

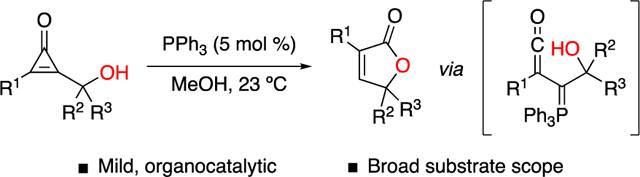
Abstract
A general method to synthesize substituted butenolides from hydroxymethylcyclopropenones is reported. Functionalized cyclopropenones undergo ring-opening reactions with catalytic amounts of phosphine, forming reactive ketene ylides. These intermediates can be trapped by pendant hydroxy groups to afford target butenolide scaffolds. The reaction proceeds efficiently in diverse solvents and with low catalyst loadings. Importantly, the cyclization is tolerant of a broad range of functional groups, yielding a variety of α- and γ-substituted butenolides.
Graphical Abstract
Butenolides are found in a variety of natural product scaffolds and possess desirable bioactive properties.1 For example, linderalactone (Figure 1A) can protect hepatocytes from oxidative damage.2 Other butenolides, including (–)-incrustoporin, are potent inhibitors of pathogenic fungi.3 These and related scaffolds4 have inspired chemists to develop efficient syntheses of α,β-unsaturated lactones. While several methods now exist,5–9 most are limited in terms of substitution pattern, functional group tolerance, or starting material accessibility. More general methods to build butenolides are therefore needed.
Figure 1.
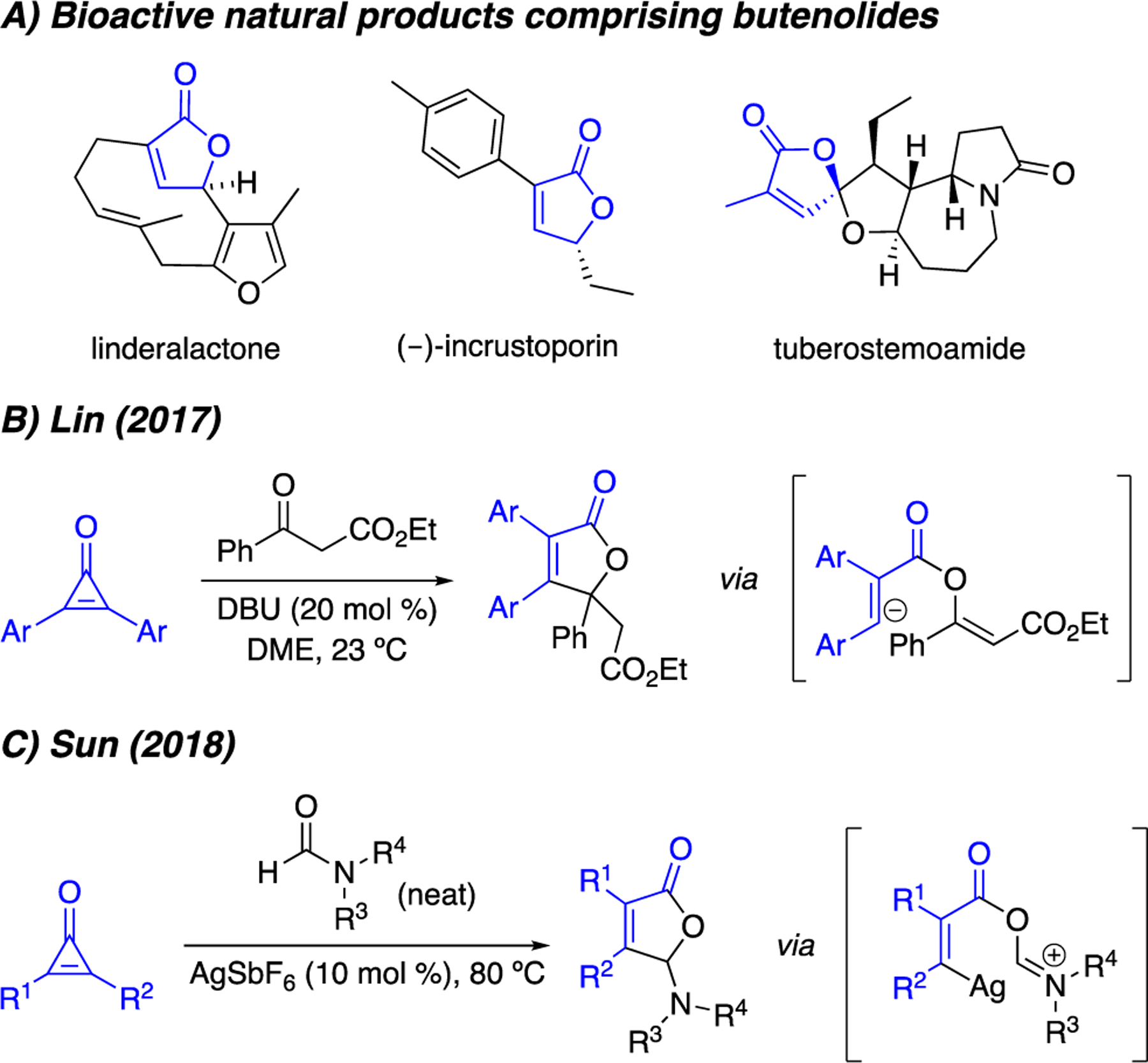
(A) Butenolides (blue) are common motifs in bioactive natural products. Recent work by (B) Lin and (C) Sun featured intermolecular reactions between CpOs and carbonyls to generate functionalized butenolides.
