

Abstract
Transection of the sural and common peroneal branches of the sciatic nerve produces cutaneous hypersensitivity at the tibial innervation territory of the mouse hindpaw that resolves within a few weeks. We report that interruption of endogenous neuropeptide Y (NPY) signaling during remission, with either conditional NPY knockdown in NPYtet/tet mice or intrathecal administration the Y1 receptor antagonist BIBO3304, reinstated hypersensitivity. These data indicate that nerve injury establishes a long-lasting latent sensitization of spinal nociceptive neurons that is masked by spinal NPY-Y1 neurotransmission. To determine whether this mechanism extends beyond the sensory component of nociception, we used conditioned place aversion and preference assays to evaluate the affective component of pain. We found that BIBO3304 produced place aversion in mice when administered during remission. Furthermore, the analgesic drug gabapentin produced place preference after NPY knockdown in NPYtet/tet but not control mice. We then used pharmacological agents and deletion mutant mice to investigate the cellular mechanisms of neuropathic latent sensitization. BIBO3304-induced reinstatement of mechanical hypersensitivity and conditioned place aversion could be prevented with intrathecal administration of an N-methyl-D-aspartate receptor antagonist (MK-801) and was absent in adenylyl cyclase type 1 (AC1) deletion mutant mice. BIBO3304-induced reinstatement could also be prevented with intrathecal administration an AC1 inhibitor (NB001) or a TRPV1 channel blocker (AMG9801) but not vehicle controls. Intrathecal administration of TRPA1 channel blocker (HC030031) prevented the reinstatement of neuropathic hypersensitivity produced either by BIBO3304 or by NPY knockdown in NPYtet/tet but not control mice. Our results confirm new mediators of latent sensitization: TRPA1 and TRPV1. We conclude that NPY acts at spinal Y1 to tonically inhibit a molecular NMDAR→AC1 intracellular signaling pathway in the dorsal horn that is induced by peripheral nerve injury and drives both the sensory and affective components of chronic neuropathic pain.
INTRODUCTION
Peripheral nerve injury sensitizes dorsal horn neurons in the spinal cord, leading to an increase in the intensity and duration of neuropathic pain (Ji et al., 2003; Latremoliere and Woolf, 2009). This central sensitization can facilitate the normally protective aspects of acute pain, but failure to resolve can facilitate the transition into a more chronic pain state. In cases of latent sensitization, this is kept in remission by opposing mechanisms of endogenous inhibition that typically include activation of inhibitory G-protein coupled receptors. For example, the neuropeptide tyrosine (NPY) Y1 receptor (Y1) is a G-protein coupled receptor that couples to inhibitory Gαi/o, leading to the inactivation of adenylyl cyclase and subsequent down regulation of cAMP (Grouzmann et al., 2001; Silva et al., 2002). Inflammation enhances NPY-mediated inhibition of hyperalgesia (Taylor et al., 2014), likely by dampening the activity of excitatory spinal interneurons (Nelson et al., 2019) and central terminals of primary afferents (Brumovsky et al., 2007) that express Y1. In the setting of latent sensitization, disinhibition of these Y1-expressing neurons or terminals causes a dramatic reinstatement of mechanical hypersensitivity (Solway et al., 2011).
Whether the latent sensitization that is masked by NPY extends beyond the sensory component of nociception is unclear. This is an important question because clinical pain patients present not only with stimulus-evoked sensory components of pain, but also spontaneous and affective components of pain (Burma et al., 2017; King and Porreca, 2014). Therefore, the current studies used conditioned place aversion (CPA) assays to evaluate the affective component of pain for the first time in the setting of neuropathic latent sensitization.
We reported that an N-methyl-d-aspartate receptor (NMDAR)-dependent Ca2+/calmodulin-dependent adenylyl cyclase type 1 (AC1) mechanism contributes to the inflammation-induced latent sensitization that is tonically inhibited either by Y1 activity likely mediated by NPY release (Fu et al., 2019; Solway et al., 2011) or by mu opioid receptor constitutive activity (MORCA) (Corder et al., 2013). We also reported that TRPA1 and TRPV1 contribute to the inflammation-induced latent sensitization that is inhibited by Y1 (Fu et al., 2019). However, the cellular and molecular mechanisms of nerve injury-induced latent sensitization remain unexplored. We used an approach involving pharmacological inhibitors and/or deletion mutant mice to evaluate the contribution of NMDAR, AC1, TRPA1 and TRPV1 to the neuropathic latent sensitization that is silenced by endogenous NPY-Y1 activity. A three-step behavioral pharmacological approach included: 1) induction of neuropathic pain; 2) allow pain resolution; 3) intrathecal injection of drug and test for pain reinstatement.
MATERIALS and METHODS
Animals
Male mice were housed 2–4 per cage in a light- (14-h light/dark cycle), temperature- (68–72° F) and humidity-controlled room with food and water provided ad libitum. Animals were allowed a minimum of one week to habituate to the facility prior to their entrance into the study. All procedures were approved by the Institutional Animal Care and Use Committee of the University of Kentucky, followed the guidelines for the treatment of animals of the International Association for the Study of Pain, and conducted in full compliance with the Association for Assessment and Accreditation of Laboratory Animal Care. Commercial mice. Male C57BL/6 mice weighing 18–20g were purchased from Charles Rivers (Indianapolis). AC1 knockout mice. As previously described (Corder et al., 2013), breeding pairs of AC1−/− mice were kindly provided by Dr. Daniel Storm (Washington University, Seattle, WA, USA), and bred at our facility using a congenic strategy onto a C57BL/6 background. Male homozygous and their WT littermates were introduced into the study at the age of 8–12 weeks. Genotype was confirmed at weaning and repeated at the end of each study by tail-snip PCR. Npytet/tet mice. As described previously (Ste Marie et al., 2005), the native NPY promotor was replaced with an artificial, tonically-active promotor containing a tetracycline/doxycycline (Dox) regulatory site. Heterozygous breeding pairs of NPY null (Erickson et al., 1996), and Npytet/tet mice (Ste Marie et al., 2005) were initially provided by Dr. Richard Palmiter (University of Washington, Seattle, WA) on a mixed 129/B6 background. We used speed congenics to generate Npytet/tet on a C57BL/6 background. The mouse line was maintained in our facility by crossing homozygous male Npytet/tet with hemizygous females, since homozygous females do not breed well. Number of progeny per litter was small (2–4 pups) but reliable. Male homozygous and their WT littermates were used in this study. Genotype was confirmed at weaning and repeated at the end of each study by tail-snip PCR.
Spared Nerve Injury Models (SNI and CpxSx) of Neuropathic Pain
Peripheral nerve injuries were performed as previously described (Decosterd and Woolf, 2000). Briefly, anesthesia was induced with 5% isoflurane and maintained with 2–3% isoflurane throughout surgery. After the fur of the left hind limb was shaved and disinfected with topical application of alcohol and then Betadine®, a skin incision was made at the level of the trifurcation of the sciatic nerve. The overlying biceps femoris muscles were retracted to expose the common peroneal, tibial, and sural nerves of the left hindpaw. Two of the three branches were ligated with 6.0 silk suture (Ethicon, Somerville, NJ), followed by transection at either end of the knot and removal of 1 mm of each nerve. In the SNI model, the common peroneal and tibial nerves were transected, with care taken to avoid perturbation of the sural branch. In the CpxSx model, the common peroneal and sural nerves were ligated and transected, sparing the tibial nerve (Shields et al., 2003). The muscle was sutured with absorbable 6–0 sutures (Ethicon) and the wound was closed with 9mm metal clips. Sham CpxSx mice were anesthetized for 10–15 mins (the typical duration of surgery) followed by skin incision and then closure with 9mm metal clips. Mice were monitored for the following 3 days, and the metal clips were removed 10 days after surgery. Sensory testing was conducted at 14–15 days post-surgery to confirm mechanical hyperalgesia.
Doxycycline Administration to NPYtet/tet Mice
7 days prior to SNI surgery, or 14 days prior to CpxSx surgery, Npytet/tet mice were provided drinking water with saccharin (5 mM; Sigma-Aldrich, St Louis, MO) or saccharin + Doxycycline hyclate (Dox, 2 mg/mL, prepared fresh every three days, Sigma-Aldrich, St Louis, MO). Although saccharin was added to enhance palatability of Dox, the change in flavor of the water led to dehydration (defined by 20 % body weight loss) within 3 days in a small number of mice, which were then removed from the study. Otherwise, delivery of Dox or saccharin in the drinking water did not change the development or intensity of behavioral signs of mechanical or cold allodynia following nerve injury, as described previously (Solway et al., 2011).
Sensory Testing
Animals were acclimated to a stainless steel grid within individual Plexiglas tubes for at least 60 min prior to behavioral testing, with the exception of all conditioned place preference and aversion tests.
Mechanical Threshold.
To evaluate sensitivity to a non-noxious mechanical stimulus, we used an incremental series of 8 von Frey filaments (Stoelting, Inc, Wood Dale, IL) of logarithmic stiffness (0.008–6 grams). The 50% withdrawal threshold was determined using the up-down method (Chaplan et al., 1994). Each filament was applied perpendicular to the plantar surface of the hindpaw skin with sufficient force to cause a slight bending of the filament. The central or lateral aspect of the paw was stimulated in the CpxSx or SNI model, respectively. A positive response was defined as a rapid withdrawal of the paw within 5 seconds, as silently counted by the experimenter.
Response to Cool Stimulation.
Using a syringe connected to PE-90 tubing, flared at the tip to a diameter of 2 mm, a drop of acetone was carefully applied to the plantar paw. The duration of paw withdrawal was recorded with a 45 sec cutoff. Three observations, collected 5 min apart, were averaged.
Conditioned Place Preference and Avoidance
As illustrated in Figure 1, conditioned place preference (CPP) and conditioned place avoidance (CPA) assays were conducted with a three-chambered apparatus fitted with a 4×16 photobeam array located 0.5cm above the flooring to quantify the time spent in each chamber using software (Place Preference, San Diego Instruments, San Diego, CA; http://www.sandiegoinstruments.com/place-preference/). The center chamber walls were neutral, while the left or right chamber walls were decorated with ¾” black and white stripes that ran horizontal or vertical, respectively. The center floor was composed of metal rods spaced 0.5cm apart while the side chambers had either smooth or textured flooring; chamber-floor pairing was counterbalanced between animals. To reduce the amount of time spent in the center chamber: 1) the inner guillotine doors were an aversive bright white; 2) an acrylic divider (3.5cm×10.5cm×33cm, 0.5cm thick; custom design, Regal Plastics https://www.regal-plastics.com) was added to reduce center chamber size; and 3) lighting of the side chambers was 20% of that of the center chamber.
