

Abstract
Aim
The aim of this study was to investigate whether the compositionality of the lower airway microbiota predicts later exacerbation risk in persons with COPD in a cohort study.
Materials and methods
We collected lower airways microbiota samples by bronchoalveolar lavage and protected specimen brushes, and oral wash samples from 122 participants with COPD. Bacterial DNA was extracted from all samples, before we sequenced the V3-V4 region of the 16S RNA gene. The frequency of moderate and severe COPD exacerbations was surveyed in telephone interviews and in a follow-up visit. Compositional taxonomy and α and β diversity were compared between participants with and without later exacerbations.
Results
The four most abundant phyla were Firmicutes, Bacteroidetes, Proteobacteria and Fusobacteria in both groups, and the four most abundant genera were Streptococcus, Veillonella, Prevotella and Gemella. The relative abundances of different taxa showed a large variation between samples and individuals, and no statistically significant difference of either compositional taxonomy, or α or β diversity could be found between participants with and without COPD exacerbations within follow-up.
Conclusion
The findings from the current study indicate that individual differences in the lower airway microbiota in persons with COPD far outweigh group differences between frequent and nonfrequent COPD exacerbators, and that the compositionality of the microbiota is so complex as to present large challenges for use as a biomarker of later exacerbations.
Short abstract
Contrary to previous reports, in this study there were no significant associations between the lung microbiota in stable COPD and COPD exacerbation frequency https://bit.ly/2ZVcNdG
Introduction
COPD exacerbations are often caused by bacterial and viral respiratory infections with common pathogens such as Haemophilus influenzae, Streptococcus pneumoniae and Moraxella catarrhalis [1]. Before next-generation sequencing was made available, these microbes were detected by growth in cultures and later PCR. PCR studies suggested the aetiology could be multi-microbial, with frequent detection of multiple bacteria and/or viruses [2–4]. However, not all COPD exacerbations show clinical or microbial signs of infection. Exacerbation-associated bacteria have been cultured from sputum obtained in clinically stable COPD, giving rise to a still unresolved debate on whether colonisation is a pathogenic factor for later exacerbation [5]. The finding of a diverse lung microbiome has led to the hypothesis that exacerbations are caused by a dysbiosis in the airway microbiome, and that differences in baseline microbiota might help explain the difference between the frequent and infrequent exacerbator type [6].
Some previous studies comparing sputum microbiota between stable and exacerbated state in COPD indicate there are differences between disease states, but the findings are inconsistent [7–12]. And, two previous retrospective studies indicated that the sputum microbiota in the stable state was different in patients prone to exacerbations compared to patients who do not exacerbate [13, 14]. However, sputum is invariably fraught by contamination of microbes from the upper airways, which is both difficult to adjust for and potentially interferes with our interpretations of the lower airway microbiota.
To address the question of whether the stable-state lower airway microbiota is predictive of later COPD exacerbation, a study should include both COPD patients known to exacerbate and not, sample the lower airways directly, and include a prospective longitudinal follow-up of exacerbation events. The current study is to our knowledge the first such study with this methodology.
Methods
Study population
COPD participants in the Bergen COPD Microbiome (MicroCOPD) study were recruited among previous participants in the Bergen COPD Cohort Study [15] and the GenKOLS Study [16], in addition to volunteers from the outpatient clinic at the Department of Thoracic Medicine, Haukeland University Hospital. The protocol for the MicroCOPD study is previously published [17].
To be included, all participants had to be >40 years of age. In the current study sample the youngest participant was 49.5 years upon inclusion. The COPD diagnosis was verified by experienced pulmonologists based on spirometry, respiratory symptoms and disease history. Participation was postponed in individuals that had been treated for a respiratory exacerbation within the last 2 weeks or if they had ongoing symptoms of exacerbation according to modified Anthonisen criteria [18]. We excluded individuals deemed not fit for bronchoscopy, namely those who had an O2 saturation <90% despite oxygen supplementation, CO2 tension >6.65 kPa in arterial blood gas analyses, increased risk of bleeding, known allergy towards the premedication or cardiac risk factors as specified in the protocol [17]. Participants were asked about their current medication, smoking history, number of exacerbations treated with systemic steroids or antibiotics and exacerbation-related hospital admissions in the preceding year. All participants underwent spirometry after inhalation of 0.4 mg salbutamol, followed by bronchoscopy. In the MicroCOPD study, a total of 130 participants with COPD were included. For the current analyses, only the 122 participants from the post-pilot phase of the MicroCOPD study were included.
The MicroCOPD study was approved by the regional ethical committee (REK Vest case number 2011/1307). All participants provided oral and written consent.
