

SUMMARY
The phosphatidylinositol 3 kinase (PI3K)-glycogen synthase kinase β (GSK3β) axis plays a central role in MYC-driven lymphomagenesis, and MYC targeting with bromodomain and extraterminal protein family inhibitors (BETi) is a promising treatment strategy in lymphoma. In a high-throughput combinatorial drug screening experiment, BETi enhance the anti-proliferative effects of PI3K inhibitors in a panel of diffuse large B cell lymphoma (DLBCL) and Burkitt lymphoma cell lines. BETi or MYC silencing upregulates several PI3K pathway genes and induces GSK3β S9 inhibitory phosphorylation, resulting in increased β-catenin protein abundance. Further-more, BETi or MYC silencing increases GSK3β S9 phosphorylation levels and β-catenin protein abundance through downregulating the E2 ubiquitin conjugating enzymes UBE2C and UBE2T. In a mouse xenograft DLBCL model, BETi decrease MYC, UBE2C, and UBE2T and increase phospho-GSK3β S9 levels, enhancing the anti-proliferative effect of PI3K inhibitors. Our study reveals prosurvival feedbacks induced by BETi involving GSK3β regulation, providing a mechanistic rationale for combination strategies.
In Brief
In this study, Derenzini et al. demonstrate that BET inhibitors enhance lymphoma vulnerability to PI3K inhibitors by inducing GSK3β feedback in a MYC-dependent manner and by downregulating E2-ubiquitin conjugating enzymes, which further enhance the feedback. These data provide the rationale for combining BET and PI3K inhibitors in lymphoma therapy.
Graphical Abstract
INTRODUCTION
Constitutive phosphoinositide 3-kinase (PI3K) activation plays a crucial role in the pathogenesis of MYC-driven lymphoma and is considered an attractive target for therapeutic intervention (Wendel et al., 2004; Sander et al., 2012; Pourdehnad et al., 2013). Furthermore, recent data highlight the function of B cell receptor (BCR)-PI3K-glycogen synthase kinase 3 (GSK3β) axis in supporting MYC-dependent transcriptional programs in lymphoma (Varano et al., 2017). GSK-3 is one of the main downstream targets of PI3K and regulates several signaling pathways involved in cellular metabolism, differentiation, immunity, and survival. GSK-3 phosphorylates a large number of proteins marking them for degradation, its activity being repressed by pro-survival phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT) signaling. When AKT is activated, it phosphorylates the two isoforms of GSK-3 on their N terminus; GSK-3α at position S21 and GSK-3β at position S9. Because GSK phosphorylation is a major target of AKT, GSK-3β S9 phosphorylation is widely used to determine the activation status of the PI3K/AKT signaling pathway (Beurel et al., 2010, McCubrey et al., 2014; Walz et al., 2017).
The MYC transcription factor plays a central role in regulating cell growth, proliferation, and metabolism and is involved in the pathogenesis of a variety of lymphomas, including diffuse large B cell lymphoma (DLBCL) and Burkitt lymphoma (BL) (Dang, 2013, 2015; Green et al., 2012; Johnson et al., 2012). Although the development of drugs that can directly inhibit MYC protein function remains challenging, MYC-dependent transcription can be pharmacologically suppressed using compounds targeting bromodomain and extraterminal (BET) proteins, such as BRD2 and BRD4 (Delmore et al., 2011; Mertz et al., 2011). Accordingly, several BET inhibitors (BETi) are being developed for the treatment of a variety of MYC-driven cancers, including lymphoma (Delmore et al., 2011; Mertz et al., 2011; Chapuy et al., 2013; Boi et al., 2015; Trabucco et al., 2015). BET inhibition depletes enhancer and promoter-bound BRD4, leading to inhibition of MYC transcription and downregulation of MYC-dependent transcriptional programs (Delmore et al., 2011; Mertz et al., 2011; Chapuy et al., 2013; Lovén et al., 2013).
While BET inhibitors have been shown to disrupt several survival pathways, including suppressing the expression of MYC, reducing nuclear factor kappa-B (NF-κB) activity (Ceribelli et al., 2014), and suppressing E2F1-dependent transcriptional programs (Chapuy et al., 2013), they predominantly exerted cytostatic effects causing G1 cell-cycle arrest (Chapuy et al., 2013). These observations prompted us to examine whether BET inhibition may induce feedback survival mechanisms preventing or attenuating its antitumor efficacy, and if so, whether blocking such feedback loops could be exploited for enhancing BET inhibitors activity through combination strategies. In this study, we report that BET inhibitors induced previously unknown MYC-dependent feedback loops involving the PI3K/GSK3 signaling. Accordingly, PI3K inhibitors (PI3Ki) enhanced the antiproliferative effects of BET inhibition in DLBCL and BL. Our findings provide mechanistic rationale for future combination strategies aimed at enhancing the efficacy of BET inhibitors in lymphoma.
RESULTS
BET Inhibitors Enhance Lymphoma Vulnerability to PI3K Inhibitors In Vitro
BET inhibitors (JQ1 and CPI-203) demonstrated a broad antiproliferative activity in a panel of 12 B cell lymphoma cell lines, including DLBCL of the germinal center B cell (GCB) and of the activated B cell (ABC) subtype, in addition to 2 BL cell lines (Figures S1A and S1B). The antiproliferative activity was observed irrespective of epigenetic modifying gene mutations (Figure S1B). The growth inhibition was predominantly due to G1 cell-cycle arrest (Figures S1C and S1D). As expected, JQ1 treatment downregulated MYC mRNA and decreased MYC protein levels (Figures S2A–S2C).
Recent investigations demonstrated the therapeutic value of targeting PI3K in ABC- and GCB-derived DLBCL (Erdmann et al., 2017; Paul et al., 2017), and several studies demonstrated oncogenic cooperation between MYC and PI3K (Wendel et al., 2004; Hoffman and Liebermann, 2008; Sander et al., 2012; Schmitz et al., 2012; Pourdehnad et al., 2013) and MYC and B cell lymphoma 2 (BCL-2) in lymphomagenesis (Strasser et al., 1990; Green et al., 2012; Johnson et al., 2012). Given the lack of effective cytotoxic activity of BET inhibitors despite their ability to downregulate MYC, we examined whether combining BET inhibitors with PI3K or BCL-2 inhibitors would produce enhanced antiproliferative effects. Using a high-throughput screening (HTS) approach, we combined BET inhibitors with drugs that inhibit PI3K/AKT/mTOR pathway activation at different signaling molecules, in addition to the selective BCL-2 inhibitor venetoclax (ABT-199). As shown in Figure 1A, inhibiting the PI3K pathway at different signaling molecules enhanced the effect of JQ1, whereas combinations with the BCL-2 inhibitor venetoclax had negligible effect on JQ1-induced antiproliferation.
Figure 1. PI3K Pathway Inhibitors Enhance BET Inhibitor Activity in Lymphoma Cell Lines.

(A) High-throughput screen of JQ1-based combinations with PI3K pathway inhibitors, the BCL2 inhibitor venetoclax (ABT-199), and the JAK2 inhibitor ruxolitinib. The boxplot graph summarizes the results of the combinatorial drug screening analyzed with the Bliss independence model in 12 cell lines treated for 72 hr with the indicated drug combinations. The y axis indicates the ratio between observed and expected inhibition in a log scale. The expected activity can be written as
where A and B are the two drugs, and Inh INLINE denote inhibited and not inhibited, respectively. Values above the “0” line indicate enhanced anti-proliferative effects for JQ1-based combinations. See Data S1 and HTS statistical analysis in the Experimental Procedures for detailed information. See also Figures S1 and S2 for single agent activity data. Error bars represent minimum and maximum measured values.
