

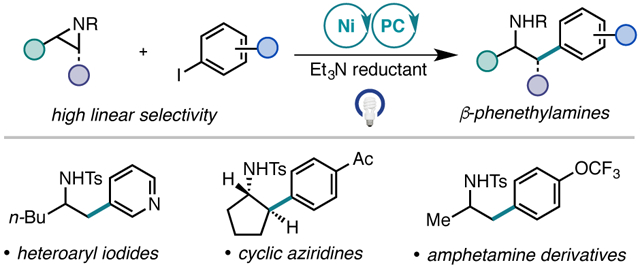
Abstract
A photo-assisted Ni-catalyzed reductive cross-coupling between tosyl-protected alkyl aziridines and commercially available (hetero)aryl iodides is reported. This mild and modular method proceeds in the absence of stoichiometric heterogeneous reductants and uses an inexpensive organic photocatalyst to access medicinally valuable β-phenethylamine derivatives. Unprecedented reactivity was achieved with the activation of cyclic aziridines. Mechanistic studies suggest that the regioselectivity and reactivity observed under these conditions are a result of nucleophilic iodide ring opening of the aziridine to generate an iodoamine as the active electrophile. This strategy also enables cross-coupling with Boc-protected aziridines.
Graphical Abstract
INTRODUCTION
The β-phenethylamine scaffold is an important nitrogen-containing motif in medicinal chemistry, with thousands of reported derivatives possessing varied pharmacological activity (Figure 1A).1 Phenethylamine itself is a monoaminergic neuromodulator. Numerous phenethylamine derivatives are naturally occurring and bioactive, such as dopamine, norepinephrine, and adrenaline.2 Synthetic derivatives belong to a range of different drug classes, such as antidepressants, anti-Parkinson agents, appetite suppressants, and decongestants. For example, amphetamine and its derivatives are important for central nervous system treatments. As a result of the strong interest in accessing the β-phenethylamine substructure, a variety of synthetic approaches have been devised.3
Figure 1.

Bioactive and pharmaceutically relevant β-phenethylamines and aziridine cross-coupling methodologies. bpp = 2,6-bis(N-pyrazolyl)-pyridine.
The arylation of aliphatic aziridines represents a particularly attractive retrosynthetic disconnection due to its modularity, enabling the installation of diverse (hetero)aryl groups and ethyl backbone substitutions in a single C─C bond-forming step. Most examples in this context rely on the use of organocuprates, generated from aryl lithium or Grignard reagents, in combination with a strong Lewis acid, which can lead to limited functional group compatibility and poor regioselectivity (Figure 1B).4 In 2013, Michael and co-workers reported a Pd-catalyzed Suzuki coupling of aliphatic aziridines with aryl boronic acids that affords β-phenethylamines in high regioselectivity.5a Recently, Zhao and co-workers reported a Pd-catalyzed C─H coupling of benzoic acids and aliphatic aziridines (Figure 1B). Although these reports significantly advanced the state of the art, both methods relied on the use of a precious metal catalyst and were only applicable to terminal aliphatic aziridines.5b, 6 Additionally, the latter methodology relied on a carboxylic acid to direct C─H activation. Therefore, the development of complementary catalytic methods is necessary to expand the generality and practicality of this approach for the synthesis of β-phenethylamines.
Over the past decade, our lab and the Jamison lab have reported Ni-catalyzed Negishi cross-coupling reactions of aziridines (Figure 1B).7 Moreover, Zhou and co-workers have reported a Ni/photoredox cross-coupling of styrenyl aziridines and potassium benzyltrifluoroborates.8 However, identification of a general and selective protocol to access β-phenethylamine products, particularly α-substituted phenethylamines, has been challenging.9,10 Recently, we reported a reductive cross-electrophile coupling of tosyl-protected (N-Ts) styrenyl aziridines with aryl iodides that selectively yielded the branched isomeric product (Figure 1C).11,12 The use of widely available and bench stable aryl iodide electrophiles, without the need to pre-generate organometallic reagents, provided a practical advantage over conventional Ni-catalyzed cross-coupling procedures with aziridines. Albeit, this strategy was still not applicable to the preparation of α-substituted β-phenethylamine products, as aliphatic aziridines were unreactive.
We report here the development of a photocatalytic Ni-catalyzed cross-electrophile coupling strategy with aliphatic aziridines and (hetero)aryl iodides that delivers a broad range of β-phenethylamine scaffolds (Figure 1D). Recent studies have demonstrated that it is possible to use a visible-light photoredox catalyst in conjunction with an amine as the reductant in Ni-catalyzed cross-electrophile coupling reactions.13 An advantage of this approach is that it addresses challenges inherent to the use of heterogenous reductants in traditional protocols. The methodology reported here highlights that a photocatalytic cross-electrophile coupling can also impart distinct and previously inaccessible reactivity compared to cross-electrophile couplings with Mn as a reductant. Mechanistic studies were performed to probe this variation in reactivity.
RESULTS AND DISCUSSION
Optimization.
