

Abstract
Neurodegenerative Diseases (NDs) are progressive multifactorial neurological pathologies related to neuronal impairment and functional loss from different brain regions. Currently, no effective treatments are available for any NDs, and this lack of efficacy has been attributed to the multitude of interconnected factors involved in their pathophysiology. In the last two decades, a new approach for the rational design of new drug candidates, also called multitarget-directed ligands (MTDLs) strategy, has emerged and has been used in the design and for the development of a variety of hybrid compounds capable to act simultaneously in diverse biological targets. Based on the polypharmacology concept, this new paradigm has been thought as a more secure and effective way for modulating concomitantly two or more biochemical pathways responsible for the onset and progress of NDs, trying to overcome low therapeutical effectiveness. As a complement to our previous review article (Curr. Med. Chem. 2007, 14 (17), 1829-1852. https://doi.org/10.2174/092986707781058805), herein we aimed to cover the period from 2008 to 2019 and highlight the most recent advances of the exploitation of Molecular Hybridization (MH) as a tool in the rational design of innovative multifunctional drug candidate prototypes for the treatment of NDs, specially focused on AD, PD, HD and ALS.
Keywords: Molecular hybridization, multi-target directed ligands, rational drug design, multifunctional drugs, neurodegenerative diseases, MTDLs
1. INTRODUCTION
Neurodegenerative Diseases (NDs) are one of the biggest challenges for the current medicine practices, medicinal chemistry and all other sciences that focus on the discovery of new drugs [1-4]. NDs are a group of severe, disabling and progressive neurological illnesses like Alzheimer’s Disease (AD), Parkinson’s Disease (PD, Huntington’s Disease (HD) and Amyotrophic Lateral Sclerosis (ALS). The number of individuals with some kind of dementia is estimated from 46.8 million people worldwide in 2015 to a number close to 50 million people in 2017. Considering the current increase in the elderly population worldwide, especially in the most developed countries, this number is thought to almost double every 20 years, reaching 75 million in 2030 and 131.5 million in 2050 [5]. In addition to other chronic inflammatory diseases (e.g. colitis, cancer, diabetes, hypertension and rheumatoid arthritis), these four main types of dementia are characterized by multiple factors involving physiological, biochemical and chemical mediators operating concurrently with, caused by the same or different Pathways [6-9]. During the last decade, profound advances in many fields of biological sciences have been established, highlighting new insights for the knowledge of how complex and multifaceted the pathophysiological hallmarks of NDs are. Currently, it is well accepted that ageing is one of the main risk factors associated with the onset of NDs. In fact, everyone ages, but only some people will be affected by an ND. The incidence of such ND patients seems to be related to neuronal cell vulnerability, once cells from all regions of the peripheral and central nervous system are affected by ageing, which explains the normal time-dependent decline in memory, motor coordination and sensory [8-10]. Meanwhile, the rate of ageing, as well as the type of neuronal cells and regions most affected during life, is individual, once these aspects are closely related to lifestyle, genetics, environmental and epigenetic factors. In fact, ageing is considered one of the most prominent risk factor for NDs and the probability that a person develops some kind of dementia, is dramatically increased with time, especially for AD patients over the 85s, PD for people older than the 70s’ and an abrupt rise to develop ALS above 40 years old. Rare cases of AD, PD and ALS are those related to mutations on specific genes, which determine the early onset of the disease, in general, around 30-40 years old. Most cases of dementia are consequence of alterations in the neuronal physiology as an effect of abnormal protein processing, oxidative stress, neuroinflammation and mitochondrial dysfunction [9-15].
The most recent progress in neurobiology has begun to clarify how specific neurons, in particular, brain regions are more susceptible to molecular and cellular changes leading to neurodegeneration. This selective neuronal vulnerability has been related to how cells in the nervous system are affected by and respond to ageing in a different manner than other cells in the body. In fact, there are enough evidences that cells in the brain experience a much more exacerbated oxidative stress, with energy supply perturbation, agglomeration and deposition of damaged proteins. During ageing, different populations of neurons seem to be more vulnerable to these deleterious biochemical changes and it determines which individual will develop or not a neurodegenerative disorder due to genetic and environmental factors [8, 10].
Given the variety of factors associated with the onset and progress of NDs, with also a pivotal role in the degree of their pathophysiological complexity, maybe it becomes clearer why current treatments based on selective-target drugs lack in therapeutic efficacy. Considering the high adaptive ability of our organism and the many concurrent biochemical windows to be modulated for a single pathology, it becomes unavoidable to adopt a new concept for the rational design of new drugs against such multifactorial disorders [1, 2, 6]. In this regard, the new strategy of multitarget directed ligands (MTDLs) is gaining special attention in the scientific community, and molecular hybridization (MH) of biophore subunits from different molecule prototypes, with different desired biological properties, has been taken as a pivotal tool for designing new molecular patterns [16]. These molecular hybrids are expected to lead to the identification of novel bioactive chemical entities with selective affinity for multiple targets, preferably in different biochemical cascades. Therefore, these innovative ligands could play a unique role in the advancement of a broader and more efficient therapy for NDs [17-19].
1.1. General Aspects of Multifactorial Pathogenesis in Neurodegenerative Diseases
Biochemical changes and neurodegeneration from different adult brain regions are typically related to specific NDs. In HD, cortical structures and striatum are the main brain regions affected, whereas substantia nigra and related cortical structures are associated with PD. Neurons from the hippocampus, frontal cortex and limbic structures are involved in the AD and spinal cord and precentral gyrus are responsible for ALS. Different genetic abnormalities are known to be related to such neuronal damage, but the molecular reasons that determine which neurons from each region will be more vulnerable to die, remain obscure. Vulnerable neurons are typically large, with myelinated axons that extend long distances, connecting different regions of the central nervous system (CNS) or from CNS to the periphery. In fact, hippocampal and cortical pyramidal neurons, upper and lower motor neurons and striatal medium spiny neurons are affected in AD, ALS and HD, respectively. Differently, in PD, smaller dopaminergic neurons in the substantia nigra are degenerated, but they have relatively long axons connecting the motor circuit of corticostriatal projections from the primary motor cortex, supplementary motor cortex, cingulate motor cortex and premotor cortex and terminating dendrites of striatal medium spiny neurons. In ALS, neurodegeneration affects long penetration neurons from the primary motor cortex in the brain to the spinal cord.
AD is the most common dementia among the population above age 65, affecting around 5.4 million people in USA, with projections of 14 million to 2050 and being the 6th leading death [11]. AD is characterized by a progressive loss of memory, cognition, task performance, speech, motor coordination and functional capacity, gradually undermining social behavior and ability to perform routine tasks such as feeding, personal care and social behavior. The origin of physiological deregulation in some brain regions remains not clearly understood, but degeneration of neurons, particularly in the basal forebrain and hippocampus, with consequential inter-neuronal interconnections and synaptic impairment, is due to aggregation of two different types of proteins. In the early onset of AD, patients are affected by memory impairment as a consequence of lower levels in neurotransmitters in the synapsis process, especially acetylcholine (ACh). Pathological changes in the neocortical availability of choline acetyltransferase (ChAT), the biosynthetic precursor of ACh, associated with reduced choline reuptake and release of ACh from the nucleus basalis of Meynert are thought to be the origin of ACh deficits and the consequent presynaptic cholinergic deficits [20]. Amyloid β (Aβ) peptide is the major constituent of extracellular deposits, known as senile or neuritic plaques, and is derived from the abnormal proteolysis of amyloid protein precursor (APP) [3]. Unclear biochemical production of APP is then abnormally cleaved by β- and γ-secretase enzymes, leading to generation of insoluble fragments of 39-43 amino acid residues. Fragments Aβ1-42, even at low concentrations, seem to be more prone to oligomerization and formation of insoluble neurotoxic aggregates organized as β-sheets structures [21-24]. In addition to the neurotoxicity of Aβ aggregates, a secondary event is the hyperphosphorylation of tau protein that plays a critical role in the stabilization of neuronal microtubules. Under pathological conditions, microtubule assembly is affected by the collapse of its structure, releasing of hyperphosphorylated tau fragments. These protein fragments undergo conformational changes in which the conversion of tau monomer to tau oligomers induces the aggregation of tau into pair helical filaments, leading to the formation of intracellular neurofibrillary tangles (NFTs) [3, 12, 18, 22]. Although most studies have emphasized the neurotoxicity of amyloid plaques, recent findings point that the increasing concentration of soluble Aβ oligomers can be also responsible for increasing neuronal injury. In addition, evidences from the treatment of AD patients with statins, have led to the hypothesis that high concentrations of cholesterol in nerve cells could be also related to Aβ production, since the activity balance of the α- and β-secretases is dependent to the lipid levels in cells [23-25].
Currently, oxidative stress (OS) has been considered a central event not only in AD onset, but in almost all NDs. In fact, a number of individual and interconnected deleterious events and biochemical changes are unified as causative and worsening factors in OS. In spite of protein deposition, excessive production of Nitrogen (NREs) and Oxygen radical species (OREs) induced by Aβ have been considered as playing a central role in microglial activation, functional alteration of mitochondria, imbalanced energy supply, production of inflammatory mediators and alteration of antioxidant defenses [10, 23-31]. The redox properties of biometals like Cu2+, Fe2+ and Zn2+ are closely related to the protein aggregation process. Changes in such ion metals concentration can increase the oxidative stress and, in turn, the ROS production. In the case of AD, APP and Aβ are able to form complexes with and reduce Cu2+, which forms a high affinity complex with Aβ, inducing its aggregation. Furthermore, Haber Weiss and Fenton reactions are involved in several cellular redox processes, but in vivo evidences have shown that Aβ neurotoxicity is dependent on the catalytic generation of H2O2 and hydroxyl radical (OH) which are enhanced by the presence of Fe+2/Fe3+, Aβ-Cu+2 and Aβ-Zn2+ complexes [28-32]. Mitochondria are the major intracellular targets of soluble Aβ oligomers (sAβ) that in excess, could interfere in the integrity of the mitochondrial membrane and in its functionality, causing overproduction of OS, inhibition of cellular respiration and ATP production [33-35]. Enough data support that sAβ interfere in mitochondria by a result of changes in the homeostasis of intracellular Ca2+ signaling, causing ion massive influx in mitochondria and neuronal apoptosis [33]. The increase in the concentration of Ca2+ in mitochondria causes the opening of the mitochondrial permeability transition pore (MPTP), allowing the uncontrolled bidirectional passage of large molecules, resulting in disintegration of organelles and functional structure [34, 35]. The associative effect of all these pathophysiological changes, including protein fragments deposition (Aβ and tau), coupled with the uncontrolled production of radical species, are crucial for the installation and progression of a complex neuroinflammatory process [36-40]. In the brain defense system, microglial cells play a macrophage-like role and seem to have pivotal importance in neuroinflammation associated with AD and other NDs. Under physiological or adverse conditions, these cells monitor their environment and regulate tissue homeostasis through scavenging functions [36]. During their regulatory functions in brain homeostasis, these cells can undergo changes in their metabolism and morphology, leading to two types, named resting and activated microglia. Resting microglia may turn into other distinct phenotypes depending on the signals received and could origin an M1 state, that releases pro-inflammatory cytokines and other cytotoxic substances, which activate astrocytes and reinforce inflammation and neurodegeneration [36, 37, 39-41].
PD is the second most common type of ND, with estimates of 10 million people affected worldwide and around 60,000 of Americans being diagnosed yearly. These numbers represent more than the combined number of people diagnosed with multiple sclerosis, muscular dystrophy and ALS [9, 42]. Although PD is known as a movement disorder, with the patient showing muscle rigidity, postural instability, resting tremor and mobility slowness, as far as the disease progress, its symptoms also include a multitude of non-motor features such as impairment in cognition, smell and sleep, autonomic dysfunction and depression. The characteristic motor impairment observed in PD is mainly attributed to the reduced level of striatal dopamine secondary to the degeneration of dopaminergic neurons in substantia nigra [43-45]. As observed for AD, ageing is the main risk factor, being rare in people younger than 50’s, but the incidence rises 5 to 10-fold between ages from 60’s to 90’s [9, 42]. The etiology of PD remains unclear, but it is a consensus about the pathophysiological hallmarks based on the loss of dopaminergic neurons in the substantia nigra and striatal projections with a widespread intracellular deposition of α-synuclein aggregates, forming the so-called Lewy bodies [43]. Overall, the current literature data support that two phenomena are differently related to the disease progression: one associated with neuronal loss as the disease progresses and another as a consequence of neurotoxicity caused by the abnormal accumulation of Lewy bodies. The second mechanism seems to be dominant in patients with late-onset PD [44]. After decades of research, a single cause for PD has not been found and is unlikely to emerge, in spite of several studies which suggest that increased neuronal α-synuclein protein levels are a primary factor in the disease. Recent findings about etiopathogenic mechanisms and interactions in the dopaminergic cells of the substantia nigra in PD have contributed decisively for a best comprehension of how biochemical and physiological changes are interconnected and contribute to the disease progression and severity. In PD, neuronal death may be caused by changes in protein processing, leading to aggregation and deposition of misfolding α-synuclein and formation of neurotoxic Lewi bodies [43-45]. For unclear reasons, brains with PD suffer a dramatic dysfunction in the proteasomal and lysosomal systems, with reduced mitochondrial activity. As described earlier for AD’s pathophysiology, an emerging concept is that homeostasis in specific brain regions is vulnerable to different genetic, cellular and environmental factors that independently or concomitantly cause neuron apoptosis over time, with important secondary changes including the balance of cellular Ca2+, excitotoxicity and inflammation [44]. Dopamine metabolism is considered a critical step for neuronal vulnerability in ventrolateral substantia nigra. Mediated by enzymes, such as monoamine oxidase (MAO), catechol-O-ethyltransferase (COMT) and aldehyde-dehydrogenase (ALDH), dopamine metabolism involves the production of highly reactive species that oxidize lipids, increases OS and contributes to mitochondrial dysfunction. Dopamine is capable to auto-oxidize at neutral pH, but its reduced sequestration into synaptic vesicles, where the environment is acidic, auto-oxidation is not possible and may represent a neuronal vulnerability factor [44-46]. Dopamine neurons with low dopamine transporter activity in the cell membrane are less susceptible to neurotoxins or dopamine-induced OS and, in turn, are less affected in PD. Although it remains unclear if OS occurs early or late during neuronal death, there is a consensus that it is present in brains under the neurodegenerative process, as a consequence of mitochondrial impairment. Nigral dopaminergic neurons have been suggested as particularly vulnerable to OS and brain metabolism due to their long unmyelinated axons, with a large number of synapses and a consequent high energy demand. In addition, these neurons exhibit a peacemaking activity involving balanced levels of cytosolic Ca2+ as the expense of energy and increased levels of dopamine and its metabolites into the cytosol could increase OS. The association of mitochondrial dysfunction and exacerbated OS could then lead to a reduction in lysosomes, impairment in lysosomal autophagy system and the consequent disturbance in the clearance process of digestion of misfolding proteins and their aggregates as well as damaged mitochondria [9]. Neuroinflammation, with T cells infiltration, microglia and astrocyte activation is another common pathophysiological aspect with great relevance in NDs, especially for AD and PD. In vivo studies strongly suggest the participation of immune system in PD pathogenesis, once high levels of glial cells activation in substantia nigra and striatum, with production of high level of pro-inflammatory mediators including tumor necrosis factor-alpha (TNF-α), interleukin-1β (IL-1β), interferon-gamma (IFN-γ) have been identified in midbrain of PD patients [9, 45]. Evidences that microglial cells accumulate around α-synuclein aggregates widespread in PD brain, and that overproduced mutants or misfolding α-synuclein are able to maintain glial activation and the releasing of inflammatory mediators have supported that neurotoxicity induced by excessive or misfolded protein may be, at least in part, caused by microglia-mediated inflammatory responses [45-47]. As the disease progress, adenosine triphosphate (ATP) and metalloproteinase-3 (MMP-3) are released by damaged dopaminergic neurons, further enhancing microglia activation and amplifying inflammatory response, establishing a vicious cycle for neurodegeneration. Acting as a neurotransmitter, ATP released by damaged neurons and activated astrocytes controls migration of microglia to injured tissue. In addition, ATP binds to P2Y receptor mainly expressed in microglia, inducing overproduction of nitric oxide (NO) and cytokines. In vitro results suggest that, in addition to ATP, MMP-3 is another protein produced by injured neurons that play important roles in microglial activation and OS exacerbation [45]. In accordance to a number of other authors, our current understanding of PD points to a multifactorial cause for cell death in the substantia nigra. The combination of all these alterations with mitochondrial dysfunction, OS, abnormal protein degradation due to alterations in the ubiquitin system or in chaperone-mediated autophagy, and to other forms of subcellular dysfunction make clear the complex multifactoriality underling the progression of PD (and AD) and the consequent challenge to a holistic understanding of its pathophysiology to support the discovery of new efficient disease modifying drugs.
ALS is a group of rare, progressive, incurable and fatal late-onset neurological diseases, which involves motor neurons, responsible for controlling voluntary muscle movement. Most cases are sporadic (90-95%), affecting 2.7 per 100,000 individuals and the other 5-10% of the cases are familial-type ALS (FALS) and are associated with a genetic dominant inheritance factor. Multiple studies have shown a 1.5-fold higher prevalence for men to women, with an average peak age at 60 years for sporadic disease and 50 years for FALS [48, 49]. The first symptoms usually appear between 50’s and 60’s, including muscle weakness, twitching and cramping, which eventually can lead to muscle impairment. As the disease progresses, patients develop symptoms of dyspnea and dysphagia. The hallmarks of ALS are the progressive loss of the upper and lower motor neurons at the spinal or bulbar level. Neuronal motor system involves signal transmissions from motor neurons in the brain (upper motor neurons) to motor neurons in the spinal cord and to motor nuclei of brain (lower motor neurons) and from the spinal cord and motor nuclei of brain to muscles [48-50]. In ALS, both types of motor neurons degenerate with a consequent interruption of signaling between the brain, spinal cord and muscles. Under this pathological condition, muscles are unable to function and gradually weaken, start to twitch and finally waste away (atrophy) [50]. Over time, all muscles get affected, and individuals are unable to speak, eat, move and even breathe. At late stages, usually ALS patients die from respiratory failure, usually within 3 to 5 years from the first symptoms [48-50]. The etiology of ALS is uncertain, but exposure to environmental toxins, chemicals (e.g. pesticides, insecticides, herbicides and fertilizers), heavy metals and cigarette fume seems to have a pivotal role in the ALS-related neurotoxicity, contributing decisively for increased OS and inflammation. Despite the molecular basis of ALS remains an intriguing issue, mutations in gene encoding copper/zinc-superoxide dismutase (Cu/Zn-SOD) are considered as the primary cause of the disease. Structurally unstable, the misfolded mutant SOD can form aggregates and deposits widespread in motor neurons in CNS. Despite apparently not directly connected to the ALS onset, the most important hypothesis for its pathogenesis points out glutamate excitotoxicity, mitochondrial dysfunction, impaired axonal transport and OS as key factors for the disease progress [48]. Glutamate is transported from presynaptic terminals, where it is synthesized, to synaptic vesicles by specific vesicular transporters. Under physiological conditions, glutamate is released in the synaptic cleft to activate postsynaptic receptors and then, be readily removed by several glial and neuronal cell transporter proteins. This continuous process of release and uptake of glutamate balance the concentration gradient and avoid induction of excitotoxicity. However, under ALS conditions, neuronal tissue has a reduced astroglial glutamate transporter, which allows the increased concentration of the neurotransmitter in the synaptic cleft and starts excitotoxicity and neurodegeneration [48]. In addition, excess of glutamate into synapsis environment causes an excessive Ca2+ influx and overactivation of glutamate receptors, such as α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) and N-methyl-D-aspartate (NMDA), formation of permeability transition pores, release of cytochrome c into the cytosol, overproduction of ROS and disturbance in the energy production [9, 48].
HD is a rare, progressive and incurable autosomal-dominant ND, characterized by a triad of motor, cognitive and psychiatric features. It is the most common monogenic neurological disorder in the developed world, with a typical onset in the prime of adult life, around a median age of 40’s, and showing irreversible progress during 10-15 years [51, 52]. Due to an apparent strong genetic component, HD is endemic to all populations, but with a higher prevalence among individuals of European ancestry. These differences are ancestry-specific, as shown in British Columbia and Canada, where HD is much more common among those people of European descent, with estimates of 17 cases per 100,000 people, whereas in the ethnically diverse remainder of the population, the incidence is around 2 cases per 100,000 inhabitants [51-53]. From these data, HD was originally thought, has migrated from North-West Europe to other parts of the world. The genetic origin of HD is related to an expanded CAG trinucleotide repeat in HTT, a gene that encodes huntingtin protein, which lies on the short arm of chromosome 4. Under mutation conditions, huntingtin is produced with abnormally long polyglutamine (PolyQ) stretch at the N-terminus of the mutated protein, which confers toxicity and predisposition for fragmentation, leading to neuronal dysfunction and apoptosis [51, 52]. Despite HD is recognized as a whole-brain disease, medium spiny neurons of the striatum are particularly vulnerable to mutant huntingtin-induced injury. Although migration is undoubtedly a major contributor to HD prevalence, recurrent mutations arising from alleles of intermediate repeat length in the general population also make a significant contribution to ethnical-dependent prevalence [51]. Huntingtin is expressed throughout the body, but depending on the cell type, its levels may vary into the nucleus or cytosol as well. Complete physiological functions of huntingtin remain unclear, but some broad biological functions are known, including a critical role in the development of the nervous system, a regulatory influence on the production and transport of brain-derived neurotrophic factor (BDNF) and in cell adhesion. Several experimental evidences suggest that structural instability and fragmentation of huntingtin is a key early step in HD pathogenesis. Depending on the HTT expression, the concentration of huntingtin fragments into the cells can be different, and its higher levels in neurons than in glial cells are likely to contribute to the prevalent neuronal pathology [53]. Regardless of the affected brain region and type of aberrant protein, oligomerization, aggregation and their deposits in the neuronal environment, HD shares a set of concomitant and interconnected protein-related biochemical events with other NDs. In a similar manner as shown earlier for Aβ and Tau (AD), α-synuclein (PD) and SOD (ALS), once aberrant huntingtin is formed in HD, it influences changes and dysfunctions in many other downstream cellular and biochemical processes, including activation of immune system, secretory pathway, mitochondrial impairment, synaptic dysfunction, OS, microglial activation and neuroinflammation leading to an extremely complex and multifaceted pathogenicity [51-53].
1.2. The MTDLs Paradigm and Molecular Hybridization as a Tool in Drug Design
In light of the variety of interconnected factors associated with the development and pathophysiological complexity of NDs, along with the inefficiency of the current chemotherapeutical alternatives, it becomes unavoidable a strategic re-think about the rational design and prospection of novel drugs against chronic multifactorial diseases such as NDs. To date, approaches in drug design are still based in the reductionist concept of “one gene-one drug-one target”, leading to a linear model for a clinical effect, by which a single molecule binds selectively to a single molecular target resulting in a desired therapeutical response and undesired adverse effects [54-56]. Conversely, chronic diseases with multiple biochemical and cellular events operating concomitantly have been focused in the light of polypharmacology. By this strategy, three therapeutical strategies are possible and two have been currently widely practiced in the clinics: i) administration of drug cocktails, with more than two drugs in different medications aiming different effects for each one and ii) the combination of two or more drugs into a same pharmaceutical formulation, each one expected to exert a particular therapeutical effect. In a third, and more recent polypharmacology-based strategy, desired multiple pharmacological effects could be reached from a single molecule, where the chemical structure is constituted by more than two different pharmacophores, could being concomitantly recognized by multiple biological targets [2, 6, 7]. In spite of the possible modulation of diverse molecular targets by a single molecule, this approach could also circumvent deleterious or inefficient effects due to the administration of multiple combined or associated drugs due to their distinct pharmacokinetics, toxicity, physic-chemical properties, bioavailability, costs and, especially, drug-drug interactions [57, 58].
Based on this new paradigm, the concept of rationally designed multitarget-directed ligands (MTDLs) gains special attention from the scientific community, especially in the academy. In this context, Molecular Hybridization (MH) has been extensively used as a keytool to design new structural patterns with potential multifunctional properties. The basis of MH in the design of a new structural architecture is the previous recognition of pharmacophoric subunits into the molecular core of two or more known bioactive prototype molecules. Then, the selected structural fragments from each selected prototype are combined through an adequate linkage, leading to new hybrid architectures that is expected to maintain the pre-selected characteristics of the original templates [2, 19, 59]. Considering that biophore substructures of known bioactive molecules are used in the MH-based design, and that original templates have been already evaluated for their toxicity, physic-chemical properties (e.g. solubility, chemical and plasmatic stabilities), pharmacological features and, perhaps, a plausible mechanism of action, it is possible that the generation of extensive chemical libraries of molecular hybrids tends to make less erratic the challenge of the discovery of effective lead-molecules [1, 2, 18, 60-62]. These molecular hybrids could bring particular innovation in pharmacodynamics, pharmacokinetics and, at the structural level, with rational selective affinity for multiple targets, preferably in different biochemical cascades. In the following section, we will discuss how MH has been exploited during the last decade as a new tool for rational drug design and discovery, especially focused on the most high-impact neurodegenerative diseases AD, PD, ALS and HD.
2. MOLECULAR HYBRIDS DESIGNED AS PROTOTYPES OF DRUG CANDIDATES FOR AD
2.1. MTDLs Inspired by Donezepil
As the current first therapeutical choice among the other few approved drugs for AD treatment, donepezil (1) has aroused great interest by researchers. Its mechanism of action is based on the specific and reversible inhibition of acetylcholinesterase (AChE), and thus inhibiting acetylcholine hydrolysis, and generating, in turn, a set of pharmacological benefits by affecting cellular and molecular processes of neurodegeneration [63]. By maintaining higher levels of ACh, donepezil may help compensation for the cholinergic deficit, one of the main hallmarks of AD [64]. Clinical studies revealed that the use of donepezil results in significant improvement in memory, concentration, language and reasoning, with no signs of toxicity, but without curative effects [18]. As a consequence, a number of examples in the literature highlight the structure of donepezil as a model in the design of new multi-target directed drug candidates by MH.
Recently, our group reported the design and synthesis of donepezil-based hybrid compounds with a multifunctional mode of action. The rationale behind the design of the new scaffold 4 (Fig. 1) was based on the combination of the N–benzylpiperidine pharmacophore from 1, with the insertion of a 3-hydroxy substituent at the heterocyclic moiety present in the AChE inhibitor (AChEI) LASSBio-767 (2) and a N-acyl-arylhydrazone functionality as an auxophore subunit present in different anti-inflammatory molecules from the literature (3a-3d, Fig. 1). Among sixteen analogues, derivatives 4a-4d showed the best selective AChE inhibitory activities, with IC50 values of 25.39, 2.71, 8.65 and 10.39 µM for AChE and 48.22, 33.86, 39.04 and 58.14 µM for BuChE, respectively. In addition, in vitro and in vivo results disclosed that compounds 4a and 4c could inhibit the Aβ-oligomers-induced neuroinflammatory response. Taking all this data, compound 4a, that also showed a better activity in neuroprotection, a donepezil-like binding mode to AChE and adequate ADME properties in computational studies, has been described as a novel MTDL drug prototype for AD [65].
Fig. (1).

