

Abstract
Purpose:
MDH2 (malate dehydrogenase 2) has recently been proposed as a novel potential pheochromocytoma/paraganglioma (PPGL) susceptibility gene, but its role in the disease has not been addressed. This study aimed to determine the prevalence of MDH2 pathogenic variants among PPGL patients and determine the associated phenotype.
Methods:
Eight hundred thirty patients with PPGLs, negative for the main PPGL driver genes, were included in the study. Interpretation of variants of unknown significance (VUS) was performed using an algorithm based on 20 computational predictions, by implementing cell-based enzymatic and immunofluorescence assays, and/or by using a molecular dynamics simulation approach.
Results:
Five variants with potential involvement in pathogenicity were identified: three missense (p.Arg104Gly, p.Val160Met and p.Ala256Thr), one in-frame deletion (p.Lys314del), and a splice-site variant (c.429+1G>T). All were germline and those with available biochemical data, corresponded to noradrenergic PPGL.
Conclusion:
This study suggests that MDH2 pathogenic variants may play a role in PPGL susceptibility and that they might be responsible for less than 1% of PPGLs in patients without pathogenic variants in other major PPGL driver genes, a prevalence similar to the one recently described for other PPGL genes. However, more epidemiological data are needed to recommend MDH2 testing in patients negative for other major PPGL genes.
Keywords: MDH2, pheochromocytoma and paraganglioma, Variants of unknown significance, Dominant-negative effect, Molecular dynamics
INTRODUCTION
Pheochromocytoma (PCCs) and paragangliomas (PGLs), abbreviated as PPGLs, are very rare neuroendocrine tumors characterized by a high degree of genetic and clinical heterogeneity.1,2
Since the description of NF1 in 1990 as the first driver gene related to PPGL development, 35 additional genes have been identified to be involved in the disease, establishing PPGLs as the human neoplasia with the highest degree of heritability. Among PPGL-associated genes, seven have been found almost exclusively mutated in the germline (SDHA, SDHB, SDHC, SDHD, SDHAF2, FH, and TMEM127), four either in the germline or somatically (RET, VHL, NF1, and MAX), one postzygotically or somatically (EPAS1), and the last one only somatically (HRAS).1,3 Furthermore, there are 22 more susceptibility genes for which the contribution to the disease remains unclear: IDH1, KIF1B, MEN1, BAP1, EGLN1/PHD2, EGLN2/PHD1, ATRX, KMT2D/MLL2, MET, TP53, BRAF, JMJD1C, KDM2B, MERTK, H3F3A, SETD2, EZH2, FGFR1, MITF, CSDE1, GOT2, and IDH3B.1,4–12 In addition, other mechanisms such as point variants in the promoter region of TERT,13,14 SDHC promoter epimutations,15 or rearrangements involving MAML3, BRAF, NGFR, and NF1 have been also described.11
Recently, our group added malate dehydrogenase 2 (MDH2) to the list of potential PPGL susceptibility genes. MDH2 encodes the mitochondrial malate dehydrogenase (MDH), essential for the conversion of malate to oxaloacetate as part of the proper functioning of the Krebs cycle. A single MDH2 PV affecting a donor splice-site (c.429+1G>A) was identified in a 55-year-old man with multiple noradrenergic PGLs associated with bone metastasis,16 and in one apparently unaffected relative with a positive biochemical diagnosis of the disease. Loss of heterozygosity (LOH) and significant reduction of MDH activity in the tumors suggested that MDH2 acts as a tumor suppressor gene. As alterations in Krebs cycle genes have been associated with a higher metastatic risk of the disease, an early genetic diagnosis of unaffected carriers in these families seems to be crucial. However, that study did not address the contribution of MDH2 to the global PPGL susceptibility or the clinical features associated with PV in this gene.
One of the main challenges for genetic screening is the classification of variants of unknown significance (VUS) to improve genetic counseling and clinical follow-up of PV carriers. The pathogenicity assessment of VUS requires taking into account the frequency reported in several databases, in silico effect prediction and functional assays.
In this international collaborative study, we determined the prevalence of germline and/or somatic MDH2 PV, and the associated phenotype in 830 unrelated PPGL index patients, negative for at least the main 13 driver PPGL susceptibility genes. Secondly, we developed a workflow of in silico predictions and simulations, and functional studies for assessing the functional impact of MDH2 VUS identified.
All MDH2 genetic changes, except one in-frame deletion and one variant affecting a donor splice-site, consisted of single-nucleotide substitutions leading to missense, synonymous, or intronic changes, for which we assessed their functional impact. Those MDH2 VUS were analyzed with one of five approaches: (1) applying 20 computational methods to predict their effect at the level of protein structure and function, implementing an enzymatic assay to assess : (2) MDH2 activity and (3) MDH2 affinity; (4) designing an immunofluorescence assay to evaluate MDH2 localization changes; and (5) using a molecular dynamics (MD) simulation approach to examine the potential changes in protein structure and dynamics for the most controversial variants. This sequential scheme aimed to categorize the vast majority of MDH2 VUS found in an extensive setting.
MATERIALS AND METHODS
Patients
Diagnosis of PPGL was established following conventional methods (including clinical, biochemical, imaging, and pathological data). A new series of 561 PPGL index cases negative for at least 13 major PPGL genes (RET, VHL, SDH genes, NF1, HRAS, EPAS1, MAX, TMEM127, and FH) and not previously tested for MDH2 were screened by Sanger (SS) or a next-generation (NGS) sequencing panel (PheoSeq17). To establish the prevalence of MDH2 PV and classify MDH2 VUS, we also considered 269 previously reported patients with negative genetic screening for the 13 PPGL genes, but including 4 carriers of MDH2 VUS.17 This outstanding series of 830 unrelated PPGL index patients was recruited through a collaborative effort from 11 participating centers: 10 of the European Network for the Study of Adrenal Tumors (ENS@T) consortium (Madrid, Paris, Liège, Würzburg, Munich, Dresden, Florence, Rotterdam, Delft, and Nijmegen), and one in the United States (Bethesda).