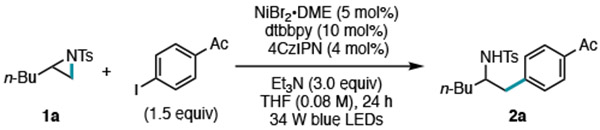
Our initial investigations focused on the coupling of aliphatic aziridine 1a with 4’-iodoacetophenone jointly catalyzed by a nickel catalyst and a visible-light photoredox catalyst (Table 1). Different amine-based reductants, solvents, catalyst loadings and reaction concentrations were investigated (see SI Optimization Studies). We found that linear isomer 2a could be obtained in 59% yield (l:b >10:1) using NiBr2•DME/4,4’-di-tert-butylbipyridine (dtbbpy) as the cross-coupling catalyst, 1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyano-benzene (4CzIPN) as the photocatalyst, and Et3N as the reductant (Table 1, entry 1).14 While homocoupling is often an issue in cross-electrophile coupling reactions, therefore requiring a large excess of aryl halide, only 15% of biaryl product was observed under these conditions using 1.5 equiv of aryl iodide.15 Aryl bromides proved to be ineffective for the transformation (Table 1, entry 2). The use of an Ir-photocatalyst that has similar redox potentials to 4CzIPN was also successful, delivering 2a in 57% yield (Table 1, entry 3). However, 4CzIPN was elected for further study since it is easily synthesized from inexpensive materials and avoids the use of a precious metal. Control studies showed that Ni, ligand, photocatalyst, light, and Et3N were all required in the reaction (Table 1, entry 4–8). Lastly, for larger scale reactions (0.5 mmol), 82% yield was obtained by switching from the use of 34 W blue LEDs to a 450-nm photoreactor (Table 1, entry 9–11).16
Table 1.
Optimization Table
 | |||
|---|---|---|---|
| Entry | Deviation from standard conditions |
conversion of 1a [%] |
Yield [%]a |
| 1 | None | 100 | 59 |
| 2 | 4'-bromoacetophenone | 82 | 0 |
| 3 | Ir(dF-CF3-ppy)2(dtbbpy)PF6 | 100 | 57 |
| 4 | No Et3N | 30 | 0 |
| 5 | No dtbbpy | 61 | 0 |
| 6 | No Ni | 100 | 0 |
| 7 | No 4CzIPN | 11 | 0 |
| 8 | No light | 0 | 0b |
| 9 | 0.5 mmol scale | 70 | 46 |
| 10 | 0.5 mmol scale for 48 h | >95 | 76 |
| 11 | Photoreactor on 0.5 mmol scale | 100 | 82 (72)c |
Reaction run on 0.1 mmol scale. Yields are the average of two runs and were determined using 1,3,5-trimethoxybenzene (1.0 equiv) as an external standard.
Reaction run at 40° C.
Isolated yield.
Substrate Scope.
The generality of the transformation was evaluated under the optimized conditions (Figure 2). In all cases examined, terminal aliphatic aziridines yielded the linear coupled product in high selectively. A wide range of para-substituted aryl iodides containing either electron-withdrawing or electron-donating functionality were tolerated, delivering products with nitrile (2b), methoxy (2c), and silyl ether (2d) groups in good yield. The reaction proceeded well with aryl iodides containing functionality that would be challenging to access with methods that use organozinc, organolithium or Grignard reagents, including chloride (2e), boronate ester (2f), acetanilide (2g), protic (2h), and carbonyl (2i) groups. Meta- and ortho-substituted aryl iodides were also tolerated (2j–m). In contrast to previously reported aziridine cross-coupling methodologies, heteroaryl iodides, a substrate class of high value in medicinal chemistry and library drug design, were competent coupling partners, providing the desired β-heteroarylethylamine products 2n and 2o.17a
Figure 2. Reaction Substrate Scopea.

a Reactions were run on 0.5 mmol scale and yields are the average of two runs. b10 mol% Ni, 20 mol% dtbbpy and 1.25 equiv Ar–I. c Reaction run for 48 h. d Product contains 10% of branched isomer, reported yield is corrected to account for this. e Aziridine 95% ee. Determined by HPLC. f Reaction run for 72 h. g Selectivity determined by 1H NMR. h b:l ratio determined by 1H NMR.
We also investigated the scope of 2-alkyl-substituted tosyl-protected aziridines. Amphetamine derivatives, which are pharmaceutically valuable motifs, were obtained in moderate to high yields (2p–t) from methyl-substituted aziridine with variation in aryl functionality such as trifluoromethoxy or 4-cyano-3-fluoro-phenyl groups. The unsubstituted ethylene-derived aziridine provided the corresponding phenethylamine 2u in 67% yield. The reaction of an enantiopure aziridine afforded product 2v with minimal erosion of enantioselectivity, suggestive of activation at the less-substituted C─N bond. Photocatalytic cross-electrophile coupling proceeded smoothly with aziridines containing synthetically versatile functional groups such as benzylic 2w, aliphatic chloride 2x, ester 2y, and phthalimide 2z. Introduction of β- and even α-branching to the aziridine substrate showed no deterioration in yield, with 2aa and 2ab generated in 64% and 53% yield, respectively. However, aliphatic 2,2-disubstituted aziridines showed limited reactivity.17b
Notably, cyclic 2,3-disubstituted aziridines were competent coupling partners. For instance, cross-coupling with cyclohexyl aziridine furnished product 2ac in 14% yield and cyclopentyl aziridine afforded product 2ad in 55% yield. Both products were formed exclusively as trans diastereomers. It is likely that the enhancement in yield of 2ad in comparison to 2ac arises from an increase in ring strain leading to more effective aziridine activation. To the best of our knowledge, no prior catalytic aziridine cross-coupling methods have been successful with this substrate class. Furthermore, whereas previous Ni-catalyzed methodologies have not been general for both aromatic and aliphatic aziridines, this photocatalytic strategy is: cross-coupling with styrenyl aziridine afforded 2ae in 53% yield as a mixture of branched and linear isomers in a 1.2:1 (b:l) ratio. The generation of the linear product under the photocatalytic coupling conditions was unexpected based on prior work. For example, under our previously reported cross-electrophile conditions with Mn, styrenyl aziridines delivered exclusively the branched isomer with bpp or dtbbpy as the ligand (Figure 1C).18
Mechanistic Investigations.