Rational design of a new series of N-benzyl-piperidinyl-aryl-acylhydrazone derivatives 4 as donepezil hybrids and compounds 4a-4d as the most active AChEIs with anti-inflammatory and neuroprotective activities.
Natural products are also important source of inspiration for the construction of promising new molecular patterns focusing on the treatment of NDs. For example, ferulic acid (FA, 5), an abundant natural phenolic constituent of the secondary metabolite spectrum of important medicinal plants, has been taken as a molecular scaffold for designing innovative MTDLs. Ferulic acid exhibits strong antioxidant and anti-inflammatory effects, besides the inhibition of Aβ fibril aggregation and prevention of Aβ-mediated toxicity both in vitro and in vivo. Recently, Li and co-authors synthesized and evaluated a series of ferulic acid-donepezil hybrids (6, Fig. 2) designed as a potential AChEI endowed of extra-properties such as metal chelation, antioxidant and inhibition of amyloid peptide (Aβ) aggregation due to the insertion of a ferulic acid structural subunit. Particularly, compound 6a exhibited the highest inhibitory potency on BuChE, with IC50 values of 3.4 μM, and moderate antioxidant activity, with a value 1.1-fold trolox equivalents. In addition, 6a showed inhibitory effects on self-induced Aβ1-42 aggregation (61.1%) and was capable to disaggregate self-induced Aβ1-42 fibrils in 53.1% at 25 µM. Furthermore, compound 6a could cross the blood–brain barrier (BBB) in vitro and showed significant neuroprotective effects against H2O2 -induced PC12 cell injury, with low toxicity in PC12 cells [66].
Fig. (2).