Microbial sampling
Prior to bronchoscopy, all participants were asked to gargle 10 mL of PBS for oral wash (OW) sampling. Bronchoscopy was performed through oral access. Topical anaesthesia (lidocaine) was delivered by oral spray formulation prior to the procedure and per-operatively through a catheter within the bronchoscope's working channel. Three protective specimen brushes were collected in the right lower lobe (rPSB). The three brushes were cut off using sterile scissors and placed in an Eppendorf tube with 1 mL of PBS. Protected bronchoalveolar lavage (BAL) was performed in all participants with FEV1>30% of predicted: two fractions each containing 50 mL of PBS were installed through a Combicath sterile catheter with a sealed tip, in the right middle lobe and aspirated through the same sterile catheter. The median retrieval was 33 mL in the group that had no exacerbations and 26 mL in the group that had exacerbations (p=0.04, Mann–Whitney U-test). For the current study we used the second fraction of BAL (BAL2), which usually had the highest yield. All samples from one participant (OW, BAL2, rPSB) used the same bottle of PBS, and for each participant we analysed one negative control sample from this bottle. A more detailed explanation of handling of negative control samples is presented in the supplementary text.
DNA extraction and 16S sequencing
The detailed protocol for laboratory processing is published [19]. Briefly, bacterial DNA was extracted by enzymatic lysis followed by processing though the FastDNA Spin Kit. Sequencing of the V3-V4 region of the 16S rRNA gene was carried out according to the Illumina 16S Metagenomic Sequencing Library Preparation guide. The V3-V4 region was PCR amplified using 45 cycles and prepared for a subsequent 8-cycle index PCR step using specific primers. The samples were pooled and diluted before 2×300 cycles of paired-end sequencing was performed using reagents from the MiSeq reagent kit v3 (Illumina Inc., San Diego, CA, USA).
Exacerbation follow-up
We collected information on health-seeking behaviour, use of antibiotics and oral steroids in structured, quarterly telephone interviews for 12 months after the bronchoscopy. All participants were also offered a follow-up examination 1–1.5 years after inclusion where we also collected exacerbation history. Exacerbations were self-reported, but only counted if the exacerbation led to any administration of antibiotics and/or oral corticosteroids or hospitalisations (moderate-to-severe exacerbations). Dates reported in the telephone interviews and the physical follow-up examination were used to confirm that the event had not already been recorded and to separate one exacerbation from another. When information on hospitalisation was contradicting, the reason for admittance was unclear (for instance hospital admittance without administration of antibiotics), or the time point for an exacerbation leading to hospitalisation was unknown, the digital hospital records were consulted.
Bioinformatic analyses
Bioinformatic analyses were performed with QIIME 2 [20] (versions 2018.8, 2018.11, 2019.4, 2019.7, 2020.2) and R (versions 3.5.0 and 3.6.0). Sequencing data from the entire MicroCOPD study (including samples not included in the current analyses) were imported batched by Illumina runs into QIIME 2 (version 2018.8) from Casava 1.8 paired-end demultiplexed fastq format. Denoising was performed using the Divisive Amplicon Denoising Algorithm version 2 (DADA2) software package [21] (via q2-dada2), and sequences were further processed with VSEARCH [22] (via q2-vsearch) for additional chimera removal and then merged to one amplicon sequence variant (ASV) table (QIIME 2, version 2018.8).
All ASVs representing <0.005% of reads (fewer than 3000 reads) were removed, as they were deemed likely to represent sequencing noise [23]. The ASV table was exported from QIIME 2 into R for bioinformatic identification and removal of contaminants with the package Decontam [24], where we used the prevalence-based method with a threshold of 0.3. A more detailed explanation of the data curation and handling of contaminants are given in the supplementary text.
The ASV table was then filtered to contain only samples included in the present study. Taxonomy was assigned using a classifier trained on the Human Oral Microbiome Database [25]. All ASVs that were unassigned to phylum level, were checked with the NCBI BLAST tool [26], and all nonbacterial ASVs were removed. Remaining ASVs were aligned with mafft (via q2-phylogeny) and a phylogenetic tree was constructed with fasttree2 [27] (via q2-phylogeny). Sequences were rarefied at a sampling depth of 1000 prior to α (within sample) and β (between samples) diversity analyses to ensure equal sampling depths (via q2-diversity).
Statistics
Differentially abundant features (i.e. genera and ASVs) and differences in compositionality were analysed using Analysis of Composition of Microbes (ANCOM) [28] (via q2-composition), balance trees [29] (via q2-gneiss), ANOVA-like differential expression analysis 2 (ALDEx2) [30] (via q2-aldex2) and differential distribution analysis [31] (with R package MicrobiomeDDA). The α diversity difference, measured with the Shannon index and Faith phylogenetic diversity, was tested with the Kruskal–Wallis test. The β diversity difference, measured with Bray–Curtis and weighted UniFrac distance, was tested with the ADONIS permutation-based test using the vegan package in R [32].
