

Abstract
Background:
Alzheimer’s disease (AD) is the most prevalent form of dementia with two pathological hallmarks of tau-containing neurofibrillary tangles and amyloid-β protein (Aβ)-containing neuritic plaques. Although Aβ and tau have been explored as potential biomarkers, levels of these pathological proteins in blood fail to distinguish AD from healthy control subjects.
Objective:
We aim to discover potential plasma proteins associated with AD pathology by performing tandem mass tag (TMT)-based quantitative proteomic analysis of proteins from peripheral and central nervous system compartments.
Methods:
We performed comparative proteomic analyses of plasma collected from AD patients and cognitively normal subjects. In addition, proteomic profiles from the inferior frontal cortex, superior frontal cortex, and cerebellum of postmortem brain tissue from five AD patients and five non-AD controls were compared with plasma proteomic profiles to search for common biomarkers. Liquid chromatography-mass spectrometry was used to analyze plasma and brain tissue labeled with isobaric TMT for relative protein quantification.
Results:
Our results showed that the proteins in complement coagulation cascade and interleukin-6 signaling were significantly altered in both plasma and brains of AD patients.
Conclusion:
Our results demonstrate the relevance in immune responses between the peripheral and central nervous systems. Those differentially regulated plasma proteins are explored as candidate biomarker profiles that illustrate chronic neuroinflammation in brains of AD patients.
Keywords: Alzheimer’s disease, biomarker, mass spectrometry, plasma, quantitative proteomics, tandem mass tag (TMT)
INTRODUCTION
The neuropathological hallmarks of Alzheimer’s disease (AD) are senile plaques of amyloid-β (Aβ) and neurofibrillary tangles made of the hyperphosphorylated form of the microtubule-binding protein tau. Genetic and neuropathologic evidence suggests that AD is caused in part by the overproduction and lack of clearance of Aβ [1–3], accompanied by enhanced neuroinflammation [4–6]. Cells bearing familial AD mutant genes (amyloid precursor protein or presenilin genes) produced higher levels of oligomeric Aβ [7], and similar increases of oligomeric Aβ were observed in plasma and postmortem brains of AD patients [8]. It has been documented that the oligomeric form of Aβ seems to be the most toxic species of Aβ as well as the precursor to the fibrillary Aβ found in senile plaques [1, 3, 9–12]. The second hallmark of AD is tau-containing neurofibrillary tangles. A common characteristic event that disrupts tau microtubule function and precedes tangle formation and neurodegeneration is increased phosphorylation of tau, especially on Ser/Thr-Pro motifs [13–20]. A combination of measurements of the 42-residue of Aβ (Aβ42), tau, and phosphorylated tau at residue 181 (pTau181) in human cerebrospinal fluids (CSF) has emerged as the best predictor for AD progression [21–23]. A decrease of CSF Aβ42 is associated with increased brain Aβ plaque load and can be used to predict Clinical Dementia Rating (CDR) scale 0 converting to CDR > 0, mild cognitive impairment (MCI) patients converting to probable AD, and diagnosis of AD [21–23]. However, peripheral biomarkers would clearly be advantageous over CSF biomarkers due to the invasive nature of CSF collection,
Both plasma Aβ [24–29] and phosphorylated tau [30–34] have been reported for their correlation with AD brain pathology, but their implementation in clinical settings needs further validation [35]. Additional plasma proteins are found to associate with AD and brain amyloid deposits [35–42]. Earlier studies in prodromal AD patients applied untargeted proteomics to compare plasma protein profiles. Participants from the Australian Imaging, Biomarkers and Lifestyle Study (AIBL) were analyzed for their brain Aβ load determined by Pittsburgh Compound B (PiB)-PET and correlated to levels of 176 plasma proteins [43]. Among all these proteins, chemokine ligand 13, immunoglobulin M-1, pancreatic polypeptide (PPY), interleukin-17, and vascular cell adhesion protein (VCAM-1) are found to be associated with people with high Aβ load. This finding was further validated in healthy control subjects enrolled in the Alzheimer’s Disease Neuroimaging Initiatives (ADNI), which indicated that those carrying the above plasma protein profile were more likely to progress to MCI or AD than non-carriers [43]. A large scale validation study using Luminex xMAP and Meso Scale Discovery platforms was performed to identify a plasma biomarker panel to be predictive of in vivo AD pathology: β2-microglobulin (B2M), cathepsin D (CTSD), ficolin-2 (FCN2), complement component 4 (C4), alpha-1 antitrypsin (A1AT), complement factor I (CFI), and apolipoprotein E (ApoE). This panel was found to be consistent in both APOE ε4 carriers and non-carriers [44].
Despite significant progress toward the discovery of plasma proteins linked to brain pathology made in the past decade, a consistent set of plasma biomarkers that segregate with AD has not been identified. Among 163 candidate plasma biomarkers identified in 21 published studies, only four common candidate biomarkers are found in five independent cohorts: A2M, complement C3, α−1-antitrypsin, and ApoE [45]. A2M is genetically associated with AD [46], and complement C3 is involved in synaptic dysfunction in AD [47]. Levels of A2M in serum or plasma were found either increased in cognitively normal people with subjective cognitive decline [48] and early stage AD patients [40, 49–51], no change [52–54], or decreased in AD subjects [40], with significant correlation found in female AD patients in one report [55].
Some of these proteins are closely associated with neuroinflammation, a process implicated by a number of AD risk factor genes. Genetic mutations found in the microglial receptor TREM2 (triggering receptors expressed on myeloid cells 2) triple a person’s risk for AD [56, 57] and increased expression of CD33, which functions to suppress Aβ clearance and modifies AD risk [58, 59]. Systems analysis of hundreds of AD brains reveals changes in networks related to immunologic molecules and microglial cells, including microglial protein TYROBP that binds TREM2 and may regulate CD33 [60]. Clearly, those proteins play critical roles in AD onset and progression, and proteomics may identify contributors and provide novel insight into AD biomarkers. Therefore, it would be ideal to combine the unbiased proteomic profiling of plasma and CSF in AD patients and control subjects with additional targeted proteomics to identify potential AD-associated biomarkers, which could be analyzed using quantitative MS and a target-specific Enzyme-Linked ImmunoSorbent Assay (ELISA).
In this study, tandem mass tag (TMT)-based quantitative proteomic analysis of proteins from peripheral and central nervous system compartments were performed to discover potential plasma proteins associated with AD pathology. A number of candidate biomarkers involving neuroinflammation, lipid transfer/metabolism, cerebral vascular function and oxidative stress were identified. By comparing the plasma and brain samples, our study reinforces the relevance between peripheral plasma biomarkers and brain pathology in AD. Among them, neuroinflammation pathways constitute the most significant candidate network that is implicated in AD pathogenesis.
MATERIALS AND METHODS
Materials and reagents
Unless indicated, all reagents used for biochemical methods and sample preparation were purchased from Sigma-Aldrich (St Louis, MO, USA). Reagents for BCA protein assay or Nano Drop-based protein concentration determination for liquid chromatography-mass spectrometry (LC-MS)/MS analysis were purchased from Thermo Scientific (Rockford, IL, USA). Pierce top two abundant protein depletion spin columns, Pierce protein concentrators (3 kDa) and TMT reagent ten-plex kits were purchased from Thermo Scientific Pierce (Rockford, IL, USA).
Subjects
Subjects in this study were recruited from the Bedford VA Hospital Dementia Care Unit. The protocol was approved by the Bedford VA Hospital Institutional Review Board and written informed consent for each participant was obtained before initiation of the study. Blood samples were donated by 36 subjects, including 18 patients diagnosed with AD (average age, 75.8 ± 9.8 years) and 18 healthy controls (average age, 72.3 ± 4.1 years). All subjects were male. Montreal Cognition Assessment (MoCA) was used to evaluate enrolled subjects, and healthy control subjects were scored over 27.
Blood and brain samples
Blood was collected in BD Vacutainer cell preparation tubes (CPT, Becton, Dickinson and Company, Franklin Lakes, NJ) and centrifuged at 1500 g relative centrifugal force (rcf) for 20 min at room temperature shortly after collection. Plasma samples were aliquoted and stored at −80°C until analysis. The postmortem brain tissue of three different regions from 5 AD patients and 5 healthy controls with pathological documentation were obtained from Bedford Brain Bank, as described before [61].
Sample preparation for MS
Plasma was first depleted using Pierce™ top two abundant protein depletion spin columns and then concentrated by Pierce™ 3 kDa protein concentrators according to manufacturer’s instructions. Preparation of tryptic peptides for TMT 10-plex labeling was carried out according to the manufacturer’s instructions. Briefly, after quantification of the protein concentrations in each plasma sample by the BCA, 100 μg of low abundant proteins from each sample was transferred into a new vial, adjusted to a final volume of 100 μL with tetraethylammonium bicarbonate (TEAB), reduced with tris (2-carboxyethyl)phosphine (TCEP) at 55°C for 1 h, and then alkylated with iodoacetamide for 30 min in the dark at room temperature. Proteins were precipitated by pre-chilled (−20°C) acetone overnight. Acetone-precipitated protein was pelleted by centrifuging at 8000 g for 10 min at 4°C. Protein pellets were re-suspended with 100 μL of 50 mM TEAB and digested with trypsin overnight at 37°C. A pooled control plasma sample (Cx) was created by combing 10 μL of plasma from each healthy participant and was prepared along with all samples from AD and healthy control subjects. Brain tissue was homogenized for TMT 10-plex labeling as described before [61].
TMT-labeling and sample clean up
TMT enables relative quantitation of proteins present in multiple samples by labeling peptides with isobaric stable isotope tags that fragment upon collision-induced dissociation into reporter ions used for quantitation. In this study, 10-plex TMT tags (Thermo Fisher) were used to label tryptic-digested peptides from plasma and brain samples. Labeling of tryptic peptides was carried out according to the manufacturer’s instructions. Briefly, the TMT reagents (0.8 mg) were dissolved in 41 μL of anhydrous acetonitrile. Aliquots of samples were incubated with TMT reagents for 1 h at room temperature. Reactions were quenched by 8 μL of 5% hydroxylamine solution for 15 min. Totally four sets of TMT 10-plex labeling kits were used. In each set of TMT 10-plex labeling procedure, reagents were divided into two portions, with half labeled with samples from AD patients and the other half labeled with samples from controls. The common control from pooled control subjects was labeled with a specific tag and was used for all tests. The combined TMT labeled samples were dried under SpeedVac and reconstituted by dilute trifluoroacetic acid solution followed by desalting by Oasis HLB 96-well μElution plate (Waters) prior to LC-MS/MS injection.
LC-MS/MS analysis
LC MS/MS was performed on a Q Exactive Orbitrap Mass Spectrometer (ThermoFisher Scientific) coupled with a Dionex Ultimate 3000 HPLC system equipped with a nano-ESI ion source. The TMT-labeled peptides were separated on a C18 reverse-phase capillary column (PepMap, 75 μm × 150 mm, ThermoFisher) with linear gradients of 2–35% acetonitrile in 0.1% formic acid, at a constant flow rate of 300 nL/min for 220 min. The instrument was operated in the positive-ion mode with the ESI spray voltage set at 1.8 kV. A full scan of MS spectra (300–1800 m/z) was acquired at a mass resolution of 70,000 with an automatic gain control target (AGC) of 3E6. Fifteen peptide ions showing the most intense signal from each scan were selected for higher energy collision-induced dissociation (HCD)-MS/MS analysis (normalized collision energy 32V) in the Orbitrap at a mass resolution of 35,000 and AGC value of 1E5. Maximal filling times were 100 ms in full scans and 120 ms (HCD) for the MS/MS scans. Ions with unassigned charge states and single charged species were rejected. The dynamic exclusion was set to 50 s and an isolation window of 1.2 m/z. The data was acquired using Thermo Xcalibur 3.0.63.
Protein identification and quantification
Raw data were processed using Proteome Discoverer (Version 2.1, ThermoFisher Scientific). Data were searched against the Homo sapiens Universal Protein Resource sequence database (UniProt, August, 2013). The searches were performed with the following guidelines: trypsin digestion with two missed cleavages allowed; fixed modification, carbamidomethyl of cysteine; variable modification, oxidation of methionine, TMT 10-plex (peptide labeled) for N-terminus and Lys; MS tolerance, 10 ppm; MS/MS tolerance, 0.02 Da; false discovery rate (FDR) at peptide and protein levels, <0.01; and required peptide length, ≥6 amino acids. Protein grouping was enabled, meaning if one protein was equal to or completely contained within the set of peptides of another protein, these two proteins were put into the same protein group. At least one unique peptide per protein group was required for identifying proteins. The relative protein abundance ratios (fold changes) were calculated between the average of ADs and the average of controls from duplicate analytical runs in each experiment. The mean and standard deviation (SD) for relative ratios of each protein were computed and a Student two-tailed t test was used to compare these measures. p-values from these tests were recorded and the protein ratios with a p < 0.05 from two independent experiments were considered significant.
Quantification of interleukin 6 (IL-6), SAA, Aβ, and tau by ELISA
To validate findings from the TMT-based proteomic profiling using an independent method, a Meso Scale Discovery (MSD) platform was used to test the same material for MS analysis. Assays for Aβ, tau, IL-6, and SAA were analyzed using V-PLEX Aβ peptide Panel 1, total Tau kit, Neuroinflammation Panel 1 Human Kit, following the manufacturer’ instructions (MSD, Gaithersburg, MD). Antibodies AT270 and HT-7 (Thermo Fisher Scientific, Waltham, MA) were used to capture/detect pTau181 using the same MSD platform.
Data analysis
The list of differentially regulated proteins was submitted to STRING (Version 11.0) to identify protein networks that are based on currently known associations among proteins indicated in the scientific literature. An enrichment analysis was also performed by submitting the list of differentially regulated proteins to open-source software Database for Annotation, Visualization, and Integrated Discovery (DAVID, v6.8) [62, 63]. DAVID extracts biological features and meanings associated with large gene lists. The gene names of the proteins were entered to analyze GO biological process (GOBP), GO molecular function (GOMF), and GO cellular components (GOCCs) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways. Statistically significant differences (p < 0.05) were identified using an EASE score, which was provided by DAVID. Metacore is a web-based bioinformatics tool and was used in this study for functional analysis of the most relevant pathways, networks, and cellular process. Metacore uses these proteins and their identifiers to navigate the curated literature database and extracts the biologically relevant information among the candidate proteins. Associated bio-functions were generated, along with a score representing the log probability of a particular process of network.