(B) Boxplot graph summarizing the results of the combinatorial drug screening analyzed with the Bliss model according to the cell of origin. Error bars represent minimum and maximum measured values. Differences between groups (BL versus ABC and ABC versus GCB) were calculated with the Student’s t test. *p < 0.05, **p < 0.01.
(C) Representative examples of combination experiments of JQ1 plus BKM-120 in TMD8 and CA-46 cells. Combination responses are examined using an 8 × 8 viability matrix which was measured after 72 hr post treatment. Full data are shown in Data S1.
(D) High-throughput results were confirmed by independent experiments using the MTS assay. Cells were incubated with increasing concentrations of BET inhibitors (JQ1 or CPI-203) and the PI3K inhibitor BKM-120 (0.25, 0.5, 0.75, 1 μM), and cell viability was assessed after 72 hr. Error bars represent SEM of triplicate experiments. Combination index data are provided in Figure S2D
The most favorable interactions between JQ1 and PI3Ki were observed in BL- and ABC-derived cell lines (Figure 1B). Detailed, individual matrices of 2 representative cell lines treated with JQ1 plus BKM-120 are shown in Figure 1C (complete data are provided in Data S1). These results were validated by independent experiments using the MTS cell proliferation assay, with two different BETi (JQ1 and CPI-203) producing similar results (Figure 1D). Combinations of BET and PI3K inhibitors showed more favorable interactions in cell lines harboring mutations of upstream components of the B cell receptor (BCR), Toll-like receptor (TLR), and PI3K pathway (CD79B, MYD88, ID3, PTEN, and SGK1) (Figure S2D).
BET Inhibitors Induce GSK3β Feedback in Lymphoma
To better understand the molecular mechanisms underlying the favorable interactions between BET and PI3K inhibitors, we examined the regulatory effect of JQ1 on the transcription of genes involved in PI3K signaling and pathways, using a dedicated PCR pathway array platform (the gene list is available in the Supplemental Experimental Procedures). We found that JQ1 treatment (0.5 μM for 24 hr) upregulated the mRNA expression of several genes in the PI3K pathway, including GSK3B and upstream components of the pathway such as PIK3CA (PI3Kα) and PIK3R1 (PI3K-p85α) (Figures 2A and S3), which was associated with an increase in protein levels in multiple cell lines (Figures 2B and 2C). Similar effects were observed using JQ1 and CPI-203, indicating a class effect (Figure 2D).These changes were associated with increased levels of phospho-GSK3β S9 in multiple cell lines (predominantly ABC and BL), as indicated by Luminex multiplex assay studies (Figure 3A). Validation of these data by western blot assay demonstrated increased pGSK3β S9 levels for up to 48 hr following treatment with JQ1 in TMD8 cells (Figure 3B). These findings were confirmed in BL cell lines and similar effects were observed using JQ1 or CPI-203 (Figure 3C). Although BET inhibitors increased total GSK3β levels in these cell lines (in line with the gene expression changes described in Figure 2A), the increased GSK3β S9 phosphorylation levels associated with increased protein abundance of its downstream target β-catenin are consistent with a “net” inhibitory effect of BET inhibitors on the GSK3β activity (Figure 3C). We could not see correlations between pAKT and pGSK3β levels (Figure 3C) suggesting that different kinases downstream of PI3K could mediate the observed GSK3β-feedback inhibition induced by BET inhibitors (reviewed in Beurel et al., 2015). Nuclear cytoplasmic fractionation experiments further corroborated our findings, as increased abundance of nuclear β-catenin was confirmed following treatment with either JQ1 or CPI-203 in ABC-derived DLBCL cell lines (Figure 3D). In line with these data, we observed an increased production of the PI3K-dependent chemokines MIP-1α (CCL3) and MIP-1β (CCL4) after treatment with BET inhibitors (Figures 3E and 3F). These effects were more pronounced in ABC-derived (TMD8 and HBL-1) and BL cell lines (CA-46), compared to GCB-derived cell lines (SUDHL-6, SUDHL-4, and LY19) (Figures 3E and 3F) and were prevented by treatment with PI3K inhibitors (Figures 4A and 4B). Cell lines showing BETi-induced increase in pGSK3β S9 and/or MIP1α (CCL3) levels demonstrated more favorable interactions with PI3K inhibitors (Figure 4C). In order to confirm the importance of the induced GSK3β inhibitory feedback in determining the favorable interactions between BET and PI3K inhibitors, we generated inducible GSK3β short hairpin RNAs (shRNAs) in TMD8 cells using a tetracycline-dependent transactivation system, with the aim of creating a model of GSK3β inhibition that could not be reversed by the addition of PI3K inhibitors. GSK3β silencing did not have significant effects on cell proliferation and resulted in increased β-catenin protein abundance (Figure S4). In line with our hypothesis, a competitive proliferation assay demonstrated a significant proliferation advantage of GSK3β-depleted cells compared to scramble shRNA transduced cells after 72 hr incubation with BETi/PI3Ki combinations (Figures 4E and 4F). These data confirm that BETi-induced GSK3β feedback plays a major role in determining the favorable interactions between BET and PI3K inhibitors in DLBCL cells.
Figure 2. Effects of BET Inhibition on PI3K Pathway Gene Expression in DLBCL Cells.

(A) Heatmap from one representative experiment showing the effect of JQ1 (0.5 μM for 24 hr) on PI3K pathway gene expression in multiple DLBCL/BL cell lines, as determined by a PCR pathway-directed array including genes belonging to PI3K pathway. MYC, and top down and upregulated genes (at least ±1.5 average fold change in 9 cell lines) are shown in the heatmap. Fold change values are depicted in a colorimetric scale from blue (low) to red (high) with respect to DMSO (control). Full data are shown in Figure S3.
(B) Representative western blot analysis confirming the effects of JQ1 (0.5 μM for 24 hr) on PI3Kα, PI3K-p85α (PIK3R1) protein levels DLBCL and BL cell lines.
(C) Scatterplots showing relative pixel density values of PI3Kα calculated versus actin. p values were calculated with the Wilcoxon rank test. Western blots images shown in (B) were analyzed with the ImageJ software.
(D) Scatterplots showing relative pixel density values of PI3K-p85α calculated versus actin. p values were calculated with the Wilcoxon rank test. Western blots images shown in (B) were analyzed with the ImageJ software.
(E) Representative western blots showing the effects of JQ1 and CPI-203 treatment (0.5 μM for 24 and 48 hr) on PI3Kα protein levels in HBL-1 and TMD8 cells, indicating similar class effects of JQ1 and CPI-203. Numbers indicate normalized PI3Kα levels relative to DMSO, calculated versus actin, and analyzed with the ImageJ software.
Figure 3. Effects of BET Inhibition on PI3K-GSK3β Signaling and Chemokine Secretion in DLBCL Cells.

(A) Effect of JQ1 treatment (0.5 μM for 24 hr) on the phosphorylation level of PI3K pathway proteins including GSK3β (S9) in DLBCL and BL cell line panel, as determined by a luminex multiplex assay.
