

Abstract
Pseudomonas aeruginosa is an opportunistic bacterium that causes life-threatening infections in immunocompromised patients. In infection, it uses heme as a primary iron source and senses the availability of exogenous heme through the heme assimilation system (Has), an extra cytoplasmic function σ-factor system. A secreted hemophore HasAp scavenges heme and, upon interaction with the outer-membrane receptor HasR, activates a signaling cascade, which in turn creates a positive feedback loop critical for sensing and adaptation within the host. The ability to sense and respond to heme as an iron source contributes to virulence. Consequently, the inhibition of this system will lead to a disruption in iron homeostasis, decreasing virulence. We have identified a salophen scaffold that successfully inhibits the activation of the Has signaling system while simultaneously targeting iron uptake via xenosiderophore receptors. We propose this dual mechanism wherein free Ga3+-salophen reduces growth through uptake and iron mimicry. A dual mechanism targeting extracellular heme signaling and uptake together with Ga3+-induced toxicity following active Ga3+salophen uptake provides a significant therapeutic advantage while reducing the propensity to develop resistance.
Keywords: antimicrobials, heme sensing, heme uptake, metallotherapeutics, Pseudomonas aeruginosa
Graphical Abstract:

Multidrug-resistant Pseudomonas aeruginosa has been classified by the CDC as a “serious threat” to public health, owing to its ability to overcome many current treatment strategies.1 It frequently infects immunocompromised patients and is a leading cause of nosocomial infections.2–4 It is particularly problematic for cystic fibrosis (CF) patients, who suffer from life-threatening chronic infection, where even direct delivery of the antibiotic tobramycin to the lung is becoming ineffective.3,4 While new antibiotics are entering the pipeline, resistance often emerges within a few years of introduction as many candidates in phase I–III trials are typically improved versions of existing inhibitors rather than new classes of antibiotics, and further, many have minimal activity against P. aeruginosa.5,6
Recently, the development of antivirulence agents that reduce the ability of bacteria to colonize or spread has gained traction with the hope that such strategies will slow resistance development by exerting less selective evolutionary pressure.7 The regulation of many virulence factors is dependent on iron acquisition from the host, which also serves as a necessary nutrient for survival.8–11 Host iron levels are kept tightly regulated, minimizing the availability of labile iron in circulation for bacterial consumption.12 This innate immune response, often referred to as “nutritional immunity”, aims to keep the availability of essential nutrients at concentrations below what would permit colonization and growth.
Though P. aeruginosa and many other pathogens secrete iron-chelating siderophores, the low abundance of labile iron shifts the preference to heme, which is more abundant in a host.13–15 Furthermore, chronic infections in CF patients show evolution toward heme as a preferred iron source along with a decrease in the production of pyoverdine, a high-affinity siderophore.16,17 We have previously characterized the nonredundant heme uptake systems in P. aeruginosa, which involve a primary heme transporter Phu (Pseudomonas heme uptake) system and the heme-sensing Has (Heme assimilation system) system.18 While the Phu system is the predominant uptake transporter, the Has system relies on the secretion of an extracellular hemophore, HasAp, to deliver exogenous heme through the outer-membrane receptor HasR. The release of heme to HasR in turn activates the ECF σ-factor HasI that binds to the has promoter, recruiting RNA polymerase and upregulating the has operon.19,20 These recent studies have further shown that the heme metabolite biliverdin IXβ (BVIX β) positively regulates HasAp translation, providing a positive feedback loop that is highly tunable and rapidly responsive to fluctuating extracellular heme levels.
The complex transcriptional and post-transcriptional regulation over the has system is consistent with its significance in infection, where it has been shown to be highly upregulated in the transcriptomic analysis of an acute mouse lung infection (∼300-fold for hasAp and ∼70-fold for hasR).21 Furthermore, the deletion of hasR significantly reduced the bacterial load, highlighting the importance of heme sensing and adaptation in infection and suggesting that HasAp is an attractive therapeutic target. Targeting the Has system combines the advantages of disrupting heme acquisition while inhibiting the ability to sense the extracellular environment. Indirectly, the reduction in heme uptake caused by the inhibition of the heme signaling cascade will lead to lower levels of BVIXβ and further repress the heme signaling ability.19,20 Moreover, HasAp is an extracellular target that circumvents typical roadblocks of penetrating Gram-negative membranes and multidrug efflux pumps that enhance antibiotic resistance.22,23
Though structural work has extensively characterized HasAp, the HasAp–HasR interaction is still under investigation, which complicates the development of molecules targeting the protein–protein interaction.24–29 Alternatively, recent reports have demonstrated that synthetic iron complexes bind in the heme site of HasAp and are coordinated by the same residues, opening the door to heme mimicry as a targeting strategy.30,31 Of note is the N,N′-disalicylal-1,2-phenylenediamine (“salophen”) complex, which is synthetically modular and more soluble than many of the larger macrocycles. The use of metallosalophen complexes has also been investigated for potential anticancer applications, establishing their bioactivity as metallotherapeutics.32–34
The design of antimicrobials that inhibit heme sensing and iron uptake have a significant advantage over traditional strategies targeting solely iron uptake. Presently, strategies aimed at targeting iron uptake have shown gallium to be an effective iron mimic due to its similar size to iron and redoxinactivity.35–40 Currently, formulations of gallium, including the FDA-approved Ganite (Ga(NO3)3), are in clinical trials for efficacy as antibiotics; however, reports of gallium-inducing virulence factor production have also emerged.41–43 Since the binding of the metallosalophens to HasAp has been confirmed, we sought to further characterize the salophen–HasAp complex and its effect on the Has system in P. aeruginosa and found that both Fe3+–salophen (FeSal) and Ga3+–salophen (GaSal) complexes inhibited the transcriptional activation of the has operon. The use of GaSal also inhibited growth as a result of uptake as a xenosiderophore through a currently unidentified receptor. Therefore, GaSal inhibits heme sensing while simultaneously causing toxicity following active uptake through the siderophore receptors. This dual mechanism of action has several advantages, not the least of which is the reduced propensity to develop resistance. We hypothesize inhibiting heme sensing and intracellular iron homeostasis will ultimately lead to the dysregulation of related metabolic and virulence pathways.
RESULTS
Salophen Complexes Bind to HasAp.
Following reports that several synthetic iron complexes with heme-like features bind to HasAp in the heme-binding site and that larger macrocycles such as phthalocyanine (Pc) could be transported through HasR, we identified the o-phenylenediaminesalicylaldehyde (“salophen”) scaffold for its synthetic accessibility and potential for derivatization.30,31 The salophen complexes were reported to bind through the same coordination and in the heme-binding site, including overlaps of the pyrrole rings of the porphyrin with the aromatic rings of the salophen.30 We synthesized the FeSal and GaSal analogues, characterized their binding affinities, and found that, while salophen binds more weakly than heme, the metal substitution from Fe3+ to Ga3+ has less of an impact on the affinity than switching the coordinating scaffold from protoporphyrin IX to salophen (Table 1).
Table 1.
Binding Affinities of Ligands to HasAp as Determined by Fluorescence Quenching
| compound | KD (nM, ± SD) |
|---|---|
| heme | 350 ± 50 |
| GaPPIX | 280 ± 30 |
| FeSal | 1050 ± 100 |
| GaSal | 2050 ± 280 |
FeSal Inhibits Has-Dependent Heme Signaling and Is a Substrate for Iron Uptake.
FeSal was previously reported as a heme uptake inhibitor.30 To test whether FeSal would affect the activation of the Has signaling cascade and further study the mechanism, we employed a β-galactosidase (β-Gal) transcriptional reporter assay previously constructed by our lab, wherein the activation of the HasR promoter following ligand release to HasR would lead to a colorimetric readout.20 P. aeruginosa PAO1 hasR-lacZ was supplemented with heme–HasAp or FeSal–HasAp (1 μM), and aliquots were harvested and assayed for β-Gal activity during the log-phase to observe maximal transcriptional activation of the Has system. Furthermore, as the signaling activation is dependent on HasAp levels, the preformed ligand–HasAp complexes were used rather than waiting for natively expressed HasAp to accumulate in the extracellular media, leading to a more effective observation of the transcriptional response. Cultures supplemented with FeSal–HasAp showed a decreased signaling activation (Figure 1A), but had no effect on the growth, indicating that FeSal could be used as an iron source (Figure 1B). Previous characterization of the Has signaling cascade revealed that transcriptional activation requires heme release to HasR and that apo–HasAp, despite its ability to interact with HasR, does not initiate the signaling cascade.20 The FeSal–HasAp complex had improved overall growth relative to the unbound salophen ligand, most likely due to increased bioavailability or solubility (Figure 1B). We next tested the growth in the presence of either FeSal– or FeSal–HasAp on the PAO1 ΔhasRΔphuR strain that lacks both heme receptors and observed no effect on the growth, whereas supplementation with heme, or holo–HasAp is highly dependent on the presence of HasR and PhuR (Figure 1B, Figure S2). The lack of transport through the heme receptors, suggests that the FeSal scaffold is recognized as a xenosiderophore and taken in through such receptors. It is not uncommon for bacteria to scavenge the iron-siderophores they do not secrete, as such xenosiderophore acquisition or piracy provides a viable survival and virulence trait.44 For instance, the recently FDA-approved iron-chelating cephalosporin, cefiderocol, is taken up by the P. aeruginosa ferric iron transport systems; however, it is linked to an artificial iron-chelating group and not any native or exogenous siderophore.45
Figure 1.