RESULTS
Upregulation of plasma proteins involved in immune response and downregulation of proteins involved in pallet degranulation and lipid transport in AD
A comparative proteomic analysis of human plasma from 18 AD subjects and 18 healthy controls was performed using TMT labeling and LC-MS/MS. After eliminating those lacking unique peptides, we quantified a total of 373 plasma proteins with at least one unique peptide, with an FDR of <1% and TMT labeling efficiency at 99%. Among these proteins, we identified 42 up-(AD/control ratio >1) and downregulated (ratio <1) plasma proteins from AD patients (Table 1).
Table 1.
Significantly up- and downregulated human plasma proteins from AD patients compared to control subjects
| Accession | Description | Score Sequest HT | Coverage | # Unique Peptides | MW [kDa] | Ratio |
|---|---|---|---|---|---|---|
| P06727 | Apolipoprotein A-IV (APOA4) | 1764 | 82 | 31 | 45.4 | 0.74 |
| P02656 | Apolipoprotein C-III (APOC3) | 304 | 56 | 5 | 10.8 | 0.80 |
| P43652 | Afamin (AFM) | 1748 | 51 | 32 | 69 | 0.80 |
| P01023 | Alpha-2-macroglobulin (A2M) | 31728 | 73 | 74 | 163.2 | 0.82 |
| P02655 | Apolipoprotein C-II (APOC2) | 670 | 52 | 5 | 11.3 | 0.82 |
| P02787 | Serotransferrin (TF) | 21279 | 77 | 65 | 77 | 0.83 |
| P02749 | Beta-2-glycoprotein 1 (APOH) | 1180 | 65 | 15 | 38.3 | 0.83 |
| P02765 | Alpha-2-HS-glycoprotein (AHSG) | 2827 | 53 | 14 | 39.3 | 0.84 |
| Q96PD5 | N-acetylmuramoyl-L-alanine amidase (PGLYRP2) | 961 | 47 | 15 | 62.2 | 0.85 |
| Q96KN2 | Beta-Ala-His dipeptidase (CNDP1) | 266 | 34 | 14 | 56.7 | 0.86 |
| P51884 | Lumican (LUM) | 877 | 41 | 12 | 38.4 | 0.86 |
| P05452 | Tetranectin (CLEC3B) | 180 | 45 | 7 | 22.5 | 0.87 |
| O95445 | Apolipoprotein M (APOM) | 300 | 49 | 7 | 21.2 | 0.87 |
| P02647 | Apolipoprotein A-I (APOA1) | 6515 | 90 | 46 | 30.8 | 0.88 |
| P49908 | Selenoprotein P (SEPP1) | 65 | 20 | 7 | 43.2 | 0.89 |
| P29622 | Kallistatin (SERPINA4) | 705 | 42 | 15 | 48.5 | 0.89 |
| P05154 | Plasma serine protease inhibitor (SERPINA5) | 156 | 19 | 8 | 45.6 | 0.89 |
| P04196 | Histidine-rich glycoprotein (HRG) | 1345 | 38 | 17 | 59.5 | 0.90 |
| P00748 | Coagulation factor XII (F12) | 590 | 37 | 20 | 67.7 | 0.90 |
| P00488 | Coagulation factor XIII A chain (F13A1) | 251 | 21 | 13 | 83.2 | 0.90 |
| P80108 | Phosphatidylinositol-glycan-specific phospholipase D (GPLD) | 312 | 25 | 15 | 92.3 | 0.90 |
| P36955 | Pigment epithelium-derived factor (SERPINF1) | 1246 | 48 | 16 | 46.3 | 0.90 |
| Q6UXB8 | Peptidase inhibitor 16 (PI16) | 42 | 8 | 4 | 49.4 | 0.90 |
| Q04756 | Hepatocyte growth factor activator (HGFAC) | 85 | 20 | 11 | 70.6 | 0.91 |
| P43251 | Biotinidase OS = Homo sapiens (BTD) | 168 | 16 | 7 | 61.1 | 0.91 |
| O75882 | Attractin (ATRN) | 459 | 16 | 23 | 158.4 | 0.93 |
| O75636 | Ficolin-3 (FCN3) | 340 | 41 | 11 | 32.9 | 0.93 |
| P07360 | Complement component C8 gamma chain (C8G) | 288 | 53 | 8 | 22.3 | 0.95 |
| P02743 | Serum amyloid P-component (APCS) | 274 | 33 | 8 | 25.4 | 1.09 |
| P0C0L4 | Complement C4-A (C4A) | 11032 | 66 | 5 | 192.7 | 1.12 |
| P02748 | Complement component C9 (C9) | 1232 | 44 | 19 | 63.1 | 1.12 |
| P35542 | Serum amyloid A-4 protein (SAA4) | 149 | 28 | 4 | 14.7 | 1.14 |
| P61626 | Lysozyme C (LYZ) | 19 | 16 | 3 | 16.5 | 1.17 |
| P02750 | Leucine-rich alpha-2-glycoprotein (LRG1) | 1069 | 46 | 11 | 38.2 | 1.22 |
| P00738 | Haptoglobin (HP) | 12490 | 76 | 21 | 45.2 | 1.23 |
| P01011 | Alpha-1-antichymotrypsin (SERPINA3) | 7219 | 55 | 22 | 47.6 | 1.27 |
| P11226 | Mannose-binding protein C (MBL2) | 23 | 13 | 3 | 26.1 | 1.30 |
| P02763 | Alpha-1-acid glycoprotein 1 (ORM1) | 3668 | 56 | 8 | 23.5 | 1.31 |
| Q02985 | Complement factor H-related protein 3 (CFHR3) | 79 | 9 | 2 | 37.3 | 1.35 |
| P0DJI9 | Serum amyloid A-2 protein (SAA2) | 542 | 60 | 3 | 13.5 | 2.12 |
| P02741 | C-reactive protein (CRP) | 156 | 25 | 6 | 25 | 2.43 |
| P0DJI8 | Serum amyloid A-1 (SAA1) | 705 | 68 | 5 | 13.5 | 2.52 |
Up- and downregulated plasma proteins in AD group (n = 18) relative to the control group (n = 18) with p < 0.05, two-tailed Student’s T test from two independent studies.
To get a general overview of plasma protein changes in AD and search for the most affected processes and potential interactions among the candidate proteins, GO term enrichment analysis was performed. All differentially regulated (including both up- and downregulated) plasma proteins were submitted for analysis using STRING (Fig. 1). Several clusters were enriched, including the GO biological process of acute-phase response and acute inflammatory response (FDR = 2.25E-14), the KEGG pathways of complement and coagulation cascades (FDR = 2.46E-10), the KEGG pathway of cholesterol metabolism (FDR = 1.13E-6) and the Reactome pathway of platelet degranulation (FDR = 4.94E-13).
Fig. 1.
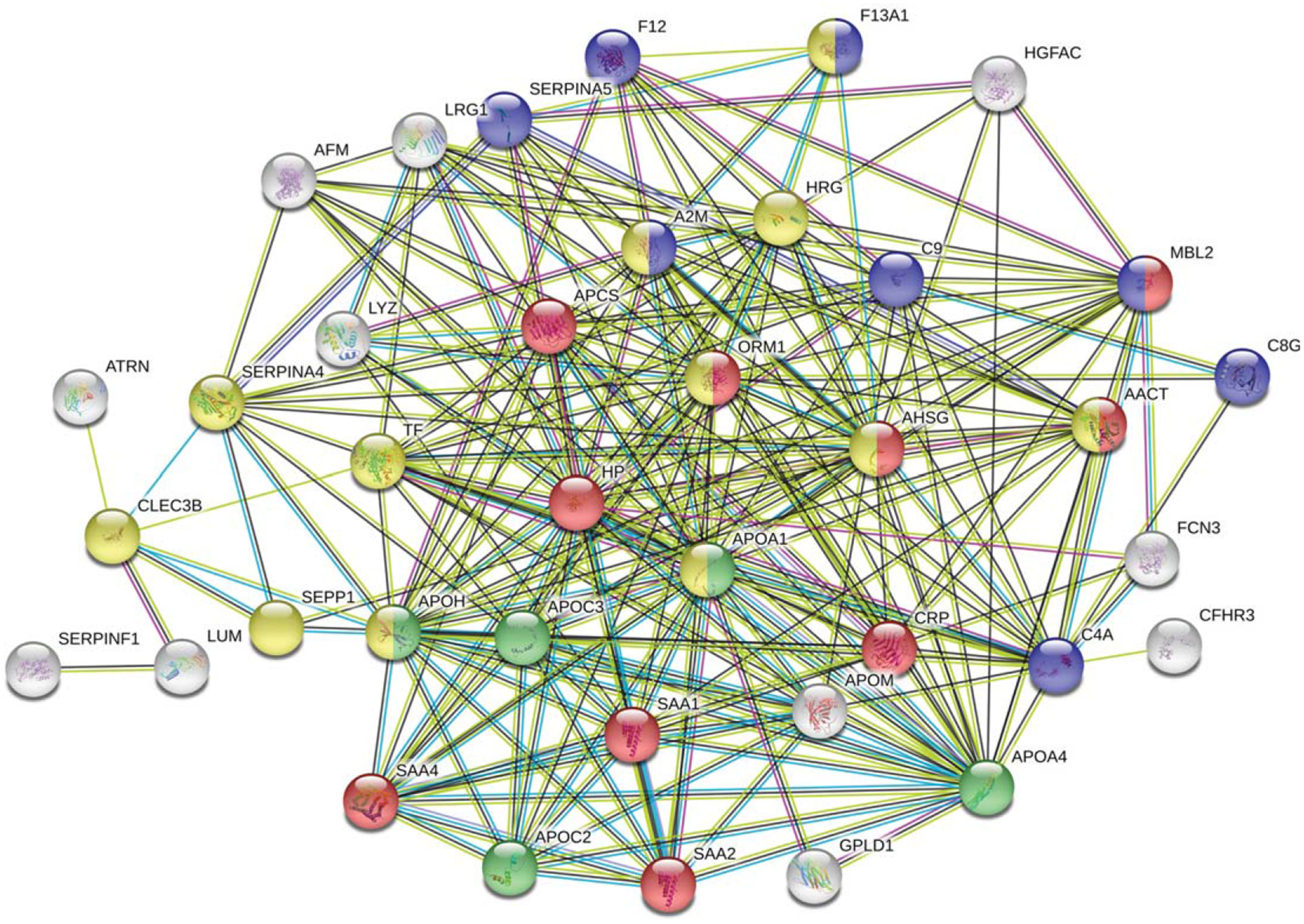
Functional enrichment in the networks of up/downregulated human plasma proteins from 18 AD patients using STRING. Results were obtained from two independent experiments. Biological Process (GO) of acute-phase response and acute inflammatory response (red), KEGG pathways of complement and coagulation cascades (blue), cholesterol metabolism (green), and Reactome pathways of platelet degranulation (yellow) were indicated on the plot.
Next, DAVID functional enrichment analysis was applied to up- and downregulated plasma proteins separately (Fig. 2). The identified plasma proteins were classified into different groups according to biological process (Fig. 2A), molecular function (Fig. 2B), and cell components (Fig. 2C). In the category of biological process, the plasma proteins from AD patients revealed enhanced, upregulated immune responses, whereas platelet degranulation and lipid transfer and metabolism were significantly downregulated (Fig. 2A). The top two enriched biological processes were acute-phase response (64% of regulated proteins were enriched in this process with p = 1.9E-19 (64%, 1.9E-19)) and inflammatory response (36%, 1.1E-04). Nine out of 14 upregulated proteins were involved in acute-phase response, including C-Reactive Protein (CRP), serum amyloid P-component (APCS), haptoglobin (HP), mannose-binding protein C (MBL2), alpha-1-acid glycoprotein 1 (ORM1), alpha-1-antichymotrypsin (SERPINA3), serum amyloid A-1 (SAA1), serum amyloid A-2 (SAA2), and serum amyloid A-4 (SAA4). Proteins involved in the inflammatory response included CRP, ORM1, SERPINA3, complement C4-A (C4A), and lysozyme C (LYZ). Other top enriched biological processes included the activation of the classic pathway of the complement cascade. Upregulated C4A, complement component C9 (C9) and MBL2 were enriched in this complement activation process.
Fig. 2.
