

Abstract
Exposure of mononuclear phagocytes to β-glucan, a naturally occurring polysaccharide, contributes to the induction of innate immune memory, which is associated with long-term epigenetic, metabolic, and functional reprogramming. Although previous studies have shown that innate immune memory induced by β-glucan confers protection against secondary infections, its impact on autoinflammatory diseases, associated with inflammasome activation and IL-1β secretion, remains poorly understood. In particular, whether β-glucan–induced long-term reprogramming affects inflammasome activation in human macrophages in the context of these diseases has not been explored. We found that NLRP3 inflammasome–mediated caspase-1 activation and subsequent IL-1β production were reduced in β-glucan–reprogrammed macrophages. β-Glucan acted upstream of the NLRP3 inflammasome by preventing potassium (K+) efflux, mitochondrial ROS (mtROS) generation, and, ultimately, apoptosis-associated speck-like protein containing a CARD (ASC) oligomerization and speck formation. Importantly, β-glucan–induced memory in macrophages resulted in a remarkable attenuation of IL-1β secretion and caspase-1 activation in patients with an NLRP3-associated autoinflammatory disease, cryopyrin-associated periodic syndromes (CAPS). Our findings demonstrate that β-glucan–induced innate immune memory represses IL-1β–mediated inflammation and support its potential clinical use in NLRP3-driven diseases.
Keywords: Inflammation
Keywords: Innate immunity, Macrophages, Memory

Introduction
Over the past decades, multiple studies have described memory phenotypes in innate immune cells, e.g., mononuclear phagocytes. The concept of innate immune memory refers to a change in the reactivity of monocytes/macrophages previously exposed to various microbe-associated molecular patterns (MAMPs) (1–4). β-Glucan, an abundant polysaccharide in the cell wall of Candida albicans (C. albicans), has been reported to be a strong inducer of innate immune memory in mononuclear phagocytes, associated with increased immune responsiveness to secondary challenges. The enhanced responsiveness observed in vitro in cultured primary cells has been linked to the induction of metabolic reprogramming and epigenetic alterations (5–8). In addition, β-glucan can confer protection against unrelated bacterial and Leishmania infections in mice (6, 9). All of these findings have raised interest in β-glucan for the development of innovative combinational immune-modulating therapies and control of macrophage function (10, 11).
Macrophages are remarkably plastic cells that undergo diverse forms of activation in response to cytokines and microbial signals. Depending on the microenvironmental stimuli, macrophages may undergo activation or “polarization” along a wide spectrum of phenotypes (12), among which classical M1 activation (induced by granulocyte macrophage–CSF [GM-CSF], IFN-γ, and TLR ligands) and alternative M2 activation (induced by macrophage-CSF [M-CSF], IL-4, IL-13, and glucocorticoids) are the best studied (13). M1-like macrophages are classically characterized by the expression of proinflammatory cytokines, high ROS production, and microbicidal and tumoricidal activity (14). In contrast, M2-like macrophages are commonly described as an antiinflammatory cell type, promoting tissue remodeling and repair and exerting potent protumoral activity (15). Imbalance in M1/M2 activation is therefore often pathological and associated with various diseases, such as cancer, inflammatory and autoimmune disorders, and chronic infections (16–20). Therefore, understanding strategies to reprogram monocyte differentiation and macrophage polarization is crucial to further design new approaches in the context of inflammatory disorders and infectious diseases (21–25).
Although β-glucan–induced innate memory is associated with a nonspecific protective effect against infections, the role of this functional reprogramming in autoinflammatory disorders is unknown (26, 27). Because of its inherent function to increase immune function and responses, it has been suggested that innate immune memory is not always beneficial (28). As such, reprogramming of mononuclear phagocytes could lead to a non-negligible risk of inducing or enhancing systemic inflammatory pathologies (for review, see ref. 26). Interestingly, the first described monogenic autoinflammatory diseases, now classified as inflammasomopathies, are linked to mutations in inflammasome components (29, 30). Inflammasomes are molecular platforms that form to mediate the activation of caspase-1, a cysteine protease responsible for the processing and release of the proinflammatory cytokines IL-1β and IL-18 (31, 32). The NLRP3 inflammasome, the first to be described and most-studied member of inflammasomes, is activated in response to the widest array of stimuli, including microbial and nonmicrobial activators (33). Gain-of-function mutations in the NLRP3 gene, which lead to increased secretion of IL-1β and dysregulated inflammation, have been identified in patients with cryopyrin-associated periodic syndromes (CAPS). CAPS, also referred to as NLRP3-associated autoinflammatory diseases (NLRP3-AIDs) (34), include a moderate form, Muckle-Wells syndrome (MWS), and a severe form, chronic infantile neurologic cutaneous articular (CINCA) syndrome (35–38).
In this study, we deciphered the effect of β-glucan–induced innate immune memory on NLRP3 inflammasome regulation in human monocyte–derived macrophages. We report that preincubation of human monocytes with β-glucan decreased macrophage secretion of mature IL-1β in response to NLRP3-activating stimuli. This decrease was observed regardless of the M1 or M2 macrophage environmental cues provided. We also report that the inhibition of pro–IL-1β processing occurred upstream of the apoptosis-associated speck-like protein containing a CARD (ASC) oligomerization and inflammasome/caspase-1 activation. Remarkably, β-glucan–induced memory also affected macrophages from patients with CAPS, reducing the secretion of mature IL-1β through the blockade of ASC oligomerization and caspase-1 activation. Thus, the natural product β-glucan imprints anti-NLRP3 inflammatory features in macrophages and represents a potential approach for the treatment of IL-1β–mediated diseases.
Results
β-Glucan attenuates IL-1β release upon NLRP3 inflammasome activation.
Macrophages derived from human primary monocytes cultured in human serum only are reprogrammed upon preincubation of monocytes for 24 hours with highly purified β-glucan from C. albicans (7). β-Glucan–reprogrammed macrophages are characterized by an increased expression of inflammatory cytokines and decreased ROS production upon restimulation 6 days later with heterologous stimuli, as well as by changes in morphology and metabolism (6, 7, 39). This innate immune memory effect is associated with a nonspecific protective effect against infections. We sought to determine whether preincubation of human monocytes with C. albicans–β-glucan particles also results in different degrees of inflammasome activation and IL-1β production in the derived reprogrammed macrophages. The innate immune memory model with human monocytes that we used is depicted in Figure 1A. Purified monocytes were maintained in GM-CSF– or M-CSF–supplemented culture medium to obtain M1-like and M2-like macrophages, respectively (Supplemental Figure 1; supplemental material available online with this article; https://doi.org/10.1172/JCI134778DS1). Supplementation with these exogenous growth factors allowed for a significant increase in the number of live cells compared with classical culture conditions with human or fetal bovine serum (Supplemental Figure 2). During the first 24 hours, isolated monocytes were preincubated with culture medium containing either GM-CSF or M-CSF, in the presence or absence of β-glucan. Importantly, we observed no difference in cell numbers or viability between macrophages derived from β-glucan–treated or untreated monocytes (Supplemental Figure 2). After 24 hours, the cells were washed, and complete medium (without β-glucan) was added for 5 days. On day 6, the GM-CSF or M-CSF monocyte–derived macrophages were primed with LPS, and the NLRP3 inflammasome was activated with nigericin or ATP to induce IL-1β release by macrophages (40). As expected, we did not detect secreted IL-1β cytokine in the supernatant of macrophages treated with LPS alone, in the absence of nigericin supplementation (Figure 1B), whereas IL-6 and TNF-α, which do not require proteolytic processing, were released (Supplemental Figure 3, A and B). By contrast, costimulation of LPS-primed cells with nigericin induced the release of IL-1β from both GM-CSF– and M-CSF–derived macrophages. This release was significantly higher in the M-CSF–derived macrophage subtype, similar to findings reported by Budai et al. (41). Most important, preincubation of monocytes with β-glucan surprisingly did not increase the inflammatory response but rather dramatically reduced the amount of IL-1β found in the supernatant of both macrophage subsets (Figure 1B), suggesting that β-glucan–induced monocyte/macrophage reprogramming affects the ability of macrophages to secrete the proinflammatory cytokine IL-1β. We observed a comparable downregulatory effect of β-glucan–induced monocyte/macrophage reprogramming on the secretion of IL-18 (Supplemental Figure 4), another IL-1 family cytokine that is activated by the inflammasome via proteolytic processing (42). Of note, unlike IL-1β, IL-18 is constitutively expressed in mononuclear phagocytes under steady state (42, 43), highlighting the importance of β-glucan in proteolytic processing rather than in cytokine expression in the context of inflammasome. β-Glucan–induced monocyte/macrophage reprogramming also reduced IL-1β and IL-18 secretion from LPS-primed macrophages upon ATP stimulation, similar to what was observed upon nigericin treatment (Figure 1C and Supplemental Figure 4). ATP and nigericin are 2 canonical NLRP3 activators that facilitate primary potassium (K+) efflux via mechanistically distinct pathways. Nigericin is an exogenous bacterial ionophore, whereas ATP activates the endogenous plasma membrane P2X7 purinergic receptor to induce K+ efflux (44). This implies that the effect of β-glucan on IL-1β or IL-18 secretion by macrophages was independent of the inflammasome-activating agent, because β-glucan inhibited both nigericin- and ATP-induced IL-1β release. In addition, as expected from previous work on β-glucan–induced immune memory (7), we found that M-CSF–derived macrophages preincubated with β-glucan had increased secretion of the proinflammatory cytokines IL-6 and TNF-α compared with the nontreated control (Supplemental Figure 3). In contrast, this is the first study to our knowledge showing that GM-CSF–derived macrophages, which are initially more proinflammatory than M-CSF–derived macrophages in terms of IL-6 and TNF-α release, were also imprinted by β-glucan, resulting in decreased expression of IL-6 and TNF-α, two cytokines that do not require proteolytic processing. Because preincubation of monocytes with β-glucan dramatically reduced the amount of IL-1β found in the supernatant of both M-CSF– and GM-CSF–derived macrophage subsets, the effect of β-glucan on IL-1β secretion by macrophages was mediated by a mechanism independent of what was previously described in innate immune–mediated memory.