Cyclopropenones (CpOs) are attractive synthons for butenolide formation. These microcycles map readily onto the target structures and are easily accessible from alkyne precursors.10 CpOs have only recently been exploited for lactone synthesis, though.11–13 The groups of Lin and Sun developed methods to convert symmetric CpOs to functionalized butenolides (Figures 1B and 1C). Both transformations proceed via intermolecular 1,2-addition of carbonyl groups into the CpO scaffold, followed by ring opening. The resulting vinyl anion intermediates subsequently cyclize to deliver the desired lactones. While robust, both methods require electron-rich carbonyl fragments. The vinyl anion intermediates are also not compatible with a range of functional groups, limiting the scope of these chemistries. Furthermore, the intermolecular nature of the reactions can present regioselectivity challenges when unsymmetrical CpOs are employed.
We hypothesized that an intramolecular reaction could convert CpOs to functionalized butenolides and potentially broaden the scope of accessible products. Toward this end, we drew inspiration from our previous work on bioorthogonal CpOs.14,15 These motifs undergo conjugate addition reactions with bioorthogonal phosphines, producing ketene-ylide intermediates upon ring fragmentation.16 The electrophiles can be readily trapped by pendant nucleophiles on the phosphine (Figure 2A) to afford covalent adducts. We surmised that pendant nucleophiles on the CpO – rather than the phosphine – could also trap the ketene (Figure 2B). If the nucleophile was part of a hydroxymethyl tether, the products would comprise lactones. Subsequent ylide protonation and phosphine elimination could ultimately deliver functionalized butenolides. Since the proposed method involves bioorthogonal reagents, it would likely be compatible with a variety of functional groups. The proximity of the hydroxy group on the CpO tether would also promote intramolecular cyclization, outcompeting any exogenous nucleophile in the trapping step.15
Figure 2.
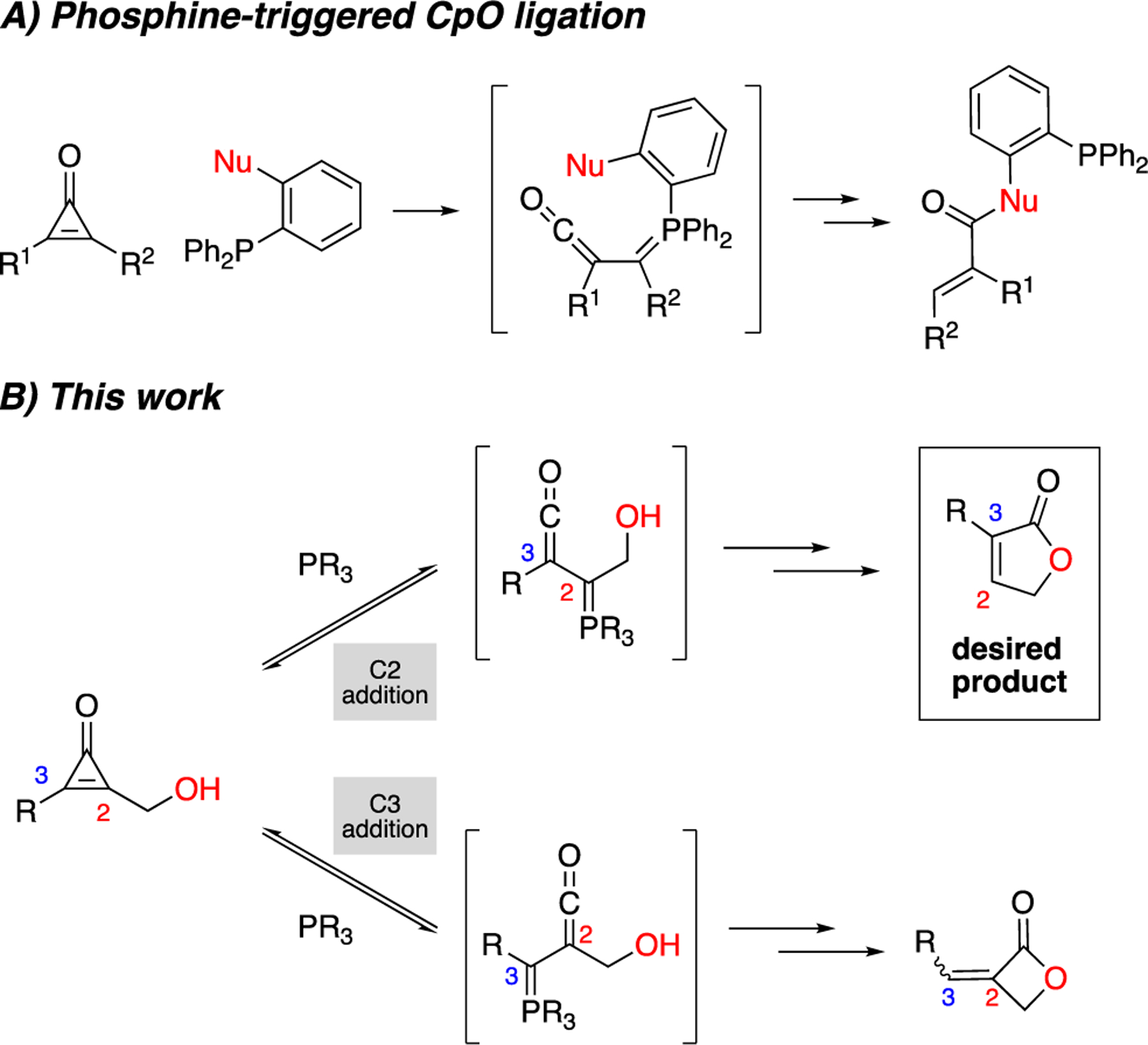
CpOs react with phosphines to reveal ketene ylides. These intermediates can be trapped with (A) pendant nucleophiles on bioorthogonal phosphines or (B) hydroxy group nucleophiles on CpO scaffolds. In the latter case, phosphine addition at C2 affords butenolides (top). Phosphine addition at C3 could provide undesired β-lactones (bottom).