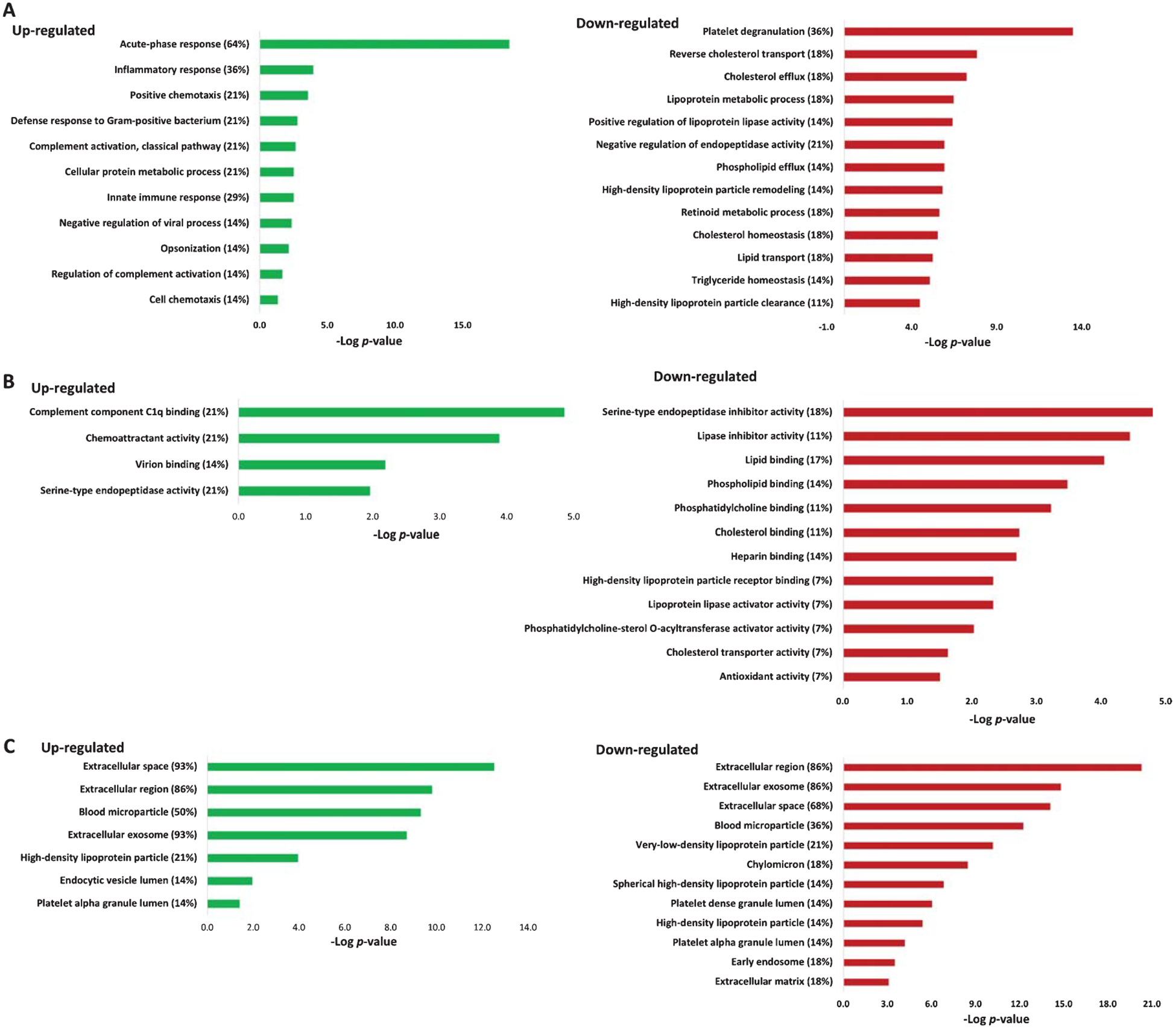
Gene ontology enrichment analysis of up- and downregulated plasma proteins in three categories: A) biological processes, B) molecular function, and C) cellular components. The percentages indicate the enriched proteins among all up- or downregulated proteins. The enrichment analysis was performed using DAVID.
The functional analysis of downregulated plasma proteins from AD patients showed that more than one third of the downregulated proteins were enriched in the biological process of platelet degranulation (36%, 3.7E-14), followed by lipid transport and metabolism including reverse cholesterol transport/cholesterol efflux (18%, 6.6E-08), lipoprotein metabolism (18%, 3.68E-07), and positive regulation of lipoprotein lipase activity (14%, 4.4E-07) (Fig. 2A). Proteins enriched in platelet degranulation included tetranectin (CLEC3B), alpha-2-HS-glycoprotein (AHSG), A2M, apolipoprotein A-I (APOA1), beta-2-glycoprotein 1 (APOH), coagulation factor XIII A chain (F13A1), histidine-rich glycoprotein (HRG), selenoprotein P (SELENOP, or SEPP1), kallistatin (SERPINA4), and serotransferrin (TF). Proteins enriched in the processes of lipid transfer and metabolism included APOA1, apolipoprotein A-IV (APOA4), apolipoprotein C-II (APOC2), apolipoprotein C-III (APOC3), apolipoprotein M (APOM), and beta-2-glycoprotein 1 (APOH). Other biological processes, such as phospholipid efflux, high-density lipoprotein (HDL) remodeling, cholesterol/triglyceride homeostasis, were also enriched.
The molecular function analysis showed that among the upregulated plasma proteins, 22% were associated with complement component C1q binding (including CRP, APCS, and C4A), followed by 22% in chemoattractant activity (SAA1, SAA2, and SAA4), virion binding, and serine-type endopeptidase activity (Fig. 2B). Whereas in the downregulated plasma proteins, 18% were associated with serine protease/proteinase inhibitor activity, including A2M, HRG, SERPINA4, plasma serine protease inhibitor (SERPINA5), and pigment epithelium-derived factor (SERPINF1), followed by lipase inhibitor activity and lipid binding (including APOA1, APOA4, APOC2, APOC3, and APOH).
The cellular component analysis of the up- and downregulated plasma proteins were enriched in the extracellular region and extracellular exosome, some were also enriched in blood microparticle, high-density lipoprotein particles as well as in platelet alpha granule lumen (Fig. 2C). Unlike upregulated proteins, downregulated proteins were enriched in very-low-density lipoprotein particles (21%) and chylomicron (18%).
The biological network of altered plasma proteins from AD patients reveals enriched IL-6 pathway
We have used the KEGG pathway analysis to examine our proteomic profiles, and we found that the complement and coagulation cascades pathway was enriched. When upregulated (p = 3.0E-4) and downregulated plasma proteins (p = 4.3E-6) were analyzed separately, or all plasma proteins were analyzed together (p = 8.0E-11), KEGG pathway analysis revealed the common complement and coagulation cascade pathway, with minimum FDR via Benjamini-Hochberg procedure (Benjamini) (Table 2).
Table 2.
KEGG and Reactome pathway analysis of up- and downregulated plasma proteins
| AD Plasma | Pathway Analysis | Terms | Accession | Gene Name | p | Benjamini |
|---|---|---|---|---|---|---|
| Up- & downregulated | KEGG-Pathway | |||||
| Complement and coagulation cascades | P01023 | Alpha-2-macroglobulin(A2M) | 8.00E-11 | 1.70E-09 | ||
| P00748 | Coagulation factor XII(F12) | |||||
| P00488 | Coagulation factor XIII A chain(F13A1) | |||||
| P0C0L4 | Complement C4A (Rodgers blood group)(C4A) | |||||
| P07360 | Complement C8 gamma chain(C8G) | |||||
| P02748 | Complement C9(C9) | |||||
| P11226 | Mannose binding lectin 2(MBL2) | |||||
| P05154 | Serpin family A member 5(SERPINA5) | |||||
| Upregulated | KEGG-Pathway | Complement and coagulation cascades | P0C0L4 | Complement C4A (Rodgers blood group)(C4A) | 3.00E-04 | 2.40E-03 |
| P02748 | Complement C9(C9) | |||||
| P11226 | Mannose binding lectin 2(MBL2) | |||||
| Reactome-Pathway R-HSA-977606 |
Regulation of complement cascade | P0C0L4 | Complement C4A (Rodgers blood group)(C4A) | 4.10E-04 | 7.30E-03 | |
| P02748 | Complement C9(C9) | |||||
| Q02985 | Complement factor H related 3(CFHR3) | |||||
| R-HSA-977225 | Amyloid fiber formation | P02743 | Amyloid P component, serum(APCS) | 4.90E-03 | 4.30E-02 | |
| P61626 | Lysozyme(LYZ) | |||||
| P0DJI8 | Serum amyloid A1(SAA1) | |||||
| Downregulated | KEGG-Pathway | Complement and coagulation cascades | P01023 | Alpha-2-macroglobulin(A2M) | 4.30E-06 | 7.30E-05 |
| P00748 | Coagulation factor XII(F12) | |||||
| P00488 | Coagulation factor XIII A chain(F13A1) | |||||
| P07360 | Complement C8 gamma chain(C8G) | |||||
| P05154 | Serpin family A member 5(SERPINA5) | |||||
| Vitamin digestion and absorption | P02647 | apolipoprotein A1(APOA1) | 6.30E-04 | 5.40E-03 | ||
| P06727 | apolipoprotein A4(APOA4) | |||||
| P43251 | biotinidase(BTD) | |||||
| Reactome-Pathway R-HSA-114608 |
Platelet degranulation | P05452 | C-type lectin domain family 3 member B(CLEC3B) | 1.60E-12 | 4.90E-11 | |
| P02765 | Alpha 2-HS glycoprotein(AHSG) | |||||
| P01023 | Alpha-2-macroglobulin(A2M) | |||||
| P02647 | Apolipoprotein A1(APOA1) | |||||
| P02749 | Apolipoprotein H(APOH) | |||||
| P00488 | Coagulation factor XIII A chain(F13A1) | |||||
| P04196 | Histidine rich glycoprotein(HRG) | |||||
| P49908 | Selenoprotein P(SELENOP) | |||||
| P29622 | Serpin family A member 4(SERPINA4) | |||||
| P02787 | Transferrin(TF) | |||||
| R-HSA-975634 | Retinoid metabolism and transport | P02647 | apolipoprotein A1(APOA1) | 1.60E-06 | 2.50E-05 | |
| P06727 | apolipoprotein A4(APOA4) | |||||
| P02655 | apolipoprotein C2(APOC2) | |||||
| P02656 | apolipoprotein C3(APOC3) | |||||
| O95445 | apolipoprotein M(APOM) |
Three regulators of complement activation, C4A, C9, and complement factor H-related protein 3 (CFHR3), were found upregulated. Reactome pathway analysis revealed 21% of the upregulated plasma proteins were enriched in regulation of complement cascade (p = 4.1E-4) and another 21% were enriched in amyloid fiber formation (p = 4.9E-3), a key pathological event of abnormal accumulation of amyloid. KEGG and Reactome pathway analysis of downregulated proteins showed that platelet degranulation (p = 1.6E-12), vitamin digestion and absorption (p = 6.3E-4), and retinoid metabolism and transport (p = 1.6E-6) were all enriched.
To investigate any changes in biological network, we uploaded the up- and downregulated plasma proteins from AD patients as the input list for generation of these networks using model canonical pathways from Metacore. Among the top eight most strongly connected networks, four were related to the IL-6 pathway which mainly involved SAA1, APCS, HP, IL-6, ACT, HP, STAT3, IL-6 receptor, and JAK2 (p = 5.1E-42, z Score ≥ 187) (Fig. 3), two networks were enriched in acute-phase response involving LRG, lysozyme, G-CSF, STAT3, STAT5, and JAK1 (p = 6.9E-46, z Score = 181) (Fig. 4), and one network was enriched in regulation of inflammatory response involving C4A, NF-κb, C3Ar, A2M receptor, and CR1.
Fig. 3.
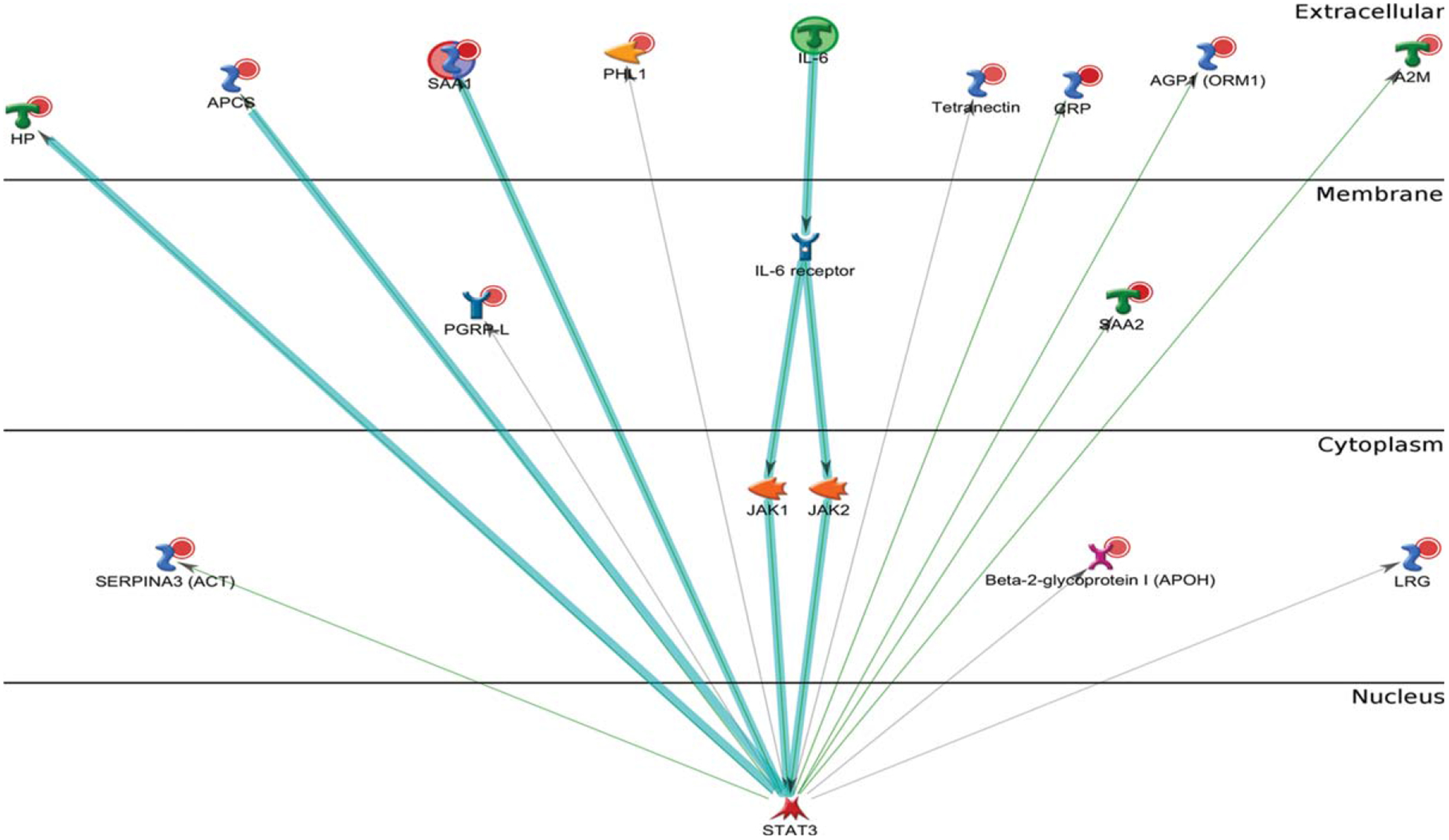
Metacore analysis reveals the top scored network of IL-6 pathway. Thick cyan lines indicate the fragments of canonical pathways. The acute-phase response containing IL-6 pathway was enriched.
