

Abstract
Activated estrogen receptor (ERα) plays a critical role in breast cancer development and is a major target for drug treatment. Serine phosphorylation within the N-terminal domain (NTD) contributes to ERα activation and may also cause drug resistance. Previous biochemical identification of phosphorylated ERα residues was limited to protein artificially overexpressed in transfected cell lines. We report mass spectrometric methods that have allowed the identification of a new site within the NTD of ERα isolated from cultured human breast cancer cells. Immunoprecipitation, trypsin digestion, and analysis by nano-LC-ESI-MS/MS (Q-STAR, MDS Sciex) and vMALDI-MSn (Finnigan™ LTQ™, Thermo-Electron) identified peptides containing 8 of 14 serine residues within the NTD, one being partially phosphorylated Ser-167, known but not previously reported by MS. Chymotrypsin digestion revealed other known sites at Ser-102/104/106 and 118. Tandem methods developed for the peptide containing Ser-118 and the use of hypothesis-driven experiments—i.e., the assumption that an intact phosphopeptide showing no molecular ion might yield fragment ions including loss of phosphoric acid in vMALDI-MS/MS—allowed the identification of a novel site at Ser-154. Quantitation by selected reaction monitoring demonstrated 6-fold and 2.5-fold increases in Ser-154 phosphorylation in estradiol- and EGF-treated cells, respectively, compared to controls, confirmed by immunoblotting with a novel rabbit polyclonal antibody. Thus, the protein isolation and MS strategies described here can facilitate discovery of novel phosphorylation sites within low abundance, clinically important cancer targets like ERα, and may thereby contribute to our understanding of the role of phosphorylation in the development of breast cancer.
Estrogen receptor alpha (ERα) belongs to the superfamily of nuclear receptors displaying a characteristic modular domain structure (Figure 1) consisting of N-terminal domain (NTD), DNA binding, hinge and C-terminal ligand binding domains [1–3]. Of ERα’s two distinct, non-acidic activation function domains (AF-1, AF-2), the C-terminal AF-2 domain is highly conserved among species and other nuclear hormone receptors and becomes activated only in response to ligand binding, whereas the N-terminal AF-1 domain is less well conserved [2–6] and displays an unfolded structure yet becomes activated in response to both ligand-dependent and -independent mechanisms [1, 7]. The distribution and site occupancy of posttranslational modifications (PTMs), particularly serine phosphorylation, are emerging as important indicators of ERα functional status [2]. With five (Ser-102, Ser-104, Ser-106, Ser-118, Ser-167) of the NTD’s 14 serine residues known to be targets of phosphorylation determining recruitment of various ERα cofactors, this domain clearly coordinates many aspects of ERα activation [2, 3, 7–10]. In vitro and clinical studies also suggest that molecular cross talk involving various growth-factor signaling pathways, particularly those leading to phosphorylation of ERα-NTD, contribute to de novo and acquired endocrine resistance, a major problem in breast cancer treatment [2, 8, 11–21].
Figure 1.
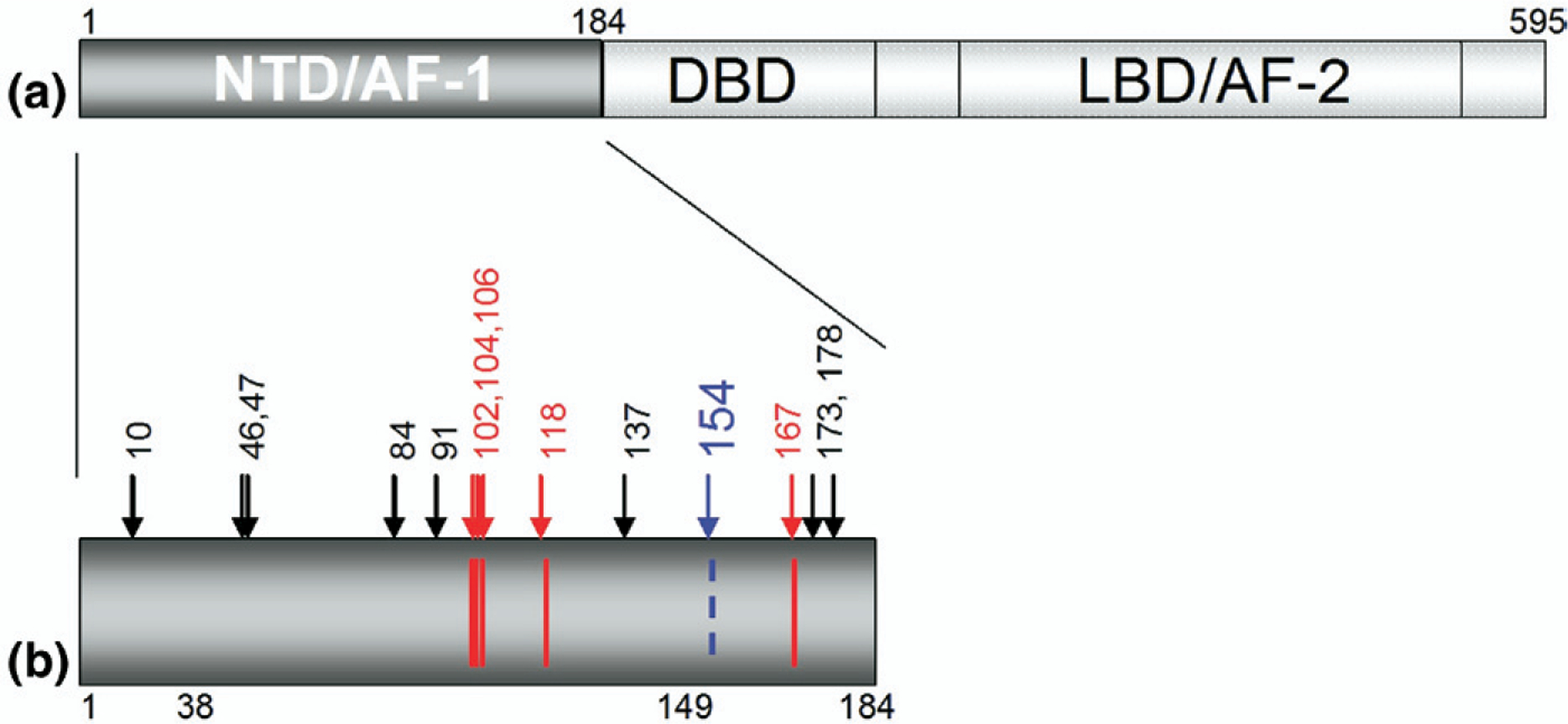
(a) The domain structure of ERα, including the largely unstructured N-terminal domain containing AF-1 (NTD/AF-1), the DNA-binding domain (DBD), and the ligand binding domain containing AF-2 (LBD/AF-2). (b) Detail of the NTD of ERα. Arrows indicate the 14 serine residues within the NTD. Phosphorylation has been reported for the 5 serine residues shown in red (102, 104, 106, 118, and 167). Residue 154 (shown in blue) is identified here as a novel serine phosphorylation site.
A number of groups have reported a major role for Ser-118 phosphorylation in the ligand-induced activation of ERα, given that mutation of this serine to alanine results in a diminished transcriptional response to estradiol activation compared with wild-type ERα [7]. Also, ligand-induced phosphorylation of Ser-167, although initially a contested issue, is now clearly recognized as regulating both the transactivating activity of ERα-NTD and ERα DNA-binding [2, 22–25]. In the context of ligand-independent ERα stimulation, as with EGF growth induction under serum deprivation, both of the aforementioned NTD serine residues become phosphorylated [7]. However, although many of the candidate kinases directing phosphorylation of NTD serine residues have been described, it is still unknown what constellation of phosphorylated serine residues determines the various activated states of ERα.
To date, no phosphorylation of endogenous ERα has been reported to be observed by mass spectrometry. Nevertheless, in keeping with suspicions that critical serine phosphorylation sites within ERα-NTD might remain undetected [26, 27], the present study interrogated the phosphorylation status of endogenous ERα-NTD using mass spectrometry (MS) and tandem MS (MS/MS) after estradiol (E2) or growth factor (EGF) stimulation of ER-positive human breast cancer cells (MCF-7). MS procedures have become an increasingly important analytical means of detecting phosphorylated serine/threonine and tyrosine residues [28, 29] due to the high level of sensitivity and unbiased ability of MS to scan proteolytic digests for various PTMs; and of particular utility given focus on the ERα-NTD was the hypothesis-driven multi-stage mass spectrometric approach described by Chang et al. [30]. Earlier studies also pointed to the advantages of using different MS platforms to optimize protein coverage and peptide fragment sensitivity, since proteolytic peptides can exhibit differential responses to alternative ionization conditions, exemplified by the use of both ESI-MS/MS and vacuum (v)MALDI-MSn platforms to confirm known and identify new phosphorylation sites within the key protein (huntingtin) involved in Huntington’s disease [31]. In the present MS study of endogenous ERα extracted and purified from ligand-dependent (E2) versus ligand-independent (EGF) stimulated human breast cancer cells, we sought to avoid potential artifacts associated with analysis of recombinant synthetic protein or ectopically overexpressed intracellular ERα, but this was reliant on the development of efficient and MS-compatible immunocapture and ERα purification steps that would be insensitive to the presence or absence of possible PTMs, particularly phosphorylation. Furthermore, because we sought to carry out relative quantitative measurements it was essential to preserve the ratios of modified to unmodified peptides that would be representative of any phosphorylation within the original protein. A phospho-enrichment procedure would negate this and would likely result in differential enrichment of the various phosphorylated species; therefore we avoided using enrichment procedures such as IMAC (immobilized metal-ion affinity chromatography) [32–35], strong cation exchange [36,37], or phospho-specific immunoprecipitation [38]. Using an optimized MS approach developed by monitoring known phosphorylation sites, Ser-118 and Ser-167, we demonstrated that Ser-154 in ERα-NTD, long suspected but unconfirmed as being phosphorylated under endogenous intracellular conditions [27], is indeed partially phosphorylated under both ligand-dependent and ligand-independent human breast cancer growth conditions.