Clinical data were collected as previously described.18 Table 1 summarizes the clinical characteristics of patients, who provided informed consent to collect clinical and genetic data, in accordance with institutional ethical–approved protocols for each center. In addition, tumor tissues from the Erasmus MC (Rotterdam, The Netherlands) and the Radboud University Medical Centre (Nijmegen, The Netherlands) were used according to the code of conduct: “Proper Secondary Use of Human Tissue” established by the Dutch Federation of Medical Scientific Societies.
Table 1.
Clinical characteristics of the patients included in the study
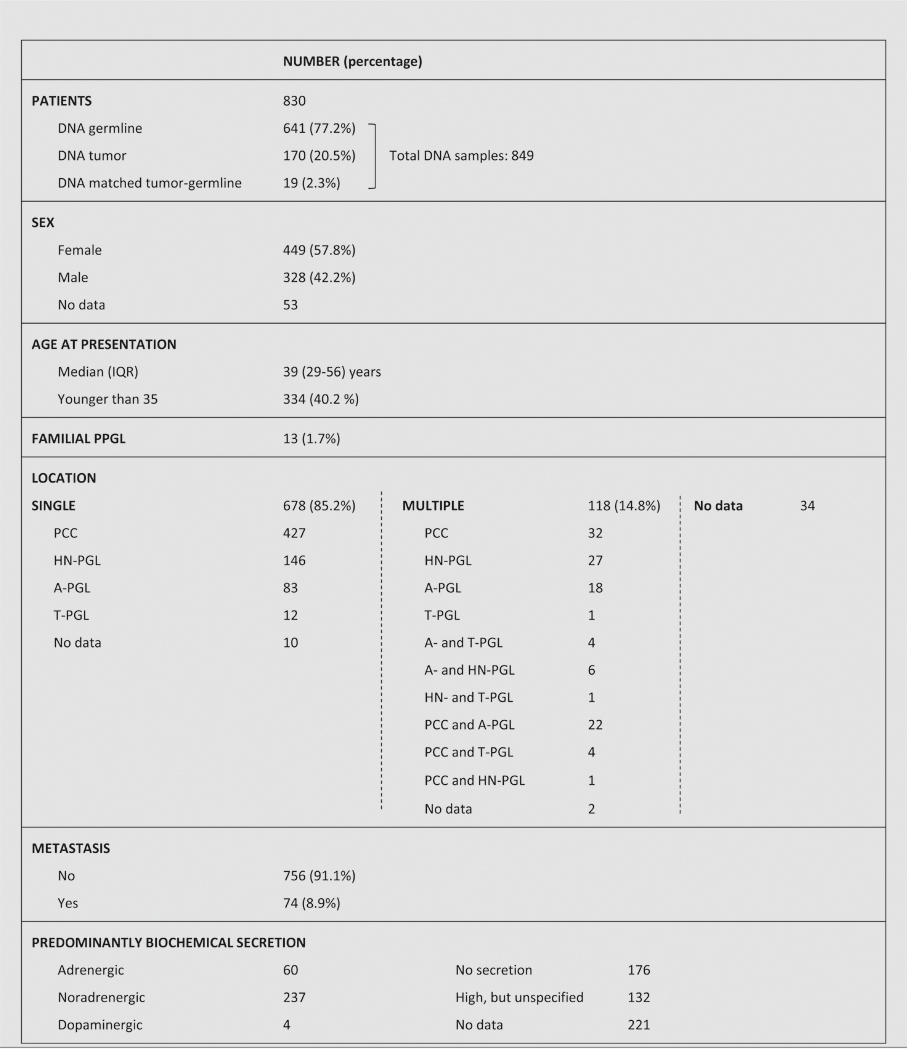 |
IQR interquartile range, PCC pheochromocytoma, PGL paraganglioma, HN head and neck, A abdominal, T thoracic, PPGL pheochromocytoma/paraganglioma
Samples
A total of 849 DNA samples from the 830 patients were available for this study. DNA was obtained exclusively from blood in 641 patients, tumor in 170 patients, and matched tumor-blood in 19 patients (Table 1). Tumor samples were formalin-fixed paraffin-embedded (FFPE) tissues in 80 (42.3%) and frozen tissues in 109 (57.7%) of the cases.
Germline DNA was extracted from peripheral blood samples following a standard method (FlexiGene DNA Kit, Qiagen). DNeasy Blood & Tissue Kit (Qiagen) and truXTRAC FFPE DNA microTUBE Kit (Covaris, MA, USA) were used to extract DNA from frozen tumor tissue and from FFPE tumor samples, respectively, following the manufacturer’s instructions.
Molecular genetic analyses
MDH2 genetic screening was performed in the CNIO (Madrid) and in the HEGP (Paris), either by NGS panel or SS. Primers sequences spanning the nine exons and intron–exon boundaries of MDH2 were those previously described.16
MDH2 gross deletions were tested in 216 cases with good germline DNA quality using a semiquantitative multiplex polymerase chain reaction (PCR) method with labeled primers, as previously described for other genes.16,19
The variant calling was based on the MDH2 transcript ENST00000315758.9. The frequency of MDH2 variants was investigated in public databases: dbSNP (https://www.ncbi.nlm.nih.gov/SNP), COSMIC (http://cancer.sanger.ac.uk/cosmic), and gnomAD (http://gnomad.broadinstitute.org/).
MDH2 variants identified in <0.1% of the population and without homozygotes described were included in the study and used for further analyses. LOH of the MDH2 variants in tumor DNA was assessed by direct sequencing (if material was available).
MDH2 expression by immunohistochemistry
Immunohistochemistry was performed as described in Supplemental Methods.
Computational prediction of functional impact
Three distinct in silico approaches were used to assess the functional and three-dimensional (3D) structural effect of the missense variants: (1) measure of the evolutionary conservation in the genome of vertebrate and mammalian species (i.e., phyloP, phastCons, GERP++), (2) prediction of the impact of amino acid substitutions in protein function (i.e., SIFT, Polyphen2, LRT, MutationAssessor, fathmm-MKL, PROVEAN, MetaSVM, MetaLR, MutationTaster), and (3) prediction of protein 3D destabilization (i.e., PoPMu-SiCv3.1, CUPSAT, I-Mutant v3.0, MAESTRO, INPS-3D).