Fig. 4.
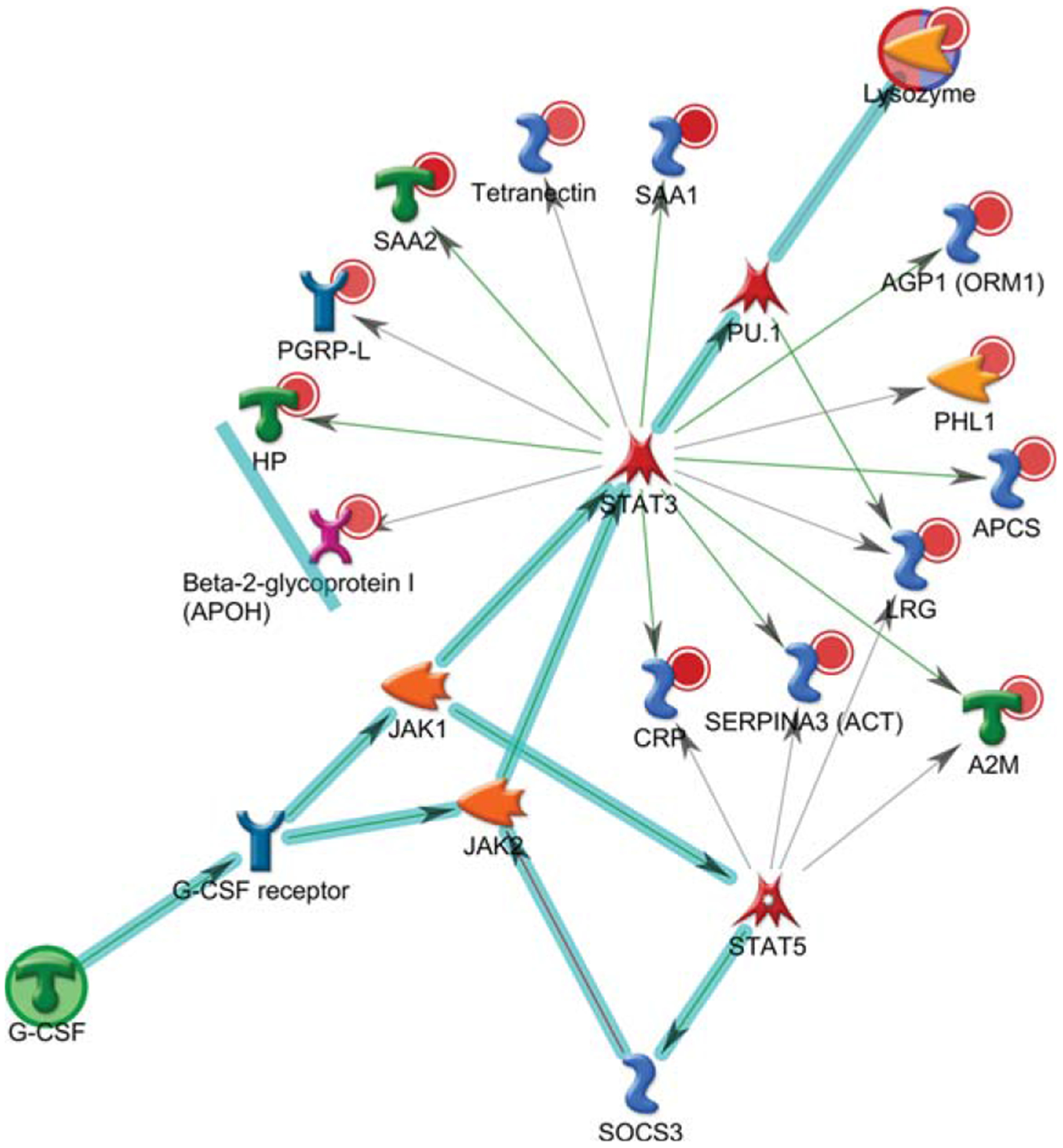
Metacore analysis reveals the top scored network of acute-phase response pathways. Thick cyan lines indicate the fragments of canonical pathways. The acute-phase response containing LRG, lysozyme, and G-CSF pathways was enriched.
Therefore, the IL-6 signaling represents one of the major pathways altered in AD patients.
We next examined whether those proteins were also implicated in other diseases besides AD. Further enrichment analysis using Metacore showed that the downregulated plasma proteins from AD (p = 1.2E-25) were enriched in disease markers related to cerebrovascular disorders, and those upregulated proteins (p = 1.7E-18) were enriched in markers related to macular degeneration (Table 3). Collectively, these up- or downregulated proteins are involved in macular degeneration or cerebrovascular disorders, which include a spectrum of pathological conditions of impaired blood flow in the eye and brain.
Table 3.
Enrichment of the up- and downregulated plasma proteins related to other diseases
| Differentially regulated | Diseases | p | Proteins enriched |
|---|---|---|---|
| Down- | Cerebrovascular Disorders | 1.2E-25 | A2M, AFM, AHSG, APOA1, APOA4, APOC2, APOC3, TF, APOH, C8G, CNDP1, Coagulation factor XII (F12), Coagulation factor XIII A, Ficolin, SERPINF1, SERPINA4 |
| Up- | Macular Degeneration | 1.7E-18 | C4A, C9, CRP, CFHR3, HP, MBL2 |
Proteomic profiling of AD postmortem brain tissue reveals differentially regulated IL-6 signaling
Our previous studies have reported that enhanced inflammation and complement activation were found in the postmortem brain tissues from five AD patients, with complement and coagulation cascade pathway being significantly enriched in two out of five AD subjects (p < 0.05) [61]. To determine the relevance of proteomic profiles between plasma and brain tissue, all differentially regulated plasma proteins identified from 18 AD patients were compared to the proteins identified from three brain regions of the postmortem brain tissues from five AD patients, including the superior frontal cortex (SF), inferior frontal cortex (IF), and cerebellum (CRLM); CRLM has the least AD pathology compared to SF and IF regions. As expected, levels of Aβ40, Aβ42, and pTau181 were increased in postmortem brain homogenates from AD patients, compared to those from control subjects (Supplementary Figure 1). Results from the comparative enrichment analysis revealed commonly enriched GO processes between up/downregulated proteins in plasma and those from three brain regions of AD patients (Fig. 5A). Besides platelet degranulation, most proteins were related to immune or inflammatory response. Positive regulation of myelination and cellular oxidant detoxification were also commonly enriched. Our analysis also revealed the commonly enriched GO molecular functions of the up/downregulated plasma and brain proteins. The top five enriched molecular activities of up and downregulated plasma proteins were highly similar to those from IF and SF brain regions but not CRLM (Fig. 5B). Other molecular activities of differentially regulated proteins between plasma and three brain regions were similar, such as hemoglobin binding, antioxidant activity and lipid binding.
Fig. 5.
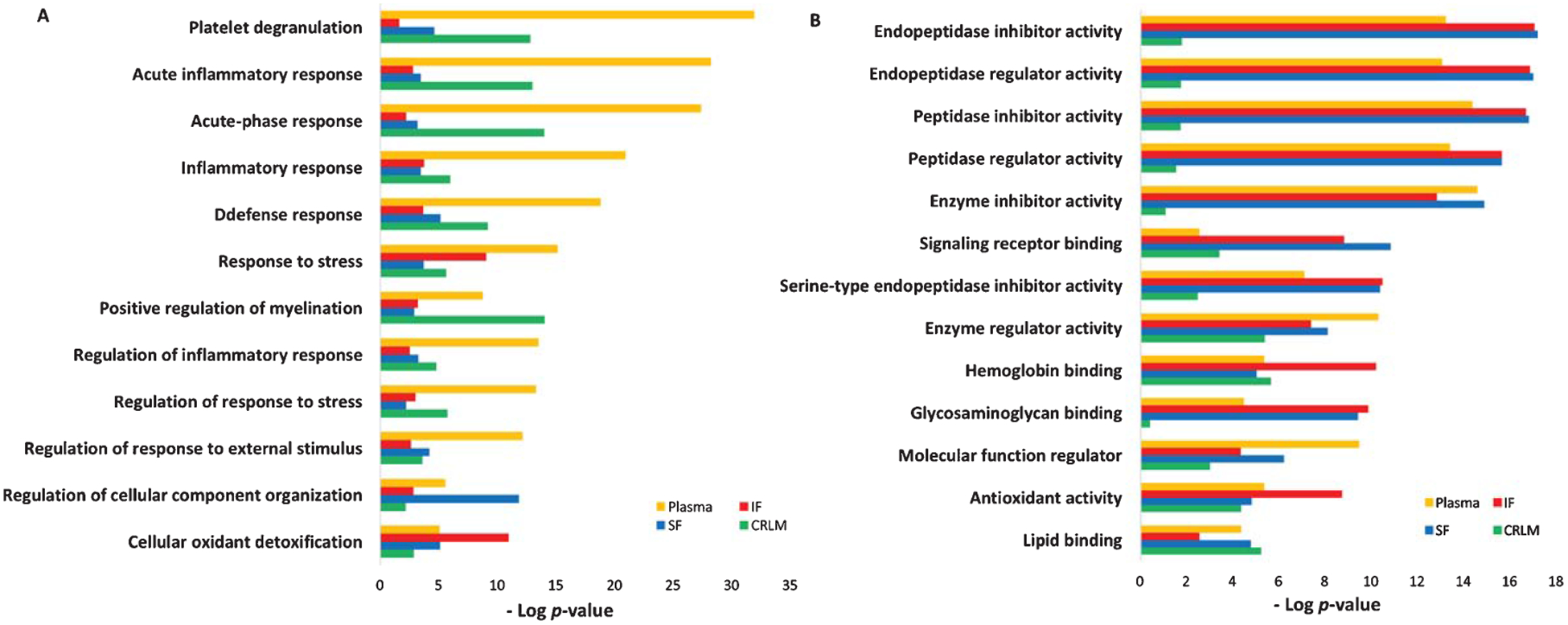
Comparison of GO functional enrichment analysis between up/downregulated plasma proteins from 18 AD patients and those from three brain regions (SF, IF, and CRLM) of 5 AD patients (p < 0.05). A) GO process. B) GO molecular function (SF, superior frontal cortex; IF, inferior frontal cortex; CRLM, cerebellum).
The enriched process networks of up- and downregulated proteins from plasma and two brain regions (IF and SF) were compared; CRLM was not included as it has the least AD pathology compared to SF and IF regions. Our results showed that inflammation/complement system and inflammation/IL-6 signaling were highly enriched and followed by immune response/phagocytosis (Fig. 6). Plasma proteins enriched in the complement system and IL-6 signaling included CRP, C4a, complement component C8 γ chain (C8γ), MBL2, Ficolin/H-Ficolin, C9, ficolin-3 (FHR-3), ORM1, SAA2, A2M, APCS, HP, ACT, SAA4, and SAA1. Proteins from plasma and SF brain region were enriched in blood coagulation (Fig. 6).
Fig. 6.
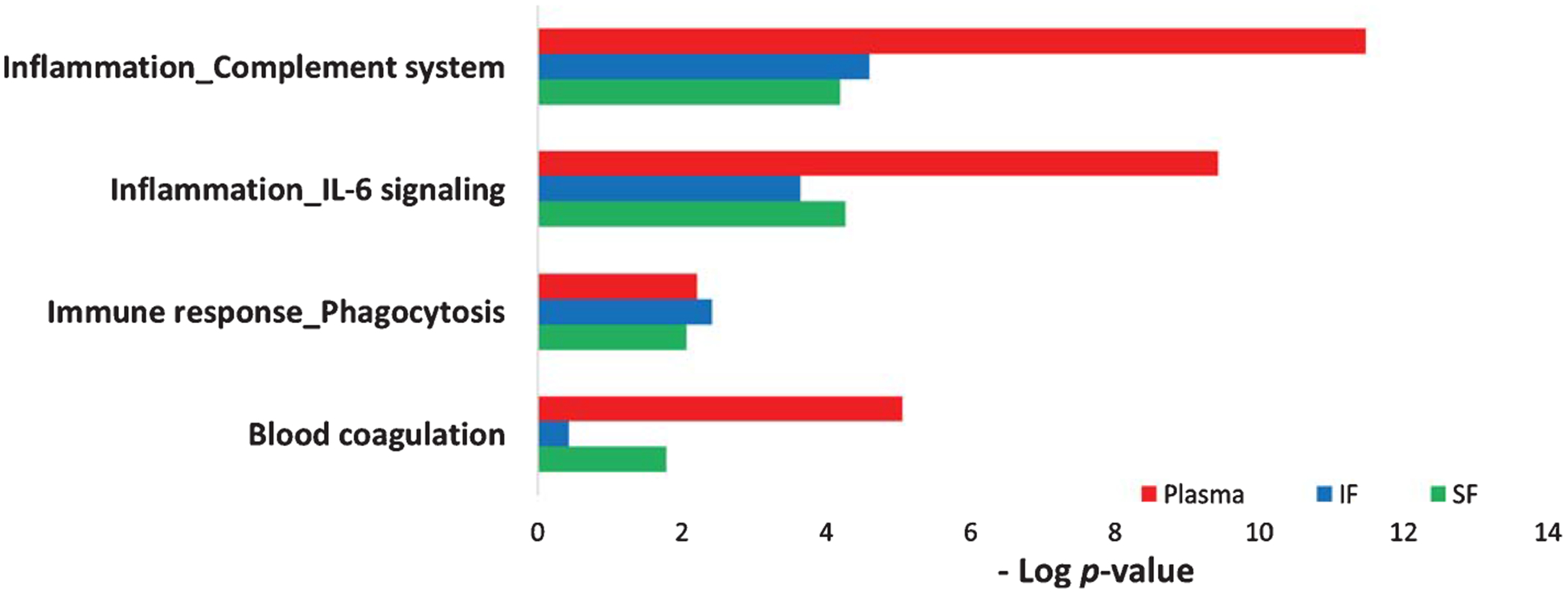
Comparison of the enriched process networks between plasma and brain proteins. Up/downregulated plasma proteins from 18 AD patients were compared to those from two AD brain regions (SF and IF) of five AD patients (p < 0.05). (SF, superior frontal cortex; IF, inferior frontal cortex). Plasma proteins enriched in the inflammation/complement system include CRP, C4a, C8γ, MBL2, Ficolin/H-Ficolin, C9, and FHR-3; IL-6 signal components include ORM1, SAA2, A2M, CRP, APCS, HP, ACT, SAA4, and SAA1; immune response/phagocytosis system include SAA2, CRP, HDL proteins, APOC3, MBL2; blood coagulation components include SAA2, A2M, Coagulation factor XII, Protein C inhibitor, SAA1, and Coagulation factor XIII A.