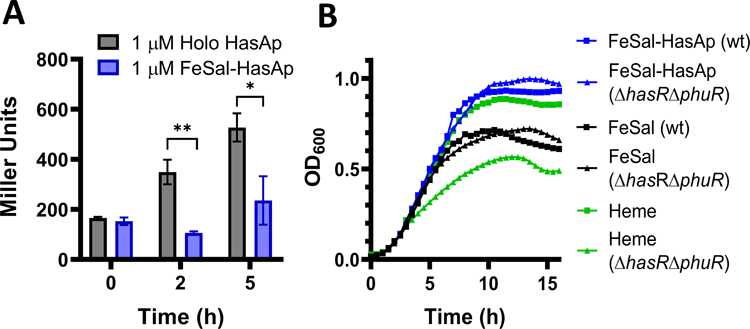
(A) Transcriptional Activation of the hasR Promoter with holo– and FeSal–HasAp. FeSal shows decreased transcriptional activation relative to heme. Both cultures were supplemented with preformed HasAp complexes and data represent the average of three independent experiments*, p < 0.05; **, p < 0.005 by t test between holo and FeSal–HasAp. (B) Growth of PAO1 Wild-Type and the ΔhasRΔphuR Strain with FeSal and FeSal–HasAp. Cultures were grown in a 96-well plate with 1 μM supplements, as described in the experimental section.
GaSal Inhibits Has Signaling and Growth.
The salophen complex was then synthesized to include a gallium metal center to prevent its utility as an iron source and increase the inhibitory potential based on the utility of gallium salts and nanoparticles as antibacterial agents.46,47 We tested GaSal–HasAp and found a similar inhibition profile for the has transcriptional activation relative to that of the holo–HasAp complex (Figure 2A). In contrast to FeSal, GaSal was inhibitory to growth when added on its own as well as when complexed with HasAp (Figure 2B). To determine if GaSal toxicity is independent of the signaling cascade and the results from the active uptake by a siderophore-dependent mechanism, we employed the P. aeruginosa ΔhasAp strain that does not produce the HasAp protein. As the maximum OD600 of the P. aeruginosa strains in iron-deplete media (M9 alone) is low, we measured the effect of GaSal inhibition in cultures supplemented with 400 nM FeCl3. This concentration was selected, as it yields a maximum OD600 midway between that observed for growth in iron-deplete or iron-replete conditions (Figure S3). Under these conditions of partial iron-restriction, the concentration of GaSal required to inhibit growth by 50% was 7 μM (Figure S3). We hypothesized that, if gallium toxicity is due to the uptake of unbound GaSal, rather than Ga stripped by endogenous siderophores, then titrating HasAp to GaSal–supplemented P. aeruginosa ΔhasAp cultures would alleviate the inhibition of growth. Employing the previously optimized conditions, P. aeruginosa ΔhasAp cultures containing 400 nM FeCl3 and 7 μM GaSal were supplemented with increasing concentrations of the HasAp protein. As shown in Figure 2C, increasing HasAp concentrations alleviated the toxicity of GaSal, confirming that GaSal is transported intact into the cell when not bound to HasAp. The formation of the GaSal–HasAp complex in the extracellular medium was also confirmed by saturation transfer difference (STD) NMR spectroscopy (Figure 3). We further confirmed that native siderophores are not stripping the metals from the salophen by using a mutant strain that lacks the gene for pyoverdine biosynthesis (Figure S4). As pyoverdine has an incredibly high affinity for ferric iron, the sustained growth in the siderophore-deficient strain and no prevention of toxicity with GaSal shows that pyoverdine, and by extension pyochelin, are not responsible for the metal uptake.48
Figure 2.

(A) Transcriptional Activation of the hasR Promoter with holo– and GaSal–HasAp. GaSal shows decreased transcriptional activation relative to heme. Both cultures were supplemented with preformed HasAp complexes and represent the average of three independent experiments: *, p < 0.05; **, p < 0.005, by a t test between holo and GaSal–HasAp. (B) GaSal antibacterial activity in the PAO1WT and ΔhasRΔphuR strains. Data represent the average of three independent cultures with blanks (media only) subtracted. Cultures were grown in M9 minimal media with 1 μM supplements. (C) HasAp titration alleviates GaSal toxicity in PAO1 ΔhasAp. Cultures were prepared in M9 minimal media with FeCl3 (400 nM) and GaSal (7 μM), with HasAp supplemented at increasing concentrations. Growth was calculated relative to gallium-free cultures in stationary-phase growth, and data represent the average of three independent experiments: *, p < 0.05; **, p < 0.005, by a t test between 0 μM HasAp and specified concentrations.
Figure 3.

(A) 1H NMR spectrum of GaSal in M9 minimal Media. Proton peaks were assigned and labeled, as depicted for comparison in the STD spectrum. (B) STD NMR spectrum using GaSal (1 mM) and HasAp (10 μM) in PAO1 ΔhasAp supernatant.
In the STD experiment, a selective saturation pulse is delivered exclusively to HasAp and the saturation distributes throughout the protein by intramolecular spin diffusion. The saturation is transferred to any bound ligands, which are detected upon dissociation from the protein. In the resultant difference spectrum, only ligand 1H peaks receiving saturation from the protein are observed and their intensities are proportional to their proximity to the protein, which is a useful observation of ligand binding epitopes.49 The top spectrum (Figure 3A) is a 1H NMR spectrum of GaSal in M9 minimal media to serve as a reference spectrum for the STD experiment. The bottom spectrum (Figure 3B) is GaSal in PAO1 ΔhasAp supernatant with added HasAp. This result shows saturation transfer to GaSal and confirms formation of the GaSal–HasAp complex in the extracellular media. Protons from the diamine ring (a and b) buried in the back of the heme binding site are clearly represented. Protons e and f are also distinguishable and could be useful sites to build outward from the salophen core to enhance binding affinity as they are pointed away from the binding site in the crystal structure of FeSal–HasAp.30 The planarity and symmetry of GaSal also enhances the STD signals and permits binding from either face of the molecule. While these experiments show that GaSal binds to HasAp, they do not explicitly show the binding site. On the basis of the binding of GaPPIX and GaPc in combination with the crystallized FeSal–HasAp structure, GaSal is likely coordinated by the same ligand set in the heme binding site.30,31
Has Cell-Surface Signaling is Scaffold-Dependent.
Gallium protoporphyrin IX (GaPPIX) is a common heme mimic with antibacterial properties, owing to the redox-inactive nature of Ga3+ versus Fe3+.37,38,50 However, unlike GaSal, GaPPIX activates the Has signaling cascade despite causing toxicity to cultures (Figure 4). This result confirms that, despite GaPPIX being toxic, the protoporphyrin IX scaffold permits signaling and transport through HasR, which limits the potential of GaPPIX as a therapeutic strategy, as the signal activation will likely overcome the initial toxicity and increase HasAp levels while exerting selective pressure toward siderophore-based iron acquisition to circumvent GaPPIX uptake. A gallium-based strategy that targets iron-uptake pathways while inhibiting heme sensing is therefore less likely to develop resistance. The use of the salophen scaffold for signaling inhibition not observed with the porphyrin scaffold is therefore a starting point for further development but also for understanding the mechanism of signaling activation between HasAp and HasR.
Figure 4.