(B) Western blot confirming sustained effects of JQ1 on c-MYC, GSK3β (S9) and total GSK3β levels in TMD8 cells for up to 48 hr.
(C) Confirmatory western blots showing similar effects of JQ1 and CPI-203 (1 μM, 24 hr) on p-GSK3β S9, total GSK3β, and β-catenin levels in 2 BL cell lines (RAJI and CA-46). Numbers below β-catenin blots indicate fold-change of protein expression versus DMSO (normalized to the relative loading controls) evaluated by densitometry analysis using the ImageJ software.
(D) Nuclear-cytoplasmic fractionation experiments confirming increased nuclear β-catenin levels in ABC-derived DLBCL cell lines (TMD8, HBL-1) following treatment with BET inhibitors (JQ1 or CPI-203 0.5 μM for 24 hr). Vinculin and lamin B1 were used as loading controls for cytoplasmic and nuclear fractions, respectively. Numbers indicate fold-change of nuclear β-catenin protein expression versus DMSO (normalized to the relative loading controls), evaluated by densitometry analysis, using the ImageJ software. N, nuclear protein fractions; C, cytoplasmic protein fractions.
(E) Effect of JQ1 treatment (0.5 μM for 24 hr) on cytokine-chemokine levels as measured by a multiplex assay in 9 representative DLBCL cell lines of ABC and GCB origin (SUDHL-4, SUDHL-6, SUDHL-8, DB, BJAB, TMD8, HBL-1, and U2932), and 1 BL cell line (CA-46). As shown, JQ1 upregulated the PI3K-dependent chemokines MIP-1α and MIP1β mostly in ABC and BL cell lines. In some cell lines, cytokine levels were below detection. Results are shown as average fold change value of cytokine-chemokine concentration in cell culture supernatants (versus DMSO) of 3 independent experiments.
(F) Standard ELISA confirming significant MIP-1α upregulation in ABC-derived DLBCL cell lines (TMD8, HBL-1) and BL cell lines (CA-46) after treatment with JQ1 or CPI-203 0.5 μM for 24 hr. Error bars represent SEM of triplicate experiments. Differences between groups (JQ1 or CPI versus DMSO) were calculated with the Student’s t test. *p < 0.05, **p < 0.01. BD (below detection).
Figure 4. Effects of BET/PI3Ki Combinations on PI3K-GSK3 Signaling.

(A) Effect of JQ1, BKM-120, and the combination on PI3K pathway activation in DLBCL cells. TMD8 and HBL-1 cells were incubated with JQ1 0.5 μM, BKM-120 0.5 μM, and the combination for 24 hr, and the effects on p-GSK3β S9 (top) and on MIP1α (CCL3) chemokine production (bottom) were assessed with Luminex multiplex assays. Error bars represent SEM of triplicate experiments. Differences between groups were calculated with the Student’s t test. *p < 0.05, **p < 0.01.
(B) Representative western blot showing the effects of JQ1, CAL-101, BKM-120, and the combinations on β-catenin levels in TMD8 cells. Cells were incubated with JQ1 0.5 μM, CAL-101 1 μM, BKM-120 0.5 μM, and the combinations for 24 hr. Numbers indicate fold-change of β-catenin protein expression versus DMSO (normalized to actin levels), evaluated by densitometry analysis, using the ImageJ software.
(C) Boxplot graph summarizing the results of the combinatorial drug screening analyzed with the Bliss model according to on-treatment regulation of pGSK3β S9 phosphorylation by JQ1 (top) (as shown in the experiments represented in Figure 3A) and MIP1α (CCL3) production by JQ1 (bottom) (as shown in the experiments represented in Figures 3E and 3F). Error bars represent minimum and maximum measured values. Differences between groups were calculated with the Student’s t test. *p < 0.05, **p < 0.01.
(D) Experimental design of the GSK3β shRNA cell growth completion assay experiments. 1:1 mixtures of wild-type (WT, GFP—) cells and lentiviral shRNA-transduced (GFP+) cells (WT+scramble [SCR], WT+Sh#1, and WT+Sh#2) were plated (250,000 cells/mL), pretreated with doxycycline (1 μg/mL) for 72 hr, and then treated with combinations of BET inhibitors (JQ1 and CPI-203) plus BKM-102 (0.5 μM). After 72 hr, the proportion of GFP+ cells in each condition was evaluated by flow cytometry analysis.
(E) Western blot assay showing effective GSK3β silencing in TMD8 cells using 2 different inducible GSK3β shRNAs. Cells transduced with scramble (SCR), GSK3β Sh#1, or GSK3β Sh#2 were incubated with doxycycline (1 μg/mL) for 72 hr.
(F) Bar graphs showing increased proportion of GFP+ cells following GSK3β depletion in TMD8 cells treated with combinations of BET and PI3K inhibitors, indicating a relative proliferative advantage of GSK3β -depleted cells in the presence of combinations of BET and PI3K inhibitors. Error bars represent SEM of triplicate experiments. Differences between groups (WT versus GFP+ cells) were calculated with the Student’s t test. *p < 0.05, **p < 0.01. Extended data are shown in Figure S4.
MYC Depletion Induces GSK3β Feedback in Lymphoma
To determine the role of MYC in the observed JQ1-induced PI3K/GSK3 feedback, we examined the effect of MYC silencing on PI3K pathway gene expression in the P-4936 B cell line, which carries a conditional, tetracycline-regulated (TET-OFF) MYC promoter (Pajic et al., 2000; Zeller et al., 2006). MYC silencing with doxycycline resulted in the time-dependent upregulation of several PI3K pathway genes, including PIK3CA, PIK3CD, and GSK3β (Figures 5A, 5B, and S5A). These changes were associated with increased levels of GSK3β and phospho-GSK3β S9 and with increased β-catenin protein abundance indicative of “net” inhibition of GSK3β activity, in addition to an increase in MIP1α levels in cell culture supernatants (Figures 5C, 5D, and S5B), therefore mirroring the effects of pharmacologic BET inhibition. Similarly, MYC silencing using small interfering RNA (siRNA) upregulated PI3K pathway gene expression in DLBCL cells (Figures 5E and 5F), and enhanced the antiproliferative effects of the PI3K inhibitor BKM-120 (Figure 5G). In line with these findings, a deep c-MYC downregulation correlated with a more favorable combinatory activity of JQ1 with PI3K inhibitors in DLBCL cell lines (Figure 5H). In summary, our data demonstrate that feedback upregulation of the PI3K pathway with consequent GSK3β feedback is a MYC-dependent event and provide a mechanistic rationale for combining BET inhibitors with PI3K inhibitors in DLBCL.
Figure 5. Effects of MYC Depletion on PI3K Pathway Gene Expression and Activation in Lymphoma Cells.

(A) Effects of MYC silencing (MYC-OFF) for 24 hr on PI3K pathway-related genes, as determined by a PCR pathway-directed array in P-4936 cells. Fold change values obtained in 2 independent experiments are represented in a colorimetric scale from blue (low) to red (high). See also Figure S5A.
(B) Representative time course experiment showing changes in PIK3CA, PIK3CD, PI3KR1, PDPK1, and GSK3B gene expression levels in relation to MYC mRNA levels in P-4396 cells treated with doxycycline (1 μg/mL), assessed by PI3K pathway PCR-directed array.