Results
Of the 122 participants, 105 had complete follow-up of exacerbations for a full year after bronchoscopy. Participants who experienced one or more exacerbations within follow-up had significantly lower lung function, higher symptom score, more frequent exacerbations in the preceding year and more use of inhaled corticosteroids (ICSs) (table 1). Median time to first exacerbation was 146 days. Exacerbations were evenly distributed across seasons of the year.
TABLE 1.
Participant characteristics at baseline by exacerbation status during follow-up
| Variable | Exacerbation group | No exacerbation group | Exacerbation unknown | Comparison |
| Subjects n | 46 | 59 | 17 | NS |
| Female | 45.7% | 47.5% | 29.4% | NS |
| Age years | 67.2±7.2 | 68.5±7.3 | 64.3±7.4 | NS |
| Smoking status | NS | |||
| Daily | 19.6% | 27.1% | 23.5% | |
| Ex-smoker | 78.3% | 72.9% | 76.5% | |
| Never | 2.2% | 0% | 0% | |
| FEV1 % pred | 46.1±17.0 | 60.4±17.3 | 68.3±22.6 | p<0.01 |
| FEV1/FVC | 0.40±0.12 | 0.49±0.11 | 0.45±0.12 | p<0.01 |
| CAT-score# | 19.4±8.1 | 15.5±7.5 | 12.5±6.4 | p=0.01 |
| PaO2 kPa¶ | 9.4±1.2 | 9.8±1.2 | 9.6±1.2 | NS |
| Exacerbation ≥2 previous year | 32.6% | 10.2% | 5.9% | p<0.01 |
| Daily use of inhaled corticosteroids | 69.6% | 39.0% | 35.3% | p≤0.01 |
| Daily use of oral steroids | 6.5% | 3.4% | 5.9% | NS |
Data are presented as % or mean±sd, unless otherwise stated. The comparisons were between the exacerbation and no exacerbation groups only, and were tested using t-tests and Chi-squared tests. NS: not significant; FEV1: forced expiratory volume in 1 s; FVC: forced vital capacity; CAT: COPD Assessment Test; PaO2: arterial oxygen tension. #: Two missing values; ¶: three missing values.
For the current analyses, we selected the 327 samples of BAL2, OW and rPSB from the 122 participants, of which three samples were judged as only containing nonbacterial taxa and filtered out. In addition, we removed the samples from the 17 participants with unknown exacerbation status. For diversity analyses, rarefied data included 235 samples. Figure 1 shows the flow chart for the sample processing.
FIGURE 1.

Sample flow chart of data processing. DADA2: Divisive Amplicon Denoising Algorithm 2; ASV: amplicon sequence variant; BAL2: second fraction of bronchoalveolar lavage; OW: oral wash; rPSB: protected specimen brushes from the right lung; HOMD: Human oral microbiome database.
Taxonomy and differential abundance testing
Taxonomy at the phylum and genus levels for the three sample types by exacerbation status are shown in figures 2 and 3. Overall, the most abundant phyla were Firmicutes, Bacteroidetes, Proteobacteria and Fusobacteria. The most abundant genera were Streptococcus, Veillonella, Prevotella and Gemella. However, the relative abundances of different taxa showed a large variation between samples. Four participants with one or more later exacerbations and one participant without later exacerbations presented with high abundances of the Moraxella genus in rPSB samples, which was also reflected by detection of Moraxella in BAL2 for those participants who had a lavage sample. When we sorted rPSB samples by relative abundance of the most common taxon, the visual pattern differed from that in BAL2 and OW (figures 2 and 3).
FIGURE 2.