Figure 1. Experimental timeline and diagram of the apparatus for conditioned place preference (CPA).
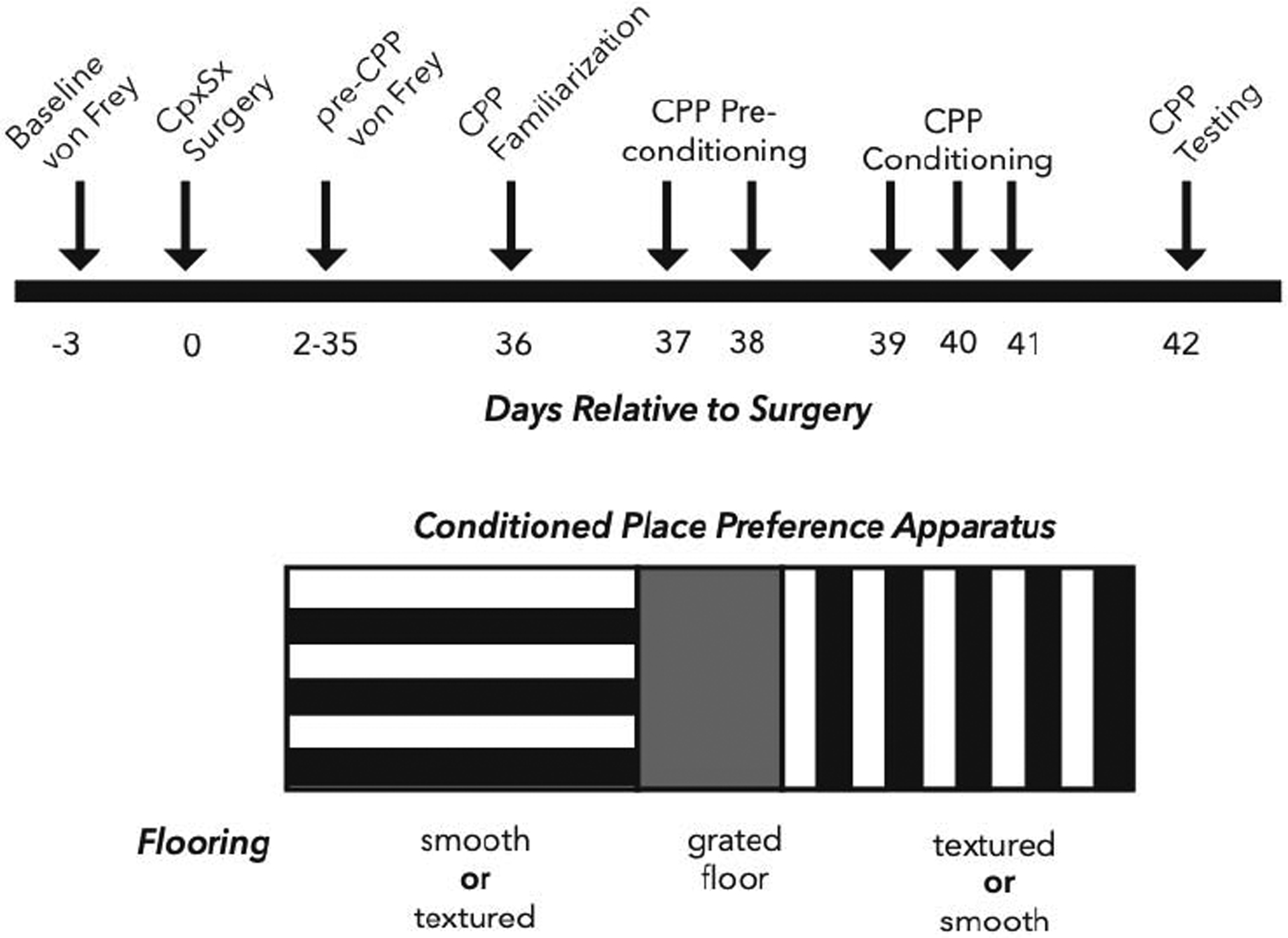
Top: Baseline von Frey thresholds were assessed 3 days prior to surgery and then at 5–7 day intervals starting on day 2 and continuing until the resolution of mechanical hypersensitivity at day 35 (Pre-CPP). The CPP assay began on day 36 with acclimation to the chambers (Familiarization), followed by two days of preconditioning, three days of conditioning, and then testing on day 42. Bottom: The CPP apparatus consisted of 3 chambers: (left) a chamber with horizontal wall paper, smooth or textured flooring (depending on counterbalancing assignment), and 20% light intensity; (center) a chamber with grey wall paper, grated bar flooring, and 100% light intensity; (right) a chamber with vertical wall paper, smooth or textured flooring (depending on counterbalancing assignment), and 20% light intensity.
Assays were conducted 35–42 days post-CpxSx surgery, after sensory thresholds had returned to baseline (Solway et al. 2011). Similar to previous studies (Corder et al., 2013), the CPA/CPP paradigms were conducted in three phases: preconditioning, conditioning, and testing. The preconditioning phase lasted three days. On preconditioning Day 1, mice were placed in the center chamber and allowed free access to all chambers for 30 min. On preconditioning Day 2 and Day 3, mice were allowed free access to all chambers for 15min. Preconditioning scores were calculated as the mean time spent in each chamber, averaged across Days 2–3. Predetermined criteria for exclusion due to chamber bias were applied as follows: mice spending less than 20% or greater than 80% of time in any one chamber were eliminated from the study. The conditioning phase began after the final preconditioning day, and lasted either one day (intrathecal route of administration, BIBO3304 and MK801, CPA paradigm) or three days (intraperitoneal route of administration, gabapentin, CPP paradigm). In the morning (between 9–10am), mice were administered vehicle and returned to their home cage. After 10 min, mice were placed in one of the side chambers (with the guillotine door closed) for 30 min with no activity recorded. 4 hours (between 1–2pm) after vehicle administration, mice were administered with the drug of interest and returned to their home cage. After 10 min, mice were placed in the opposite side chamber (with door closed) for 30 min. The post-conditioning testing phase) began the morning (between 9–10am) after the conditioning phase was completed. Mice were placed in the center chamber with open access to all chambers for 15 min with activity recorded. Differences scores were calculated by subtracting preconditioning scores from postconditioning (testing day) scores. Negative difference scores indicated place aversion, whereas positive difference scores indicated place preference.
Intrathecal (i.t.) Drug Administration
Intrathecal injection was performed in lightly restrained, unanesthetized mice as previously described (Fairbanks, 2003). Briefly, a 30 G needle attached to a Hamilton microsyringe was inserted between L5/L6 vertebrae, puncturing the dura (confirmed by presence of reflexive paw or tail flick). A 5 μl volume of vehicle or drug was then administered. The data of Figure 4A–B and 6A–E include animals that were injected twice using a cross-over design with a 7-day separation between two injections. Animals received vehicle for the first injection and then drug for the second injection, or vice versa. In all cases, group means of vehicle and drug did not differ on either injection day, and so were combined for final analysis. The data of Figure 3A–B were obtained on Day 28 after two i.t. injections, each separated by 15 min. For Figures 3C–D and 4C–D, animals were injected twice during the CPP conditioning phase: vehicle in the morning (approximately 9am) and then drug 4hrs later (approximately 1pm).
Figure 4. Nerve injury-induced latent pain sensitization requires spinal AC1.
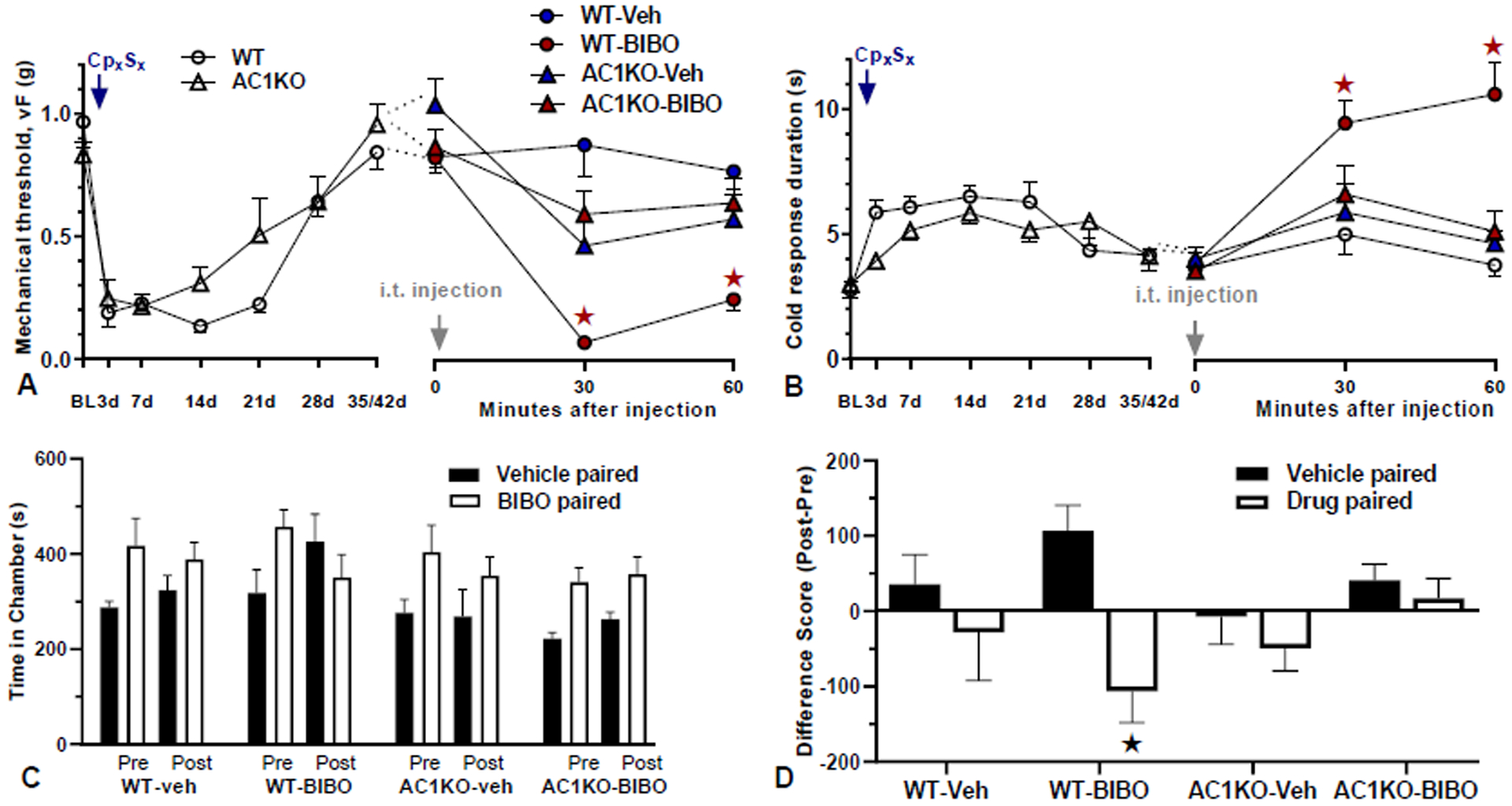
A–B) Time course of mechanical (A) and cold (B) thresholds in AC1KO mice and their WT littermates at pre-surgery baseline, d3, d28 after CpxSx surgery, and 30 and 60 min after intrathecal (i.t.) injection. A–B, Left) Progression and resolution of CpxSx-induced mechanical and cold hypersensitivity. n=11 (AC1KO); n=12 (WT). ★P < 0.05. A–B, Right) During pain remission 28–35 days after CpxSx surgery, BIBO3304 (BIBO, 5 μg/5 μl) or vehicle was injected. All animals were injected twice using a cross-over design with 7-day separation between the two i.t. injections. Post-injection n=12 (WT-Veh); n=12 (WT-BIBO); n=13 (AC1KO-Veh); n=13 (AC1KO-BIBO). ★P < 0.05 (AC1KO-BIBO vs WT-BIBO). C) time spent in side chambers during the preconditioning and postconditioning test phases of a CPA assay conducted 40–43 days after CpxSx surgery. D) difference score of postconditioning minus preconditioning values. BIBO3304 produces place aversion in WT but not AC1KO mice. n=4–6 per group, ★P < 0.05 (WT-BIBO: vehicle vs drug chamber). Values represent mean ± SEM.