Design strategy for the new series of ferulic acid-donepezil hybrids (6).
This strategy was also used recently by Sang and co-workers that combined the 4-benzylpiperidine pharmacophore of donepezil (1) with ferulic acid (5) and secondary amines 7 (Fig. 3). The presence of secondary amines was justified by previous results demonstrating that the tertiary amino group linked to a lipophilic moiety represents a key requirement for a good AChE inhibition. In vitro studies displayed that all target-compounds showed excellent selective inhibition of butyrylcholinesterase (BuChE). Particularly, compound 8a was identified as the most potent BuChE inhibitor with IC50= 0.021 µM for EqBuChE, 8.63 µM for rat BuChE and0.07 µM for hBuChE. Molecular docking studies of 8a revealed a weaker affinity for AChE, explaining the high selectivity for BuChE. Compound 8a also showed modest antioxidant activity (0.55 eq of Trolox), good protective effect against H2O2-induced PC12 cell injury, with low toxicity. Moreover, compound 8a proved to be an excellent inhibitor of self-induced Aβ1-42 aggregation (50.8%), counteracting self-induced Aβ1-42 aggregation (38.7%) and good in vitro ability to cross the BBB [67].
Fig. (3).

Design and structure of a new series of ferulic acid-O-alkylamine hybrids (8) and the most potent derivative 8a.
Another new family of multi-target molecules able to interact with both AChE and BuChE was synthetized by Sang and co-workers with combination of the structure of JMC49 (9) and donepezil (1, Fig. 4). The most promising 3,4-dihydro-2(1H)-quinoline-O-alkylamine derivative 10, showed potent and balanced inhibitory activities toward AChE, BuChE, and human MAO-A and MAO-B with IC50 values of 0.56 µM, 2.3 µM, 0.3 µM and 1.4 µM, respectively, but with low selectivity. Both kinetic analysis of AChE inhibition and molecular modeling study suggested that 10 binds simultaneously to the catalytic active site and peripheral anionic site of AChE. Compound 10 was thought to be an attractive starting point for further lead optimization in the drug discovery process against AD [68].
Fig. (4).

MH of JMC49 (9) and donepezil (1) in the design of the most active 3,4-dihydro-2(1H)-quinoline-O-alkylamine derivative 10.
In another approach, our group worked in the design of compounds with an innovative structural framework that could act as strong modulators of the neuroinflammatory process associated with NDs, combining a selective AChEI activity with a concomitant antioxidant, biometal chelation and neuroprotective properties. For this goal, a series of feruloyl-donepezil hybrid compounds were designed by the combination of the N-benzylpiperidine pharmacophore from 1 and the antioxidant feruloyl fragment present in curcumin and ferulic acid (Fig. 5). In the investigation of cholinesterase inhibition, compounds 12a-c showed to be the best AChEIs, with 12a as the most potent derivative with an IC50 of 0.46 μM. Further studies revealed significant neuroprotective activity for compound 12a, counteracting oxidative-induced damage in human neuronal cells. In different animal models, 12a also exhibited anti-inflammatory effects, probably by inhibiting cyclooxygenase (COX) 1 and 2. These sets of multiple potential effects on neuroprotection, oxidative stress and inflammation, may suggest that this compound could be an interesting prototype for development of innovative drug candidates for, not only AD, but also for other NDs, such as PD and HD [69].
Fig. (5).

Design strategy and chemical structures for the most potent ferulic acid-donepezil hybrids 11a-c and 12a-c.
Mishra and co-workers also reported the synthesis and evaluation of a novel series of donepezil-based multi-functional agents to inhibit AChE and Aβ aggregation (Fig. 5). The strategy consisted of joining an active part of the donepezil to a curcumin fragment, with the insertion of a piperazine moiety within a single molecule. The authors installed a piperazine scaffold aiming to provide the best anti-AD efficacy, due to earlier findings that piperazine and its derivatives exhibit a broad spectrum of CNS activities. Biological results revealed compounds 11a (IC50 = 0.045 µM), 11b (IC50= 0.034 µM), and 11c (IC50 = 0.025 µM) as the most active ChEs inhibitors, also shown excellent inhibition of Aβ-aggregation in 78.2, 80.4 and 81.6%, respectively. These three compounds also successfully diminished the H2O2-induced oxidative stress in SH-SY5Y cells and displayed excellent neuroprotective activities [70].
In the hippocampus and cerebral cortex, the NO/sGC/cGMP signaling pathway plays a pivotal role in the processing of learning and memory. Recent studies have shown that the inhibition of the NO/sGC pathway alters the expression and activity of NOS, sGC, and phosphodiesterase (PDE) [71]. PDEs belongs to one super enzyme family in charge of hydrolyzing cGMP and contributes to Aβ neuropathology. Phosphodiesterase-9 (PDE9) inhibitors could regulate the NO/sGC/cGMP signaling pathway, which plays a very central role in synaptic transmission and plasticity. Therefore, new PDE9 inhibitors, such as compound 13 (Fig. 6), have been developed for the treatment of AD. Based on this findings, Hu and co-workers designed a series of pyrazolopyrimidinone-donepezil hybrids 14, by a combination of a pyrazolopyrimidinone moiety from 13 and a benzylpiperidine subunit (Fig. 6), as novel MTDLs for the treatment of AD. Among all tested derivatives, compounds 14a (IC50 = 0.048 µM for AChE and IC50 = 0.530 µM for PDE9A) and 14b (IC50 = 0.223 µM for AChE and IC50 = 0.285 µM for PDE9A) exhibited the best balanced dual-target AChE/PDE9A inhibitory activities. Meanwhile, these two compounds exhibited good BBB permeability and low
Fig. (6).

Design strategy of a series of donepezil-PDE9A hybrids (14).
neurotoxicity. In special, compounds 14a and 14b could ameliorate learning deficits induced by scopolamine. Moreover, 14a could also improve cognition and spatial memory in mice with Aβ25-35 induced cognitive deficits in the Morris water-maze test. In view of these results, compound 14a was considered a potential drug candidate for AD [72].
The structure of donepezil (1) was also used as inspiration by Hiremathad and co-workers in the drawing of a hybrid donepezil-like scaffold (15, Fig. 7), endowed with inhibitory properties of AChE, β-amyloid aggregation in addition to metal chelation and radical scavenging activities. The design strategy was based on the conjugation of a benzyl-hydroxypyridinone and benzofuran moieties as analogues of the main molecular fragments of 1, the benzylpiperidine and the indanone moieties, connected to each other through alkyl linkers of different lengths. Docking studies revealed that the target-compounds were able to interact with both binding sites, the catalytic anionic site (CAS) and peripheral anionic site (PAS) of the AChE. In addition to good AChE inhibition, these results suggested a possible inhibition of the formation of the senile plaques, since the PAS allosteric site of AChE is associated with Aβ-aggregation [73]. Based on this premise, a series hydroxypyridinone-benzofuran hybrid (HP-BF (16), Fig. 7) was prepared and evaluated towards AChE inhibition, anti-Aβ-aggregation, metal chelating ability, radical scavenging activity and neuroprotection. Compounds 16a-c showed a significant multiple-action profile, exhibiting moderate inhibition of AChE (IC50= 76, 246 and 408 µM, respectively) and Aβ-aggregation (36.1, 49.8 and 59.3%, respectively). In particular, derivatives 16b and 16c also showed radical scavenging activity with EC50 values of 199 and 227 µM, respectively and good metal chelation ability for Fe3+ and Cu2+ [73].
Fig. (7).

Design strategy of the series of hydroxypyridinone-benzofuran (HP-BF) hybrids (16).
Considering the structures of the representative indanone 17 and the benzylidene-indanone derivatives such as compound 18, previously reported as promising AChE and Aβ-aggregation inhibitors, respectively, Huang and co-workers designed a new series of indanone hybrid derivatives (19, Fig. 8). With the goal of identifying novel dual AChE and Aβ aggregation inhibitors, fourteen indanone derivatives were obtained and evaluated, revealing that most of them were potent and effective in the inhibition of AChE, with IC50 values in the nanomolar range. In particular, the most potent compounds 19a (IC50= 14.8 nM) and 19b (IC50= 18.6 nM) exhibited a similar potency to donepezil and a markedly higher inhibitory activity than tacrine https://www.sciencedirect.com/topics/chemistry/tacrine. Further investigations showed that compounds 19a-b also exhibited antioxidant activities and were capable to similarly inhibit Aβ aggregation in 85.5% and 83.8%, respectively, and catalyzed the disaggregation of Aβ fibrils generated by self-induced Aβ aggregation. In the PAMPA assay, both compounds showed good ability to cross BBB, which highlighted them as potential ligands for further development of multifunctional drug candidates for AD [74].
Fig. (8).

Design of the optimized multifunctional indanone hybrids 19a-b by MH of the indanone derivative 17 and benzylideneindanone 18, with AChEI, antioxidant, anti-Aβ aggregation and Aβ-disaggregation properties.
Two novel series of compounds (21 and 22, Fig. 9) were obtained as MTDLs against AD by Yan and co-authors as a result of a rational combination of structural fragments from 1 and curcumin (20), aiming novel ligands as AChEIs, metal chelators, antioxidants and inhibitors of Aβ-aggregation. From the series 22, compound 22a displayed a potent AChE inhibition (IC50= 187 nM), with the highest 66.3-fold BuChE/AChE selectivity, and a 45.3% of inhibition of Aβ1-42 self-aggregation at 20 µM. Besides, 22a displayed remarkable antioxidant effect, a significant metal-chelating ability for Cu (II) and an excellent BBB permeability [75].
Fig. (9).

Design of new donepezil-curcumin hybrid series 21 and 22, and the structure of the most active MTDL 22a.
The monoaminoxidases (MAO) A and B could be considered as interesting molecular targets for the development of drugs against NDs, once these enzymes catalyze the deamination of a variety of endogenous amines with the concomitant production of H2O2. Hydrogen peroxide is the reagent for Fenton reaction that produces ROS and, in turn, exacerbates the oxidative stress and neuronal damage. Based on these findings, and seeking for innovative multifunctional ligands acting simultaneously in the enzymatic inhibition of the MAO and ChEs, Wang and co-workers elected the propargylamine as pharmacophore for MAO-A and B inhibition. This structural fragment was rationally coupled to an 8-hydroxyquinoline subunit aiming to improve the metal chelating ability from the prototype M30 (25), combined to an N-benzylpiperidine as AChEI subunit, present in donepezil (1) and the MAO/AChEI ASS234 (24), as reported previously by the same group. As a result, the authors obtained another series of molecular hybrids with the general scaffold 25 (Fig. 10). Among them, the racemic compound 25a showed the best multi-target profile, being capable to inhibit MAO-A and B (IC50= 6.2 and 10.2 μM, respectively), AChE and BuChE in an almost non-selective way (IC50= 1.81 and 1.6 μM, respectively. Kinetic studies with 25a evidenced an irreversible inhibition of MAO A/B and a mixed-type inhibitory mode of AChE, with also metal-chelating ability. In silico studies suggested that both enantiomers of 25a could accommodate in a similar manner in the cavity of MAO-A, MAO-B and ChEs, with comparable affinities in each case, which may explain the low selectivity in both enzyme inhibition. In vivo assays confirmed that 25a was capable to revert induced-impairment in memory and cognition, which coupled to the adequate predicted ADMET properties, less toxicity than donepezil at high concentrations and a similar cell viability at low concentrations, make this compound an interesting disease-modifying candidate for future development for AD and PD [76].
Fig. (10).

General structure of multifunctional DPH hybrids and the most promising 25, designed by MH of the structures of donepezil (1), M30 (23) and ASS234 (24).
Wu and co-workers planned another series of donepezil-based hybrids searching for novel ligands capable of acting as multifunctional inhibitors of AChE and MAO, with additional antioxidant properties. The new scaffold 28 was devised by combination of the N-benzylpiperidine, as an AChEI pharmacophore, with a metal chelating moiety represented by the 8-hydroxyquinoline system (26) and a propargylamine functionality (27, Fig. 11). Among all compounds synthesized, derivative 28a showed a selective MAO inhibition with IC50 values of 10.1 and 100 μM for MAO-A and MAO-B, respectively, and an interesting, but non-selective, AChE inhibition (IC50= 0.029 μM) and BuChE (IC50= 0.039 μM). In addition, compound 28a showed a significant ability for chelating Zn2+ and Cu2+ ions and antioxidant activity with 1.12 trolox equivalents in the ORAC assay [77].
Fig. (11).

Design of new series donepezil-8-hydroxyquinoline-propargylamine hybrids (28) and compound 28a, a potent ChE and MAO inhibitor, with antioxidant and metal chelating properties.
In another attempt for the discovery of new AChEIs for AD, the arylsulfonylhydrazone scaffold 31 (Fig. 12) was explored by Fernandes and co-workers. The authors used MH between a previous reported donepezil-based piperazine-acylhydrazone derivative 29 and acylhydrazone-4-quinolone derivatives 30, with moderate activity on AChE, BuChE and Aβ-aggregation to generate a novel structural pattern with a bioisosteric exchange of the acylhydrazone moiety in 29 for the sulfonylhydrazone in 31. Biological results disclosed that among fifteen of these hybrids, twelve showed IC50 values in the range of 0.64 µM to 51.09 µM. A SAR analysis suggested that the methylcatechol moiety and arylsulfonyl substituents were responsible for the best AChE inhibition than both the benzodioxole and alkylsulfonyl chains [78].
Fig. (12).