The regioselectivity of the proposed reaction was further considered. Phosphine addition at C2 (i.e., the carbon bearing the hydroxymethyl tether) would likely provide the desired products. However, phosphine addition at C3 could give undesired β-lactones. Cyclization en route to the β-lactone (4-exo-dig) is disfavored according to Baldwin’s rules, but notable examples exist.17–19 We reasoned that butenolide formation could still predominate in the reaction, even if phosphine attacked C3. Ketene-ylide formation is reversible in the absence of trapping nucleophiles,14,20 and cyclization en route to the β-lactone would likely be slower than the reverse reaction to reform the CpO. Thus, the reaction could funnel to a butenolide product, regardless of the initial site of phosphine addition.
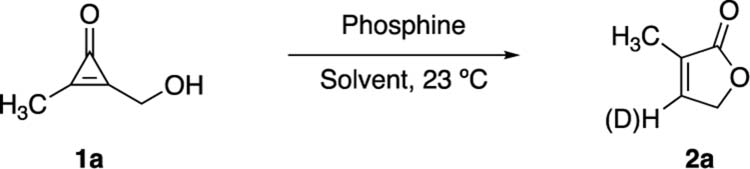
To examine the overall strategy, model CpO 1a was synthesized and treated with a panel of phosphines. The reactions were performed in various solvents and monitored using 1H NMR spectroscopy (Tables 1 and S1, Figures S1–S15). 1,3,5-Triaza-7-phosphaadamantane (PTA) was initially selected due to its potent reactivity and unique stability in protic solvents. When combined stoichiometrically with 1a, PTA afforded rapid conversion to the desired butenolide (entry 1). The reaction slowed significantly when the catalyst loading was reduced to 10 mol % (entry 2). Notably, no intermolecular solvent trapping was observed even at the longer reaction times. Sluggish reactivity was not general to all alkyl-substituted phosphines. When 1a was treated with cyclohexyldiphenylphosphine (CyDPP), rapid formation of 2a was observed even at low catalyst loadings (entry 3).
Table 1.
Optimization of butenolide cyclization
 | ||||
|---|---|---|---|---|
| entry | phosphine (mol %) | solvent | time | conversion (%)b |
| 1 | PTA (100) | CD3OD | 10 min | >95 |
| 2 | PTA (10) | CD3OD | 2 h | 67 |
| 3 | CyDPP (5) | CD3OD | 2.5 h | 90 |
| 4 | P(o-tolyl)3 (5) | CD3OD | 2.5 h | 0 |
| 5 | PPh3 (100) | CD3OD | 10 min | >95 |
| 6 | PPh3 (100) | DMSO-d6 | 22 h | >95 |
| 7 | PPh3 (5) | DMSO-d6 | 24 h | 16 |
| 8 | PPh3 (100) | C6D6 | 1 h | >95 |
| 9 | PPh3 (5) | C6D6 | 24 h | 94 |
| 10 | PPh3(10) | CD3OD | 1.5 h | >95 |
| 11 | PPh3 (5) | CD3OD | 2.5 h | >95 |
Reaction conditions: CpO (15 μmol), TMS-acetylene (3 μmol), solvent (600 μL)
NMR conversion, calculated from integral ratios between starting CpO and butenolide product
Anticipating that alkyl phosphines would be prone to oxidation and thus less general, we further investigated triarylphosphines. We were particularly drawn to tri(o-tolyl)phosphine, as this reagent would likely be sufficiently nucleophilic to add into the CpO core, but be reasonably air stable. When CpO 1a was treated with tri(o-tolyl)phosphine, though, no conversion to butenolide 2a was observed (entry 4). No butenolide was formed even with longer reaction times or elevated temperatures (Table S1, Figures S14–S15). The increased steric bulk surrounding the phosphine likely precluded efficient conjugate addition. Indeed, this phosphine is rarely used in Michael-type reactions and is primarily employed as a metal ligand.21–23
We next tested a less sterically encumbered reagent, triphenylphosphine (PPh3). PPh3 is commercially available, inexpensive, and bench stable, making it attractive for methods development. When 1a was treated with stoichiometric amounts of PPh3 in CD3OD, rapid conversion to butenolide 2a was observed (entry 5). Efficient cyclization occurred even at reduced catalyst loadings (entries 10–11) with no evidence of intermolecular solvent trapping. Additionally, in the realm of phosphine organocatalysis, few reactions feature catalyst loadings below 10 mol %, and even fewer use air-stable, commercially available reagents.24 We were also surprised that low catalyst loadings afforded rapid butenolide formation, considering the diminished nucleophilicity of PPh3. We hypothesized that the polar protic solvent accelerated the reaction via hydrogen-bond activation of the starting CpO. Similar observations were made when CpOs were tuned for bioorthogonal ligation.15 When the cyclizations were performed in DMSO-d6 or C6D6, longer reaction times or stoichiometric amounts of PPh3 were required for full conversion (entries 6–9).
