. Recombinant synthesis of hybrid lipid-peptide polymer fusions that self-assemble and encapsulate hydrophobic drugs. Angew. Chem. Int. Ed. Engl. 2017;56:13979–13984. doi: 10.1002/anie.201704625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Huang W., Rollett A., Kaplan D.L. Silk-elastin-like protein biomaterials for the controlled delivery of therapeutics. Expert Opin. Drug Deliv. 2015;12:779–791. doi: 10.1517/17425247.2015.989830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Xia X.-X., Xu Q., Hu X., Qin G., Kaplan D.L. Tunable self-assembly of genetically engineered silk--elastin-like protein polymers. Biomacromolecules. 2011;12:3844–3850. doi: 10.1021/bm201165h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Xia X.-X., Wang M., Lin Y., Xu Q., Kaplan D.L. Hydrophobic drug-triggered self-assembly of nanoparticles from silk-elastin-like protein polymers for drug delivery. Biomacromolecules. 2014;15:908–914. doi: 10.1021/bm4017594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Liu W., MacKay J.A., Dreher M.R., Chen M., McDaniel J.R., Simnick A.J., Callahan D.J., Zalutsky M.R., Chilkoti A. Injectable intratumoral depot of thermally responsive polypeptide-radionuclide conjugates delays tumor progression in a mouse model. J. Control. Release. 2010;144:2–9. doi: 10.1016/j.jconrel.2010.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Liu W., McDaniel J., Li X., Asai D., Quiroz F.G., Schaal J., Park J.S., Zalutsky M., Chilkoti A. Brachytherapy Using Injectable Seeds That Are Self-Assembled from Genetically Encoded Polypeptides <em>In Situ</em>. Cancer Res. 2012;72 doi: 10.1158/0008-5472.CAN-12-2127. 5956 LP – 5965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Schaal J.L., Li X., Mastria E., Bhattacharyya J., Zalutsky M.R., Chilkoti A., Liu W. Injectable polypeptide micelles that form radiation crosslinked hydrogels in situ for intratumoral radiotherapy. J. Control. Release. 2016;228:58–66. doi: 10.1016/j.jconrel.2016.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366.Wang Z., Guo J., Ning J., Feng X., Liu X., Sun J., Chen X., Lu F., Gao W. One-month zero-order sustained release and tumor eradication after a single subcutaneous injection of interferon alpha fused with a body-temperature-responsive polypeptide. Biomater. Sci. 2019;7:104–112. doi: 10.1039/C8BM01096J. [DOI] [PubMed] [Google Scholar]
- 367.Amiram M., Luginbuhl K.M., Li X., Feinglos M.N., Chilkoti A. Injectable protease-operated depots of glucagon-like peptide-1 provide extended and tunable glucose control. Proc. Natl. Acad. Sci. U. S. A. 2013;110:2792–2797. doi: 10.1073/pnas.1214518110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368.Luginbuhl K.M., Schaal J.L., Umstead B., Mastria E.M., Li X., Banskota S., Arnold S., Feinglos M., D'Alessio D., Chilkoti A. One-week glucose control via zero-order release kinetics from an injectable depot of glucagon-like peptide-1 fused to a thermosensitive biopolymer. Nat. Biomed. Eng. 2017;1:78. doi: 10.1038/s41551-017-0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Gilroy C.A., Roberts S., Chilkoti A. Fusion of fibroblast growth factor 21 to a thermally responsive biopolymer forms an injectable depot with sustained anti-diabetic action. J. Control. Release. 2018;277:154–164. doi: 10.1016/j.jconrel.2018.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Wang W., Jashnani A., Aluri S.R., Gustafson J.A., Hsueh P.-Y., Yarber F., McKown R.L., Laurie G.W., Hamm-Alvarez S.F., MacKay J.A. A thermo-responsive protein treatment for dry eyes. J. Control. Release. 2015;199:156–167. doi: 10.1016/j.jconrel.2014.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Adams S.B., Jr., Shamji M.F., Nettles D.L., Hwang P., Setton L.A. Sustained release of antibiotics from injectable and thermally responsive polypeptide depots. J Biomed Mater Res B Appl Biomater. 2009;90:67–74. doi: 10.1002/jbm.b.31254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372.Shamji M.F., Betre H., Kraus V.B., Chen J., Chilkoti A., Pichika R., Masuda K., Setton L.A. Development and characterization of a fusion protein between thermally responsive elastin-like polypeptide and interleukin-1 receptor antagonist: sustained release of a local antiinflammatory therapeutic. Arthritis Rheum. 2007;56:3650–3661. doi: 10.1002/art.22952. [DOI] [PubMed] [Google Scholar]
- 373.PhaseBio Announces Global License of PB1023 for the Treatment of Sarcopenia-Related Diseases to ImmunoForge Nasdaq:PHAS, (n.d.). https://www.globenewswire.com/news-release/2019/04/09/1799631/0/en/PhaseBio-Announces-Global-License-of-PB1023-for-the-Treatment-of-Sarcopenia-Related-Diseases-to-ImmunoForge.html (accessed August 13, 2020).
- 374.GlobeNewswire, PhaseBio Announces Case Study Highlighting PB1046 Hemodynamic Data Presented at the 14th Pulmonary Vascular Research Institute World Congress, (2020). https://investors.phasebio.com/news-releases/news-release-details/phasebio-announces-case-study-highlighting-pb1046-hemodynamic (accessed August 13, 2020).