Design of sulfonylhydrazone analogues and compound 31a.
In silico studies evidenced that, in fact, the most potent AChEI 31a could interact with the PAS of AChE in a similar manner of donepezil. Furthermore, compound 31a showed antioxidant activity, with no significant toxicity on LL24 cells and adequate predicted oral absorption and brain penetration, which was considered a promising profile for further development against AD [78].
Coumarins (2H-chromen-2-one) are natural occurring benzopyrones from plants with several reported biological activities, including antioxidant and anti-inflammatory. Coumarin is considered a privileged ring system present as a structural fragment in a variety of AChEIs, such as hymecromone (32a), umbellipherone (32b), and ensaculin (33), which are capable to interact with PAS site of AChE. Based on these findings, and considering that amine functional group is also necessary for interactions with CAS of AChE, as elicited by donepezil, compounds 32 were used as structural models in the design of a novel hybrid family of umbellipherone and benzylamine scaffolds 34 (Fig. 13), using a triazole ring as linker subunit. Thus, varying the nature of substituents in the benzyl system, a series of benzyl-triazole-umbellipherone hybrids was synthesized and evaluated for their activities on AChE and BuChE, as well as neuroprotection against H2O2-induced cell death in PC12 neurons. Compound 34a (Fig. 13), bearing 3-methoxy substituent on benzyl moiety was identified as the best inhibitor of AChE (IC50= 3.4 µM), with a 3-fold higher selectivity than for BuChE (IC50= 1.1 μM). Neuroprotective evaluation against hydrogen peroxide-induced damage in neuronal cells disclosed 34a as the most promising derivative with 72.5, 76.4 and 83.3% of cell viability when tested at the concentrations of 1, 10 and 100 µM, respectively, in comparison to quercetin used as reference drug with 90.8% of cell viability at 10 µM. These results are suggestive that coumarin-based ligands may be further investigated for the development of new ChE inhibitors targeting AD treatment [79].
Fig. (13).

Structure of representative coumarins 32 and 33 and structural design of a series of multifunctional hybrids 34, with compound 34a as the most active.
In another work, Valencia and cols. described the synthesis and evaluation of donepezil-flavonoid hybrids (36), designed by MH between the structure of donepezil (1) and a flavonoid nucleus, represented by luteolin (35a) and apigenin (35b, Fig. 14). Compound 36a was identified as the best AChEI (IC50= 46 nM), with moderate inhibition of 5-LOX (IC50= 74.3 μM), MAO-A (IC50= 15.3μM) and MAO-B (IC50= 5.2μM), and a good BBB permeability of 5.9x10-6 cm/s [80].
Fig. (14).

Donepezil_Flavonoid hybrids 36 with inhibition of AChE, MAOs, 5-LOX and blood-brain permeable permeability.
The N-benzylpiperidine pharmacophore subunit of donepezil (1) was combined with the coumarin framework 37 into a single molecule, in the design of hybrid ligands with expected inhibitory activity on ChEs and MAO-B. This work represents a tentative optimization of an earlier series of tacrine-coumarin hybrids as MTDLs for the treatment of AD, represented by compound 38 (Fig. 15) with good enzyme-inhibitory activity, but with tacrine-like hepatotoxicity [81]. In that previous study, tacrine was used as a structural pattern aiming for the inhibition of ChEs, while the coumarin moiety was though a pharmacophore for MAOs inhibition. In another attempt, the authors decided for the replacement of tacrine fragment for the N-benzylpiperidine, once donepezil is a potent AChEI, without significant toxicity. Thus, a novel series of donepezil-coumarin hybrids was synthesized and evaluated for their activity on ChEs and MAO inhibition, disclosing that most of the compounds displayed potent inhibitory activity for AChE and BuChE, besides highly selective inhibition of MAO-B. Compound 38a was identified as the most potent non-selective inhibitor for eeAChE and eqBuChE (IC50= 0.87 μM and 0.93 μM, respectively), with additional good and balanced inhibition of human ChEs and MAO-B, with IC50 values of 1.37 μM for hAChE, 1.98 μM for hBuChE and 2.62 μM for hMAO-B. Kinetic and in silico studies evidenced that 38a is a mixed-type inhibitor, could binding simultaneously to CAS, PAS and mid-gorge sites of AChE, and showed a competitive mode of inhibition related to MAO-B. Results from membrane permeability assay (PAMPA) and toxicity on SH-SY5Y human neuroblastoma cells, evidenced that compound 38a has a good ability to cross BBB, with no cytotoxicity. As per these biological data, 38a might be considered a promising multifunctional lead candidate suitable to further investigation and development towards AD treatment [82].
Fig. (15).

Design strategy for the optimized donepezil-coumarin hybrids 38, the compound 38a, as the most promising AChE and MAO-B inhibitor.
A combination of structural fragments of donepezil (1) and butylated hydroxytoluene (BHT, 39) was the strategy adopted by Cai and cols. in the design of the donepezil-BHT core 40 (Fig. 16). Among all target-compounds, derivative 40a was highlighted due to its best and balanced multifunctional profile of action, inhibiting AChE (IC50= 0.75 μM) and MAO-B (IC50= 7.4 μM), with additional excellent antioxidant activity (IC50= 71.7 μM in the DPPH assay; 0.82 and 1.62 trolox equivalents in the ABTS and ORAC methods, respectively) and significant inhibition of AChE-induced Aβ aggregation. In addition, compound 40a displayed neuroprotective activity towards H2O2-induced oxidative damage in PC12 cells and lipopolysaccharide (LPS) stimulated inflammation in BV2 cells, with adequate BBB penetration elicited in PAMPA assay [83].
Fig. (16).

Donepezil-BHT hybrids (40) with anti-cholinesterase, anti-MAO-B, neuroprotective and antioxidant properties.
Ebselen (41), an organoselenium glutathione peroxidase mimic, with antioxidant and anti-inflammatory activities, has recently undergone clinical trials as a drug candidate for AD. Based on these data, its structure was used as a model in combination with a donepezil-like N-benzyl-aza-heterocycle subunit, as AChEI fragment, to furnish the new structural architecture 42 (Fig. 17). The resultant donepezil-ebselen hybrids were evaluated for their in vitro inhibitory effects on AChE, BuChE and H2O2-induced oxidative damage. Compounds 42a and 42b exhibited good, but not selective, AChE and BuChE inhibition with IC50 values of 0.76 and 0.46 μM, respectively. In relation to GPx-like activity, most of the compounds were able to catalytically reduce peroxide production, and particularly 42a and 42b exhibited higher activities than ebselen used as reference drug. In the H2O2-sequestering activity, compounds 42a and 42b showed a similar effect to 41, suggesting that these organoselenium hybrids could represent interesting innovation in the search for new multifunctional agents for AD treatment [84].
Fig. (17).

Molecular planning to obtain the hybrids 42a and 42b in the search for multi-target compounds for the treatment of AD.
Geniposide (43), an abundant bioactive iridoid from plants, can be hydrolyzed by β-glucosidase to generate genipin (43a, Fig. 18). These two natural metabolites and some other analogues have been reported for their ability in the relief of AD symptoms. Based on these findings, the structure of 44 was used in combination with the N-benzylpiperidine moiety of 1 for the design of a series of donepezil-genipin hybrids (45, Fig. 18). In the structural design strategy, the N-benzylpiperidine fragment from donepezil was modified by incorporating a bioisosteric substituted arylmethyl-piperazine portion at the C-8 position of genipin. Besides, the C-1 hydroxyl group of 44 was replaced by a methoxy group, aiming to contribute to chemical stability of the target-compounds. The biological evaluation revealed that most of the compounds had higher anti-AChE activity than 44, with derivative 45b (IC50= 218 nM) showing a comparable potency to donepezil. Interestingly, in the MTT test, used to investigate the neuroprotective activity in
Fig. (18).

MH of donepezil (1) and denipin (44) to generate hybrid compounds 45a-b.
Aβ-injured PC12 cells, compound 45a showed a higher neuroprotective effect than donepezil, suggesting that further studies should be performed in order to elucidate its neuroprotective mechanism [85].
Samadi and co-works synthesized a family of 6-chloro-pyridonepezils (47) planned to be new dual AChEIs as potential drugs for the treatment of AD. The structural architecture of the 6-Chloro-pyridonepezil scaffold was envisaged as a hybrid of chloropyridines and donepezil (1) hybrids in which the 2-chloropyridine-3,5-dicarbonitrile heterocyclic ring system of the 2-chloropyridine (46) is connected by an adequate polymethylene linker with the donepezil-pharmacophore N-benzylpiperidine. The resultant hybrid series was evaluated against human AChE and BuChE, revealing that most of the compounds were strong ChE inhibitors. The most potent derivative was 6-chloro-pyridonepezil (47a, Fig. 19) with IC50 values of 0.013 µM and 8.13 µM for hAChE and hBuChE, respectively, exhibiting a 625-fold higher selectivity AChE, and a potency comparable to that of donepezil (IC50= 0.01 µM). Results from PAMPA assay suggested that all tested compounds are able to penetrate BBB by passive diffusion. In addition, docking studies corroborated the AChE selectivity observed in vitro, showing that these 6-chloro-pyridonepezil hybrids, and especially derivative 47a, behave as dual hAChEIs, binding simultaneously to the CAS and PAS and could be suitable to the development for AD treatment [86].
Fig. (19).

Chemical structure of the promising 6-chloropyridine-donepezil hybrid 47, designed as dual ChEIs based on the structure of donepezil (1) and 2-choropyridine (46).
In an ambitious project for searching new potent cholinesterase inhibitors as drug candidates for AD, Jin and his research group designed and synthesized more than sixty hybrid molecules based on acridine, tetrahydroacridine and quinoline structural scaffold. Among all target-compounds tested, the most highlighted derivative was the N-benzyl-piperidinyl-6-chloro-tetrahydroacridine 49a (Fig. 20), a hybrid molecule based on the structures of tacrine (48) and donepezil (1). Compound 49a (dual binding inhibitor) was able of inhibiting AChE (IC50= 5.6 nM, Ki= 1.8 nM) and BuChE (IC50= 321 nM, Ki= 36.3 nM) with high selectivity. These findings make clear that the low selectivity of tacrine (a single site binder) for cholinesterase could be modulated by the insertion of the N-benzyl-piperidine pharmacophore from donepezil [87].
Fig. (20).
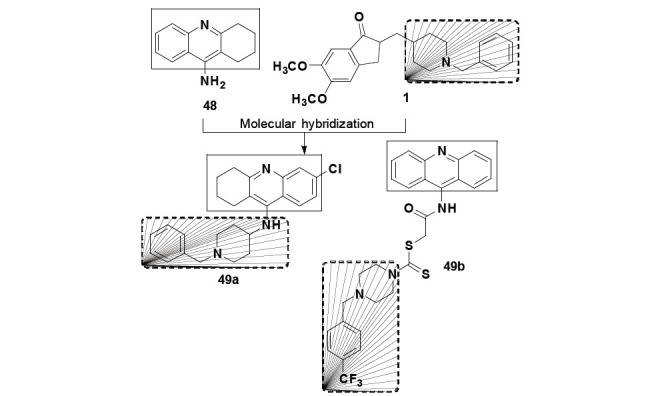
Chemical structure of compound 49a and 49b, the most promising derivative among a series of tacrine-donepezil hybrids.
More recently, Hussein and co-authors also used tacrine and donepezil as structural models for searching new AChE and BuChE inhibitors, proposing a novel tacrine-donepezil hybrid (Fig. 20). Among twelve hybrid derivatives, eight showed selective inhibitory activity for BuChE and no significant cytotoxicity towards NIH3T3 cells. In addition, both docking studies and in vitro evaluation pointed to compound 49b as the most promising and highly selective BuChE inhibitor (IC50= 41.129 and 0.014 µM for AChE and BuChE, respectively), with an adequate prediction for BBB permeability [88]. However, the results obtained on the inhibition of ChEs were no better than the reference compounds, donepezil and tacrine, and the authors did not discuss about the biological activity. Following the same strategy, Camps and co-workers developed another family of donepezil-tacrine hybrids using a methylene chain of different lengths as spacer subunit (Fig. 21). Among the new ligands obtained, compounds 50a and 50b should be highlighted for their very similar activity profile. Both compounds 50a and 50b were able to selectively inhibit hAChE with IC50 values of 0.67 and 0.27 nM, respectively. In addition to AChE inhibition, these were able to inhibit hAChE-induced βA aggregation by 37.6% and 46.1%, respectively, at the concentration of 100 µM [89]. Taking these results into account, Viayna and co-workers synthesized a second-generation of hybrid ligands inspired in the structure of 50a-b, varying the tacrine-related substructure (Fig. 21). Among a number of new ligands synthesized, the authors highlighted compounds 50c and 50d due to their good and selective AChE inhibition (IC50= 5.37 and 6.32 nM, respectively), although less potent than the previous prototypes. However, when evaluated for their effects on hAChE-induced βA aggregation, compounds 50c and 50d showed to be capable to inhibit 55.5% and 64.3% of oligomers aggregation at 100 µM, respectively. In addition, both compounds were able to inhibit βA self-aggregation in 71.5 and 83.7%, respectively, at 10 µM. Finally, compounds 50c and 50d were also capable to inhibit BACE-1 activity in
Fig. (21).
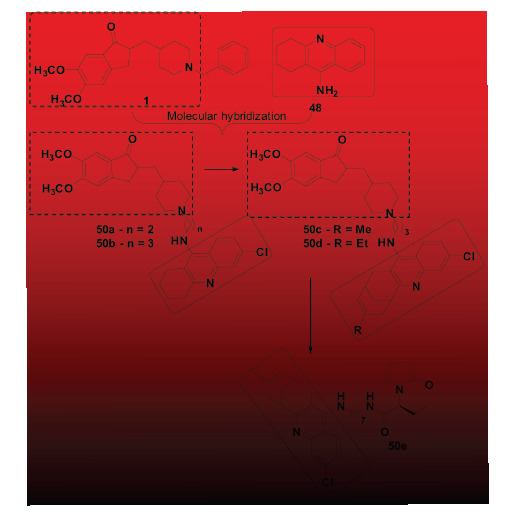
Design of a novel series of tacrine-donepezil hybrids (50).
29.1% and 19.2%, respectively, at a concentration of 5 µM [90]. A few years later, Sola and co-workers used these previous results to explore additional changes in the structure of the prototype 50c to generate the third series of optimized hybrids, preserving the tacrine-like fragment of 50c and modifying the remainder core (Fig. 21). From this new molecular architecture, the authors identified ligand 50e, as the most promising hybrid, which remains potent and selective hAChE inhibition (IC50 = 4.2 nM), also inhibiting βA aggregation (36.4% inhibition at 20 µM). In addition, 50e was also able to inhibit 54.1% of tau protein aggregation, at a concentration of 20 µM, showing a wider optimized multifunctional profile of action, could additionally act on the formation of intracellular neurofibrillary tangles [91].
2.2. MTDLs Inspired by Tacrine
Several tacrine-based compounds have been reported in the literature boosted by their inhibitory profile and high affinity for CAS and PAS of AChE. Thus, a number of tacrine hybrids have been rationally designed by the combination with a number of pharmacophores related to radical scavenging, antioxidant and anti Aβ-aggregation activities with the goal of modulation of oxidative stress and fibril deposition, besides ChE inhibition [92-99].
In this context and seeking for novel potent dual AChEIs, capable to bind concomitantly to CAS and PAS of AChE, Camps and co-workers designed a new series of hybrid compounds (53, Fig. 22), based on the structure of the selective AChEI 6-chlorotacrine (51) and the quinoline-ester derivative (52), a non-selective and weak AChE/BuChE inhibitor (IC50 > 10 µM). Although 52 has not shown significant ChE inhibitory activity, computational studies suggested that the quinoline scaffold could be capable to establish important π-π interactions with the aromatic residues Trp286 and Tyr72 at the PAS of AChE. In fact, the quinoline-6-chlorotacrine hybrid 53 was identified as the most potent compound of that series with IC50 values of 19 and 223 nM for hAChE and hBuChE, respectively. Despite their selective cholinesterase inhibitory properties, derivative 53 showed a weak anti Aβ-aggregating activity (29% of AChE-induced Aβ aggregation at 100 µM and 21% of self-induced Aβ-42 aggregation at 50 µM) [100].
Fig. (22).