We aimed to test the optimized reaction conditions with a variety of hydroxymethyl CpOs. Such probes can be readily accessed via appropriately functionalized alkynes.10 We thus prepared a variety of alkyl- and aryl-substituted alkynes via acetylide addition to formaldehyde or Sonogashira cross-coupling reactions, respectively (Scheme S1). Each alkyne also comprised a THP-protected hydroxymethyl tether. The alkyne products were subjected to difluorocarbene, a reagent generated in situ via the conditions of Olah25 (Scheme 1). The resulting difluorocyclopropenes were then hydrolyzed and deprotected to furnish the desired hydroxymethyl CpOs (1a-u, Scheme 1). The carbene insertion and hydrolysis sequence was not compatible with alkynes bearing nitrogen heterocycles, and attempts to isolate the corresponding CpOs resulted in decomposition. These results are in stark contrast to heterocyclic alkenes, which undergo robust difluorocarbene insertion.26,27
Scheme 1.
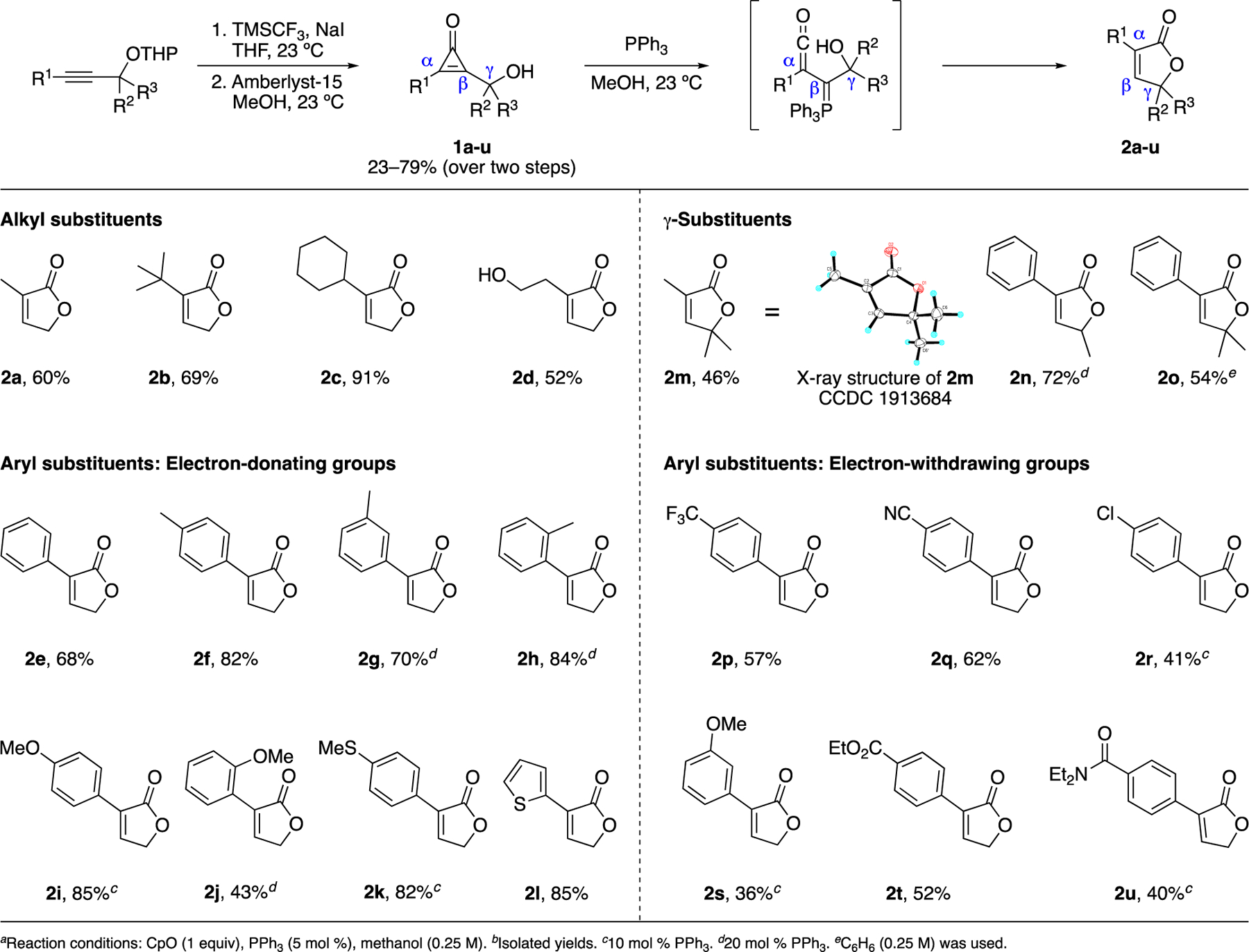
Diverse butenolides were synthesized from substituted CpOsa,b
The panel of hydroxymethyl-tethered CpOs was subjected to butenolide formation conditions (Scheme 1). As anticipated, the cyclization was tolerant of a broad range of functional groups. α-Alkyl-substituted CpOs (2a-d), including branched (2b), and cyclic (2c) substrates, were efficiently converted. The reaction also proceeded in the presence of competing nucleophiles (2d), albeit with lower yields. α-Aryl substituted CpOs also generated butenolides in the presence of PPh3. Both electron-donating (2e-2l) and electron-withdrawing (2p-2u) groups were examined, along with thiophene heterocycles (2l). Notably, robust product formation was observed even in the presence of electrophilic esters (2t) and cyano groups (2q). The reaction was also tolerant of different substitution patterns on the aryl ring (2f-2j, 2s).