Experimental
Materials
2-Methylnaphthalene-1,4-dione (Menadione), 17-β-estradiol, dithiothreitol (DTT), iodoacetamide (IAA), formic acid (FA), and trifluoroacetic acid (TFA) were purchased from Sigma–Aldrich (St. Louis, MO, USA). The recombinant α isoform of ER (66.2 kDa) was obtained from Pan Vera (Madison, WI, USA). Agarose conjugated (sc8002AC) and free anti-ERα F10 antibodies (sc8002) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Phospho-S104/106, phospho-SER-118, and phospho-SER-167 antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). Sequencing-grade modified porcine trypsin was from Promega (Madison, WI, USA) and sequencing-grade modified bovine chymotrypsin was from Princeton Separations (Adelphia, NJ, USA). HPLC solvents, acetonitrile (ACN), and water were obtained from Honeywell Burdick and Jackson (Muskegon, MI, USA), 2,5-dihydroxybenzoic acid (DHB) from LaserBio Labs (Sophia Antipolis, France), and α-cyano-4-hydroxycinnamic acid (CHCA) from Agilent Technologies (Santa Clara, CA, USA).
Cell Culture
Recombinant ERα was used to quantitate the yield of protein derived from the immunoprecipitation (IP) step and to optimize digestion and peptide analysis by MS but all the data presented here were obtained on ERα extracted from the MCF-7 human breast cancer cell line. This was obtained from the American Type Culture Collection (Manassas, VA, USA) and maintained in Dulbecco’s modified Eagle’s medium (Mediatech, Herndon, VA, USA) supplemented with 10% fetal bovine serum (Mediatech), 1% penicillin/streptomycin (Mediatech), and 10 μg/ml insulin (Sigma). Treatment conditions for all cell lines involved plating approximately 3 × 106 cells in normal media onto 15-cm dishes and attachment and growth for 24 h. Before estradiol (E2) treatment (10 nM, 30 min), cells were grown in estrogen-free culture conditions (phenol red-free Dulbecco’s modified Eagle’s medium-H-16 supplemented with 10% charcoal-stripped serum, 1% penicillin/streptomycin, and 10 μg/ml insulin) for 24 h to eliminate any artifactual estrogen stimulatory influences or estrogen metabolism to catechols and quinones. Before EGF treatment (50 ng/ml, 10 min) cells were grown in serum-free conditions for 24 h to eliminate any artifactual growth factor influences present in the fetal calf serum.
IP, SDS-PAGE, and In-Gel Proteolytic Digestion
After aspiration of culture media and one room-temperature phosphate-buffered saline (PBS) wash, cells from 20 × 15-cm plates were harvested on ice using a cell scraper and 0.8 mL of ice-cold cell lysis buffer [100 mM NaCl, 20 mM Tris (pH 7.5), 0.5% IGEPAL, 100 mM NaF, 10 mM Na3VO4, 50 mM Na2MoO4, 10 μL/mL phosphatase inhibitor cocktail I (Sigma–Aldrich), 320 nM Okadaic acid, 100 U/mL deoxyribonuclease I and 1 tablet/10 mL Roche mini-complete protease inhibitor cocktail]. Cellular contents were transferred to 1.5-mL Eppendorf tubes, sonicated (10 s) twice per tube, then centrifuged at 16,000 rpm (30 min, 4 °C) and collected into one 50-mL universal tube (~25 mL total). Agarose-conjugated F-10 ERα antibody (160 μL, prewashed in 0.5% IGEPAL/PBS buffer) was added to the cell lysate and allowed to incubate by gently rotating in a cold room overnight. The immune complex was centrifuged at 1000 rpm at 4 °C for 1 min, supernatant removed, and the complex transferred to a 1.5-mL Eppendorf tube and washed with a PBS-based buffer (PBS, 0.5% IGEPAL) for 10 min at room temperature. The wash, centrifugation, and elution of supernatant were repeated three more times and the resultant pellet was re-suspended in 20 μL 4× protein elution buffer [100 mM NaCl, 4% (wt/vol) SDS, 25 mM Tris (pH 7.5)]. Samples were heated to 92 °C for 7 min then the elution buffer was removed from the beads using a gel-loading tip and stored in a separate Eppendorf tube. The heating, removal, and storage of elution buffer were repeated twice, first after adding 15 μL of 4× elution buffer and, second, after adding 10 μL of 4× elution loading buffer. A razor blade was used to remove the tip of the Eppendorf tube (containing the Ab-beads and inaccessible elution buffer) placed into a 100 MW Microcon (Millipore, Bedford, MA, USA) centrifugal filter device (tip pointing up) and centrifuged at 11,000 g for 5 min to remove all remaining elution buffer and proteins from the Ab-beads. This too was added to the separate Eppendorf tube containing the previously eluted proteins (~55 μL final volume). Loading buffer [10 μL; 100 mM NaCl, 25 mM Tris (pH 7.5), 30% glycerol, 0.5% IGEPAL, 4% SDS, bromophenol blue] was added to the eluted protein solution and heated at 92 °C for a further 5 min. All 65 μL of sample was added to one well on a1.5-mm-thick 10-well Nu-PAGE 4 to 12% Bis-Tris gel (Invitrogen, Carlsbad, CA, USA). After electrophoresis, gels were stained with the Imperial protein dye (Pierce, IL, USA), then destained in water. Bands at 66 kDa (ERα) were excised, chopped, destained, and dehydrated with ACN. Proteins were subsequently reduced with 10 mM dithiothreitol in 25 mM NH4HCO3 at 56 °C for 1 h and alkylated with 55 mM iodoacetamide in 25 mM NH4HCO3 at room temperature for 45 min. Samples were then washed, dried, rehydrated on ice for 10 min in 125 ng of sequence-grade trypsin or 200 ng chymotrypsin, reconstituted in 25 mM NH4HCO3, covered with an additional 20 μL NH4HCO3 solution, and incubated overnight, either at 37 °C or room temperature, respectively. The resulting proteolytic peptides were subjected to aqueous (30 μL NH4HCO3, 20 min vortex) and hydrophobic extraction (2 × 30 μL of 50% ACN, 5% formic acid, 20 min vortex, 10 min sonication), concentrated under vacuum to 20 μL, and stored at −80 °C until used for mass spectrometry.
Mass Spectrometry
Unseparated proteolytic peptide mixtures were analyzed by MALDI-TOF-MS on a Voyager DESTR Plus instrument (Applied Biosystems, Foster City, CA, USA). The peptide mass fingerprints confirmed the identity of both commercial recombinant ERα and that extracted from the MCF-7 cells by IP. The latter samples were further analyzed by reverse-phase nano-HPLCMS/MS using an Ultimate HPLC (Dionex, Sunnyvale, CA, USA) with the C18 analytical column directly connected to a Q-STAR Pulsar I quadrupole orthogonal TOF mass spectrometer (MDS Sciex, Concorde, Canada) as previously described [31]. In addition, unseparated peptide mixtures were analyzed by MALDI-MS, MS/MS, MS3, and MS4 on a vMALDI-LTQ™ linear ion trap (Thermo Fisher, San Jose, CA, USA) with 2,5-dihydroxybenzoic acid (DHB; LaserBio Labs) in 10% ACN/0.5% TFA as matrix. All ions observed in the vMALDI-MS spectrum were subjected to MS/MS to probe for the neutral loss of phosphoric acid (98 Da)[30]. Species that showed this loss were interrogated further by MS3 by selecting the (M + H − 98)+ peak as precursor ion. For the hypothesis-driven investigation, all ERα-NTD peptides observed to contain unmodified serine residues were interrogated for possible phosphorylation by carrying out MS/MS at a mass 80 m/z units greater than the observed molecular ion, irrespective of whether any ions could be observed at this mass [28, 29].
For relative quantitation, vMALDI-LTQ in selected reaction monitoring (SRM) mode was used to monitor the levels of Ser-154 phosphorylation in the peptide corresponding to residues 143–158 (EAGPPAFYRPNSDNRR) between different treatment samples, compared with the untreated control sample. A method was developed to carry out three scan events: (1) an MS scan (1000–2000 m/z), (2) an SRM MS/MS scan at 1927.00 m/z ([H11006]1.5 m/z) monitoring 1215.0–1221.0 m/z to measure the intensity of the c11 fragment ion (1217.6 m/z) from the phosphorylated peptide, and (3) an SRM MS/MS scan at 1847.00 m/z (±1.5 m/z), again monitoring 1215.0–1221.0 m/z to measure the intensity of the same c11 fragment ion from the corresponding unmodified peptide. This trio of scan events was carried out 30 times for each sample spot and repeated 4 times for each treatment sample. There were two biological repeats. The unmodified to phosphorylated c11 ion intensity ratio was then used to represent the level of Ser-154 phosphorylation in each treatment sample as a fold induction relative to the untreated control sample. The following formula was used to calculate any treatment induced increase in Ser-154 phosphorylation from the vMALDI data:
Bioinformatics and Protein Database Searches
MALDI-TOF-MS data were analyzed using Data Explorer 4.0™ (Applied Biosystems) and peptide mass fingerprints were searched against the Swiss-Prot mammalian database using Mascot (Matrix Sciences, London, UK). For ESI-MS/MS datasets, tandem mass spectra were submitted to our Mascot in-house database search engine (version 2.1) [39]. vMALDI-MSn data were analyzed using Xcalibur 2.0 SR2 software and database searches to identify phosphopeptides were performed using Bioworks software version 3.2 (Turbo SEQUEST search engine) allowing for mass accuracies of 2 Da (for vMALDI-MS precursor ions) and 1 Da (for vMALDI-MSn fragment ions) [40]. For the QSTAR LC-MS/MS datasets, three-dimensional images of the total ion chromatogram were generated using MSight 1.0 (Swiss Institute of Bioinformatics, http://www.expasy.org/MSight/).