- 375.Qin G., Hu X., Cebe P., Kaplan D.L. Mechanism of resilin elasticity. Nat. Commun. 2012;3 doi: 10.1038/ncomms2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376.Li L., Tong Z., Jia X., Kiick K.L. Resilin-like polypeptide hydrogels engineered for versatile biological functions. Soft Matter. 2013;9:665–673. doi: 10.1039/C2SM26812D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Li L., Kiick K.L. Resilin-based materials for biomedical applications. ACS Macro Lett. 2013;2:635–640. doi: 10.1021/mz4002194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378.Ardell D.H., Andersen S.O. Tentative identification of a resilin gene in Drosophila melanogaster. Insect Biochem. Mol. Biol. 2001;31:965–970. doi: 10.1016/S0965-1748(01)00044-3. [DOI] [PubMed] [Google Scholar]
- 379.Elvin C.M., Carr A.G., Huson M.G., Maxwell J.M., Pearson R.D., Vuocolo T., Liyou N.E., Wong D.C.C., Merritt D.J., Dixon N.E. Synthesis and properties of crosslinked recombinant pro-resilin. Nature. 2005;437:999–1002. doi: 10.1038/nature04085. [DOI] [PubMed] [Google Scholar]
- 380.Lyons R.E., Nairn K.M., Huson M.G., Kim M., Dumsday G., Elvin C.M. Comparisons of recombinant Resilin-like proteins: repetitive domains are sufficient to confer Resilin-like properties. Biomacromolecules. 2009;10:3009–3014. doi: 10.1021/bm900601h. [DOI] [PubMed] [Google Scholar]
- 381.Balu R., Knott R., Cowieson N.P., Elvin C.M., Hill A.J., Choudhury N.R., Dutta N.K. Structural ensembles reveal intrinsic disorder for the multi-stimuli responsive bio-mimetic protein Rec1-resilin. Sci. Rep. 2015;5:1–12. doi: 10.1038/srep10896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382.Lyons R.E., Elvin C.M., Taylor K., Lekieffre N., Ramshaw J.A.M. Purification of recombinant protein by cold-coacervation of fusion constructs incorporating resilin-inspired polypeptides. Biotechnol. Bioeng. 2012;109:2947–2954. doi: 10.1002/bit.24565. [DOI] [PubMed] [Google Scholar]
- 383.Dutta N.K., Truong M.Y., Mayavan S., Roy Choudhury N., Elvin C.M., Kim M., Knott R., Nairn K.M., Hill A.J. A genetically engineered protein responsive to multiple stimuli. Angew. Chemie - Int. Ed. 2011;50:4428–4431. doi: 10.1002/anie.201007920. [DOI] [PubMed] [Google Scholar]
- 384.Truong M.Y., Dutta N.K., Choudhury N.R., Kim M., Elvin C.M., Hill A.J., Thierry B., Vasilev K. A pH-responsive interface derived from resilin-mimetic protein Rec1-resilin. Biomaterials. 2010;31:4434–4446. doi: 10.1016/j.biomaterials.2010.02.019. [DOI] [PubMed] [Google Scholar]
- 385.Li L., Luo T., Kiick K.L. Temperature-triggered phase separation of a hydrophilic resilin-like polypeptide. Macromol. Rapid Commun. 2015;36:90–95. doi: 10.1002/marc.201400521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386.Canning S.L., Neal T.J., Armes S.P. pH-responsive schizophrenic Diblock copolymers prepared by polymerization-induced self-assembly. Macromolecules. 2017;50:6108–6116. doi: 10.1021/acs.macromol.7b01005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387.Maeda Y., Mochiduki H., Ikeda I. Hydration changes during Thermosensitive Association of a Block Copolymer Consisting of LCST and UCST blocks. Macromol. Rapid Commun. 2004;25:1330–1334. doi: 10.1002/marc.200400062. [DOI] [Google Scholar]
- 388.Arotçaréna M., Heise B., Ishaya S., Laschewsky A. Switching the inside and the outside of aggregates of water-soluble block copolymers with double Thermoresponsivity. J. Am. Chem. Soc. 2002;124:3787–3793. doi: 10.1021/ja012167d. [DOI] [PubMed] [Google Scholar]
- 389.Weitzhandler I., Dzuricky M., Hoffmann I., Garcia Quiroz F., Gradzielski M., Chilkoti A. Micellar self-assembly of recombinant Resilin−/elastin-like block Copolypeptides. Biomacromolecules. 2017;18:2419–2426. doi: 10.1021/acs.biomac.7b00589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390.Charati M.B., Ifkovits J.L., Burdick J.A., Linhardt J.G., Kiick K.L. Hydrophilic elastomeric biomaterials based on resilin-like polypeptides. Soft Matter. 2009;5:3412–3416. doi: 10.1039/B910980C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391.Li L., Teller S., Clifton R.J., Jia X., Kiick K.L. Tunable mechanical stability and deformation response of a Resilin-based elastomer. Biomacromolecules. 2011;12:2302–2310. doi: 10.1021/bm200373p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392.Renner J.N., Cherry K.M., Su R.S.-C., Liu J.C. Characterization of Resilin-based materials for tissue engineering applications. Biomacromolecules. 2012;13:3678–3685. doi: 10.1021/bm301129b. [DOI] [PubMed] [Google Scholar]
- 393.D'Andrea L.D., Iaccarino G., Fattorusso R., Sorriento D., Carannante C., Capasso D., Trimarco B., Pedone C. Targeting angiogenesis: Structural characterization and biological properties of a de novo engineered VEGF mimicking peptide. Proc. Natl. Acad. Sci. U. S. A. 2005;102 doi: 10.1073/pnas.0505047102. 14215 LP – 14220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394.Qin G., Lapidot S., Numata K., Hu X., Meirovitch S., Dekel M., Podoler I., Shoseyov O., Kaplan D.L. Expression, cross-linking, and characterization of recombinant chitin binding Resilin. Biomacromolecules. 2009;10:3227–3234. doi: 10.1021/bm900735g. [DOI] [PubMed] [Google Scholar]
- 395.Su R.S.C., Galas R.J., Lin C.Y., Liu J.C. Redox-responsive Resilin-like hydrogels for tissue engineering and drug delivery applications. Macromol. Biosci. 2019;19:1–9. doi: 10.1002/mabi.201900122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 396.Li L., Tong Z., Jia X., Kiick K.L. Resilin-like polypeptide hydrogels engineered for versatile biological function. Soft Matter. 2013;9:665–673. doi: 10.1039/C2SM26812D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397.Bracalello A., Santopietro V., Vassalli M., Marletta G., Del Gaudio R., Bochicchio B., Pepe A. Design and production of a chimeric Resilin-, elastin-, and collagen-like engineered polypeptide. Biomacromolecules. 2011;12:2957–2965. doi: 10.1021/bm2005388. [DOI] [PubMed] [Google Scholar]