The CpO-phosphine reaction further enabled access to more highly substituted butenolides. As noted above, the cyclization is tolerant of numerous α-substituents (Scheme 1). These groups are positioned away from the ketene, and thus minimally interfere with trapping. Additional substituents on the hydroxymethyl tether provided access to α,γ-disubstituted butenolides (2m-2o). Compound 2n, in particular, comprises the α,γ-substitution pattern present in incrustoporin (Figure 1A) and related natural products (Figure 1A). While the CpO reaction cannot provide β-substituted butenolides, such scaffolds are readily accessible post-cyclization. Cycloadditions28,29 and Heck couplings30 can be used in this regard, along with several other methods.31,32 Notably, functionalized butenolides can also serve as gateways to butyrolactones33–36 and other interesting scaffolds.37,38
Our collective results demonstrated that butenolides are favored in the hydroxymethyl CpO-phosphine reaction. All but one of the cyclizations proceeded with no competing β-lactone formation. Such side products were observed only when CpO 1m was subjected to low catalyst loadings in meth-anol (Figures 3A, S16). The gem-dimethyl groups on 1m likely promoted phosphine addition to the more accessible C3 position and accelerated β-lactone cyclization to provide 3a-b.39 An appreciable amount of product 2m was still formed under these challenging conditions, supporting the original hypothesis that butenolide formation can predominate, despite the potential for competing pathways.
Figure 3.
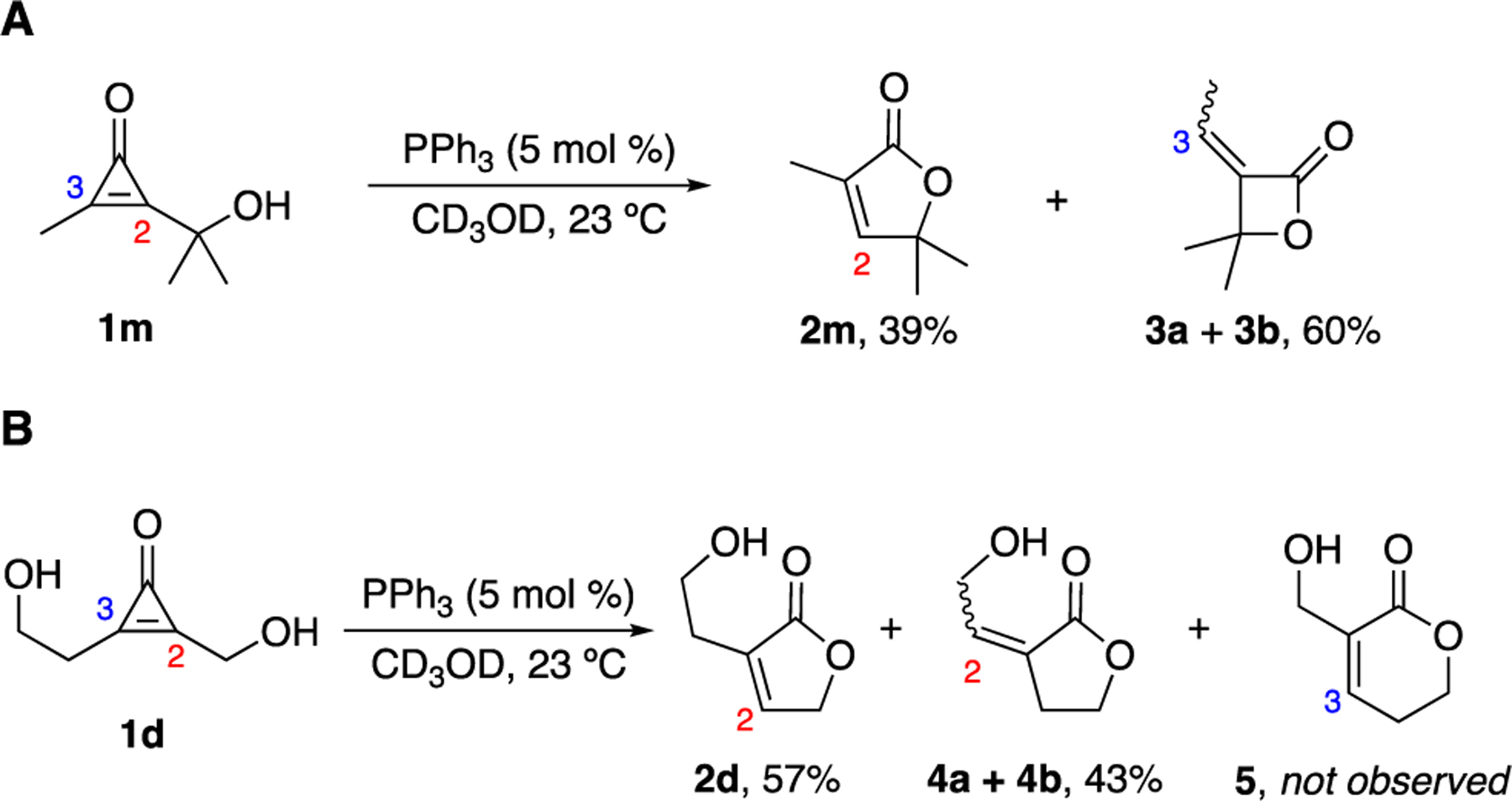
Mechanistic studies involving butenolide formation. (A) Upon phosphine treatment, CpO 1m formed β-lactone products 3a-b. The additional steric bulk at C2 likely disfavored phosphine addition. (B) δ-Lactone 5 was not observed when diol-CpO 1d was treated with PPh3. This reaction produced a mixture of γ-lactones (4a-b), in addition to the desired butenolide 2d. For (A)-(B), percent conversion values (from NMR analyses) are reported.