Phospho-Ser-154 Antibody Production
A 14 amino acid peptide containing phosphorylated Ser-154 with an N-terminal cysteine residue added for KLH coupling was used to immunize two rabbits (Anaspec, San Jose, CA, USA). Following boostings with ELISA assays confirming antibodies specific for the phospho-Ser-154 peptide relative to the non-phosphorylated peptide, serum from both rabbits was affinity purified on a phospho-Ser-154 peptide column. Antibodies eluting from the phospho-Ser-154 column were reselected using a non-phosphorylated Ser-154 peptide column with the flow through fraction constituting the phospho-Ser-154 antibody pool used for Western analysis.
Results and Discussion
Phosphorylation Status of ERα Purified from MCF-7 Human Breast Cancer Cells
Because the protocol used to isolate ERα from MCF-7 whole cell lysates involved 12 h of immunopurification using an agarose-coupled ERα antibody, it was essential to ensure complete inactivation of endogenous phosphatases (see Experimental section) and full preservation of ERα phosphorylation status. Using commercially available antibodies to detect phosphorylated ERα serine residues, including phospho-Ser-104/106, phospho-Ser-118, and phospho-Ser-167, aliquots of the immunopurified ERα from E2-treated, control E2 (charcoal-stripped serum, phenol red-free media), EGF-treated, and control EGF (phenol red-free media) MCF-7 cells were analyzed on Western blots. As shown in Figure 2a, the phosphorylation status of the immunopurified ERα recapitulated known results [2, 7, 24], with Ser-118 exhibiting the greatest phosphorylation in response to E2, although still robustly phosphorylated following EGF treatment, in which Ser-167 was the dominantly phosphorylated serine in response to EGF, although still significantly phosphorylated in response to E2, and in which Ser-104/106 showed the greatest phosphorylation with E2 treatment while becoming less phosphorylated with EGF treatment.
Figure 2.
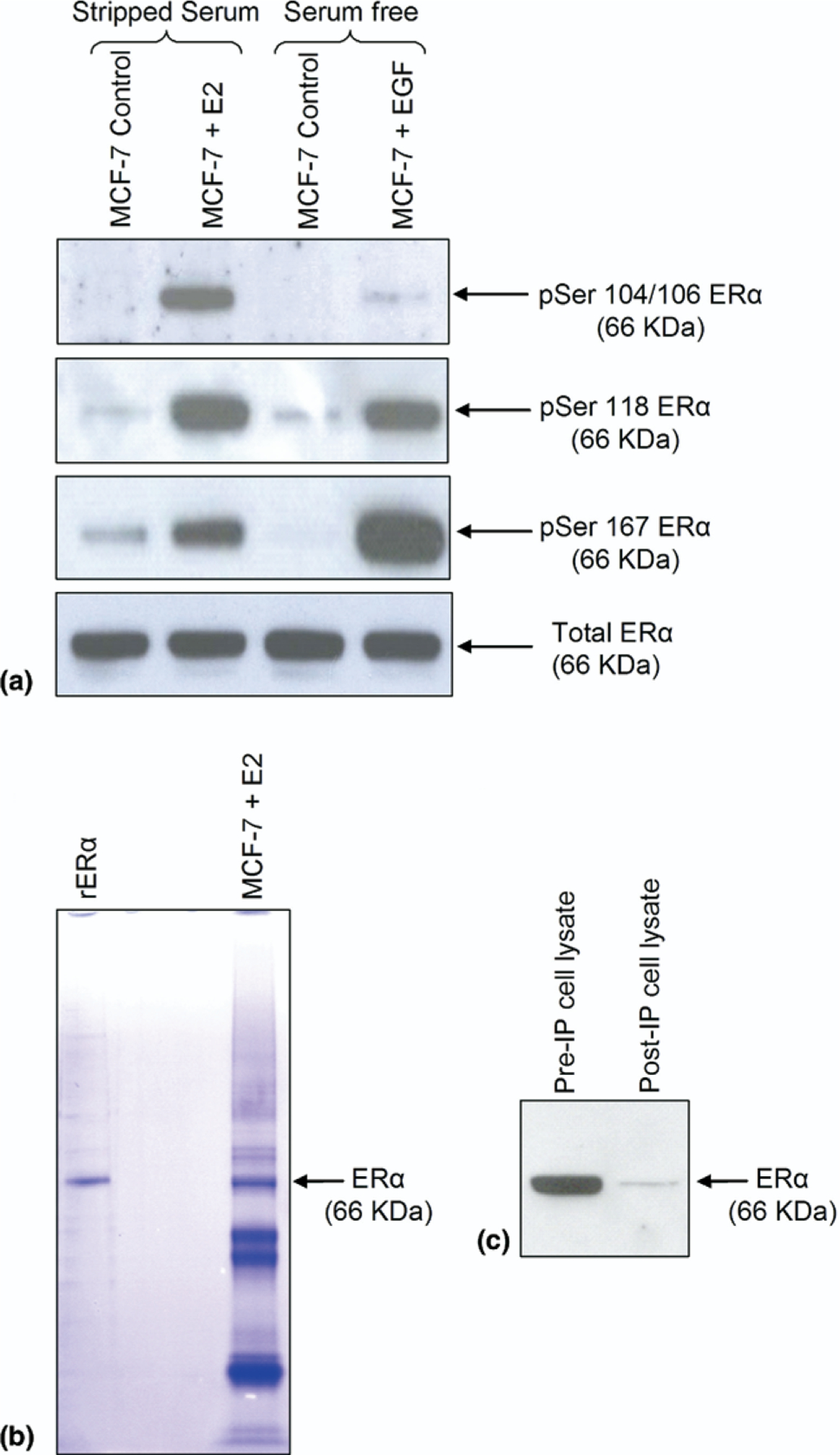
(a) Phospho-ER. Western analysis of total and phosphorylated ERα at serine residues 104/106, 118, and 167 following immunoprecipitation from MCF-7 cells treated with E2 (10 nM), EGF (50 ng/mL), or appropriate control. (b) Immunoprecipitation and one-dimensional SDS-PAGE. Coomassie-stained ERα immunoprecipitated from MCF-7 cellular lysates using the agarose conjugated anti-ERα antibody (F-10), versus commercially obtained recombinant ERα (rERα). (c) Western blot analysis; 10 μL aliquots from the same MCF-7 cell lysate before and after immunoprecipitation, probed with the F-10 antibody.
Having confirmed the preservation of ERα phosphorylation following its immunopurification, large-scale isolation of ERα for MS analysis was performed using approximately 108 human breast cancer MCF-7 cells per treatment condition. Using commercially available recombinant human ERα (rERα) as a standard, the immunoprecipitated ERα was subjected to one-dimensional SDS gel electrophoresis. As shown in Figure 2b, following gel staining, a clear protein band at 66 kDa was apparent from the immunoprecipitated ERα preparation, whose mobility was consistent with the rERα band. Based on the relative intensity of this band derived from 6 pmol rERα, it was estimated that approximately 6–10 pmol of ERα was extracted from 108 MCF-7 cells, which is consistent with the estimated MCF-7 content of about 80 fmol ERα/106 unstimulated cells. Additionally, Western analysis (Figure 2c) and densitometry of the cell lysate before and after ERα immunoprecipitation demonstrated that the ERα immunocapture procedure was roughly 95% efficient.
E2-Induced Phosphorylation of Ser-167
The region of the ERα-NTD spanning residues 121–174 was previously suspected to contain unidentified phosphorylated serine residues [26, 27]. This was predicted to yield informative peptides upon separate tryptic and chymotryptic digestion that would be optimal for MS analysis. Following proteolysis of the immunocaptured ERα band purified from E2-treated MCF-7 cells, the resulting peptides were separated by on-line nano-HPLC reversed-phase C18 chromatography and analyzed by a hybrid quadrupole time-of-flight mass spectrometer (Q-STAR). Selected peptide ions were fragmented by collision-induced dissociation (CID) to obtain peptide sequence information. The ESI-MS/MS spectra were searched with Mascot (Matrix Science, Boston, MA, USA) using a custom-designed ERα database. Trypsin digestion resulted in the detection of peptides covering 8 of the 14 ERα-NTD serine residues in unmodified form (Ser-10, 46, 47, 137, 154, 167, 173, and 178). In addition, we were able to detect partial phosphorylation of the known ERα-NTD phosphorylation site Ser-167, the phosphopeptide giving a Mascot score of 50, validated by manual inspection and assignment of the fragment ions. Figure 3a shows the ESI-MS/MS spectrum of the triply protonated precursor ion [M + 3H]3+ at m/z 584.9 (M = 1751.7) of the mono-phosphorylated tryptic peptide LApS167TNDKGSM*AM*ESAK, residues 165–180, with M* corresponding to mono-oxidized methionine (i.e., +16 Da per modified residue). This CID spectrum is consistent with the presence of a phosphate group at Ser-167. Fragment ions corresponding to y- and b-ions were observed, including several featuring the loss of 98 Da,i.e., y15−98 and y14−98, attributable to the elimination of a neutral phosphoric acid moiety (represented as −P). Significantly, MS/MS analysis of this mono-phosphorylated peptide produced no evidence for phosphorylation of the two other serine residues situated within this peptide fragment, Ser-173 or Ser-178. Although the fragmentation pattern did not discount threonine-168 as a phosphorylated residue on this peptide, the Western blotting data using the phospho-Ser-167 antibody and previous studies provide unequivocal evidence that the phosphorylated residue on this proteolytic peptide is Ser-167.
Figure 3.