(C) Western blot assay showing increased GSK3β protein abundance, enhanced p-GSK3β S9 phosphorylation, and increased β-catenin levels in P-4936 cells after 24 hr of MYC depletion (MYC-OFF). See also Figure S5B.
(D) Bar graph showing the effect of 24-hr MYC silencing (MYC-OFF) on MIP1α (CCL3) concentrations in cell culture supernatants of P-4936 cells, as detected by standard ELISA. Error bars represent SEM of triplicate experiments. Differences between groups were calculated with the Student’s t test. *p < 0.05, **p < 0.01.
(E) Western blot showing the effects of the 2 different MYC siRNAs on MYC protein levels after 24 hr of incubation in TMD8 cells.
(F) Effects of MYC silencing by RNA interference (siRNA#1) on PI3K pathway gene expression in TMD8 cells. Fold change values obtained in 3 independent experiments with respect to scramble (SCR) siRNA (control) are shown.
(G) MYC silencing (with 2 different siRNAs) enhances the antiproliferative effect of the PI3K inhibitor BKM-120 (0.1, 0.25, 0.5 μM) in TMD8 cells at 48 hr. Viability data were normalized to the effect of MYC siRNA alone. Error bars represent SEM of triplicate experiments.
(H) Boxplot graph summarizing the results of the combinatorial drug screening analyzed with the Bliss model according to on-treatment regulation of MYC protein abundance at 24 hr by JQ1, as shown in the experiments represented in Figure S2B. The cut-off used in this plot (35% downregulation) corresponds to the median value of MYC downregulation calculated across all cell lines at 24 hr (Figure S2B). Error bars represent minimum and maximum measured values. Differences between groups were calculated with the Student’s t test. *p < 0.05, **p < 0.01.
BET Inhibitors Regulate Protein Ubiquitination
Stable isotope labeling of amino acids in cell culture (SILAC)-based quantitative mass spectrometry was recently used to clarify the mechanism of the action of drugs that have complex biologic functions, such as lenalidomide (Krönke et al., 2014). Here, we applied SILAC-based quantitative mass spectrometry to two representative DLBCL cell lines treated with 0.5 μM JQ1 for 24 hr (HBL-1 of ABC origin and SUDHL-6 of GCB origin) (Figure 6A; Data S2). Pathway analysis using Ingenuity software showed that cell cycle, DNA damage response, nucleotide synthesis, and protein ubiquitination were among the top dysregulated cellular processes in both cell lines (Figures 6A and S6A). Of the 4,041 proteins that were detectable in both cell lines, only 10 proteins were commonly increased or decreased after treatment with JQ1 (Figures 6A and 6B). Three of top 5 downregulated proteins (UBE2C, UBE2T, and CDC20) belonged to the protein ubiquitination machinery (Figure 6B). These proteins are known to regulate the anaphase promoting complex/cyclosome (APC/C) ubiquitin E3 ligase (UBE2C and CDC20) and DNA damage response (UBE2T) (Rape and Kirschner, 2004; Buschhorn and Peters, 2006; Machida et al., 2006; Ueki et al., 2009; Williamson et al., 2009; Zhang et al., 2014). The effect of BET inhibitors (JQ1 and CPI-203) on UBE2C and UBE2T protein abundance was confirmed using western blotting in ABC (HBL-1 and TMD8) and GCB (SUDHL-6 and SUDHL-8) cell lines (Figure 6C).
Figure 6. BETi-Mediated Regulation of the Ubiquitin System.

(A) Scatterplot of quantitative proteomics data showing protein changes in HBL-1 (x axis) and SUDHL6-cells (y axis) in a log2 scale. Average log2 fold change of one forward and reverse experiment in HBL-1 and SUDHL-6 cells were used to generate the scatterplot. See also Figure S6A and Data S2.
(B) Venn diagram showing overlap of significantly up and downregulated proteins in HBL-1 (red) and SUDHL-6 cells (blue), and bar graph showing fold change in the top 10 commonly up- and downregulated proteins in HBL-1 (red) and SUDHL-6 cells (blue).
(C) Representative western blot showing marked decrease in UBE2C and UBE2T protein levels in 4 representative cell lines after 24 and 48 hr of incubation with 0.5 μM JQ1.
(D) Effects of JQ1 (0.5 μM for 24 hr) on ubiquitination pathways gene expression in HBL-1 and SUDHL-6 cells, as determined by PCR pathway-directed array. Genes commonly regulated in HBL-1 and SUDHL-6 cells by at least ±1.5-fold change are shown. Fold change values of JQ1-treated cells versus DMSO obtained in 2 independent experiments are represented in a colorimetric scale from blue (low) to red (high). See also Figure S6B.
(E) Significantly regulated ubiquitination pathway genes (at least ±1.5-fold change) in P-4936 cells treated with doxycycline (1 μg/mL) (MYC-OFF) for 24 hr, as detected by PCR pathway-directed array. Fold change values obtained in 2 independent experiments are represented in a colorimetric scale from blue (low) to red (high).
(F) Representative western blot showing the effects of UBE2C, UBE2T, and combined UBE2C plus UBE2T silencing on GSK3β/β-catenin axis in lymphoma cells (2 ABC-derived DLBCL cell lines [TMD8 and HBL-1]) at 48 hr.
(G) Bar graphs showing the antiproliferative effects of Scramble, UBE2C, UBE2T, and combined UBE2C/T siRNAs plus or minus BKM-120 in HBL-1 cells. Combined UBE2C/T silencing significantly enhanced the antiproliferative effect of the PI3K inhibitor BKM-120 (0.5 μM) at 48 hr. Error bars represent SEM of triplicate experiments. Differences between groups were calculated with the Student’s t test. *p < 0.05, **p < 0.01.
To better understand the mechanisms and extent of regulation of protein ubiquitination machinery by BET inhibitors, we investigated the effect of JQ1 on gene expression levels of 84 genes spanning E1, E2, and E3 enzymes (Figure 6D; gene list available in the Supplemental Experimental Procedures). JQ1 treatment suppressed the expression of a wide range of E2-conjugating and E3-ligase enzymes (Figures 6D and S6B). Again, using the P-4936 human B cell lymphoma cell line, we found that doxycycline-induced MYC silencing resulted in downregulation of key E2 and E3 genes, including UBE2C, UBE2T, SKP2, and UBE2S (Figure 6E). The functional consequences of these findings were further investigated using gene silencing experiments. RNA interference for UBE2C and/or UBE2T in 2 ABC DLBCL cell lines resulted in increased GSK3β, phospho-GSK3β S9, and β-catenin protein levels (Figure 6F). Interestingly the effects of combined UBE2C/UBE2T silencing on GSK3β S9 phosphorylation were more pronounced compared to the effects of the single knockdowns, suggesting non-redundant functions of UBE2C and UBE2T in PI3K pathway regulation. In line with this data, combined UBE2C and UBE2T depletion enhanced the anti-lymphoma activity of the PI3K inhibitor BKM-120 in DLBCL cells, thus recapitulating the effects of BET inhibitors (Figure 6G).
Taken together, these data indicate that BET inhibitors may control the levels of GSK3β phosphorylation and β-catenin protein abundance by regulating ubiquitination pathways, and downregulation of E2 conjugating enzymes could be a relevant mechanism underlying the favorable interactions between BET and PI3K inhibitors.