Design, synthesis and optimization of hybrids 53 and 55 with multifunctional inhibitory activity of AChE and Aβ-Aggregates.
In another work, considering that increased basicity of the Nitrogen atom from the pyridine system could lead to its protonation at the physiological pH and that the resulting cationic nitrogen atom could establish more affective cation-π interactions with aromatic residues located at the PAS, the authors decided for a bioisosteric change of oxygen atoms in the aliphatic ring and ester subunits of 52 by an amine and amide nitrogen atoms, respectively. These simple structural modifications led the AChEI 54 (IC50= 65 and 920 nM for human AChE and BuChE, respectively, (Fig. 22) with a remarkable increase in the selectivity for AChE [101].
Taking into account all these previous findings, and aiming for an improved multifunctional activity on AChE and Aβ-aggregation inhibition, the authors additionally investigated how a MH of the optimized PAS-binding pharmacophore 54 and the 6-cholorotacrine moiety, linked by a tether of different lengths in the methylene chain, could influence in concomitant interactions of each subunit with CAS and PAS of AChE. Thus, a new series of tetrahydrobenzo [1,6]naphthyridine-6-chlorotacrine hybrid homologues 55 (Fig. 22) was synthesized and evaluated, leading to the discovery of compound 55a as a picomolar hAChEI (IC50= 0.006 nM), with additional high hBuChE inhibitory activity (IC50= 120 nM) and moderate dual effect on Aβ (52.5% at 10 µM) and tau (40.7% at 10 µM) aggregation [102].
The structural pattern of scutellarin 56 inspired Spilovska and co-authors to explore its structural combination with 6-chlorotacrine (51), leading to a family of six novel scutellarin-tacrine hybrids. The rationale of this strategy considered 6-chlorotacrine 51, as an AChEI with higher potency than tacrine and comparable hepatotoxicity. As a result, a series of scutellarin-6-clorotacrine 57 (Fig. 23) was prepared and evaluated for ChE inhibition, antioxidant, BBB permeability and hepatotoxicity. Biological results highlighted compound 57a, bearing two methylene ether between two basic scaffolds, as a potent and selective hAChEI (IC50= 1.63 nM), with a probable mixed non-competitive mode of enzyme inhibition, but without free scavenging ability. SAR analysis suggested that the lack of antioxidant activity could be due to the substitution of methoxy groups in the scutellarin moiety instead of free phenolic groups, which presumably confer higher antioxidant potential. Moreover, in the hepatotoxicity assay towards HepG2 cells, compound 57a showed a comparable hepatotoxic profile to that of the 6-Cl-THA (51) [103].
Fig. (23).

Structural design of Scutellarin-6-clorotacrine hybrids 57 and the most active derivative 57a.
Liao and colleagues used a similar strategy to develop some multi-target hybrids capable of acting as antioxidants, AChEIs and neuroprotective agents (Fig. 24). The new hybrid series was designed by a combination of 5,6,7-trimethoxyflavone (58) with 6-chlorotacrine (51). Among all compounds synthesized, derivative 59 showed the highest inhibitory activity for AChE (IC50= 12.85 nM), exhibiting also a significant antioxidant activity and inhibiting self-induced Aβ aggregation in 33.8% at 25 μM. In addition, compound 59 showed a good neuroprotective effect against H2O2-induced oxidative stress, maintaining cell viability a rate of 45.3% at 5 μM and an adequate BBB penetration ability of 7.92 x 10-6 cm/s [104].
Fig. (24).

Strategy for design of the 5,6,7-trimethoxyflavone–6-chlorotacrine hybrid compound 59.
In general, substituted 4-Hydroxy-cynnamic acids (60) are known for their antioxidant, neuroprotective and hepatoprotective properties. 2,3,5,6-Tetramethylpyrazine (TMP, 61) also known as ligustrazine is an alkaloid isolated from fermented cocoa beans and natto that has been described for its multiple pharmacological activities, including anti-inflammatory, neuroprotective, enhancement of mitochondrial biosynthesis, blockade of Ca2+-channel and prevention of oxidative stress‐induced apoptosis, which are all related to NDs pathological changes, especially in AD and PD. Considering this information, two series of tacrine-di-TMP-phenolic acids (62) and tacrine-p-coumaric acid hybrids (63, Fig. 25) were designed as potential drug candidates for AD. All six hybrid compounds were evaluated for their in vitro inhibitory activities of AChE, BuChE and self-mediated Aβ aggregation, antioxidant and neuroprotective properties, as well as their in vivo hepatotoxicity. Most compounds exhibited significant inhibition of ChEs, with compound 63a as the most promising selective AChEI with IC50 values of 3.9 and 65.2 nM for eeAChE and hAChE, respectively, and IC50 of 24.3 and 48.8 nM for eqBuChE and hBuChE, respectively. This compound also showed an effective ability to block self Aβ-aggregation and antioxidant activity against PC12 cell damage and very low hepatotoxicity in vitro and in vivo compared to tacrine [105].
Fig. (25).

Strategy for design of the tacrine hybrid 62 and 63.
Novel tacrine-ferulic acid hybrids were synthesized and evaluated by Fu and co-workers (Fig. 26) in the search for multifunctional ChE inhibitors with additional inhibitory activity of self-induced Aβ aggregation and Cu2+-chelating ability. Compounds 64a (IC50 AChE= 52.7 nM, IC50 BuChE= 215.4 nM) and 64b (IC50 AChE= 61.7 nM, IC50 BuChE= 106.9 nM) were identified as promising MTDLs, due to their high selectivity for AChE inhibition over BuChE with SI= 4.09 and 1.73, respectively. Moreover, compound 64b also showed significant inhibition of self-Aβ aggregation (37.2% at 20 µM), Cu2+-chelating activity and the best activity against Aβ-induced neurotoxicity in neuro-2A cells [106].
Fig. (26).

Rational design strategy for tacrine_Ferulic acid hybrid compounds 64 based on the MH of tacrine (48) and ferulic acid (5).
Taking tacrine (48) as AChEI prototype, Dgachi and co-workers explored the combination with an antioxidant motif for the design of a new family of hybrid multitarget ligands aiming AChEI activity along with antioxidant and neuroprotective properties, without the severe hepatotoxicity of tacrine. The antioxidant motif was selected from the structure of kojic acid (65), a naturally occurring fungal metabolite, with remarkable ability in scavenging ROS species and consequent antioxidant properties. Thus, MH of 48 and 65 resulted in a new kojo-tacrine scaffold (66, Fig. 27), with a variety of substituents at the aryl portion leading to 12 hybrid homologues. Their evaluation towards human hepatocytes, inhibition of AChE/BuChE, antioxidant, neuroprotective and in silico prediction of ADMET properties highlighted compound 66a as the most promising hybrid, exhibiting a 6-fold lower hepatotoxicity than tacrine, with a moderate and selective inhibitory activity against AChE (IC50= 4.52 mM), significant antioxidant and neuroprotective activities against Aβ1-40-induced damage in human neuroblastoma (SH-SY5Y) cells [107].
Fig. (27).

MH of tacrine (48) and kojic acid (65) in the search for new multi-target ligands for AD treatment.
In another approach, Hamulakova and co-workers explored the combination of the structures of tacrine (48) and 7-hydroxy-coumarine (67) to generate a new series of hybrid compounds using alkylenediamines or alkylidene polyamine as tethers. The target-compounds were designed as potential inhibitors of AChE, BuChE and Aβ-aggregation resulted from the tacrine substructure with additional antioxidant activity due to the presence of 7-hydroxycoumarine fragment. Biological evaluation for cholinesterase inhibition, ability for Cu(II) complexation, DNA protection and inhibition of Aβ aggregation led to the identification of compound 68 (Fig. 28) as the most active and promising multitarget compound, inhibiting AChE with an IC50 of 38 nM, 58,3% of Aβ aggregation and a good radical scavenging activity [108].
Fig. (28).

MH of tacrine (48) and 7-hydroxycoumarine (67) in the design of compound 68, a multitarget inhibitor of AChE, BuChE, Aβ-aggregation and the chelating ability for Cu2+.
In another work, MH of 48 and tianeptine (69) was used by Ceschi and co-workers in the structural design of a new series of tacrine-tianeptine hybrids 70 (Fig. 29) with potential neuroprotective and antidepressant activities. The target-compounds were assayed against AChE and BuChE, revealing compound 70a as the most promising ligand, showing a good selectivity and high potency in the inhibition of AChE with an IC50 of 6.79 nM. Molecular docking studies corroborated this high inhibitory potency and selectivity, evidencing that the tacrine moiety in 70a interacts with CAS in the bottom of the gorge of AChE active site, whereas tianeptine subunit binds to the peripheral anionic site (PAS). Beyond that, compounds 70b and 70c showed a significant neuroprotective profile and ability to reduce in vitro basal secretion of S100B in 67.4 and 72.7%, respectively, at the concentration of 0.03 mM. Compound S100B is a calcium-binding protein expressed and secreted by astrocytes, able to play a neurotrophic role in neighboring cells and to protect against glutamate-induced excitotoxicity. Thus, these results support the promising neuroprotective therapeutic potential of compounds 70b and 70c, which could be used as adjuvant drugs in the management of NDs [109].
Fig. (29).

Design strategy for a new series tacrine-tianeptine 70 and structures of the promising neuroprotective hybrids 70a, 70b and 70c.
In a recent study, 3-substituted-5-anilinio-1,2,4-thiadiazole derivatives (71) were identified as potent radical-scavenging agents. As per these findings, a series of tacrine-1,2,4-thiadiazole hybrids (72, Fig. 30) was designed in the expectation that the combination of the 9-amino-1,2,3,4-tetraidroacridine subunit from tacrine (48) could assure AChE and BuChE inhibitory properties, with additional antioxidant and NMDA-antagonist profile resultant from the presence of the thiadiazole subunit, using a 5-methyleneamino fragment as a tether. Pharmacological evaluation revealed compound 72a as 17-fold more active in the inhibition of BuChE (IC50= 0.073 µM) than AChE (IC50= 1.24 µM), with significant ability to block both MK-801 and ifenprodil binding sites at NMDA receptor. In addition, all tested compounds exhibited higher radical-scavenging activity (IC50 values of 12.3-14.5 µM) than Trolox used as a reference. It is important to note that in a healthy brain, acetylcholine (ACh) is primarily hydrolyzed by AChE, but BuChE plays an auxiliary role. However, during AD progression, the activity of BuChE gradually increases as the AChE activity slows down, which makes BuChE inhibitors interesting target-prototypes for AD treatment. Taken together, all these findings suggest compound 72a as an interesting multitarget-directed ligand, with remarkable selectivity for BuChE, with affinity by NMDA receptor binding sites and antioxidant activity, for further investigation as a potential multifunctional drug candidate, not only for AD, but for other NDs in which OS and excitotoxicity play relevant pathological role [110].
Fig. (30).