The b:l mixture observed for styrenyl aziridine, and the reactivity with cyclic aziridines are unprecedented in Ni-catalyzed cross-coupling reactions. This prompted us to examine the mechanism of the photo-assisted cross-coupling.
Previous Ni-catalyzed aziridine cross-coupling methodologies were proposed to proceed via aziridine oxidative addition to Ni, an elementary step that has been studied across various ligand and aziridine classes (Figure 3A). For example, Hillhouse and Jamison have shown that oxidative addition of bpy- or phen-ligated Ni0 to a N-Ts aliphatic aziridine occurs at the terminal C─N bond to yield 3-Me.19 In contrast, our group has shown that for N-Ts styrenyl aziridines, oxidative addition occurs through cleavage of the internal C─N bond with [bpy]Ni(COD) to form 3-Ph.20 The nearly equal b:l product regioselectivity for N-Ts styrenyl aziridine observed under the photo-assisted reaction conditions is inconsistent with this regiochemical outcome of aziridine oxidative addition. To provide additional evidence against this aziridine activation mechanism, we conducted a competition experiment between Ar–I (5.0 equiv) and aziridine 1a (5.0 equiv) with [dtbbpy]Ni0(COD)(4) (1.0 equiv) (Figure 4A). Exclusive reactivity of the Ar–I was observed in less than 10 mins, suggesting that aziridine activation by Ni0 is unlikely under these reaction conditions.
Figure 3.

Possible pathways for aziridine activation. SET = single electron transfer.
Figure 4.

Mechanistic experiments. Standard conditions are the same as in Figure 1, entry 1. a Yields were determined by 1H NMR using 1,3,5-tri-methoxybenzene (1.0 equiv) as an external standard. b Isolated yield.
For these reasons, we considered several alternative aziridine activation mechanisms. First, direct single-electron reduction of the aziridine by the photocatalyst could afford a radical intermediate that is intercepted by Ni in cross-coupling (Figure 3B).8b Alternatively, the aziridine could undergo in situ ring opening by iodide to generate a β-iodoamine, followed by photocatalytic Ni-catalyzed cross-electrophile coupling to render the product (Figure 3C).
As a mechanistic probe for direct reduction of the aziridine by the photocatalyst, the reaction of aziridine 1a, Et3N and 4CzIPN was evaluated in the presence of light. However, no consumption of aziridine was observed. Furthermore, single-electron reduction of 1a by either the photocatalyst or Ni would favor the generation of a 2° versus 1° radical, ultimately affording the branched cross-coupled product or racemization of enantioenriched 2v if ring opening was reversible. Since neither of these are observed, this suggests that single-electron reduction of the aziridine is not taking place. Nevertheless, use of aziridine 6 under the reaction conditions afforded cyclized product 7 in 43% yield as a 54:46 mixture of cis:trans diastere-oisomers (Figure 4B). This provides support for the involvement of a carbon-centered radical in cross-coupling.
In evaluating an iodide ring opening as a mechanism for aziridine activation (Figure 3C), we noted that the b:l selectivity observed for the photocatalytic styrenyl and aliphatic aziridine coupling reactions is similar to that reported for halide ring opening of aziridines.21 The expansion in scope to include cyclic aliphatic aziridines is also consistent with this proposal since these aziridines have not been observed to undergo oxidative addition with Ni or Pd, but secondary alkyl iodides are competent in cross-electrophile coupling reactions.22 Additionally, Weix and co-workers have reported Ni-catalyzed epoxide cross-electrophile coupling reactions that are proposed to proceed through in situ formation of an iodohydrin using Et3N•HCl and NaI as exogenous additives alongside Zn as a reductant.23
To further evaluate the iodide ring-opening proposal, we sought evidence for the generation of β-iodoamine under the reaction conditions. Monitoring the reaction of 1a by 1H NMR, we observed formation of β-iodoamine 8 within the first 2 h (Figure 4C). As the reaction proceeded, 8 was present in approximately 15% yield, while a decrease of aziridine 1a and an increase in cross-coupled product 2a occurred. Detection of 8 in this reaction cannot be taken as direct evidence for its involvement in cross-coupling since the mass balance is such that it could be involved in unproductive chemistry. Evidence in favor of the direct involvement of the iodoamine in cross-coupling was obtained by subjecting a catalytic amount of 8 (0.1 equiv) in combination with aziridine 1b (0.9 equiv) to the reaction conditions (Figure 4D).24 In this case, product 2a was observed in 10% yield (quantitative yield relative to β-iodoamine 8) as well as product 2p in 71% yield.