Figure 1. Effect of β-glucan on IL-1β release upon NLRP3 inflammasome activation.

(A) Schematic overview of innate immune memory methodology. Monocytes were preincubated with β-glucan or left untreated in a medium containing either GM-CSF or M-CSF. After 24 hours, the stimulus was washed away, and the cells were differentiated for an additional 5 days, after which the macrophages were restimulated. d6, day 6. (B) Macrophages were primed for 3 hours with LPS and then stimulated for 1 hour with nigericin or vehicle as a control. Culture supernatants were collected, and the concentration of secreted IL-1β was determined by ELISA. (C) Macrophages were primed for 3 hours with LPS and stimulated for 1 hour with ATP. Culture supernatants were collected, and the concentration of secreted IL-1β was determined by ELISA. Note that the highest IL-1β values in control (None) GM-CSF and M-CSF macrophages were from different healthy donors. (D) Real-time quantitative PCR (RT-qPCR) of IL1B mRNA in macrophages primed for 3 hours with LPS. Results were normalized to β-actin expression levels. The results obtained for the cells differentiated with GM-CSF in the absence of β-glucan were arbitrarily set at 1 to express the results as fold change. (B–D) Graphs show the mean ± SEM of at least 3 independent experiments. For B, n = 10; for C and D, n = 7; *P < 0.05 and **P < 0.01, by Wilcoxon matched-pairs, signed-rank test.
IL-1β expression in macrophages is a 2-step regulated process, whereby a pattern recognition receptor–mediated signal induces transcription of IL1B into cytoplasmic pro–IL-1β protein, and a second signal activates the inflammasome to process the cytoplasmic pro–IL-1β into a mature, biologically active and secretable IL-1β form (40). To investigate the mechanism underlying the downregulation of IL-1β by β-glucan, we first assessed the transcriptional regulation of its gene. We quantified the mRNA levels of IL1B following LPS stimulation of macrophages. As shown in Figure 1D, β-glucan treatment decreased the amount of IL1B mRNA found in GM-CSF–derived macrophages but induced increased expression (P = 0.0469; n = 7 donors) when the cells were differentiated in the presence of M-CSF. This opposing effect on gene expression depending on the macrophage type is reminiscent of what we observed for the nonproteolytically activated IL-6 and TNF-α cytokines (Supplemental Figure 3), emphasizing that the mechanism of β-glucan imprinting on inflammasome-dependent IL-1β is unique. Hence, although the strong decrease in secreted IL-1β could be partly attributed to the decrease in IL1B mRNA in GM-CSF–derived macrophages, the decreased protein levels observed in M-CSF–derived macrophages could not be a consequence of altered IL1B transcription.
β-Glucan prevents NLRP3-dependent pyroptosis and attenuates IL-1β secretion via dectin-1.
Inflammasome activation may also lead to a proinflammatory programmed cell death process called pyroptosis (45). To assess whether β-glucan–induced innate immune memory also affects pyroptosis, we measured the release of the enzyme lactate dehydrogenase (LDH). As shown in Figure 2A, LDH release paralleled the secretion of IL-1β upon LPS-plus-nigericin stimulation of GM-CSF and M-CSF monocyte–derived macrophages. Importantly, we found that both IL-1β release and pyroptosis were significantly reduced by β-glucan–induced monocyte/macrophage reprogramming (Figure 2A). Similarly, LDH and IL-1β release induced by nigericin was significantly blocked by the specific NLRP3 inhibitor MCC950 (Figure 2A), suggesting that β-glucan attenuates NLRP3-dependent IL-1β release and pyroptosis.
Figure 2. Effect of β-glucan on pyroptosis upon NLRP3 inflammasome activation and the importance of the dectin-1 receptor.

(A) Monocytes were preincubated or not with β-glucan and then differentiated with either GM-CSF or M-CSF. Macrophages were primed for 3 hours with LPS and then stimulated with nigericin or vehicle as a control. For some experiments, LPS-primed macrophages were preincubated with a NLRP3 inhibitor for 30 minutes before addition of nigericin. Culture supernatants were collected, and the concentration of secreted IL-1β was determined by ELISA. Cell lysis was determined by LDH release, and results were normalized to the maximum (Max) LDH release (100%) induced by lysing the cells with 1% Triton X-100. (B) Monocytes were pretreated for 30 minutes with anti–dectin-1 mAb (α–Dectin-1) or an isotype control before incubation with β-glucan (or vehicle) and differentiation with either GM-CSF or M-CSF. Macrophages were primed for 3 hours with LPS and then stimulated for 1 hour with nigericin or were left untreated. Culture supernatants were collected, and the concentration of secreted IL-1β was determined by ELISA. (A and B) Graphs show the mean ± SEM of at least 3 independent experiments. (A) n = 4 and n = 6; *P < 0.05, by paired, 2-tailed Student’s t test. (B) n = 5; *P < 0.05, by Wilcoxon matched-pairs, signed-rank test.
Innate immune memory of monocytes is dependent on the recognition of β-glucan by the dectin-1 receptor (7). To ascertain a role for dectin-1 in the attenuation of IL-1β release by β-glucan–induced memory macrophages, we treated cells with a specific anti–dectin-1 mAb. Although incubation of monocytes with a nonspecific control IgG mAb did not affect the reduced secretion of IL-1β induced by β-glucan reprogramming (Figure 2B), the specific anti–dectin-1 mAb incubated during the 24-hour β-glucan stimulation period restored the production of IL-1β by LPS and nigericin-stimulated macrophages (Figure 2B), suggesting that the β-glucan effect was mediated via the dectin-1 pathway.
β-Glucan attenuates NLRP3 inflammasome assembly and activation.
The NLRP3 inflammasome is composed of multiple proteins, and their expression affects its activity. We assessed the impact of β-glucan–induced reprogramming on the expression of IL-1β and inflammasome components in the stimulated cell lysates of either GM-CSF– or M-CSF–derived macrophages. As shown in Figure 3, A and B, the expression of NLRP3, ASC, pro–IL-1β, and pro–caspase-1 in the stimulated cell lysates was not affected by β-glucan–induced reprogramming. We observed a significant increase in the levels of pro–IL-1β protein only in the β-glucan/M-CSF–derived cells (Figure 3B), which is consistent with the mRNA results (Figure 1D). Of note, although we observed a decrease in IL1B mRNA in β-glucan/GM-CSF–derived macrophages (Figure 1D), it was not reflected in the amount of pro–IL-1β protein present (Figure 3B). Therefore, the decreased levels of secreted mature IL-1β protein detected in both GM-CSF– and M-CSF–derived macrophages upon β-glucan reprogramming cannot be explained by the modulation of IL1B transcription and subsequent translation. In addition, and in line with the decreased IL-1β production by macrophages we observed by ELISA (Figure 1B), β-glucan inhibited both caspase-1 processing and IL-1β maturation induced by the NLRP3 agonist nigericin, as evidenced by reduced active p20 caspase-1 and active p17 IL-1β levels in cell supernatants, respectively (Figure 3, A and C).
Figure 3. Effect of β-glucan on NLRP3 inflammasome–mediated activation of caspase-1.

(A) Monocytes were preincubated or not with β-glucan and differentiated with either GM-CSF or M-CSF. The resulting macrophages were primed for 3 hours with LPS and then stimulated for 1 hour with nigericin or vehicle as a control. Caspase-1 (pro form and cleaved p20), NLRP3, ASC, and IL-1β (pro form and mature IL-1β) expression was assessed by Western blotting. Mature IL-1β and the p20 fragment of caspase-1 were also detected in cell culture supernatants. β-Actin was used as the loading control. Blots are representative of 5 independent experiments. (B) Densitometric analysis of the blots shown in A. Immunoreactive bands in cell lysates were normalized to β-actin. (C) Densitometric analysis of the blots done on supernatants shown in A. Data represent the mean ± SEM of the analysis of 4 independent experiments. n = 5. *P < 0.05, by Wilcoxon matched-pairs, signed-rank test. casp-1, caspase 1.