Figure 6. Nerve injury-induced latent pain sensitization requires spinal TRPA1.
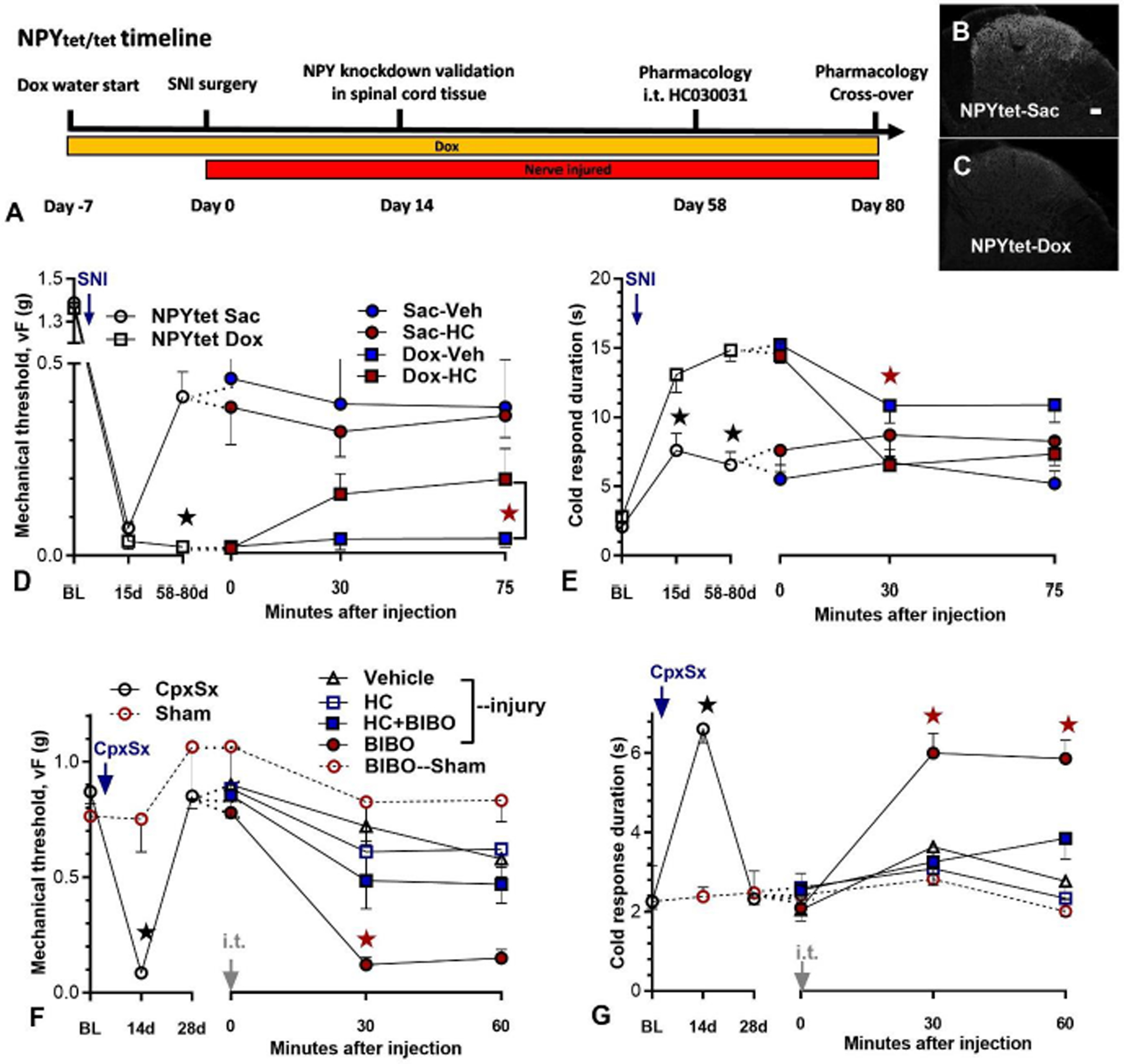
A–E) NPYtet/tet mice were provided with doxycycline (Dox) and saccharin (Sac) or just saccharin in their drinking water beginning 7 days prior to spared nerve injury (SNI) surgery. A) Timeline of an NPY knockdown validation study and a behavioral pharmacology study in NPYtet/tet mice. B–C) NPY immunoreactivity in lumbar dorsal horn on the side ipsilateral to SNI at Day 14 after surgery. Scale bar: 50 μm. D–E) Time course of mechanical and cold thresholds at pre-surgery baseline, after SNI surgery, and 30 and 75 min after intrathecal (i.t.) injection. D–E Left) Progression of SNI-induced mechanical and cold (E) hypersensitivity in NPYtet/tet mice fed with doxycycline (n=7) or without doxycycline (n=6). Doxycycline reduced mechanical and cold hypersensitivity (★P < 0.05). D, E Right) Effect of HC030031 (10 μg) when intrathecally administered several weeks after SNI surgery in mice treated with or without doxycycline. HC030031 attenuated mechanical (D) and cold (E) hypersensitivity in NPY knockdown mice, but not in control mice. All animals were injected twice using a cross-over design with 7-day separation between two i.t. injections. Post-injection n=6 (Sac-Veh); n=6 (Sac-HC); n=7 (Dox-Veh); n=7 (Dox-HC). ★P < 0.05 (NPYtet/tet Dox HC vs NPYtet/tet Dox veh). F–G). Time course of mechanical and cold thresholds at pre-surgery baseline, after CpxSx or Sham surgery, and 30 and 60 min after i.t. injection. After the induction (14 days) and resolution (28 days) of CpxSx-induced hyperalgesia [n=32 (CpxSx); n=6 (sham)], co-administration of HC030031 (HC, 10 μg) attenuated BIBO3304-evoked reinstatement of mechanical and cold hypersensitivity. Post-injection n=10 (CpxSx-Veh); n=6 (CpxSx-HC); n=9 (CpxSx-BIBO); n=7 (CpxSx-HC+BIBO); n=6 (sham-BIBO). ★P < 0.05 (HC+BIBO vs BIBO). Values represent mean ± SEM.
Figure 3. Nerve injury-induced latent pain sensitization requires spinal NMDAR.
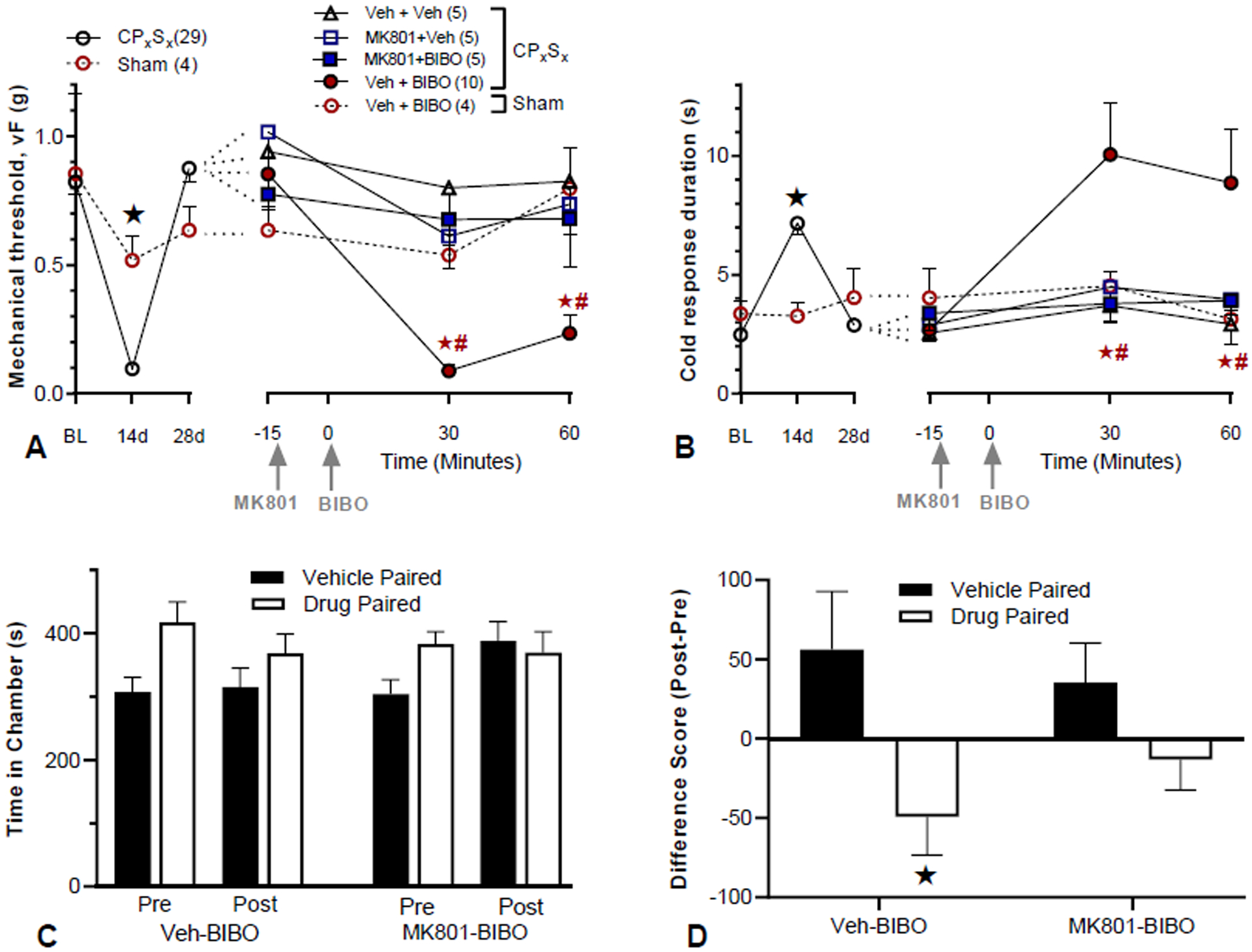
A–B) Time course of mechanical (A) and cold (B) thresholds at pre-surgery baseline, d3, d28 after CpxSx surgery, and 30 and 60 min after the 2nd i.t. injection. A–B, Left) Progression and resolution of CpxSx-induced mechanical and cold hypersensitivity. Pre-injection: n=5 (sham); n=29 (CpxSx). ★P < 0.05 (CpxSx vs sham). A–B, Right) 28 days after CpxSx surgery, MK801 (MK, 1 μg/5 μl) or vehicle (Veh) was followed 15 min later with injection of BIBO3304 (BIBO, 5 μg/5 μl) or vehicle. Post-injection n=5 (CpxSx, Veh+Veh); n=5 (CpxSx, MK801+Veh); n=10 (CpxSx, Veh+BIBO); n=8 (CpxSx, MK801+Veh); n=5 (sham, Veh+Veh). MK-801 prevented BIBO3304-evoked reinstatement of mechanical and cold hypersensitivity. ★P < 0.05 (MK801 + BIBO vs Veh + BIBO), #P < 0.05 (Veh + BIBO vs Veh + Veh). C) time spent in side chambers during the preconditioning and postconditioning test phases of a CPA assay conducted 28 days after CpxSx surgery. D) difference score of postconditioning minus preconditioning values. MK801 prevented BIBO3304-evoked affective pain [difference scores as postconditioning minus preconditioning values; n=10 per group, ★P < 0.05 (BIBO+Veh: vehicle vs drug chamber)]. vs drug chamber. CPA and CPP: #P < 0.05, Pre vs Post; ★P < 0.05 vehicle vs drug chamber. Some data in A and B are the same as shown in Fig 2A and 2B, respectively. Values represent mean ± SEM.