A consensus interpretation was established according to the number of tools predicting a damaging effect versus a benign or tolerated one. The consensus was given if at least 70% (functional impact: 7 of 10; 3D stability: 4 of 5) of the predictors agreed in the variant classification. Otherwise, the results were considered as inconclusive.
Moreover, we included functional annotations (e.g., ligand binding sites, catalytic residues, posttranslational modifications of proteins, residues in protein–protein interaction interfaces) retrieved by the Structure-PPi system.20 Structure-PPi also considers residues in physical proximity (at a distance of 5 Å) to amino acid changes found in other type of cancers.
ESEFinder and RESCUE-ESE using Alamut Visual software version 2.7 (Interactive Biosoftware, Rouen, France) were used to predict splicing changes in synonymous and intronic variants.
RBP1 gene expression analysis
The low expression of retinol binding protein 1 (RBP1) is a marker of Krebs cycle disruption.21 Its assessment is detailed in Supplemental Methods.
Plasmids and cell culture
QuickChange Lightning Site-Directed Mutagenesis Kit (Agilent) was used to generate missense variants in pCMV6-AC-MDH2 (Origene), a plasmid containing the full complementary DNA (cDNA) sequence (NM_005918) of the human MDH2 gene. We generated a polymorphism with a minor allele frequency (MAF) = 0.037 in gnomAD (rs10256: p.Lys301Arg), to be used as control. Primers used to generate mutated vectors are described in Table S1. The introduction of the PV of interest was confirmed by SS (Fig. S1).
Sh8561 MDH2 knockdown Hela cells16 (MDH2 KD cells) culture conditions are specified in Supplemental Methods.
Microscopic analysis
For immunofluorescence imaging, 300,000 cells were seeded 24 h before transfection on coverslips in 12-well plates. Cells were transiently transfected with 2 μg of plasmids mutated with each variant using Lipofectamine 2000 (Invitrogen) following the vendor’s manual. Twenty-four hours after transfection cells were stained as described in Supplemental Methods and analyzed by confocal microscopy (Leica TCS SP5 X).
In silico molecular dynamics (MD) simulations
The initial model of the system used the crystal structure of a tetramer of oxaloacetate and NADH-bound MDH2 (PDBid: 4wlo). The structure contains coordinates for residues 24–337 of each monomer. The dimer formed by monomers A and B was extracted and ligands and crystal waters were removed. Missing hydrogen atoms were added using the H++server,22 and in the case of the mutants, the point variants were introduced using Pymol.23 The systems were placed in dodecahedral boxes, with the minimal distance between the protein and the borders of the box being 10 Å. The systems were solvated with TIP3P waters,24 and Na+/CL− was added to neutralize them to obtain a physiological salt concentration of 0.15 mol/l. First, the systems were minimized during 500 steps, applying the steepest descent algorithm. Consequently, the solvent was equilibrated for 100 ps in the NVT ensemble, followed by another 100 ps in the isobaric–isothermal (NpT) ensemble. The resulting configurations were used for MD production runs of 400 ns for each system. The trajectories were analyzed by principal component analysis (PCA) (see Supplemental Methods).
Simulations and analyses of the trajectories were performed as described in Supplemental Methods25–31.
Enzymatic activity assay
MDH2 KD cells were seeded in T150 flasks at 16·106 cells/flask 24 h before transfection. Each flask was transiently transfected with 20 μg of each variant plasmid using Lipofectamine 2000 (Invitrogen), according to the manufacturer’s recommendations. MDH2 KD cells transfected either with wild-type (WT) cDNA (pCMV6-AC-MDH2), MDH2 KD cells with pCMV6-AC empty vector (EV), or MDH2 KD cells with pCMV6-AC-MDH2-c.902A>G vector were used as controls. To mimic the heterozygous state of p.Arg104Gly patient, we cotransfected 10 μg pCMV6-AC-MDH2 and 10 μg pCMV6-AC-MDH2-c.310A>G plasmids; as control, we used cells cotransfected with 10 μg pCMV6-AC-MDH2 and 10 μg pCMV6-AC EV, and 20 μg EV alone. Twenty-four hours after transfection, enzymatic activity assay was performed as described in Supplemental Methods.
RESULTS
MDH2 variants and computational analyses
Twelve MDH2 heterozygous variants (Fig. 1) were found, demonstrating a germline origin for 11 of them (only tumor DNA was available in the remaining case). Clinical data of the MDH2 carriers are detailed in Table S3. None of the patients had family history of the disease.
Fig. 1. Graphical representation of the variants identified in MDH2 and the residues of the protein affected.

The PV previously reported16 is indicated with a star. Missense variants are framed in continuous line; in-frame deletion is framed in discontinuous line; synonymous variant is shown in the upper part of the figure; intronic variants are shown in the lower part of the figure
Five of the 12 were missense (41.7%), 1 synonymous (8.3%), 4 were located in the intronic region (33.3%), 1 was an in-frame deletion (8.3%), and 1 affected a donor splice-site (8.3%) (Fig. 1). Five of them were unreported variants (3 missense, 1 intronic, and 1 in-frame deletion), six showed a low allele frequency (<1·10−3) and no homozygotes in gnomAD, and the donor splice-site variant had been previously suggested to be pathogenic16 (Table 2; Table S5). The two probands with the splice-site MDH2 PV are not related.
Table 2.