(A) Transcriptional activation of the hasR promoter with holo– and GaPPIX–HasAp. GaPPIX shows increased (2 h) or similar transcriptional activation to heme. Cultures were supplemented with preformed HasAp complexes, and data represent the average of three independent experiments: **, p < 0.005, by a t test between holo– and GaPPIX–HasAp. (B) Growth of PAO1 Supplemented with either 1 μM Heme or GaPPIX. Supplementation with GaPPIX inhibits growth in M9 minimal media. Cultures were grown in a 96-well plate, as described in Methods.
As the signaling inhibition is dependent on the scaffold and not the metal, we sought to test whether GaSal–HasAp prevented the interaction with HasR and thus signaling. As signaling activation requires a protein–protein interaction, conformational changes, ligand transfer, and energy-dependent uptake, we recognize that any in vitro method is not a physiological characterization of the HasAp–HasR interaction but rather a useful determination of the relative associations of the HasAp complexes with HasR. We measured the binding affinities of holo–, apo–, and GaSal–HasAp to HasR by surface plasmon resonance (SPR) detection and found that the holo– and apo–HasAp bind HasR with a similar affinity (Table 2). In contrast, the binding of GaSal–HasAp to HasR was weaker; however, we could only estimate the KD value to be greater than 1 μM, as at higher concentrations, we observed nonspecific binding most likely originating from the heterogeneity introduced as a consequence of the aggregation of the HasR lipid nanodiscs. The KD values were determined by steady-state association measurements at various analyte concentrations (Figure S6). This result indicates that the HasAp–HasR interaction, while weaker for the GaSal–HasAp complex, is not completely inhibited and suggests the nature of the scaffold is critical in facilitating the correct protein–protein interaction for ligand release and transcriptional activation.
Table 2.
Binding Affinities of HasAp to HasR as Measured by SPR
| ligand | KD (nM, ± SD) |
|---|---|
| apo–HasAp | 810 ± 50 |
| holo–HasAp | 479 ± 20 |
| GaSal–HasAp | >1000 |
Structural Studies of the Salophen–HasAp Complex Reveal Differences in Solution Conformations that May Affect Signaling.
The salophen–HasAp complex effectively inhibits the activation of the Has signaling cascade without completely inhibiting the binding of HasAp to HasR, indicating that there must be differences in the ligand release that prevent the transfer and uptake. We next sought to investigate if differences in the solution conformations of HasAp could lead to the inability to initiate ligand transfer despite nearly superimposable crystal structures. HasAp undergoes a large conformational rearrangement of the H32 loop upon heme binding (Figure 5A; RMSD between apo and holo = 5.7 Å; PDB 3MOK and 3ELL).51 In the FeSal–HasAp crystal structure, this loop is also in the closed position and reveals less significant differences in the overall fold between the holo–HasAp and FeSal–HasAp complexes (Figure 5B; RMSD = 0.81 Å; PDB 3ELL and 3W8M).30 We expect that if the GaSal–HasAp complex is more conformationally flexible relative to holo–HasAp and is otherwise identical to FeSal–HasAp structurally then these differences may be responsible for the impaired ligand release despite the maintained HasR binding. We analyzed the solution structures of apo–, holo–, and GaSal–HasAp by HDX-MS. Differences in the deuterium uptake over time are reflective of how readily backbone amide hydrogens of a protein exchange with the solvent hydrogens or deuterons in a manner that is dependent on the structure, solvent-exposure, and flexibility. We have previously used this technique to describe ligand-induced structural and dynamic changes of the heme-binding proteins PhuS and HemO.52,53 Here, heme binding resulted in significant protection from deuteration throughout the heme binding site, consistent with loop closure and decreased solvent exposure (Figure 5C). Similar protection of the heme site was observed upon GaSal binding, further suggesting that GaSal binds to HasAp in the heme site and is coordinated with the same ligands (Figure 5D). As GaSal–HasAp is not crystallized, we have used the FeSal–HasAp structure as a representative example for mapping structural changes and visualizing the bound ligand.
Figure 5.

Conformational changes of HasAp upon ligand binding. (A) Alignment of apo–, holo–, and FeSal–HasAp, highlighting the closure of the H32 loop from the apo– (orange, PDB 3MOK) to the holo– (blue, PDB 3ELL) and FeSal–HasAp (red, PDB 3W8M) forms. (B) Alignment of holo– and FeSal–HasAp, highlighting the closed-loop conformation with minimal structural differences. Relative deuteration of HasAp bound to heme (C) and GaSal (D). Individual peptides are plotted from the N- to C-terminus based on the first residue number. For each peptide, differences in the percent deuteration at each time point, color coded according to the legend, with the sum of all differences integrated over time are represented in gray bars. Also, 98% confidence intervals are represented as dashed (for individual time points) and solid (for total sums) lines. Peptides exceeding both confidence intervals were considered to display a statistically significant difference in deuterium uptake between the apo and ligand bound form and were mapped onto the crystal structure of holo–HasAp (panel C, inset; PDB 3ELL). Positive values indicate protection from deuteration upon ligand binding, and negative values indicate increased deuteration of a region upon ligand binding. Significant regions are highlighted and color coded on the HasAp structure (panel C, inset).
In contrast with the crystallographic comparison of FeSal–HasAp and holo–HasAp, the H32 and Y75 loops contained the greatest differences in the deuterium uptake between the heme-bound and GaSal-bound states (Figure 6A). On the H32 loop, the GaSal binding increased protection from deuteration relative to the apo form but not at strongly as the heme binding (Figure 6B). GaSal is a smaller scaffold that does not have as extensive interactions with the protein, particularly those between the backbone of the H32 loop and heme (Figure 6C). Notably, in the heme-bound crystal structure, the His loop is observed to engage in several possible interactions with the porphyrin, including hydrogen bonds between the amide hydrogens of P34 and G35 with the heme propionates, or the V37 carbonyl with the heme methyl groups that are absent in the salophen-bound complex. The lack of these interactions and increased flexibility of the heme site do not prevent HasR binding but, combined with the structural differences in the salophen ligand, likely hinder the initiation of ligand release and uptake through HasR.
Figure 6.

Deuteration differences between ligand-bound forms. (A) Percent deuteration of GaSal–HasAp minus holo–HasAp. Positive peaks represent greater deuteration in the salophen-bound form, and negative peaks indicate increased deuteration in the heme-bound form. Inset: representation of protein regions that are different between all three HasAp states, using FeSal–HasAp as a representation of GaSal–HasAp. (B) Deuteration of a peptide comprising residues 26–54. (C) Heme binding site with heme (blue) and FeSal (red) overlaid. Potential contacts between the H32 loop and heme are drawn in red.
To assess whether this observation carried a thermodynamic consequence, we measured the melting temperatures of the three HasAp complexes by circular dichroism. All three HasAp spectra show similar degrees of folding at 25 °C, indicating that the overall structure is not significantly impacted by ligand binding (Figure 7A). The melting temperature of the apo form (58 °C) was significantly increased by heme binding, where the protein only began to unfold at temperatures >90 °C (Figure 7B). HasAp bound to GaSal still showed increased thermal stability (65 °C) but not nearly to the extent of heme binding. Taken together, the HDX-MS and CD data indicate significant differences between the holo– and GaSal–HasAp complexes not reflected in the FeSal–HasAp crystal structure that may be responsible for the lack of signaling. These differences in solution carry physiological consequences beyond inhibiting signaling, wherein the decreased stability of the GaSal–HasAp complex compared to the holo–HasAp complex may increase the susceptibility to proteolytic cleavage in the host.
Figure 7.

Thermal denaturation of HasAp bound to heme or GaSal. (A) Comparison of three ligand states at 25 °C. (B) Thermal denaturation profiles at 222 nm fit to a sigmoidal distribution. CD spectra were recorded as described in Methods.