BET Inhibition Increases DLBCL Vulnerability to PI3K Inhibition In Vivo
To determine whether the observed BET inhibitors-induced feedbacks are maintained in vivo, we examined the effect of CPI-203 on selected targets using a human DLBCL xenograft model. CPI-203 treatment (5 mg/kg given intraperitoneally [i.p.] twice daily) resulted in a modest inhibition of tumor growth (Figure 7A) without causing a significant weight loss (Figure S7A). At the molecular level, CPI-203 therapy resulted in downregulation of c-MYC, UBE2C, and UBE2T protein levels and in a trend toward increased GSK3β S9 phosphorylation (with 6 of 8 CPI-203-treated mice displaying relatively high p-GSK3β S9 levels versus only 3 of 8 mice in the control group) (Figures 7B–7D). In line with our findings, BET inhibition increased vulnerability to the PI3K inhibitor BKM-120 in vivo, with no significant weight loss (Figures 7E and S7B).
Figure 7. Antiproliferative Activity of BETi-PI3Ki Combinations In Vivo.

(A) Human TMD8 cells were established as subcutaneous (s.c.) tumors in NSG mice and treated by i.p. injection with vehicle (n = 8) or the BET inhibitor CPI-203 (5 μg/kg BID) (n = 8) for 18 days. Tumor volume was measured 3 times per week. Error bars represent SEM. Differences between groups were calculated with the Student’s t test. *p < 0.05, **p < 0.01.
(B) Western blot showing the in vivo effects of CPI-203 therapy on selected targets (MYC, UBE2C, and UBE2T).
(C) Scatterplots summarizing changes in expression levels of c-MYC, UBE2C, and UBE2T (as in B), analyzed in aggregate, and expressed as ratios of pixel densities between actin and protein of interest. Densitometry analysis was performed by using the ImageJ software. Differences between groups were calculated with the Wilcoxon rank test.*p < 0.05, **p < 0.01.
(D) In vivo effects of CPI-203 on p-GSK3β S9 phosphorylation in TMD8 xenografts. Proteins were extracted from tumor tissues and analyzed by Luminex multiplex assay. The expression levels of GSK3β S9 in mice bearing TMD8 xenografts treated with CPI-203 compared to vehicle were analyzed by Luminex multiplex assay and normalized to tubulin expression levels in each mouse.
(E) Combination experiment of CPI-203 and BKM-120 in TMD8 mouse xenografts. NSG mice were treated with vehicle (n = 8), the BET inhibitor CPI-203 (5 μg/kg twice daily i.p.) (n = 8), the PI3K inhibitor BKM-120 (15 μg/Kg/daily by oral gavage), and the combination (n = 8). Error bars represent SEM. Differences between groups were calculated with the Student’s t test (CPI-203 versus combination). *p < 0.05, **p < 0.01. See also Figure S7.
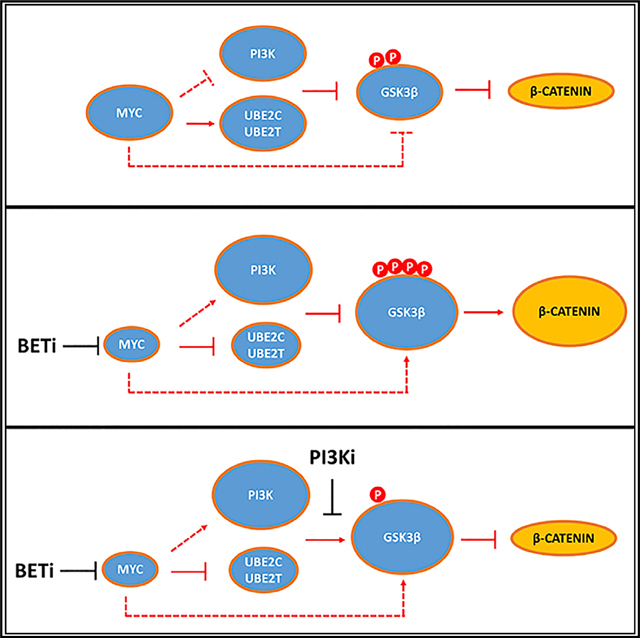
(F) Model of interaction between BET inhibitors, the PI3K/GSK3β axis and ubiquitination pathways. Upon BET inhibition, MYC downregulation triggers negative feedbacks loops involving multiple PI3K pathway nodes, resulting in increased levels of phospho-GSK3β S9 and β-catenin signaling. Transcriptional down-regulation of the E2-conjugating enzymes UBE2C and UBE2T results in increased GSK3B phosphorylation, contributing to negative feedback regulation of the PI3K pathway.
DISCUSSION
Our study provides insights on the complex biologic activity of BET inhibitors. While previous studies reported several mechanisms of anti-tumor activity of BET inhibitors in lymphoma (Chapuy et al., 2013; Ceribelli et al., 2014), our study describes prosurvival feedback mechanisms involving GSK3β. BET inhibitors downregulation of MYC leads to upregulation of PI3K pathway components and GSK3β inhibition. These molecular events were regulated by transcriptional and post-translational mechanisms (Figure 7F). Downregulation of MYC by BET inhibitors or genetic silencing of MYC resulted in the upregulation of several genes involved in regulating the PI3K pathway. This effect was associated with increased GSK3β S9 phosphorylation and beta-catenin protein abundance and with increased production of the PI3K-dependent chemokines MIP-1α and MIP-1β (Takahashi et al., 2015). Notably JQ1-induced inhibition of GSK3β and MIP1α increased production correlated with more favorable interactions between BET and PI3K inhibitors, confirming the functional relevance of these feedbacks and providing potential biomarkers for future clinical trials. These observations provided a mechanistic explanation for the favorable in vitro anti-lymphoma activity that we observed with the combination of BET inhibitors and several PI3K pathway inhibitors. Recently, small molecule inhibitors of the phosphatidylinositol 3-kinase pathway have been described to be active in DLBCL with different mechanisms according to the cell of origin (Erdmann et al., 2017; Paul et al., 2017). Our data also show differential efficacy of these combinations according to the cell of origin, with PI3Ki-based combinations being more active in the BL- and ABC-derived cell lines. Of note, recent data highlight the crucial role of BCR-dependent GSK3β inhibitory phosphorylation in maintaining fitness of MYC-driven lymphoma (Varano et al., 2017). Our observations of increased efficacy of PI3Ki-based combinations in ABC- and BL-derived cell lines harboring upstream BCR signaling mutations (CD79B, ID3, and GNA13) are in line with this model, as BETi-induced pGSK3β S9 accumulation could increase the vulnerability to PI3K inhibition in those cell lines relying on BCR-dependent GSK3β inhibition for survival. In line with this idea, shRNA-induced GSK3β depletion (that cannot be reversed by PI3K inhibitors) significantly decreased the efficacy of BET/PI3Ki combinations. Taken together, these data are consistent with a model where BET inhibition induces a GSK3β inhibitory feedback resulting in increased β-catenin signaling. At the same time, BET inhibitors, while inhibiting GSK3β activity, also increase GSK3β total protein levels. When a PI3K inhibitor is added, it reverses the GSK3β feedback unleashing a higher quantity of active GSK3β, thus resulting in enhanced antiproliferative effects. Additionally, a different mechanistic rationale for combining BETi with PI3Ki was recently reported in solid tumor preclinical models, suggesting that these mechanisms may be cell-type-specific (Stratikopoulos et al., 2015).