Summary of the in silico and in vitro analyses in MDH2 missense and deletion VUS
| VUS | V-l p.Ser3Phe | V-2 p.Argl04Gly | V-5 p.Glnl30Arg | V-6 p.Vall60Met | V-9 p.Ala256Thr | V-10 p.Lys314del | |
|---|---|---|---|---|---|---|---|
| SNP ID ExAC allele freq. gnomAD* allele freq. 1000 Genomes* | not described | not described | not described | rsl38541865 1,66–10−4; 0 hom | rsl47655350 4,69–10−5; 0 hom | not described | |
| Evolutionary conservation Interpretation (a) | Conserved position | Conserved position | Conserved position | Conserved position | Conserved position | Conserved position ¤ | |
| Functional domain | NA (Low complexity region) | Ldh_l_N domain (PF00056) | Ldh_l_N domain (PF00056) | Ldh_l_N domain (PF00056) | Ldh_l_C domain (PF02866) | Ldh_l_C domain (PF02866) | |
| Protein function prediction Interpretation (a) | Inconclusive (5/9) | Impaired (10/10) | Neutral (3/10) | Impaired (8/10) | Impaired (9/10) | NA | |
| 3D structural annotations (Structure-PPi) | Transit peptide to Mitochondria (aa 1–24) | NAD-binding site | α-helix | NA | α-helix | α-helix | |
| 3D structural prediction Interpretation (a) | No effect | Destabilize (4/5) | Inconclusive (3/5) | Destabilize (5/5) | Destabilize (5/5) | NA | |
| Neighbor positions mutated in other cancers (b) (Structure-PPi) | NA | Malignant melanoma, Ovary carcinoma, Colon carcinoma, Endometrium carcinoma | Breast carcinoma | Stomach carcinoma | Endometrium carcinoma, Head and neck squamous cell carcinoma | Endometrium carcinoma (p.Ser310Ser), Prostate carcinoma (p.Ser317Leu), Oral carcinoma (p.Ala319Ser) | |
| RBP1 expression (c) | +++ | + | NA | + | NA | + | |
| LOH | no | no | NA | no | NA | Yes | |
| MDH2 subcelular localization (d) | M | M | M | M | M | NA | |
| Enzymatic experiment | with saturating [NAD+] and [malate] (e) | +++ | + | +++ | +++ | +++ | NA |
| co-transfection experiment (e) | NA | ++ | NA | NA | NA | NA | |
| Affinity assay: with lower [NAD+] and [malate] (f) | NA | Impaired for malate and NAD+ | NA | Likely impaired for malate | Likely impaired for malate and NAD+ | NA | |
| Molecular dynamics simulations | NA | NA | NA | Substrate binding site likely affected | Dimerization likely affected | NA | |
| Classification of VUS | LIKELY BENIGN | PATHOGENIC | LIKELY BENIGN | LIKELY PATHOGENIC | LIKELY PATHOGENIC | PATHOGENIC | |
Results in bold indicate major alterations. V-variant, NA not available, Ldh_1_N domain lactate/malate dehydrogenase, NAD-binding domain (Pfam accession: PF00056), Ldh_1_C domain lactate/malate dehydrogenase, alpha/beta C-terminal domain (Pfam accession: PF02866), SNP single-nucleotide polymorphism, VUS variant of unknown significance, M mitochondrial
See Table S4 for breakdown of the different computational predictors. The number of tools predicting a damaging effect is in brackets
See Fig. S2 for mapping of the pathogenic variant onto the crystal structure
RBP1 messenger RNA (mRNA) expression relative to tumors with pathogenic variants in non-Krebs cycle genes: +++ corresponds to nonsignificant RBP1 expression reduction, + corresponds to significant (p < 0.05) RBP1 expression reduction
M: mitochondrial subcelular localization
From in vitro MDH2 enzymatic activity (Fig. 2a, b): +++ corresponds to MDH2 activity >75% compared with control, ++ 25–75%, and + 0–25%
http://gnomad.broadinstitute.org/ and http://www.internationalgenome.org/ (Last accessed 7 June 2017)
Despite the fact that the variant p.K314del is not annotated in the dbNSFP database, we infer the evolutionary conservation of this position considering the annotated variants at this position (i.e., p.K314Q, p.K314E, p.K314*, p.K314T, p.K314R, p.K314M and p.K314N). For each method, the lowest and the highest scores are included
The synonymous variant (p.Phe333Phe), found in a patient with a nonfunctional T-PGL, had no effect according to in silico splicing predictors (Table S5). Similarly, the splicing for three of the four intronic variants (c.320–26A>C, c.733 +47G>A, and c.734–5C>A) was not predicted affected, while it was anticipated as altered in the remaining intronic variant (c.319+37G>A) identified in a 48-year-old patient with a noradrenergic PCC. RNA of this patient was not available to confirm this prediction.
Of the five MDH2 missense variants identified (Table 2), only one (p.Ser3Phe) was outside the functional domains in the transit peptide to mitochondria. The variants p.Arg104Gly, p.Gln130Arg, and p.Val160Met were positioned in the lactate/malate dehydrogenase, NAD-binding domain; and p.Ala256Thr in the lactate/malate dehydrogenase, alpha/beta C-terminal domain. Furthermore, they affected conserved positions in vertebrate and mammalian species, as indicated by phyloP, phastCons, and GERP++ methods. Indeed, p.Arg104Gly is positioned in the NAD-binding site.
Three of the missense variants (p.Arg104Gly, p.Val160Met, and p.Ala256Thr) were predicted to have a damaging effect (impaired functional predictions and destabilization of the 3D structure) (Table 2; Table S4). The variant p.Ser3Phe was classified as inconclusive according to the functional impact predictions and the corresponding 3D structural predictions could not be performed (residue not present in the crystal structure [PDB ID: 2DFD]). The variant p.Gln130Arg was categorized as neutral and inconclusive according to the functional and 3D structure predictions, respectively. Moreover, the COSMIC database reported MDH2 somatic PV in other cancers in neighbor positions (d ≤ 5 Å), as indicated by Structure-PPi (Fig. S2), for all the variants except for p.Ser3Phe.
The variants p.Arg104Gly and p.Ala256Thr were found in two young patients (25 and 29 years old, respectively) with norepinephrine-producing PCC both diagnosed during pregnancy. One of them developed metachronous bone metastases. The p.Val160Met was identified in a PCC patient without biochemical data. The remaining two missense variants (p.Ser3Phe and p.Gln130Arg) were found in patients older than 45 years, diagnosed with PCC; the former involving an adrenergic tumor and the latter without evidence of excess in catecholamine production (Table S3).