Although not a model of β-glucan innate imprinting, direct extracellular activation of dectin-1 in fully differentiated human monocyte–derived DCs by large and linear β-(1,3)-glucan particles of curdlan activates caspase-8 and induces IL-1β production and maturation (46). We therefore assessed whether our model of β-(1,3-1,6)-glucan imprinting of human monocytes would have a long-lasting effect on caspase-8 activity in the resulting human-derived macrophages and determined whether caspase-8 participates in the β-glucan reprogramming of the inflammasome in macrophages, as observed for caspase-1. Caspase-8 mediates priming and activation of the canonical NLRP3 inflammasome (47), however β-glucan did not alter the amount of NLRP3 upon activation with LPS and nigericin (Figure 3B), nor did β-glucan decrease the amount of pro–IL-1β (Figure 3B). In addition, the modest caspase-8 activity detected upon activation of the canonical NLRP3 inflammasome with LPS and nigericin was not affected by β-glucan priming (Supplemental Figure 5). We therefore concluded that the effect of β-glucan on the NLRP3 inflammasome is not attributable to caspase-8 activity.
ASC oligomerization leading to ASC speck formation mediates caspase-1 activation (48, 49). To determine the mechanism of impairment of the NLRP3 inflammasome by β-glucan–induced memory in macrophages, we investigated the effect of β-glucan on ASC oligomerization and subsequent speck formation. ASC oligomerization was measured by immunoblotting of the DSS–cross-linked insoluble macrophage cell lysate fraction. As shown in Figure 4, A and B, LPS plus nigericin stimulation induced ASC oligomerization in both GM-CSF– and M-CSF–derived macrophages. Interestingly, we found that ASC oligomerization was attenuated in macrophages with β-glucan–induced memory (Figure 4, A and B). Similar results were obtained by immunofluorescence staining of endogenous ASC in macrophages. We observed ASC specks in both GM-CSF– and M-CSF–derived macrophages upon treatment with LPS and nigericin (Figure 4, C and D). Importantly, the frequency of ASC speck–containing cells significantly decreased in β-glucan–reprogrammed macrophages (Figure 4, C and D), suggesting that β-glucan regulates caspase-1 activation and IL-1β release upstream of ASC oligomerization and speck formation.
Figure 4. Effect of β-glucan on ASC oligomerization and speck formation.

(A) Monocytes were preincubated or not with β-glucan and differentiated with either GM-CSF or M-CSF. Macrophages were primed for 3 hours with LPS and then stimulated for 1 hour with nigericin or vehicle as a control. Total cell lysates were obtained in Triton X-100–containing buffer. Insoluble (pellet) fractions were cross-linked with DSS to capture ASC oligomers. The soluble (input) and insoluble fractions were analyzed by immunoblotting with an ASC Ab. β-Actin was used as a loading control. Blots are representative of 3 independent experiments. (B) Densitometric analysis of the ASC oligomers on the blots of 3 healthy donors. *P < 0.05 and **P < 0.01, by paired, 2-tailed Student’s t test. (C) Representative immunofluorescence microscopic images of ASC specks. Macrophages (generated as in A) were stimulated for 1 hour with nigericin or vehicle. DNA is stained in blue and ASC in green. Red arrowheads point to ASC specks, and the enlarged inserts show cells with an ASC speck. Original magnification, ×40. Scale bars: 100 μm. (D) Quantification of the percentage ASC specks (4 × 200 cells/nuclei [DAPI-stained], analyzed with ImageJ). Data represent the mean ± SEM of the analysis of 3 independent experiments. n = 4. **P < 0.01, by paired, 2-tailed Student’s t test.
NLRP3 is responsive to a diverse array of stimuli ranging from pathogens to ATP, pore-forming toxins, and synthetic nanoparticles (50). Because the stimuli are diverse and are structurally and chemically distinct, with apparent differences in their mode of action, some common signal or signals should converge on a single pathway to activate NLRP3.
Potassium (K+) efflux is one of the most described and proposed mechanisms for regulating activation of the NLRP3 inflammasome pathway (51). In addition, mtROS production has been linked to canonical NLRP3 activation (52) in a model in which K+ efflux results in mtROS generation to trigger the NLRP3 inflammasome (53).
To further decipher the mechanism of impairment of the NLRP3 inflammasome by β-glucan–induced memory in macrophages, we therefore first investigated the intracellular K+ content of the macrophages following activation of the NLRP3 inflammasome. As expected, using Asante Potassium Green-2 (APG-2), a fluorescence indicator of cytosolic K+, we observed a significant efflux of K+ after nigericin activation of the inflammasome in both GM-CSF– and M-CSF–derived macrophages (Figure 5A). This K+ efflux was significantly blocked in GM-CSF–derived macrophages and significantly reduced in M-CSF–derived macrophages upon β-glucan reprogramming (Figure 5A).
Figure 5. Effect of β-glucan on K+ efflux and mtROS.

(A) Monocytes were preincubated or not with β-glucan and differentiated with either GM-CSF or M-CSF. Macrophages were primed for 3 hours with LPS, stained with APG-2, and then stimulated for 20 minutes with nigericin or vehicle as a control. Results are expressed as the average K+ content (average APG-2 fluorescence intensity per cell), as measured through Icy software’s image analysis pipeline. Data represent average values ± SEM of the analysis of 3 independent experiments. n = 7 and n = 9. *P < 0.05 and **P < 0.01, by 1-way ANOVA Friedman test with Dunn’s correction for multiple comparisons. (B) Macrophages were primed for 3 hours with LPS and then stimulated for 30 minutes with nigericin. Cells were stained with MitoSOX for mtROS levels. Data are expressed as the MFI relative to unstained cells. Ratio ± SEM of the analysis of 3 independent experiments. n = 4 (GM-CSF) and n = 3 (M-CSF). *P < 0.05, by paired, 2-tailed Student’s t test.
We next assessed mtROS generation in the macrophages upon activation of the NLRP3 inflammasome using a mitochondria-specific label (MitoSOX) that distinguishes ROS-generating mitochondria (52, 54). Consistent with the decrease in K+ efflux, mtROS levels were significantly attenuated in both GM-CSF and M-CSF macrophages with β-glucan–induced memory (Figure 5B). Altogether, these data confirmed that β-glucan downregulates the K+ efflux and mtROS generation that are the early mechanisms of NLRP3 inflammasome activation described upstream of ASC oligomerization, speck formation, caspase-1 activation, and IL-1β release.
β-Glucan attenuates IL-1βrelease and caspase-1 activation in cells from patients with CAPS. In light of the attenuating effect of β-glucan on NLRP3 inflammasome activation in macrophages generated from healthy individuals, we hypothesized that β-glucan may also affect uncontrolled NLRP3 inflammasome activation in patients with gain-of-function mutations in the NLRP3 gene. Patients with CAPS harbor NLRP3 gain-of-function mutations, and a series of studies have demonstrated that mononuclear phagocytes from patients with CAPS release significantly more IL-1β than do those from healthy controls (55–57). Accordingly, our results showed that IL-1β secretion was detectable in the supernatants of untreated blood monocytes isolated from 8 patients with CAPS (Table 1) and was significantly higher than that in supernatants from the control group after 18 hours of culture (Figure 6A). Moreover, monocytes from patients with CAPS released significantly higher levels of IL-1β than did those from healthy subjects upon LPS stimulation, regardless of the mutation or treatment (Figure 6A and Supplemental Figure 6A).
Table 1. Clinical characteristics of patients with CAPS.
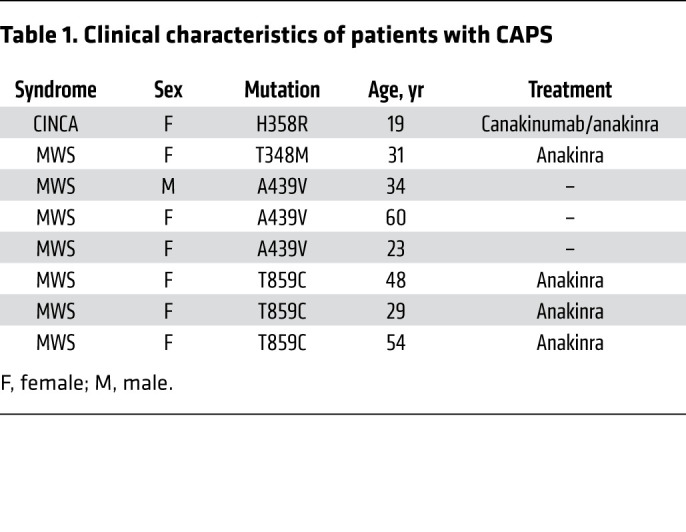
Figure 6. Effect of β-glucan on IL-1β release and caspase-1 activation in macrophages from patients with CAPS.