We carried out additional experiments to examine the propensity for hydroxymethyl CpOs to form butenolides. The observed product distributions likely reflect the faster rate of five- versus four-membered ring cyclization following ketene formation (Figure 2B). An alternative explanation is that phosphine attack is favored at C2. To investigate these possibilities, we devised a competitive trapping experiment with diol 1d. This compound comprises two hydroxy group tethers, and is capable of cyclizing to five- or six-membered rings, depending on the ketene formed. We anticipated that 1d would provide a mixture of γ- and δ-lactones upon phosphine treatment, as the steric environments around C2 and C3 are similar and unlikely to bias phosphine addition. Additionally, five- or six-membered ring formation (post-ketene generation) should be similarly facile. When diol 1d was treated with PPh3, a mixture of γ-lactones was observed, in addition to butenolide 2d (Figure S15). No δ-lactone (5) formed, though, to our surprise (Figures 3B, S17). The ketene en route to 4 was capable of forming and cyclizing, as demonstrated with a CpO probe outfitted with a single hydroxyethyl tether (Figure S18). These data suggest that phosphine addition to C2 could be preferred when CpOs are functionalized with hydroxymethyl appendages. The exact mechanism is the subject of ongoing work, but the tether could promote internal hydrogen-bond activation, preorganizing the CpO for C2 attack.
In conclusion, we developed a method to prepare substituted butenolides using mild and bioorthogonal reagents: hydroxymethyl-tethered cyclopropenones and aryl phosphines. This method features mild reaction conditions and low catalyst loadings, and can produce a variety of targets. The reaction exhibits wide functional group tolerance, typical of biocompatible reagents. The reported transformation is complementary to existing methods that furnish α,γ-substituted butenolides,40–49 but is potentially more generalizable. Importantly, the requisite hydroxymethyl CpOs can be derived from propargyl alcohols, widely used and available materials in organic synthesis. We further anticipate that CpOs and other bioorthogonal reagents will continue to inspire the development of useful methodologies.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by the U.S. National Institutes of Health (R01 GM126226 to J.A.P.) and the Alfred P. Sloan Foundation (J.A.P.). A.J.F. was supported by a George Hewitt Medical Research Postdoctoral Fellowship. We thank the Dong, Blum, Heyduk, Nowick, and Chamberlin laboratories for providing reagents and equipment. We also thank Philip Dennison (UCI) for assistance with NMR experiments, Felix Grün (UCI), Jasper Ostrom (UCI), and Benjamin Katz (UCI) for assistance with mass spectrometry experiments, and Dan Huh (UCI) and Joseph Ziller (UCI) for assistance with X-ray crystallography experiments. Last, we thank members of the Prescher laboratory for manuscript edits and helpful discussions.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.orglett.9b03298.
Experimental details and spectroscopic data for new compounds (PDF)
Crystallographic data on compound 2m (CIF)
Accession Codes
CCDC 1913684 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.cdcc.cam.ac.uk/data_request/cif, or by emailing data_request@cdcc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
REFERENCES
- 1.Karuppiah V; Sun W; Li Z Chapter 13: Natural Products of Actinobacteria Derived from Marine Organisms. Stud. Nat. Prod. Chem 2016, 48, 417–446. [Google Scholar]
- 2.Gan L-S; Zheng Y-L; Mo J-X; Liu X; Li X-H; Zhou C-X Sesquiterpene Lactones from the Root Tubers of Lindera aggregata. J. Nat. Prod 2009, 72, 1497–1501. [DOI] [PubMed] [Google Scholar]
- 3.Lu A; Wang J; Liu T; Han J; Li Y; Su M; Chen J; Zhang H; Wang L; Wang Q Small Changes Result in Large Differences: Discovery of (−)-Incrustoporin Derivatives as Novel Antiviral and Antifungal Agents. J. Agric. Food Chem 2014, 62, 8799–8807. [DOI] [PubMed] [Google Scholar]
- 4.Hou Y; Shi T; Yang Y; Fan X; Chen J; Cao F; Wang Z Asymmetric Total Syntheses and Biological Studies of Tuberostemoamide and Sessilifoliamide A. Org. Lett 2019, 21, 2952–2956. [DOI] [PubMed] [Google Scholar]
- 5.Rao YS Chemistry of Butenolides. Chem. Rev 1964, 64, 353–388. [Google Scholar]
- 6.Rao YS Recent advances in the chemistry of unsaturated lactones. Chem. Rev 1976, 76, 625–694. [Google Scholar]