Drugs
The following drugs and doses were used for intrathecal injections: BIBO 3304 trifluoroacetate (BIBO 5 μg/5 μl, Tocris Biosciences, United Kingdom); Gabapentin 100mg/10ml/kg (Sigma-Aldrich, St Louis, MO); (+)-MK-801 hydrogen maleate 1 μg/5 μl (Sigma-Aldrich, St Louis, MO), (this dose of MK801 was devoid of overt motor effects); NB001 1.5 μg/5 μl (Sigma-Aldrich, St Louis, MO); HC 030031, 10 μg/5 μl (Tocris Biosciences, United Kingdom); AMG 9810 10 nmol/5 μl (Tocris Biosciences, United Kingdom). Vehicle used for Figure 6 was ethanol: alkamuls EL-620 (Rhodia, Cranbury, NJ): saline in a volume ratio of 2:2:6; vehicle used for other experiments was ethanol: alkamuls EL-620: saline in a volume ratio of 1:1:8.
Blinding procedure
The experimenter was blinded to drug treatments in all behavioral pharmacology experiments by a laboratory colleague. Briefly, all drugs and vehicle were made in identical tubes. A lab mate color-coded the tubes. The key for coding was kept hidden in a notebook until the completion of the experiment, at which point the key was obtained for data analysis.
Immunohistochemistry
NPYtet/tet mice were deeply anesthetized with isoflurane and transcardially perfused with 100ml ice-cold 0.1 M PBS containing heparin (10,000 USP units/L) followed by 100 ml 10% phosphate buffered formalin (Fisher Scientific, Pittsburgh, PA) 14 days after SNI. Lumbar spinal cords were dissected and post-fixed in 10% formalin for 4h, cryoprotected in 30% sucrose overnight, and sectioned transversely at 30 μm (L4-L6) on a freezing microtome. The sections were washed 3 times in 0.1 M PBS and then pretreated with blocking solution [3% normal goat serum (Gemini Bio Products, Broderick, CA) and 0.3% Triton X-100 (Sigma-Aldrich, St Louis, MO) in 0.1 M PBS] for 1 hour. Sections were incubated overnight at room temperature on a slow rocker in antibody dilution solution (1% normal goat serum and 0.3% Triton X-100 in 0.1 M PBS) containing the primary antibody rabbit anti-NPY (1:1,000, Peninsula Laborotories). Sections were then washed, incubated in antibody dilution solution containing the secondary antibody (1:1000, Alexa Fluor 568-conjugated goat anti-rabbit, Invitrogen A11036, RRID:AB_143011) for 1.5h at room temperature, washed again, mounted onto Superfrost Plus slides, air dried for 45 min, and mounted using Prolong Gold with DAPI mounting medium (Molecular Probes, Eugene, OR).
Statistics
Differences between means were analyzed by two-way analysis of variance (ANOVA) with Drug, Dose, and/or Genotype as grouping factors and Time as a repeated measure. If a significant main effect was found (p < 0.05), the ANOVA was followed by Bonferroni’s (for 2-group comparison) or Turkey’s post-hoc (for multi-group comparison) tests. Conditioned place paradigm difference scores were analyzed using a two-tailed, paired t-test with α=0.05.
RESULTS
Affective neuropathic pain is tonically opposed by spinal NPY-Y1 signaling.
We could not use the convention model of spared nerve injury (SNI) because resolution does not occur (Decosterd and Woolf, 2000). So instead, we used a variant of the SNI model that entails the transection of the common peroneal and sural branches of the sciatic nerve (the CpxSx model). With this model, we previously reported that intrathecal administration of the Y1 antagonist BIBO3304 reinstated mechanical hypersensitivity when administered 21 days after surgery (Solway et al., 2011). As summarized by the pre-drug values illustrated by the left side of the x-axes of Figures 2A–B, CpxSx produced mechanical hyperalgesia and cold allodynia at Day 14 that resolved by Day 28 (see Fig 3 for results of the overall ANOVA). Consistent with our previous findings (Solway et al., 2011), intrathecal BIBO3304 changed neither mechanical (Fig 2A) nor cold (Fig 2B) thresholds in Sham mice (p > 0.05), but reinstated mechanical hypersensitivity in CpxSx mice (p<0.05) (see Fig 3 for results of the overall ANOVA). Thus, this study repeats previous results and provides further evidence for our working hypothesis that tonic Y1 activity inhibits behavioral signs of mechanical and cold hypersenstivity. Clinical pain patients, however, present not only with stimulus-evoked sensory components of pain, but also spontaneous and emotional components of pain (Burma et al., 2017; King and Porreca, 2014; Melzack and Casey, 2014). To evaluate this affective component in the CpxSx model of neuropathic latent sensitization, we determined whether BIBO3304 would produce conditional place aversion (CPA) (see Figure 1). As illustrated in Fig 2C, sham mice did not show aversion to intrathecal administration of vehicle or BIBO3304 (p>0.05). However, CpxSx mice avoided (spent less time in) the chamber paired with intrathecal administration of BIBO3304 during the postconditioning phase (p=0.009 as compared to the preconditioning phase). This is best illustrated in Fig2D showing that BIBO3304 produced CPA in CpxSx mice (p<0.05) but not sham mice (p>0.05).
Figure 2. Sensory and affective components of neuropathic pain are tonically opposed by spinal NPY-Y1 signaling.
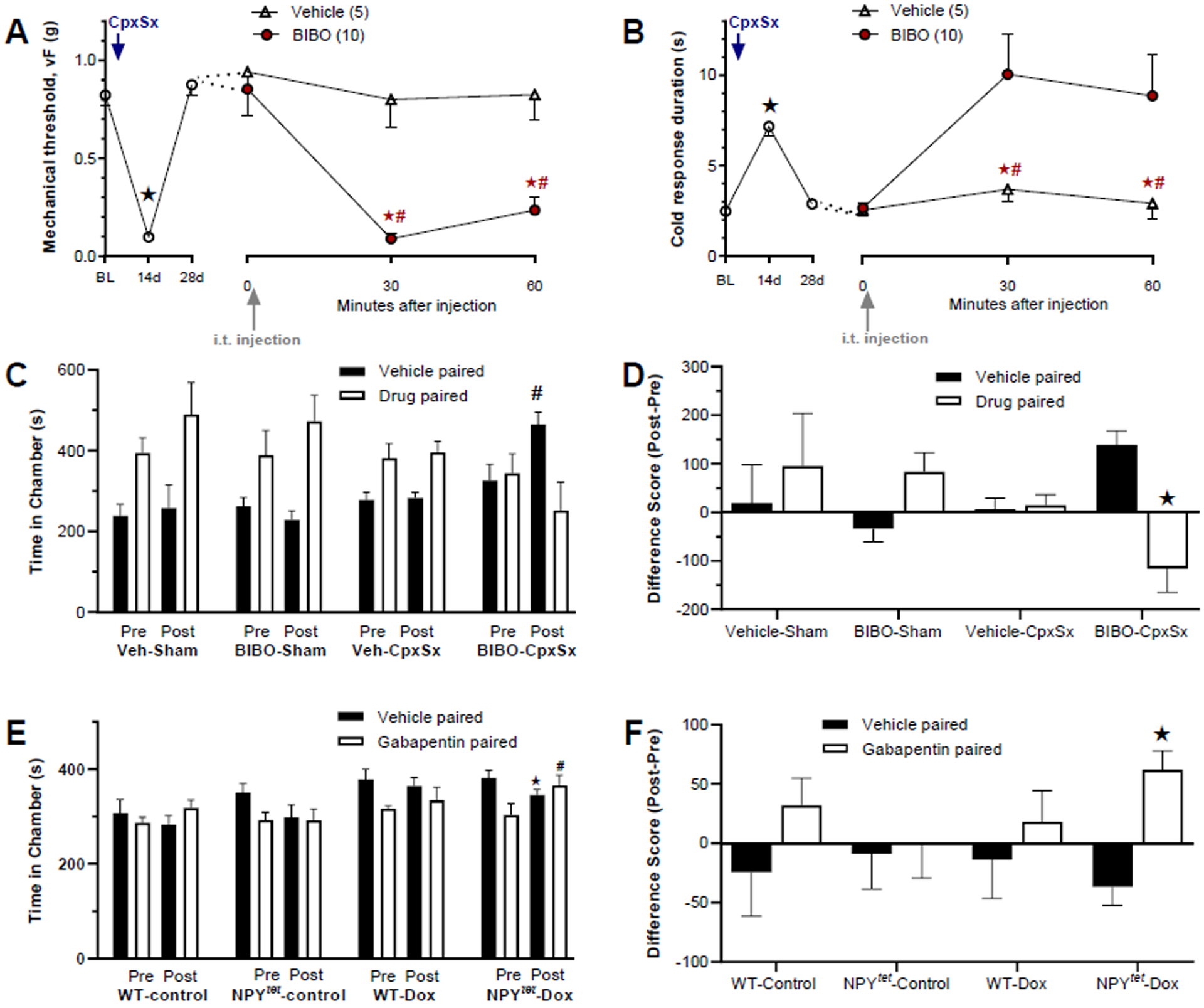
Transection of the common peroneal and sural nerves (CpxSx) was followed 28 days later with behavioral measurements. A–B) Measurement of von Frey threshold and hindpaw response duration to plantar application of a drop of acetone, n=5–10 per group. C–D) Time spent in side chambers during the preconditioning and postconditioning test phases of a conditioned place aversion (CPA) assay to the Y1 antagonist BIBO3304 (BIBO), n=5 per group. E–F) Time spent in side chambers during the preconditioning and postconditioning test phases of a conditioned place preference (CPP) assay to the antihyperalgesic drug gabapentin, n=5–7 per group. NPY-Y1 signaling was interrupted with intrathecal (i.t.) injection of 5 ug / 5 ul BIBO3304 (A–D) or with doxycycline included in the drinking water of NPYtet/tet mice (E–F). In Panels C and E, “Pre” denotes preconditioning phase values, while “Post” denotes post-conditioning test phase values. Panels D and F illustrate the difference score of postconditioning minus preconditioning values. vF and cold: #P < 0.05, BIBO vs vehicle group; ★P < 0.05, vehicle vs drug chamber. CPA and CPP: #P < 0.05, Pre vs Post; ★P < 0.05 vehicle vs drug chamber. The data in panels A and B are shown again in Fig 3A and 3B, respectively. Values represent mean ± SEM.