Previous studies focused on the transcriptional regulation by BET inhibitors of key oncogenes, such as MYC. Our quantitative proteomics experiments demonstrated that the biologic activity of BET inhibitors is also mediated through complex post-translational mechanisms involving the regulation of E2 and E3 enzymes leading to modulation of intracellular signaling pathways and increased phosphorylation of target proteins, such as GSK3β. Whether this is a cell-type-specific event is yet to be determined. We demonstrated that depletion of E2 enzymes induced by BET inhibition could contribute to the observed GSK3β feedback and to the increased β-catenin levels, and according to these observations, combined depletion of UBE2C and UBE2T enhanced the antiproliferative activity of PI3K inhibitors. However, Hu et al. (2016) described an inhibitory effect of UBE2C knockdown on PI3K activation in different tumor types, which could imply that E2 regulation of PI3K signaling is cell-type-specific. The human genome encodes 38 E2 ligase and more than 600 E3 ubiquitin ligases (Liu et al., 2015): lineage-dependent expression level of these enzymes may lead to lymphoid-restricted substrate specificity and ubiquitination mechanisms, which needs to be further explored.
In conclusion, our study described a feedback mechanism induced by BET inhibitors increasing vulnerability to PI3K inhibitors and established a framework for the development of combinatorial strategies to enhance BET inhibitors activity in DLBCL.
EXPERIMENTAL PROCEDURES
Cell Lines and Reagents
The human DLBCL-derived cell lines SUDHL-4, SUDHL-6, OCI-LY-19, and U-2932 were obtained from the DSMZ-German Collection of Microorganisms and Cell Cultures, Department of Human and Animal Cell Cultures (Braunsch-weig, Germany). DB, SUDHL-8, and SUDHL-10 and the BL cell lines RAJI and CA-46 were obtained from ATCC (American Type Culture Collection). The DLBCL-derived cell lines (HBL-1, TMD8, and BJAB) were provided by Dr. R.E. Davis (MD Anderson Cancer Center, Houston, TX). P-4936 cells (Pajic et al., 2000; Zeller et al., 2006) were provided by Dr. J. Zhang (Thompson lab, Memorial Sloan Kettering Cancer Center, New York, NY). Cell lines were fingerprinted at the Integrated Genomic Operation Core, (Memorial Sloan Kettering Cancer Center, New York, NY). Cell lines were cultured in RPMI 1640 medium supplemented with 10%–20% heat-inactivated fetal bovine serum (Hyclone, GE Healthcare Life Sciences [cat. #SH30396.03]), 1% L-glutamine, and penicillin-streptomycin in a humid environment of 5% CO2 at 37°C. Mutations were annotated according to Cancer Cell Line Encyclopedia (https://www.broadinstitute.org/ccle/home) and Davis et al. (2010), Pasqualucci et al. (2011), Ngo et al. (2011), Fontan et al. (2012), and Zhang et al. (2013).
JQ1 was purchased from BPS Biosciences (San Diego, CA). The following compounds were purchased from Selleckchem (Houston, TX): CAL-101, BKM-120, IPI-145, Everolimus, MK-2206, ABT-199, and Ruxolitinib. Doxycycline was purchased from SIGMA. CPI-203 was purchased from Adooq Biosciences (Irvine, CA). Detailed information on western blot antibodies is provided in the Supplemental Information.
High-Throughput Screening Experiments
Combination Studies
For the synergy assessment studies, a compound matrix was prepared in which compound 1 at 10 μM was titrated against compound 2 at 2.5 μM in a doubling dilution series and transferred into a 1,536-well microtiter assay plate. Internal controls for each assay plate were dispensed as previously described. To start the assay, cells were seeded at 1,000 cells per well in 8 μL of complete RPMI medium and incubated for 2 days followed by 1 μL AB for an additional day at 37°C. Plates were imaged on the LEADseeker Multimodality Imaging System (GE Healthcare, Piscataway, NJ) and resulting files were deconvoluted to obtain fluorescence values associated with each drug. Detailed information on single agent studies is available in the Supplemental Information.
HTS Statistical Analysis
To evaluate whether a drug combination shows synergy, we compared the observed activity of the combination at that level to the expected activity under Bliss independence model. By treating percent inhibition as a probability and using the product rule for the probability of independent events, the expected activity can be written as
where A and B are the two drugs, andInh INLINE denote inhibited and not inhibited, respectively (Feller, 1971; Tallarida, 2001). Detailed information is available in the Supplemental Information.
In Vitro Proliferation Assay
Cells were seeded in 96-well plates at 25,000 cells/100 μL/well with either vehicle (DMSO 0.1%) or increasing concentrations of drugs for 24, 48, and 72 hr. Cell viability was assessed by adding MTS reagent (Promega) to the culture medium at 1:5 dilution, according to manufacturer’s instructions.
Luminex Multiplex Assays
Samples were run using dedicated kits (Millipore) on a Luminex MAGPIX machine. The effects of JQ1 on MYC levels were assessed with a Luminex multiplex assay (#48–617MAG, Millipore) according to the manufacturer’s instructions. To assess the effects of JQ1 on phosphorylation levels of PI3K pathway components (PTEN S380, AKT S473, GSK3a S21, GSK3β S9, TSC2 S939, mTOR S2448, and P70S6K T412), protein lysates were examined with a Luminex multiplex assay (#48–611MAG Millipore), according to the manufacturer’s instructions.
Each sample was run in triplicate, using 20 μg of total protein per sample, using the Luminex MAGPIX machine. Results were normalized to beta-tubulin expression levels, and beta-tubulin magnetic beads, and antibody was purchased from Millipore (#46–713MAG).
Mass Spectrometry Studies
Cell Culture, Lysis, and In Situ Digestion
Cells were grown as suspension cultures in RPMI media supplemented with 10% FBS and penicillin and streptomycin either unlabeled L-arginine (Arg0) and L-lysine (Lys0) at 50 μg/L or equimolar amounts of the isotopic variants [U-13C6]-L-arginine (Arg10) and [U-13C6]-L-lysine HCl L-lysine (Lys6) (Cambridge Isotope Laboratories). After five cell doublings in suspension flask, cells were >99% labeled with the isotopes. Cells were collected, lysed in RIPA buffer, quantitated for protein BCA, mixed at a 1:1 ratio, separated by SDS/PAGE, and stained with Simply Blue (Life Technologies), and 15 gel sections were excised with in situ trypsin digestion of polypeptides in each gel slice that was performed as previously described (Shevchenko et al., 2006). Detailed methods are available in the Supplemental Information.
Maxquant
All tandem mass spectrometry (MS/MS) samples were analyzed using MaxQuant (Max Planck Institute of Biochemistry, Martinsried, Germany; version 1.3.0.3) and Scaffold Q+S (Proteomesoftware, Portland, OR, version 4.4.1). Detailed description is available in the Supplemental Information.
Mass Spectrometry Data Analysis
5,444 proteins were initially identified. Proteins with a missing value in one or both of the 2 cell lines were removed, as were proteins where one replicate fold change value was ≥ 10 times the other. 4,041 proteins were then evaluable for the final analysis (see Data S2). Only proteins changing by at least 1.5-fold change in both forward and reverse experiments were considered as significantly deregulated. Pathway analysis was performed with the Ingenuity software (IPA) (QIAGEN). Additional details on data analysis are provided in the Supplemental Information.