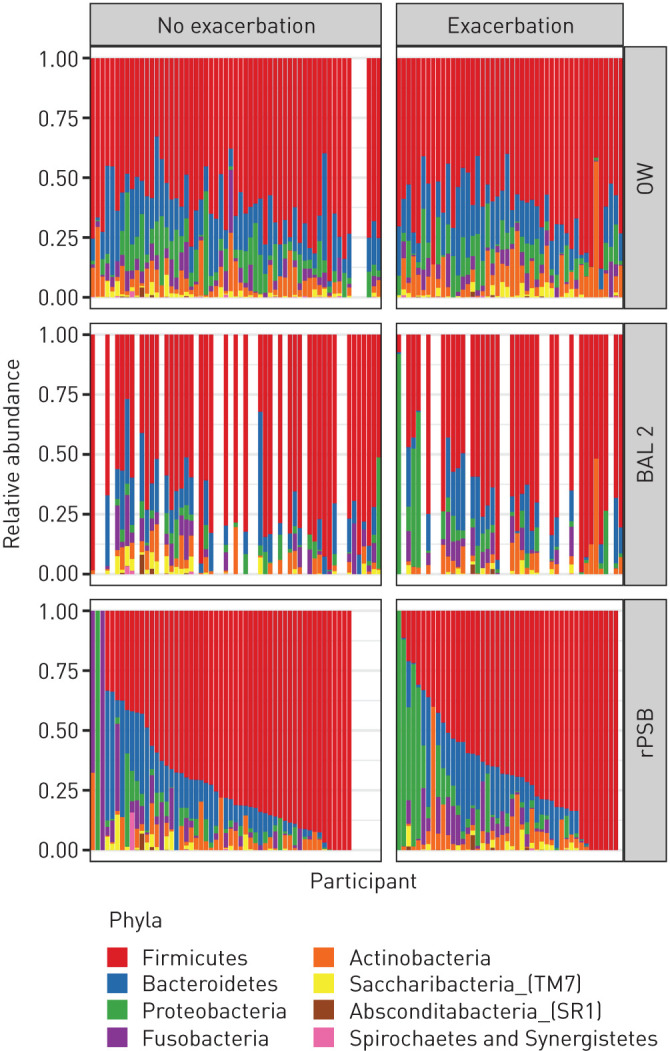
Bacterial taxonomy at the phylum level in participants with COPD with and without exacerbation during follow-up. Taxonomic groups in the legend are sorted in decreasing order based on the average relative frequency of that group in all samples. Each bar represents one participant, ordered in the same position horizontally for all three sample types according to relative abundance of Firmicutes in rPSB samples. OW: oral wash; BAL2: second fraction of bronchoalveolar lavage; rPSB: right protected specimen brushes.
FIGURE 3.

Bacterial taxonomy at the genus level in participants with COPD with and without exacerbation during follow-up. Taxonomic groups in the legend are sorted in decreasing order based on the average relative frequency of that group in all samples. Each bar represents one participant, ordered in the same position horizontally for all three sample types according to relative abundance of Streptococcus in rPSB samples. OW: oral wash; BAL2: second fraction of bronchoalveolar lavage; rPSB: right protected specimen brushes.
The visual impression from both figure 2 (phylum level) and figure 3 (genus level) was that participants without later exacerbations had more Firmicutes (figure 2), and more streptococci (figure 3), than participants with later exacerbations. However, when testing for statistical significance in difference of abundance between the two groups, few were found. ANCOM, balance trees (gneiss) and ALDEx2 performed on BAL2 and rPSB did not identify any differentially abundant ASVs or genera between the group with and without exacerbations. Differential distribution analysis identified two ASVs as differentially expressed between the groups in rPSB samples (supplementary table 1). These two ASVs were classified as Capnocytophaga gingivitis (more abundant, less prevalent and less dispersed in group with exacerbations (adjusted p=0.011)) and Prevotella pallens (less abundant, more prevalent and more dispersed in group with exacerbations (adjusted p=0.041)).
To see whether patients who had two or more exacerbations during follow-up differed from those who had zero or one, we repeated the above analyses on this outcome (supplementary table 2). Again, ANCOM, balance trees (gneiss) and ALDEx2 performed on BAL2 and rPSB did not identify any differentially abundant ASVs or genera between the groups. However, differential distribution analysis identified two ASVs (Capnocytophaga leadbetteri, adjusted p=0.008, and Prevotella oris, adjusted p=0.045) and one genus (Moraxella, adjusted p=0.026) as differentially expressed between the groups in BAL samples.
Next, we checked whether the taxonomy by sample type and exacerbation status varied by use of ICSs. The bacterial composition did not appear altered by ICS use, as illustrated in a heat map of genera in BAL2 (figure 4). This impression was strengthened by differential abundance testing by the same four tests as above, revealing no significant differences in taxa between COPD patients who used ICSs and COPD patients who did not (supplementary table 3).
FIGURE 4.

Taxonomy in bronchoalveolar lavage. Heat map of 20 most common taxa at genus level. Divided into exacerbation and inhaled corticosteroid categories. All features with a relative abundance above 10% are adjusted white in order to better visualise the less common features. Within each category, samples are ordered according to increasing abundance of Streptococcus.
Finally, we examined whether taxonomy differed by previous exacerbations the last year before bronchoscopy (supplemental table 4). Again, only the differential distribution analysis yielded a potential difference, with Actinomyces graevenitzii being less abundant and more prevalent in patients having previous exacerbations (adjusted p=0.011).
Overall the main message from the differential abundance tests was that very few differences were found.
Diversity
The α diversity in BAL 2 and rPSB, measured by Faith phylogenetic diversity and Shannon diversity index, did not significantly differ between the group with and without later exacerbations. Box plots of the α diversity are presented in the supplementary material (supplementary figures 5 and 6).