- 398.Megeed Z., Cappello J., Ghandehari H. Genetically engineered silk-elastinlike protein polymers for controlled drug delivery. Adv. Drug Deliv. Rev. 2002;54:1075–1091. doi: 10.1016/S0169-409X(02)00063-7. [DOI] [PubMed] [Google Scholar]
- 399.Maeda H. The enhanced permeability and retention (EPR) effect in tumor vasculature: the key role of tumor-selective macromolecular drug targeting. Adv. Enzym. Regul. 2001;41:189–207. doi: 10.1016/S0065-2571(00)00013-3. [DOI] [PubMed] [Google Scholar]
- 400.Parker R.N., Wu W.A., McKay T.B., Xu Q., Kaplan D.L. Design of Silk-Elastin-like Protein Nanoparticle Systems with Mucoadhesive properties. J. Funct. Biomater. 2019;10:49. doi: 10.3390/jfb10040049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 401.Cappello J., Crissman J.W., Crissman M., Ferrari F.A., Textor G., Wallis O., Whitledge J.R., Zhou X., Burman D., Aukerman L., Stedronsky E.R. In-situ self-assembling protein polymer gel systems for administration, delivery, and release of drugs. J. Control. Release. 1998;53:105–117. doi: 10.1016/S0168-3659(97)00243-5. [DOI] [PubMed] [Google Scholar]
- 402.Dinerman A.A., Cappello J., Ghandehari H., Hoag S.W. Solute diffusion in genetically engineered silk–elastinlike protein polymer hydrogels. J. Control. Release. 2002;82:277–287. doi: 10.1016/S0168-3659(02)00134-7. [DOI] [PubMed] [Google Scholar]
- 403.Megeed Z., Cappello J., Ghandehari H. Controlled release of plasmid DNA from a genetically engineered silk-elastinlike hydrogel. Pharm. Res. 2002;19:954–959. doi: 10.1023/a:1016406120288. [DOI] [PubMed] [Google Scholar]
- 404.Megeed Z., Haider M., Li D., O'Malley B.W., Cappello J., Ghandehari H. In vitro and in vivo evaluation of recombinant silk-elastinlike hydrogels for cancer gene therapy. J. Control. Release. 2004;94:433–445. doi: 10.1016/j.jconrel.2003.10.027. [DOI] [PubMed] [Google Scholar]
- 405.Hatefi A., Cappello J., Ghandehari H. Adenoviral gene delivery to solid tumors by recombinant silk–Elastinlike protein polymers. Pharm. Res. 2007;24:773–779. doi: 10.1007/s11095-006-9200-5. [DOI] [PubMed] [Google Scholar]
- 406.Price R., Gustafson J., Greish K., Cappello J., McGill L., Ghandehari H. Comparison of silk-elastinlike protein polymer hydrogel and poloxamer in matrix-mediated gene delivery. Int. J. Pharm. 2012;427:97–104. doi: 10.1016/j.ijpharm.2011.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407.Gustafson J.A., Price R.A., Greish K., Cappello J., Ghandehari H. Silk-elastin-like hydrogel improves the safety of adenovirus-mediated gene-directed enzyme−Prodrug therapy. Mol. Pharm. 2010;7:1050–1056. doi: 10.1021/mp100161u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408.Jung S.-H., Choi J.-W., Yun C.-O., Kim S.H., Kwon I.C., Ghandehari H. Direct observation of interactions of silk-Elastinlike protein polymer with adenoviruses and Elastase. Mol. Pharm. 2015;12:1673–1679. doi: 10.1021/acs.molpharmaceut.5b00075. [DOI] [PubMed] [Google Scholar]
- 409.Jensen M.M., Jia W., Schults A.J., Isaacson K.J., Steinhauff D., Green B., Zachary B., Cappello J., Ghandehari H., Oottamasathien S. Temperature-responsive silk-elastinlike protein polymer enhancement of intravesical drug delivery of a therapeutic glycosaminoglycan for treatment of interstitial cystitis/painful bladder syndrome. Biomaterials. 2019;217:119293. doi: 10.1016/j.biomaterials.2019.119293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410.Jensen M.M., Jia W., Isaacson K.J., Schults A., Cappello J., Prestwich G.D., Oottamasathien S., Ghandehari H. Silk-elastinlike protein polymers enhance the efficacy of a therapeutic glycosaminoglycan for prophylactic treatment of radiation-induced proctitis. J. Control. Release. 2017;263:46–56. doi: 10.1016/j.jconrel.2017.02.025. [DOI] [PubMed] [Google Scholar]
- 411.Schellenberger V., Wang C., Geething N.C., Spink B.J., Campbell A., W. To, Scholle M.D., Yin Y., Yao Y., Bogin O., Cleland J.L., Silverman J., Stemmer W.P.C. A recombinant polypeptide extends the in vivo half-life of peptides and proteins in a tunable manner. Nat. Biotechnol. 2009;27:1186–1190. doi: 10.1038/nbt.1588. [DOI] [PubMed] [Google Scholar]
- 412.Ding S., Song M., Sim B.-C., Gu C., Podust V.N., Wang C.-W., McLaughlin B., Shah T.P., Lax R., Gast R., Sharan R., Vasek A., Hartman M.A., Deniston C., Srinivas P., Schellenberger V. Multivalent antiviral XTEN–peptide conjugates with long in vivo half-life and enhanced solubility. Bioconjug. Chem. 2014;25:1351–1359. doi: 10.1021/bc500215m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413.Podust V.N., Balan S., Sim B.-C., Coyle M.P., Ernst U., Peters R.T., Schellenberger V. Extension of in vivo half-life of biologically active molecules by XTEN protein polymers. J. Control. Release. 2016;240:52–66. doi: 10.1016/j.jconrel.2015.10.038. [DOI] [PubMed] [Google Scholar]
- 414.JL K.M. Cleland, Aronson R., Humphriss E., Shore C., Zhou R. Safety, pharmacokinetics, and pharmacodynamics of a single subcutaneous dose of VRS-859 in patients with type 2 diabetes. Am. Diabetes Assoc. 2012 [Google Scholar]