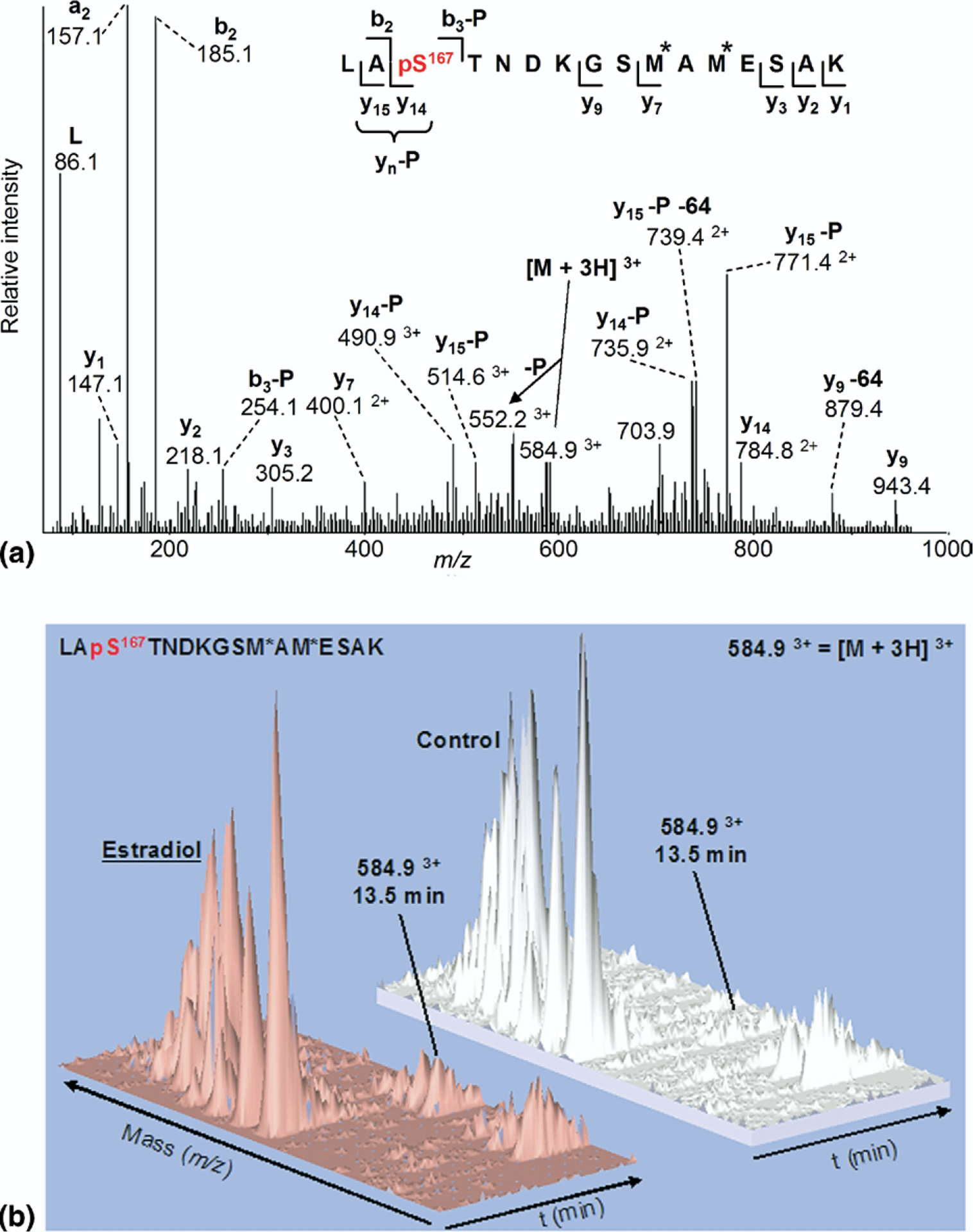
(a) ESI-MS/MS spectrum of triply charged monophosphorylated peptide LApS167TNDKGSM*AM*ESAK (residues 165–180) at m/z 584.9 (M = 1751.72) from tryptic digestion of ERα isolated from MCF-7 cells treated with E2 (M* = mono-oxidized methionine.) (b) three-dimensional plots of ESI-MS representing m/z, retention time, and ion intensity on the x, y, and z axes, respectively, showing increased ion current for phosphorylated peptide m/z = 584.9, eluting at 13.5 min, from cells treated with estradiol compared to untreated control.
Three-dimensional plots illustrated in Figure 3b compare MS data for the Ser-167 phosphopeptide from cells grown in the absence (control) or presence of E2. The plots were generated using MSight 1.0, which displays the Q-STAR total ion chromatogram with m/z, chromatographic retention time, and ion intensity plotted on the x, y, and z axes, respectively. When normal ized to the unmodified peaks, following E2 induction this demonstrates an approximate 8:1 increase in Ser-167 phosphorylation relative to control, consistent with the previous Western blots, thus corroborating the immuno-affinity results in Figure 2. All other peaks remain approximately the same intensity.
Chymotrypsin Digestion Revealed Other Known Phosphorylation Sites
A substantial section of the NTD is devoid of basic residues and is therefore resistant to trypsin digestion. Chymotrypsin was used in an alternative digestion scheme to probe the phosphorylation status of other serine residues not seen in the tryptic digest. These include Ser-102/104/106 and 118, previously identified as being phosphorylated, although not reported by mass spectrometry. Analysis of the unseparated peptide mixture derived from chymotryptic digestion of the 66-kDa band again confirmed the identity of ERα from the MCF-7 cells. To validate our method of detection of protein phosphorylation, the known phosphorylation site, Ser-118, was probed using an adaptation of the hypothesis-driven multiple stage mass spectrometric approach used by Chang et al. [30]. Briefly, a 3-Da window was selected for vMALDI-MS/MS at 80 m/z higher than the unmodified Ser-118–containing peptide for the presence (or absence) of phospho-Ser-118, based on the preferential neutral loss of phosphoric acid (−98 Da, or −P). Subsequent MS3 was carried out on the corresponding (MH−P)+ peak to provide sequence data to confirm or reject the presence of the phosphorylated residue. Figure 4a shows the vMALDI-MS/MS spectrum of peptide M*LLHPPPQLpS118PF (* = methionine sulfoxide). When searched with Sequest using the Bioworks 3.2 engine this gave an XCorr score of 2.05 (scores >1.5 are significant). An abundant ion at m/z 1374.58 confirmed the 98-Da neutral loss from the predicted [M + H]+ precursor ion at m/z 1472.60. The loss of 64 Da from the molecular ion also characterized the presence of methionine sulfoxide. Further fragmentation was indicated by the presence of the y8, y8−P, and b10−P ions. The fragment ion in the MS/MS spectrum corresponding to the 98-Da neutral loss was selected for MS3 analysis (Figure 4b) and Sequest gave an XCorr score of 2.53. This spectrum revealed a characteristic set of sequence ions (y6,7,8, b9,11), the internal ion PPPQL−28 and again the loss of 64 Da. The sequence ions confirmed the phosphorylation site as Ser-118, now converted to dehydro-alanine (shown as dS), which validated this technique for phosphopeptide identification in ERα. It is noteworthy that this peptide gave almost no signal by ESI-MS, so in this case the use of MALDI was essential, whereas the reverse was true for the Ser-167-phosphopeptide.
Figure 4.
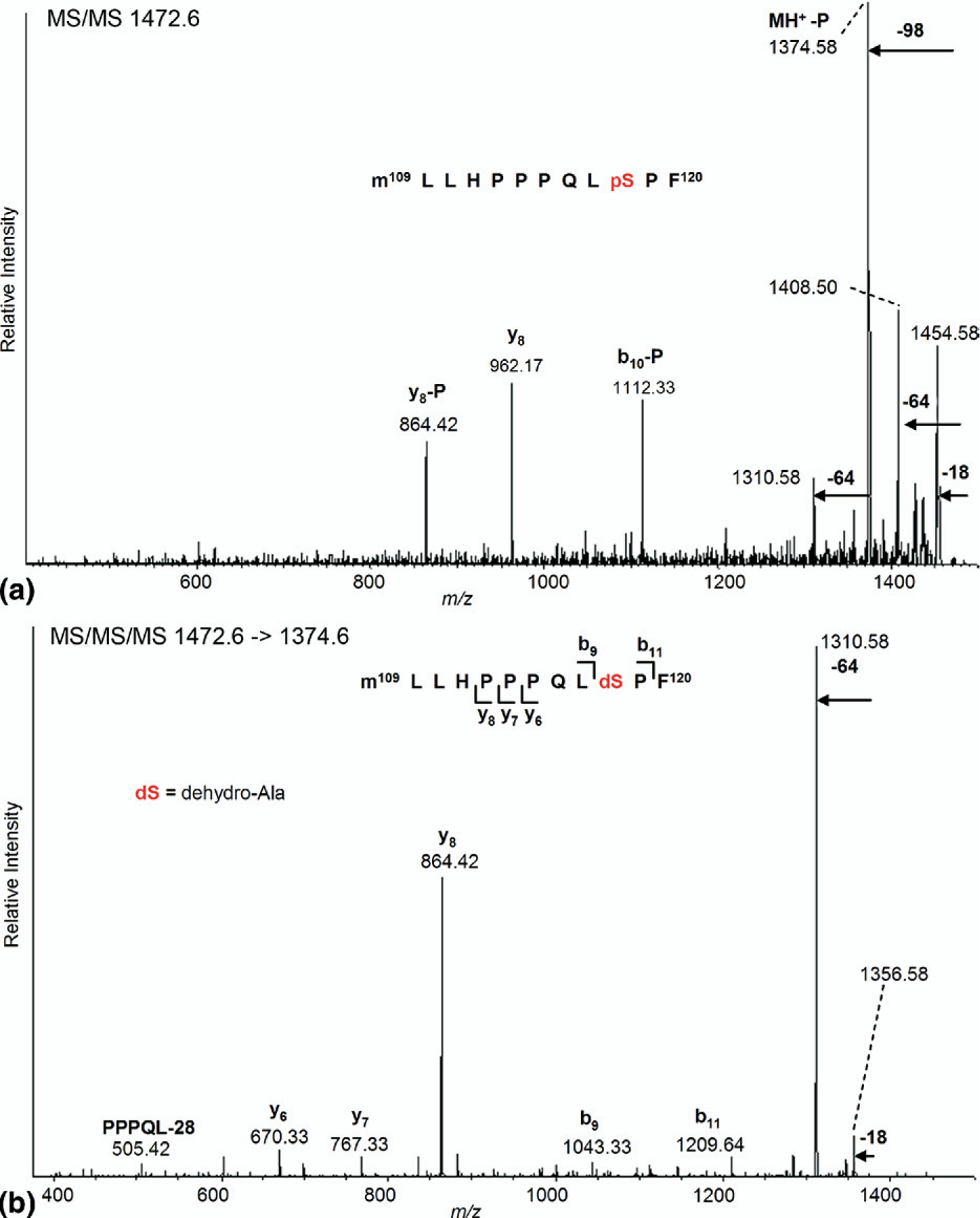
vMALDI-MS/MS and MS3 identification of phosphorylated peptide M*LLHPPPQLpS118PF (residues 109–120) from chymotrypsin-digested endogenous ERα. (a) MS/MS of the putative precursor ion at m/z 1472.60 showing a characteristic loss of 98 Da. (b) MS3 of the neutral loss fragment at m/z 1374.58, confirming the conversion of phospho-Ser-118 to dehydro-alanine.