Neither β diversity, measured by Bray–Curtis distance (nonphylogenetic) and weighted UniFrac distance (phylogenetic), differed by exacerbation status (supplementary figures 7 and 8). The four rPSB samples and three BAL2 samples that clustered separately (based on weighted UniFrac distance, supplementary figure 9) were the samples with high abundances of Moraxella (figure 3). Both unadjusted and adjusted (age, sex, FEV1, ICS use) PERMANOVA analyses showed nonsignificant differences in weighted UniFrac distances between patients with and without later exacerbations (supplementary tables 5 and 6 and supplementary figure 9).
Discussion
This study did not show any statistically significant characteristics of the lung microbiota in stable COPD, neither in differential abundance of taxonomy, nor α or β diversity, that could predict whether the participants would experience moderate or severe exacerbations in the subsequent follow-up. These findings challenge the few existing previous reports.
Pragman et al. [14] reported lower α diversity in sputum from patients with frequent exacerbations, and a significant difference in ASV counts between frequent (n=11) and infrequent (n=11) exacerbators, where Actinomyces was found more abundant in patients with infrequent exacerbations. In another recent study, there was an increased abundance of Pseudomonas, Selenomonas and Anaerococcus, in sputum from patients with frequent exacerbations (n=31) compared with patients without exacerbations (n=23). There was also a significant difference in β diversity between patients with frequent and infrequent exacerbations [13]. A study of the microbiota in BAL from patients with stable COPD retrospectively recorded the exacerbation history of 21 patients and suggested that Streptococcus and Rothia species might be protective and that Pseudomonas might be predictive of exacerbations [33].
The current study has superior methodology with a 2–5 times larger sample size than previous studies, prospective follow-up of exacerbations and protected sampling of the lower airways. However, other potential causes of the differences in results between our study and previous studies are worthy of consideration. Two of the previous studies suggest that Pseudomonas is associated with more frequent exacerbations, whereas we did not report Pseudomonas in any of our samples. Some Pseudomonas species are known colonisers in patients with severe disease or impaired immunity, but Pseudomonas is ubiquitous in the environment, and there is a high potential for Pseudomonas to be a contaminant. The differences between our study and the two previous reports could reflect differences between study populations, but is more likely caused by Pseudomonas being identified as a contaminant by the Decontam algorithm in our study, and therefore removed. In the filtered data, Pseudomonas was completely removed. In unfiltered data, Pseudomonas was identified in high abundances in samples from participants both with and without later exacerbations.
The overall abundance of Streptococcus in our study appeared by visual inspection higher in those participants without later exacerbation (figures 2 and 3), perhaps supporting the suggestion of this taxa being protective as suggested by Ren et al. [33]. However, none of the four differential abundance tests we performed found Streptococcus to be differentially abundant. The compositionality of data, the lack of quantitative measures, and the low taxonomic resolution of 16S sequencing for Streptococcus are challenges that may explain why no association was detected, in addition to the obvious possibility that no association actually exists. We also found that the differences in abundance within groups were bigger than between groups. To date, there is no consensus on what is to be considered a true clinically significant difference in compositionality. For instance, it may be that a select few pathogens have a large influence in some individuals, but not in others. This may be undetected when examining differences on group levels.
An intriguing finding in our study was that Moraxella, a pathogen known to be involved in COPD exacerbations, was present in high abundance in stable-state COPD in five participants. Four of these participants were in the exacerbation group. This was also reflected by differences in β diversity (weighted UniFrac distance) separating these samples from the remaining samples. The participant presenting with Moraxella without exacerbation in the follow-up was not part of the diversity analyses due to low sampling depth (below 1000). Although the presentation of Moraxella is not useful for predicting exacerbations in the group as a whole, we might speculate that this alteration in the microbiota can have a clinical impact on these individuals.
Overall, our results challenge previous findings of alterations in the stable microbiota in patients with different exacerbation frequencies. However, results are difficult to compare across studies due to variations in sampling technique (induced or spontaneous sputum or bronchoscopically sampled BAL or brushes), DNA extraction, choice of primers and hypervariable region, number of PCR cycles, management of negative controls, bioinformatic processing and statistical analysis. Each step of the microbiome analysis workflow can introduce bias, for instance by over-identifying certain bacterial taxa. This limits comparison of results from different protocols, and even misrepresents the true distribution within a study [34].
There are some methodological limitations in our study as well: 1) We have relied on self-reported information regarding exacerbations and medication use. Even though participants were called four times throughout the 1-year follow-up period, this can be prone to recall bias. 2) We do not present quantitative results. Quantitative PCR was performed on a subset of samples in MicroCOPD as part of a method paper on laboratory contamination [35], but the extent of this analysis was not sufficient to evaluate whether bacterial load was different between groups in this analysis. Pragman et al. [14] found no difference in bacterial load in groups of different exacerbation frequencies. However, it is biologically plausible that differences in bacterial load by itself impacts the immune response, and our study cannot assess this. 3) Our analyses are based on one sampling of the microbiota, at one time point for each participant. Each sample's representativeness for an individual's microbiota is uncertain, as we do not know how stable the microbiota is. Mayhew et al. [8] reported that stability of sputum microbiota over time was more likely to be decreased in individuals with higher exacerbation frequencies. Due to the cross-sectional sampling, this was not possible to assess in the current study. 4) The MicroCOPD study included many samples from a large number of participants. Although handled by only three well-trained laboratory technicians at our centre, samples had to be run in different batches. We looked for batch effects by examining principal-component analysis plots of different β diversity metrics, which showed no clustering based on run (see supplementary material). Samples from participants with exacerbations versus without were not unevenly distributed across runs.