To understand whether iodoamine formation is reliant on Ni, aziridine 1a and 4’-iodoacetophone were subjected to the photocatalytic conditions without Ni.25 Complete consumption of the aziridine was observed, but the iodide-opened intermediate 8 was not detected (Figure 5). Instead, sultam 9 was obtained in 45% yield. While unexpected, the generation of sultam 9 provides indirect support for the intermediacy of iodoamine 8 and suggests that iodide ring opening can occur independent of Ni. While sultam is present in highest yields in the absence of Ni, it was also detected in trace amounts under the standard catalytic conditions (for example, see SI page S12). Sultam 9 likely arises from aziridine 1a by iodide ring opening to form 8, followed by photocatalytic alkyl iodide reduction and intramolecular radical cyclization.26, 27 Consistent with this proposal, addition of a catalytic amount of tetrabutylammonium iodide (TBAI) to the Ni-free reaction resulted in an increase in yield of 9 (Figure 5, entry 2). Moreover, subjecting iodoamine 8 directly to the photocatalytic conditions in the absence of Ni afforded 9 in 18% yield (see SI page S34). In addition to providing support for the mechanistic proposal, the photocatalytic isomerization of N-Ts aziridines to sultams represents an attractive synthetic approach to these medicinally valuable heterocycles.28
Figure 5.

Photocatalytic reactions in the absence of Ni. a Yields are the average of two runs and were determined by 1H NMR using 1,3,5-trimethoxybenzene (1.0 equiv) as an external standard. ET/PT = electron transfer/proton transfer.
Discussion.
These data suggest that iodide ring opening is a likely mechanism for aziridine activation in the photocatalytic cross-electrophile coupling. A catalytic cycle that incorporates an iodoamine and radical intermediate is proposed (Figure 6). First, facile oxidative addition of the aryl iodide with Ni0 generates B. Concomitantly, nucleophilic iodide ring opening of 1a is mediated by the in situ generation of HI (see SI page S37) to form 8. SET of 8 (with either [dtbbpy]NiI─I E or 4CzIPN−•) or halogen atom abstraction (HAA) from E generates C.29 This radical intermediate can be trapped with B to generate D. The formation of D allows for reductive elimination to yield the cross-coupled product and intermediate E, which can be reduced by 4CzIPN−• (PC/PC−• = −1.21 vs SCE in MeCN, NiI/ Ni0 = −1.17 vs SCE in THF).30 While a Ni0/NiII/NiIII/NiI cycle is proposed, a mechanism that involves selective addition of NiI─I E to the aryl iodide, followed by single-electron reduction and reaction of the resulting NiI–aryl intermediate with iodoamine 8, is also consistent with the data presented and a recent mechanistic study by Diao and co-workers (see SI page S38).31, 32
Figure 6.

Proposed catalytic cycle. HAA = halogen atom abstraction
As previously noted, aliphatic aziridines were not effective substrates in the Mn-mediated Ni-catalyzed reductive coupling. In light of the above mechanistic studies, we hypothesized that the difference in scope between the Mn and photocatalytic protocol was due to the generation of HI under the photocatalytic conditions, which facilitates aziridine ring opening and subsequent cross-coupling. While iodide is also a byproduct of the Mn-mediated coupling, it is likely more strongly sequestered as the MnI2 salt, preventing the formation of 8. Indeed, addition of MnI2 under our previously reported Ni/Mn conditions (1.0 equiv) with aziridine 1a did not result in any detectable cross-coupled product nor iodoamine 8. However, the use of 8 (1.0 equiv) instead of aziridine 1a generated 2a in 27% yield with bpp as ligand and 60% yield using dtbbpy (see SI S35–S36).
Prior mechanistic studies on C(sp3)–C(sp2) cross-electrophile coupling reactions have implicated the importance of NiI intermediates for selective activation of the C(sp3) electrophile. Whereas our competition experiments between aziridine and aryl iodide indicate that oxidative addition of Ni0 is selective for iodoarene over aziridine, NiI–I E or NiI–Ar (as in Figure S13) is likely selective for aziridine activation.15b, 29, 31 Under the Mn-reductive coupling conditions, NiI or Mn could activate the aziridine via SET. Due to the stability of benzylic radicals, styrenyl aziridines are expected to be more effective reaction partners than aliphatic aziridines, thereby explaining why only styrenyl aziridines are reactive under the Mn conditions. Under the photocatalytic conditions, in situ formation of iodoamine from the aliphatic aziridine restores the ability of this substrate class to participate in cross-electrophile coupling through SET or HAA with NiI or photocatalyst.
Expansion of the Methodology Based on Mechanistic Findings.
The new mechanistic inference led us to hypothesize that the scope could be expanded to previously unsuccessful substrates. For example, aryl bromides did not provide the cross-coupled product presumably because bromoamine is an ineffective electrophile under these conditions. Consistent with this hypothesis, addition of TBAI (10 mol%) to the aziridine coupling reaction with 4’-bromoacetophenone restored reactivity to give 2a in 45% yield (Figure 7A).
Figure 7.

Expansion in scope based on mechanistic findings. Standard conditions are the same as in Figure 1, entry 1. a Yields are the average of two runs and were determined by 1H NMR using 1,3,5-trimethoxybenzene (1.0 equiv) as an external standard. b l:b ratio determined by 1H NMR. cYields are the average of two isolated runs.