(A) Monocytes from healthy donors (HD) and patients with CAPS were left untreated or were stimulated with LPS for the indicated durations. The concentration of secreted IL-1β was determined by ELISA in culture supernatants. (B–E) Monocytes were preincubated with or without β-glucan and differentiated into macrophages with either GM-CSF or M-CSF. Macrophages were primed with LPS for 3 hours and then activated with nigericin for 1 hour. (B) The amount of secreted IL-1β was determined by ELISA in culture supernatants. (C) Cells plus supernatants from patients with CAPS were assayed for caspase-1 activity and normalized to the activity measured in LPS-plus-vehicle–stimulated macrophages. Ac-YVAD-CHO was used as a specific caspase-1 inhibitor (Inh.). (D) Caspase-1 (pro form and cleaved p20) expression and activation were assessed by Western blotting of cell lysates. The p20 fragment of caspase-1 was also detected in cell culture supernatants. β-Actin was used as the loading control. Blots show results for cell lysates from 2 patients with CAPS: MWS T859C (left panel) and MWS A439V (right panel). (E) Total cell lysates were obtained in Triton X-100–containing buffer. The insoluble (pellet) fraction was cross-linked with DSS to capture ASC oligomers. The soluble (input) and insoluble fractions were analyzed by immunoblotting with an ASC Ab. β-Actin was used as the loading control. Blot results are for cell lysates from 1 patient with CAPS: MWS A439V. (A–C) Graphs show the mean ± SEM of at least 3 independent experiments; n = 8 (healthy donors) and n = 8 (patients with CAPS). *P < 0.05, by Wilcoxon matched-pairs, signed-rank test. #P < 0.05, ##P < 0.01, and ###P < 0.001, for statistical differences between healthy donors and patients with CAPS, by unpaired Mann-Whitney U test.
To investigate the effect of β-glucan–induced memory on macrophages with gain-of-function mutations in the NLRP3 gene, monocytes isolated from the same 8 CAPS patients were differentiated with either GM-CSF or M-CSF after a 24-hour preincubation in the presence or absence of β-glucan. Interestingly, we observed a significantly higher release of IL-1β from macrophages of patients with CAPS compared with those of healthy control donors only when the monocytes were differentiated into a proinflammatory macrophage subset by GM-CSF (Figure 6B). Importantly, however, IL-1β secretion by either GM-CSF– or M-CSF–derived macrophages from patients with CAPS was significantly inhibited by β-glucan, independently of treatment or disease state (Figure 6B and Supplemental Figure 6B). Next, to understand whether the low levels of extracellular IL-1β present in supernatants of LPS-plus-nigericin–stimulated, β-glucan–derived reprogrammed macrophages from CAPS patients were dependent on caspase-1, we analyzed caspase-1 activity. As shown in Figure 6C, caspase-1 activity was detected following stimulation with LPS and nigericin in both GM-CSF– and M-CSF–derived macrophages, and its specific activity was demonstrated by the use of a caspase-1 inhibitor. Importantly, preincubation of monocytes with β-glucan significantly reduced caspase-1 activity in both subsets of macrophages obtained from patients with CAPS (Figure 6C). In addition, we observed that β-glucan inhibited caspase-1 cleavage in both GM-CSF– and M-CSF–derived macrophages from patients with CAPS (Figure 6D). Finally, LPS and nigericin stimulation induced ASC oligomerization in both GM-CSF– and M-CSF–derived macrophages from patients with CAPS, which was attenuated in macrophages with β-glucan–induced memory (Figure 6E). In summary, β-glucan–induced memory strongly reduced ASC oligomerization, caspase-1 activation, and IL-1β release in patients with CAPS.
Discussion
NLRP3 is the best-described inflammasome sensor and represents an attractive drug target. Upon infection or cellular damage, NLRP3 assembles into a multiprotein inflammasome complex leading to the release of IL-1β. However, NLRP3 inflammasome activity can also be detrimental to the host, and its aberrant chronic activation is associated with severe pathologies and the rare monogenic CAPS (or NLRP3-AID) (58). β-Glucan, a polysaccharide of the fungal cell wall of the yeast C. albicans, has been widely described as a potent mediator of innate immune memory, which confers an enhanced immune function to mononuclear phagocytes (6–8). Although the literature focuses on the beneficial reprogramming effects of β-glucans against infection, to our knowledge, no work has addressed the effect of β-glucan–induced innate memory on macrophages and its possible impact on the inflammasome and associated autoinflammatory diseases.
In this study, using a model of innate immune memory, we investigated how preincubation of human monocytes with particulate β-glucan affects the biological response of macrophages following NLRP3 inflammasome activation. In macrophages, activation of the NLRP3 inflammasome relies on a 2-signal model. First, microbial components prime NLRP3 through the NF-κB pathway, and second, NLRP3 is activated for inflammasome assembly by a wide range of stimuli. Our results show that innate immune memory induced by β-glucan strongly reduced IL-1β release by macrophages upon NLRP3 priming by LPS and activation by either nigericin or ATP. The decrease in biologically active IL-1β secretion we observed in macrophages with β-glucan–derived memory was not controlled at the first priming step of this process. Indeed, our results demonstrate that β-glucan reduced the secretion of active IL-1β by macrophages without significantly downregulating NLRP3 and pro–IL-1β protein levels. By contrast, β-glucan reduced K+ efflux and production of mtROS and subsequently attenuated the formation of ASC pyroptosomes and activation of caspase-1, indicating that β-glucan can target NLRP3 to exert its antiinflammatory activity upstream of ASC oligomerization. Remarkably, the attenuating effect of β-glucan early in the process of NLRP3 activation was independent of the quality of the second signal. Indeed, the effect was observed with ATP or with the bacterial ionophore nigericin, both facilitators of K+ efflux by mechanistically distinct pathways, which excludes a simple modulation of the endogenous ATP-gated P2X7 receptor channels and rather implies a very general mechanism. In this respect, another important observation resides in the fact that the NLRP3 inflammasome–modulating properties of β-glucan did not change when the differentiation of monocytes was driven by either GM-CSF or M-CSF and were, therefore, independent of microenvironmental cues and the functional polarization of macrophages.
Despite increasing interest in the field, the mechanisms underlying NLRP3 activation remain a controversial topic (59). Activation of primed NLRP3 is induced by a plethora of stimuli that are structurally and chemically unrelated (e.g., pore-forming toxins, activators of ion channels, uric acid crystals, β-amyloid protein), suggesting that NLRP3 senses downstream cytosolic stress signals such as ion imbalances or oxidized mitochondrial DNA, the latter being proposed as an NLRP3 ligand (60). Potassium (K+) efflux is one of the most-described and proposed mechanisms for regulating activation of the NLRP3 inflammasome pathway (51) and is modelized as the first event resulting in mtROS generation to trigger the NLRP3 inflammasome (53). In addition, coordination between priming and activation signals may be orchestrated by trafficking of NLRP3 to other organelles in the cell (61). Whether organelle-linked mechanisms control NLRP3 is still a matter under investigation (62). Furthermore, several mechanisms of posttranslational modifications of NLRP3 have been suggested to regulate NLRP3 inflammasome activation (63), and of these modifications, the ubiquitination and phosphorylation of NLRP3 are the best characterized.
We report here that β-glucan targets NLRP3 inflammasome activation upstream of ASC oligomerization, at the level of ion efflux and mtROS, attenuating pyroptosis and IL-1β release induced by either the plasma membrane ATP-gated P2X7 purinergic receptor or the exogenous ionophore nigericin. Our data suggest that the first controlled event by β-glucan reprogramming is the reduction of K+ efflux from macrophages, yet the upstream underlying mechanisms remain to be determined. One might postulate that β-glucan blocks NLRP3 inflammasome activation by controlling an unknown upstream event that reduces K+ efflux from macrophages, as suggested by others (64, 65). Of note, because of the reduced inflammasome-mediated pyroptosis induced by β-glucan priming of macrophages, the current study cannot exclude the fact that the K+ efflux is accompanied by a passive K+ release across leaky pores from damaged cells. Finally, further investigation is required to determine whether microtubule dynamics and posttranslational modification of NLRP3 are also key regulatory mechanisms in β-glucan–mediated control of the NLRP3 inflammasome.
CAPS, the autoinflammatory disease due to NLRP3 dysfunction, is rare and is found in approximately 1 of 360,000 individuals in France and in 1 to 2 per 1,000,000 persons in the United States (66). In this study, we have examined the biological effects of β-glucan–induced memory on monocyte-derived macrophages from a large cohort of 8 patients with CAPS. Monocytes from patients with CAPS were more inflammatory than healthy donor–derived cells, and macrophages generated from monocytes of CAPS patients presented a similar pattern. The secretion of mature IL-1β upon exogenous NLRP3 activation in macrophages from CAPS patients was higher than in their healthy counterparts’ cells. Despite this extreme inflammatory context, β-glucan–induced reprogramming of monocytes strongly reduced the secretion of IL-1β in both GM-CSF– and M-CSF–derived macrophages from patients with CAPS, to the same level as that seen in macrophages from healthy donors. Remarkably, the β-glucan effect was independent of the NRLP3 mutation, clinical classification, or pharmacological treatment of patients with CAPS. Additionally, we confirmed that the β-glucan–induced attenuation of IL-1β release by CAPS macrophages was dependent on the inhibition of ASC oligomerization and caspase-1 cleavage and activity. The reduced caspase-1 activity induced by β-glucan in CAPS-derived macrophages was comparable to that induced by the synthetic caspase-1 inhibitor Ac-YVAD-CHO.