Our studies illustrated a list of differentially regulated proteins that were commonly presented in both plasma and brain regions (Table 4). Haptoglobin (HP) was commonly upregulated in plasma and IF and SF, but downregulated in the CRLM region. Both A2M and TF were commonly downregulated in plasma and CRLM. Since HP and A2M were among the plasma proteins enriched in the IL-6 signaling pathway, it was apparent that IL-6 signaling was affected in both plasma and three brain regions from patients with AD.
Table 4.
The common up- and downregulated plasma and brain proteins from AD subjects
| Samples | Accession | Description | Ratio |
|---|---|---|---|
| Plasma | P00738 | Haptoglobin | 1.19 |
| Brain IF | 1.82 | ||
| Brain SF | 1.94 | ||
| Brain CRLM | 0.63 | ||
| Plasma | P01023 | Alpha-2-macroglobulin (A2M) | 0.79 |
| Brain CRLM | 0.70 | ||
| Plasma | P02787 | Serotransferrin (TF) | 0.79 |
| Brain CRLM | 0.74 |
The up- and downregulated brain proteins were from three brain areas in at least 2 out of 5 AD patients when comparing to the average levels of proteins from 5 control subjects. Up- and downregulated plasma proteins in AD group (n = 18) were compared to the control group (n = 18); with p < 0.05, two-tailed Student’s T test from two independent studies. IF, inferior frontal; SF, superior frontal; CRLM, cerebellum.
To confirm the MS-based quantitative proteomics, we analyzed two key proteins IL-6 and SAA in plasma from AD and control subjects by ELISA. Levels of IL-6 and SAA showed a significant (p < 0.05) increase at 5.3-fold and 3.7-fold in plasma from AD patients, compared to those from control subjects (Fig. 7). The detection of significantly upregulated plasma IL-6 in AD patients by ELISA is consistent with the activation of IL-6 signaling in plasma samples from AD patients, which was demonstrated by MS-based proteomic profiling.
Fig. 7.
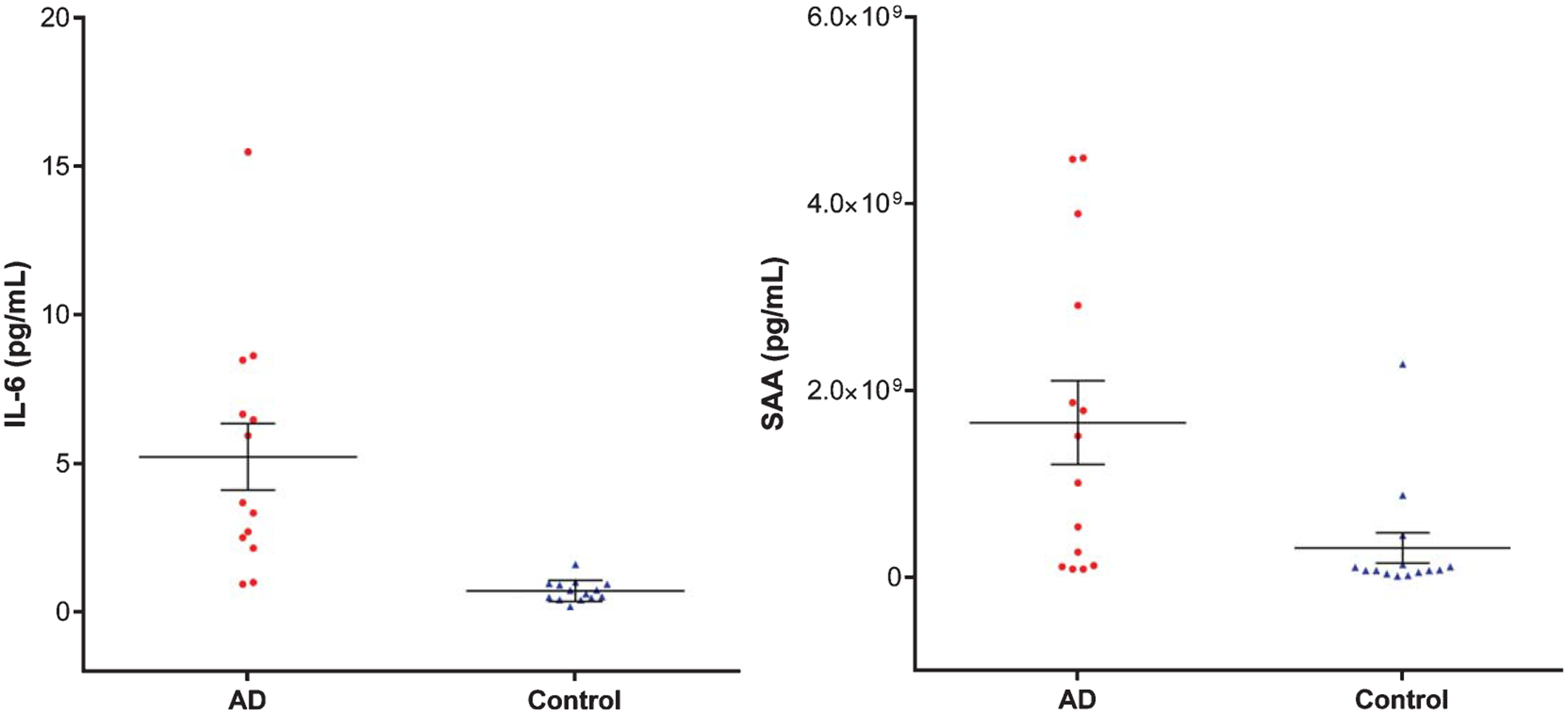
ELISA-based quantification of IL-6 and SAA in plasma from AD and control subjects. The standard error of means (short lines) and means (long middle line) are illustrated for each group. The differences between AD and control subjects are statistically significant (p < 0.05).
The increase of SAA quantified by ELISA (Fig. 7) confirmed MS-based quantification of SAA1, SAA2, and SAA4, which were increased in AD subjects (Table 1). Taken together, this study demonstrated the advantage of MS over immunoassay in its ability to quantitatively differentiate SAA-1 and SAA-2 from SAA4 (Table 1).
DISCUSSION
In this study we compared plasma samples with brain tissue, a design which illustrates an effective approach to identify candidate biomarkers for future validation in a larger number of plasma and postmortem brain samples. Given a lack of a single fluidic biomarkers for AD diagnosis, a composite protein profile may have greater sensitivity and specificity to meet the criteria for an AD biomarker. Furthermore, candidate plasma biomarkers can be easily applied in clinical settings to predict the disease state if they are validated in postmortem brain tissue from AD patients.
Blood-brain barrier leakage and vascular dysfunction in AD
In the current study, we found that several proteins involved in amyloid fiber formation were upregulated in AD. Blood biomarkers potentially reflect pathological alteration in brains partly due to a leaky blood-brain barrier (BBB) [64]. Aβ deposits are found in damaged brain vessels that are a characteristic feature of cerebral amyloid angiopathy (CAA). If this occurs at the early stage of AD, it could induce CAA [65]. The accumulation of Aβ deposits likely disrupts the vessel wall, sometimes leading to microaneurysm formation and fibrinoid necrosis [66]. Since the majority of AD patients have coexisting CAA, and one study reported its presence in up to 80% to 90% of AD patients [67], pathological proteins in brains of AD patients are likely enriched in blood compared to those from control subjects.
This study revealed alteration in platelet degranulation or the release of granule contents [68], which is relevant to Aβ-induced clotting after minor BBB leakage activates blood platelets to repair any damage [69]. Activation of platelets at the lesion sites leads to the healing cascade [70], and deficiency in this step causes subsequent inflammation processes. Vascular lesions increase during AD progression, and platelets gradually become dysfunctional in repairing these lesions. In this study, we found dysregulated platelet function based on proteomic analysis of both plasma and postmortem brain tissues from AD patients. We also identified a number of differentially regulated proteins involved in platelet degranulation process such as A2M and TF (Table 2).
IL-6 inflammation pathway in AD
KEGG and Reactome pathway analyses of our proteomic dataset revealed alteration of complement and coagulation cascades, which is consistent with previous plasma proteomic studies [40, 53, 54]. The complement and coagulation systems shared certain activators and inhibitors and can be reciprocally activated, and they have been linked with AD pathogenesis [71]. Several studies have reported that complement activation is a key component of neuroinflammation process in AD [72–74]. The activation of complement systems in AD promotes an Aβ-induced neuroinflammatory response in microglia [75] despite its protective effects toward Aβ clearance. Using iTRAQ-based comparative proteomics analysis, several complement proteins, such as C4b-binding α protein chain, complement factor B, and complement factor H proteins were found up- and downregulated in AD plasma [40, 53, 54]. In our study, several regulators of complement cascade proteins including C4A, C9, and CFHR3, were found upregulated in plasma from ADs in our study. This is consistent with a previous genetic report of significant increases in the number of copies for both C4A and C4B in AD patients compared with healthy controls [76]. Complement factor H regulates the formation of C3 and C5 convertase and is present in amyloid plaques [77]. The elevated level in plasma correlated with hippocampal metabolic abnormalities in AD patients [49].
In brain samples, the pro-inflammatory cytokines and oxygen radicals released by glial cells are neurotoxic and cause tissue damage and neuronal death [78]. Some studies suggest that inflammatory cytokines such as IL-1β, IL-6, IL-18, and their related receptors were increased in the AD brain [79, 80], other studies suggest that inflammatory markers (including IL-6) were not found to be significantly different among early- and late-onset AD subjects and normal controls, but were found to be associated with age [81]. In our age matched plasma samples, we have identified upregulated IL-6 inflammation signaling pathway in plasma samples from AD patients, supporting the activation of inflammatory processes in brains. Among them, IL-6 is a key player during the transition between acute and chronic inflammation and a main stimulator of the production of acute phase proteins [82]. IL-6 production in cultured glial cells can be triggered by adding Aβ peptides [83]. IL-6 enhances neuronal damage induced by Aβ in cultured rat cortical neurons [84], and facilitates hyperphos-phorylation of tau in neurons [85]. Genetic analysis of polymorphisms in specific alleles of IL-6 revealed an association between IL-6 haplotype-tagging single nucleotide polymorphisms (SNPs) and reduced risk of late-onset AD [86].
Our proteomic profiling revealed that elevated IL-6 and several important proteins associated with IL-6 signaling were upregulated, suggesting that IL-6 plays a pivotal role in brain inflammation and AD pathogenesis. IL-6 activates Janus Kinases (JAK) and subsequently a number of proteins including Signal Transducers and Activators of Transcription (STAT) factors like STAT3. In microglial cell lines, JAK/STAT3 signaling mediates neuroinflammatory responses induced by Aβ42 and complement protein C5a. Blocking this pathway reduces the expression of TNF-α, IL-1β, and IL-6 [75]. In an APP/Presenilin1 (PS1) transgenic AD mouse model, knocking out STAT3 caused decreased activation of pro-inflammatory cytokines, reduced Aβ levels and plaque burden, upregulated amyloid clearance pathways, and even improved spatial learning and reduced memory decline [87]. STAT3 is one of the main regulators of the lysozyme pathway. Lysozyme plays an important role in the innate immune system and has been shown to prevent Aβ aggregation in vitro [88]. As increased lysozyme levels have been found in CSF from AD patients, we found increased plasma levels of lysozyme in AD patients, indicating the activation of G-CSF and lysozyme pathways that could be as a compensatory response to Aβ aggregation. Therefore, our human plasma proteomic results suggest that levels of STAT3 and lysozyme could be potential indicators for pathological status of AD in brains.
SAA and immune response in AD
IL-6 as well as IL-1 and TNF-α regulate SAA-1 and SAA-2. Similar to another acute phase protein CRP, SAA1, and SAA2 increase within hours after inflammatory stimulus [89]. While SAA1 and SAA2 are inducible during acute-phase response, SAA4 is constitutively expressed in a variety of tissues and cells [90]. SAA1, 2, and 4 are the main components of SAA family [91]; SAA3, which was not analyzed here, is a pseudogene and detected in human gland epithelial cell lines [90]. Our LC-MS/MS analysis combines quadruple precursor ion selection with high-resolution and accurate mass, which allows us to identify proteins with high specificity and investigate more precisely the MS/MS spectra generated from the plasma samples of AD patients. In the case of SAA1 and SAA2, which are small proteins with 104 amino acids in length, they are >90% identical to each other [89]. Using LC-MS/MS analysis, we were able to specifically discriminate between SAA1 and SAA2 and identify different isoforms of SAA, including SAA1, SAA2 and SAA4. We found that SAA1 and SAA2 were upregulated to a greater extent as compared with SAA4 (Table 1). This shows the high specificity of the mass-based methods over antibody-based immunoassays. Furthermore, quantitative LC-MS/MS produces consistent outcomes with good reproducibility from multiple experiments (Supplementary Table 1).
Elevated SAA has been reported in several chronic diseases, including amyloidosis, atherosclerosis, and rheumatoid arthritis [92], and expression of SAA was identified in AD brains [93, 94] and predominantly localized to neuritic plaques, myelin sheaths, and axonal membrane [95]. In CSF, the SAA concentration is much higher in samples from AD subjects than in those from controls [93]. Besides its role in recruiting immune cells to inflammatory sites, SAA directly regulates lipoprotein metabolism in the plasma, transports cholesterol to the liver for secretion into the bile, and induces CSF-HDL remodeling which may impair Aβ clearance [96].