DISCUSSION
As an extracellular protein, HasAp must be secreted in high amounts to counteract both the diffusion from the site of infection and the host defense mechanisms such as proteolysis to effectively scavenge heme. Its relative abundance and role in virulence coupled with the advantage of an extracellular target make HasAp an attractive therapeutic target that can lead to the dysregulation of heme sensing and host adaptation. Though GaPPIX is a potent redox-inactive heme mimic that enters the cell through the Has and Phu systems where it causes toxicity, the levels of transcriptional activation of the Has system are not decreased. This will lead to a downstream increase in HasAp levels, which will, over time, counteract the inhibitory effect and lead to increased heme-scavenging abilities. Additionally, the lack of iron obtained through this pathway will also lead to adaptation toward siderophore-based iron acquisition as the heme systems lead to gallium toxicity, limiting the potential of GaPPIX as a long-term therapeutic option. Structurally, GaPPIX is identical to heme, which also increases the potential for off-target effects and inhibition of human heme binding proteins such as heme oxygenase, highlighting a need for new scaffolds with greater selectivity toward HasAp.54
Previous heme mimetic approaches reported that FeSal and FePhthalocyanine (FePc) were inhibitors of heme uptake when cultures were not supplied with holo–HasAp as an iron source.30 However, in the presence of holo–HasAp, only FePc was inhibitory, suggesting that the larger phthalocyanine macrocycle blocks HasR and inhibits heme uptake. More recent studies showed that GaPc activity results from active transport through HasR and not inhibition of heme uptake, as previously described.31 On the basis of these conflicting reports, we sought to revisit the mechanism of inhibition by the salophen complexes as potential therapeutics due to their synthetic accessibility and modularity. While the salophen scaffold binds to HasAp, unlike the PPIX scaffold, it is less likely that these molecules will be generic inhibitors of heme proteins, given the significance of both the extensive hydrophobic and salt bridge interactions afforded by the propionate groups of PPIX.55,56
We found that, unlike GaPPIX, both FeSal and GaSal inhibited the activation of the cell surface signaling cascade. We have previously demonstrated that heme-dependent activation of the signaling cascade requires the release of heme from HasAp to HasR.20 The lack of signaling suggests that FeSal and GaSal are either not released to HasR or, if they are released, do not induce the requisite conformational change in the HasR signaling domain required to trigger the cascade or to be transported. We also found that the metallosalophens retained activity in the ΔhasRΔphuR mutant, indicating that the uptake is independent of the heme receptors. As metal chelates, the salophen complexes are likely a substrate for xenosiderophore receptors expressed by P. aeruginosa to utilize siderophores from other bacteria, a strategy that has also been used to develop siderophore–drug conjugates.44,57,58 The development of cefiderocol, an FDA-approved iron-chelating cephalosporin that differs from typical siderophore–drug conjugates, was found to be transported despite containing no natural siderophore and only included iron-chelating features linked to a cephalosporin antibiotic.44,45 The current data would support a similar mechanism of uptake that will be explored in future studies. As GaSal displayed antimicrobial activity consistent with previous reports of gallium-based therapies, this strategy conveniently combines the use of gallium salts and a porphyrin mimetic into a single compound that targets both iron and heme pathways without leading to the upregulation of the heme-sensing system.41,59
We further characterized the mechanism through which GaSal is inhibitory to better understand what features could be explored in later metallosalophen derivatives. While the salophen complexes have weaker binding affinities than the PPIX counterparts, we found that, while GaSal–HasAp has a weaker binding affinity than holo–HasAp to HasR, the interaction was not completely inhibited. To further investigate the weaker binding and decreased transcriptional activation by the metallosalophen complexes, we studied the HasAp complexes by HDX-MS. These experiments showed increased conformational flexibility of the H32 loop with GaSal–HasAp relative to holo–HasAp, which is consistent with the loss of stabilizing interactions with the PPIX macrocycle.25 The lack of these interactions is also consistent with the decreased binding affinity of the metallosalophens relative to their PPIX counterparts. The increased flexibility of the H32 loop may therefore prevent HasR binding in a manner that triggers the conformational change and concerted ligand release. The increased flexibility of the loop is also reflected in the lower Tm values. Since the melting temperature of GaSal–HasAp is also much closer to apo–HasAp than holo–HasAp, GaSal binding leads to a less-stable and more proteolytically susceptible HasAp complex, further diminishing the ability of P. aeruginosa to acquire and sense heme.
With the utility of GaSal, we envision that future derivatives will have an increased HasAp affinity. New designs could also extend molecules anchored to the heme site into the HasAp–HasR interaction region, with the goal of blocking binding to HasR entirely. Conveniently, approaches that either block the HasR interaction entirely or prevent ligand release from HasAp are likely to have merit. On the basis of our findings, the development of an inhibitor that is transported through HasR should be avoided, as this will lead to the activation of the signaling cascade. Additionally, these inhibitors should maintain affinity to iron uptake pathways to not target the heme-sensing pathway exclusively, which would be overcome by adaptation to iron acquisition.
CONCLUSION
Various gallium formulations have been employed for antimicrobial applications. The use of GaSal represents a more specific approach that targets the extracellular hemophore HasAp and the heme-sensing ability of P. aeruginosa in addition to targeting iron uptake for antimicrobial activity. The biophysical consequences of salophen versus heme binding build on the structural characterization of the Has system previously reported. This work provides a platform for the development of new heme mimics that can target bacterial hemophore signaling as well as iron uptake. These dual mechanisms have the propensity to slow the development of resistance as the simultaneous evolution of mutations in separate pathways is likely to lead to reduced fitness and survival of the bacteria. Furthermore, the fact that these mechanisms are critical for infection but not survival outside of the host further lowers selective pressure for the development of resistance.
METHODS
Bacterial Strains.
Pseudomonas aeruginosa (PAO1 and mutants) strains were stored as glycerol stocks in lysogeny broth (LB) at −80 °C and were freshly streaked on Pseudomonas isolation agar (BD Biosciences) before transferring to liquid culture media. PAO1 (wild type) was used as reported.60 PAO1 LacZ fusions and hasAp deletion strains were constructed as described previously.20 The heme receptor deletion strain (PAO1 ΔhasRΔphuR) was constructed as reported.18 The pyoverdine biosynthesis deletion strain (PAO1 ΔpvdA) was constructed as reported.17
Expression and Purification of WT HasAp.
HasAp was prepared from freshly transformed E. coli BL21(DE3) competent cells harboring the pET11a plasmid with the full-length hasAp gene, as previously described with an additional purification step.20 Briefly, a single colony was cultured for 16 h in a LB medium (50 mL) containing 100 μg/mL ampicillin. The cells were harvested by centrifugation, resuspended in M9 media, and divided evenly among four 1 L cultures in M9 containing 100 μg/mL ampicillin and grown to an A600 of ∼1.0. The cells were once again pelleted and resuspended in four fresh 1 L M9 cultures, induced with a 1 mM final concentration of isopropyl-β-D-thiogalactopyranoside, and grown for 16 h at 30 °C. Cells were harvested by centrifugation; resuspended in 40 mL of lysis buffer (20 mM Tris-HCl (pH 7.5), 20 mM NaCl, 1 mM EDTA) containing a protease inhibitor cocktail tablet (cOmplete EDTA-Free, Roche Applied Science), 1 mg/mL DNase, and 25 mg/mL lysozyme; and passed through an LM-20 microfluidizer at 20000 psi. The suspension was centrifuged at 25000 rpm for 1 h to separate the cell debris. The supernatant was applied to a Q-Sepharose column (2.6 × 10 cm) pre-equilibrated with 20 mM Tris-HCl (pH 7.5) and 20 mM NaCl. The column was washed (3–5 column volumes) with buffer, and the protein was eluted over a gradient from 20 to 600 mM NaCl in 20 mM Tris-HCl (pH 7.5). The purity of the eluted fractions was determined by SDS-PAGE, and those containing HasAp were pooled. To separate the apoprotein, the pooled fractions were concentrated to ∼5 mL and exchanged into a 50 mM sodium phosphate buffer with 0.7 M ammonium sulfate (pH 7.0). The concentrate was loaded onto a butyl sepharose fast flow (GE Healthcare, 2.6 × 10 cm) column equilibrated with the same buffer. Weakly bound contaminants and holo–HasAp were eluted with 2–3 bed volumes of 50 mM sodium phosphate containing 0.5 M ammonium sulfate. Apo–HasAp was then eluted with a linear gradient of sodium phosphate (50 to 20 mM) containing ammonium sulfate (0.5 to 0 M). Fractions were once again analyzed by SDS-PAGE, and the apo–HasAp was pooled, concentrated (Spin-X UF 10k MWCO, Corning), and exchanged into a 20 mM sodium phosphate buffer.
Expression and Purification of HasR.
HasR was prepared as reported with modifications to prepare the protein in lipid discs.61,62 Briefly, a single colony of freshly transformed Escherichia coli BL21 (DE3) cells harboring the pHasR22b plasmid was selected to inoculate 50 mL of noninducing MDAG-135 media containing 100 μg/mL Amp and grown overnight at 37 °C and 225 rpm. This culture was used to inoculate four 1 L flasks of autoinducing MDA-5052 media containing 100 μg/mL Amp and grown for 10 h at 25 °C. Cells were harvested by centrifugation for 15 min at 7,000 rpm at 4 °C.