A previously unreported in-frame deletion (p.Lys314del) was found in a 55-year-old patient, with multiple noradrenergic PGLs. It affected a conserved residue, for which PV in neighboring residues have been described in several cancers (Table 2; Fig. S2); LOH was demonstrated in the tumor.
Furthermore, the c.429+1G>T variant, previously described,16 was found in a 57-year-old patient diagnosed with a PCC and liver metastases.
MDH2 immunohistochemistry
MDH2 immunohistochemistry it is not useful to classify VUS or select patients for MDH2 screening (see Supplemental Results).
RBP1 expression in MDH2 variants
RBP1 measurement was performed in four available tumors (p.Ser3Phe, p.Arg104Gly, p.Val160Met, and p.Lys314del-tumor), observing a reduced RBP1 expression in three tumors compared with controls: 93.86 ± 1.83% (p = 0.007), 83.18 ± 0.65% (p = 0.007), and 82.44 ± 19.72% (p = 0.030) for p.Lys314del-, p.Arg104Gly-, and p.Val160Met-tumor, respectively (Fig. S3).
MDH2 localization
None of the variants was associated with an altered MDH2 localization, or mitochondrial quantity and morphology (see Supplemental Results).
Enzymatic activity characterization
Only variant p.Arg104Gly displayed a significant lower MDH2 enzymatic activity at saturating concentration of substrates (p < 0.0001) compared with WT, comparable with the activity detected in the KD cells not expressing MDH2 (Fig. 2a). On the other hand, citrate synthase activity, present exclusively in the mitochondria, was similar for all variants (Table S2), suggesting that none of them produced an increased mitochondrial biogenesis to compensate the possible aberrant MDH2 variant. Thus, this functional assay only supported pathogenicity for p.Arg104Gly, having an incomplete functional proof of in silico predictions.
Fig. 2. MDH2 activity measured in MDH2 KD cells transfected with different vectors containing the variant of unknown significance (VUS) in MDH2.

a Enzymatic activities at saturating concentrations of malate (25 mM) and NAD+ (2 mM) in p.R104G (p.Arg104Gly), p.Q130R (p.Gln130Arg), p.V160M (p.Val160Met), and p.S3F (p.Ser3Phe) variants, plus C (p.K301R-p.Lys301Arg) which is a polymorphism with minor allele frequency of 0.037, knockdown (KD) and wild-type (WT) as controls, are expressed as mean (nmol/min/mg) ± SD of 3 paired independent experiments in quadruplicate. Different shadings indicate the only variants tested in experiments in c–f. b Enzymatic activities at saturating concentrations of cells cotransfected with WT, empty vector (EV), and/or p.R104G plasmids expressed as mean of fold-change over activity in cells cotransfected with WT and EV (control) ± SD of 2 paired independent experiments at least in quadruplicate; μg of plasmid DNA transfected for each condition are showed in the figure. c–f Enzymatic activities in p.A256T, p.V160M, p.R104G, and control (p.K301R) expressed as mean of fold-change over control ± SD of at least 2 paired independent experiments in triplicate. Assays performed with reduced concentrations of malate or NAD+: c 5-fold reduction malate (5 mM), d 10-fold reduction malate (2.5 mM), e 4-fold reduction of NAD+ (0.5 mM), and f 20-fold reduction of NAD+ (0.1 mM). *****p < 0.0001, ****p< 0.001, ***p < 0.01, **p < 0.03, and *p < 0.05 based on a two-sided Mann–Whitney U test
Enzymatic activity assay to check a dominant-negative effect
LOH was not detected in any of the tumors carrying the missense variants. To evaluate if MDH2 variants could exert a dominant-negative effect on MDH2 WT, we took as a model the p.Arg104Gly variant. We cotransfected WT plasmid, p.Arg104Gly plasmid, and a combination of both to mimic the heterozygous state of the mutated patient. EV was used to achieve the same amount of total transfected plasmid (20 μg in each cotransfection). Cells cotransfected with both WT and p.Arg104Gly plasmids exhibited lower enzymatic activity in comparison with those cotransfected with WT and EV ones (27.8% ± 22.6; p = 0.0002) (Fig. 2b).
MDH2 missense variants affinity characterization
Another assay was designed to evaluate if p.Val160Met and p.Ala256Thr variants affected the affinity of the enzyme for the substrates, instead of the maximal activity. We used the p.Arg104Gly variant as positive control. A tendency of reduced enzymatic activity when decreasing malate concentration was observed for p.Val160Met, significant at 5 mM (5-fold reduction to malate saturating concentration, p = 0.0256) (Fig. 2c) and at 2.5 mM (10-fold reduction to malate saturating concentration, p = 0.0047) (Fig. 2d). Furthermore, a subtle decrease in the activity was observed for p.Ala256Thr when diminishing concentration of NAD+ (Fig. 2f) and malate (Fig. 2d) to 20-fold (p = 0.0464) and 10-fold (p = 0.0366), respectively. No significant changes were observed at higher NAD+ concentrations (0.5 mM) (Fig. 2e).
Molecular dynamics (MD) simulations
Simulations of the dimers of the WT apoenzyme, and the p.Ala256Thr and p.Val160Met variants revealed differences in their principal motions. In the WT, we observed movements of the two monomers relative to each other, and large conformational changes in one of the helices of the substrate-binding site and its adjacent loop (Fig. S5A). In the p.Ala256Thr mutant, the character of the main motions was conserved (Fig. S5B), whereas in the p.Val160Met mutant the relative movement of the monomers was strongly reduced (Fig. S5C).
Although the dynamics of the p.Ala256Thr variant were similar to that of the WT, closer inspection revealed conformational changes. In the WT and p.Val160Met variant, the side chain of Phe260, neighboring Ala/Thr256, switches between two orientations, while in the p.Ala256Thr mutant it remains immobile (Fig. 3a). On a larger scale, the opening between the two monomers becomes slightly enlarged (Fig. 3b).