- 7.Knight DW Synthetic approaches to butenolides. Contemp. Org. Synth 1994, 1, 287–315. [Google Scholar]
- 8.Carter NB; Nadany AE; Sweeney JB Recent developments in the synthesis of furan-2(5H)-ones. J. Chem. Soc., Perkin Trans 1 2002, 2324–2342. [Google Scholar]
- 9.Mao B; Fañanás-Mastral M; Feringa BL Catalytic Asymmetric Synthesis of Butenolides and Butyrolactones. Chem. Rev 2017, 117, 10502–10566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Komatsu K; Kitagawa T Cyclopropenylium Cations, Cyclopropenones, and Heteroanalogues–Recent Advances. Chem. Rev 2003, 103, 1371–1427. [DOI] [PubMed] [Google Scholar]
- 11.Li X; Han C; Yao H; Lin A Organocatalyzed [3 + 2] Annulation of Cyclopropenones and β‑Ketoesters: An Approach to Substituted Butenolides with a Quaternary Center. Org. Lett 2017, 19, 778–781. [DOI] [PubMed] [Google Scholar]
- 12.Ren J-T; Wang J-X; Tian H; Xu J-L; Hu H; Aslam M; Sun M Ag(I)-Catalyzed [3 + 2]-Annulation of Cyclopropenones and Formamides via C–C Bond Cleavage. Org. Lett 2018, 20, 6636–6639. [DOI] [PubMed] [Google Scholar]
- 13.Xu J; Cao J; Fang C; Lu T; Du D Organocatalytic C–C bond activation of cyclopropenones for ring-opening formal [3 + 2] cycloaddition with isatins. Org. Chem. Front 2017, 4, 560–564. [Google Scholar]
- 14.Shih H-W; Prescher JA A Bioorthogonal Ligation of Cyclopropenones Mediated by Triarylphosphines. J. Am. Chem. Soc 2015, 137, 10036–10039. [DOI] [PubMed] [Google Scholar]
- 15.Row RD; Shih H-W; Alexander AT; Mehl RA; Prescher JA Cyclopropenones for Metabolic Targeting and Sequential Bioorthogonal Labeling. J. Am. Chem. Soc 2017, 139, 7370–7375. [DOI] [PubMed] [Google Scholar]
- 16.Hamada A; Takizawa T Synthesis of phosphorane having ketene group at α-position. Tetrahedron Lett. 1972, 13, 1849–1850. [Google Scholar]
- 17.Cooke MP Jr. Metal-Halogen Exchange-Initiated Intramolecular Conjugate Addition Reactions of Conjugated Acetylenic Esters. J. Org. Chem 1993, 58, 6833–6837. [Google Scholar]
- 18.Cooke MP Jr. Boron-Activation of Acetylenes to Nucleophilic Addition Reactions. J. Org. Chem 1994, 59, 2930–2931. [Google Scholar]
- 19.Alabugin IV; Gilmore K; Manoharan M Rules for Anionic and Radical Ring Closure of Alkynes. J. Am. Chem. Soc 2011, 133, 12608–12623. [DOI] [PubMed] [Google Scholar]
- 20.Wei Y; Zhao WT; Yang YL; Zhang Z; Shi M Allenic Esters from Cyclopropenones by Lewis Base Catalysis: Substrate Scope, the Asymmetric Variant from the Dynamic Kinetic Asymmetric Transformation, and Mechanistic Studies. ChemCatChem 2015, 7, 3340–3349. [Google Scholar]
- 21.Ziegler CB Jr.; Heck RF Palladium-catalyzed vinylic substitution with highly activated aryl halides. J. Org. Chem 1978, 43, 2941–2946. [Google Scholar]
- 22.Hartwig JF Transition Metal Catalyzed Synthesis of Arylamines and Aryl Ethers from Aryl Halides and Triflates: Scope and Mechanism Angew. Chem. Int. Ed 1998, 37, 2046–2067. [DOI] [PubMed] [Google Scholar]
- 23.Wolfe JP; Wagaw S; Marcoux J-F; Buchwald SL Rational Development of Practical Catalysts for Aromatic Carbon–Nitrogen Bond Formation. Acc. Chem. Res 1998, 31, 805–818. [Google Scholar]
- 24.Guo H; Fan YC; Sun Z; Wu Y; Kwon O Phosphine Organocatalysis. Chem. Rev 2018, 118, 10049–10293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang F; Luo T; Hu J; Wang Y; Krishnan HS; Jog PV; Ganesh SK; Prakash GKS; Olah GA Synthesis of gem‐Difluorinated Cyclopropanes and Cyclopropenes: Trifluoromethyltrimethylsilane as a Difluorocarbene Source Angew. Chem. Int. Ed 2011, 50, 7153–7157. [DOI] [PubMed] [Google Scholar]
- 26.Zhou J; Campbell-Conroy EL; Silina A; Uy J; Pierre F; Hurley DJ; Hilgraf N; Frieman BA; DeNinno MP Synthesis of Fused Bicyclic Piperidines: Potential Bioactive Templates for Medicinal Chemistry. J. Org. Chem 2015, 80, 70–79. [DOI] [PubMed] [Google Scholar]
- 27.Nosik PS; Ryabukhin SV; Pashko MO; Grabchuk GP; Grygorenko OO; Volochnyuk DM Synthesis of 1-hetaryl-2,2-difluorocyclopropane-derived building blocks: The case of pyrazoles. J. Fluorine Chem 2019, 217, 80–89. [Google Scholar]
- 28.Hanessian S; Murray PJ Stereochemical control of nature’s biosynthetic pathways: A general strategy for the synthesis of polypropionate-derived structural units from a single chiral progenitor. Tetrahedron 1987, 43, 5055–5072. [Google Scholar]
- 29.Boukouvalas J; Maltais F; Lachance N Furanolate-Based Strategy for Sequential 2,3,4-Trisubstitution of Butenolide: Total Synthesis of Nostoclides I and II. Tetrahedron Lett 1994, 35, 7897–7900. [Google Scholar]