For previous Ni-catalyzed aziridine cross-coupling reactions, the use of N-Ts protected aziridines was necessary to facilitate oxidative addition of Ni to the aziridine.9,19,20 Since cross-coupling is not mediated by Ni-aziridine oxidative addition under the photocatalytic conditions, we wondered whether alternative protecting groups that are easier to deprotect but were previously unsuccessful in Ni-catalyzed aziridine cross-coupling reactions could be used.33 Gratifyingly, employment of N-Boc aziridine 10a yielded product 10b in 28% yield as a 3.4:1 l:b mixture (Figure 7B). These results further highlight the opportunities available with this new strategy which were previously not possible with Ni-catalyzed aziridine cross-coupling reactions.
CONCLUSIONS
In summary, we have described a Ni-photoredox protocol that enables the modular assembly of a large scope of β-phenethylamine derivatives, allowing access to high value substrate classes for medicinal chemists. Many of the substrates have previously proven to be challenging for conventional aziridine cross-coupling methodologies. More generally, the results highlight for cross-electrophile couplings that a photocatalytic protocol can enable distinct scope compared to that using heterogeneous reductants. Mechanistic studies indicate that formation of an iodoamine through nucleophilic iodide ring opening is key for successful reactivity. This aziridine activation mode differs in comparison to those in previous TM-catalyzed methods that are reliant on oxidative C─N bond activation. Based on the described mechanistic details, ongoing studies are focused on utilizing this activation strategy for other strained ring systems with photocatalytic Ni cross-electrophile coupling catalysis.
Supplementary Material
ACKNOWLEDGMENT
Financial support was provided by NIGMS R35 GM126986 and an F32 Ruth L. Kirschstein NRSA Fellowship under Award No. F32 EB027587-01 (T.J.S). The authors gratefully acknowledge Dr. Brian P. Woods for preliminary investigations and Dr. Christoph Heinz for initial substrate scope isolations. We thank Jesus I. Martinez Alvarado for help with emission quenching studies. Dr. Phillip D. Jeffrey is acknowledged for X-ray crystallography. Lotus Separations is thanked for assistance with the purification of 7.
Funding Sources
No competing financial interests have been declared.
Footnotes
Supporting Information.
The Supporting Information is available free of charge on the ACS Publications website.
Experimental details, optimization studies and characterization data (PDF)
Crystallographic data (CIF)
REFERENCES
- (1).(a) Lewin A; Navarro H; Mascarella S Structure–activity correlations for β-phenethylamines at human trace amine receptor 1. Bioorg. & Med. Chem 2008, 16, 7415–7423. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Irsfeld M; Spadafore M; Pruess B β-phenylethylamine, a small molecule with a large impact. WebmedCentral. 2013. 4. [PMC free article] [PubMed] [Google Scholar]; (c) Mosnaim AD; Wolf ME Trace Amines Neurological Disorder. 1st Ed., Elsevier, San Diego, 2017. [Google Scholar]
- (2).(a) Sabelli H; Fink P; Fawcett J; Tom C Sustained antidepressant effect of PEA replacement. J. Neuropsychiatry Clin. Neurosci 1996, 8, 168–171. [DOI] [PubMed] [Google Scholar]; (b) Parker M; Marona-Lewicka D; Kurrasch D; Shulgin A; Nichols D Synthesis and Pharmacological Evaluation of Ring-Methylated Derivatives of 3,4-(Methylenedioxy)amphetamine (MDA). J. Med. Chem 1998, 41, 1001–1005. [DOI] [PubMed] [Google Scholar]; (c) Gallardo-Godoy A; Fierro A; McLean T; Castillo M; Cassels B; Reyes-Parada M; Nichols D Sulfur-Substituted α-Alkyl Phenethylamines as Selective and Reversible MAO-A Inhibitors: Biological Activities, CoMFA Analysis, and Active Site Modeling. J. Med. Chem 2005, 48, 2407–2419. [DOI] [PubMed] [Google Scholar]; (d) Vitaku E; Smith DT; Njardarson JT Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem 2014, 57, 10257–10274. [DOI] [PubMed] [Google Scholar]