Pellets were resuspended in 40 mL of lysis buffer and passed through a LM-20 microfluidizer at 20000 psi. The debris was removed by centrifugation at 12000 rpm for 15 min, and the supernatant was centrifuged for 1 h at 25000 rpm to pellet the cellular membranes. Pelleted membranes were resuspended in 30 mL of lysis buffer with an added EDTA-free protease inhibitor tablet overnight. The cytoplasmic membrane proteins were solubilized by the addition of 2% (v/v) Triton X-100 (Sigma) and 0.5% (v/v) N-lauroylsarcosine sodium salt (Sigma). The membrane fractions were stirred at room temperature for 1 h and pelleted at 25000 rpm for 1 h at 4 °C. The resulting supernatant containing only the cytoplasmic membrane proteins was discarded. The pelleted outer membrane (OM) fraction was then resuspended in 30 mL of lysis buffer containing an EDTA-free protease inhibitor cocktail tablet and stirred at 4 °C overnight.
The RC DC protein assay (Bio-Rad) was used to determine the total protein concentration. The OM fragments were diluted to at least 10 mg/mL final concentration. The styrene–maleic acid copolymer (Xiran SL30010 P20) was then added to the OM suspension to achieve a final concentration of 2.5% (v/v) and inverted continuously at room temperature (RT) for 1 h. The suspension was frozen in liquid nitrogen and thawed at 42 °C a total of 5 times, followed by passage through a microfluidizer at 20000 psi. This cycle was then repeated once more. The final suspension was centrifuged at 25000 rpm for 1 h at 4 °C, and the supernatant containing the HasR in lipid nanodisc (HasR-smalp) was collected. The supernatant was concentrated, and the HasR concentration was determined using an extinction coefficient of 126 mM/cm after subtracting the blank containing the filtrate (to account for the absorption of residual lipids and polymer).
Chemical Synthesis.
All the reagents and solvents were purchased from commercial sources and used as received, unless otherwise stated. The 1H and 13C NMR spectra were obtained in DMSO d6 on a 400 MHz spectrometer with chemical shifts referenced to tetramethylsilane (TMS). The purities (>95%, HPLC) of FeSal and GaSal were confirmed by HPLC, as only singular peaks were detected.
Synthesis of 2,2′-((1 E, 1′E)-(1,2-Phenylenebis-(azaneylylidene))bis(methaneylylidene))diphenol (Salophen).
O-Phenylenediamine (5.4 g, 50 mmol) was dissolved in dichloromethane (100 mL). To the resulting solution was added salicylaldehyde (12.2 g, 100 mmol). The mixture was stirred at room temperature for 8 h before removing the solvent via rotary evaporation to afford an orange solid (10.5 g, 99%). 1H NMR (400 MHz, DMSOd6): δ 12.94 (s, 2 H), 8.941 (s, 2 H), 7.68−7.66 (d, 2 H), 7.48−7.40 (m, 6 H), 7.00−6.95 (t, 4 H).63
Synthesis of FeSal Chloride.
To a solution of salophen (0.316 g, 1 mmol) in ethanol (3 mL) was added ferric chloride (0.16 g, 1 mmol). The reaction mixture was heated at 65 °C in a sealed pressure tube overnight. A black precipitate was washed with cold ether and collected (70 mg, 17%). NMR peaks were broadened beyond detection due to the para-magnetic effects of iron. HRMS (ESI) m/z: [M − Cl]+ calcd for C20H14N2O2Fe 370.0405, found 370.0443.
Synthesis of GaSalophen Nitrate.
To a solution of salophen (0.316 g, 1 mmol) in ethanol (10 mL) was added gallium(III) nitrate hydrate (0.274 g, 1 mmol). The reaction mixture was refluxed for 2 h and cooled to room temperature. The final product was collected by filtration and washed with ethanol and diethyl ether to afford yellow crystals (0.2 g, 50%). 1H NMR (400 MHz, DMSOd6): δ 9.37 (s, 2 H), 8.16−8.13 (m, 2 H), 7.68−7.66 (dd, 2 H), 7.66−7.51 (m, 4 H), 7.02−6.99 (d, 2 H), 6.90−6.86 (t, 2 H). 13C NMR (400 MHz, DMSO d6): δ 167.9, 162.9, 137, 136.6, 135.4, 129, 122.3, 118.3, 117.1, 117. HRMS (ESI) m/z: [M -NO3]+ calcd for C20H14N2O2Ga 383.0311, found 383.0301.
Preparation of HasAp Complexes.
Heme and GaPPIX were purchased from Frontier Scientific. Heme solutions were prepared as described previously immediately before use, and concentrations were determined via a pyridine hemochrome assay.64 GaPPIX solutions were prepared identically; except, concentrations were determined using the extinction coefficient at 404 nm (90.6 mM/cm). Apo–HasAp was prepared at a desired concentration in a 20 mM sodium phosphate buffer (pH 7.4, 25 °C). To this solution was added a 3-fold molar excess of ligand prepared in the same buffer solution. Excess unbound ligand was removed using 7K MWCO centrifugal desalting columns (Zeba Spin, Thermo Fisher Scientific) according to the manufacturer’s recommendations.
Binding Affinity Determination by Fluorescence Quenching.
Binding affinities were determined on a ISS K2 multifrequency fluorometer in the L-format using 1 cm quartz cuvettes. To a 1 μM solution of apo–HasAp in a 20 mM sodium phosphate buffer (pH 7.4, 25 °C) was titrated the ligand of interest. Emission spectra were recorded (300–500 nm) following excitation at 295 nm. The decrease in the maximum emission was plotted against the ligand concentration (accounting for dilution) and fit to the one-site binding using GraphPad Prism 8.
Growth Curves.
P. aeruginosa strains were grown overnight in 50 mL LB Broth with shaking at 37 °C. Cells were harvested by centrifugation and resuspended in M9 minimal medium. Strains were then inoculated at A600 = 0.05 in 10 mL M9 minimal medium and grown for 3 h to deplete bacterial iron stores.20 Cultures were then plated in 96-well plates (Costar clear, flat-bottom with lid, Corning Inc.) at A600 ≈ 0.05 to a final volume of 200 μL in M9 media. Supplements (2 μL) were added as 100× stock solutions. Growth curves were recorded at 600 nm with a Biotek Synergy HT plate reader over 16 h of growth with shaking at 37 °C. To account for background absorbance, blank wells were prepared with the same supplements but only with M9 media without culture. The blank wells were subtracted from the average of three growth wells per condition.
Transcriptional Reporter Assay.
The chromosomal fusions of the PhasR–lacZ and promoterless lacZ gene were previously constructed and used to report on the transcriptional activation of the Has signaling cascade.20 For all β-gal assays, strains were grown overnight in 50 mL of LB with shaking at 37 °C. Cells were harvested by centrifugation and resuspended in M9 media. Strains were then inoculated at A600 = 0.05 in triplicate in 25 mL of M9 minimal media and cultured for 3 h to deplete bacterial iron stores.20 Growth cultures were then supplemented with 1 μM of either holo–HasAp, GaPPIX–HasAp, FeSal–HasAp, or GaSal–HasAp. Aliquots (1 mL) at 0 (just prior to supplementation), 2, and 5 h post supplementation were harvested and assayed for β-gal activity as described previously.65
Circular Dichroism.
Circular dichroism experiments were performed on a Jasco J-810 spectropolarimeter using 1 mm quartz cuvettes. All samples were recorded with 10 μM HasAp in 10 mM potassium phosphate (pH 7.4) at 25 °C from 190 to 260 nm at a scan rate of 50 nm/min, with each spectrum representing 5 accumulations. Data were acquired at a 0.2 mm resolution and a 10 nm bandwidth. The mean residue ellipticity (degree cm2/dmol) was calculated using CDPRO software as recommended by Jasco. Thermal denaturation studies were performed over a temperature range of 20–90 °C at 222 nm. Melting temperatures were determined by fitting denaturation data to a Boltzmann–Sigmoidal distribution in GraphPad Prism 8.
Saturation Transfer Difference NMR.