Fig. 3. Molecular dynamics simulations in MDH2 p.Ala256Thr and p.Val160Met variants of unknown significance (VUS) versus wild-type (WT).

a Distance between the Cζ atom of F260 and the Cβ atom of A/T256 for both monomers of WT MDH2 (magenta, upper panels), Ala256Thr (blue, middle panels), and V160M (green, lower panels) during the simulations. A short distance corresponds to the F260 down conformation, and a long distance to the F260 up conformation (inset). b and c Distribution of the distance between the Cα atoms of K269 of the two monomers of the a Ala256Thr and b Val160Met variants from snapshots from the simulation trajectories, compared with WT
Even on a short time scale, the MD simulations revealed changes in conformation and dynamics of the variants, compared with the WT. The results suggest that the p.Ala256Thr variant affects the conformation of the neighboring residues, which contribute to the dimeric interface. This implies that the p.Ala256Thr variant may affect dimerization of MDH2. In the p.Val160Met variant, changes in the dynamics of the substrate-binding site may affect substrate affinity.
DISCUSSION
After the identification of major susceptibility PPGL genes, the list of other genes with modest contributions to the disease has kept growing and it is likely that this number will continue to increase over the near future.3 Examples include SDHA,32 TMEM127,33 MAX,34 or FH.35 Through the use of NGS panels36 to offer a comprehensive genetic diagnosis, multiple VUS are identified, for which the functional interpretation represents a crucial challenge in an accurate genetic counseling session. Herein, we aimed to determine the prevalence and the clinical characteristics of MDH2 PV carriers in 830 patients with PPGL without PV in major PPGL susceptibility genes and to investigate the potential pathogenicity of every identified MDH2 variant. We were able to classify 2 MDH2 variants as pathogenic and provide evidence that suggests an altered molecular function of MDH2 in 2 others (which have been designated as likely PV), following the criteria established by the American College of Medical Genetics and Genomics and the Association for Molecular Pathology guidelines.37 Furthermore, a new patient, carrying the already reported c.429+1G>T MDH2 variant, was identified.
RBP1 expression in the tumor, bioinformatics predictions, and functional assays suggested that p.Arg104Gly is a PV located in the highly conserved NAD-binding site and significantly impairing MDH2 activity. This variant was found in a young patient with a noradrenergic PCC. PV in neighboring positions are reported in different cancers, supporting the implication of PV at this residue in the neoplastic process. However, neither LOH nor any other somatic PV was found in the corresponding tumor sample. Enzymatic assays performed by cotransfection of WT and p.Arg104Gly plasmids resembling the heterozygous character of this PV suggested a dominant-negative effect of the p.Arg104Gly-mutant.
The two other missense variants, p.Ala256Thr and p.Val160Met, reported as rare SNPs and located in conserved residues, were predicted to produce protein 3D structure destabilization and impaired the MDH2 molecular function. The p.Val160Met was detected in a 54-year-old PCC patient, whose tumor showed low RBP1 expression. The p.Ala256Thr tumor sample was not available, so the interpretation of this variant was based exclusively on functional assays. This latter variant was found in a 29-year-old female with a noradrenergic metastatic PCC. The substitutions at residues Val160 and Ala256 could also affect the substrate binding affinity and protein 3D stability, and therefore, neither the enzymatic assay nor the immunofluorescence experiments are able to evaluate the effect. Because of that, we conducted MD simulations only with these variants, which suggested that p.Val160Met could be modifying malate binding to the catalytic site, and consequently affecting MDH2 affinity for its substrate. This was demonstrated in vitro, as MDH2 activity decreased when we reduced malate concentration, pointing to a lower affinity of the mutated enzyme for malate. For variant p.Ala256Thr, MD simulations predicted that it could be affecting enzyme dimerization. We observed a slight decrease in MDH2 activity when reducing both malate and NAD+ down to low concentrations, which could be related to impaired dimerization. A second somatic hit was not observed in the tumor of p.Val160Met-related patient and p.Ala256Thr-related tumor was not available. A dominant-negative effect for p.Val160Met and p.Ala256Thr variants might be the underlying mechanism as occurs with the p. Arg104Gly variant, although this has not been tested in this study. Thus, the p.Ala256Thr variant could be classified as likely pathogenic. Regarding the p.Val160Met variant, although most of our analyses suggested a potential pathogenic role as well, it was also classified as likely pathogenic due to the high number of alleles found (46/277206) in the general population.
For the two other novel missense variants (p.Ser3Phe and p.Gln130Arg) identified, computational analyses did not reach a consensus. In addition, patients carrying these variants had predominant adrenaline production or nonfunctional tumors, which is in discordance with MDH2-mutated patient16 and other Krebs cycle genes. The high RBP1 expression in the p.Ser3Phe-tumor supported that this variant is not pathogenic. Thus, we classified these two variants as likely neutral.
The four intronic variants and the synonymous one were not classified, as no RNA was available. The computational tools indicated no agreement, highlighting the relevance of having access to a tissue tumor sample to study at least the RBP1 and/or MDH2 expression.
The p.Lys314del, identified in a patient with multiple noradrenergic PGLs, was classified as pathogenic, as it affects a conserved amino acid, and LOH and low RBP1 expression in the tumor sample were found.
Finally, a 57-year-old patient with a metastatic pheochromocytoma, clinical phenotype similar to the patient reported,16 was identified to carry the same variant affecting a donor splice-site (c.429+1G>A).
In summary, taking into account only those MDH2 variants identified that display characteristics supportive of a pathogenicity potential, we provide more evidence that suggests the potential role of MDH2 in PPGL predisposition, and indicates that MDH2 germline PV could be responsible for 0.6% of PPGL cases, prevalence comparable with that reported for other recently described PPGL genes. The apparent lack of family history in four pedigrees investigated suggests an incomplete penetrance of MDH2, similar to the one observed in other Krebs cycle genes, such as SDHA or FH. Furthermore, there are other similarities worthy to mention. In this regard, families affected with encephalopathy due to recessive MDH2 deficiency have been described.38 One could expect to find PPGL patients in these families, but as it happens in pedigrees affected with the Leigh syndrome (OMIM 256000) associated with autosomal recessive PV in SDHA,39 their members do not develop either these tumors.