The mentioned shortcomings are widely shared with published studies in the field. However, we have also applied some methods that strengthen the integrity of our results. First, MicroCOPD is the largest bronchoscopy study on the COPD microbiota. Second, we performed prospective follow-up of exacerbations. Third, we argue that protected sampling by bronchoscopy is superior to sputum collection [36]. Sputum is likely contaminated as it passes through the mouth. In addition, we present OW samples in this analysis. Fourth, we sequenced negative control samples from every examination and then used the validated Decontam method [24] to identify contaminants. A paper describing the use and implications of Decontam in the MicroCOPD study is recently published [35]. We consider this statistical approach an improvement compared to other strategies in this field such as removal of all ASVs found (consistently) in negative controls [37], removal based on lists of “known” contaminants [10, 38, 39], figure presentation of negative control taxa [33], or ruling out contamination if negative samples yield no band on gel electrophoresis [40]. Other publications do not address contamination [11, 12].
Thus, in the largest study to date of the potential predictive value of the lower airway microbiota, a significant prognostic factor for later exacerbations was not found. This study highlights the difficulties in examining microbial dysbiosis of the low biomass airway microbiota in participants where the between-individual variation is potentially much larger than the between-clinical group variation. New statistical methods, or much larger sample sizes, are likely needed to overcome this problem.
Supplementary material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material 00168-2020.supp (527.1KB, pdf)
Acknowledgements
The authors wish to thank Eli Nordeide, Hildegunn Bakke Fleten and Lise Østgård Monsen (Haukeland University Hospital, Bergen, Norway) for conducting the telephone interviews, and Ingvild Haaland (University of Bergen/Haukeland University Hospital, Bergen, Norway) and Tharmini Kalananthan (Haukeland University Hospital, Bergen, Norway) for preparation of bronchoalveolar lavage samples.
Footnotes
This article has supplementary material available from openres.ersjournals.com
The sequencing data and relevant metadata are available at the DRYAD data repository from https://doi.org/10.5061/dryad.tqjq2bvw3
Author contributions: E.O. Leiten, R. Nielsen, H.G. Wiker, P.S. Bakke, S. Tangedal and T.M.L. Eagan participated in planning of the study. R. Nielsen, G.R. Husebø, E.O. Leiten, E.M.H. Martinsen and T.M.L. Eagan participated in the data collection. H.G. Wiker and C. Drengenes planned and performed the sequencing analyses. T.M.L. Eagan and R. Nielsen performed the bioinformatic pre-processing. E.O. Leiten, E.M.H. Martinsen and T.M.L. Eagan performed the bioinformatic and statistical analyses. E.O. Leiten and T.M.L. Eagan drafted the manuscript. All authors participated in the revision of the manuscript and approved the final version.
Conflict of interest: E.O. Leiten has nothing to disclose.
Conflict of interest: R. Nielsen reports grants from the Timber Merchant Delphins Endowment (The Norwegian Medical Association), GlaxoSmithKline and Boehringer Ingelheim, during the conduct of the study; grants and personal fees from AstraZeneca, personal fees from GlaxoSmithKline, grants and personal fees from Boehringer Ingelheim, and grants from Novartis, outside the submitted work.
Conflict of interest: H.G. Wiker has nothing to disclose.
Conflict of interest: P.S. Bakke reports an advisory board fee from GlaxoSmithKline, and advisory board and lecture fees from Chiesi, AstraZeneca and Boehringer Ingelheim, outside the submitted work.
Conflict of interest: E.M.H. Martinsen has nothing to disclose.
Conflict of interest: C. Drengenes has nothing to disclose.
Conflict of interest: S. Tangedal has nothing to disclose.
Conflict of interest: G.R. Husebø has nothing to disclose.
Conflict of interest: T.M.L. Eagan reports a grant for data collection in the MicroCOPD study from Helse Vest (regional health authority research board) during the conduct of the study and lecture fee from Boehringer Ingelheim outside the submitted work.
Support statement: The MicroCOPD study was funded by unrestricted grants and fellowships from Helse Vest, Bergen Medical Research Foundation, the Endowment of Timber Merchant A. Delphin and Wife through the Norwegian Medical Association and GlaxoSmithKline through the Norwegian Respiratory Society. Funding information for this article has been deposited with the Crossref Funder Registry.