Other apolipoproteins for lipid transport and their dysfunction in AD
In our study, several Apolipoproteins, such as APOA1, APOA4, APOC2, and APOC3, were found downregulated in the plasma of AD patients. Apolipoproteins transport lipids through the circulatory system and play a crucial role in neuronal development and maintenance of synaptic plasticity. Several dysregulated apolipoproteins and lipid metabolism have been reported in the previous proteomic studies, such as downregulated APOA1, APOA2, and APOB in plasma or serum from AD subjects as compared with the controls [97]. Another report suggests that the expression of APOA1, APOC3, and APOA4 is associated with risk of AD, and low serum levels of these three apolipoproteins are correlated with development of AD based on CDR scores [98]. The apoA1, apoA4, and apoC3 genes are closely linked in the human genome, and the apoA1-apoA4-apoC3 gene cluster may synergistically affect lipid profiles related to AD [98]. ApoE is another protein altered in AD plasma samples [44]. The APOE ε4 allele is the strongest genetic risk factor for the development of late-onset AD, and multiple regulatory elements in the region of APOE locus influence expression levels of APOE [99]. The levels of regulatory element methylation in the extended TOMM40-apoE-apoC2 region of the long arm of chromosome 19 were correlated with AD status [100].
APOC3, a component of very low-density lipoprotein particles (VLDL), binds Aβ, decreases in plasma from AD patients, and negatively correlates with MMSE scores [101]. APOA4 has anti-oxidant, anti-inflammatory, and anti-atherosclerotic actions in vivo [102], and its deficiency increases Aβ deposition and cognitive damage in an AD animal model [103]. It appears that alteration of plasma lipid profile (e.g., hyperlipidemia) may cause subtle changes in brain lipid metabolism and composition [104]. Our results suggest these plasma apolipoproteins, as they can be measured directly, can potentially be used as biomarkers to evaluate status of brain lipid profiles related to AD.
Oxidative markers specific to AD
Neuroinflammation and oxidative stress are closely associated, and oxidative stress is a classic aging marker. Oxidative biomarkers in peripheral system for AD diagnosis is difficult to identify, even though they play a significant role in the pathogenesis of AD [105]. In this study, we found several proteins with anti-oxidation function downregulated in plasma from AD patients.
Haptoglobin is an acute-phase protein released in the peripheral blood as a marker for inflammation, and it carries antioxidant and anti-inflammatory activities. In this study, we observed that HP was commonly elevated in both plasma and IF and SF brain regions from AD subjects, consistent with elevated HP levels among AD patients compared to healthy controls observed in previous findings [106, 107].
An essential trace element for the antioxidant defense is Selenium (Se) and its binding protein selenoprotein. Selenoprotein protects cells against oxidative stress damage, and Selenoprotein P is a heparin-binding glycoprotein transporting Se to neurons, functioning as an antioxidant and a contributor to regulation and interaction with redox-active metals (copper, iron, and mercury). Decrease in plasma selenium over time is associated with cognitive decline and AD, as compared with healthy controls [108]. Selenoprotein P plays various roles in cytoskeleton assembly and disassembly with misfolded proteins (e.g., hyperphosphorylated tau-protein and Aβ) [109].
A similar iron-binding protein, Transferrin (TF, or serotransferrin), transports iron from sites of absorption and heme degradation to those of storage and utilization, thus maintaining a relatively non-reactive state to prevent iron from stimulating oxygen radical formation as an important anti-oxidant defense mechanism [110]. While decreased TF levels were found across a wide range of disorders including common infection, inflammation, liver diseases, cancer, and other chronic disease states, lower levels of TF were also found in the blood of AD patients [111] and in the white matter of various cortical regions [112], compared to control subjects. Consistent with these early reports, we found decreased levels of TF in AD plasma and brain samples in this study.
Anti-oxidation agents like Kallistatin have been administered to reduce cardiovascular damage [113], but their role in AD pathogenesis is not clear. Similar to TF, reduced Kallistatin was found in the serum of patients with diabetes, hypertension, or cancer [114]. Experimentally, high H2O2 concentration inhibited Kallistatin expression, while low H2O2 levels induced its expression [114]. In this study, we found that the plasma level of Kallistatin was downregulated among AD patients. Plasma antioxidants like TF and Kallistatin may have neuroprotective effects against neurodegenerative disease like AD, where enhanced oxidative stress and lowered antioxidant defense damage organelles such as the mitochondria and impair brain function. Therefore, biomarkers for oxidative stress could be accompanying components for predicting AD status and ongoing loss of synapses and neuronal death.
Conclusions
The current study presents a novel approach to discovering concordant biomarker profiles indicative of specific pathological processes in both peripheral and central nervous systems. Since the BBB is disrupted in AD, specific proteins that pass between the brain and the bloodstream can be detected and quantified to predict brain pathology. In this study, a large body of candidate proteins in plasma which reflect certain aspects of the disease status of AD were revealed and could be further investigated and validated as peripheral biomarkers for AD. The results suggest that specific plasma proteins in the complement coagulation cascade and IL-6/JAK/STAT3 signaling can be candidate biomarkers to indicate neuroinflammation of AD. Additional proteins involved in lipid processes, cerebral vascular dysfunction, and oxidative stress can also be explored as candidate biomarkers for AD diagnosis.
Supplementary Material
ACKNOWLEDGMENTS
We thank Drs. Stephen Wong, Clara Velazquez Sanchez, Abby Gelb, Jianting Sheng, and Yuliang Cao for helpful discussions, and Sarah Daley for editorial assistance. This study was supported by the award I21BX002215 and IO1 BX003527 from the Biomedical Laboratory Research and Development Service of the Veterans Affairs Office of Research and Development, RF1AG063913 from National Institute of Aging, and the Cure Alzheimer’s Fund (WX). The views expressed in this article are those of the authors and do not represent the views of the US Department of Veterans Affairs or the US Government.
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/20-0110r1).
Footnotes
SUPPLEMENTARY MATERIAL
The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JAD-200110.
REFERENCES
- [1].Selkoe DJ (2004) Alzheimer disease: Mechanistic understanding predicts novel therapies. Ann Intern Med 140, 627–638. [DOI] [PubMed] [Google Scholar]
- [2].Xia W (2001) Amyloid metabolism and secretases in Alzheimer’s disease. Curr Neurol Neurosci Rep 1, 422–427. [DOI] [PubMed] [Google Scholar]
- [3].Selkoe DJ, Hardy J (2016) The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med 8, 595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Shi Y, Holtzman DM (2018) Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight. Nat Rev Immunol 18, 759–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Heppner FL, Ransohoff RM, Becher B (2015) Immune attack: The role of inflammation in Alzheimer disease. Nat Rev Neurosci 16, 358–372. [DOI] [PubMed] [Google Scholar]
- [6].Castellano JM, Kim J, Stewart FR, Jiang H, DeMattos RB, Patterson BW, Fagan AM, Morris JC, Mawuenyega KG, Cruchaga C, Goate AM, Bales KR, Paul SM, Bateman RJ, Holtzman DM (2011) Human apoE isoforms differentially regulate brain amyloid-beta peptide clearance. Sci Transl Med 3, 89ra57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Xia W, Zhang J, Kholodenko D, Citron M, Podlisny MB, Teplow DB, Haass C, Seubert P, Koo EH, Selkoe DJ (1997) Enhanced production and oligomerization of the 42-residue amyloid beta-protein by Chinese hamster ovary cells stably expressing mutant presenilins. J Biol Chem 272, 7977–7982. [DOI] [PubMed] [Google Scholar]
- [8].Xia W, Yang T, Shankar G, Smith IM, Shen Y, Walsh DM, Selkoe DJ (2009) A specific enzyme-linked immunosorbent assay for measuring beta-amyloid protein oligomers in human plasma and brain tissue of patients with Alzheimer disease. Arch Neurol 66, 190–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Klein WL (2002) Abeta toxicity in Alzheimer’s disease: Globular oligomers (ADDLs) as new vaccine and drug targets. Neurochem Int 41, 345–352. [DOI] [PubMed] [Google Scholar]
- [10].Walsh DM, Selkoe DJ (2004) Deciphering the molecular basis of memory failure in Alzheimer’s disease. Neuron 44, 181–193. [DOI] [PubMed] [Google Scholar]
- [11].Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ (2002) Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416, 535–539. [DOI] [PubMed] [Google Scholar]
- [12].Xia W (2010) Brain amyloid beta protein and memory disruption in Alzheimer’s disease. Neuropsychiatr Dis Treat 6, 605–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Bancher C, Brunner C, Lassmann H, Budka H, Jellinger K, Wiche G, Seitelberger F, Grundke-Iqbal I, Iqbal K, Wisniewski HM (1989) Accumulation of abnormally phosphorylated tau precedes the formation of neurofibrillary tangles in Alzheimer’s disease. Brain Res 477, 90–99. [DOI] [PubMed] [Google Scholar]
- [14].Preuss U, Mandelkow EM (1998) Mitotic phosphorylation of tau protein in neuronal cell lines resembles phosphorylation in Alzheimer’s disease. Eur J Cell Biol 76, 176–184. [DOI] [PubMed] [Google Scholar]
- [15].Vincent I, Zheng JH, Dickson DW, Kress Y, Davies P (1998) Mitotic phosphoepitopes precede paired helical filaments in Alzheimer’s disease. Neurobiol Aging 19, 287–296. [DOI] [PubMed] [Google Scholar]
- [16].Lu KP, Liou YC, Vincent I (2003) Proline-directed phosphorylation and isomerization in mitotic regulation and in Alzheimer’s disease. BioEssays 25, 174–181. [DOI] [PubMed] [Google Scholar]
- [17].Lee VM, Balin BJ, Otvos L Jr., Trojanowski JQ (1991) A68: A major subunit of paired helical filaments and derivatized forms of normal Tau. Science 251, 675–678. [DOI] [PubMed] [Google Scholar]
- [18].Goedert M, Spillantini MG, Cairns NJ, Crowther RA (1992) Tau proteins of Alzheimer paired helical filaments: Abnormal phosphorylation of all six brain isoforms. Neuron 8, 159–168. [DOI] [PubMed] [Google Scholar]
- [19].Greenberg SG, Davies P, Schein JD, Binder LI (1992) Hydrofluoric acid-treated tau PHF proteins display the same biochemical properties as normal tau. J Biol Chem 267, 564–569. [PubMed] [Google Scholar]
- [20].Matsuo ES, Shin RW, Billingsley ML, Van deVoorde A, O’Connor M, Trojanowski JQ, Lee VM (1994) Biopsy-derived adult human brain tau is phosphorylated at many of the same sites as Alzheimer’s disease paired helical filament tau. Neuron 13, 989–1002. [DOI] [PubMed] [Google Scholar]
- [21].Fagan AM, Roe CM, Xiong C, Mintun MA, Morris JC, Holtzman DM (2007) Cerebrospinal fluid tau/beta amyloid(42) ratio as a prediction of cognitive decline in nondemented older adults. Arch Neurol 64, 343–349. [DOI] [PubMed] [Google Scholar]
- [22].Hansson O, Zetterberg H, Buchhave P, Londos E, Blennow K, Minthon L (2006) Association between CSF biomarkers and incipient Alzheimer’s disease in patients with mild cognitive impairment: A follow-up study. Lancet Neurol 5, 228–234. [DOI] [PubMed] [Google Scholar]
- [23].Shaw LM, Vanderstichele H, Knapik-Czajka M, Figurski M, Coart E, Blennow K, Soares H, Simon AJ, Lewczuk P, Dean RA, Siemers E, Potter W, Lee VM, Trojanowski JQ (2011) Qualification of the analytical and clinical performance of CSF biomarker analyses in ADNI. Acta Neuropathol 121, 597–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Nakamura A, Kaneko N, Villemagne VL, Kato T, Doecke J, Dore V, Fowler C, Li QX, Martins R, Rowe C, Tomita T, Matsuzaki K, Ishii K, Ishii K, Arahata Y, Iwamoto S, Ito K, Tanaka K, Masters CL, Yanagisawa K (2018) High performance plasma amyloid-beta biomarkers for Alzheimer’s disease. Nature 554, 249–254. [DOI] [PubMed] [Google Scholar]
- [25].Teunissen CE, Chiu MJ, Yang CC, Yang SY, Scheltens P, Zetterberg H, Blennow K (2018) Plasma amyloid-beta (Abeta42) correlates with cerebrospinal fluid Abeta42 in Alzheimer’s disease. J Alzheimers Dis 62, 1857–1863. [DOI] [PubMed] [Google Scholar]