- (3).(a) Haak E; Siebeneicher H; Doye S An Ammonia Equivalent for the Dimethyltitanocene-Catalyzed Intermolecular Hydroamination of Alkynes. Org. Lett 2000, 2, 1935–1937. [DOI] [PubMed] [Google Scholar]; (b) Bentley K β-Phenylethylamines and the isoquinoline alkaloids. Nat. Prod. Reports, 2006, 23, 444–463. [DOI] [PubMed] [Google Scholar]; (c) Kindt S; Heinrich M Recent Advances in Meerwein Arylation. Synthesis, 2016, 48, 1597–1606. [Google Scholar]; (d) Wang D; Wu L; Wang F; Wan X; Chen P; Lin Z; Liu G Asymmetric Copper-Catalyzed Intermolecular Aminoarylation of Styrenes: Efficient Access to Optical 2,2-Diarylethylamines. J. Am. Chem. Soc 2017, 139, 6811–6814. [DOI] [PubMed] [Google Scholar]; (e) Monos T; McAtee R; Stephenson C Arylsulfonylacetamides as bifunctional reagents for alkene aminoarylation. Science, 2018, 361, 1369–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).(a) Eis MJ; Ganem B BF3-Etherate Promoted Alkylation of Aziridines with Organocopper Reagents: A New Synthesis of Amines. Tetrahedron Lett. 1985, 26, 1153–1156. [Google Scholar]; (b) Baldwin JE; Farthing CN; Russell AT; Schofield CJ; Spivey AC Use of (S)-N-tert-butoxycarbonylaziridine-2carboxylate derivatives for α-amino acid synthesis. Tetrahedron Lett. 1996, 37, 3761–3764. [Google Scholar]; (c) Sweeney J Aziridines : epoxides ‘ ugly cousins? Chem. Soc. Rev 2002, 31, 247–258. [DOI] [PubMed] [Google Scholar]; (d) Michaelis D; Dineen T Ring opening of aziridines with ortho-bromophenyl metal reagents: synthesis of 2-substituted indolines. Tetrahedron Lett. 2009, 50, 1920–1923. [Google Scholar]
- (5).(a) Duda ML; Michael FE Palladium-Catalyzed Cross-Coupling of N-Sulfonylaziridines with Boronic Acids. J. Am. Chem. Soc. 2013, 135, 18347–18349. [DOI] [PubMed] [Google Scholar]; (b) For catalytic methods where β-phenethylamines have been synthesized from styrenyl aziridines: Takeda Y, Kuroda A, Sameera W, Morokuma K, Minakata S Palladium-catalyzed regioselective and stereo-invertive ring-opening borylation of 2-arylaziridines with bis(pinacolato)diboron: experimental and computational studies, Chem. Sci, 2016, 7, 6141–6152. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Takeda Y, Ikeda Y, Kuroda A, Tanaka S, Minakata S Pd/NHC-Catalyzed Enantiospecific and Regioselective Suzuki–Miyaura Arylation of 2-Arylaziridines: Synthesis of Enantioenriched 2-Arylphenethylamine Derivatives, J. Am Chem. Soc, 2014, 136, 8544–8547. [DOI] [PubMed] [Google Scholar]
- (6).Zhou K; Zhu Y; Fan W; Chen Y; Xu X; Zhang J; Zhao Y Late-Stage Functionalization of Aromatic Acids with Aliphatic Aziridines: Direct Approach to Form β-Branched Arylethylamine Backbones. ACS Catal. 2019, 9, 6738–6743. [Google Scholar]
- (7).Huang C-Y; Doyle AG The Chemistry of Transition Metals with Three-Membered Ring Heterocycles. Chem. Rev 2014, 114, 8153–8198. [DOI] [PubMed] [Google Scholar]
- (8).(a) Yu X-Y; Zhou Q-Q; Wang P-Z; Liao C-M; Chen J-R; Xiao W-J Dual Photoredox/Nickel-Catalyzed Regioselective Cross-Coupling of 2-Arylaziridines and Potassium Benzyltrifluoroborates: Synthesis of β-Substituted Amines. Org. Lett 2018, 20, 421–424. [DOI] [PubMed] [Google Scholar]; (b) For another example of aziridines used as substrates in photoredox catalysis Larraufie M; Pellet R; Fensterbank L; Goddard J; Lacôte E; Malacria M; Ollivier C Visible-Light-Induced Photoreductive Generation of Radicals from Epoxides and Aziridines. Angew. Chem. Int. Ed 2011, 50, 4463–4466. [DOI] [PubMed] [Google Scholar]
- (9).Jensen KL; Standley EA; Jamison TF Highly Regioselective Nickel-Catalyzed Cross-Coupling of N -Tosylaziridines and Alkylzinc Reagents. J. Am. Chem. Soc 2014, 136, 11145–11152. [DOI] [PubMed] [Google Scholar]
- (10).Nielsen DK; Huang C-Y; Doyle AG Directed Nickel-Catalyzed Negishi Cross Coupling of Alkyl Aziridines. J. Am. Chem. Soc 2013, 135, 13605–13609. [DOI] [PubMed] [Google Scholar]
- (11).Woods BP; Orlandi M; Huang C-Y; Sigman MS; Doyle AG Nickel-Catalyzed Enantioselective Reductive Cross-Coupling of Styrenyl Aziridines. J. Am. Chem. Soc 2017, 139, 5688–5691. [DOI] [PubMed] [Google Scholar]
- (12).(a) For examples of Ni-catalyzed cross-coupling of C─N bonds : Maity P, Shacklady-McAtee D, Yap G, Sirianni E, Watson M Nickel-Catalyzed Cross Couplings of Benzylic Ammonium Salts and Boronic Acids: Stereospecific Formation of Diarylethanes via C─N Bond Activation, J. Am. Chem. Soc, 2012, 135, 280–285. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Martin-Montero R, Yatham V, Yin H, Davies J, Martin R Ni-catalyzed Reductive Deaminative Arylation at sp3 Carbon Centers, Org. Lett, 2019, 21, 2947–2951. [DOI] [PubMed] [Google Scholar]; (c) Moragas T, Gaydou M, Martin R Nickel-Catalyzed Carboxylation of Benzylic C─N Bonds with CO2, Angew. Chem. Int. Ed, 2016, 55, 5053–5057. [DOI] [PubMed] [Google Scholar]; (d) Basch C, Liao J, Xu J, Piane J, Watson M Harnessing Alkyl Amines as Electrophiles for Nickel-Catalyzed Cross Couplings via C─N Bond Activation, J. Am. Chem. Soc 2017, 139, 5313–5316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Paul A; Smith MD; Vannucci AK Photoredox-Assisted Reductive Cross-Coupling: Mechanistic Insight into Catalytic Aryl-Alkyl Cross-Couplings. J. Org. Chem 2017, 82, 1996–2003. [DOI] [PubMed] [Google Scholar]