- 415.Geething N.C., W. To, Spink B.J., Scholle M.D., Wang C., Yin Y., Yao Y., Schellenberger V., Cleland J.L., Stemmer W.P.C., Silverman J. Gcg-XTEN: An improved glucagon capable of preventing hypoglycemia without increasing baseline blood glucose. PLoS One. 2010;5 doi: 10.1371/journal.pone.0010175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416.Alters S.E., McLaughlin B., Spink B., Lachinyan T., Wang C., Podust V., Schellenberger V., Stemmer W.P.C. GLP2-2G-XTEN: a pharmaceutical protein with improved serum half-life and efficacy in a rat Crohn's disease model. PLoS One. 2012;7 doi: 10.1371/journal.pone.0050630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417.Haeckel A., Appler F., Figge L., Kratz H., Lukas M., Michel R., Schnorr J., Zille M., Hamm B., Schellenberger E. XTEN-Annexin A5: XTEN allows complete expression of long-circulating protein-based imaging probes as recombinant alternative to PEGylation. J. Nucl. Med. 2014;55:508–514. doi: 10.2967/jnumed.113.128108. [DOI] [PubMed] [Google Scholar]
- 418.Moore W.V., Nguyen H.J., Kletter G.B., Miller B.S., Rogers D., Ng D., Moore J.A., Humphriss E., Cleland J.L., Bright G.M. A Randomized Safety and Efficacy Study of Somavaratan (VRS-317), a Long-Acting rhGH, in Pediatric Growth Hormone Deficiency. J. Clin. Endocrinol. Metab. 2016;101:1091–1097. doi: 10.1210/jc.2015-3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419.Leutjen T., Di Trapani K., Ng D., Humphriss E., Charlton W. 017–Safety and Efficacy of Somavaratan (VRS-317), a Long-Acting Growth Hormone, in Children with Growth Hormone Deficiency (GHD): 2.5-Year Results from the VISTA Trial. J. Pediatr. Nurs. Nurs. Care Child. Fam. 2017;34:107–108. doi: 10.1016/j.pedn.2017.02.026. [DOI] [Google Scholar]
- 420.van der Flier A., Liu Z.L., Liu Z., Mercury O., Ismail A., Seth-Chhabra E., Kulman J., Schellenberger V., Light D.R., Peters R. BLOOD Coagul. FIBRINOLYTIC FACTORS POSTER II. 2015. Prolonged Half-Life and Improved Recovery of Recombinant Factor IX-XTEN Fusion Proteins in Hemophilia B Mouse Model. [Google Scholar]
- 421.Powell J.S., Josephson N.C., Quon D., Ragni M.V., Cheng G., Li E., Jiang H., Li L., Dumont J.A., Goyal J., Zhang X., Sommer J., McCue J., Barbetti M., Luk A., Pierce G.F. Safety and prolonged activity of recombinant factor VIII Fc fusion protein in hemophilia A patients. Blood. 2012;119:3031–3037. doi: 10.1182/blood-2011-09-382846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 422.Nolan B., Mahlangu J., Perry D., Young G., Liesner R., Konkle B., Rangarajan S., Brown S., Hanabusa H., Pasi K.J., Pabinger I., Jackson S., Cristiano L.M., Li X., Pierce G.F., Allen G. Long-term safety and efficacy of recombinant factor VIII Fc fusion protein (rFVIIIFc) in subjects with haemophilia A. Haemophilia. 2016;22:72–80. doi: 10.1111/hae.12766. [DOI] [PubMed] [Google Scholar]
- 423.Chhabra E. Seth, Liu T., Kulman J., Patarroyo-White S., Yang B., Lu Q., Drager D., Moore N., Liu J., Holthaus A.M., Sommer J.M., Ismail A., Rabinovich D., Liu Z., van der Flier A., Goodman A., Furcht C., Tie M., Carlage T., Mauldin R., Dobrowsky T.M., Liu Z., Mercury O., Zhu L., Mei B., Schellenberger V., Jiang H., Pierce G.F., Salas J., Peters R. BIVV001, a new class of factor VIII replacement for hemophilia A that is independent of von Willebrand factor in primates and mice. Blood. 2020;135:1486–1496. doi: 10.1182/blood.2019001292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 424.Haeckel A., Appler F., Ariza de Schellenberger A., Schellenberger E. XTEN as biological alternative to PEGylation allows complete expression of a protease-Activatable Killin-based cytostatic. PLoS One. 2016;11 doi: 10.1371/journal.pone.0157193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 425.Cattaruzza F., Nazeer A., Lange Z., Hammond M., Koski C., Dao-Pick T., Henkensiefken A., Derynck M.K., Irving B.A., Schellenberger V. AACR Virtual Annu. Meet. II. 2020. HER2-XPAT and EGFR-XPAT: Pro-Drug T Cell Engagers (TCEs) Engineered to Address On-Target, Off-Tumor Toxicity With Potent Efficacy in vitro and in vivo and Large Safety Margins in NHP. [Google Scholar]
- 426.Schlapschy M., Binder U., Börger C., Theobald I., Wachinger K., Kisling S., Haller D., Skerra A. PASylation: a biological alternative to PEGylation for extending the plasma half-life of pharmaceutically active proteins. Protein Eng. Des. Sel. 2013;26:489–501. doi: 10.1093/protein/gzt023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427.Brandl F., Merten H., Zimmermann M., Béhé M., Zangemeister-Wittke U., Plückthun A. Influence of size and charge of unstructured polypeptides on pharmacokinetics and biodistribution of targeted fusion proteins. J. Control. Release. 2019;307:379–392. doi: 10.1016/j.jconrel.2019.06.030. [DOI] [PubMed] [Google Scholar]
- 428.Strohl W.R. Fusion proteins for half-life extension of biologics as a strategy to make biobetters. BioDrugs. 2015;29:215–239. doi: 10.1007/s40259-015-0133-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429.Harari D., Kuhn N., Abramovich R., Sasson K., Zozulya A.L., Smith P., Schlapschy M., Aharoni R., Köster M., Eilam R., Skerra A., Schreiber G. Enhanced in vivo efficacy of a type I interferon superagonist with extended plasma half-life in a mouse model of multiple sclerosis. J. Biol. Chem. 2014;289:29014–29029. doi: 10.1074/jbc.M114.602474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 430.Zvonova E.A., Ershov A.V., Ershova O.A., Sudomoina M.A., Degterev M.B., Poroshin G.N., Eremeev A.V., Karpov A.P., Vishnevsky A.Y., Goldenkova-Pavlova I.V., Petrov A.V., Ruchko S.V., Shuster A.M. PASylation technology improves recombinant interferon-β1b solubility, stability, and biological activity. Appl. Microbiol. Biotechnol. 2017;101:1975–1987. doi: 10.1007/s00253-016-7944-3. [DOI] [PubMed] [Google Scholar]