Detection of a Novel ERα Phosphorylation Site by Neutral Loss Scanning
Despite earlier studies suggesting that Ser-154 might be a phosphorylation site, neither the ESI-MS/MS data nor vMALDI-MS of the tryptic peptides from cells that had not been treated with estradiol or EGF showed peaks that could correspond to this species, even though vMALDI-MS showed a significant peak for an unphosphorylated peptide containing Ser-154, EAGPPAFYRPNS154DNRR (m/z 1847.0). Previous studies using a vMALDI-LTQ quadrupole linear ion trap had demonstrated the power of applying a hypothesis-driven multiple-stage MS approach for the detection of novel phosphorylation sites on huntingtin protein previously undetected by ESI (Q-STAR) [31]. We used a modified version of this same approach (as used in Figure 4) to interrogate the peptides from trypsin digests of ERα isolated from E2-treated MCF-7 cells to search for previously undetected serine phosphorylation sites. Applying this neutral loss scanning MS approach to interrogate all MCF-7–derived ERα tryptic peptides observed with unmodified serine residues, phosphorylation on ERα Ser-154 was identified, as shown by the spectra illustrated in Figure 5. Figure 5a shows the vMALDI-MS/MS spectrum of the ERα peptide EAGPPAFYRPNpS154DNRR obtained from E2- treated MCF-7 cells with very low Sequest XCorr score(0.78, below the level of significance). However, an abundant ion at m/z 1829.1 confirmed the 98-Da neutral loss from the [M + H]+ precursor ion at m/z 1927.0+ and further fragmentation was indicated by c11 and b11 ions. The 98-Da neutral loss fragment from the MS/MS spectrum was selected for MS3 analysis (Figure 5b) and this spectrum gave an excellent Sequest XCorr (2.47); this spectrum also revealed an interesting set of sequence ions (c11, b11, b11−H2O, b15, b11−H2O, b15+H2O). The c11 fragment ion (1217.7 m/z) was further fragmented (MS4, inset Figure 5b) revealing the sequence ions (b7 and b9, y8−NH3, y10) providing further evidence that the phosphorylation site was the serine 154 residue. It is also noteworthy that the ions at 1200.5 and 1182.3 in the MS4 spectrum mimic exactly the b11 and b11−H2O ions in the MS3 spectrum.
Figure 5.
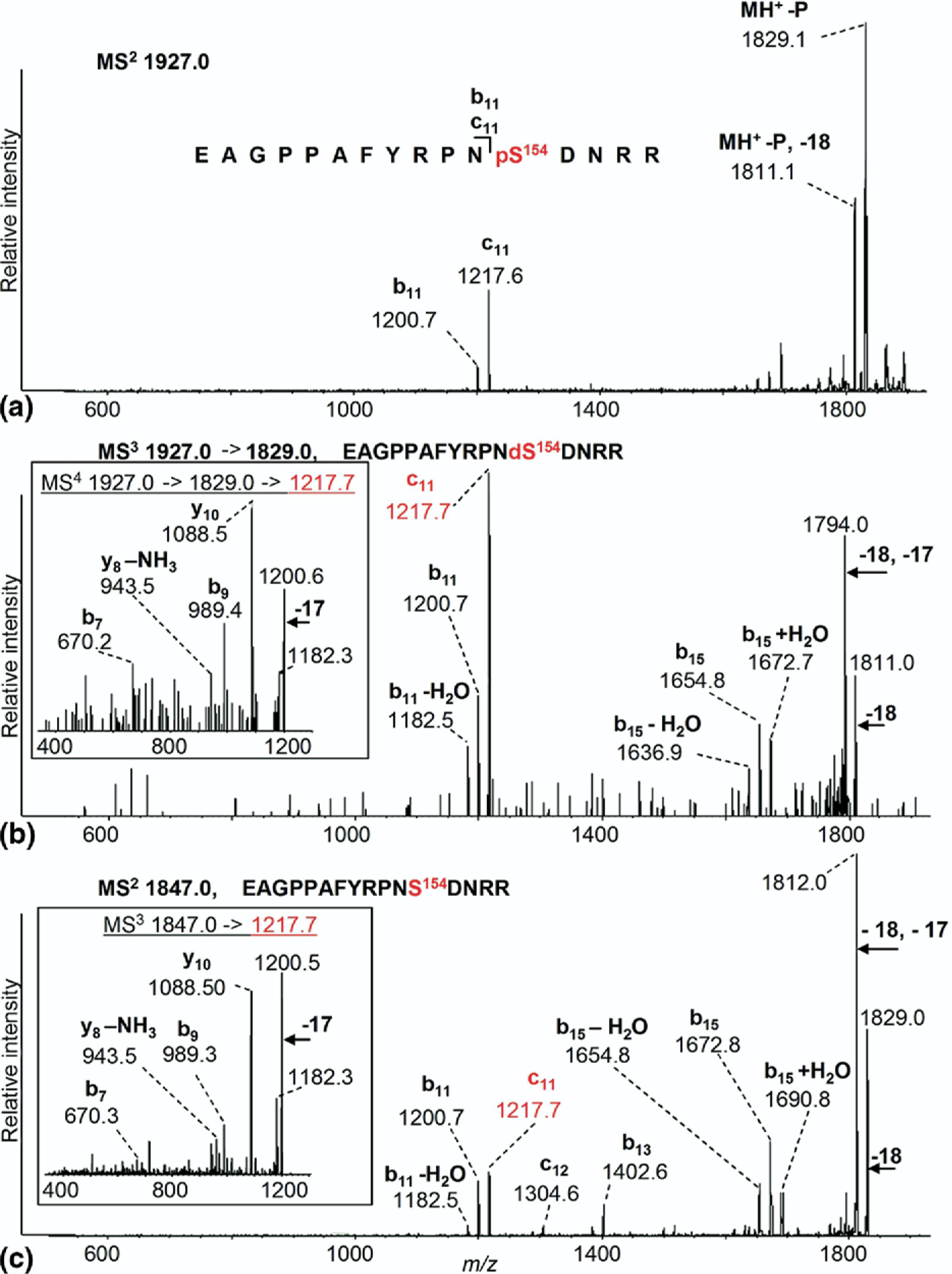
vMALDI-MS/MS, MS3 and MS4 identification of phosphorylated tryptic peptide EAGPPAFYRPNpS154DNRR (residues 143–158) from MCF-7 cells treated with E2. (a) The precursor ion [M + H]+ at m/z 1927.0 selected for MS/MS analysis. −P represents loss of phosphoric acid, i.e., loss of 98 Da. (b) MS3 spectrum of m/z 1829.0 [M+H−98]+ confirming that Ser-154 is now dehydro-alanine. The c11 fragment ion (1217.7 m/z) was fragmented (MS4; for additional sequence information see inset). (c) MS/MS of the unmodified peptide at m/z 1847.0 corresponding to EAGPPAFYRPNSDNRR. The c11 ion (1217.7 m/z) was further fragmented (MS3; see inset).
Due to the unique fragmentation and production of the relatively unusual c ions, further confirmation of this interpretation was sought. MS/MS analysis of the unmodified peptide precursor ion (m/z 1847.0; Figure 5c) gave a high XCorr score (2.78) and revealed a set of sequence ions (c11,12, b11,13,14,15, b11−H2O, b15−NH3, b15+H2O) analogous to the fragmentation of the corresponding neutral loss-derived dehydro-alanine-containing peptide (as shown in Figure 5b, MS3 of m/z 1829.0), resulting from its phosphorylated precursor. For the non-phosphorylated peptide at m/z 1847.0, further fragmentation of the c11 ion (MS3 of m/z 1217.7; inset Figure 5c) revealed the sequence ions (b7 and b9, y8−NH3, y10) and an additional internal fragment ion PPAFYR-NH3; thus the fragmentation was almost identical to that of the corresponding neutral loss derived dehydro-alanine-containing peptide (inset Figure 5b; MS4 of m/z 1217.7) derived from its phosphorylated precursor. Table 1 summarizes the tandem mass spectrometry data observed for peptides containing phosphorylated serine residues 118, 154 or 167 and the corresponding peptides for which these residues are not phosphorylated. In each case M (obs) is the molecular mass of the neutral peptide after subtraction of the mass(es) of the attached proton(s) and ΔM is the difference between the measured mass and that calculated from the known atomic composition of the peptide. Mascot scores and retention times are presented only for peptides monitored by HPLC-MS/MS, whereas XCorr values are presented for peptides monitored by vMALDI-MSn, where n was either 2 (MS/MS) or 3 (MS/MS/MS) (Table 1).