Xenograft Studies
NSG mice (Jackson Laboratory) were used for in vivo studies and were cared for in accordance with guidelines approved by the Memorial Sloan Kettering Cancer Center Institutional Animal Care and Use Committee and Research Animal Resource Center, and in line with the ARRIVE guidelines (Kilkenny et al., 2010). Detailed descriptions of xenograft studies are provided in the Supplemental Information.
Detailed descriptions of pathway arrays and qPCR, siRNA, shRNA, western blot methods, nuclear-cytoplasmic fractionation, cytokine and chemokine detection, flow cytometry, mass spectrometry studies, and HTS statistics are available in the Supplemental Information.
This study was approved by the institutional review board.
Statistical Analysis
Procedures to determine the effects of certain conditions on cell proliferation and apoptosis were performed in 3 independent experiments. The two-tailed Student’s t test and Wilcoxon rank test were used to estimate the statistical significance of differences between results from the 3 experiments. Significance was set at p < 0.05. The PRISM software was used for the statistical analyses.
Combination Index Calculation
Replicates Were Averaged for the Analysis.
The selected combinations were tested for nature of effect using the Chou-Talalay (CT) method (Chou, 2010), which enables quantification of drug outcomes as a combination index (CI); CI = 1 shows additivity, CI < 1 shows synergism, and CI > 1 shows antagonism.
Data Availability
Proteomics data generated or analyzed during this study are included in this published article (and its supplemental information files). The raw proteomics data reported in this paper are also publicly available at the Peptide Atlas website (https://db.systemsbiology.net/sbeams/cgi/PeptideAtlas/PASS_View?identifier=PASS01217). Original uncropped western blots are available at the Mendeley database: https://doi.org/10.17632/px94fdcpvz.1.
Supplementary Material
Highlights.
BET inhibitors enhance lymphoma cell vulnerability to PI3K inhibitors
BET inhibitors induce GSK3β feedback in a MYC-dependent manner
Downregulation of UBE2C and UBE2T, induced by BET inhibitors, enhances GSK3β feedback
BET inhibitors decrease MYC, UBE2C, and UBE2T and increase GSK3β S9 levels in vivo
ACKNOWLEDGMENTS
This work was supported in part by the MSK SPORE in lymphoma (P50 CA192937–01A1 to A.Y. and V.E.S.), the Vogelstein Fund for Lymphoma Research (to A.Y.), and the Memorial Sloan Kettering Cancer Center Core (P30 CA008748).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures, seven figures, and two data files and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.07.055.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Beurel E, Michalek SM, and Jope RS (2010). Innate and adaptive immune responses regulated by glycogen synthase kinase-3 (GSK3). Trends Immunol. 31, 24–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beurel E, Grieco SF, and Jope RS (2015). Glycogen synthase kinase-3 (GSK3): regulation, actions, and diseases. Pharmacol. Ther. 148, 114–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boi M, Gaudio E, Bonetti P, Kwee I, Bernasconi E, Tarantelli C, Rinaldi A, Testoni M, Cascione L, Ponzoni M, et al. (2015). The BET bromodomain inhibitor OTX015 affects pathogenetic pathways in preclinical B-cell tumor models and synergizes with targeted drugs. Clin. Cancer Res. 21, 1628–1638. [DOI] [PubMed] [Google Scholar]
- Buschhorn BA, and Peters JM (2006). How APC/C orders destruction. Nat. Cell Biol. 8, 209–211. [DOI] [PubMed] [Google Scholar]
- Ceribelli M, Kelly PN, Shaffer AL, Wright GW, Xiao W, Yang Y, Mathews Griner LA, Guha R, Shinn P, Keller JM, et al. (2014). Blockade of oncogenic IκB kinase activity in diffuse large B-cell lymphoma by bromodomain and extraterminal domain protein inhibitors. Proc. Natl. Acad. Sci. USA 111, 11365–11370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapuy B, McKeown MR, Lin CY, Monti S, Roemer MG, Qi J, Rahl PB, Sun HH, Yeda KT, Doench JG, et al. (2013). Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer Cell 24, 777–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou TC (2010). Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 70, 440–446. [DOI] [PubMed] [Google Scholar]
- Dang CV (2013). MYC, metabolism, cell growth, and tumorigenesis. Cold Spring Harb. Perspect. Med. 3, a014217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV (2015). Web of the extended Myc network captures metabolism for tumorigenesis. Cancer Cell 27, 160–162. [DOI] [PubMed] [Google Scholar]
- Davis RE, Ngo VN, Lenz G, Tolar P, Young RM, Romesser PB, Kohlhammer H, Lamy L, Zhao H, Yang Y, et al. (2010). Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature 463, 88–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, et al. (2011). BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 146, 904–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdmann T, Klener P, Lynch JT, Grau M, Vočková P, Molinsky J, Tuskova D, Hudson K, Polanska UM, Grondine M, et al. (2017). Sensitivity to PI3K and AKT inhibitors is mediated by divergent molecular mechanisms in subtypes of DLBCL. Blood 130, 310–322. [DOI] [PubMed] [Google Scholar]
- Feller W (1971). An Introduction to Probability Theory and Its Applications (New York, NY: John Wiley&Sons; ). [Google Scholar]
- Fontan L, Yang C, Kabaleeswaran V, Volpon L, Osborne MJ, Beltran E, Garcia M, Cerchietti L, Shaknovich R, Yang SN, et al. (2012). MALT1 small molecule inhibitors specifically suppress ABC-DLBCL in vitro and in vivo. Cancer Cell 22, 812–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green TM, Young KH, Visco C, Xu-Monette ZY, Orazi A, Go RS, Nielsen O, Gadeberg OV, Mourits-Andersen T, Frederiksen M, et al. (2012). Immunohistochemical double-hit score is a strong predictor of outcome in patients with diffuse large B-cell lymphoma treated with rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone. J. Clin. Oncol. 30, 3460–3467. [DOI] [PubMed] [Google Scholar]
- Hoffman B, and Liebermann DA (2008). Apoptotic signaling by c-MYC. Oncogene 27, 6462–6472. [DOI] [PubMed] [Google Scholar]
- Hu W, Xiao L, Cao C, Hua S, and Wu D (2016). UBE2T promotes nasopharyngeal carcinoma cell proliferation, invasion, and metastasis by activating the AKT/GSK3β/β-catenin pathway. Oncotarget 7, 15161–15172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson NA, Slack GW, Savage KJ, Connors JM, Ben-Neriah S, Rogic S, Scott DW, Tan KL, Steidl C, Sehn LH, et al. (2012). Concurrent expression of MYC and BCL2 in diffuse large B-cell lymphoma treated with rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone. J. Clin. Oncol. 30, 3452–3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne WJ, Cuthill IC, Emerson M, and Altman DG (2010). Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol. 8, e1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krönke J, Udeshi ND, Narla A, Grauman P, Hurst SN, McConkey M, Svinkina T, Heckl D, Comer E, Li X, et al. (2014). Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 343, 301–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Shaik S, Dai X, Wu Q, Zhou X, Wang Z, and Wei W (2015). Targeting the ubiquitin pathway for cancer treatment. Biochim. Biophys. Acta 1855, 50–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovén J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, and Young RA (2013). Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 153, 320–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machida YJ, Machida Y, Chen Y, Gurtan AM, Kupfer GM, D’Andrea AD, and Dutta A (2006). UBE2T is the E2 in the Fanconi anemia pathway and undergoes negative autoregulation. Mol. Cell 23, 589–596. [DOI] [PubMed] [Google Scholar]