Although intrathecal BIBO3304 was directed to Y1 at the spinal cord, pharmacological approaches do not rule out off-target sites at receptor systems other than those driven by NPY. To control for this, we interrupted spinal NPY neuromodulation with a genetic approach using NPY tet/tet mice (Ste Marie et al., 2005). We confirmed previous studies showing that exposure to doxycline (with saccarhin for palatability) in the drinking water dramatically decreased nerve injury-induced NPY expression in DRG and dorsal horn (Solway et al., 2011). To evaluate affective neuropathic pain, we performed CpxSx, waited several weeks for hyperalgesia to resolve, and then administered gabapentin. The control experiments of Figs 2E–F illustrate that systemic administration of gabapentin did not change place preference in saccharin-exposed wildtype (p=0.234) or NPYtet/tet mice (p=0.162), nor in doxycycline-exposed wildtype mice (p=0.234 and p=0.162, respectively). By contrast, gabapentin produced conditioned place preference (CPP) in doxycycline-exposed NPYtet/tet mice: they spent significantly more time in the gabapentin-paired chamber during the postconditioning as compared to the preconditioning phase (p=0.001). This is consistent with gabapentin-induced CPP described in the rat SNI model of neuropathic pain (Griggs et al., 2015), and indicates that conditional knock-down of NPY produces an aversive state. In summary, either Y1 antagonism of endogenous NPY at the spinal cord or genetic NPY knockdown reinstates not only the sensory but also the affective component of neuropathic pain, indicating that spinal NPY tonically inhibits affective neuropathic pain.
Nerve injury-induced latent pain sensitization requires spinal NMDAR activation.
NMDAR-Ca2+-AC1-cAMP signaling is required to maintain latent sensitization after inflammation (Corder et al., 2013; Fu et al., 2019). To test the hypothesis that similar mechanisms suppress latent sensitization after peripheral nerve injury, we used the CpxSx model. As summarized by the pre-drug values illustrated by the left side of the x-axes of Figs 3A–B and consistent with Figs 2A–B, CpxSx produced mechanical hyperalgesia (ANOVA Surgery × Time; F2, 60 = 19.6; p = 0.0025) and cold allodynia (ANOVA Surgery × Time; F2, 60 = 10.4; p = 0.0001) at Day 14 that resolved by Day 28. The large cohort of CpxSx mice were then segregated into 4 groups and administered first with either vehicle or the activity-dependent NMDAR blocker MK-801 and then either vehicle or the Y1 antagonist BIBO3304. This pharmacology data is plotted on the right side of the x-axes of Figs 3A–B, and two-way ANOVA revealed main effects of Group × Time for mechanical thresholds (F4, 27 = 4.20; p = 0.009) and cold responses (F4, 27 = 2.79; p = 0.046). Consistent with our previous findings (Solway et al., 2011), intrathecal BIBO3304 changed neither mechanical (Fig 3A) nor cold (Fig 3B) thresholds in Sham mice (p > 0.05), but reinstated mechanical hypersensitivity in CpxSx mice (p<0.05). MK801 but not vehicle prevented BIBO3304-induced reinstatement of mechanical and cold hypersensitivity (p<0.05).
To assess the influence of NMDARs on affective pain, we determined determined whether MK801 would block BIBO3304-induced conditioned place aversion. As illustrated in Figs 3C–D and consistent with the results of Figs 2C–D, intrathecal BIBO3304 produced place aversion in CpxSx mice (p=0.027). This was blocked by intrathecal MK801 (p=0.141).
Nerve injury-induced latent sensitization requires spinal AC1
To test the hypothesis that AC1 is required for the latent sensitization that is masked by Y1, we next used AC1 deletion mutant mice. Fig 4 illustrates that both baseline mechanical thresholds (Fig 34, P > 0.05) and cold withdrawal responses (Fig 4B, P > 0.05) were similar between AC1KO and WT mice. For the first several weeks after CpxSx, AC1KO did not change mechanical (Fig 4A, Gene × Time, F1, 20 = 1.391; p = 0.2520) or cold (Fig 4B, Gene × Time, F1, 20 = 2.148; p = 0.1583) hypersensitivity. After the resolution of mechanical and cold hypersensitivity, we next injected BIBO3304. AC1 deletion prevented the ability of BIBO3304 to reinstate mechanical hyperalgesia (Fig 4A, Gene × Time, F1, 23 = 11.72, p = 0.0023) and cold allodynia (Fig 4B Gene × Time, F1, 26 = 8.31; p = 0.0018).
We used CPA assays to evaluate the effect of spinally-directed BIBO3304 on behavioral signs of affective pain. As illustrated in Figs 4C–D, intrathecal administration of BIBO3304 produced place aversion in wildtype (p=0.004) but not AC1KO mice (p=0.497). There was no effect of vehicle in wildtype (p=0.426) or AC1KO mice (p=0.406), and no difference in difference scores between drug paired chambers in AC1KO mice (AC1KO: afternoon vehicle-paired chamber vs afternoon BIBO3304-paired chamber; p=0.136).
To determine whether the site of action of AC1 is at the spinal cord, we next used the intrathecal route to deliver the selective AC1 inhibitor, NB001 (Wang et al., 2011) to the dorsal horn. NB001 abolished the ability of BIBO3304 to reinstate mechanical hyperalgesia (Fig 5A, Drug × Time, F1, 12 = 22.81, p = 0.0005) and cold allodynia (Fig 5B, Drug × Time F1, 26 = 18.75; p = 0.001).
Figure 5. Nerve injury-induced latent sensitization requires spinal AC1.
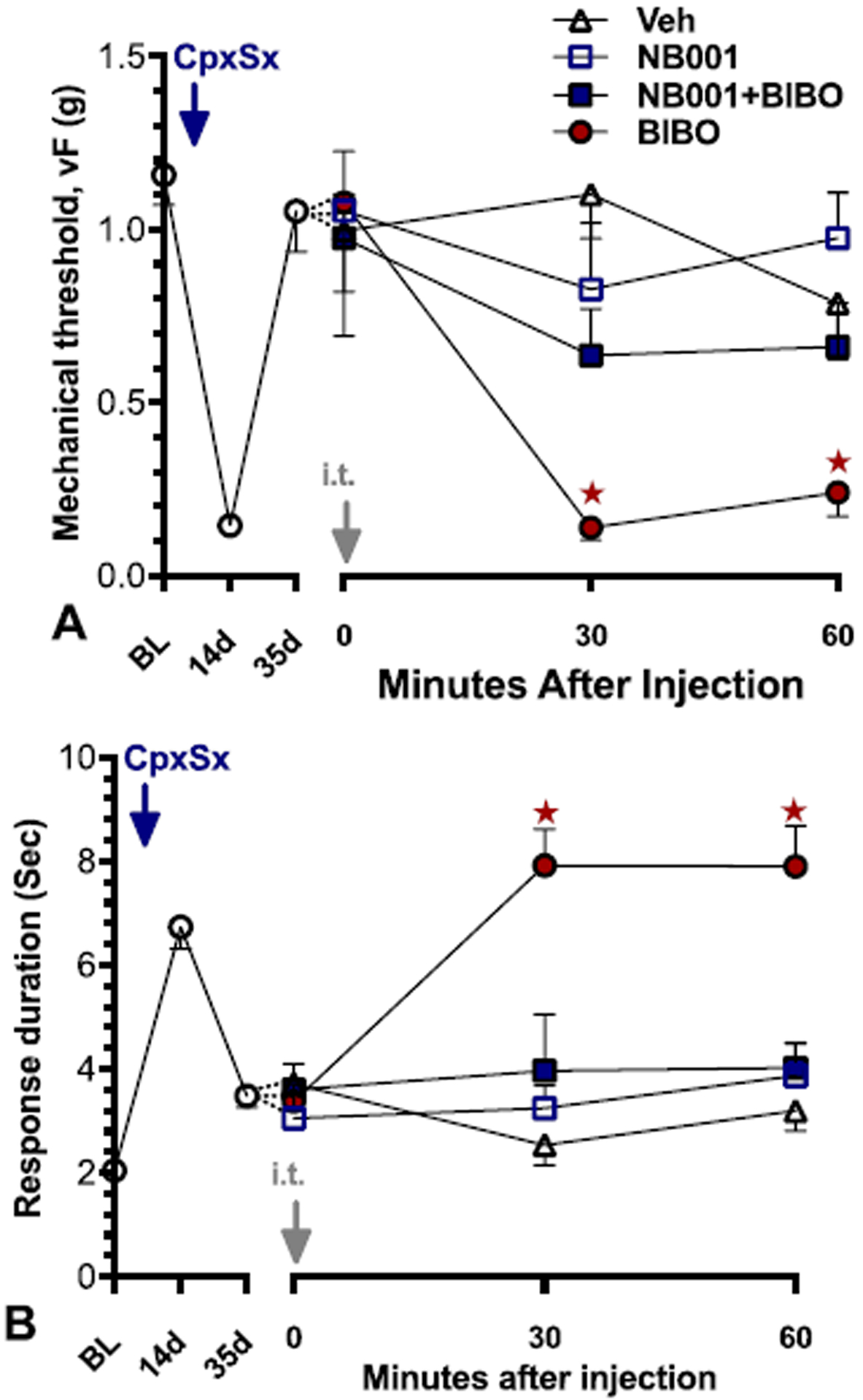
Time course of mechanical (A) and cold (B) thresholds at pre-surgery baseline, after CpxSx surgery, and 30 and 60 min after intrathecal (i.t.) injection. After the induction (14 days) and resolution (35 days) of CpxSx-induced hyperalgesia (n=27), co-administration of NB001 (1.5 μg) prevented BIBO3304-evoked reinstatement of mechanical and cold hypersensitivity. Post-injection n=7 (Veh); n=6 (NB001); n=7 (BIBO); n=7 (NB001+BIBO). ★P < 0.05 (NB001+BIBO vs BIBO). Values represented as mean ± SEM.