Table 1.
Phospho-serine peptides and corresponding unmodified serine peptides detected
| M (obs) | ΔM (Da) | Proteolytic peptides | Protease | P-site | Mascot score (ESI-MS/MS) | XCorr value vMALDI-MS”) | LC tret |
|---|---|---|---|---|---|---|---|
| 1751.63 | −0.07 | LApS167TNDKGS(ox)MA(ox)MESAK | Trypsin | Ser-167 | 50 | — | 13.5 min |
| 1671.80 | 0.06 | LAS167TNDKGS(ox)MA(ox)MESAK | Trypsin | — | 61 | — | 12.9 min |
| 1472.60 | −0.10 | (ox)MLLHPPPQLpS118PF | Chymotrypsin | Ser-118 | — | 2.05 (MS2), | — |
| 2.53 (MS3) | |||||||
| 1392.60 | −0.13 | (ox)MLLHPPPQLS118PF | Chymotrypsin | — | — | 2.61 (MS2) | — |
| 1926.30 | 0.44 | EAGPPAFYRPNpS154DNRR | Trypsin | Ser-154 | — | 0.78 (MS2), | — |
| 2.47 (MS3) | |||||||
| 1846.00 | 0.11 | EAGPPAFYRPNS154DNRR | Trypsin | - | 29 | 2.78 (MS2) | 16.5 min |
Relative Quantitation by SRM of Ser-154 Phosphorylation Induction
To assess the degree of E2- or EGF-induced ERα Ser-154 phosphorylation relative to unstimulated ERα from MCF-7 cells, the ability of the vMALDI-LTQ to carry out selected reaction monitoring (SRM) was used. This enables the measurement of the presence and intensity of a specific predetermined fragment ion after CID. The MALDI mode was selected rather than LC-ESI because it allowed peptide fragments to be monitored over a longer period of time, whereas the LC-MS/MS experiments with the Q-STAR gave only transitory signals that were not conducive to collecting multiple measurements. Given that the c11 ion was common to both phosphorylated and unmodified peptides we were able to normalize for variability in total peptide levels observed between the different samples. This ion was preferable to the b11 ion because there was overlap between b11 and the c11−NH3 ion. An SRM method was developed that compared the intensity of the c11 fragment ion (1217.6 m/z) from the phosphorylated peptide containing ERα residues 143–158 (EAGPPAFYRPNpSDNRR) with that of the same c11 fragment ion from the corresponding unmodified ERα peptide. The data obtained from multiple biological replicate experiments shown in Figure 6a revealed that E2 induced a 6-fold increase in ERα phosphorylation at Ser-154 relative to untreated control, whereas EGF induced a weaker intracellular response with 2.5-fold induction in ERα Ser-154 phosphorylation over untreated control.
Figure 6.
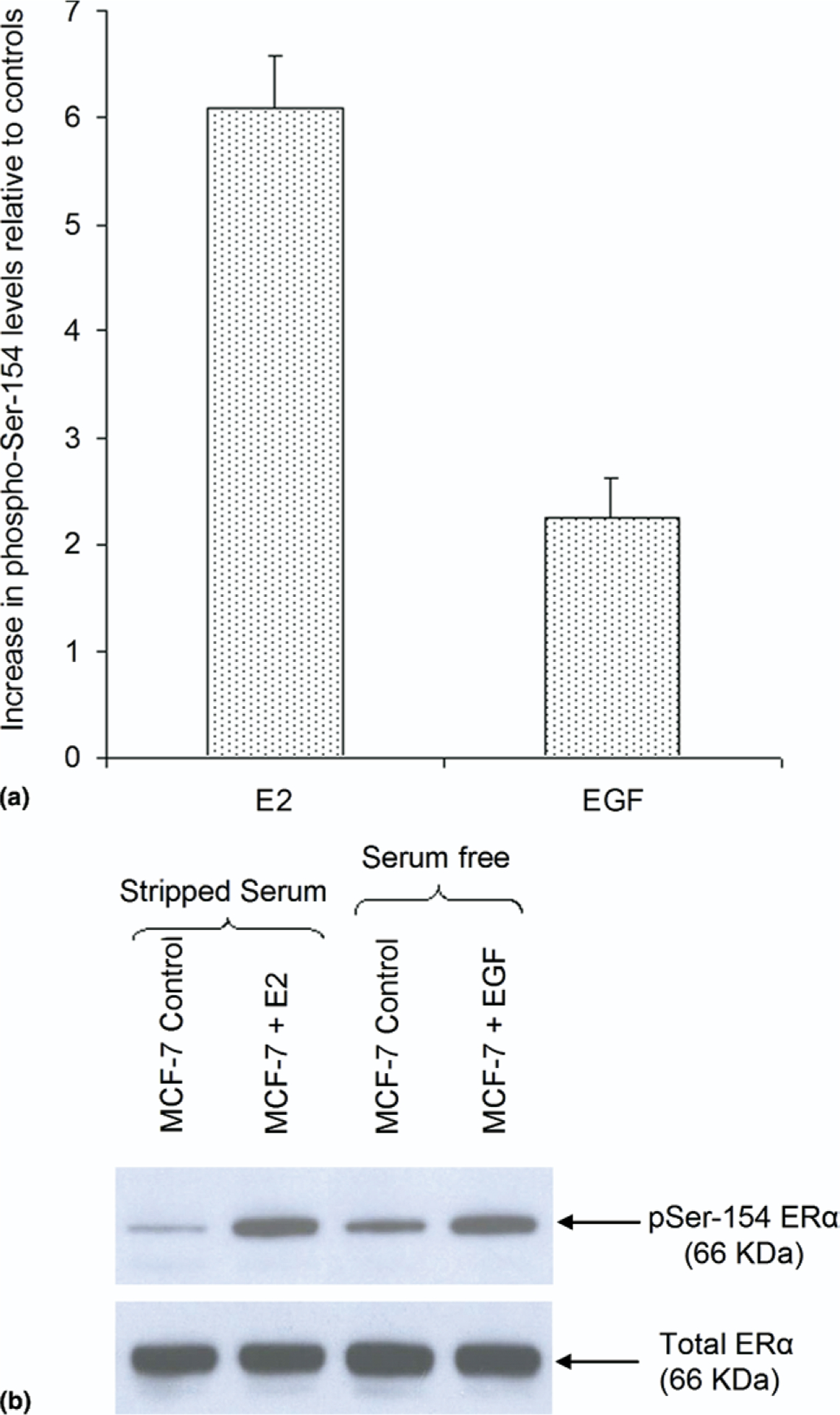
Relative quantitation of phospho-Ser-154 levels by MS and immunoblotting. (a) vMALDI-LTQ in SRM mode demonstrates levels of Ser-154 phosphorylation after E2 (10 nM) or EGF (50 ng/mL) treatment relative to their respective controls. (b) Western analysis of total and phosphorylated ERα at Ser-154 following immunoprecipitation from MCF-7 cells treated with E2 (10 nM), EGF (50 ng/mL), or appropriate control.
Although this report illustrates the analytical power of modern MS approaches to discover previously undetected PTMs in such low abundance and clinically relevant endogenous proteins like ERα, it is also important to note the contribution made by using different MS instrument platforms in the identification and quantification of specific ERα PTMs like phospho-Ser-167 and phospho-Ser-154. In particular, it is still not fully understood why ESI preferentially ionized the phospho-Ser-167 peptide (Figure 3), whereas MALDI preferentially ionized the phospho-Ser-118 and phospho-Ser-154 peptides (Figures 4 and 5) from the same ERα digests, underscoring the benefits of using different MS platforms to achieve full protein coverage and phospho-peptide detection. Likewise, vMALDI-LTQ provided an advantage over the Q-STAR because we used its SRM mode to quantitate relative phospho-peptide levels, allowing us to measure differential induction of phospho-Ser-154 following E2 and EGF stimulation of MCF-7 cells. The complementary application of different MS platforms as demonstrated in this study also serves to emphasize further advantages that may be gained by applying emerging MS innovations such as electron-transfer dissociation (ETD) fragmentation along with our present MS approaches. The use of ETD has recently been suggested to be superior to CID for identification of phospho-peptides, although the two methods could be effectively combined in alternating ETD and CID modes for a truly comprehensive analysis of phosphopeptides [41].
Antibody Specific to ERα Phospho-Ser-154 Confirms MS Findings
Informed by the MS detection of phospho-Ser-154, a rabbit polyclonal antibody specific to ERα phospho-Ser-154 was produced and affinity purified as described earlier under the Experimental section. As shown in Figure 6b, Western analysis with this ERα phospho-Ser-154 antibody performed on the same immunopurified ERα material used for MS analysis (Figure 2a) demonstrated that E2 and EGF induced phosphorylation of Ser-154 consistent with levels determined by MS analysis. With regard to E2-induced phosphorylation of Ser-154, it is also interesting to note that its nearly 6-fold induction is quantitatively similar to the E2-induced Ser-167 phosphorylation response (Figure 2a).
A competition experiment confirmed the specificity of the antibody to ERα phosphorylated at serine-154. The phosphopeptide used to produce the antibody was mixed with the phospho-Ser-154 antibody at a 1000:1 ratio in the TBS-Tween/milk solution. A blot containing multiple samples of ER immunoprecipitated from control and E2-treated MCF-7 cells was then incubated in the peptide/antibody mix and compared to a repeat blot that was incubated in phospho-Ser-154 antibody alone. The competing peptide completely prevented binding of the antibody to ERα, as was shown by the lack of signal from the 66-kDa region for all samples incubated in the peptide/antibody mix (data not shown).