- (14).(a) Uoyama H; Goushi K; Shizu K; Nomura H; Adachi C Highly efficient organic light-emitting diodes from delayed fluorescence. Nature 2012, 492, 234–238. [DOI] [PubMed] [Google Scholar]; (b) Luo J; Zhang J Donor-Acceptor Fluorophores for Visible-Light-Promoted Organic Synthesis: Photoredox/Ni Dual Catalytic C(sp3)─C(sp2) Cross-Coupling. ACS Catal. 2016, 6, 873–877. [Google Scholar]
- (15).(a) Hassan J; Sévignon M; Gozzi C; Schulz E; Lemaire M Aryl–Aryl Bond Formation One Century after the Discovery of the Ullmann Reaction. Chem. Rev 2002, 102, 1359–1470. [DOI] [PubMed] [Google Scholar]; (b) Everson DA; Weix DJ Cross-Electrophile Coupling: Principles of Reactivity and Selectivity. J. Org. Chem 2014, 79, 4793–4798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Le C; Wismer MK; Shi Z-C; Zhang R; Conway DV; Li G; Vachal P; Davies IW; MacMillan DW A General Small-Scale Reactor to Enable Standardization and Acceleration of Photocatalytic Reactions. ACS Central Sci. 2017, 3, 647–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Preliminary studies show that (a) trans-1-iodo-1-octene is a competent coupling partner and (b) cross-coupling is observed with 2,2-dimethyl-1-tosylaziridine. See supporting information page S26.
- (18).Under the Mn conditions, the bpp ligand gave a higher yield of the cross-coupled product compared to dtbbpy. The ligand was not shown to have an effect on the b:l selectivity. Using styrenyl aziridine (1.0 equiv) and 4’-iodoacetophenone (2.0 equiv), bpp afforded the cross-coupled product in 90% yield (ref. 11) and dtbbpy afforded the product in 19% yield (see SI page S36).
- (19).Oxidative addition of terminal aliphatic aziridines is proposed to occur via a concerted SN2 mechanism leading to high linear selectivity: Lin BL; Clough CR; Hillhouse GL Interactions of Aziridines with Nickel Complexes: Oxidative-Addition and Reductive-Elimination Reactions That Break and Make C─N Bonds. J. Am. Chem. Soc 2002, 124, 2890–2891. [DOI] [PubMed] [Google Scholar]
- (20).Oxidative addition of styrenyl aziridines is proposed to occur through a radical intermediate, in which the more stable branched/benzylic isomer is favored: Huang C-Y; Doyle AG Nickel-Catalyzed Negishi Alkylations of Styrenyl Aziridines. J. Am. Chem. Soc 2012, 134, 9541–9544. [DOI] [PubMed] [Google Scholar]
- (21).(a) Hu X Nucleophilic ring opening of aziridines. Tetrahedron, 2004, 60, 2701–2743. [Google Scholar]; (b) Wu J; Sun X; Xia H Ring Opening of Aziridines with Silylated Nucleophiles under Neutral Conditions. Eur. J. Org. Chem 2005, 22, 4769–4772. [Google Scholar]; (c) Minakata S; Hotta T; Oderaotoshi Y; Komatsu M Ring Opening and Expansion of Aziridines in a Silica–Water Reaction Medium. J. Org. Chem 2006, 71, 7471–7472. [DOI] [PubMed] [Google Scholar]; (d) Kumar M; Pandey S; Gandhi S; Singh V PPh3/halogenating agent-mediated highly efficient ring opening of activated and non-activated aziridines Tetrahedron Lett. 2009, 50, 363–365. [Google Scholar]; (e) Sabir S; Kumar G; Verma V; Jat J Aziridine Ring Opening: An Overview of Sustainable Methods. Chemistry Select, 2018, 3, 3702–3711. [Google Scholar]
- (22).(a) For examples of reactions between Pd and aziridines: Ney JE; Wolfe JP Synthesis and Reactivity of Azapalladacyclobutanes. J. Am. Chem. Soc 2006, 128, 15415–15422. [DOI] [PubMed] [Google Scholar]; (b) Matsukawa S; Takahashi H; Harada T TBD-Catalyzed Ring Opening of Aziridines with Silylated Nucleophiles. Synth. Comm 2012, 43, 406–414. [Google Scholar]; (c) Takeda Y; Shibuta K; Aoki S; Tohnai N; Minakata S Catalyst-controlled regiodivergent ring-opening C(sp3)–Si bond-forming reactions of 2-arylaziridines with silylborane enabled by synergistic palladium/copper dual catalysis. Chem. Sci 2019, 10, 8642–8647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).(a) Zhao Y; Weix DJ Nickel-Catalyzed Regiodivergent Opening of Epoxides with Aryl Halides: Co-Catalysis Controls Regioselectivity. J. Am. Chem. Soc 2014, 136, 48–51. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lu X; Yang C; Liu J; Zhang Z; Lu X; Lou X; Xiao B; Fu Y Cu-Catalyzed cross-coupling reactions of epoxides with organoboron compounds. Chem. Comm 2014, 51, 2388–2391. [DOI] [PubMed] [Google Scholar]
- (24).For further details on these experiments please see supporting information page S31-S34.