Tacrine-1,2,4-thiadiazole hybrid compounds 106 designed as multifunctional antioxidants, NMDA-antagonists and cholinesterase inhibitors.
In the search for new compounds that inhibit Aβ aggregation, one has discovered that multialkoxybenzene (73), in which the benzene ring is highly enriched by electron density, was an interesting pharmacophore subunit. Based on these findings and considering the well-known tacrine (48) affinity for AChE and BuChE, Luo and co-workers developed a series of tacrine-multialkoxybenzene as drug candidates for AD (74). Biological data and in silico studies for inhibition of ChEs and kinetic study of AChE inhibition, self-induced Aβ-aggregation revealed compounds 74a (Fig. 31) as the most active inhibitors of cholinesterase, with higher effects on BuChE. The most potent compound 74a, showed a 4-fold higher selectivity for BuChE (IC50= 5.19 nM) than for AChE (IC50= 20.52 nM), followed by 74b (IC50= 9.73 and 18.43 nM for AChE and BuChE, respectively) and 74c and 74d as almost non-selective BuChE inhibitors (IC50= 7.94 and 11.36 nM, respectively). All compounds, including the less active cholinesterase inhibitors, were capable of inhibiting Aβ1-42 self-induced aggregation in 63.1 to 68.5% at 20 µM [111].
Fig. (31).

Hybridization model of tacrine-multialkoxybenzene.
2.3. MTDLs Inspired by Clioquinol
Metal-Protein Attenuation Compounds (MPACs) have been reported to disrupt the interaction between biometals and Aβ peptide in the brain [112]. Successful trials with 5-chloro-7-iodo-8-hydroxyquinoline (clioquinol, 75) for AD treatment have aroused the interest in assessing whether its therapeutic action could be related to the coordination of neurotoxic trace metals, such as Cu (II) and Zn (II). One hypothesis is that the coordination of Zn (II) and its displacement from low-affinity binding sites of the Aβ protein in the brain would result in the clearance of Aβ tangles. On the other hand, concomitant Cu (II) complexation would lead to a net decrease in neurotoxic H2O2 production [113].
In 2019, Hu and co-authors reported the synthesis pharmacologic evaluation of a new series of clioquinol-rolipram hybrids (77, Fig. 32) as promising MTDLs for AD treatment, designed by the combination of the structures of 75 and rolipram (76), a phosphodiesterase-4 (PDE4) inhibitor, reported for its ability in enhancing memory performance and contextual learning. From this series, compound 77a demonstrated the best PDE4D inhibitory activity (IC50= 0.4 μM) with 0.60 ORAC activity, comparable to clioquinol, and a good permeability (12.92 x 10-6 cm/s) in the PAMPA assay. Furthermore, compound 77a showed a concentration-dependent protective effect against t-BuOOH-induced intracellular oxidative stress, with no effect on cell viability at a concentration of 10 μM, enhancing memory capacity and cognitive functions in animal models using Morris water-maze test (MWM) [114].
Fig. (32).

New hybrids 111 designed like multi-target compound.
Bearing in mind that the imbalance in radical species generated by impaired brain metabolism contributes decisively for increasing OS, cellular damage and inflammation related to NDs pathogenesis, phosphodiesterases (PDEs) have been considered a promising molecular target for the design of novel disease-modifying neuroactive drugs. PDEs are a superfamily of enzymes responsible to hydrolyze phosphodiester bonds in 3’,5’-cyclic adenosine monophosphate (cAMP) and 3’,5’-cyclic guanosine monophosphate (cGMP), two intracellular second messengers that play capital roles in various extracellular signals and biological processes. With this information in mind, Song and co-workers also considered the inhibition of PDE4 as a suitable target to modulate the neuroinflammation associated to AD pathogenesis. Moreover, the authors looked at the importance of modulating biometals to counteract OS and improve neuroprotection and designed a novel series of clioquinol-roflumilast/rolipram hybrids (79), which scaffold is resultant of the MH of substructures of 75, 76 and roflumilast (78, Fig. 33). The biological evaluation revealed compounds 79a and 79b as the most promising multifunctional hybrids, with high BBB permeability and potent inhibitory activity of PDE4 with IC50 values of 0.49 and 0.40 µM, respectively. In addition, the in vivo Morris Water Maze test evidenced their significant improvement of cognitive memory, but compound 79b showed better behavioral performance, probably due to its higher ability to protect Aβ-induced neuronal damage [115].
Fig. (33).

New clioquinol-roflumilast/rolipram hybrids 79 designed as multi-target ligands for AD and structures of compounds 79a and 79b as the most active PDE4 inhibitors with memory improving properties.
A series of chromone derivatives (82) was designed by Li and co-workers using clioquinol (75) and chromone (80, Fig. 34) as suitable structural models for MH. To search new ligands with potential effects against AD, a family of seven molecular hybrids was screened for MAO inhibition, metal-chelating ability and Aβ-lowering and antioxidant activities. Compound 82a was highlighted due to its better multifunctional profile, inhibiting both MAO isoforms with IC50 values of 5.12 and 0.816 µM for hMAO-A and hMAO-B, respectively, along with a moderate inhibition of Aβ aggregation (75.1% at 20 µM) and Cu+2, Zn+2, Fe+3 and Fe+2-chelating ability. In addition, 82a was also able to control ROS generation, showed a strong antioxidant activity (ORAC = 3.62) and reduced PC12 cells death induced by OS, with adequate BBB permeability [116].
Fig. (34).

Design of a new chromone derivative 82a with antioxidant, metal-chelating, and selective inhibition of AChE and MAOs properties.
Focusing MAO-B inhibition as a strategy to discover new disease-modifying agents for AD, Xie and co-workers used MH of the pharmacophores of selegiline (83) and 75 to design a new selegiline-clioquinol hybrid scaffold (84) with potential multifunctional properties (Fig. 35). The choice of 83 as a structural model was based on its activity as an irreversible and selective inhibitor of MAO-B, which acts as a neuroprotective agent in cellular and animal models of AD. Biological results pointed out compound 84 as the best selective MAO-B inhibitor (IC50= 0.21 μM), with additional strong antioxidant activity (ORAC = 4.20) and biometal chelating ability, which effectively inhibited Cu(II)-induced Aβ1–42 aggregation [117].
Fig. (35).

Design strategy and chemical structures for the most potent selegiline-clioquinol hybrids 84.
2.4. MTDLs Inspired by Melatonin
Structurally, melatonin or N-acetyl-5-methoxytryptamine (86) is an indolamine that is produced by almost every unicellular and multicellular organism, including plants and animals, and in human being, it is biosynthesized mainly by the pineal gland. Decreased levels of melatonin in serum and in cerebrospinal fluid (CSF), are observed in patients with AD. Melatonin is a hormone that regulates sleep-wake cycles and as the individual ages or in some age-related diseases, the endogenous secretion of melatonin drops markedly, and melatonin supplementation can ameliorate sleep disorders [118].
In recent studies, carried out by Cheng and co-authors, a series of novel (-)-meptazinol–melatonin hybrids (87, Fig. 36) was designed in the basis of the structures of (-)-meptazinol (85) and melatonin (86) as potential multi-target agents against AD. Compound 87a was identified as the antioxidant activity evidenced by its ORAC 7.2-fold higher than trolox, with additional dual inhibitory capability of both hAChE (IC50= 0.31 µM) and hBuChE (IC50= 0.29 µM), but with low selectivity. Furthermore, the results of ThT assay, performed for identification and quantification of amyloid fibrils formation and analysis by transmission electron microscopy (TEM) suggested that 87a could effectively retard Aβ1–40 and Aβ1–42 self-aggregation in a dose-dependent manner. Taken all these data together, compound 87a, that targets ChEs, Aβ aggregation and OS was highlighted as a valuable candidate worthy of further development for drug candidate in the AD treatment [119].
Fig. (36).

Design strategy for (-)-MEP-melatonin hybrids 87.
In 2014, López-Iglesias and co-workers reported a new series of melatonin−N,N-dibenzyl(N-methyl)amine hybrids (89, Fig. 37) with a multifunctional profile against targets related to AD pathogenesis. The melatonin-based hybrids were designed by a rational combination of two pharmacophoric subunits potentially responsible for anticholinergic, antioxidant and neuroprotective properties. Firstly, the indol-ethylamide subunit from melatonin was thought to interact with AChE-PAS due to its character aromatic that ensures π-staking interactions. The second selected fragment was the basic N,N-dibenzyl-(N-methyl)-amine, which is present in the AChEI AP2238 (88). As a result, compounds 86a-c (Fig. 37) have stood out for their high potency in hAChE inhibition with IC50 values of 1.9, 2.5 and 2.1 µM, respectively and hBuChE with IC50 values of 3.7, 17.0 and >10 µM, respectively. These compounds also showed potent peroxyl radical absorbance, ranging from 1.5 to 4.3 fold higher than Trolox, with low cytotoxicity and adequate BBB permeability, which led them to be considered as a potent antioxidant and selective AChEIs suitable for further consideration in drug development against AD [120].
Fig. (37).

Strategy for the design of the melatonin-based hybrids 89a-c with remarkable AChEI and antioxidant activities.
2.5. MTDLs Inspired by Coumarin
The coumarin (90) is a natural product isolated for the first time from Coumarouna odorata Aube, an original French plant. This compound has been used for 200 years in traditional medicine preparations for many different diseases. Nowadays, it is well-known that coumarin is an abundant secondary metabolite in a number of different natural sources and is responsible for a wide range of biological properties [121]. Its chemical structure is characterized by a 1,2-benzopyrone-oxa-heterocycle system and, alongside with different natural and synthetic derivatives, have been reported in the literature by their MAO and ChE inhibitory activities, with some of them exhibiting a multi-target profile [122].
Based on these findings, Jiang and co-authors develop a series of new coumarin-dithiocarbamate hybrid compounds (92, Fig. 38) that were evaluated as potential multifunctional agents for the treatment of AD. Biological results evidenced their potent and selective inhibition of AChE, also inhibiting self-induced Aβ aggregation. Compound 92a showed the highest ability for hAChE inhibition (IC50= 0.027 µM), being 1.5-fold more potent than donepezil, and good inhibition of Aβ aggregation (40.19% at 25 µM). In addition, it also expressed specific metal-chelating ability for Fe3+ and did not showed significant toxicity in mice at doses up to 1000 mg/kg. The authors highlighted 92a as the first reported dithiocarbamate derivative with multi-targets activities related to AD treatment [123].
Fig. (38).

MH between coumarin (90) and dithiocarbamate 91 in the design strategy for novel coumarin-dithiocarbamate hybrids 92 with multipotent activities.
Another series of new coumarin-dithiocarbamate hybrids 95 (Fig. 39) were designed and synthesized by He and co-authors as multitarget agents for AD treatment. Pharmacological evaluation of that series against ChEs and MAOs led to the identification of compound 95a as the best balanced multipotent hybrid, with high potency in the inhibition of AChE with IC50 values of 6.8 and 8.9 nM for eeAChE and hAChE, respectively. Moreover, compound 95b exhibited the best inhibition of hMAO-B, showing a good balanced dual inhibitory activity of both hAChE and hMAO-B with IC50= 0.114 μM for hAChE and 0.101 μM for hMAO-B. Due to these promise multitarget activities, compound 95b was further evaluated and showed adequate BBB permeability, as well as no significant toxicity towards human neuroblastoma cells, acute toxicity and neuroprotective effects in scopolamine-induced cognitive impairment in mice. These results highlighted compound 95b as a potential multitarget agent for AD treatment and were considered as a starting point for the design of new multitarget AChE/MAO-B inhibitors based on dithiocarbamate 94 scaffold [124].
Fig. (39).

Design strategy for new coumarin-dithiocarbamate hybrids 95a-b.
Vafadarnejad and co-authors synthesized a novel series of coumarin-pyridinium hybrids (98) obtained by the fusion of pharmacophoric subunits of Ensaculin (96) and 1-(4-fluorobenzyl)-pyridinium-(±)-XJP (97, Fig. 40) in the search for new ChE inhibitors. From this series, compound 98a showed the best AChEI activity, with an IC50 value of 10.14 µM, whereas compounds 98b and 98c depicted the best potency in the inhibition of BuChE with IC50 values of 0.32 and 0.43 mM, respectively. Moreover, compound 98b exhibited the best selectivity for BuChE with a selective index (SI) of 101.18. Compounds 98a and 98b were also evaluated for the inhibition of BACE-1, both showed only a poor activity [125].
Fig. (40).

Design strategy for the series of coumarin-pyridinium hybrids (98) using ensaculin (96) and 1-(4_Fluorobenzyl)-pyridinium-(±)-XJP (97) as structural prototypes.
Baleh and co-workers took inspiration from de natural lipoic acid (99), which is known for its therapeutic potential as a direct scavenger of ROS, and 3-phenylcoumarin derivative AP2469 (100) that has been reported to show numerous valuable biological activities, especially for NDs, in the draw of two new hybrid scaffolds represented by compounds 101a and 101b (Fig. 41). The pharmacophore fragments from each structural pattern 99 and 100 were connected by the application of click reaction to construct a 1,2,3-triazole tether. In this case, the use of MH led rise to a series of lipoic acid-coumarin hybrids 101 with an expected multi-target directed profile of action. Among a number of hybrid analogues, varying the length of the spacer subunit and the substituents on the coumarin system, only compounds with four methylene units in the oxamethylene-triazole spacer showed significant inhibitory activity on AChE. Compounds 101a and 101b showed the highest inhibitory activities for AChE, with 101b exhibiting in vitro and in vivo multiple desirable effects, consistent with the intended MTDLs profile. This compound was the most potent AChEI (IC50= 7.3 µM) and displayed the highest antioxidant activity in neuronal PC12 cells at the 50 µM (43.7%), aside of a good biometal-chelator for Mg2+, Ca2+, Al3+, Cu2+, Fe2+ and Zn2+. Furthermore, this compound showed 52.6% (at 10 µM) of neuroprotection in human neuroblastoma cells [126].
Fig. (41).

Design strategy for compounds 101.
In another approach involving lipoic acid (99) and coumarin scaffold, Jalili-Baleh and collaborators designed a novel series of coumarin-lipoic acid hybrids (103, Fig. 42). A click cycloaddition reaction was used for the construction of a 1.2.3-triazole spacer subunit, linking the substructure of chromone to the lipoic acid, furnishing a new architecture for potential MTDLs against AD. Compound 103a was highlighted for its best multifunctional properties, inhibiting AChE with IC50 of 16.4 μM, besides maintaining 67.8% of viable neuronal cells when treated with H2O2, clearly evidencing a neuroprotective activity. This compound was also capable to inhibit 51.2% of Aβ aggregation, decrease in intracellular ROS generation, reduction by Fe3+ and selective metal chelation ability [127].
Fig. (42).