- [26].Shahpasand-Kroner H, Klafki HW, Bauer C, Schuchhardt J, Huttenrauch M, Stazi M, Bouter C, Wirths O, Vogelgsang J, Wiltfang J (2018) A two-step immunoassay for the simultaneous assessment of Abeta38, Abeta40 and Abeta42 in human blood plasma supports the Abeta42/Abeta40 ratio as a promising biomarker candidate of Alzheimer’s disease. Alzheimers Res Ther 10, 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Hanon O, Vidal JS, Lehmann S, Bombois S, Allinquant B, Treluyer JM, Gele P, Delmaire C, Blanc F, Mangin JF, Buee L, Touchon J, Hugon J, Vellas B, Galbrun E, Benetos A, Berrut G, Paillaud E, Wallon D, Castelnovo G, Volpe-Gillot L, Paccalin M, Robert PH, Godefroy O, Dantoine T, Camus V, Belmin J, Vandel P, Novella JL, Duron E, Rigaud AS, Schraen-Maschke S, Gabelle A (2018) Plasma amyloid levels within the Alzheimer’s process and correlations with central biomarkers. Alzheimers Dement 14, 858–868. [DOI] [PubMed] [Google Scholar]
- [28].Ferreri AJ, Illerhaus G, Zucca E, Cavalli F (2010) Flows and flaws in primary central nervous system lymphoma. Nat Rev Clin Oncol 7, doi: 10.1038/nrclinonc.2010.1039-c1031; author reply doi:1010:1038/nrclinonc.2010.1039-c1032. [DOI] [PubMed] [Google Scholar]
- [29].Nabers A, Perna L, Lange J, Mons U, Schartner J, Guldenhaupt J, Saum KU, Janelidze S, Holleczek B, Rujescu D, Hansson O, Gerwert K, Brenner H (2018) Amyloid blood biomarker detects Alzheimer’s disease. EMBO Mol Med 10, e8763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Janelidze S, Mattsson N, Palmqvist S, Smith R, Beach TG, Serrano GE, Chai X, Proctor NK, Eichenlaub U, Zetterberg H, Blennow K, Reiman EM, Stomrud E, Dage JL, Hansson O (2020) Plasma P-tau181 in Alzheimer’s disease: Relationship to other biomarkers, differential diagnosis, neuropathology and longitudinal progression to Alzheimer’s dementia. Nat Med 26, 379–386. [DOI] [PubMed] [Google Scholar]
- [31].Thijssen EH, La Joie R, Wolf A, Strom A, Wang P, Iaccarino L, Bourakova V, Cobigo Y, Heuer H, Spina S, VandeVrede L, Chai X, Proctor NK, Airey DC, Shcherbinin S, Duggan Evans C, Sims JR, Zetterberg H, Blennow K, Karydas AM, Teunissen CE, Kramer JH, Grinberg LT, Seeley WW, Rosen H, Boeve BF, Miller BL, Rabinovici GD, Dage JL, Rojas JC, Boxer AL; Advancing Research and Treatment for Frontotemporal Lobar Degeneration (ARTFL) investigators (2020) Diagnostic value of plasma phosphorylated tau181 in Alzheimer’s disease and frontotemporal lobar degeneration. Nat Med 26, 387–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Tatebe H, Kasai T, Ohmichi T, Kishi Y, Kakeya T, Waragai M, Kondo M, Allsop D, Tokuda T (2017) Quantification of plasma phosphorylated tau to use as a biomarker for brain Alzheimer pathology: Pilot case-control studies including patients with Alzheimer’s disease and down syndrome. Mol Neurodegener 12, 63–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Yang C-C, Chiu M-J, Chen T-F, Chang H-L, Liu B-H, Yang S-Y (2018) Assay of plasma phosphorylated tau protein (Threonine 181) and total tau protein in early-stage Alzheimer’s disease. J Alzheimers Dis 61, 1323–1332. [DOI] [PubMed] [Google Scholar]
- [34].Mielke MM, Hagen CE, Xu J, Chai X, Vemuri P, Lowe VJ, Airey DC, Knopman DS, Roberts RO, Machulda MM, Jack CR Jr., Petersen RC, Dage JL (2018) Plasma phospho-tau181 increases with Alzheimer’s disease clinical severity and is associated with tau- and amyloid-positron emission tomography. Alzheimers Dement 14, 989–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Zetterberg H, Burnham SC (2019) Blood-based molecular biomarkers for Alzheimer’s disease. Mol Brain 12, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Ray S, Britschgi M, Herbert C, Takeda-Uchimura Y, Boxer A, Blennow K, Friedman LF, Galasko DR, Jutel M, Karydas A, Kaye JA, Leszek J, Miller BL, Minthon L, Quinn JF, Rabinovici GD, Robinson WH, Sabbagh MN, So YT, Sparks DL, Tabaton M, Tinklenberg J, Yesavage JA, Tibshirani R, Wyss-Coray T (2007) Classification and prediction of clinical Alzheimer’s diagnosis based on plasma signaling proteins. Nat Med 13, 1359–1362. [DOI] [PubMed] [Google Scholar]
- [37].Llano DA, Devanarayan V, Simon AJ (2013) Evaluation of plasma proteomic data for Alzheimer disease state classification and for the prediction of progression from mild cognitive impairment to Alzheimer disease. Alzheimer Dis Assoc Disord 27, 233–243. [DOI] [PubMed] [Google Scholar]
- [38].Hu WT, Holtzman DM, Fagan AM, Shaw LM, Perrin R, Arnold SE, Grossman M, Xiong C, Craig-Schapiro R, Clark CM, Pickering E, Kuhn M, Chen Y, Van Deerlin VM, McCluskey L, Elman L, Karlawish J, Chen-Plotkin A, Hurtig HI, Siderowf A, Swenson F, Lee VM, Morris JC, Trojanowski JQ, Soares H (2012) Plasma multianalyte profiling in mild cognitive impairment and Alzheimer disease. Neurology 79, 897–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Soares HD, Potter WZ, Pickering E, Kuhn M, Immermann FW, Shera DM, Ferm M, Dean RA, Simon AJ, Swenson F, Siuciak JA, Kaplow J, Thambisetty M, Zagouras P, Koroshetz WJ, Wan HI, Trojanowski JQ, Shaw LM (2012) Plasma biomarkers associated with the apolipoprotein E genotype and Alzheimer disease. Arch Neurol 69, 1310–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Song F, Poljak A, Kochan NA, Raftery M, Brodaty H, Smythe GA, Sachdev PS (2014) Plasma protein profiling of mild cognitive impairment and Alzheimer’s disease using iTRAQ quantitative proteomics. Proteome Sci 12, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Blennow K, Zetterberg H (2018) Biomarkers for Alzheimer’s disease: Current status and prospects for the future. J Intern Med 284, 643–663. [DOI] [PubMed] [Google Scholar]
- [42].Hye A, Riddoch-Contreras J, Baird AL, Ashton NJ, Bazenet C, Leung R, Westman E, Simmons A, Dobson R, Sattlecker M, Lupton M, Lunnon K, Keohane A, Ward M, Pike I, Zucht HD, Pepin D, Zheng W, Tunnicliffe A, Richardson J, Gauthier S, Soininen H, Kloszewska I, Mecocci P, Tsolaki M, Vellas B, Lovestone S (2014) Plasma proteins predict conversion to dementia from prodromal disease. Alzheimers Dement 10, 799–807 e792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Burnham SC, Faux NG, Wilson W, Laws SM, Ames D, Bedo J, Bush AI, Doecke JD, Ellis KA, Head R, Jones G, Kiiveri H, Martins RN, Rembach A, Rowe CC, Salvado O, Macaulay SL, Masters CL, Villemagne VL (2014) A blood-based predictor for neocortical Abeta burden in Alzheimer’s disease: Results from the AIBL study. Mol Psychiatry 19, 519–526. [DOI] [PubMed] [Google Scholar]
- [44].Westwood S, Baird AL, Anand SN, Nevado-Holgado AJ, Kormilitzin A, Shi L, Hye A, Ashton NJ, Morgan AR, Bos I, Vos SJB, Baker S, Buckley NJ, Ten Kate M, Scheltens P, Teunissen CE, Vandenberghe R, Gabel S, Meersmans K, Engelborghs S, De Roeck EE, Sleegers K, Frisoni GB, Blin O, Richardson JC, Bordet R, Molinuevo JL, Rami L, Wallin A, Kettunen P, Tsolaki M, Verhey F, Lléo A, Sala I, Popp J, Peyratout G, Martinez-Lage P, Tainta M, Johannsen P, Freund-Levi Y, Frölich L, Dobricic V, Legido-Quigley C, Bertram L, Barkhof F, Zetterberg H, Morgan BP, Streffer J, Visser PJ, Lovestone S (2020) Validation of plasma proteomic biomarkers relating to brain amyloid burden in the EMIF-Alzheimer’s disease multimodal biomarker discovery cohort. J Alzheimers Dis 74, 213–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Kiddle SJ, Sattlecker M, Proitsi P, Simmons A, Westman E, Bazenet C, Nelson SK, Williams S, Hodges A, Johnston C, Soininen H, Kloszewska I, Mecocci P, Tsolaki M, Vellas B, Newhouse S, Lovestone S, Dobson RJ (2014) Candidate blood proteome markers of Alzheimer’s disease onset and progression: A systematic review and replication study. J Alzheimers Dis 38, 515–531. [DOI] [PubMed] [Google Scholar]
- [46].Blacker D, Wilcox MA, Laird NM, Rodes L, Horvath SM, Go RC, Perry R, Watson B Jr., Bassett SS, McInnis MG, Albert MS, Hyman BT, Tanzi RE (1998) Alpha-2 macroglobulin is genetically associated with Alzheimer disease. Nat Genet 19, 357–360. [DOI] [PubMed] [Google Scholar]
- [47].Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, Merry KM, Shi Q, Rosenthal A, Barres BA, Lemere CA, Selkoe DJ, Stevens B (2016) Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352, 712–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Hall JR, Wiechmann AR, Johnson LA, Edwards ML, O’Bryant SE (2019) Levels of alpha-2 Macroglobulin in cognitively normal Mexican-Americans with subjective cognitive decline: A HABLE Study. Curr Neurobiol 10, 22–25. [PMC free article] [PubMed] [Google Scholar]
- [49].Thambisetty M, Hye A, Foy C, Daly E, Glover A, Cooper A, Simmons A, Murphy D, Lovestone S (2008) Proteome-based identification of plasma proteins associated with hippocampal metabolism in early Alzheimer’s disease. J Neurol 255, 1712–1720. [DOI] [PubMed] [Google Scholar]
- [50].Varma VR, Varma S, An Y, Hohman TJ, Seddighi S, Casanova R, Beri A, Dammer EB, Seyfried NT, Pletnikova O, Moghekar A, Wilson MR, Lah JJ, O’Brien RJ, Levey AI, Troncoso JC, Albert MS, Thambisetty M (2017) Alpha-2 macroglobulin in Alzheimer’s disease: A marker of neuronal injury through the RCAN1 pathway. Mol Psychiatry 22, 13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Hye A, Lynham S, Thambisetty M, Causevic M, Campbell J, Byers HL, Hooper C, Rijsdijk F, Tabrizi SJ, Banner S, Shaw CE, Foy C, Poppe M, Archer N, Hamilton G, Powell J, Brown RG, Sham P, Ward M, Lovestone S (2006) Proteome-based plasma biomarkers for Alzheimer’s disease. Brain 129, 3042–3050. [DOI] [PubMed] [Google Scholar]
- [52].Licastro F, Parnetti L, Morini MC, Davis LJ, Cucinotta D, Gaiti A, Senin U (1995) Acute phase reactant alpha 1-antichymotrypsin is increased in cerebrospinal fluid and serum of patients with probable Alzheimer disease. Alzheimer Dis Assoc Disord 9, 112–118. [DOI] [PubMed] [Google Scholar]
- [53].Shen L, Liao L, Chen C, Guo Y, Song D, Wang Y, Chen Y, Zhang K, Ying M, Li S, Liu Q, Ni J (2017) Proteomics analysis of blood serums from Alzheimer’s disease patients using iTRAQ labeling technology. J Alzheimers Dis 56, 361–378. [DOI] [PubMed] [Google Scholar]
- [54].Muenchhoff J, Poljak A, Song F, Raftery M, Brodaty H, Duncan M, McEvoy M, Attia J, Schofield PW, Sachdev PS (2015) Plasma protein profiling of mild cognitive impairment and Alzheimer’s disease across two independent cohorts. J Alzheimers Dis 43, 1355–1373. [DOI] [PubMed] [Google Scholar]
- [55].Yang H, Lyutvinskiy Y, Herukka SK, Soininen H, Rutishauser D, Zubarev RA (2014) Prognostic polypeptide blood plasma biomarkers of Alzheimer’s disease progression. J Alzheimers Dis 40, 659–666. [DOI] [PubMed] [Google Scholar]
- [56].Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, Cruchaga C, Sassi C, Kauwe JS, Younkin S, Hazrati L, Collinge J, Pocock J, Lashley T, Williams J, Lambert JC, Amouyel P, Goate A, Rademakers R, Morgan K, Powell J, St George-Hyslop P, Singleton A, Hardy J (2013) TREM2 variants in Alzheimer’s disease. N Engl J Med 368, 117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, Bjornsson S, Huttenlocher J, Levey AI, Lah JJ, Rujescu D, Hampel H, Giegling I, Andreassen OA, Engedal K, Ulstein I, Djurovic S, Ibrahim-Verbaas C, Hofman A, Ikram MA, van Duijn CM, Thorsteinsdottir U, Kong A, Stefansson K (2013) Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med 368, 107–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Griciuc A, Serrano-Pozo A, Parrado AR, Lesinski AN, Asselin CN, Mullin K, Hooli B, Choi SH, Hyman BT, Tanzi RE (2013) Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron 78, 631–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Bradshaw EM, Chibnik LB, Keenan BT, Ottoboni L, Raj T, Tang A, Rosenkrantz LL, Imboywa S, Lee M, Von Korff A, Morris MC, Evans DA, Johnson K, Sperling RA, Schneider JA, Bennett DA, De Jager PL (2013) CD33 Alzheimer’s disease locus: Altered monocyte function and amyloid biology. Nat Neurosci 16, 848–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Zhang B, Gaiteri C, Bodea LG, Wang Z, McElwee J, Podtelezhnikov AA, Zhang C, Xie T, Tran L, Dobrin R, Fluder E, Clurman B, Melquist S, Narayanan M, Suver C, Shah H, Mahajan M, Gillis T, Mysore J, MacDonald ME, Lamb JR, Bennett DA, Molony C, Stone DJ, Gudnason V, Myers AJ, Schadt EE, Neumann H, Zhu J, Emilsson V (2013) Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell 153, 707–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Chen M, Lee HK, Moo L, Hanlon E, Stein T, Xia W (2018) Common proteomic profiles of induced pluripotent stem cell-derived three-dimensional neurons and brain tissue from Alzheimer patients. J Proteomics 182, 21–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Huang da W, Sherman BT, Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4, 44–57. [DOI] [PubMed] [Google Scholar]