- 431.Morath V., Bolze F., Schlapschy M., Schneider S., Sedlmayer F., Seyfarth K., Klingenspor M., Skerra A. PASylation of murine Leptin leads to extended plasma half-life and enhanced in vivo efficacy. Mol. Pharm. 2015;12:1431–1442. doi: 10.1021/mp5007147. [DOI] [PubMed] [Google Scholar]
- 432.Bolze F., Bast A., Mocek S., Morath V., Yuan D., Rink N., Schlapschy M., Zimmermann A., Heikenwalder M., Skerra A., Klingenspor M. Treatment of diet-induced lipodystrophic C57BL/6J mice with long-acting PASylated leptin normalises insulin sensitivity and hepatic steatosis by promoting lipid utilisation. Diabetologia. 2016;59:2005–2012. doi: 10.1007/s00125-016-4004-6. [DOI] [PubMed] [Google Scholar]
- 433.Hedayati M.H., Norouzian D., Aminian M., Teimourian S., Ahangari Cohan R., Sardari S., Khorramizadeh M.R. Molecular design, expression and evaluation of PASylated human recombinant erythropoietin with enhanced functional properties. Protein J. 2017;36:36–48. doi: 10.1007/s10930-017-9699-9. [DOI] [PubMed] [Google Scholar]
- 434.Kuhn N., Schmidt C.Q., Schlapschy M., Skerra A. PASylated Coversin, a C5-specific complement inhibitor with extended pharmacokinetics, shows enhanced anti-hemolytic activity in vitro. Bioconjug. Chem. 2016;27:2359–2371. doi: 10.1021/acs.bioconjchem.6b00369. [DOI] [PubMed] [Google Scholar]
- 435.Binder U., Skerra A. PASylation®: a versatile technology to extend drug delivery. Curr. Opin. Colloid Interface Sci. 2017;31:10–17. doi: 10.1016/j.cocis.2017.06.004. [DOI] [Google Scholar]
- 436.Falvo E., Tremante E., Arcovito A., Papi M., Elad N., Boffi A., Morea V., Conti G., Toffoli G., Fracasso G., Giacomini P., Ceci P. Improved doxorubicin encapsulation and pharmacokinetics of ferritin–fusion protein Nanocarriers bearing Proline, serine, and alanine elements. Biomacromolecules. 2016;17:514–522. doi: 10.1021/acs.biomac.5b01446. [DOI] [PubMed] [Google Scholar]
- 437.Fracasso G., Falvo E., Colotti G., Fazi F., Ingegnere T., Amalfitano A., Doglietto G.B., Alfieri S., Boffi A., Morea V., Conti G., Tremante E., Giacomini P., Arcovito A., Ceci P. Selective delivery of doxorubicin by novel stimuli-sensitive nano-ferritins overcomes tumor refractoriness. J. Control. Release. 2016;239:10–18. doi: 10.1016/j.jconrel.2016.08.010. [DOI] [PubMed] [Google Scholar]
- 438.Morys S., Krhac Levacic A., Urnauer S., Kempter S., Kern S., Rädler J.O., Spitzweg C., Lächelt U., Wagner E. Influence of Defined Hydrophilic Blocks within Oligoaminoamide Copolymers: Compaction versus Shielding of pDNA Nanoparticles. Polym. 2017;9 doi: 10.3390/polym9040142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 439.Bietz J.A., Rothfus J.A. Comparison of peptides from wheat gliadin and glutenin. Cereal Chem. 1970;47 [Google Scholar]
- 440.Arangoa M.A., Ponchel G., Orecchioni A.M., Renedo M.J., Duchêne D., Irache J.M. Bioadhesive potential of gliadin nanoparticulate systems. Eur. J. Pharm. Sci. 2000;11:333–341. doi: 10.1016/S0928-0987(00)00121-4. [DOI] [PubMed] [Google Scholar]
- 441.Duclairoir C., Orecchioni A.M., Depraetere P., Osterstock F., Nakache E. Evaluation of gliadins nanoparticles as drug delivery systems: a study of three different drugs. Int. J. Pharm. 2003;253:133–144. doi: 10.1016/S0378-5173(02)00701-9. [DOI] [PubMed] [Google Scholar]
- 442.Arangoa M.A., Campanero M.A., Renedo M.J., Ponchel G., Irache J.M. Gliadin nanoparticles as carriers for the oral administration of lipophilic drugs. Relationships between bioadhesion and pharmacokinetics. Pharm. Res. 2001;18:1521–1527. doi: 10.1023/A:1013018111829. [DOI] [PubMed] [Google Scholar]
- 443.Gulfam M., Kim J.E., Lee J.M., Ku B., Chung B.H., Chung B.G. Anticancer drug-loaded gliadin nanoparticles induce apoptosis in breast cancer cells. Langmuir. 2012;28:8216–8223. doi: 10.1021/la300691n. [DOI] [PubMed] [Google Scholar]
- 444.Umamaheshwari R.B., Ramteke S., Jain N.K. Anti-Helicobacter pylori effect of mucoadhesive nanoparticles bearing amoxicilin in experimental gerbils model. AAPS PharmSciTech. 2004;5 doi: 10.1208/pt050232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 445.Teng Z., Liu C., Yang X., Li L., Tang C., Jiang Y. Fractionation of soybean globulins using Ca2+ and Mg 2+: a comparative analysis, JAOCS. J. Am. Oil Chem. Soc. 2009;86:409–417. doi: 10.1007/s11746-009-1367-6. [DOI] [Google Scholar]
- 446.Li X., Li Y., Hua Y., Qiu A., Yang C., Cui S. Effect of concentration, ionic strength and freeze-drying on the heat-induced aggregation of soy proteins. Food Chem. 2007;104:1410–1417. doi: 10.1016/j.foodchem.2007.02.003. [DOI] [Google Scholar]
- 447.Hong C.K., Wool R.F. Development of a bio-based composite material from soybean oil and keratin fibers. J. Appl. Polym. Sci. 2005;95:1524–1538. doi: 10.1002/app.21044. [DOI] [Google Scholar]
- 448.Teng Z., Luo Y., Wang Q. Nanoparticles synthesized from soy protein: preparation, characterization, and application for nutraceutical encapsulation. J. Agric. Food Chem. 2012;60:2712–2720. doi: 10.1021/jf205238x. [DOI] [PubMed] [Google Scholar]
- 449.Ding X., Yao P. Soy protein/soy polysaccharide complex nanogels: folic acid loading, protection, and controlled delivery. Langmuir. 2013;29:8636–8644. doi: 10.1021/la401664y. [DOI] [PubMed] [Google Scholar]
- 450.Chen L., Hébrard G., Beyssac E., Denis S., Subirade M. In vitro study of the release properties of soy-zein protein microspheres with a dynamic artificial digestive system. J. Agric. Food Chem. 2010;58:9861–9867. doi: 10.1021/jf101918w. [DOI] [PubMed] [Google Scholar]
- 451.Chen L., Subirade M. Elaboration and characterization of soy/zein protein microspheres for controlled nutraceutical delivery. Biomacromolecules. 2009;10:3327–3334. doi: 10.1021/bm900989y. [DOI] [PubMed] [Google Scholar]