Design strategy for the synthesis of coumarin-lipoic acid hybrids 103.
Yang and co-workers described the synthesis and biological evaluation of series of coumarin-pargyline hybrids (105, Fig. 43) as novel dual inhibitors of AD. Pargyline 104 is an irreversible selective MAO-B inhibitor drug. The pargyline fragment could be a selective inhibitor of MAO-B playing an important role in the neuroprotective properties of the new derivatives, as well as inhibitors of Aβ1-42 aggregation. A biological evaluation revealed compounds 105 as a good inhibitor of MAO-B and MAO-A, with IC50 values of 0.027 and 3.275 μM, respectively, Aβ1-42 aggregation (54.0% at 25 μM), and showed low in vitro cytotoxicity [128]. Additional new coumarin-N-benzyl pyridinium hybrid 107 was designed as a multipotent inhibitor of ChEs and MAO (Fig. 43). This strategy focused on the structure of benzyl pyridinium salts (106), which could interact with the CAS of AChE, and the coumarin scaffold 102 was used for MAO-B inhibition. The biological screening results indicated that compound 107 displayed potent mixed-type inhibitory activity for hMAO-B (IC50= 1.57 μM) and balanced inhibitor to ChEs (IC50= 0.037 and 2.32 μM for eeAChE and eqBuChE, respectively). In addition, compound 107 showed a good ability to inhibit Aβ1–42 self-aggregation and cross the BBB with no toxicity on PC12 neuroblastoma cells [129].
Fig. (43).

Design strategy and chemical structures for the most potent coumarin-pargyline hybrids 105 and coumarin-N-benzyl pyridinium hybrids 107.
2.6. MTDLs Inspired by Curcumin
Curcumin (20) is a natural compound extracted from Curcuma longa, a tropical turmeric plant, extensively described in traditional Chinese medicine as an anti-inflammatory. Curcumin has a symmetric chemical structure, with two ferulic acid (5) subunits internalized in its framework and the literature is rich in reports of its potent anti-inflammatory, antioxidant, cytoprotective and ability to modulate several different cellular processes. These sets of biological properties have been considered as potentially useful towards chronic diseases such as cancer, NDs, hepatic disorders and autoimmune diseases [130, 131].
Due to its multiple activities, curcumin has been taking as inspiration in the design of novel hybrid chemical entities with potential disease-modifying properties for 0NDs, especially AD and PD. In the work of Lan and co-workers, a novel family of curcumin-cinnamic acid hybrids was designed as potential multifunctional ChE inhibitors for AD. The rationale of this new molecular architecture was based on fusing the N-benzyl pyridinium moiety 108 with a variety of substituted cinnamic acids that could mimic the structure of curcumin and, in turn, of ferulic acid, to produce the singular molecular pattern 109 (Fig. 44). Evaluation of these new family of hybrid compounds led to the identification of 109a as a very potent and selective AChE inhibitor (IC50= 12.1 nM for eeAChE, 8.6 nM for hAChE, 2.6 µM for eqBuChE and 4.4 µM for hBuChE). It also showed good inhibition of Aβ1-42 aggregation (64.7% at 20 µM) and neuroprotective activity on PC12 cells against Aβ-induced cell toxicity [132].
Fig. (44).

Design strategy for the synthesis of curcumin-cinnamic acid hybrids 109.
9H-Carbazole (110) and its derivatives are well-known inhibitors of Aβ-aggregation. Some 9H-carbazole derivatives with substituents in the positions 2 and 8 have been described for their capability for inhibiting AChE and neuroprotection [133].
Based on these findings, Lei Fang and co-workers developed a series of molecular hybrids (111), designed by a combination of the carbazole pharmacophore subunit and 4-hydorxy-3-methoxy-cinnamoyl fragment from ferulic acid (5). The target compounds were expected to inhibit AChE, with antioxidant and neuroprotective additional properties, showing a promising multifunctional attribute as drug candidates for AD. As a result, a new series of amide hybrids of ferulic acid and 9H-carbazole was designed, and compound 111a (Fig. 45) identified as the best AChEI (IC50= 1.9 µM), with higher potency than galantamine, with significant antioxidant and neuroprotective properties [133].
Fig. (45).

Compound 111a, an AChEI with antioxidant and neuroprotective properties, designed by MH of 9H-carbazole derivatives and ferulic acid.
A series of hybrids compounds of curcumin (20) and melatonin (86, Fig. 46) were designed for the treatment of AD. The curcumin-melatonin hybrids 112 were evaluated by neuroprotective activity in MC65 cells, a cellular model of AD that is associated with the Aβ-induced OS in cells. In particular, compound 112a showed a significant neuroprotection in a nanomolar potency (IC50= 27.6 nM). In vitro assays with 112a evidenced its inhibitory activity in the production of Aβ oligomers along with important antioxidant properties and an adequate ability to transpose BBB in vivo [134].
Fig. (46).

Design of the curcumin-melatonin hybrid scaffold 112 and the most promising derivative 112a as a potential multi-target drug candidate prototype for AD.
2.7. MTDLs Inspired by Resveratrol
Resveratrol (113) is a natural polyphenol found in red grapes, red wines and some berries. This compound displays potent antioxidant activity due to its ability to scavenge free radicals and chelate metals. Resveratrol has been reported for its wide range of biological properties, including antitumor, antioxidant, neuroprotective and antiviral. In addition, 113 inhibits Aβ aggregation and modulates diverse intracellular effectors involved in neuronal cell survival/death, as well as increases PP2A activity and dephosphorylation of tau protein, a key feature of AD [135-137].
Oxidative stress is one of the main causes of neuronal death in AD and other NDs. By considering these data, 113 was elected by Cheng and co-workers as a structural model for MH with maltol (114), another phenolic compound with antioxidant and metal-chelating properties, in the design of a series of resveratrol-maltol hybrids (115, Fig. 47), expected to lead to the discovery of novel molecules with the neuroprotective and anti-inflammatory effects. Biological evaluation pointed out compounds 115a and 115b for their Aβ self-aggregation inhibition with IC50 values of 7.20 and 8.29 μM, radical scavenging activity with IC50 values of 1.94 and 1.18 μM, respectively [138].
Fig. (47).

Structural design of a series of resveratrol-maltol hybrids 115 and the most potent ROS scavengers and Aβ-aggregation inhibitors 115a and 115b.
Xia Yang and collaborators investigated the therapeutic potential of a series of pyridoxine-resveratrol hybrids. The rational concept of such hybrid compounds was based, in part, on the role of the two isoforms of the flavin adenine dinucleotide (FAD) containing enzymes MAO-A and MAO-B in the generation of ROS as a consequence of oxidation of neurotransmitters. Thus, inhibition of MAO-B into the damaged neuronal tissue could be of great value in therapeutics of AD. On the other hand, resveratrol is a known potent antioxidant, natural stilbenoid, with anti-inflammatory and neuroprotective properties. Taking these findings into account, the authors designed a new family of molecular hybrids by a combination of pyridoxine moiety and the pharmacophore stilbene fragment from resveratrol to devise a single molecular scaffold with potential multifunctional action. As a result, sixteen pyridoxine-resveratrol hybrid tertiary amine derivatives were synthesized and tested for inhibition of ChEs, MAO-A and B, as well as for antioxidant and metal chelating properties. Among them (Fig. 48), the piperidine and diethylamine analogues 118a and 119 exhibited the highest potency in selective AChE inhibition with IC50 values of 2.11 and 1.56 μM, respectively, while the morpholine derivative 118b exhibited the best selective MAO-B inhibition with an IC50 value of 2.68 μM. All these compounds also showed significant antioxidant activities and metal chelating ability for Cu2+ and Fe2+. All this biological data suggest that these compounds could be considered for further development of MTDLs, not only for AD, but also for PD [139].
Fig. (48).

MH of pyridoxine-resveratrol 118 and 119.
A series of deferiprone-resveratrol hybrids 121 (Fig. 49), was planned as multitarget-directed ligands to combat the pathogenesis of AD. The structural design strategy used resveratrol (113) as a model for combination with 3-hydroxypyridin-4-one moiety from deferiprone (120) to furnish a novel hybrid scaffold series of 120 into the structure of resveratrol. Most of the synthesized analogues exhibited higher antioxidant and metal chelating properties than the parent compounds, probably due to the stilbene-mimic and 3-hydroxypyridin-4-one fragments, respectively. In addition, compounds 121a and 121b exhibited significant inhibition of Aβ1–42 aggregation with IC50 values of 10.70 and 8.94 μM, respectively, suggesting that both could be considered as potentially effective agents against AD [140].
Fig. (49).

Design strategy and chemical structures for the most potent deferiprone-resveratrol hybrids 121.
A series of hydroxylated resveratrol dimers (124, Fig. 50) was planned by MH of structural fragments of the isoprenyl-resveratrol derivative 122 and the compound 123 in the search for bioactive ligands with potential use against AD. Huang and co-workers evaluated a series of such hybrid compounds and identified 124a and 124b as better antioxidants and inflammatory than resveratrol-derivative 122. Moreover, both hybrids showed moderate inhibitory activity against hMAO-B with IC50= 3.91 and 0.90 μM, respectively, with a significant antioxidant profile in the DPPH, ABTS and ferric reducing ability of plasma (FRAP) assays. In cellular assays, 124a and 124b showed low toxicity towards PC12 and BV2 cells, besides remarkable neuroprotective effects against ROS and H2O2-induced apoptosis and a significant in vitro anti-inflammatory activity [141].
Fig. (50).

Design of resveratrol-based isoprenylated hybrid compounds 124 and chemical structures of the most active hybrids.
In the search for novel agents with an MTDL profile for AD, Xie and co-workers used the MH of resveratrol (113), described in the literature as an inhibitor of Aβ protein binding, and indazole 125, a potent MAO inhibitor, to design a new family of compounds capable of acting on both molecular targets (Fig. 51). Thus, a new family of hybrid indazole-resveratrol compounds was synthesized and evaluated for its possible MTDL profile by joining both biological activities of 113 and 125 patterns. Initially, all compounds were tested for their ability to inhibit MAO-A and MAO-B, revealing 126 as the most potent and selective MAO-B inhibitor (IC50= 1.14 µM), but with a moderate inhibition of MAO-A (IC50= 30.4 µM). Interestingly, this dual inhibitory property towards MAOs is considered an interesting action profile for the treatment of NDs normally associated with depression as a secondary symptom of behavioral and cognition impairments. Next, the inhibition of βA self-induced aggregation was evaluated and, again, compound 126 (IC50= 19.5 µM) was highlighted for its best activity, comparable to resveratrol (IC50= 17.9 µM). Finally, the lead-compound 126 was subjected to neurotoxicity evaluation in PC12 cell line at concentrations of 6.25, 12.5, 25, 50 and 100 µM, with no toxic effects observed [142].
Fig. (51).

MH of resveratrol 113 and indazole 125 in the design of a new family of indazole-resveratrol hybrids, with compound 126 as the most promising MTDL.
2.8. MTDLs Inspired by in other Bioactive Molecules
In another recent work, Cao and co-workers decided to explore the structure of flurbiprofen and chalcones in the design of new hybrid molecules with affinity for AD-related multiple targets [143]. For this attempt, non-steroidal anti-inflammatory agents seem to be of relevant therapeutic value in the clinics of AD, PD and HD, once it is well characterized the capital role of neuroinflammation in the progress and severity of such NDs. Particularly, flurbiprofen (127, Fig. 52) was selected as an interesting structural prototype, not only for its anti-inflammatory properties, but also due to recent evidences of its ability for selective lowering the production of Aβ, reducing the pathological levels of tau hyperphosphorylation and glutamine and, in turn, modulating the cellular signaling of neuroinflammation and microglia activation [144, 145].
Fig. (52).

MH of flurbiprofen 127 and chalcones 128 in the design of a new series of 4'-OH_Flurbiprofen-chalcone hybrids, with compounds 129a and 129b, as the most active as potential MTDLs.
On the other hand, chalcones are considered privileged structures in the design of ligands with a wide range of biological activities, such as anti-inflammatory and antioxidants, with special importance for neuroactive compounds. Their flexible structure, composed of two aromatic rings inter-connected by an α,β-unsaturated ketone system, is suggested to ensure an adequate brain permeability and its neuroprotective and anti-neuroinflammatory activities. In a recent review, we discussed a special role that chalcones could play in the activation of the glutathione defense system due to its capability in acting as Michael acceptors. These molecules could activate the Keap1/Nrf2/ARE pathway as a result of a nucleophilic attack of cysteine residues from the Keap1 domain to the Michael acceptor site of a chalcone structure. As a result, Nrf2 dissociates from the cytoplasmic complex with Keap1 and is translocated to the nucleus and bound to the antioxidant response element (ARE), promoting the activation of the expression of several detoxification, antioxidant and anti-inflammatory genes, as well as genes involved in the clearance of damaged proteins like Aβ and tau in AD, or α-synuclein and huntingtin in PD and HD, respectively [146].
Taken all the above-mentioned arguments, flurbiprofen was selected to combine with chalcone scaffold to generate a series of novel flurbiprofen-chalcone hybrids planned to act by a multitarget mode in brain tissue as inhibitors of Aβ-aggregation, MAO, antioxidants, metal-chelating and anti-neuroinflammatory agents. Eighteen 4'-hydroxy-flurbiprofen-chalcone hybrid derivatives were synthesized and screened for antioxidant potential, with compounds 129 (Fig. 52) showing the best ORAC-FL values of 3.4, 3.5, 3.1 and 3.3 trolox equiv., respectively. In addition, the two dialkylamino homologues 129a and 129b showed the best inhibition of self-induced Aβ1-42 aggregation (60.0 and 78.2%, respectively) and Cu2+-induced Aβ1-42 aggregation (52.4 and 95.0%, respectively), with good cell viability until the dose of 40 µM. Moreover, these two representative compounds inhibited MAO-A selectively in 73.1 and 75.5% (at 10 µM) in comparison to MAO-B (47.5 and 48.5, respectively), showing selective metal-chelating abilities for Cu2+ and Al3+ and significant anti-neuroinflammatory activities in vitro, which was evidenced by the inhibition of induced-NO release and production of TNF-α. Taken together, these results suggest that compounds 129a and 129b, and particularly 129b which results from PAMPA assay indicated an appropriate BBB permeability, could be considered as promising innovative drug candidate prototypes for AD [143].
In another work, Telpoukhovskaia and co-workers employed the molecular structure of thioflavin-T (130), deferiprone (131, Fig. 53) and rivastigmine (132) in the design of multifunctional molecules as potential antioxidant and metal chelating AChEIs. Based on the structure of 130 and rivastigmine (132) the authors designed hybrid compound 133a, a direct hybrid, and 133b, an oxa-isoster hybrid dimethylcarbamate thioflavin-based orto-phenol derivative. A third hybrid compound 133c was obtained by a combination of the dimethylcarbamate moiety from rivastigmine (132) and phenyl-deferiprone analogue 131. A biological evaluation revealed a significant metal chelating ability for all three hybrids 133a-c, with low toxicity against neuronal cells. However, all three compounds showed only a moderate AChE inhibitory activity, with 133a being the most potent AChEI with IC50= 20.9 µM, followed by derivatives 133b and 133c with IC50 values of 58.2 and 48.8 µM, respectively [147].
Fig. (53).