- (25).Zhang W; Hu W; Su L; Liu L Hydrated Nickel (II) Halides Mediated Ring Opening Reaction with N-Tosylaziridines. Chinese Chem. Lett 2012, 23, 285–288. [Google Scholar]
- (26).(a) In the absence of Ni, iodide could be generated by the reduction of Ar–I by 4CzIPN. Although the reduced form of 4CzIPN is not sufficiently reducing to react with primary aliphatic iodides, the reduction of aliphatic and aromatic iodides is known to be facilitated by the presence of amines and light.Lautenberger W; Jones E; Miller J Reaction of amines with halo alkanes. I. Photochemical reaction of butylamine with carbon tetrachloride. J. Am. Chem. Soc 1968, 90, 1110–1115.Kropp P; Adkins R Photochemistry of alkyl halides. 12. Bromides vs. iodides J. Am. Chem. Soc 1991, 113, 2709–2717.Tucker J; Nguyen J; Narayanam J; Krabbe S; Stephenson C Tin-free radical cyclization reactions initiated by visible light photoredox catalysis. Chem. Comm 2010, 46, 4985–4987.Kim H; Lee C Visible-Light-Induced Photocatalytic Reductive Transformations of Organohalides. Angew. Chem.; Int. Ed, 2012, 51, 12303–12306.Shen Y; Cornella J; Julia-Hernandez F; Martin R Visible Light-Promoted Atom Transfer Radical Cyclization of Unactivated Alkyl Iodides. ACS Catal. 2017, 7, 409–412.Böhm A; Bach T Radical Reactions Induced by Visible Light in Dichloromethane Solutions of Hünig's Base: Synthetic Applications and Mechanistic Observations. Chem. Eur. J 2016, 22, 15921–15928.
- (27).The formation of sultam 9 also suggests that direct reduction of the aziridine is not operative. If direct reduction of aziridine were to occur, the regioisomer of 8 would be favored since reduction would favor the generation of a 2° radical prior to intramolecular cyclization.
- (28).(a) Scozzafava A; Owa T; Mastrolorenzo A; Supuran C Anticancer and Antiviral Sulfonamides. Curr. Med. Chem 2003, 10, 925–953. [DOI] [PubMed] [Google Scholar]; (b) Rassadin V; Grosheva D; Tomashevskii A; Sokolov V Methods of Sultam Synthesis. Chem. Heterocyc. Compd 2013, 49, 39–65. [Google Scholar]
- (29).Diccianni JB; Katigbak J; Hu C; Diao T Mechanistic Characterization of (Xantphos)Ni(I)-Mediated Alkyl Bromide Activation: Oxidative Addition, Electron Transfer, or Halogen-Atom Abstraction. J. Am. Chem. Soc 2019, 141, 1788–1796 [DOI] [PubMed] [Google Scholar]
- (30).Shields BJ; Doyle AG Direct C(sp3)–H Cross Coupling Enabled by Catalytic Generation of Chlorine Radicals. J. Am. Chem. Soc 2016, 138, 12719–12722.(b) If HAA is operative, E would react with 8 to generate NiI2 which can be reduced by the photocatalyst from NiII to Ni0.
- (31).(a) Previous reports related to a Ni0/NiII/NiIII/NiI mechanism: Tasker S; Standley E; Jamison T Recent advances in homogeneous nickel catalysis. Nature, 2014, 509, 299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gutierrez O; Tellis J; Primer D; Molander G; Kozlowski M Nickel-Catalyzed Cross-Coupling of Photoredox-Generated Radicals: Uncovering a General Manifold for Stereoconvergence in Nickel-Catalyzed Cross-Couplings. J. Am. Chem. Soc 2015, 137, 4896–4899. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Beromi M; Brudvig G; Hazari N; Lant H; Mercado B Synthesis and Reactivity of Paramagnetic Nickel Polypyridyl Complexes Relevant to C(sp2)─C(sp3) Coupling Reactions. Angew. Chem. Int. Ed 2019, 58, 6094–6098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).(a) For studies that implicate alternative mechanisms for Ni-catalyzed reductive coupling: Lin Q; Diao T Mechanism of Ni-Catalyzed Reductive 1,2-Dicarbofunctionalization of Alkenes. J. Am. Chem. Soc 2019, 141, 17937–17948. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shu W; García-Domínguez A; Quirós M; Mondal R; Cárdenas D; Nevado C Ni-Catalyzed Reductive Dicarbofunctionalization of Nonactivated Alkenes: Scope and Mechanistic Insights. J. Am. Chem. Soc 2019, 141, 13812–13821. [DOI] [PubMed] [Google Scholar]
- (33).Wuts PGM; Greene TW Greene's Protective Groups in Organic Synthesis. 4th Ed. John Wiley & Sons, Inc., Hoboken: 2006. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.