- 452.Hadzieva J., Mladenovska K., Simonoska Crcarevska M., Glavaš Dodov M., Dimchevska S., Geškovski N., Grozdanov A., Popovski E., Petruševski G., Chachorovska M., Petreska Ivanovska T., Petruševska-Tozi L., Ugarkovic S., Goracinova K. Lactobacillus casei loaded soy protein-alginate microparticles prepared by spray-drying. Food Technol. Biotechnol. 2017;55:173–186. doi: 10.17113/ftb.55.02.17.4991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 453.Peles Z., Binderman I., Berdicevsky I., Zilberman M. Soy protein films for wound-healing applications: antibiotic release, bacterial inhibition and cellular response. J. Tissue Eng. Regen. Med. 2013;7:401–412. doi: 10.1002/term. [DOI] [PubMed] [Google Scholar]
- 454.Song F., Zhang L.M. Gelation modification of soy protein isolate by a naturally occurring cross-linking agent and its potential biomedical application. Ind. Eng. Chem. Res. 2009;48:7077–7083. doi: 10.1021/ie801372f. [DOI] [Google Scholar]
- 455.Chen L., Remondetto G., Rouabhia M., Subirade M. Kinetics of the breakdown of cross-linked soy protein films for drug delivery. Biomaterials. 2008;29:3750–3756. doi: 10.1016/j.biomaterials.2008.05.025. [DOI] [PubMed] [Google Scholar]
- 456.Vaz C.M., van Doeveren P.F.N.M., Reis R.L., Cunha A.M. Soy matrix drug delivery systems obtained by melt-processing techniques. Biomacromolecules. 2003;4:1520–1529. doi: 10.1021/bm034050i. [DOI] [PubMed] [Google Scholar]
- 457.Xu W., Yang Y. Drug sorption onto and release from soy protein fibers. J. Mater. Sci. Mater. Med. 2009;20:2477–2486. doi: 10.1007/s10856-009-3821-2. [DOI] [PubMed] [Google Scholar]
- 458.Liu X., Sun Q., Wang H., Zhang L., Wang J.Y. Microspheres of corn protein, zein, for an ivermectin drug delivery system. Biomaterials. 2005;26:109–115. doi: 10.1016/j.biomaterials.2004.02.013. [DOI] [PubMed] [Google Scholar]
- 459.Paliwal R., Palakurthi S. Zein in controlled drug delivery and tissue engineering. J. Control. Release. 2014;189:108–122. doi: 10.1016/j.jconrel.2014.06.036. [DOI] [PubMed] [Google Scholar]
- 460.P. Usa, P. Argos, K. Pedersenfl, M. David Marksl, B.A. Larkinsflll, A Structural Model for Maize Zein Proteins*, J. Biol. Chem. 257 (1982) 9984–9990. [PubMed]
- 461.Matsushima N., Danno G.I., Takezawa H., Izumi Y. Three-dimensional structure of maize α-zein proteins studied by small-angle X-ray scattering. Biochim. Biophys. Acta - Protein Struct. Mol. Enzymol. 1997;1339:14–22. doi: 10.1016/S0167-4838(96)00212-9. [DOI] [PubMed] [Google Scholar]
- 462.Wongsasulak S., Puttipaiboon N., Yoovidhya T. Fabrication, gastromucoadhesivity, swelling, and degradation of Zein-chitosan composite ultrafine fibers. J. Food Sci. 2013;78:926–935. doi: 10.1111/1750-3841.12126. [DOI] [PubMed] [Google Scholar]
- 463.Liu L.S., Fishman M.L., Hicks K.B., Kende M., Ruthel G. Pectin/zein beads for potential colon-specific drug delivery: synthesis and in vitro evaluation. Drug Deliv. 2006;13:417–423. doi: 10.1080/10717540500394935. [DOI] [PubMed] [Google Scholar]
- 464.Gao Z., Ding P., Zhang L., Shi J., Yuan S., Wei J., Chen D. Study of a Pingyangmycin delivery system: Zein/Zein-SAIB in situ gels. Int. J. Pharm. 2007;328:57–64. doi: 10.1016/j.ijpharm.2006.07.048. [DOI] [PubMed] [Google Scholar]
- 465.Cao X., Geng J., Su W., Zhang L., Xu Q., Zhang L., Xie Y., Wu S., Sun Y., Gao Z. Doxorubicin-loaded zein in situ gel for interstitial chemotherapy. Chem. Pharm. Bull. 2012;60:1227–1233. doi: 10.1248/cpb.c12-00270. [DOI] [PubMed] [Google Scholar]
- 466.Matsuda Y., Suzuki T., Sato E., Sato M., Koizumi S., Unno K., Kato T., Nakai K. Novel preparation of Zein microspheres conjugated with PS-K available for Cancer immunotherapy. Chem. Pharm. Bull. 2002;2091 doi: 10.1248/cpb.37.757. [DOI] [PubMed] [Google Scholar]
- 467.Suzuki T., Sato E., Matsuda Y., Tada H., Unno K., Kato T. Preparation of zein microspheres conjugated with antitumor drugs available for selective cancer chemotherapy and development of a simple colorimetric determination of drugs in microspheres. Chem. Pharm. Bull. 2002;57:364–370. doi: 10.1248/cpb.37.1051. [DOI] [PubMed] [Google Scholar]
- 468.Fu J.X., Wang H.J., Zhou Y.Q., Wang J.Y. Antibacterial activity of ciprofloxacin-loaded zein microsphere films. Mater. Sci. Eng. C. 2009;29:1161–1166. doi: 10.1016/j.msec.2008.09.031. [DOI] [Google Scholar]
- 469.Lau E.T.L., Johnson S.K., Mikkelsen D., Halley P.J., Steadman K.J. Preparation and in vitro release of zein microparticles loaded with prednisolone for oral delivery. J. Microencapsul. 2012;29:706–712. doi: 10.3109/02652048.2012.686527. [DOI] [PubMed] [Google Scholar]
- 470.Muthuselvi L., Dhathathreyan A. Simple coacervates of zein to encapsulate Gitoxin. Colloids Surfaces B Biointerfaces. 2006;51:39–43. doi: 10.1016/j.colsurfb.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 471.Lai L.F., Guo H.X. Preparation of new 5-fluorouracil-loaded zein nanoparticles for liver targeting. Int. J. Pharm. 2011;404:317–323. doi: 10.1016/j.ijpharm.2010.11.025. [DOI] [PubMed] [Google Scholar]
- 472.Dong F., Dong X., Zhou L., Xiao H., Ho P.Y., Wong M.S., Wang Y. Doxorubicin-loaded biodegradable self-assembly zein nanoparticle and its anti-cancer effect: preparation, in vitro evaluation, and cellular uptake. Colloids Surfaces B Biointerfaces. 2016;140:324–331. doi: 10.1016/j.colsurfb.2015.12.048. [DOI] [PubMed] [Google Scholar]