- [63].Huang DW, Sherman BT, Tan Q, Collins JR, Alvord WG, Roayaei J, Stephens R, Baseler MW, Lane HC, Lempicki RA (2007) The DAVID Gene Functional Classification Tool: A novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol 8, R183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Sweeney MD, Sagare AP, Zlokovic BV (2018) Blood–brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat Rev Neurol 14, 133–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Attems J, Jellinger K, Thal DR, Van Nostrand W (2011) Review: Sporadic cerebral amyloid angiopathy. Neuropathol Appl Neurobiol 37, 75–93. [DOI] [PubMed] [Google Scholar]
- [66].Keable A, Fenna K, Yuen HM, Johnston DA, Smyth NR, Smith C, Al-Shahi Salman R, Samarasekera N, Nicoll JA, Attems J, Kalaria RN, Weller RO, Carare RO (2016) Deposition of amyloid beta in the walls of human leptomeningeal arteries in relation to perivascular drainage pathways in cerebral amyloid angiopathy. Biochim Biophys Acta 1862, 1037–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Arvanitakis Z, Leurgans SE, Wang Z, Wilson RS, Bennett DA, Schneider JA (2011) Cerebral amyloid angiopathy pathology and cognitive domains in older persons. Ann Neurol 69, 320–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Rex S, Beaulieu LM, Perlman DH, Vitseva O, Blair PS, McComb ME, Costello CE, Freedman JE (2009) Immune versus thrombotic stimulation of platelets differentially regulates signalling pathways, intracellular protein-protein interactions, and alpha-granule release. Thrombosis Haemostasis 102, 97–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Donner L, Falker K, Gremer L, Klinker S, Pagani G, Ljungberg LU, Lothmann K, Rizzi F, Schaller M, Gohlke H, Willbold D, Grenegard M, Elvers M (2016) Platelets contribute to amyloid-beta aggregation in cerebral vessels through integrin alphaIIbbeta3-induced outside-in signaling and clusterin release. Sci Signal 9, ra52. [DOI] [PubMed] [Google Scholar]
- [70].Davis VL, Abukabda AB, Radio NM, Witt-Enderby PA, Clafshenkel WP, Cairone JV, Rutkowski JL (2014) Platelet-rich preparations to improve healing. Part II: Platelet activation and enrichment, leukocyte inclusion, and other selection criteria. J Oral Implantol 40, 511–521. [DOI] [PubMed] [Google Scholar]
- [71].Thambisetty M, Simmons A, Hye A, Campbell J, Westman E, Zhang Y, Wahlund LO, Kinsey A, Causevic M, Killick R, Kloszewska I, Mecocci P, Soininen H, Tsolaki M, Vellas B, Spenger C, Lovestone S (2011) Plasma biomarkers of brain atrophy in Alzheimer’s disease. PLoS One 6, e28527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Shen Y, Meri S (2003) Yin and Yang: Complement activation and regulation in Alzheimer’s disease. Prog Neurobiol 70, 463–472. [DOI] [PubMed] [Google Scholar]
- [73].Emmerling MR, Watson MD, Raby CA, Spiegel K (2000) The role of complement in Alzheimer’s disease pathology. Biochim Biophys Acta 1502, 158–171. [DOI] [PubMed] [Google Scholar]
- [74].Tenner AJ (2001) Complement in Alzheimer’s disease: Opportunities for modulating protective and pathogenic events. Neurobiol Aging 22, 849–861. [DOI] [PubMed] [Google Scholar]
- [75].An XQ, Xi W, Gu CY, Huang X (2018) Complement protein C5a enhances the beta-amyloid-induced neuro-inflammatory response in microglia in Alzheimer’s disease. Med Sci (Paris) 34 Focus issue F1, 116–120. [DOI] [PubMed] [Google Scholar]
- [76].Zorzetto M, Datturi F, Divizia L, Pistono C, Campo I, De Silvestri A, Cuccia M, Ricevuti G (2017) Complement C4A and C4B gene copy number study in Alzheimer’s disease patients. Curr Alzheimer Res 14, 303–308. [DOI] [PubMed] [Google Scholar]
- [77].Strohmeyer R, Shen Y, Rogers J (2000) Detection of complement alternative pathway mRNA and proteins in the Alzheimer’s disease brain. Brain Res Mol Brain Res 81, 7–18. [DOI] [PubMed] [Google Scholar]
- [78].Veerhuis R, Nielsen HM, Tenner AJ (2011) Complement in the brain. Mol Immunol 48, 1592–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Sastre M, Klockgether T, Heneka MT (2006) Contribution of inflammatory processes to Alzheimer’s disease: Molecular mechanisms. Int J Dev Neurosci 24, 167–176. [DOI] [PubMed] [Google Scholar]
- [80].Ojala J, Alafuzoff I, Herukka SK, van Groen T, Tanila H, Pirttila T (2009) Expression of interleukin-18 is increased in the brains of Alzheimer’s disease patients. Neurobiol Aging 30, 198–209. [DOI] [PubMed] [Google Scholar]
- [81].Elahi FM, Casaletto KB, La Joie R, Walters SM, Harvey D, Wolf A, Edwards L, Rivera-Contreras W, Karydas A, Cobigo Y, Rosen HJ, DeCarli C, Miller BL, Rabinovici GD, Kramer JH (2020) Plasma biomarkers of astrocytic and neuronal dysfunction in early- and late-onset Alzheimer’s disease. Alzheimers Dement 16, 681–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Gabay C (2006) Interleukin-6 and chronic inflammation. Arthritis Res Ther 8 Suppl 2, S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Chong Y (1997) Effect of a carboxy-terminal fragment of the Alzheimer’s amyloid precursor protein on expression of proinflammatory cytokines in rat glial cells. Life Sci 61, 2323–2333. [DOI] [PubMed] [Google Scholar]
- [84].Qiu Z, Gruol DL (2003) Interleukin-6, beta-amyloid peptide and NMDA interactions in rat cortical neurons. J Neuroimmunol 139, 51–57. [DOI] [PubMed] [Google Scholar]
- [85].Quintanilla RA, Orellana DI, Gonzalez-Billault C, Maccioni RB (2004) Interleukin-6 induces Alzheimer-type phosphorylation of tau protein by deregulating the cdk5/p35 pathway. Exp Cell Res 295, 245–257. [DOI] [PubMed] [Google Scholar]
- [86].Chen WT, Hong CJ, Lin YT, Chang WH, Huang HT, Liao JY, Chang YJ, Hsieh YF, Cheng CY, Liu HC, Chen YR, Cheng IH (2012) Amyloid-beta (Abeta) D7H mutation increases oligomeric Abeta42 and alters properties of Abeta-zinc/copper assemblies. PLoS One 7, e35807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Reichenbach N, Delekate A, Plescher M, Schmitt F, Krauss S, Blank N, Halle A, Petzold GC (2019) Inhibition of Stat3-mediated astrogliosis ameliorates pathology in an Alzheimer’s disease model. EMBO Mol Med 11, e9665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Helmfors L, Boman A, Civitelli L, Nath S, Sandin L, Jane-fjord C, McCann H, Zetterberg H, Blennow K, Halliday G, Brorsson AC, Kagedal K (2015) Protective properties of lysozyme on beta-amyloid pathology: Implications for Alzheimer disease. Neurobiol Dis 83, 122–133. [DOI] [PubMed] [Google Scholar]
- [89].De Buck M, Gouwy M, Wang JM, Van Snick J, Opdenakker G, Struyf S, Van Damme J (2016) Structure and expression of different serum amyloid A (SAA) variants and their concentration-dependent functions during host insults. Curr Med Chem 23, 1725–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Larson MA, Wei SH, Weber A, Weber AT, McDonald TL (2003) Induction of human mammary-associated serum amyloid A3 expression by prolactin or lipopolysaccharide. Biochem Biophys Res Commun 301, 1030–1037. [DOI] [PubMed] [Google Scholar]
- [91].Uhlar CM, Whitehead AS (1999) Serum amyloid A, the major vertebrate acute-phase reactant. Eur J Biochem 265, 501–523. [DOI] [PubMed] [Google Scholar]
- [92].Ye RD, Sun L (2015) Emerging functions of serum amyloid A in inflammation. J Leukocyte Biol 98, 923–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Kindy MS, Yu J, Guo JT, Zhu H (1999) Apolipoprotein serum amyloid A in Alzheimer’s disease. J Alzheimers Dis 1, 155–167. [DOI] [PubMed] [Google Scholar]
- [94].Liang JS, Sloane JA, Wells JM, Abraham CR, Fine RE, Sipe JD (1997) Evidence for local production of acute phase response apolipoprotein serum amyloid A in Alzheimer’s disease brain. Neurosci Lett 225, 73–76. [DOI] [PubMed] [Google Scholar]
- [95].Chung TF, Sipe JD, McKee A, Fine RE, Schreiber BM, Liang JS, Johnson RJ (2000) Serum amyloid A in Alzheimer’s disease brain is predominantly localized to myelin sheaths and axonal membrane. Amyloid 7, 105–110. [DOI] [PubMed] [Google Scholar]
- [96].Miida T, Yamada T, Seino U, Ito M, Fueki Y, Takahashi A, Kosuge K, Soda S, Hanyu O, Obayashi K, Miyazaki O, Okada M (2006) Serum amyloid A (SAA)-induced remodeling of CSF-HDL. Biochim Biophys Acta 1761, 424–433. [DOI] [PubMed] [Google Scholar]
- [97].Kitamura Y, Usami R, Ichihara S, Kida H, Satoh M, Tomimoto H, Murata M, Oikawa S (2017) Plasma protein profiling for potential biomarkers in the early diagnosis of Alzheimer’s disease. Neurol Res 39, 231–238. [DOI] [PubMed] [Google Scholar]
- [98].Lin Q, Cao Y, Gao J (2015) Decreased expression of the APOA1-APOC3-APOA4 gene cluster is associated with risk of Alzheimer’s disease. Drug Des Devel Ther 9, 5421–5431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Cervantes S, Samaranch L, Vidal-Taboada JM, Lamet I, Bullido MJ, Frank-Garcia A, Coria F, Lleo A, Clarimon J, Lorenzo E, Alonso E, Sanchez-Juan P, Rodriguez-Rodriguez E, Combarros O, Rosich M, Vilella E, Pastor P (2011) Genetic variation in APOE cluster region and Alzheimer’s disease risk. Neurobiol Aging 32, 2107.e2107–2117. [DOI] [PubMed] [Google Scholar]
- [100].Shao Y, Shaw M, Todd K, Khrestian M, D’Aleo G, Barnard PJ, Zahratka J, Pillai J, Yu CE, Keene CD, Leverenz JB, Bekris LM (2018) DNA methylation of TOMM40-APOE-APOC2 in Alzheimer’s disease. J Hum Genet 63, 459–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Shih YH, Tsai KJ, Lee CW, Shiesh SC, Chen WT, Pai MC, Kuo YM (2014) Apolipoprotein C-III is an amyloid-beta-binding protein and an early marker for Alzheimer’s disease. J Alzheimers Dis 41, 855–865. [DOI] [PubMed] [Google Scholar]
- [102].Recalde D, Ostos MA, Badell E, Garcia-Otin AL, Pidoux J, Castro G, Zakin MM, Scott-Algara D (2004) Human apolipoprotein A-IV reduces secretion of proinflammatory cytokines and atherosclerotic effects of a chronic infection mimicked by lipopolysaccharide. Arterioscler Thromb Vasc Biol 24, 756–761. [DOI] [PubMed] [Google Scholar]
- [103].Cui Y, Huang M, He Y, Zhang S, Luo Y (2011) Genetic ablation of apolipoprotein A-IV accelerates Alzheimer’s disease pathogenesis in a mouse model. Am J Pathol 178, 1298–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Migrenne S, Le Foll C, Levin BE, Magnan C (2011) Brain lipid sensing and nervous control of energy balance. Diabetes Metabol 37, 83–88. [DOI] [PubMed] [Google Scholar]
- [105].Persson T, Popescu BO, Cedazo-Minguez A (2014) Oxidative stress in Alzheimer’s disease: Why did antioxidant therapy fail? Oxid Med Cell Longev 2014, 427318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Zhu CJ, Jiang GX, Chen JM, Zhou ZM, Cheng Q (2018) Serum haptoglobin in Chinese patients with Alzheimer’s disease and mild cognitive impairment: A case-control study. Brain Res Bull 137, 301–305. [DOI] [PubMed] [Google Scholar]
- [107].Song IU, Kim YD, Chung SW, Cho HJ (2015) Association between serum haptoglobin and the pathogenesis of Alzheimer’s disease. Intern Med 54, 453–457. [DOI] [PubMed] [Google Scholar]
- [108].Akbaraly TN, Hininger-Favier I, Carriere I, Arnaud J, Gourlet V, Roussel AM, Berr C (2007) Plasma selenium over time and cognitive decline in the elderly. Epidemiology 18, 52–58. [DOI] [PubMed] [Google Scholar]
- [109].Solovyev N, Drobyshev E, Bjorklund G, Dubrovskii Y, Lysiuk R, Rayman MP (2018) Selenium, selenoprotein P, and Alzheimer’s disease: Is there a link? Free Radic Biol Med 127, 124–133. [DOI] [PubMed] [Google Scholar]
- [110].Olanow CW (1993) A radical hypothesis for neurodegeneration. Trends Neurosci 16, 439–444. [DOI] [PubMed] [Google Scholar]
- [111].Fischer P, Gotz ME, Danielczyk W, Gsell W, Riederer P (1997) Blood transferrin and ferritin in Alzheimer’s disease. Life Sci 60, 2273–2278. [DOI] [PubMed] [Google Scholar]
- [112].Connor JR, Snyder BS, Beard JL, Fine RE, Mufson EJ (1992) Regional distribution of iron and iron-regulatory proteins in the brain in aging and Alzheimer’s disease. J Neurosci Res 31, 327–335. [DOI] [PubMed] [Google Scholar]
- [113].Gao L, Yin H, Smith SRJ, Chao L, Chao J (2008) Role of kallistatin in prevention of cardiac remodeling after chronic myocardial infarction. Lab Invest 88, 1157–1166. [DOI] [PubMed] [Google Scholar]
- [114].Chao J, Guo Y, Li P, Chao L (2017) Opposing effects of oxygen regulation on kallistatin expression: Kallistatin as a novel mediator of oxygen-induced HIF-1-eNOS-NO pathway. Oxid Med Cell Longev 2017, 5262958. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.