To date, treatment of patients with CAPS relies on targeting the symptomatic oversecretion of IL-1β. Three anti–IL-1β–targeted therapies have been approved for patients with CAPS, including the IL-1 receptor antagonist anakinra, the soluble decoy receptor rilonacept, and the anti–IL-1β–neutralizing mAb canakinumab (67). Because of the administration frequency and conservation needs, IL-1β–targeted therapy is invasive, complex, and also costly (68). In addition, IL-1β, an acute-phase protein, is crucial for effective immune responses to infection (69), and inhibitors targeting IL-1β may lead to unintended immunosuppressive effects in addition to preventing NLRP3 inflammasome activity in itself. Therefore, targeting the origin of the disease, i.e., NLRP3, would represent the best therapeutic strategy. Remarkably, although a number of NLRP3 inhibitors have been reported, none of them has been formally approved for the treatment of NLRP3 inflammasome–driven diseases (70). Of interest, most of these candidate drugs directly interact with NLRP3 (70), but none seems to regulate the early activation events upstream of NLRP3 inflammasome assembly, although regulation of K+ efflux was suggested by others in the context of cryopyrinopathies (65). β-Glucan is a safe molecule present in food products and already widely used in food supplementation. As such, β-glucan is already applied as a food additive to enhance immune responsiveness in animals (71, 72). In humans, the reported clinical trials on the effects of oral intake of β-glucan on immune markers are too limited to draw conclusions at this time (73, 74). Of note, parenteral injection of β-glucan in rodent animal models induces protective immunity (6, 7, 75), and encouraging protective effects have been reported in HIV and high-risk surgical patients (76–80). In the present study, β-glucan affected NLRP3 activation to reduce the secretion of IL-1β in both GM-CSF– and M-CSF–derived macrophages from patients with CAPS, regardless of the NRLP3 mutation or pharmacological treatment. Therefore, provided the appropriate supplementation dose and route are established, β-glucan supplementation could represent a potential new therapeutic approach.
In conclusion, in the present study, we demonstrate that β-glucan–induced reprogramming of human monocytes, through a dectin-1–dependent mechanism, resulted in reduced NLRP3 inflammasome activation and subsequent IL-1β secretion by macrophages. In addition, the ability of β-glucan to block NLRP3 activation in patients with CAPS makes it an attractive, low-cost, and well-tolerated new candidate for the treatment of NLRP3-driven disorders.
Methods
Cell isolation.
Human PBMCs were separated using Ficoll-Paque media (GE Healthcare) by density centrifugation of heparinized blood from healthy donors (at the Etablissement Français du Sang [EFS], Paris, France). Monocytes were purified by positive selection using human CD14 MicroBeads (Miltenyi Biotec). The purity evaluated by flow cytometry was routinely greater than 90%. Cells were seeded at 5 × 105 cells/mL in RPMI 1640, supplemented with 2 mM GlutaMax, 50 μg/mL gentamicin, and 1 mM sodium pyruvate (Gibco, Invitrogen, Thermo Fisher Scientific). Cells were maintained in a humidified atmosphere of 5% CO2 at 37°C.
Stimulation experiments.
Monocytes were incubated for 24 hours with RPMI 1640 containing either GM-CSF (25 ng/mL, 130-093-865, Miltenyi Biotec) or M-CSF (25 ng/mL, 216-MC-025, R&D Systems) in the presence or absence of purified β-glucan from C. albicans (10 μg/mL; depyrogenated). After the incubation period, the cells were washed once with 200 μL warm PBS and incubated for 6 days in RPMI containing 10% FBS (F7524, MilliporeSigma) and either GM-CSF or M-CSF (Figure 1A), and the medium was changed once on day 3. For inflammasome activation, macrophages were primed with LPS (250 ng/mL, catalog code tlrl-3pelps, InvivoGen) in RPMI containing 10% FBS. After 3 hours, LPS-primed macrophages were given fresh media (RPMI 1640 without FBS) in the presence of adenosine triphosphate (ATP) (5 mM, catalog code tlrl-atpl, InvivoGen), nigericin (10 μM, catalog code tlrl-nig, InvivoGen), or vehicle control for 1 hour.
For inhibition, before addition of inflammasome activation stimuli, macrophages were preincubated for 30 minutes with 1 μM MCC950 (NLRP-3 inhibitor, CP-456773, MilliporeSigma) or vehicle control. In some experiments, the anti–dectin-1 mAbs (10 μg/mL, ab82888, Abcam) or IgG1 isotype control Abs (10 μg/mL, 401402, BioLegend) were added 30 minutes before β-glucan stimulation and maintained in the culture medium for 24 hours.
Patients.
Eight patients with CAPS (1 with CINCA and 7 with MWS, 3 different mutations) were enrolled in the study (Table 1). Healthy controls were studied in parallel. Blood samples were obtained from the patients, and PBMCs were separated using Ficoll-Paque media by density centrifugation of heparinized blood as described above. Cells were suspended in RPMI at 5 × 106/mL and seeded at 0.5 × 106/cells per well in 96-well flat-bottomed white plates in a total of 100 μL RPMI for 1 hour. Nonadherent cells were discarded, and the adherent monocytes were treated and differentiated according to the experimental setup described above (Figure 1A).
Cytokine assay.
IL-1β, IL-6, TNF-α, IL-18, IL-12 (all from R&D Systems), and IL-10 (BioLegend) levels in cell culture supernatants were determined by ELISA according to the manufacturers’ instructions. Absorbance was measured with a Synergy H1 Microplate Reader (BioTek Instruments).
RNA isolation and real-time PCR analysis.
Total RNA was isolated from macrophages using TriZOL Reagent (Invitrogen, Thermo Fisher Scientific). RNA quality was monitored by spectrophotometric analysis. Total RNA (1 μg) from each sample was used for reverse transcription with the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Life Technologies, Thermo Fisher Scientific). Real-time PCR was performed in an ABI Step One Plus Real-Time PCR System (Applied Biosystems, Life Technologies) using TaqMan 2× Universal Master Mix and the TaqMan Gene Expression Assay for IL-1β (assay ID: Hs01555410, Applied Biosystems, Life Technologies, Thermo Fisher Scientific). Each sample was assayed in triplicate. Thermal cycling conditions were as follows: denaturation at 95°C for 10 minutes and 40 cycles at 95°C for 15 seconds and 60°C for 60 seconds. The fold change of the cytokine genes in the samples was calculated using the 2−ΔΔCt method. All values were normalized to endogenous control β-actin (Hs99999903; Applied Biosystems, Life Technologies) and were expressed in arbitrary units.
Western blotting.
Total cellular protein extracts were obtained by lysing cells for 30 minutes at 4°C in lysis buffer (Pierce RIPA Buffer, Thermo Fisher Scientific) in the presence of protease inhibitors (cOmplete Protease Inhibitor Cocktail, Roche). Samples were centrifuged for 15 minutes at 4°C, 12,000 g and supernatants collected in new tubes. Cell culture supernatants were collected and concentrated (×10) using Amicon Ultra Centrifugal Filters (MilliporeSigma). Proteins from cell extracts and supernatants were suspended in Laemmli Buffer 1×, boiled at 95°C for 10 minutes and resolved by SDS-PAGE. Then, proteins were transferred with an iBlot instrument (Life Technologies, Thermo Fisher Scientific). Immunoblotting was performed using the following primary Abs: anti-NLRP3 (clone D4D8T, 15101), anti–caspase-1 (clone D7F10, 3866), anti–IL-1β (clone 3A6, 12242) (all from Cell Signaling Technology), and anti-ASC (clone B-3, sc-514414, Santa Cruz Biotechnology). Anti–mouse or anti–rabbit IgG, HRP-linked Abs (NA931 and NA934, respectively, GE Healthcare) were used as secondary Abs, and protein bands were visualized with ECL Prime (GE Healthcare) or SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific) and a myECL Imager (Thermo Fisher Scientific). Proteins were normalized to β-actin (clone C4, sc-47778, Santa Cruz Biotechnology) using ImageJ software (NIH).
For the ASC oligomerization assay, cells were directly lysed in the well by the addition of cold PBS containing 0.5% Triton X-100 and protease inhibitor mixture (cOmplete Protease Inhibitor Cocktail, Roche). Cell lysates were separated into a Triton X-100 soluble fraction and an insoluble fraction by centrifugation at 6000 × g for 20 minutes at 4°C. The soluble fraction of cell lysates was subsequently used for immunoblotting, whereas the Triton X-100 insoluble pellets were washed twice with PBS and then chemically cross-linked with 2 mM disuccinimidyl suberate (DSS) (Thermo Fisher Scientific) for 30 minutes at room temperature. Cross-linked pellets were further spun down at 6000 × g for 20 minutes and eluted in Laemmli buffer for SDS-PAGE.