- 30.Khoobi M; Alipour M; Zarei S; Jafarpour F; Shafiee A A facile route to flavone and neoflavone backbones via a regioselective palladium catalyzed oxidative Heck reaction. Chem. Commun 2012, 48, 2985–2987. [DOI] [PubMed] [Google Scholar]
- 31.Almirante N; Cerri A Synthesis of Digitoxigenin from 3β-[(tert-Butyldimethylsilyl)oxy]-17α-iodo-5β-androstan-14β-ol via 17β Stereoselective Free-Radical Introduction of γ-Butyrolactone Moiety. J. Org. Chem 1997, 62, 3402–3404. [DOI] [PubMed] [Google Scholar]
- 32.Mukai C; Moharram SM; Azukizawa S; Hanaoka M Total Syntheses of (+)-Secosyrins 1 and 2 and (+)-Syributins 1 and 2. J. Org. Chem 1997, 62, 8095–8103. [DOI] [PubMed] [Google Scholar]
- 33.Feringa BL; de Lange B Asymmetric 1,4-Additions to 5-Alkoxy-2(5H)-furanones. An Efficient Synthesis of (R)- and (S)-3,4-Epoxy-1-butanol. Tetrahedron 1988, 44, 7213–7222. [Google Scholar]
- 34.de Lange B; van Bolhuis F; Feringa BL Asymmetric 1,4-additions to 5-alkoxy-2(5H)-furanones enantioselective synthesis and absolute configuration determination of β-amino-γ-butyrolactones and amino diols. Tetrahedron 1989, 45, 6799–6818. [Google Scholar]
- 35.Sweidan A; Chollet-Krugler M; van de Weghe P; Chokr A; Tomasi S; Bonnaure-Mallet M; Bousarghin L Design, synthesis and biological evaluation of potential antibacterial butyrolactones. Bioorg. Med. Chem 2016, 24, 5823–5833. [DOI] [PubMed] [Google Scholar]
- 36.Goldfogel MJ; Roberts CC; Manan RS; Meek SJ Diastereoselective Synthesis of γ‑Substituted 2‑Butenolides via (CDC)-Rh-Catalyzed Intermolecular Hydroalkylation of Dienes with Silyloxyfurans. Org. Lett 2017, 19, 90–93. [DOI] [PubMed] [Google Scholar]
- 37.Chu CK; Beach JW; Ullas GV; Kosugi Y An efficient total synthesis of 3’-azido-3’-deoxythymidine (AZT) and 3’-azido-2’,3’-dideoxyuridine (AZDDU, CS-87) from D-mannitol. Tetrahedron Lett. 1988, 29, 5349–5352. [Google Scholar]
- 38.Okabe M; Sun R-C; Tam SY-K; Todaro LJ; Coffen DL Synthesis of the Dideoxynucleosides ddC and CNT from Glutamic Acid, Ribonolactone, and Pyrimidine Bases. J. Org. Chem 1988, 53, 4780–4786. [Google Scholar]
- 39.Beesley RM; Ingold CK; Thorpe JF CXIX.—The formation and stability of spiro-compounds. Part I. spiro-Compounds from cyclohexane. J. Chem. Soc., Trans 1915, 107, 1080–1106. [Google Scholar]
- 40.Cowell A; Stille JK Synthesis of Lactones by the Palladium-Catalyzed Carbonylation of Halo Alcohols. J. Am. Chem. Soc 1980, 102, 4193–4198. [Google Scholar]
- 41.Buchwald SL; Fang Q; King SM A new method for the preparation of 3,5-disubstituted butenolides. Tetrahedron Lett. 1988, 29, 3445–3448. [Google Scholar]
- 42.DeShong P; Sidler DR; Rybczynski PJ; Slough GA; Rheingold AL A general method for the preparation of carbonyl compounds and butenolides from organomanganese pentacarbonyl complexes. J. Am. Chem. Soc 1988, 110, 2575–2585. [Google Scholar]
- 43.Tiecco M; Testaferri L; Tingoli M; Bagnoli L; Santi C Selenium Catalysed Conversion of β,γ-Unsaturated Acids into Butenolides. Synlett 1993, 798–800. [Google Scholar]
- 44.Yoneda E; Zhang S-W; Zhou D-Y; Onitsuka K; Takahashi S Ruthenium-Catalyzed Cyclocarbonylation of Allenyl Alcohols and Amines: Selective Synthesis of Lactones and Lactams. J. Org. Chem 2003, 68, 8571–8576. [DOI] [PubMed] [Google Scholar]
- 45.Oh CH; Park SJ; Ryu JH; Gupta AK Regioselective Pd-catalyzed alkylative lactonizations of 4-hydroxy-2-alkynecarboxylates with organoboronic acids. Tetrahedron Lett. 2004, 45, 7039–7042. [Google Scholar]
- 46.Kang J-E; Lee E-S; Park S-I; Shin S Gold-catalyzed cyclization of tert-butyl allenoate: general synthesis of 2,4-functionalized butenolides. Tetrahedron Lett. 2005, 46, 7431–7433. [Google Scholar]
- 47.Takii K; Kanbayashi N; Onitsuka K Modular synthesis of optically active lactones by Ru-catalyzed asymmetric allylic carboxylation and ring-closing metathesis reaction. Chem. Commun 2012, 48, 3872–3874. [DOI] [PubMed] [Google Scholar]
- 48.Matsushita K; Komori T; Oi S; Inoue Y Carbonylation of Tertiary Propargylic Alcohols Catalyzed by a Cationic Palladium (II) Complex: Synthesis of 2(5H)-Furanones. Tetrahedron Lett. 1994, 35, 5889–5890. [Google Scholar]
- 49.Yu W-Y; Alper H Palladium-Catalyzed Cyclocarbonylation of Terminal and Internal Alkynols to 2(5H)-Furanones. J. Org. Chem 1997, 62, 5684–5687. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.