PAO1 ΔhasAp was cultured overnight from a single colony in LB. The cells were harvested by centrifugation and resuspended in M9 minimal media. Then, 10 mL of M9 were inoculated at A600 ≈ 0.05 and grown for 3 h. The cell-free supernatant was obtained by centrifugation and subsequent filter sterilization by a 0.22 μM syringe filter. The supernatant was then used as the solvent for subsequent STD experiments. The final sample volume contained 600 μL of the supernatant with or without 10 μM HasAp (as a negative control) and 5% D2O for solvent locking. The STD experiments were performed at 25 °C on an Agilent DD2 500 MHz spectrometer. The vendor-supplied pulse sequence, dpfgse_satxfer.c, was used, and the on- and off-resonance fids were subtracted in-place through phase-cycling to yield only the difference fid. Selective saturation was performed for 2.5 s and consisted of 50 ms Gaussian pulses separated by a 1 ms delay at a field strength of 50 Hz. A spectral width of 6000 Hz (12 ppm), a 90-degree pulse of 9.6 μs, and 16384 points were used to collect the data with a 0.5 s delay between transients. Transmitter offset was on the water signal. Solvent suppression was achieved via excitation sculpting. The selective irradiation on-resonance with the protein was at 1.5 ppm, and the off-resonance irradiation was at 25 ppm.
Surface Plasmon Resonance.
Apo–HasAp, GaSal–HasAp, and holo–HasAp were covalently bound to the surface of flow cells 2, 3, and 4 of a CM5 chip to a final level of 50 RU using the NHS-EDC kit (GE Life Sciences, Piscataway, New Jersey). Flow cell 1 was used as the blank. HasR-his (0–1000 nM) in 120 μL of HBS-EP buffer (GE Life Sciences) was injected into flow cells 1–4 at 25 °C until the signal reached saturation. The surface was then washed with the buffer for 3 min, and the dissociation of analyte–ligand complexes was followed over time. The flow cells were regenerated by injecting 15 μL aliquots of 10 mM glycine (pH 1.5) followed by 15 μL aliquots of 10 mM NaOH, and the process was repeated. Values from the reference flow cell were subtracted to obtain the values for specific binding. The maximum response units during the steady-state phase were plotted as a function of the HasR concentration, and data were fitted to a 1:1 binding model using BIAeval 4.1 software (Biacore).
Hydrogen–Deuterium Exchange Mass Spectrometry.
The coverage maps for all proteins were obtained from undeuterated controls as follows: 3 μL of the 20 μM sample in a 20 mM sodium phosphate buffer (pH 7.4, 25 °C) was diluted with 27 μL of an ice-cold quench (100 mM glycine, 5.5 M guanidine–HCl, pH 2.4). After 5 min, 120 μL of the 50 mM glycine buffer (pH 2.4) was added prior to the injection. Then, 50 μL of the quenched samples was injected into a Waters HDX nanoAcquity UPLC (Waters, Milford, MA) with in-line digestion (NovaBioAssays immobulized protease type XVIII/pepsin column). Peptic fragments were trapped on an Acquity UPLC BEH C18 peptide trap and separated on an Acquity UPLC BEH C18 column. A 7 min, 5% to 35% acetonitrile (0.1% formic acid) gradient was used to elute peptides directly into a Waters Synapt G2-Si mass spectrometer (Waters, Milford, MA). MSE data were acquired with a 20–30 V ramp trap CE for high-energy acquisition of the product ions as well as continuous lock-mass (Leu-Enk) for mass-accuracy corrections. Peptides were identified using the ProteinLynx Global Server 3.0.3 (PLGS) from Waters. Further filtering of 0.3 fragments per residues was applied in DynamX 3.0.
For each state (i.e., apo and ligand-bound), the HD exchange reactions and controls were acquired using a LEAP autosampler controlled by Chronos software. The reactions were performed as follows: 2 μL of 20 μM of apo–HasAp or in complex with the ligand in a 20 mM sodium phosphate buffer (pH 7.4, 25 °C) was incubated in 18 μL of a 20 mM sodium phosphate buffer containing 30 μM ligand (99.99% D2O, pD 7.4). All reactions were performed at 25 °C. Prior to injection, deuteration reactions were quenched at various times (10 s, 1 min, 10 min, or 1 and 2 h) with 60 μL of a 100 mM glycine buffer and 5.5 M guanidine–HCl, pH 2.4, followed 1 min later by a post-quench dilution of 170 μL of a 50 mM glycine buffer, pH 2.4. The resulting sample volume was injected. Back-exchange correction was performed against fully deuterated controls acquired by incubating 2 μL of 20 μM apo–HasAp containing 6.0 M guanidine–HCl in a 18 μL 20 mM sodium phosphate buffer (99.99% D2O, pD 7.4) for 24 h at 25 °C prior to data collection. All deuteration time points and controls were acquired in triplicates.
The deuterium uptake for all identified peptides with increasing deuteration times and for the fully deuterated control was determined using Water’s DynamX 3.0 software. The normalized percentage of deuterium uptake (%Dt) at an incubation time t for a given peptide was calculated as follows:
where mt is the centroid mass at incubation time t, m0 is the centroid mass of the undeuterated control, and mf is the centroid mass of the fully deuterated control. Percent deuteration difference plots, Δ%Dt(Apo − Liganded), displaying the difference in the percent deuteration between the apo and ligand bound HasAp for all identified peptides at all deuterium incubation times probed, were generated. Confidence intervals for the Δ%D plots were determined using the method outlined by Houde et al., adjusted to the percent deuteration using the fully deuterated controls.66 Confidence intervals (98%) were plotted on the Δ%D plots as horizontal dashed lines and used to determined peptides with statistically significant differences in deuterium uptake between the apo and complexed state.
Supplementary Material
ACKNOWLEDGMENTS
SPR experiments were performed in the Biosensor Core facility at the University of Maryland, Baltimore. The PAO1 ΔpvdA strain was provided by Dr. Amanda Oglesby. G.C. acknowledges the Chemistry/Biology Interface Training Program (NIGMS/NIH T32GM066706) and a Department of Pharmaceutical Sciences Merit Award.
ABBREVIATIONS USED:
- Has
heme assimilation system
- Phu
Pseudomonas heme uptake
- PPIX
protoporphyrin IX
- Sal
salophen
- GaPc
gallium phthalocyanine
- STD NMR
saturation transfer difference NMR
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsinfecdis.0c00138.
Figure S1: fluorescence quenching graphs for KD determination. Figure S2: heme receptor mutant controls. Figure S3: FeCl3 and GaSal optimization results/IC50 determination. Figure S4: pyoverdine biosynthesis deletion growth curves with FeSal and GaSal. Figure S5: coverage map for wild-type full-length HasAp used for HDX-MS. Figure S6: surface plasmon resonance traces for HasAp/HasR (PDF)
The authors declare no competing financial interest.