References
- 1.Ko FW, Chan KP, Hui DS, et al. Acute exacerbation of COPD. Respirology 2016; 21: 1152–1165. doi: 10.1111/resp.12780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shimizu K, Yoshii Y, Morozumi M, et al. Pathogens in COPD exacerbations identified by comprehensive real-time PCR plus older methods. Int J Chron Obstruct Pulmon Dis 2015; 10: 2009–2016. doi: 10.2147/COPD.S82752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Curran T, Coyle PV, McManus TE, et al. Evaluation of real-time PCR for the detection and quantification of bacteria in chronic obstructive pulmonary disease. FEMS Immunol Med Microbiol 2007; 50: 112–118. doi: 10.1111/j.1574-695X.2007.00241.x [DOI] [PubMed] [Google Scholar]
- 4.Perotin JM, Dury S, Renois F, et al. Detection of multiple viral and bacterial infections in acute exacerbation of chronic obstructive pulmonary disease: a pilot prospective study. J Med Virol 2013; 85: 866–873. doi: 10.1002/jmv.23495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lopez-Campos JL, Agusti A. Heterogeneity of chronic obstructive pulmonary disease exacerbations: a two-axes classification proposal. Lancet Respir Med 2015; 3: 729–734. doi: 10.1016/S2213-2600(15)00242-8 [DOI] [PubMed] [Google Scholar]
- 6.Dickson RP, Martinez FJ, Huffnagle GB. The role of the microbiome in exacerbations of chronic lung diseases. Lancet 2014; 384: 691–702. doi: 10.1016/S0140-6736(14)61136-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tangedal S, Nielsen R, Aanerud M, et al. Sputum microbiota and inflammation at stable state and during exacerbations in a cohort of chronic obstructive pulmonary disease (COPD) patients. PLoS One 2019; 14: e0222449. doi: 10.1371/journal.pone.0222449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mayhew D, Devos N, Lambert C, et al. Longitudinal profiling of the lung microbiome in the AERIS study demonstrates repeatability of bacterial and eosinophilic COPD exacerbations. Thorax 2018; 73: 422–430. doi: 10.1136/thoraxjnl-2017-210408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jubinville E, Veillette M, Milot J, et al. Exacerbation induces a microbiota shift in sputa of COPD patients. PLoS One 2018; 13: e0194355. doi: 10.1371/journal.pone.0194355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Z, Bafadhel M, Haldar K, et al. Lung microbiome dynamics in COPD exacerbations. Eur Respir J 2016; 47: 1082–1092. doi: 10.1183/13993003.01406-2015 [DOI] [PubMed] [Google Scholar]
- 11.Haldar K, Bafadhel M, Lau K, et al. Microbiome balance in sputum determined by PCR stratifies COPD exacerbations and shows potential for selective use of antibiotics. PLoS ONE 2017; 12: e0182833. doi: 10.1371/journal.pone.0182833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang YJ, Sethi S, Murphy T, et al. Airway microbiome dynamics in exacerbations of chronic obstructive pulmonary disease. J Clin Microbiol 2014; 52: 2813–2823. doi: 10.1128/JCM.00035-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Millares L, Pascual S, Monton C, et al. Relationship between the respiratory microbiome and the severity of airflow limitation, history of exacerbations and circulating eosinophils in COPD patients. BMC Pulm Med 2019; 19: 112. doi: 10.1186/s12890-019-0867-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pragman AA, Knutson KA, Gould TJ, et al. Chronic obstructive pulmonary disease upper airway microbiota alpha diversity is associated with exacerbation phenotype: a case–control observational study. Respir Res 2019; 20: 114. doi: 10.1186/s12931-019-1080-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eagan TM, Ueland T, Wagner PD, et al. Systemic inflammatory markers in COPD: results from the Bergen COPD Cohort Study. Eur Respir J 2010; 35: 540–548. doi: 10.1183/09031936.00088209 [DOI] [PubMed] [Google Scholar]
- 16.Sorheim IC, Johannessen A, Grydeland TB, et al. Case-control studies on risk factors for chronic obstructive pulmonary disease: how does the sampling of the cases and controls affect the results? Clin Respir J 2010; 4: 89–96. doi: 10.1111/j.1752-699X.2009.00154.x [DOI] [PubMed] [Google Scholar]
- 17.Grønseth R, Haaland I, Wiker HG, et al. The Bergen COPD microbiome study (MicroCOPD): rationale, design, and initial experiences. Eur Clin Respir J 2014; 1: 10.3402/ecrj.v1.26196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Anthonisen NR, Manfreda J, Warren CP, et al. Antibiotic therapy in exacerbations of chronic obstructive pulmonary disease. Ann Intern Med 1987; 106: 196–204. doi: 10.7326/0003-4819-106-2-196 [DOI] [PubMed] [Google Scholar]
- 19.Hoang T, Wiker HG, Eagan T, et al. 16S Amplicon PCR for the V3-V4 region for the MicroCOPD samples. Protocols.io 2019; protocol [ 10.17504/protocols.io.2sygefw]. [DOI]