Nerve injury-induced latent sensitization requires spinal TRPA1 and TRPV1
The non-selective cation channels TRPA1 and TRPV1 are expressed on the central terminals of C and Aδ nociceptors and detect noxious somatosensory stimuli (Julius, 2013). Our previous findings demonstrate that a NMDAR-AC1-PKA-TRPA1/V1 signaling pathway drives inflammatory-induced latent sensitization (Fu et al., 2019), and this is under the inhibitory control of the endogenous NPY-Y1 activity. To test the hypothesis that TRPA1 contributes to the latent sensitization generated by nerve injury as well, we evaluated the contribution of AC1 in a model of tonic neuropathic pain driven by conditional NPY knockdown using NPYtet/tet mice with SNI surgery (Solway et al, 2011). In contrast to the C57Bl/6 mouse strain that typically exhibits an essentially permanent mechanical hypersensitivity following SNI, this resolves after several weeks in control NPYtet/tet mice (Solway et al, 2011). The timeline of this study is illustrated in Fig 6A. As illustrated in Figs 6B–C, continuous exposure to Dox in the drinking water essentially silenced artificial NPY promotor-driven expression in dorsal horn, consistent with our previous observations (Solway et al., 2011). As illustrated by the open symbols within left sides of the x-axes in Figs 6D–E, NPY knockdown increased the duration of both SNI-induced mechanical (Gene × Time, F1, 11 = 18.68; p = 0.0012) and cold (Gene × Time, F1, 11 = 44.15; p < 0.0001) hypersensitivity. Next, as illustrated by the closed symbols within right sides of the x-axes in Figs 6D–E, we intrathecally delivered the TRPA1 inhibitor HC030031 after mechanical and cold hypersensitivity had decreased (58/80 days). We found that HC030031 but not vehicle attenuated mechanical (Drug × Time, F1, 12 = 4.872; p = 0.0475) and reversed cold (Drug × Time, F1, 12 = 9.872; p = 0.0085) hypersensitivity in NPY knockdown mice. HC030031 did not change mechanical or cold sensitivity in control NPYtet/tet mice that were given drinking water without doxycycline.
The NPYtet/tet study supports the idea that endogenous NPY-Y1 signaling tonically inhibits a TRPA1-mediated latent sensitization. To extend this, we intrathecally injected a TRPA1 channel inhibitor, HC030031. We chose an HC030031 dose that was comparable to the 1–10 nmol doses used to reduce referred allodynia in a model of visceral pain and mechanical hyperalgesia in a latent sensitization model of chronic inflammatory pain (Fu et al., 2019; Pitcher et al., 2007). We avoided doses higher than 10 nmol as they can produce motor deficits (W. Fu, unpublished observations). DAs illustrated in Figs 6F–G, HC030031 attenuated BIBO3304-evoked reinstatement of mechanical hyperalgesia (Fig 6F, Group × Time, F4, 33 = 13.8; p < 0.0001) and cold allodynia (Fig 6G, Group × Time, F4, 34 = 10.21; p < 0.0001).
To test the hypothesis that TRPV1 contributes to the latent sensitization that is masked by endogenous NPY-Y1 activity, we intrathecally injected BIBO3304 together with AMG9810, a TRPV1 antagonist. We chose an AMG9810 dose that was slightly higher previously shown to reduce inflammation-induced hyperalgesia and latent sensitization (da Costa et al., 2010; Fu et al., 2019)As illustrated in Fig 7, we found that, when during pain remission in CpxSx mice, AMG9810 attenuated the BIBO3304-induced reinstatement of mechanical hyperalgesia (Fig 7A, Group × Time, F4, 34 = 9.964; p < 0.0001) but not cold allodynia (Fig 7B, p > 0.05).
Figure 7. TRPV1 antagonism attenuates Y1-antagonist-induced reinstatement of neuropathic pain.
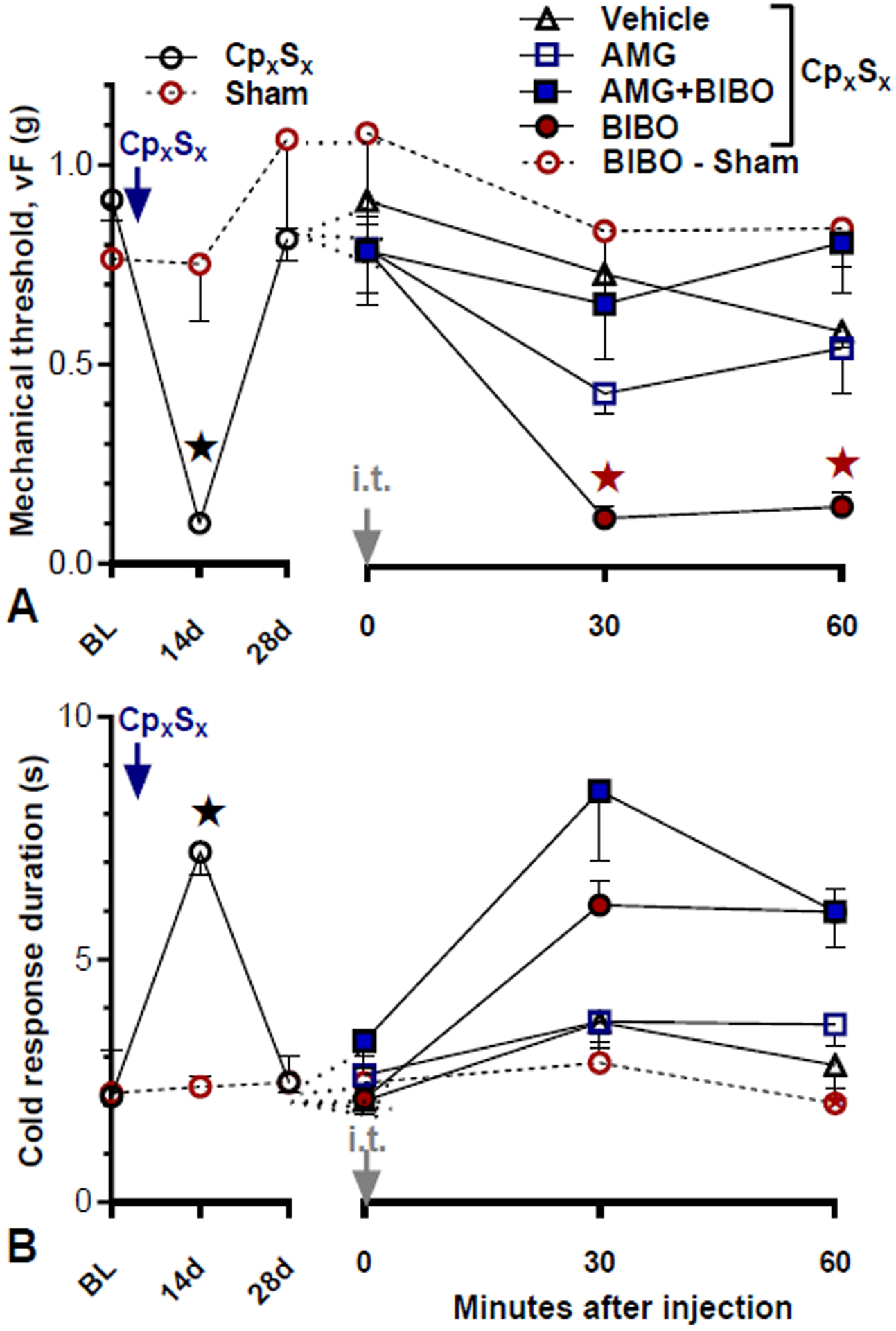
Time course of mechanical (A) and cold (B) thresholds at pre-surgery baseline, after CpxSx surgery, and 30 and 60 min after intrathecal (i.t.) injection. After the induction (14 days) and resolution (28 days) of CpxSx-induced hyperalgesia [n=6 (sham); n=33 (CpxSx)], Co-administration of AMG 9810 (AMG, 10 nmol) attenuated BIBO3304-evoked reinstatement of mechanical, but not cold hypersensitivity. Post-injection n=10 (CpxSx-Veh); n=7 (CpxSx-AMG); n=9 (CpxSx-BIBO); n=7 (CpxSx-AMG+BIBO); n=6 (sham-BIBO). ★P < 0.05 (AMG+BIBO vs BIBO). Values represent mean ± SEM.
DISCUSSION
Affective neuropathic pain is tonically opposed by spinal NPY-Y1 signaling.
We previously reported that NPY tonically inhibits mechanical and cold hypersensitivity associated with tissue or nerve injury (Solway et al., 2011)(Fu et al., 2019). We used von Frey hairs and cool sensations that engage pain circuits and evoke reflexive measures of pain acute nociceptive pain. However, reflexive measures of pain can be observed in decerebrated animals (Woolf, 1984) and are unlikely to capture the more clinically relevant features of neuropathic pain (King and Porreca, 2014). Indeed, pain in humans is a multidimensional experience with not only sensory components, but also cognitive and motivational components (Melzack and Casey, 2014). In particular, clinical chronic pain has a strong emotional component that is fundamentally aversive, or unpleasant. To determine whether the NPY-Y1 system regulates this affective component of chronic pain, we used CPA to evaluate the aversive nature of BIBO3304 or NPY knockdown, and we used CPP to evaluate the negative reinforcement associated with removal of aversion (e.g. upon administration of the analgesic gabapentin) and corresponding reward of pain relief (King and Porreca, 2014). We found that BIBO3304 produced place aversion in mice when administered during remission. Furthermore, produced place preference after NPY knockdown-induced pain in NPYtet/tet but not control mice. We conclude that NPY acts at spinal Y1 to tonically inhibit not only the sensory but also the affective components of chronic neuropathic pain.