REFERENCES
- 1.CDC. (2019) in Antibiotic Resistance Threats in the United States, 2019; Center of Disease Control and Prevention, Atlanta, GA. [Google Scholar]
- 2.Kerr KG, and Snelling AM (2009) Pseudomonas aeruginosa: A Formidable and Ever-Present Adversary. J. Hosp. Infect 73, 338–344. [DOI] [PubMed] [Google Scholar]
- 3.Costerton JW (2001) Cystic Fibrosis Pathogenesis and the Role of Biofilms in Persistent Infection. Trends Microbiol 9, 50–52. [DOI] [PubMed] [Google Scholar]
- 4.Brüssow H (2012) Pseudomonas Biofilms, Cystic Fibrosis, and Phage: A Silver Lining? mBio 3, e00061–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burrows LL (2018) The Therapeutic Pipeline for Pseudomonas aeruginosa Infections. ACS Infect. Dis 4, 1041–1047. [DOI] [PubMed] [Google Scholar]
- 6.Lee HH, and Collins JJ (2012) Microbial Environments Confound Antibiotic Efficacy. Nat. Chem. Biol 8, 6–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dickey SW, Cheung GYC, and Otto M (2017) Different Drugs for Bad Bugs: Antivirulence Strategies in the Age of Antibiotic Resistance. Nat. Rev. Drug Discovery 16, 457–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Runyen-Janecky LJ (2013) Role and Regulation of Heme Iron Acquisition in Gram-Negative Pathogens. Front. Cell. Infect. Microbiol 3, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fetherston JD, Bertolino VJ, and Perry RD (1999) YbtP and YbtQ: Two ABC Transporters Required for Iron Uptake in Yersinia pestis. Mol. Microbiol 32, 289–299. [DOI] [PubMed] [Google Scholar]
- 10.Schryvers AB, and Stojiljkovic I (1999) Iron Acquisition Systems in the Pathogenic Neisseria. Mol. Microbiol 32, 1117–1123. [DOI] [PubMed] [Google Scholar]
- 11.Wandersman C, and Stojiljkovic I (2000) Bacterial Heme Sources: The Role of Heme, Hemoprotein Receptors and Hemophores. Curr. Opin. Microbiol 3, 215–220. [DOI] [PubMed] [Google Scholar]
- 12.Skaar EP (2010) The Battle for Iron between Bacterial Pathogens and Their Vertebrate Hosts. PLoS Pathog 6, e1000949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Takase H, Nitanai H, Hoshino K, and Otani T (2000) Impact of Siderophore Production on Pseudomonas aeruginosa Infections in Immunosuppressed Mice. Infect. Immun 68, 1834–1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Skaar EP, Humayun M, Bae T, DeBord KL, and Schneewind O (2004) Iron-Source Preference of Staphylococcus aureus Infections. Science (Washington, DC, U. S.) 305, 1626–1628. [DOI] [PubMed] [Google Scholar]
- 15.Konings AF, Martin LW, Sharples KJ, Roddam LF, Latham R, Reid DW, and Lamont IL (2013) Pseudomonas aeruginosa Uses Multiple Pathways To Acquire Iron during Chronic Infection in Cystic Fibrosis Lungs. Infect. Immun 81, 2697–2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marvig RL, Sommer LM, Molin S, and Johansen HK (2015) Convergent Evolution and Adaptation of Pseudomonas aeruginosa within Patients with Cystic Fibrosis. Nat. Genet 47, 57–64. [DOI] [PubMed] [Google Scholar]
- 17.Nguyen AT, O’Neill MJ, Watts AM, Robson CL, Lamont IL, Wilks A, and Oglesby-Sherrouse AG (2014) Adaptation of Iron Homeostasis Pathways by a Pseudomonas aeruginosa Pyoverdine Mutant in the Cystic Fibrosis Lung. J. Bacteriol 196, 2265–2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith AD, and Wilks A (2015) Differential Contributions of the Outer Membrane Receptors PhuR and HasR to Heme Acquisition in Pseudomonas aeruginosa. J. Biol. Chem 290, 7756–7766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mouriño S, Giardina BJ, Reyes-Caballero H, and Wilks A (2016) Metabolite-Driven Regulation of Heme Uptake by the Biliverdin IXβ/δ-Selective Heme Oxygenase (HemO) of Pseudomonas aeruginosa. J. Biol. Chem 291, 20503–20515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dent AT, Mouriño S, Huang W, and Wilks A (2019) Post-Transcriptional Regulation of the Pseudomonas aeruginosa Heme Assimilation System (Has) Fine-Tunes Extracellular Heme Sensing. J. Biol. Chem 294, 2771–2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Damron FH, Oglesby-Sherrouse AG, Wilks A, and Barbier M (2016) Dual-Seq Transcriptomics Reveals the Battle for Iron during Pseudomonas aeruginosa Acute Murine Pneumonia. Sci. Rep 6, 39172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Winkler ML, Papp-Wallace KM, Hujer AM, Domitrovic TN, Hujer KM, Hurless KN, Tuohy M, Hall G, and Bonomo RA (2015) Unexpected Challenges in Treating Multidrug-Resistant Gram-Negative Bacteria: Resistance to Ceftazidime-Avibactam in Archived Isolates of Pseudomonas aeruginosa. Antimicrob. Agents Chemother 59, 1020–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Braz VS, Furlan JPR, Fernandes AFT, and Stehling EG (2016) Mutations in NalC Induce MexAB-OprM Overexpression Resulting in High Level of Aztreonam Resistance in Environmental Isolates of Pseudomonas aeruginosa. FEMS Microbiol. Lett 363, fnw166. [DOI] [PubMed] [Google Scholar]
- 24.Létoffé S, Redeker V, and Wandersman C (1998) Isolation and Characterization of an Extracellular Haem-Binding Protein from Pseudomonas aeruginosa That Shares Function and Sequence Similarities with the Serratia marcescens HasA Haemophore. Mol. Microbiol 28, 1223–1234. [DOI] [PubMed] [Google Scholar]
- 25.Jepkorir G, Rodriguez JC, Rui H, Im W, Lovell S, Battaile KP, Alontaga AY, Yukl ET, Moenne-Loccoz P, and Rivera M (2010) Structural, NMR Spectroscopic, and Computational Investigation of Hemin Loading in the Hemophore HasAp from Pseudomonas aeruginosa. J. Am. Chem. Soc 132, 9857–9872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kumar R, Qi Y, Matsumura H, Lovell S, Yao H, Battaile KP, Im W, Moënne-Loccoz P, and Rivera M (2016) Replacing Arginine 33 for Alanine in the Hemophore HasA from Pseudomonas aeruginosa Causes Closure of the H32 Loop in the Apo-Protein. Biochemistry 55, 2622–2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kumar R, Matsumura H, Lovell S, Yao H, Rodríguez JC, Battaile KP, Moënne-Loccoz P, and Rivera M (2014) Replacing the Axial Ligand Tyrosine 75 or Its Hydrogen Bond Partner Histidine 83 minimally Affects Hemin Acquisition by the Hemophore HasAp from Pseudomonas aeruginosa. Biochemistry 53, 2112–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Caillet-Saguy C, Piccioli M, Turano P, Izadi-Pruneyre N, Delepierre M, Bertini I, and Lecroisey A (2009) Mapping the Interaction between the Hemophore HasA and Its Outer Membrane Receptor HasR Using CRINEPT–TROSY NMR Spectroscopy. J. Am. Chem. Soc 131, 1736–1744. [DOI] [PubMed] [Google Scholar]
- 29.Wojtowicz H, Prochnicka-Chalufour A, de Amorim GC, Roudenko O, Simenel C, Malki I, Pehau-Arnaudet G, Gubellini F, Koutsioubas A, Pérez J, Delepelaire P, Delepierre M, Fronzes R, and Izadi-Pruneyre N (2016) Structural Basis of the Signalling through a Bacterial Membrane Receptor HasR Deciphered by an Integrative Approach. Biochem. J 473, 2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shirataki C, Shoji O, Terada M, Ozaki S, Sugimoto H, Shiro Y, and Watanabe Y (2014) Inhibition of Heme Uptake in Pseudomonas aeruginosa by Its Hemophore (HasA p) Bound to Synthetic Metal Complexes. Angew. Chem., Int. Ed 53, 2862–2866. [DOI] [PubMed] [Google Scholar]
- 31.Shisaka Y, Iwai Y, Yamada S, Uehara H, Tosha T, Sugimoto H, Shiro Y, Stanfield JK, Ogawa K, Watanabe Y, and Shoji O (2019) Hijacking the Heme Acquisition System of Pseudomonas aeruginosa for the Delivery of Phthalocyanine as an Antimicrobial. ACS Chem. Biol 14, 1637–1642. [DOI] [PubMed] [Google Scholar]
- 32.Mandal SS (2011) Metallo-Salen Complexes Show Promise towards Treatment of Leukemia. Leuk. Res 35, 571–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vančo J, Šindelář Z, Dvořák Z, and Trávníceěk Z (2015) Iron-Salophen Complexes Involving Azole-Derived Ligands: A New Group of Compounds with High-Level and Broad-Spectrum in Vitro Antitumor Activity. J. Inorg. Biochem 142, 92–100. [DOI] [PubMed] [Google Scholar]
- 34.Sagasser J, Ma BN, Baecker D, Salcher S, Hermann M, Lamprecht J, Angerer S, Obexer P, Kircher B, and Gust R (2019) A New Approach in Cancer Treatment: Discovery of Chlorido[ N, N ′-Disalicylidene-1,2-Phenylenediamine]Iron(III) Complexes as Ferroptosis Inducers. J. Med. Chem 62, 8053–8061. [DOI] [PubMed] [Google Scholar]
- 35.Kaneko Y, Thoendel M, Olakanmi O, Britigan BE, and Singh PK (2007) The Transition Metal Gallium Disrupts Pseudomonas aeruginosa Iron Metabolism and Has Antimicrobial and Antibiofilm Activity. J. Clin. Invest 117, 877–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Apseloff G (1999) Therapeutic Uses of Gallium Nitrate: Past, Present, and Future. Am. J. Ther 6, 327–339. [DOI] [PubMed] [Google Scholar]
- 37.Chang D, Garcia RA, Akers KS, Mende K, Murray CK, Wenke JC, and Sanchez CJ (2016) Activity of Gallium Meso- and Protoporphyrin IX against Biofilms of Multidrug-Resistant Acinetobacter baumannii Isolates. Pharmaceuticals (Basel) 9 (1), 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hijazi S, Visca P, and Frangipani E (2017) Gallium-Protoporphyrin IX Inhibits Pseudomonas aeruginosa Growth by Targeting Cytochromes. Front. Cell. Infect. Microbiol 7, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Banin E, Lozinski A, Brady KM, Berenshtein E, Butterfield PW, Moshe M, Chevion M, Greenberg EP, and Banin E (2008) The Potential of Desferrioxamine-Gallium as an Anti-Pseudomonas Therapeutic Agent. Proc. Natl. Acad. Sci. U. S. A 105, 16761–16766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pandey A, Savino C, Ahn SH, Yang Z, Van Lanen SG, and Boros E (2019) Theranostic Gallium Siderophore Ciprofloxacin Conjugate with Broad Spectrum Antibiotic Potency. J. Med. Chem 62, 9947–9960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goss CH, Kaneko Y, Khuu L, Anderson GD, Ravishankar S, Aitken ML, Lechtzin N, Zhou G, Czyz DM, McLean K, Olakanmi O, Shuman HA, Teresi M, Wilhelm E, Caldwell E, Salipante SJ, Hornick DB, Siehnel RJ, Becker L, Britigan BE, and Singh PK (2018) Gallium Disrupts Bacterial Iron Metabolism and Has Therapeutic Effects in Mice and Humans with Lung Infections. Sci. Transl. Med 10, eaat7520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Silva P CF Foundation Grant to Support Testing of Bacterial Lung Infection Treatment https://cysticfibrosisnewstoday.com/2017/01/12/aridis-pharmaceuticals-receives-award-from-cystic-fibrosis-foundation-therapeutics-to-advance-panaecin/ (accessed April 2, 2020).