The AF1 and AF2 domains of ERα (Figure 1), both serving to mediate gene induction by ERα, do so by different mechanisms, with AF2 activation brought about by ligand-dependent steric recruitment of cofactors to the C-terminal domain, whereas AF1 activation and cofactor recruitment are mediated by induction of an undefined constellation of different serine phosphorylation sites within the N-terminal domain of ERα [4–6, 42, 43]. With the present MS analysis establishing Ser-154 as a phosphorylated target upon both ligand-dependent (E2) and ligand-independent (EGF) ERα activation, 6 of 14 NTD serine residues are now known to be subject to endogenous phosphorylation and may thereby be considered potential regulators of AF1 function. Furthermore, our MS analyses also indicate that Ser-10, 46, 47, 137, 173, and 178 are not subject to endogenous phosphorylation, as determined by our experimental conditions and within current instrumentation and detection limits. Studies using other pro-teases to improve coverage and interrogate peptides containing the remaining serine residues within the ERα-NTD are currently under way. Given such important clinical issues as endocrine resistance, thought to be mediated by ligand-independent NTD phosphorylation [2, 8, 11–21], developing a complete catalogue of endogenous ERα PTMs, now feasible using modern MS approaches, would seem to be an important and achievable endeavor.
Although the functional consequences of Ser-154 phosphorylation remain to be explored, it is interesting to note that Scansite (scansite.mit.edu), a kinase motif search engine, classified Ser-154, like Ser-167, as embedded in a phosphorylation motif similar to that targeted by the growth factor activated serine/threonine kinase, AKT. Since EGF treatment of MCF-7 cells induced substantial phospho-Ser-167 levels along with a modest increase in endogenous phospho-Ser-154 levels, recent reports implicating AKT-stimulated phosphorylation of Ser-167 in association with tamoxifen resistance now raise an interesting question about the possible clinical relevance of phospho-Ser-154 induction in ER-positive breast cancers, alone or in concert with phospho-Ser-167 induction [44–46]. Such mechanistic and clinical questions can now be more readily addressed with the availability of our ERα phospho-Ser-154–specific antibody, developed as a result of the MS analytical approach and findings described here.
Conclusion
Activated ERα plays a critical role in the development of most human breast cancers while also serving as the molecular target for life saving endocrine therapies [4, 6, 47]. Many aspects of ERα transcriptional activation are modulated by phosphorylation of key serine residues within its NTD as a result of either ligand-dependent or -independent influences [2, 7]. Initial studies examining phosphorylation within the ERα-NTD relied on ERα overexpressed in metabolically labeled mammalian (COS) and insect (Sf9) cells [26, 27, 48]. Although these studies successfully identified several serine residues within the NTD targeted for phosphorylation, including the identification of Ser-118 as a key target residue, technical limitations left other phosphorylated resides contested or unidentified [26, 27, 48]. MS analysis, with its great advances in precision and sensitivity, now plays an increasingly important role in the detection of in vivo phosphorylated protein residues [49]. In the present study endogenous ERα, immunocaptured and purified from MCF-7 human breast cancer cells shortly after either ligand-dependent or -independent ERα activating stimulation, was analyzed by MS using two complementary platforms, ESI-MS/MS and vMALDI-MSn, allowing for the detection of known ERα-NTD phosphorylation sites, Ser-118 and Ser-167, as well as identification and induction of a previously unrecognized NTD phosphorylation site, phospho-Ser-154. MS analysis demonstrated that endogenous phospho-Ser-154 induction occurs more so in response to E2 (6-fold) than to EGF (2.5-fold) stimulation; and the presence and relative levels of these induction responses were confirmed using a custom-produced polyclonal antibody specific for ERα phospho-Ser-154.
This present report describes modifications in the N-terminal domain of ERα, a region that can be monitored using chymotrypsin digestion. A future study in preparation will address the issue of overall sequence coverage using several different enzymes and will attempt a comprehensive analysis of all the posttranslational modifications of this protein (Astriku, Britton et al., in preparation). For the current work there is no formal measure of sensitivity but, as described in the text, all measurements were carried out on endogenous protein extracted from a human breast cancer cell line in the low picomole range. The site occupancy for phosphorylation is typically low, so the techniques used here can be considered very sensitive. Thus, the protein isolation and MS strategies described here can facilitate discovery of novel phosphorylation sites within low-abundance, clinically important cancer targets like ERα, and may thereby contribute to our understanding of the role of phosphorylation in the development of breast cancer.
Acknowledgments
We thank Crystal Berger for her laboratory assistance and Rosa Viner (Thermo) for her vMALDI-LTQ operations advice. This work was supported in part by California Breast Cancer Research Program Grant 10YB-0125 and National Cancer Institute Grant R01-CA-71468. The Mass Spectrometry Core at the Buck Institute is a Nathan Shock Center of Excellence for Basic Mechanisms of Aging and Age-related Diseases and is partially supported by National Institute on Aging Grant P30-AG-025708.
Footnotes
The authors declare no conflict of interest.
References
- 1.Joel PB; Traish AM; Lannigan DA Estradiol-Induced Phosphorylation of Serine 118 in the Estrogen Receptor Is Independent of p42/p44 Mitogen-Activated Protein Kinase. J. Biol. Chem 1998, 273, 13317–13323. [DOI] [PubMed] [Google Scholar]
- 2.Likhite VS; Stossi F; Kim K; Katzenellenbogen BS; Katzenellenbogen JA Kinase-Specific Phosphorylation of the Estrogen Receptor Changes Receptor Interactions with Ligand, Deoxyribonucleic Acid, and Coregulators Associated with Alterations in Estrogen and Tamoxifen Activity. Mol. Endocrinol 2006, 20, 3120–3132. [DOI] [PubMed] [Google Scholar]
- 3.McInerney EM; Katzenellenbogen BS Different Regions in Activation Function-1 of the Human Estrogen Receptor Required for Antiestrogen- and Estradiol-dependent Transcription Activation. J. Biol. Chem 1996, 271, 24172–24178. [DOI] [PubMed] [Google Scholar]
- 4.Jensen EV; Jordan VC The Estrogen Receptor: A Model for Molecular Medicine. Clin. Cancer Res 2003, 9, 1980–1989. [PubMed] [Google Scholar]
- 5.Shiau AK; Barstad D; Loria PM; Cheng L; Kushner PJ; Agard DA; Greene GL The Structural Basis of Estrogen Receptor/ Coactivator Recognition and the Antagonism of This Interaction by Tamoxifen. Cell 1998, 95, 927–937. [DOI] [PubMed] [Google Scholar]
- 6.Levenson AS; Jordan VC Selective Oestrogen Receptor Modulation: Molecular Pharmacology for the Millennium. Eur. J. Cancer 1999, 35, 1628–1639. [DOI] [PubMed] [Google Scholar]
- 7.Lannigan DA Estrogen Receptor Phosphorylation. Steroids 2003, 68, 1–9. [DOI] [PubMed] [Google Scholar]
- 8.Britton DJ; Hutcheson IR; Knowlden JM; Barrow D; Giles M; McClelland RA; Gee JMW; Nicholson RI Bidirectional Cross Talk between ERα and EGFR Signalling Pathways Regulates Tamoxifen-Resistant Growth. Breast Cancer Res. Treat 2006, 96, 131–146. [DOI] [PubMed] [Google Scholar]
- 9.Lange CA Making Sense of Cross-Talk between Steroid Hormone Receptors and Intracellular Signaling Pathways: Who Will Have the Last Word? Mol. Endocrinol 2004, 18, 269–278. [DOI] [PubMed] [Google Scholar]
- 10.Lavery DN; McEwan IJ Structure and Function of Steroid Receptor AF1 Transactivation Domains: Induction of Active Conformations. Biochem. J 2005, 391, 449–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gee JM; Robertson JF; Gutteridge E; Ellis IO; Pinder SE; Rubini M; Nicholson RI Epidermal Growth Factor Receptor/HER2/ Insulin-like Growth Factor Receptor Signalling and Oestrogen Receptor Activity in Clinical Breast Cancer. Endocr. Relat. Cancer 2005, 12, S99–S111. [DOI] [PubMed] [Google Scholar]
- 12.Glaros S; Atanaskova N; Zhao C; Skafar DF; Reddy KB Activation Function-1 Domain of Estrogen Receptor Regulates the Agonistic and Antagonistic Actions of Tamoxifen. Mol. Endocrinol 2006, 20, 996–1008. [DOI] [PubMed] [Google Scholar]
- 13.Gutierrez MC; Detre S; Johnston S; Mohsin SK; Shou J; Allred DC; Schiff R; Osborne CK; Dowsett M Molecular Changes in Tamoxifen-Resistant Breast Cancer: Relationship between Estrogen Receptor, HER-2, and p38 Mitogen-Activated Protein Kinase. J. Clin. Oncol 2005, 23, 2469–2476. [DOI] [PubMed] [Google Scholar]