Surface markers.
On day 6, macrophages were detached from the plate using Accutase Solution (MilliporeSigma) at 37°C for 15 minutes followed by manual shaking and repeated pipetting. After blocking of Fc receptors (FcR Blocking Reagent, Miltenyi Biotec), staining was performed using anti–CD163 phycoerythrin (PE) (clone REA812, 130-112-128, Miltenyi Biotec), and anti–CD206 PE-Vio770 (clone DCN228, 130-100-152, Miltenyi Biotec). Cells were also stained with fluorescence minus one (FMO) mixes containing the relevant isotype control: REA control coupled with PE (clone REA293, 130-113-438, Miltenyi Biotec) or mouse IgG1 PE-Vio770 (clone IS5-21F5, 130-113-202, Miltenyi Biotec). Acquisition was performed on a LSR Fortessa (BD Biosciences) after fixation of cells with 4% formaldehyde solution at room temperature, protected from light. Analysis was performed using FlowJo software, version 9.9.5 (Tree Star).
ASC immunofluorescence.
Treated macrophages were fixed in 4% formaldehyde solution for 10 minutes at 37°C and then washed with PBS, and excess formaldehyde solution was quenched for 10 minutes with 0.1 M glycine. Cells were permeabilized with saponin 0.2% BSA 3% for 20 minutes at 4°C. Anti-ASC Ab (1:500, clone B-3, sc-514414, Santa Cruz Biotechnology) was incubated at 4°C in the saponin-BSA solution overnight. The next day, samples were incubated with an Alexa Fluor 488–conjugated anti–mouse IgG secondary Ab (1:500; A11001, Invitrogen, Thermo Fisher Scientific). DAPI (Invitrogen, Thermo Fisher Scientific) was used at 1:1000 for nuclear staining. Coverslips were mounted using VectaShield Mounting Medium (Vector Laboratories) and sealed. Cells were imaged using an EVOS FL Cell Imaging system and a ×40 objective (Thermo Fisher Scientific). Images were analyzed with ImageJ software, and brightness was adjusted with Adobe Photoshop CC 2018.
Intracellular K+ measurements.
LPS-primed macrophages in 96-well, flat, clear-bottomed black plates (Costar) were loaded with Asante Potassium Green-2 (APG-2) (Abcam), a fluorescence indicator of intracellular K+. More specifically, cells were labeled with a staining solution of APG-2 AM 5 μM (ab142806, Abcam) plus Pluronic F-127 0.1% (P6867, Invitrogen, Thermo Fisher Scientific) for 30 minutes at 37°C. Cells were then stimulated with 10 μM nigericin for 20 minutes. After washing with PBS twice, cells were imaged using an EVOS FL Cell Imaging System and a ×10 objective (Thermo Fisher Scientific). Quantification of the average fluorescence intensity per cell was performed through an image analysis pipeline. The pipeline consists of HK-mean segmentation in Icy software (81, 82). Qualitative evaluation was done to confirm that the pipeline identified all of the cells (average of 600 cells) in the field of view with an error rate of 0.2% ± 0.2%.
mtROS.
LPS-primed macrophages were detached using Accutase (A6964, MilliporeSigma) for 15 minutes at 37°C, followed by manual shaking and repeated pipetting. Cells were then stimulated with 10 μM nigericin in RPMI 1640 for 30 minutes at 37°C in 96-well round-bottomed plates. mtROS levels were measured by staining cells with MitoSOX (M36008, Thermo Fisher Scientific) at 5 mM for 15 minutes at 37°C in HBSS (11550456, Gibco, Thermo Fisher Scientific). Cells were then washed, resuspended with HBSS, and acquired on a LSR Fortessa (BD Biosciences). Fluorescence was measured using a 561-nm laser and a 585/15 filter for the PE channel. Data were analyzed using FlowJo software, version 9.9.5 (Tree Star).
Cell viability.
Fluorescein diacetate (FDA) is a cell-permeant esterase substrate used to measure both enzymatic activity (to activate its fluorescence) and cell-membrane integrity (required for intracellular retention of the fluorescent product). Cell cultures were performed in 96-well, flat, clear-bottomed black plates. Monocytes were incubated for 24 hours with RPMI 1640 alone (human serum and FBS conditions) (Supplemental Figure 2). In parallel, monocytes were incubated for 24 hours with RPMI 1640 containing either GM-CSF or M-CSF in the presence or absence of purified β-glucan from C. albicans. After the 24-hour incubation, the cells were washed once with warm PBS and incubated for another 5 days in RPMI supplemented with 10% human serum (Human Serum Type AB [male], MilliporeSigma), 10% FBS, and 10% FBS plus GM-CSF or M-CSF, and the medium was changed once on day 3. The amount of remaining viable cells was evaluated on day 6 by loading cells with FDA. FDA (F7378, MilliporeSigma) was dissolved in acetone to a stock concentration of 5 mg/mL. A final staining solution (8 μg/mL) of FDA was prepared in serum-free RPMI. Cells were stained for 3 minutes at room temperature and then washed, and viability was assessed by measuring the fluorescence on a Synergy H1 Microplate Reader (excitation: 490, emission: 526; BioTek Instruments).
LDH release assay.
LDH release was measured using the Cytotoxicity Detection Kit (Roche) according to the manufacturer’s instructions. Macrophages in 96-well, flat, clear-bottomed white plates were pretreated with the indicated inhibitors for 30 minutes (see Stimulation experiments for the various concentrations used) and primed with LPS for 3 hours. Macrophages were then stimulated with vehicle or nigericin for 1.5 hours (M-CSF macrophages) or 3 hours (GM-CSF macrophages) before cell-free culture supernatants were collected and incubated with LDH assay solution at 25°C for 30 minutes. The optical density values were analyzed at 490 nm by subtracting the reference value at 620 nm. The experiment was repeated at least 3 times, and the results are expressed as a percentage of the maximum LDH release, obtained by lysing the cells in 1% Triton X‑100.
Caspase activity assay.
LPS-primed macrophages were incubated with either vehicle or nigericin for 1 hour, and caspase-1 or caspase-8 activity was then measured using the Caspase-Glo 1 Inflammasome Assay (G9951, Promega) or the Caspase-Glo 8 Assay System (G8201, Promega), respectively, according to the manufacturer’s instructions. Briefly, the white 96-well plate containing cells was removed from the incubator, and 100 μL of the supernatant in each well was collected and used for IL-1β cytokine detection. An aliquot of 100 μL of the Caspase-Glo 1 Reagent or Caspase-Glo 8 was then added to each well containing cells and the remaining 100 μL supernatant and gently mixed on a plate shaker for 30 seconds. The mixture was then incubated for 1 hour at room temperature before measuring the luminescence on a Synergy H1 microplate reader (BioTek Instruments). Ac-YVAD-CHO (G9951, Promega) was used as a specific caspase-1 inhibitor, and z-IETD-fmk (1148, BioVision) was used as a specific caspase-8 inhibitor.
Statistics.
Experiments were performed at least 3 times, and the results were analyzed using a Wilcoxon matched-pairs, signed-rank test unless otherwise stated. A P value of less than 0.05 was considered statistically significant. All data were analyzed using GraphPad Prism, version 5.0 (GraphPad Software). Data are shown as the mean ± SEM.
Study approval.
Blood samples from healthy donors were obtained from the EFS (Paris, France, habilitation HS-2015-25101), and written informed consent from was obtained from the volunteers. Blood samples from patients with CAPS were obtained with informed consent of the patients. The informed consent protocol was approved by the local ethics board (CPP Ile de France 1).
Author contributions
GC conducted the experiments, acquired and analyzed data, and wrote the manuscript. MB, ACP, and RL acquired and analyzed data. DLW provided the β-glucan used in this study. BN consulted on the study and edited the manuscript. GG and SGL obtained informed consent and collected samples from patients with CAPS. JQ designed the research study, analyzed data, and wrote the manuscript.
Supplementary Material
Acknowledgments
We gratefully acknowledge valuable discussions with Molly Ingersoll, Elisa Gomez-Perdiguero, and Melanie Hamon (Institut Pasteur). The authors also wish to thank Serge Amselem, Irina Giurgea, and Laurence Cuisset for sequencing NLRP3 in patients with CAPS, and Léa Savey for recruiting 1 CAPS patient (all from Sorbonne Université). We also acknowledge the help of the Image Analysis Hub of the Institut Pasteur for this work. GC, MB, RL, ACP, and JQ were supported by an Agence Nationale de la Recherche (ANR) JCJC grant (ANR-16-CE15-0014-01) and by a grant from the Institut Carnot–Microbes et Santé grant (no. 11 CARN-017-01, to JQ). The authors also acknowledge funding from CAPES (finance code 001). This work was also supported, in part, by NIH grants GM119197, GM53522, and GM083016 (to DLW).