- 473.Alhakamy N.A., Ahmed O.A.A., Aldawsari H.M., Alfaifi M.Y., Eid B.G., Abdel-Naim A.B., Fahmy U.A. Encapsulation of lovastatin in zein nanoparticles exhibits enhanced apoptotic activity in hepg2 cells. Int. J. Mol. Sci. 2019;20:2–15. doi: 10.3390/ijms20225788. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 474.Mehta S.K., Kaur G., Verma A. Fabrication of plant protein microspheres for encapsulation, stabilization and in vitro release of multiple anti-tuberculosis drugs. Colloids Surfaces A Physicochem. Eng. Asp. 2011;375:219–230. doi: 10.1016/j.colsurfa.2010.12.014. [DOI] [Google Scholar]
- 475.Müller V., Piai J.F., Fajardo A.R., Fávaro S.L., Rubira A.F., Muniz E.C. Preparation and characterization of zein and zein-chitosan microspheres with great prospective of application in controlled drug release. J. Nanomater. 2011;2011 doi: 10.1155/2011/928728. [DOI] [Google Scholar]
- 476.Holt C., Carver J.A., Ecroyd H., Thorn D.C. Invited review: caseins and the casein micelle: their biological functions, structures, and behavior in foods1. J. Dairy Sci. 2013;96:6127–6146. doi: 10.3168/jds.2013-6831. [DOI] [PubMed] [Google Scholar]
- 477.Głąb T.K., Boratyński J. Potential of casein as a carrier for biologically active agents. Top. Curr. Chem. 2017;375:71. doi: 10.1007/s41061-017-0158-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 478.Pereira P.C. Milk nutritional composition and its role in human health. Nutrition. 2014;30:619–627. doi: 10.1016/j.nut.2013.10.011. [DOI] [PubMed] [Google Scholar]
- 479.M.H. ABD EL-SALAM, S. EL-SHIBINY Formation and potential uses of milk proteins as nano delivery vehicles for nutraceuticals: a review. Int. J. Dairy Technol. 2012;65:13–21. doi: 10.1111/j.1471-0307.2011.00737.x. [DOI] [Google Scholar]
- 480.Graveland-Bikker J.F., de Kruif C.G. Unique milk protein based nanotubes: food and nanotechnology meet. Trends Food Sci. Technol. 2006;17:196–203. doi: 10.1016/j.tifs.2005.12.009. [DOI] [Google Scholar]
- 481.Giroux H.J., Houde J., Britten M. Preparation of nanoparticles from denatured whey protein by pH-cycling treatment. Food Hydrocoll. 2010;24:341–346. doi: 10.1016/j.foodhyd.2009.10.013. [DOI] [Google Scholar]
- 482.Sauer A., Moraru C.I. Heat stability of micellar casein concentrates as affected by temperature and pH. J. Dairy Sci. 2012;95:6339–6350. doi: 10.3168/jds.2012-5706. [DOI] [PubMed] [Google Scholar]
- 483.Semo E., Kesselman E., Danino D., Livney Y.D. Casein micelle as a natural nano-capsular vehicle for nutraceuticals. Food Hydrocoll. 2007;21:936–942. doi: 10.1016/j.foodhyd.2006.09.006. [DOI] [Google Scholar]
- 484.Sahu A., Kasoju N., Bora U. Fluorescence study of the curcumin−casein micelle complexation and its application as a drug nanocarrier to cancer cells. Biomacromolecules. 2008;9:2905–2912. doi: 10.1021/bm800683f. [DOI] [PubMed] [Google Scholar]
- 485.Shapira A., Assaraf Y.G., Livney Y.D. Beta-casein nanovehicles for oral delivery of chemotherapeutic drugs. Nanomed. Nanotechnol. Biol. Med. 2010;6:119–126. doi: 10.1016/j.nano.2009.06.006. [DOI] [PubMed] [Google Scholar]
- 486.Shapira A., Markman G., Assaraf Y.G., Livney Y.D. β-casein–based nanovehicles for oral delivery of chemotherapeutic drugs: drug-protein interactions and mitoxantrone loading capacity. Nanomed. Nanotechnol. Biol. Med. 2010;6:547–555. doi: 10.1016/j.nano.2010.01.003. [DOI] [PubMed] [Google Scholar]
- 487.Gandhi S., Roy I. Doxorubicin-loaded casein nanoparticles for drug delivery: preparation, characterization and in vitro evaluation. Int. J. Biol. Macromol. 2019;121:6–12. doi: 10.1016/j.ijbiomac.2018.10.005. [DOI] [PubMed] [Google Scholar]
- 488.Penalva R., Esparza I., Agüeros M., Gonzalez-Navarro C.J., Gonzalez-Ferrero C., Irache J.M. Casein nanoparticles as carriers for the oral delivery of folic acid. Food Hydrocoll. 2015;44:399–406. doi: 10.1016/j.foodhyd.2014.10.004. [DOI] [Google Scholar]
- 489.Elzoghby A.O., Helmy M.W., Samy W.M., Elgindy N.A. Novel ionically crosslinked casein nanoparticles for flutamide delivery: formulation, characterization, and in vivo pharmacokinetics. Int. J. Nanomedicine. 2013;8:1721–1732. doi: 10.2147/IJN.S40674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 490.Motoki M., Aso H., Seguro K., Nio N. αs1-Casein Film Prepared Using Transglutaminase. Agric. Biol. Chem. 1987;51:993–996. doi: 10.1271/bbb1961.51.993. [DOI] [Google Scholar]
- 491.Abu Diak O., Bani-Jaber A., Amro B., Jones D., Andrews G.P. The manufacture and characterization of casein films as novel tablet coatings. Food Bioprod. Process. 2007;85:284–290. doi: 10.1205/fbp07030. [DOI] [Google Scholar]
- 492.Song F., Zhang L.-M., Yang C., Yan L. Genipin-crosslinked casein hydrogels for controlled drug delivery. Int. J. Pharm. 2009;373:41–47. doi: 10.1016/j.ijpharm.2009.02.005. [DOI] [PubMed] [Google Scholar]
- 493.Song F., Zhang L.-M., Shi J.-F., Li N.-N. Novel casein hydrogels: formation, structure and controlled drug release. Colloids Surfaces B Biointerfaces. 2010;79:142–148. doi: 10.1016/j.colsurfb.2010.03.045. [DOI] [PubMed] [Google Scholar]
- 494.Millar F.C., Corrigan O.I. Influence of sodium Caseinate on the dissolution rate of hydrochlorothiazide and Chlorothiazide. Drug Dev. Ind. Pharm. 1991;17:1593–1607. doi: 10.3109/03639049109057310. [DOI] [Google Scholar]
- 495.Millar F.C., Corrigan O.I. Dissolution mechanism of ibuprofen-casein compacts. Int. J. Pharm. 1993;92:97–104. doi: 10.1016/0378-5173(93)90268-K. [DOI] [Google Scholar]
- 496.Gubbins R.H., O'Driscoll C.M., Corrigan O.I. The effects of casein on diclofenac release from hydroxypropylmethylcellulose (HPMC) compacts. Int. J. Pharm. 2003;260:69–76. doi: 10.1016/S0378-5173(03)00235-7. [DOI] [PubMed] [Google Scholar]