- McCubrey JA, Steelman LS, Bertrand FE, Davis NM, Abrams SL, Montalto G, D’Assoro AB, Libra M, Nicoletti F, Maestro R, et al. (2014). Multifaceted roles of GSK-3 and Wnt/β-catenin in hematopoiesis and leukemogenesis: opportunities for therapeutic intervention. Leukemia 28, 15–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertz JA, Conery AR, Bryant BM, Sandy P, Balasubramanian S, Mele DA, Bergeron L, and Sims RJ 3rd. (2011). Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc. Natl. Acad. Sci. USA 108, 16669–16674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo VN, Young RM, Schmitz R, Jhavar S, Xiao W, Lim KH, Kohlhammer H, Xu W, Yang Y, Zhao H, et al. (2011). Oncogenically active MYD88 mutations in human lymphoma. Nature 470, 115–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajic A, Spitkovsky D, Christoph B, Kempkes B, Schuhmacher M, Staege MS, Brielmeier M, Ellwart J, Kohlhuber F, Bornkamm GW, et al. (2000). Cell cycle activation by c-myc in a Burkitt lymphoma model cell line. Int. J. Cancer 87, 787–793. [DOI] [PubMed] [Google Scholar]
- Pasqualucci L, Dominguez-Sola D, Chiarenza A, Fabbri G, Grunn A, Trifonov V, Kasper LH, Lerach S, Tang H, Ma J, et al. (2011). Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature 471, 189–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul J, Soujon M, Wengner AM, Zitzmann-Kolbe S, Sturz A, Haike K, Keng Magdalene KH, Tan SH, Lange M, Tan SY, et al. (2017). Simultaneous inhibition of PI3Kd and PI3Kα induces ABC-DLBCL regression by blocking BCR-dependent and -independent activation of NF-κB and AKT. Cancer Cell 31, 64–78. [DOI] [PubMed] [Google Scholar]
- Pourdehnad M, Truitt ML, Siddiqi IN, Ducker GS, Shokat KM, and Ruggero D (2013). Myc and mTOR converge on a common node in protein synthesis control that confers synthetic lethality in Myc-driven cancers. Proc. Natl. Acad. Sci. USA 110, 11988–11993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rape M, and Kirschner MW (2004). Autonomous regulation of the anaphase-promoting complex couples mitosis to S-phase entry. Nature 432, 588–595. [DOI] [PubMed] [Google Scholar]
- Sander S, Calado DP, Srinivasan L, Köchert K, Zhang B, Rosolowski M, Rodig SJ, Holzmann K, Stilgenbauer S, Siebert R, et al. (2012). Synergy between PI3K signaling and MYC in Burkitt lymphomagenesis. Cancer Cell 22, 167–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz R, Young RM, Ceribelli M, Jhavar S, Xiao W, Zhang M, Wright G, Shaffer AL, Hodson DJ, Buras E, et al. (2012). Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature 490, 116–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevchenko A, Tomas H, Havlis J, Olsen JV, and Mann M (2006). In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 1, 2856–2860. [DOI] [PubMed] [Google Scholar]
- Strasser A, Harris AW, Bath ML, and Cory S (1990). Novel primitive lymphoid tumours induced in transgenic mice by cooperation between myc and bcl-2. Nature 348, 331–333. [DOI] [PubMed] [Google Scholar]
- Stratikopoulos EE, Dendy M, Szabolcs M, Khaykin AJ, Lefebvre C, Zhou MM, and Parsons R (2015). Kinase and BET inhibitors together clamp inhibition of PI3K signaling and overcome resistance to therapy. Cancer Cell 27, 837–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Sivina M, Hoellenriegel J, Oki Y, Hagemeister FB, Fayad L, Romaguera JE, Fowler N, Fanale MA, Kwak LW, et al. (2015). CCL3 and CCL4 are biomarkers for B cell receptor pathway activation and prognostic serum markers in diffuse large B cell lymphoma. Br. J. Haematol. 171, 726–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallarida RJ (2001). Drug synergism: its detection and applications. J. Pharmacol. Exp. Ther. 298, 865–872. [PubMed] [Google Scholar]
- Trabucco SE, Gerstein RM, Evens AM, Bradner JE, Shultz LD, Greiner DL, and Zhang H (2015). Inhibition of bromodomain proteins for the treatment of human diffuse large B-cell lymphoma. Clin. Cancer Res. 21, 113–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueki T, Park JH, Nishidate T, Kijima K, Hirata K, Nakamura Y, and Katagiri T (2009). Ubiquitination and downregulation of BRCA1 by ubiquitin-conjugating enzyme E2T overexpression in human breast cancer cells. Cancer Res. 69, 8752–8760. [DOI] [PubMed] [Google Scholar]
- Varano G, Raffel S, Sormani M, Zanardi F, Lonardi S, Zasada C, Perucho L, Petrocelli V, Haake A, Lee AK, et al. (2017). The B-cell receptor controls fitness of MYC-driven lymphoma cells via GSK3β inhibition. Nature 546, 302–306. [DOI] [PubMed] [Google Scholar]
- Walz A, Ugolkov A, Chandra S, Kozikowski A, Carneiro BA, O’Halloran TV, Giles FJ, Billadeau DD, and Mazar AP (2017). Molecular pathways: revisiting glycogen synthase kinase-3β as a target for the treatment of cancer. Clin. Cancer Res. 23, 1891–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendel HG, De Stanchina E, Fridman JS, Malina A, Ray S, Kogan S, Cordon-Cardo C, Pelletier J, and Lowe SW (2004). Survival signalling by Akt and eIF4E in oncogenesis and cancer therapy. Nature 428, 332–337. [DOI] [PubMed] [Google Scholar]
- Williamson A, Wickliffe KE, Mellone BG, Song L, Karpen GH, and Rape M (2009). Identification of a physiological E2 module for the human anaphase-promoting complex. Proc. Natl. Acad. Sci. USA 106, 18213–18218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeller KI, Zhao X, Lee CW, Chiu KP, Yao F, Yustein JT, Ooi HS, Or-lov YL, Shahab A, Yong HC, et al. (2006). Global mapping of c-Myc binding sites and target gene networks in human B cells. Proc. Natl. Acad. Sci. USA 103, 17834–17839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Wan L, Dai X, Sun Y, and Wei W (2014). Functional characterization of Anaphase Promoting Complex/Cyclosome (APC/C) E3 ubiquitin ligases in tumorigenesis. Biochim. Biophys. Acta 1845, 277–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Grubor V, Love CL, Banerjee A, Richards KL, Mieczkowski PA, Dunphy C, Choi W, Au WY, Srivastava G, et al. (2013). Genetic heterogeneity of diffuse large B-cell lymphoma. Proc. Natl. Acad. Sci. USA 110, 1398–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Proteomics data generated or analyzed during this study are included in this published article (and its supplemental information files). The raw proteomics data reported in this paper are also publicly available at the Peptide Atlas website (https://db.systemsbiology.net/sbeams/cgi/PeptideAtlas/PASS_View?identifier=PASS01217). Original uncropped western blots are available at the Mendeley database: https://doi.org/10.17632/px94fdcpvz.1.