- 20.Bolyen E, Rideout JR, Dillon MR, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol 2019; 37: 852–857. doi: 10.1038/s41587-019-0209-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Callahan BJ, McMurdie PJ, Rosen MJ, et al. DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods 2016; 13: 581–583. doi: 10.1038/nmeth.3869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rognes T, Flouri T, Nichols B, et al. VSEARCH: a versatile open source tool for metagenomics. PeerJ 2016; 4: e2584. doi: 10.7717/peerj.2584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Navas-Molina JA, Peralta-Sánchez JM, González A, et al. Advancing our understanding of the human microbiome using QIIME. Meth Enzymol 2013; 531: 371–444. doi: 10.1016/B978-0-12-407863-5.00019-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Davis NM, Proctor DM, Holmes SP, et al. Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome 2018; 6: 226. doi: 10.1186/s40168-018-0605-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen T, Yu WH, Izard J, et al. The Human Oral Microbiome Database: a web accessible resource for investigating oral microbe taxonomic and genomic information. Database (Oxford) 2010; 2010: baq013. doi: 10.1093/database/baq013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Madden T. The BLAST sequence analysis tool In: McEntyre J, O'stell J, eds. The NCBI Handbook. Bethesda, National Center for Biotechnology Information, 2002. [Google Scholar]
- 27.Price MN, Dehal PS, Arkin AP. FastTree 2--approximately maximum-likelihood trees for large alignments. PLoS ONE 2010; 5: e9490. doi: 10.1371/journal.pone.0009490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mandal S, Van Treuren W, White RA, et al. Analysis of composition of microbiomes: a novel method for studying microbial composition. Microb Ecol Health Dis 2015; 26: 27663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morton JT, Sanders J, Quinn RA, et al. Balance trees reveal microbial niche differentiation. mSystems 2017; 2: e00162-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fernandes AD, Reid JN, Macklaim JM, et al. Unifying the analysis of high-throughput sequencing datasets: characterizing RNA-seq, 16S rRNA gene sequencing and selective growth experiments by compositional data analysis. Microbiome 2014; 2: 15. doi: 10.1186/2049-2618-2-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen J, King E, Deek R, et al. An omnibus test for differential distribution analysis of microbiome sequencing data. Bioinformatics 2018; 34: 643–651. doi: 10.1093/bioinformatics/btx650 [DOI] [PubMed] [Google Scholar]
- 32.Oksanen J, Blanchet FG, Friendly M, et al. vegan: Community Ecology Package. R package version 2.5-6 www.CRAN.R-project.org/package=vegan Date last accessed: March 30, 2020. Date last updated: September 01, 2019. 2019.
- 33.Ren L, Zhang R, Rao J, et al. Transcriptionally active lung microbiome and its association with bacterial biomass and host inflammatory status. mSystems 2018; 3: e00199-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brooks JP. Challenges for case–control studies with microbiome data. Ann Epidemiol 2016; 26: 336–341.e331. doi: 10.1016/j.annepidem.2016.03.009 [DOI] [PubMed] [Google Scholar]
- 35.Drengenes C, Wiker HG, Kalananthan T, et al. Laboratory contamination in airway microbiome studies. BMC Microbiol 2019; 19: 187. doi: 10.1186/s12866-019-1560-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grønseth R, Drengenes C, Wiker HG, et al. Protected sampling is preferable in bronchoscopic studies of the airway microbiome. ERJ Open Res 2017; 3: 00019-2017. doi: 10.1183/23120541.00019-2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leitao Filho FS, Alotaibi NM, Ngan D, et al. Sputum microbiome is associated with 1-year mortality following COPD hospitalizations. Am J Respir Crit Care Med 2018; 199: 1205–1213. [DOI] [PubMed] [Google Scholar]
- 38.Wang Z, Singh R, Miller BE, et al. Sputum microbiome temporal variability and dysbiosis in chronic obstructive pulmonary disease exacerbations: an analysis of the COPDMAP study. Thorax 2018; 73: 331–338. doi: 10.1136/thoraxjnl-2017-210741 [DOI] [PubMed] [Google Scholar]
- 39.Wang Z, Maschera B, Lea S, et al. Airway host-microbiome interactions in chronic obstructive pulmonary disease. Respir Res 2019; 20: 113. doi: 10.1186/s12931-019-1085-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Millares L, Perez-Brocal V, Ferrari R, et al. Functional metagenomics of the bronchial microbiome in COPD. PLoS ONE 2015; 10: e0144448. doi: 10.1371/journal.pone.0144448 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material 00168-2020.supp (527.1KB, pdf)