Nerve injury-induced latent pain sensitization requires spinal NMDAR→AC1 activation.
The non-selective cation channel NMDAR is universally expressed in the superficial dorsal horn. NMDAR-driven Ca2+ increases are linked to adenylyl cyclase activation and downstream cAMP signaling pathways (Chetkovich and Sweatt, 1993; Wong et al., 1999), leading to the induction and maintenance of central sensitization (Latremoliere and Woolf, 2009). In addition, AC1 contributes to high frequency stimulation-induced LTP (Wang et al., 2011). We used pharmacological agents and deletion mutant mice to investigate the contribution of NMDAR and AC1 to neuropathic latent sensitization. We used the AC1 deletion mutant mouse originally described by Wong et al (Wong et al., 1999) and shown to exhibit deficits in long-term memory and behavioral signs of inflammatory pain (Wei et al., 2002). We found that BIBO3304-induced reinstatement of mechanical hypersensitivity and conditioned place aversion could be prevented with intrathecal administration of an N-methyl-D-aspartate receptor antagonist (MK-801) and was absent in adenylyl cyclase type 1 (AC1) deletion mutant mice. Furthermore, BIBO3304-induced reinstatement could be prevented with intrathecal administration an AC1 inhibitor (NB001). These results in a model of chronic neuropathic pain are consistent with our previous reports that an NMDAR→AC1-dependent latent sensitization in a model of chronic inflammatory pain is kept in remission by MORCA (Corder et al., 2013) or the NPY-Y1 axis (Fu et al., 2019) in the dorsal horn. Based on these results we conclude that 1) Nerve injury induces an NMDAR- and AC1-dependent latent sensitization in the dorsal horn; and 2) this NMDAR→AC1 pathway is kept in remission by endogenous NPY-Y1 signaling.
Nerve injury-induced latent pain sensitization requires spinal TRPA1 and TRPV1.
In DRG neurons, Y1 is co-expressed with TRPV1 (Gibbs et al., 2004), which in turn is largely co-expressed with TRPA1 (Story et al., 2003). Y1 activation down-regulates TPRV1- or TRPA1-induced Ca2+ mobilization (Xu et al., 2010; Xu et al., 2008) or calcitonin-gene related peptide release (Chen et al., 2011; Demartini et al., 2018; Gibbs et al., 2004; Gibbs et al., 2006; Gibbs and Hargreaves, 2008). Studies focused on relatively early timepoints after nerve injury (e.g. 1–2 weeks) indicate that both TRPA1 and TRPV1 contribute to mechanical hypersensitivity (Chen et al., 2011; Demartini et al., 2018). Our studies extend these findings to much longer timepoints using a model of latent neuropathic pain. We used the intrathecal route of administration to target the central terminals (with the caveat that drug might diffuse from the intrathecal space to the dorsal root ganglion). The results demonstrate that intrathecal administration of a TRPA1 channel blocker (HC030031) prevented the reinstatement of neuropathic hypersensitivity produced either by BIBO3304 or by NPY knockdown in NPYtet/tet but not control mice. Similarly, intrathecal administration of a TRPV1 channel blocker (AMG9801) but not vehicle prevented the reinstatement of neuropathic hypersensitivity produced by BIBO3304. In summary, we conclude that both TRPA1 and TRPV1 contribute to the nerve injury-induced latent sensitization that is masked by endogenous Y1 activity. This conclusion would be bolstered with additional studies using conditional TRPA1 and TRPV1 knockout mice.
Tonic inhibition of latent sensitization by endogenous NPY-Y1 activity is ligand-dependent
Initial findings from our laboratory suggest that a ligand-independent mechanism, MORCA, tonically keeps latent sensitization in remission to the long-lasting endogenous anti-hyperalgesia mediated (Corder et al., 2013). This conclusion supported by more recent studies that reported that mu opioid receptor knockout mice display normal latent sensitization when challenged with naltrexone (Walwyn et al., 2016). Because both Y1 and MOR belong to Class I GPCRs that couple to Gαi/o, it is reasonable to propose that injury produces Y1 constitutive activity. We think that this is unlikely, however, for two reasons: 1) Unlike MOR, Y1 does not exhibit ligand-independent activation in vitro, due to its third intracellular loop that can stabilize the inactive state of the receptor (Chee et al., 2008); and 2) the present studies clearly demonstrate that genetic knockdown of ligand (NPY) produces reinstatement of hyperalgesia. This is consistent with previous studies indicating that doxycycline administration to NPY tet/tet mice that experienced inflammation or nerve injury produced mechanical and cold hypersensitivity in a reversible and repeatable manner (Solway et al., 2011). Because models of peripheral nerve injury, including spared nerve injury, are associated with robust increases in NPY levels in large sensory neurons and in the dorsal horn (Intondi et al., 2010; Ossipov et al., 2002; Wakisaka et al., 1992), future studies are needed to test the idea that the tonic inhibition of injury-induced latent sensitization by NPY-Y1 relies on a long-lasting upregulation of spinal NPY release from the central terminals of primary afferents or from intrinsic NPY-expressing spinal interneurons. Such studies might be accomplished with a recently-developed method to indirectly assess NPY release as a function of clathrin-coated pit internalization of Y1 in the dorsal horn (Marvizon et al., 2019).
Summary
Our overall summary is illustrated in Fig 8. Based on current results and the literature, we conclude that peripheral nerve injury establishes a molecular NMDAR→AC1→TRPA1/TRPV1 intracellular signaling pathway in the dorsal horn that drives both the sensory and affective components of chronic neuropathic pain, but this is kept in check by a compensatory pain inhibitory system that includes tonic NPY release and Y1 activation of the pre and/or postsynaptic terminals of Y1-expressing DRG neurons and dorsal horn interneurons, respectively.
Figure 8. Working hypotheses of two opposing mechanisms that concomitantly drive and silence nerve injury-induced latent sensitization.
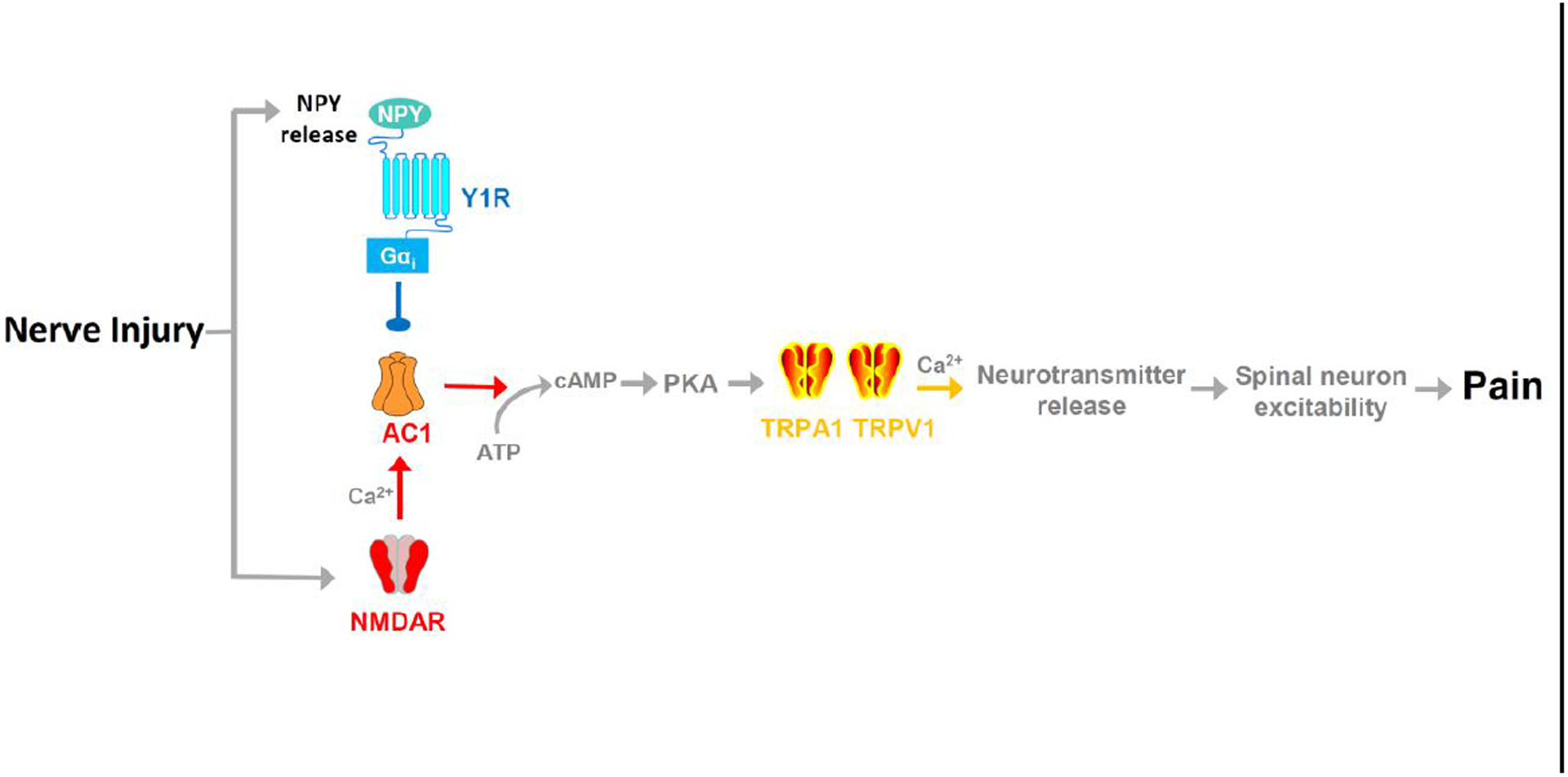
Nerve injury sensitizes a pronociceptive signaling pathway that is tonically silenced by endogenous pain inhibitory NPY-Y1 activity: NMDAR → AC1 → TRPA1/V1. In this model, a Y1 receptor antagonist or NPY knockdown suppresses NPY-Y1, leading to disinhibition of pronociceptive neurotransmitter release, spinal neuron excitability and, ultimately, pain. Line Arrows denote activation/production; Line Dot denotes inhibition.
Highlights.
Mouse models of latent pain sensitization reflect the extended duration of chronic neuropathic pain.
Peripheral nerve injury establishes a latent pain sensitization the requires NMDAR, AC1, TRPA1, and TRPV1.
NPY – Y1 tonically inhibits the cognitive / emotional components of latent neuropathic pain sensitization.