- 497.Lambert L.A., Perri H., Halbrooks P.J., Mason A.B. Evolution of the transferrin family: conservation of residues associated with iron and anion binding. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2005;142:129–141. doi: 10.1016/j.cbpb.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 498.Shen Y., Li X., Dong D., Zhang B., Xue Y., Shang P. Transferrin receptor 1 in cancer: a new sight for cancer therapy. Am. J. Cancer Res. 2018;8:916–931. [PMC free article] [PubMed] [Google Scholar]
- 499.Paterson J., Webster C.I. Exploiting transferrin receptor for delivering drugs across the blood-brain barrier. Drug Discov. Today Technol. 2016;20:49–52. doi: 10.1016/j.ddtec.2016.07.009. [DOI] [PubMed] [Google Scholar]
- 500.Szwed M., Kania K.D., Jozwiak Z. Relationship between therapeutic efficacy of doxorubicin-transferrin conjugate and expression of P-glycoprotein in chronic erythromyeloblastoid leukemia cells sensitive and resistant to doxorubicin. Cell. Oncol. (Dordr). 2014;37:421–428. doi: 10.1007/s13402-014-0205-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 501.Szwed M., Laroche-Clary A., Robert J., Jozwiak Z. Efficacy of doxorubicin-transferrin conjugate in apoptosis induction in human leukemia cells through reactive oxygen species generation. Cell. Oncol. (Dordr). 2016;39:107–118. doi: 10.1007/s13402-015-0256-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 502.Saxena M., Delgado Y., Sharma R.K., Sharma S., Guzmán S.L.P.D.L., Tinoco A.D., Griebenow K. Inducing cell death in vitro in cancer cells by targeted delivery of cytochrome c via a transferrin conjugate. PLoS One. 2018;13 doi: 10.1371/journal.pone.0195542. e0195542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 503.Wang K., Zhang Y., Wang J., Yuan A., Sun M., Wu J., Hu Y. Self-assembled IR780-loaded transferrin nanoparticles as an imaging, targeting and PDT/PTT agent for cancer therapy. Sci. Rep. 2016;6:27421. doi: 10.1038/srep27421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 504.Kim B.-J., Zhou J., Martin B., Carlson O.D., Maudsley S., Greig N.H., Mattson M.P., Ladenheim E.E., Wustner J., Turner A., Sadeghi H., Egan J.M. Transferrin fusion technology: a novel approach to prolonging biological half-life of insulinotropic peptides. J. Pharmacol. Exp. Ther. 2010;334:682–692. doi: 10.1124/jpet.110.166470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 505.Schmidt S.R. John Wiley & Sons, Incorporated, Somerset; UNITED STATES: 2013. Fusion Protein Technologies for Biopharmaceuticals : Applications and Challenges. [Google Scholar]
- 506.Chen X., Lee H.-F., Zaro J.L., Shen W.-C. Effects of receptor binding on plasma half-life of bifunctional transferrin fusion proteins. Mol. Pharm. 2011;8:457–465. doi: 10.1021/mp1003064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 507.Bialasek M., Kubiak M., Gorczak M., Braniewska A., Kucharzewska-Siembieda P., Krol M., Taciak B. Exploiting iron-binding proteins for drug delivery. J. Physiol. Pharmacol. 2019;70 doi: 10.26402/jpp.2019.5.03. [DOI] [PubMed] [Google Scholar]
- 508.Mann S., Williams J.M., Treffry A., Harrison P.M. Reconstituted and native iron-cores of bacterioferritin and ferritin. J. Mol. Biol. 1987;198:405–416. doi: 10.1016/0022-2836(87)90290-7. [DOI] [PubMed] [Google Scholar]
- 509.de Turris V., Cardoso Trabuco M., Peruzzi G., Boffi A., Testi C., Vallone B., Celeste Montemiglio L., Des Georges A., Calisti L., Benni I., Bonamore A., Baiocco P. Humanized archaeal ferritin as a tool for cell targeted delivery. Nanoscale. 2017;9:647–655. doi: 10.1039/c6nr07129e. [DOI] [PubMed] [Google Scholar]
- 510.Pontillo N., Pane F., Messori L., Amoresano A., Merlino A. Cisplatin encapsulation within a ferritin nanocage: a high-resolution crystallographic study. Chem. Commun. (Camb). 2016;52:4136–4139. doi: 10.1039/c5cc10365g. [DOI] [PubMed] [Google Scholar]
- 511.Wang Q., Zhang C., Liu L., Li Z., Guo F., Li X., Luo J., Zhao D., Liu Y., Su Z. High hydrostatic pressure encapsulation of doxorubicin in ferritin nanocages with enhanced efficiency. J. Biotechnol. 2017;254:34–42. doi: 10.1016/j.jbiotec.2017.05.025. [DOI] [PubMed] [Google Scholar]
- 512.Fan K., Jia X., Zhou M., Wang K., Conde J., He J., Tian J., Yan X. Ferritin nanocarrier traverses the blood brain barrier and kills Glioma. ACS Nano. 2018;12:4105–4115. doi: 10.1021/acsnano.7b06969. [DOI] [PubMed] [Google Scholar]
- 513.Liang M., Fan K., Zhou M., Duan D., Zheng J., Yang D., Feng J., Yan X. H-ferritin–nanocaged doxorubicin nanoparticles specifically target and kill tumors with a single-dose injection. Proc. Natl. Acad. Sci. 2014;111 doi: 10.1073/pnas.1407808111. 14900 LP – 14905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 514.Mazzucchelli S., Truffi M., Baccarini F., Beretta M., Sorrentino L., Bellini M., Rizzuto M.A., Ottria R., Ravelli A., Ciuffreda P., Prosperi D., Corsi F. H-Ferritin-nanocaged olaparib: a promising choice for both BRCA-mutated and sporadic triple negative breast cancer. Sci. Rep. 2017;7:7505. doi: 10.1038/s41598-017-07617-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 515.Jiang B., Yan L., Zhang J., Zhou M., Shi G., Tian X., Fan K., Hao C., Yan X. Biomineralization synthesis of the cobalt Nanozyme in SP94-ferritin Nanocages for prognostic diagnosis of hepatocellular carcinoma. ACS Appl. Mater. Interfaces. 2019;11:9747–9755. doi: 10.1021/acsami.8b20942. [DOI] [PubMed] [Google Scholar]
- 516.Li L., Muñoz-Culla M., Carmona U., Lopez M.P., Yang F., Trigueros C., Otaegui D., Zhang L., Knez M. Ferritin-mediated siRNA delivery and gene silencing in human tumor and primary cells. Biomaterials. 2016;98:143–151. doi: 10.1016/j.biomaterials.2016.05.006. [DOI] [PubMed] [Google Scholar]