- 14.Sarwar N; Jiang J; Shousha S; Coombes RC; Ali S Investigation of Estrogen Receptor alpha Phosphorylation in Breast Cancer. J. Clin. Oncol. Meeting Abstr 2006, 24, 20046. [Google Scholar]
- 15.Sarwar N; Kim JS; Jiang J; Peston D; Shousha S; Coombes RC; Ali S Phosphorylation of Estrogen Receptor alpha at Serine 118 in Human Breast Tumours. J. Clin. Oncol. Meeting Abstr 2005, 23, 9576. [Google Scholar]
- 16.Schiff R; Massarweh SA; Shou J; Bharwani L; Mohsin SK; Osborne CK Cross-Talk between Estrogen Receptor and Growth Factor Pathways as a Molecular Target for Overcoming Endocrine Resistance. Clin. Cancer Res 2004, 10, 331S–336S. [DOI] [PubMed] [Google Scholar]
- 17.Shou J; Massarweh S; Osborne CK; Wakeling AE; Ali S; Weiss H; Schiff R Mechanisms of Tamoxifen Resistance: Increased Estrogen Receptor-HER2/neu Cross-Talk in ER/HER2-Positive Breast Cancer.J. Natl. Cancer Inst 2004, 96, 926–935. [DOI] [PubMed] [Google Scholar]
- 18.Swaby RF; Huang M; Ruth KJ; Ross EA; Gong Y; Page RE; Freedman GM; Goldstein LJ; Di Cristofano A Retrospective Analysis of Phosphorylation Status of the Estrogen Receptor in Patients with Early Stage Disease. J. Clin. Oncol. Meeting Abstr 2007, 25, 21034. [Google Scholar]
- 19.Massarweh S; Schiff R Unraveling the Mechanisms of Endocrine Resistance in Breast Cancer: New Therapeutic Opportunities. Clin. Cancer Res 2007, 13, 1950–1954. [DOI] [PubMed] [Google Scholar]
- 20.Benz CC; Scott GK; Sarup JC; Johnson RM; Tripathy D; Coronado E; Shepard HM; Osborne CK Estrogen-Dependent, Tamoxifen-Resistant Tumorigenic Growth of MCF-7 Cells Transfected with HER2/neu. Breast Cancer Res. Treat 1992, 24, 85–95. [DOI] [PubMed] [Google Scholar]
- 21.Hutcheson IR; Knowlden JM; Madden T-A; Barrow D; Gee JMW; Wakeling AE; Nicholson RI Oestrogen Receptor-Mediated Modulation of the EGFR/MAPK Pathway in Tamoxifen-Resistant MCF-7 Cells. Breast Cancer Res. Treat 2003, 81, 81–93. [DOI] [PubMed] [Google Scholar]
- 22.Joel PB; Smith J; Sturgill TW; Fisher TL; Blenis J; Lannigan DA pp90rsk1 Regulates Estrogen Receptor-Mediated Transcription through Phosphorylation of Ser-167. Mol. Cell. Biol 1998, 18, 1978–1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arnold SF; Obourn JD; Jaffe H; Notides AC Phosphorylation of the Human Estrogen Receptor by Mitogen-Activated Protein Kinase and Casein Kinase II: Consequence on DNA Binding. J. Steroid Biochem. Mol. Biol 1995, 55, 163–172. [DOI] [PubMed] [Google Scholar]
- 24.Clark DE; Poteet-Smith CE; Smith JA; Lannigan DA Rsk2 Allosterically Activates Estrogen Receptor α by Docking to the Hormone-Binding Domain. EMBO J. 2001, 20, 3484–3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tzeng DZ; Klinge CM Phosphorylation of Purified Estradiol-Liganded Estrogen Receptor by Casein Kinase II Increases Estrogen Response Element Binding but Does Not Alter Ligand Stability. Biochem. Biophys. Res. Commun 1996, 223, 554–560. [DOI] [PubMed] [Google Scholar]
- 26.Joel PB; Traish AM; Lannigan DA Estradiol and Phorbol Ester Cause Phosphorylation of Serine 118 in the Human Estrogen Receptor. Mol. Endocrinol 1995, 9, 1041–1052. [DOI] [PubMed] [Google Scholar]
- 27.Le Goff P; Montano MM; Schodin DJ; Katzenellenbogen BS Phosphorylation of the Human Estrogen Receptor. Identification of Hormone-Regulated Sites and Examination of Their Influence on Transcriptional Activity. J. Biol. Chem 1994, 269, 4458–4466. [PubMed] [Google Scholar]
- 28.Moran MF; Tong J; Taylor P; Ewing RM Emerging Applications for Phospho-Proteomics in Cancer Molecular Therapeutics. Biochim. Biophys. Acta 2006, 1766, 230–241. [DOI] [PubMed] [Google Scholar]
- 29.Collins MO; Yu L; Choudhary JS Analysis of Protein Phosphorylation on a Proteome-Scale. Proteomics 2007, 7, 2751–2768. [DOI] [PubMed] [Google Scholar]
- 30.Chang EJ; Archambault V; McLachlin DT; Krutchinsky AN; Chait BT Analysis of Protein Phosphorylation by Hypothesis-Driven Multiple-Stage Mass Spectrometry. Anal. Chem 2004, 76, 4472–4483. [DOI] [PubMed] [Google Scholar]
- 31.Schilling B; Gafni J; Torcassi C; Cong X; Row RH; LaFevre-Bernt MA; Cusack MP; Ratovitski T; Hirschhorn R; Ross CA; Gibson BW; Ellerby LM Huntingtin Phosphorylation Sites Mapped by Mass Spectrometry: Modulation of Cleavage and Toxicity. J. Biol. Chem 2006, 281, 23686–23697. [DOI] [PubMed] [Google Scholar]
- 32.Cao P; Stults JT Phosphopeptide Analysis by On-line Immobilized Metal-Ion Affinity Chromatography-Capillary Electrophoresis-Electrospray Ionization Mass Spectrometry. J. Chromatogr. A 1999, 853, 225–235. [DOI] [PubMed] [Google Scholar]
- 33.Corthals GL; Aebersold R; Goodlett DR; Burlingame AL Identification of Phosphorylation Sites Using Microimmobilized Metal Affinity Chromatography In: Methods in Enzymology. San Diego, CA: Academic Press, 2005; pp 66–81. [DOI] [PubMed] [Google Scholar]
- 34.Figeys D; Gygi SP; Zhang Y; Watts J; Gu M; Aebersold R Electrophoresis Combined with Novel Mass Spectrometry Techniques: Powerful Tools for the Analysis of Proteins and Proteomes. Electrophoresis 1998, 19, 1811–1818. [DOI] [PubMed] [Google Scholar]
- 35.Neville DCA; Rozanas CR; Price EM; Gruis DB; Verkman AS; Townsend RR Evidence for Phosphorylation of Serine 753 in CFTR Using a Novel Metal-Ion Affinity Resin and Matrix-Assisted Laser Desorption Mass Spectrometry. Protein Sci. 1997, 6, 2436–2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ballif BA; Villen J; Beausoleil SA; Schwartz D; Gygi SP Phosphoproteomic Analysis of the Developing Mouse Brain. Mol. Cell. Proteomics 2004, 3, 1093–1101. [DOI] [PubMed] [Google Scholar]
- 37.Beausoleil SA; Jedrychowski M; Schwartz D; Elias JE; Villen J; Li J; Cohn MA; Cantley LC; Gygi SP Large-Scale Characterization of HeLa Cell Nuclear Phosphoproteins. Proc. Natl. Acad. Sci. U.S.A 2004, 101, 12130–12135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gronborg M; Kristiansen TZ; Stensballe A; Andersen JS; Ohara O; Mann M; Jensen ON; Pandey A A Mass Spectrometry-Based Proteomic Approach for Identification of Serine/Threonine-Phosphorylated Proteins by Enrichment with Phospho-Specific Antibodies: Identification of a Novel Protein, Frigg, as a Protein Kinase A Substrate. Mol. Cell. Proteomics 2002, 1, 517–527. [DOI] [PubMed] [Google Scholar]
- 39.Perkins DN; Pappin DJC; Creasy DM; Cottrell JS Probability-Based Protein Identification by Searching Sequence Database Using Mass Spectrometry Data. Electrophoresis 1999, 20, 3551–3567. [DOI] [PubMed] [Google Scholar]
- 40.Eng JK; McCormack AL; Yates JR An Approach to Correlate Tandem Mass Spectral Data of Peptides with Amino Acid Sequences in a Protein Database. J. Am. Soc. Mass Spectrom 1994, 5, 976–989. [DOI] [PubMed] [Google Scholar]
- 41.Molina H; Horn DM; Tang N; Mathivanan S; Pandey A Global Proteomic Profiling of Phosphopeptides Using Electron Transfer Dissociation Tandem Mass Spectrometry. Proc. Natl. Acad. Sci. U.S.A 2007, 104, 2199–2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Warnmark A; Wikstrom A; Wright APH; Gustafsson J-A; Hard T The N-terminal Regions of Estrogen Receptor alpha and beta Are Unstructured In Vitro and Show Different TBP Binding Properties. J. Biol. Chem 2001, 276, 45939–45944. [DOI] [PubMed] [Google Scholar]
- 43.Kumar RAJ; Thompson EB Transactivation Functions of the N-Terminal Domains of Nuclear Hormone Receptors: Protein Folding and Coactivator Interactions. Mol. Endocrinol 2003, 17, 1–10. [DOI] [PubMed] [Google Scholar]
- 44.Campbell RA; Bhat-Nakshatri P; Patel NM; Constantinidou D; Ali S; Nakshatri H Phosphatidylinositol 3-Kinase/AKT-Mediated Activation of Estrogen Receptor alpha. A New Model for Anti-Estrogen Resistance. J. Biol. Chem 2001, 276, 9817–9824. [DOI] [PubMed] [Google Scholar]
- 45.deGraffenried LA; Friedrichs WE; Fulcher L; Fernandes G; Silva JM; Peralba JM; Hidalgo M Eicosapentaenoic Acid Restores Tamoxifen Sensitivity in Breast Cancer Cells with High Akt Activity. Ann. Oncol 2003, 14, 1051–1056. [DOI] [PubMed] [Google Scholar]
- 46.Jordan VC Targeting Antihormone Resistance in Breast Cancer: A Simple Solution. Ann. Oncol 2003, 14, 969–970. [DOI] [PubMed] [Google Scholar]
- 47.Sommer S; Fuqua SAW Estrogen Receptor and Breast Cancer. Semin. Cancer Biol 2001, 11, 339–352. [DOI] [PubMed] [Google Scholar]
- 48.Arnold SF; Obourn JD; Jaffe H; Notides AC Serine 167 Is the Major Estradiol-induced Phosphorylation Site on the Human Estrogen Receptor. Mol. Endocrinol 1994, 8, 1208–1214. [DOI] [PubMed] [Google Scholar]
- 49.Surti TS; Huang L; Jan YN; Jan LY; Cooper EC Identification by Mass Spectrometry and Functional Characterization of Two Phosphorylation Sites of KCNQ2/KCNQ3 Channels. Proc. Natl. Acad. Sci. U.S.A 2005, 102, 17828–17833. [DOI] [PMC free article] [PubMed] [Google Scholar]