On the other hand, it is worthy to note that MDH2 variants were found in metastatic cases, as two of five patients (three of six, if we include the reported MDH2 patient)16 developed metastases. Taking into account the low prevalence of MDH2 PV, as well as the low penetrance, its genetic testing could be considered in a research direction manner until providing further epidemiological and segregation data that would confirm the implication of MDH2 PV in PPGL susceptibility.
NGS is becoming the rational tool to apply to PPGL genetic diagnosis, and unavoidably it leads to an increasing number of VUS reported. The task of classifying VUS for genetic counseling is especially complex when considering genes scarcely analyzed, which exhibit a high ratio of missense variants, as previously shown for MAX.40 In this study, we were able to demonstrate a functional impact for two variants (p.Arg104Gly and p.Lys314del) and suggested an altered molecular function for other two (p.Val160Met and p.Ala256Thr), but there was insufficient evidence to consider them pathogenic even after applying up to five approaches to classify them. Although, it is likely that this rationale is unapproachable in the clinical setting when tumor tissue is unavailable, we demonstrated that MDH2 variants could be classified by a multidisciplinary approach.
Supplementary Material
ACKNOWLEDGEMENTS
We gratefully acknowledge Diego Megias and all Confocal Microscopy Core Unit (CNIO) team for their technical support and advice, as well as Santiago Ramón-Maiques from Structural Bases of Genome Integrity Group (CNIO) for fruitful discussions. This work was supported by the Instituto de Salud Carlos III (ISCIII), Acción Estratégica en Salud, (projects PI14/00240 and PI17/01796), cofinanced by Fondo Europeo de Desarrollo Regional (FEDER), GETNE (Grupo Español de Tumores Neuroendocrinos), the European Union Seventh Framework Programme (FP7/2007-2013) under grant agreement no. 259735, the Paradifference foundation, and the Intramural Research Program of the National Institutes of Health (NIH) and National Institute of Child Health and Human Development (NICHD). Bruna Calsina is supported by the Becas de excelencia Rafael del Pino 2017, Maria Currás-Freixes was supported by the Severo Ochoa Excellence Programme (project SEV-2011-0191), Laura Contreras by a CIBERER contract, Alexandre Buffet received financial support from ITMO Cancer AVIESAN (Alliance Nationale pour les Sciences de la Vie et de la Santé, National Alliance for Life Sciences & Health) within the framework of the Cancer Plan, and Susan Richter is supported by the Deutsche Forschungsgemeinschaft (RI 2684/1–1).
Footnotes
ELECTRONIC SUPPLEMENTARY MATERIAL
The online version of this article (https://doi.org/10.1038/s41436–018-0068–7) contains supplementary material, which is available to authorized users.
DISCLOSURE
The authors declare no conflicts of interest.
REFERENCES
- 1.Dahia PLM. Pheochromocytoma and paraganglioma pathogenesis: learning from genetic heterogeneity. Nat Rev Cancer. 2014;14:108–19. [DOI] [PubMed] [Google Scholar]
- 2.Crona J, Backman S, Maharjan R, et al. Spatiotemporal heterogeneity characterizes the genetic landscape of pheochromocytoma and defines early events in tumorigenesis. Clin Cancer Res. 2015;21:4451–4460. [DOI] [PubMed] [Google Scholar]
- 3.Favier J, Amar L, Gimenez-Roqueplo A-P. Paraganglioma and phaeochromocytoma: from genetics to personalized medicine. Nat Rev Endocrinol. 2014;11:101–11. [DOI] [PubMed] [Google Scholar]
- 4.Gaal J, Burnichon N, Korpershoek E, et al. Isocitrate dehydrogenase mutations are rare in pheochromocytomas and paragangliomas. J Clin Endocrinol Metab. 2010;95:1274–1278. [DOI] [PubMed] [Google Scholar]
- 5.Yang C, Zhuang Z, Fliedner SMJ, et al. Germ-line PHD1 and PHD2 mutations detected in patients with pheochromocytoma/paragangliomapolycythemia. J Mol Med. 2014;93:93–104. [DOI] [PubMed] [Google Scholar]
- 6.Fishbein L, Khare S, Wubbenhorst B, et al. Whole-exome sequencing identifies somatic ATRX mutations in pheochromocytomas and paragangliomas. Nat Commun. 2015;6:6140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Castro-Vega LJ, Letouze E, Burnichon N, et al. Multi-omics analysis defines core genomic alterations in pheochromocytomas and paragangliomas. Nat Commun. 2015;6:6044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Luchetti A, Walsh D, Rodger F, et al. Profiling of somatic mutations in phaeochromocytoma and paraganglioma by targeted next generation sequencing analysis. Int J Endocrinol. 2015;2015:138573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Toledo RA, Qin Y, Cheng ZM, et al. Recurrent Mutations of Chromatin-Remodeling Genes and Kinase Receptors in Pheochromocytomas and Paragangliomas. Clin Cancer Res. 2016;22:2301–2310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Castro-Vega LJ, Kiando SR, Burnichon N, Buffet A, Amar L, Simian C. The MITF, p.E318K variant as a risk factor for pheochromocytoma and paraganglioma. J Clin Endocrinol Metab. 2016;101: 4764–4768. [DOI] [PubMed] [Google Scholar]
- 11.Fishbein L, Leshchiner I, Walter V, et al. Comprehensive molecular characterization of pheochromocytoma and paraganglioma. Cancer Cell. 2017;31:181–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Remacha L, Comino-Méndez I, Richter S, et al. Targeted Exome Sequencing of Krebs Cycle Genes Reveals Candidate Cancer–Predisposing Mutations in Pheochromocytomas and Paragangliomas. Clin Cancer Res. 2017;23: 6315–6324. [DOI] [PubMed] [Google Scholar]