Acknowledgements
Funding: This work was supported by the National Institute of Health (grant numbers R01-NS045954, R01-DA037621, and R01-NS062306 to BKT.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors have no financial or otherwise relationships that might lead to a conflict of interest.
No human subjects
REFERENCES
- Brumovsky P, Shi TS, Landry M, Villar MJ, Hokfelt T, 2007. Neuropeptide tyrosine and pain. Trends Pharmacol Sci 28, 93–102. [DOI] [PubMed] [Google Scholar]
- Burma NE, Leduc-Pessah H, Fan CY, Trang T, 2017. Animal models of chronic pain: Advances and challenges for clinical translation. J Neurosci Res 95, 1242–1256. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL, 1994. Quantitative assessment of tactile allodynia in the rat paw. Journal of neuroscience methods 53, 55–63. [DOI] [PubMed] [Google Scholar]
- Chee MJ, Morl K, Lindner D, Merten N, Zamponi GW, Light PE, Beck-Sickinger AG, Colmers WF, 2008. The third intracellular loop stabilizes the inactive state of the neuropeptide Y1 receptor. J Biol Chem 283, 33337–33346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Yang C, Wang ZJ, 2011. Proteinase-activated receptor 2 sensitizes transient receptor potential vanilloid 1, transient receptor potential vanilloid 4, and transient receptor potential ankyrin 1 in paclitaxel-induced neuropathic pain. Neuroscience 193, 440–451. [DOI] [PubMed] [Google Scholar]
- Chetkovich DM, Sweatt JD, 1993. nMDA receptor activation increases cyclic AMP in area CA1 of the hippocampus via calcium/calmodulin stimulation of adenylyl cyclase. J Neurochem 61, 1933–1942. [DOI] [PubMed] [Google Scholar]
- Corder G, Doolen S, Donahue RR, Winter MK, Jutras BL, He Y, Hu X, Wieskopf JS, Mogil JS, Storm DR, Wang ZJ, McCarson KE, Taylor BK, 2013. Constitutive mu-opioid receptor activity leads to long-term endogenous analgesia and dependence. Science 341, 1394–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Costa DS, Meotti FC, Andrade EL, Leal PC, Motta EM, Calixto JB, 2010. The involvement of the transient receptor potential A1 (TRPA1) in the maintenance of mechanical and cold hyperalgesia in persistent inflammation. Pain 148, 431–437. [DOI] [PubMed] [Google Scholar]
- Decosterd I, Woolf CJ, 2000. Spared nerve injury: an animal model of persistent peripheral neuropathic pain. Pain 87, 149–158. [DOI] [PubMed] [Google Scholar]
- Demartini C, Greco R, Zanaboni AM, Francesconi O, Nativi C, Tassorelli C, Deseure K, 2018. Antagonism of Transient Receptor Potential Ankyrin Type-1 Channels as a Potential Target for the Treatment of Trigeminal Neuropathic Pain: Study in an Animal Model. Int J Mol Sci 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson JC, Clegg KE, Palmiter RD, 1996. Sensitivity to leptin and susceptibility to seizures of mice lacking neuropeptide Y. Nature 381, 415–421. [DOI] [PubMed] [Google Scholar]
- Fairbanks CA, 2003. Spinal delivery of analgesics in experimental models of pain and analgesia. Advanced drug delivery reviews 55, 1007–1041. [DOI] [PubMed] [Google Scholar]
- Fu W, Nelson TS, Santos DF, Doolen S, Gutierrez JJP, Ye N, Zhou J, Taylor B, 2019. An NPY Y1 receptor antagonist unmasks latent sensitization and reveals the contribution of Protein Kinase A and EPAC to chronic inflammatory pain. Pain. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs J, Flores CM, Hargreaves KM, 2004. Neuropeptide Y inhibits capsaicin-sensitive nociceptors via a Y1-receptor-mediated mechanism. Neuroscience 125, 703–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs JL, Flores CM, Hargreaves KM, 2006. Attenuation of capsaicin-evoked mechanical allodynia by peripheral neuropeptide Y Y1 receptors. Pain 124, 167–174. [DOI] [PubMed] [Google Scholar]
- Gibbs JL, Hargreaves KM, 2008. Neuropeptide Y Y1 receptor effects on pulpal nociceptors. Journal of dental research 87, 948–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griggs RB, Bardo MT, Taylor BK, 2015. Gabapentin alleviates affective pain after traumatic nerve injury. Neuroreport 26, 522–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grouzmann E, Meyer C, Burki E, Brunner H, 2001. Neuropeptide Y Y2 receptor signalling mechanisms in the human glioblastoma cell line LN319. Peptides 22, 379–386. [DOI] [PubMed] [Google Scholar]
- Intondi AB, Zadina JE, Zhang X, Taylor BK, 2010. Topography and time course of changes in spinal neuropeptide Y immunoreactivity after spared nerve injury. Neuroscience 165, 914–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Kohno T, Moore KA, Woolf CJ, 2003. Central sensitization and LTP: do pain and memory share similar mechanisms? Trends Neurosci 26, 696–705. [DOI] [PubMed] [Google Scholar]
- Julius D, 2013. TRP channels and pain. Annu Rev Cell Dev Biol 29, 355–384. [DOI] [PubMed] [Google Scholar]
- King T, Porreca F, 2014. Preclinical assessment of pain: improving models in discovery research. Curr Top Behav Neurosci 20, 101–120. [DOI] [PubMed] [Google Scholar]
- Latremoliere A, Woolf CJ, 2009. Central sensitization: a generator of pain hypersensitivity by central neural plasticity. The journal of pain : official journal of the American Pain Society 10, 895–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marvizon JC, Chen W, Fu W, Taylor BK, 2019. Neuropeptide Y release in the rat spinal cord measured with Y1 receptor internalization is increased after nerve injury. Neuropharmacology 158, 107732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melzack R, Casey K, 2014. Melzack & Casey Determinants of Pain 1968 from original. [Google Scholar]
- Nelson TS, Fu W, Donahue RR, Corder GF, Hokfelt T, Wiley RG, Taylor BK, 2019. Facilitation of neuropathic pain by the NPY Y1 receptor-expressing subpopulation of excitatory interneurons in the dorsal horn. Sci Rep 9, 7248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossipov MH, Zhang ET, Carvajal C, Gardell L, Quirion R, Dumont Y, Lai J, Porreca F, 2002. Selective mediation of nerve injury-induced tactile hypersensitivity by neuropeptide Y. The Journal of neuroscience : the official journal of the Society for Neuroscience 22, 9858–9867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitcher MH, Price TJ, Entrena JM, Cervero F, 2007. Spinal NKCC1 blockade inhibits TRPV1-dependent referred allodynia. Mol Pain 3, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields SD, Eckert WA 3rd, Basbaum AI, 2003. Spared nerve injury model of neuropathic pain in the mouse: a behavioral and anatomic analysis. The journal of pain : official journal of the American Pain Society 4, 465–470. [DOI] [PubMed] [Google Scholar]
- Silva AP, Cavadas C, Grouzmann E, 2002. Neuropeptide Y and its receptors as potential therapeutic drug targets. Clinica chimica acta; international journal of clinical chemistry 326, 3–25. [DOI] [PubMed] [Google Scholar]
- Solway B, Bose SC, Corder G, Donahue RR, Taylor BK, 2011. Tonic inhibition of chronic pain by neuropeptide Y. Proceedings of the National Academy of Sciences of the United States of America 108, 7224–7229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ste Marie L, Luquet S, Cole TB, Palmiter RD, 2005. Modulation of neuropeptide Y expression in adult mice does not affect feeding. Proceedings of the National Academy of Sciences of the United States of America 102, 18632–18637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Story GM, Peier AM, Reeve AJ, Eid SR, Mosbacher J, Hricik TR, Earley TJ, Hergarden AC, Andersson DA, Hwang SW, McIntyre P, Jegla T, Bevan S, Patapoutian A, 2003. ANKTM1, a TRP-like channel expressed in nociceptive neurons, is activated by cold temperatures. Cell 112, 819–829. [DOI] [PubMed] [Google Scholar]
- Taylor BK, Fu W, Kuphal KE, Stiller CO, Winter MK, Chen W, Corder GF, Urban JH, McCarson KE, Marvizon JC, 2014. Inflammation enhances Y1 receptor signaling, neuropeptide Y-mediated inhibition of hyperalgesia, and substance P release from primary afferent neurons. Neuroscience 256, 178–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakisaka S, Kajander KC, Bennett GJ, 1992. Effects of peripheral nerve injuries and tissue inflammation on the levels of neuropeptide Y-like immunoreactivity in rat primary afferent neurons. Brain research 598, 349–352. [DOI] [PubMed] [Google Scholar]
- Walwyn WM, Chen W, Kim H, Minasyan A, Ennes HS, McRoberts JA, Marvizon JC, 2016. Sustained Suppression of Hyperalgesia during Latent Sensitization by mu-, delta-, and kappa-opioid receptors and alpha2A Adrenergic Receptors: Role of Constitutive Activity. J Neurosci 36, 204–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Xu H, Wu LJ, Kim SS, Chen T, Koga K, Descalzi G, Gong B, Vadakkan KI, Zhang X, Kaang BK, Zhuo M, 2011. Identification of an adenylyl cyclase inhibitor for treating neuropathic and inflammatory pain. Sci Transl Med 3, 65ra63. [DOI] [PubMed] [Google Scholar]
- Wei F, Qiu CS, Kim SJ, Muglia L, Maas JW, Pineda VV, Xu HM, Chen ZF, Storm DR, Muglia LJ, Zhuo M, 2002. Genetic elimination of behavioral sensitization in mice lacking calmodulin-stimulated adenylyl cyclases. Neuron 36, 713–726. [DOI] [PubMed] [Google Scholar]
- Wong ST, Athos J, Figueroa XA, Pineda VV, Schaefer ML, Chavkin CC, Muglia LJ, Storm DR, 1999. Calcium-stimulated adenylyl cyclase activity is critical for hippocampus-dependent long-term memory and late phase LTP. Neuron 23, 787–798. [DOI] [PubMed] [Google Scholar]
- Woolf CJ, 1984. Long term alterations in the excitability of the flexion reflex produced by peripheral tissue injury in the chronic decerebrate rat. Pain 18, 325–343. [DOI] [PubMed] [Google Scholar]
- Xu J, Li M, Shen P, 2010. A G-protein-coupled neuropeptide Y-like receptor suppresses behavioral and sensory response to multiple stressful stimuli in Drosophila. J Neurosci 30, 2504–2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Sornborger AT, Lee JK, Shen P, 2008. Drosophila TRPA channel modulates sugar-stimulated neural excitation, avoidance and social response. Nature neuroscience 11, 676–682. [DOI] [PubMed] [Google Scholar]