- 43.García-Contreras R, Pérez-Eretza B, Lira-Silva E, Jasso-Chávez R, Coria-Jiménez R, Rangel-Vega A, Maeda T, and Wood TK (2014) Gallium Induces the Production of Virulence Factors in Pseudomonas aeruginosa. Pathog. Dis 70, 95–98. [DOI] [PubMed] [Google Scholar]
- 44.Luscher A, Moynié L, Auguste P. Saint, Bumann D, Mazza L, Pletzer D, Naismith JH, and Köhler T (2018) TonB-Dependent Receptor Repertoire of Pseudomonas aeruginosa for Uptake of Siderophore-Drug Conjugates. Antimicrob. Agents Chemother 62, e00097–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ito A, Nishikawa T, Matsumoto S, Yoshizawa H, Sato T, Nakamura R, Tsuji M, and Yamano Y (2016) Siderophore Cephalosporin Cefiderocol Utilizes Ferric Iron Transporter Systems for Antibacterial Activity against Pseudomonas aeruginosa. Antimicrob. Agents Chemother 60, 7396–7401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bonchi C, Imperi F, Minandri F, Visca P, and Frangipani E (2014) Repurposing of Gallium-Based Drugs for Antibacterial Therapy. BioFactors 40, 303–312. [DOI] [PubMed] [Google Scholar]
- 47.Rangel-Vega A, Bernstein LR, Mandujano-Tinoco EA, Garcia-Contreras SJ, and Garcia-Contreras R (2015) Drug Repurposing as an Alternative for the Treatment of Recalcitrant Bacterial Infections. Front. Microbiol 6, 282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Braud A, Hannauer M, Mislin GLA, and Schalk IJ (2009) The Pseudomonas aeruginosa Pyochelin-Iron Uptake Pathway and Its Metal Specificity. J. Bacteriol 191, 3517–3525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mayer M, and Meyer B (2001) Group Epitope Mapping by Saturation Transfer Difference NMR To Identify Segments of a Ligand in Direct Contact with a Protein Receptor. J. Am. Chem. Soc 123, 6108–6117. [DOI] [PubMed] [Google Scholar]
- 50.Stojiljkovic I, Kumar V, and Srinivasan N (1999) Non-Iron Metalloporphyrins: Potent Antibacterial Compounds That Exploit Haem/Hb Uptake Systems of Pathogenic Bacteria. Mol. Microbiol 31, 429–442. [DOI] [PubMed] [Google Scholar]
- 51.Wolff N, Izadi-Pruneyre N, Couprie J, Habeck M, Linge J, Rieping W, Wandersman C, Nilges M, Delepierre M, and Lecroisey A (2008) Comparative Analysis of Structural and Dynamic Properties of the Loaded and Unloaded Hemophore HasA: Functional Implications. J. Mol. Biol 376, 517–525. [DOI] [PubMed] [Google Scholar]
- 52.Deredge DJ, Huang W, Hui C, Matsumura H, Yue Z, Moënne-Loccoz P, Shen J, Wintrode PL, and Wilks A (2017) Ligand-Induced Allostery in the Interaction of the Pseudomonas aeruginosa Heme Binding Protein with Heme Oxygenase. Proc. Natl. Acad. Sci. U. S. A 114, 3421–3426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Heinzl GA, Huang W, Robinson E, Xue F, Moënne-Loccoz P, and Wilks A (2018) The Asp99–Arg188 Salt Bridge of the Pseudomonas aeruginosa HemO Is Critical in Allowing Conformational Flexibility during Catalysis. JBIC, J. Biol. Inorg. Chem 23, 1057–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wong RJ, Vreman HJ, Schulz S, Kalish FS, Pierce NW, and Stevenson DK (2011) In Vitro Inhibition of Heme Oxygenase Isoenzymes by Metalloporphyrins. J. Perinatol 31, S35–S41. [DOI] [PubMed] [Google Scholar]
- 55.Thöny-Meyer L (2009) in Heme Transport and Incorporation into Proteins In Tetrapyrroles (Warren MJ, and Smith AG, Eds.), pp 149–159, Springer; New York, New York, NY. [Google Scholar]
- 56.Sudhamsu J, Kabir M, Airola MV, Patel BA, Yeh S-R, Rousseau DL, and Crane BR (2010) Co-Expression of Ferrochelatase Allows for Complete Heme Incorporation into Recombinant Proteins Produced in E. coli. Protein Expression Purif 73, 78–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cornelis P, and Dingemans J (2013) Pseudomonas aeruginosa Adapts Its Iron Uptake Strategies in Function of the Type of Infections. Front. Cell. Infect. Microbiol 3, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Moynié L, Milenkovic S, Mislin GLA, Gasser V, Malloci G, Baco E, McCaughan RP, Page MGP, Schalk IJ, Ceccarelli M, and Naismith JH (2019) The Complex of Ferric-Enterobactin with Its Transporter from Pseudomonas aeruginosa Suggests a Two-Site Model. Nat. Commun 10, 3673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Choi S-R, Britigan BE, and Narayanasamy P (2019) Iron/Heme Metabolism-Targeted Gallium(III) Nanoparticles Are Active against Extracellular and Intracellular Pseudomonas aeruginosa and Acinetobacter baumannii. Antimicrob. Agents Chemother 63, e02643–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Holloway BW (1955) Genetic Recombination in Pseudomonas aeruginosa. Microbiology 13, 572–581. [DOI] [PubMed] [Google Scholar]
- 61.Smith AD, Modi AR, Sun S, Dawson JH, and Wilks A (2015) Spectroscopic Determination of Distinct Heme Ligands in Outer-Membrane Receptors PhuR and HasR of Pseudomonas aeruginosa. Biochemistry 54, 2601–2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Studier FW (2005) Protein Production by Auto-Induction in High Density Shaking Cultures. Protein Expression Purif 41, 207–234. [DOI] [PubMed] [Google Scholar]
- 63.Samuelsen SV, Santilli C, Ahlquist MSG, and Madsen R (2019) Development and Mechanistic Investigation of the Manganese(Iii) Salen-Catalyzed Dehydrogenation of Alcohols. Chem. Sci 10, 1150–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fuhrop JH, and Smith KM, Eds. (1975) in Porphyrins and Metalloporphyrins, Elsevier, Amsterdam, The Netherlands. [Google Scholar]
- 65.Miller JH (1992) in A Short Course in Bacterial Genetics: A Laboratory Manual and Handbook for Escherichia coli and Related Bacteria, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. [Google Scholar]
- 66.Houde D, Berkowitz SA, and Engen JR (2011) The Utility of Hydrogen/Deuterium Exchange Mass Spectrometry in Biopharmaceutical Comparability Studies. J. Pharm. Sci 100, 2071–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.