- 13.Papathomas TG, Oudijk L, Zwarthoff EC, et al. Telomerase reverse transcriptase promoter mutations in tumors originating from the adrenal gland and extra-adrenal paraganglia. Endocr Relat Cancer. 2014;21: 653–661. [DOI] [PubMed] [Google Scholar]
- 14.Liu T, Brown TC, Juhlin CC, et al. The activating TERT promoter mutation C228T is recurrent in subsets of adrenal tumors. Endocr Relat Cancer. 2014;21:427–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Richter S, Klink B, Nacke B, et al. Epigenetic mutation of the succinate dehydrogenase C promoter in a patient with two paragangliomas. J Clin Endocrinol Metab. 2016;101:359–363. [DOI] [PubMed] [Google Scholar]
- 16.Cascón A, Comino-Méndez I, Currás-Freixes M, et al. Whole-exome sequencing identifies MDH2 as a new familial paraganglioma gene. J Natl Cancer Inst. 2015;107:1–5. [DOI] [PubMed] [Google Scholar]
- 17.Currás-Freixes M, Piñeiro-Yañez E, Montero-Conde C, et al. PheoSeq: a targeted next-generation sequencing assay for pheochromocytoma and paraganglioma diagnostics. J Mol Diagn.2017;19:575–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Currás-Freixes M, Inglada-Pérez L, Mancikova V, et al. Recommendations for somatic and germline genetic testing of single pheochromocytoma and paraganglioma based on findings from a series of 329 patients. J Med Genet. 2015;52:647–656. [DOI] [PubMed] [Google Scholar]
- 19.Cascón A, Montero-Conde C, Ruiz-Llorente S, et al. Gross SDHB deletions in patients with paraganglioma detected by multiplex PCR: a possible hot spot? Genes Chromosomes Cancer. 2006;45:213–219. [DOI] [PubMed] [Google Scholar]
- 20.Vázquez M, Valencia A, Pons T. Structure-PPi: a module for the annotation of cancer-related single-nucleotide variants at protein-protein interfaces. Bioinformatics. 2015;31:2397–2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chou AP, Chowdhury R, Li S, et al. Identification of retinol binding protein 1 promoter hypermethylation in isocitrate dehydrogenase 1 and 2 mutant gliomas. J Natl Cancer Inst. 2012;104:1458–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gordon JC, Myers JB, Folta T, et al. H++: a server for estimating pKas and adding missing hydrogens to macromolecules. Nucleic Acids Res. 2005;33:W368–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.DeLano WL. The PyMOL molecular graphics system. Schrödinger LLC. Version 1. 2002. http://www.pymol.org needs an acces date
- 24.Mahoney MW, Jorgensen WL. A five-site model for liquid water and the reproduction of the density anomaly by rigid, nonpolarizable potential functions. J Chem Phys. 2000;112:8910. [Google Scholar]
- 25.Van Der Spoel D, Lindahl E, Hess B et al. GROMACS: fast, flexible, and free. J Comput Chem. 2005;26:1701–1718. [DOI] [PubMed] [Google Scholar]
- 26.Hess B, Kutzner C, Van Der Spoel D, Lindahl E. GRGMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J Chem Theory Comput. 2008;4:435–447. [DOI] [PubMed] [Google Scholar]
- 27.Abraham MJ, Murtola T, Schulz R et al. Gromacs: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1–2:19–25. [Google Scholar]
- 28.Lindorff-Larsen K, Piana S, Palmo K et al. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins. 2010;78: 1950–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Best RB, Hummer G. Optimized molecular dynamics force fields applied to the helix-coil transition of polypeptides. J Phys Chem B. 2009;113: 9004–9015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bussi G, Donadio D, Parrinello M. Canonical sampling through velocity rescaling. J Chem Phys. 2007;126:014101. [DOI] [PubMed] [Google Scholar]
- 31.Parrinello M, Rahman A. Polymorphic transitions in single crystals: a new molecular dynamics method. J Appl Phys. 1981;52:7182–7190. [Google Scholar]
- 32.Korpershoek E, Favier J, Gaal J, et al. SDHA immunohistochemistry detects germline SDHA gene mutations in apparently sporadic paragangliomas and pheochromocytomas. J Clin Endocrinol Metab.2011;96:E1472–1476 [DOI] [PubMed] [Google Scholar]
- 33.Qin Y, Yao L, King EE, et al. Germline mutations in TMEM127 confer susceptibility to pheochromocytoma. Nat Genet. 2010;42:229–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Comino-Méndez I, Gracia-Aznárez FJ, Schiavi F, et al. Exome sequencing identifies MAX mutations as a cause of hereditary pheochromocytoma. Nat Genet. 2011;43:663–667. [DOI] [PubMed] [Google Scholar]
- 35.Castro-Vega LJ, Buffet A, De Cubas AA, et al. Germline mutations in FH confer predisposition to malignant pheochromocytomas and paragangliomas. Hum Mol Genet. 2014;23:2440–2446. [DOI] [PubMed] [Google Scholar]
- 36.Toledo RA, Burnichon N, Cascon A, et al. Consensus statement on next-generation-sequencing-based diagnostic testing of hereditary phaeochromocytomas and paragangliomas. Nat Rev Endocrinol. 2016;13:233–247. [DOI] [PubMed] [Google Scholar]
- 37.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ait-El-Mkadem S, Dayem-Quere M, Gusic M, et al. Mutations in MDH2, encoding a Krebs cycle enzyme, cause early-onset severe encephalopathy. Am J Hum Genet. 2017;100:151–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Renkema GH, Wortmann SB, Smeets RJ, et al. SDHA mutations causing a multisystem mitochondrial disease: novel mutations and genetic overlap with hereditary tumors. Eur J Hum Genet. 2015;23:202–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Comino-Méndez I, Leandro-García LJ, Montoya G, et al. Functional and in silico assessment of MAX variants of unknown significance. J Mol Med. 2015;93:1247–1255. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.