Version 1. 07/27/2020
Electronic publication
Version 2. 09/01/2020
Print issue publication
Funding Statement
ANR JCJC grant
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Copyright: © 2020, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2020;130(9):4561–4573.https://doi.org/10.1172/JCI134778.
Contributor Information
Giorgio Camilli, Email: g.camilli85@gmail.com.
Mathieu Bohm, Email: mathieu.bohm@pasteur.fr.
Rachel Lavenir, Email: rachel.lavenir@pasteur.fr.
David L. Williams, Email: williamd@etsu.edu.
Gilles Grateau, Email: gilles.grateau@aphp.fr.
Sophie Georgin-Lavialle, Email: sophie.georgin-lavialle@aphp.fr.
Jessica Quintin, Email: jessica.quintin@pasteur.fr.
References
- 1.Kleinnijenhuis J, et al. Bacille Calmette-Guerin induces NOD2-dependent nonspecific protection from reinfection via epigenetic reprogramming of monocytes. Proc Natl Acad Sci USA. 2012;109(43):17537–17542. doi: 10.1073/pnas.1202870109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kokoshis PL, Williams DL, Cook JA, Di Luzio NR. Increased resistance to Staphylococcus aureus infection and enhancement in serum lysozyme activity by glucan. Science. 1978;199(4335):1340–1342. doi: 10.1126/science.628841. [DOI] [PubMed] [Google Scholar]
- 3.Santecchia I, et al. Innate immune memory through TLR2 and NOD2 contributes to the control of Leptospira interrogans infection. PLoS Pathog. 2019;15(5):e1007811. doi: 10.1371/journal.ppat.1007811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bauer M, Weis S, Netea MG, Wetzker R. Remembering Pathogen Dose: Long-Term Adaptation in Innate Immunity. Trends Immunol. 2018;39(6):438–445. doi: 10.1016/j.it.2018.04.001. [DOI] [PubMed] [Google Scholar]
- 5.Arts RJ, et al. Glutaminolysis and fumarate accumulation integrate immunometabolic and epigenetic programs in trained immunity. Cell Metab. 2016;24(6):807–819. doi: 10.1016/j.cmet.2016.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheng SC, et al. mTOR- and HIF-1α-mediated aerobic glycolysis as metabolic basis for trained immunity. Science. 2014;345(6204):1250684. doi: 10.1126/science.1250684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Quintin J, et al. Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe. 2012;12(2):223–232. doi: 10.1016/j.chom.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saeed S, et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science. 2014;345(6204):1251086. doi: 10.1126/science.1251086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dos Santos JC, et al. β-Glucan-induced trained immunity protects against Leishmania braziliensis infection: a crucial role for IL-32. Cell Rep. 2019;28(10):2659–2672.e6. doi: 10.1016/j.celrep.2019.08.004. [DOI] [PubMed] [Google Scholar]
- 10.Sánchez-Ramón S, Conejero L, Netea MG, Sancho D, Palomares Ó, Subiza JL. Trained immunity-based vaccines: a new paradigm for the development of broad-spectrum anti-infectious formulations. Front Immunol. 2018;9:2936. doi: 10.3389/fimmu.2018.02936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Töpfer E, Boraschi D, Italiani P. Innate immune memory: the latest Frontier of adjuvanticity. J Immunol Res. 2015;2015:478408. doi: 10.1155/2015/478408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xue J, et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity. 2014;40(2):274–288. doi: 10.1016/j.immuni.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 2014;6:13. doi: 10.12703/P6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5(12):953–964. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- 15.Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122(3):787–795. doi: 10.1172/JCI59643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Falleni M, et al. M1 and M2 macrophages’ clinicopathological significance in cutaneous melanoma. Melanoma Res. 2017;27(3):200–210. doi: 10.1097/CMR.0000000000000352. [DOI] [PubMed] [Google Scholar]
- 17.Jiang Z, Zhu L. Update on the role of alternatively activated macrophages in asthma. J Asthma Allergy. 2016;9:101–107. doi: 10.2147/JAA.S104508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lewis CE, Pollard JW. Distinct role of macrophages in different tumor microenvironments. Cancer Res. 2006;66(2):605–612. doi: 10.1158/0008-5472.CAN-05-4005. [DOI] [PubMed] [Google Scholar]
- 19.Moore KJ, Sheedy FJ, Fisher EA. Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol. 2013;13(10):709–721. doi: 10.1038/nri3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112(12):1796–1808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Charo IF. Macrophage polarization and insulin resistance: PPARgamma in control. Cell Metab. 2007;6(2):96–98. doi: 10.1016/j.cmet.2007.07.006. [DOI] [PubMed] [Google Scholar]
- 22.Fujita E, et al. Statin attenuates experimental anti-glomerular basement membrane glomerulonephritis together with the augmentation of alternatively activated macrophages. Am J Pathol. 2010;177(3):1143–1154. doi: 10.2353/ajpath.2010.090608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol. 2017;14(7):399–416. doi: 10.1038/nrclinonc.2016.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Poh AR, Ernst M. Targeting macrophages in cancer: from bench to bedside. Front Oncol. 2018;8:49. doi: 10.3389/fonc.2018.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takeda Y, et al. Macrophage skewing by Phd2 haplodeficiency prevents ischaemia by inducing arteriogenesis. Nature. 2011;479(7371):122–126. doi: 10.1038/nature10507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arts RJW, Joosten LAB, Netea MG. The potential role of trained immunity in autoimmune and autoinflammatory disorders. Front Immunol. 2018;9:298. doi: 10.3389/fimmu.2018.00298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Netea MG, et al. Trained immunity: a program of innate immune memory in health and disease. Science. 2016;352(6284):aaf1098. doi: 10.1126/science.aaf1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Włodarczyk M, Druszczyńska M, Fol M. Trained innate immunity not always amicable. Int J Mol Sci. 2019;20(10):E2565. doi: 10.3390/ijms20102565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moghaddas F, Masters SL. The classification, genetic diagnosis and modelling of monogenic autoinflammatory disorders. Clin Sci. 2018;132(17):1901–1924. doi: 10.1042/CS20171498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Broderick L, De Nardo D, Franklin BS, Hoffman HM, Latz E. The inflammasomes and autoinflammatory syndromes. Annu Rev Pathol. 2015;10:395–424. doi: 10.1146/annurev-pathol-012414-040431. [DOI] [PubMed] [Google Scholar]
- 31.Bauernfeind F, et al. Inflammasomes: current understanding and open questions. Cell Mol Life Sci. 2011;68(5):765–783. doi: 10.1007/s00018-010-0567-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Franchi L, Eigenbrod T, Muñoz-Planillo R, Nuñez G. The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol. 2009;10(3):241–247. doi: 10.1038/ni.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sutterwala FS, Haasken S, Cassel SL. Mechanism of NLRP3 inflammasome activation. Ann N Y Acad Sci. 2014;1319:82–95. doi: 10.1111/nyas.12458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ben-Chetrit E, et al. Consensus proposal for taxonomy and definition of the autoinflammatory diseases (AIDs): a Delphi study. Ann Rheum Dis. 2018;77(11):1558–1565. doi: 10.1136/annrheumdis-2017-212515. [DOI] [PubMed] [Google Scholar]
- 35.Aganna E, et al. Association of mutations in the NALP3/CIAS1/PYPAF1 gene with a broad phenotype including recurrent fever, cold sensitivity, sensorineural deafness, and AA amyloidosis. Arthritis Rheum. 2002;46(9):2445–2452. doi: 10.1002/art.10509. [DOI] [PubMed] [Google Scholar]
- 36.Aksentijevich I, et al. De novo CIAS1 mutations, cytokine activation, and evidence for genetic heterogeneity in patients with neonatal-onset multisystem inflammatory disease (NOMID): a new member of the expanding family of pyrin-associated autoinflammatory diseases. Arthritis Rheum. 2002;46(12):3340–3348. doi: 10.1002/art.10688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aksentijevich I, et al. The clinical continuum of cryopyrinopathies: novel CIAS1 mutations in North American patients and a new cryopyrin model. Arthritis Rheum. 2007;56(4):1273–1285. doi: 10.1002/art.22491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat Genet. 2001;29(3):301–305. doi: 10.1038/ng756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bekkering S, Blok BA, Joosten LA, Riksen NP, van Crevel R, Netea MG. In vitro experimental model of trained innate immunity in human primary monocytes. Clin Vaccine Immunol. 2016;23(12):926–933. doi: 10.1128/CVI.00349-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Netea MG, et al. Differential requirement for the activation of the inflammasome for processing and release of IL-1beta in monocytes and macrophages. Blood. 2009;113(10):2324–2335. doi: 10.1182/blood-2008-03-146720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Budai MM, Tőzsér J, Benkő S. Different dynamics of NLRP3 inflammasome-mediated IL‑1β production in GM-CSF- and M-CSF-differentiated human macrophages. J Leukoc Biol. 2017;101(6):1335–1347. doi: 10.1189/jlb.3A0716-300RR. [DOI] [PubMed] [Google Scholar]
- 42.Puren AJ, Fantuzzi G, Dinarello CA. Gene expression, synthesis, and secretion of interleukin 18 and interleukin 1beta are differentially regulated in human blood mononuclear cells and mouse spleen cells. Proc Natl Acad Sci USA. 1999;96(5):2256–2261. doi: 10.1073/pnas.96.5.2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stoll S, et al. Production of functional IL-18 by different subtypes of murine and human dendritic cells (DC): DC-derived IL-18 enhances IL-12-dependent Th1 development. Eur J Immunol. 1998;28(10):3231–3239. doi: 10.1002/(SICI)1521-4141(199810)28:10<3231::AID-IMMU3231>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 44.Mariathasan S, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440(7081):228–232. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- 45.Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. 2009;7(2):99–109. doi: 10.1038/nrmicro2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gringhuis SI, et al. Dectin-1 is an extracellular pathogen sensor for the induction and processing of IL-1β via a noncanonical caspase-8 inflammasome. Nat Immunol. 2012;13(3):246–254. doi: 10.1038/ni.2222. [DOI] [PubMed] [Google Scholar]
- 47.Gurung P, et al. FADD and caspase-8 mediate priming and activation of the canonical and noncanonical Nlrp3 inflammasomes. J Immunol. 2014;192(4):1835–1846. doi: 10.4049/jimmunol.1302839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fernandes-Alnemri T, et al. The pyroptosome: a supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ. 2007;14(9):1590–1604. doi: 10.1038/sj.cdd.4402194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Srinivasula SM, Poyet JL, Razmara M, Datta P, Zhang Z, Alnemri ES. The PYRIN-CARD protein ASC is an activating adaptor for caspase-1. J Biol Chem. 2002;277(24):21119–21122. doi: 10.1074/jbc.C200179200. [DOI] [PubMed] [Google Scholar]
- 50.Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140(6):821–832. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 51.Muñoz-Planillo R, Kuffa P, Martínez-Colón G, Smith BL, Rajendiran TM, Núñez G. K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity. 2013;38(6):1142–1153. doi: 10.1016/j.immuni.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469(7329):221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 53.Yaron JR, et al. K(+) regulates Ca(2+) to drive inflammasome signaling: dynamic visualization of ion flux in live cells. Cell Death Dis. 2015;6:e1954. doi: 10.1038/cddis.2015.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ahmad T, et al. Computational classification of mitochondrial shapes reflects stress and redox state. Cell Death Dis. 2013;4:e461. doi: 10.1038/cddis.2012.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Carta S, et al. Cell stress increases ATP release in NLRP3 inflammasome-mediated autoinflammatory diseases, resulting in cytokine imbalance. Proc Natl Acad Sci USA. 2015;112(9):2835–2840. doi: 10.1073/pnas.1424741112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Goldbach-Mansky R, et al. Neonatal-onset multisystem inflammatory disease responsive to interleukin-1beta inhibition. N Engl J Med. 2006;355(6):581–592. doi: 10.1056/NEJMoa055137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tassi S, et al. Altered redox state of monocytes from cryopyrin-associated periodic syndromes causes accelerated IL-1beta secretion. Proc Natl Acad Sci USA. 2010;107(21):9789–9794. doi: 10.1073/pnas.1000779107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Masters SL, Simon A, Aksentijevich I, Kastner DL. Horror autoinflammaticus: the molecular pathophysiology of autoinflammatory disease (*) Annu Rev Immunol. 2009;27:621–668. doi: 10.1146/annurev.immunol.25.022106.141627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yang Y, Wang H, Kouadir M, Song H, Shi F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. 2019;10(2):128. doi: 10.1038/s41419-019-1413-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhong Z, et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature. 2018;560(7717):198–203. doi: 10.1038/s41586-018-0372-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Misawa T, et al. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat Immunol. 2013;14(5):454–460. doi: 10.1038/ni.2550. [DOI] [PubMed] [Google Scholar]
- 62.Hamilton C, Anand PK. Right place, right time: localisation and assembly of the NLRP3 inflammasome. F1000Res. 2019;8:F1000 Faculty Rev-676. doi: 10.12688/f1000research.18557.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yang J, Liu Z, Xiao TS. Post-translational regulation of inflammasomes. Cell Mol Immunol. 2017;14(1):65–79. doi: 10.1038/cmi.2016.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yin H, et al. Curcumin suppresses IL-1β secretion and prevents inflammation through inhibition of the NLRP3 inflammasome. J Immunol. 2018;200(8):2835–2846. doi: 10.4049/jimmunol.1701495. [DOI] [PubMed] [Google Scholar]
- 65.Youm YH, et al. The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat Med. 2015;21(3):263–269. doi: 10.1038/nm.3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kuemmerle-Deschner JB, Haug I. Canakinumab in patients with cryopyrin-associated periodic syndrome: an update for clinicians. Ther Adv Musculoskelet Dis. 2013;5(6):315–329. doi: 10.1177/1759720X13502629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dinarello CA, Simon A, van der Meer JW. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat Rev Drug Discov. 2012;11(8):633–652. doi: 10.1038/nrd3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lorenzoni V, et al. The cost of illness of ultra-rare cryopyrin associated periodic syndromes (CAPS) in Italy: preliminary results from the Radicea Project. Value in Health. 2016;19(7):A585–A586. [Google Scholar]
- 69.Sahoo M, Ceballos-Olvera I, del Barrio L, Re F. Role of the inflammasome, IL-1β, and IL-18 in bacterial infections. ScientificWorldJournal. 2011;11:2037–2050. doi: 10.1100/2011/212680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. 2019;19(8):477–489. doi: 10.1038/s41577-019-0165-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Moon SH, Lee I, Feng X, Lee HY, Kim J, Ahn DU. Effect of dietary beta-glucan on the performance of broilers and the quality of broiler breast meat. Asian-australas J Anim Sci. 2016;29(3):384–389. doi: 10.5713/ajas.15.0141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vetvicka V, Vannucci L, Sima P. The effects of β-glucan on pig growth and immunity. Open Biochem J. 2014;8:89–93. doi: 10.2174/1874091X01408010089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Demir G, Klein HO, Mandel-Molinas N, Tuzuner N. Beta glucan induces proliferation and activation of monocytes in peripheral blood of patients with advanced breast cancer. Int Immunopharmacol. 2007;7(1):113–116. doi: 10.1016/j.intimp.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 74.Leentjens J, Quintin J, Gerretsen J, Kox M, Pickkers P, Netea MG. The effects of orally administered beta-glucan on innate immune responses in humans, a randomized open-label intervention pilot-study. PLoS ONE. 2014;9(9):e108794. doi: 10.1371/journal.pone.0108794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Liang J, et al. Enhanced clearance of a multiple antibiotic resistant Staphylococcus aureus in rats treated with PGG-glucan is associated with increased leukocyte counts and increased neutrophil oxidative burst activity. Int J Immunopharmacol. 1998;20(11):595–614. doi: 10.1016/S0192-0561(98)00007-1. [DOI] [PubMed] [Google Scholar]
- 76.Gordon M, et al. A phase II controlled study of a combination of the immune modulator, lentinan, with didanosine (ddI) in HIV patients with CD4 cells of 200-500/mm3. J Med. 1995;26(5-6):193–207. [PubMed] [Google Scholar]
- 77.Gordon M, et al. A placebo-controlled trial of the immune modulator, lentinan, in HIV-positive patients: a phase I/II trial. J Med. 1998;29(5-6):305–330. [PubMed] [Google Scholar]
- 78.Babineau TJ, et al. A phase II multicenter, double-blind, randomized, placebo-controlled study of three dosages of an immunomodulator (PGG-glucan) in high-risk surgical patients. Arch Surg. 1994;129(11):1204–1210. doi: 10.1001/archsurg.1994.01420350102014. [DOI] [PubMed] [Google Scholar]
- 79.Babineau TJ, Marcello P, Swails W, Kenler A, Bistrian B, Forse RA. Randomized phase I/II trial of a macrophage-specific immunomodulator (PGG-glucan) in high-risk surgical patients. Ann Surg. 1994;220(5):601–609. doi: 10.1097/00000658-199411000-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dellinger EP, et al. Effect of PGG-glucan on the rate of serious postoperative infection or death observed after high-risk gastrointestinal operations. Betafectin Gastrointestinal Study Group. Arch Surg. 1999;134(9):977–983. doi: 10.1001/archsurg.134.9.977. [DOI] [PubMed] [Google Scholar]
- 81.de Chaumont F, et al. Icy: an open bioimage informatics platform for extended reproducible research. Nat Methods. 2012;9(7):690–696. doi: 10.1038/nmeth.2075. [DOI] [PubMed] [Google Scholar]
- 82. Rigaud S, Quintin J. Cell Fluorescence quantification (Version v1). Zenodo. https://zenodo.org/record/3706554 Updated March 11, 2020.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.