Structural design of the hybrid compounds 133a and 133b, two rivastigmine-thioflavin T-based derivatives, and 133c, a rivastigmine-deferiprone derivative with multifunctional properties.
A series of salicylamide derivatives (136) was designed and evaluated as potential multifunctional agents for AD by Song and co-authors (Fig. 54). The target compounds were designed by MH of salicylic acid (134) and chromone-2-carboxamido-alkylbenzylamine (135). In vitro evaluation evidenced compound 136a as the most promising multifunctional ligand, exhibiting moderate AChE inhibitory activity (IC50= 15.2 μM), with high selectivity in relation to BuChE, besides a strong antioxidant activity expressed by 2.46 trolox equivalent. In addition, 136a showed significant metal-chelating ability, aside from anti-neuroinflammatory and Aβ-aggregation inhibitory activities (42.5% at 25μM), with a good BBB permeability (6.27x10-6 cm/s) [148].
Fig. (54).

Design strategy for a series of N-alkylbenzylamine salicylamide derivatives 136.
Memoquin (137), the first pre-clinical phase tested MTDL drug candidate for AD [149], capable to interact with the three molecular targets related to AD pathology: AChE, Aβ and BACE-1, was selected by Pan and co-authors as a structural pattern to an MH approach in the design of novel memoquin-ferulic acid hybrids. The rationale underlying this strategy was based on the combination of the N-alkyl-methoxyphenyl side chain of 137 with the 4-hydroxy-5-methoxy-cynnamoyl fragment of ferulic acid (5) in the drawing of a novel family of memoquin-ferulic acid hybrids 138 (Fig. 55). Aiming to obtain innovative multifunctional compounds, with additional strong antioxidant and metal-chelating
Fig. (55).

Design strategy for memoquin_Ferulic acid hybrids 138.
abilities and better brain permeability than the pattern 137. The hybrid 138a was identified as a lead-compound with the best biological results, exhibiting the significant inhibitory activity of ChEs (IC50= 3.2 and 36.2 µM, for AChE and BuChE, respectively) and Aβ1-42 aggregation (49.3% at 50 µM, 38.1% at 25 µM and 26.8% at 10 µM). Compound 138a also showed excellent in vitro antioxidant and neuroprotective properties, with adequate BBB permeability being reported as promising therapeutical multifunctional agent for AD [150].
In 2017, Wang and co-workers reported a novel series of cinnamamide-dibenzylamine hybrids, with potential therapeutic use for AD treatment (Fig. 56). The structural design involved the fusion of the N,N-dibenzyl-(N-methyl)-amine pharmacophore from AP2238 (139) and cinnamamide subunit 140, in a new chemical entity, with the goal of discovering inhibitors of ChEs and Aβ self-induced aggregation. The selection of these two molecular patterns was based on earlier reports that compound 139 have been designed dual AChEI, capable of binding both CAS and PAS of hAChE and that cinnamamide derivative 140 has been identified as an inhibitor of self-induced Aβ aggregation. Among all synthetized hybrid homologues, compound 141 exhibited the best biological results, with good inhibitory activity of AChE (IC50= 4.64 μM) and BuChE (IC50= 9.72 μM), Aβ1–42 self-induced aggregation (56.2% at 20 μM) and metal-chelating properties. Moreover, 141 did not show significant cytotoxicity against PC12 cells [151].
Fig. (56).

Design strategy and chemical structures for the most potent Cinnamamide-dibenzylamine hybrid 141.
Taking into account the pivotal role of OS and the recent findings concerning the overexpression of histone deacetylases (HDAC), mainly HDAC3 and HDAC6, in the hippocampus of AD brains, Hu and co-workers proposed novel molecular hybrids capable of acting concomitantly as antioxidants and inhibitors of HDAC3 and HDAC6. For such purpose, a hybrid scaffold was planned from the MH of the structure of tubastatin A (142), a known HDAC inhibitor, and ebselen (41), a potent antioxidant (Fig. 57). Biological results revealed compound 143 as a potent inhibitor of HDAC3 (IC50= 1.3 µM) and HDAC6 (IC50= 0.04 µM), with additional neuroprotective activities in both in vitro H2O2 and 6-OHDA-induced neuronal damage models at 5 and 10 µM, and no apparent neuronal cytotoxicity on PC12 cells [152].
Fig. (57).

Molecular hybridization between tubastatin A (142) and ebselen (41) in the design of the MTDL 143.
Hydroxybenzoic acids are known to exhibit multiple biological properties, including antioxidant, neuroprotective, anti-inflammatory, antimicrobial and anticancer. Protocatechuic acid (144) and gallic acid (145) are two representative examples of such bioactive class, but due to their low bioavailability, they have not been clinically effective as antioxidants.
With the aim of targeting mitochondrial impairment and the consequent ROS imbalance and deficits in energy supply associated with NDs pathogenesis, in addition to ChE inhibition, novel molecular hybrids were envisaged by Li and co-workers as a result of the overlap of the structures of 144 and 145 in a single molecule, with the insertion of an alkyl-triphenylphosphonium cation (TPP) functionality (Fig. 58). Pharmacological results led to the identification of the most promising hybrids 146a and 146d with the highest antioxidant potential and highly potent BuChE inhibition with IC50 values of 85 and 106 nM, respectively and compounds 146b and 146c with inhibition of AChE (IC50 = 7.7 and 7.2 µM). Further cytotoxicity assays showed no significant effects on human neuroblastoma (SH-SY5Y) and hepatocarcinoma (HepG2) cell lines [153].
Fig. (58).

Synthetic strategy for the development of gallic acid-protocateic acid hybrids 146.
Sang and co-workers reported compounds used the ferulic acid derivatives EJMC-7f (147) and LDDD-3 (148) to draw a novel hybrid molecular architecture, represented by compound 149 with multifunctional action against AD. In previous studies, derivative EJMC-7f (147, Fig. 59) was identified as a potent BuChE inhibitor (IC50= 0.021 µM), and a moderate Aβ-aggregation inhibitor and antioxidant, whereas LDDD-3 (148) has been reported as a high selective BuChE inhibitor. Face to these findings, and in the search for innovative and improved compounds with concomitant antioxidant, anti-Aβ aggregation and BuChE inhibitory activities, Sang and co-workers used the structures of the ferulic acid derivatives 147 and 148 as models to draw a novel hybrid molecular architecture, represented by compound 149 (Fig. 59). The biological screening revealed the hybrid derivative 149 with the best MTDL profile, being capable to selectively inhibit BuChE in a nanomolar range (IC50= 8.9 nM), in comparison to AChE inhibition (IC50= 12.1 µM), but without relevant antioxidant activity via ORAC method. However, 149 exhibited the inhibitory activity of MAO-A and MAO-B with IC50 values of 6.3 and 8.6 µM, respectively, with the additional ability for inhibiting Aβ-aggregation in 53.9% at the concentration of 25 µM and disassemble the well-structured Aβ fibrils. Besides its higher potency in BuChE inhibition and better MTDL profile in relation to the parent prototypes 147 and 148, compound 149 also showed neuroprotective effects against Aβ-induced damage in SH-SY5Y cells, with very significant BBB permeability (Pe= 16.6 x 10-6 cm/s). Additionally, 149 was tested in an AlCl3-induced zebrafish AD model, showing significantly improved dyskinesia recovery rate and potent protective effect on Aβ1-40-induced vascular injury, with very low in vivo acute toxicity. To our knowledge, these sets of results highlight compound 149 as one of the most advanced MTDL drug candidate prototypes reported in the recent literature [154].
Fig. (59).

The design strategy by MH for the novel ferulic acid derivative 149.
Malawska and co-workers found that compounds having β-hydroxylated amine groups, such as GRL-8234 (150, Fig. 60), had the ability to inhibit BACE-1 by interacting specifically with the amino acid residues Asp32 and Asp228. Considering this information and searching for new disease-modifying compounds for AD, they planned the synthesis of the new BACE-1 hybrid inhibitors (151) based on the MH of the AChEI phtalimide-type donepezil derivative 152 and the β-hydroxy-N-benzylamine subunit from 151. A family of synthetic hybrids was screened and led to the identification of compound 152 (Fig. 60) with the desired multi-target action, selectively inhibiting AChE with IC50 value of 3.32 µM, 47.3% of BACE-1 (at 50 µM) and 24.9% of Aβ aggregation (at 10 µM) [155].
Fig. (60).

Design strategy by MH for multitarget anti-Alzheimer’s agents 152.
Scutellarin or 4,5,6-trihydroxyflavone-7-glucuronide (56), is a natural plant metabolite with remarkable antioxidant and anti-inflammatory effects, metal-chelating ability and inhibitory activity on Aβ fibrils formation. For these sets of interesting properties related to NDs pathobiology, 56 has been described as a useful prototype for MH in the design of novel compounds capable to act as multifunctional neuroprotective agents. In this regard, Sang and colleagues exploited the structure of 56 in combination with N,N-dialkyl carbamate pharmacophore from rivastigmine (132) aiming to obtain novel hybrid scutellarin-based AChEIs with antioxidant and neuroprotective properties, represented by compound 153 (Fig. 61). Thus, a new series of hybrids with a flavone-carbamate scaffold was synthetized and screened for antioxidant activity, revealing that almost all compounds exhibited moderate to high radical scavenging activities and neuroprotective effects. Further in vitro and in vivo assays highlighted the N,N-diethyl carbamate derivative 153a as the most balanced multiple active hybrid compound, with a selective AChE inhibitory activity with an IC50 value of 0.57 µM, acting as a mixed-type inhibitor, binding simultaneously to CAS and PAS of AChE as expected for a rivastigmine-based hybrid. Moreover, 153a exhibited antioxidant activity (1.3-fold of Trolox), metal-chelating ability for Cu2+, Zn2+, Al3+ and Fe2+, aside of a protective activity against Aβ1-42- induced injury in human neuroblastoma cells (SH-SY5Y) with a cell viability of 78.5%. In vivo results corroborated the neuroprotective effects of compound 153a that was able to revert the scopolamine-induced cognitive impairment in mice [156].
Fig. (61).

Design strategy for new series scutellarin-carbamate hybrid derivatives represented by the MTDL 153a.
In another approach, Xiao and co-workers exploited the MH of the carbamate subunit from 132 and the chalcone scaffold present in the derivative 154 to furnish a novel molecular hybrid architecture of potential multifunctional ChE inhibitors for AD. The selection of the 4’-aminochalcone fragment from 154 as a pharmacophore was justified by evidence that this subunit could be responsible for interactions with Aβ plaques. As a result, the authors obtained a family of eighteen novel aminochalcone-rivastigmine hybrid compounds (Fig. 62), which biological screening revealed compound 155 with the most promising multitarget profile. The lead-molecule 155 inhibited selectively MAO-B (IC50= 0.29 µM) and AChE (IC50= 4.91 µM) in a selective and mixed-type mode of ChE inhibition, displaying the ability of binding simultaneously to the CAS and PA of AChE. In addition, 155 exhibited antioxidant activity 2.83-fold higher than trolox, metal chelation ability and inhibited self-induced Aβ1-42 aggregation and Cu2+-induced Aβ1-42 aggregation by 89.5 and 79.7% at 25 μM respectively, with in vitro adequate BBB transposition properties [157].
Fig. (62).

Design of 4’-aminochalcone-revastigmine hybrid 155, identified as MAO-B, AChE inhibitor, with additional antioxidant and Aβ aggregation inhibitory activities.
As described earlier, PDEs are considered interesting target-enzymes in the controlling of OS and unbalanced production of radical species during the progress of NDs. PDE10 is one of these enzymes that is responsible for the hydrolysis of both cAMP and cGMP highly expressed in brain tissue. Recent findings evidenced that inhibition of PDE10 could lead to increased levels of cGMP in striatum and phosphorylated cAMP-response element binding protein (CREB), which is a downstream biomarker of cAMP production, and could represent potential advances in the modulation of pathobiology related to NDs [158-160].
In this regard, Li and co-workers planned two series of molecular hybrids (160 and 161) by combination of a PDE-inhibitor pharmacophore and antioxidants based on the structure of PDE10A inhibitors, in particular, the quinazoline derivatives 156 and 157 (Fig. 63). The antioxidant pharmacophore, linked to 4- or 6-position at the quinazoline fragment, was idealized from structural subunits present in the known antioxidants melatonin (158), p-coumaric acid (159) and lipoic acid (99, Fig. 63). From the first series, compound 160a was highlighted due to its significant PDE10 inhibitory activity, but lesser than papaverine, and a strong antioxidant activity with ORAC of 3.3, in comparison to ferulic acid (ORAC= 1.6). Conversely, in the other series, compounds 161a and 161b showed the best PDE10 inhibitory activities with IC50 values of 0.33 and 0.24 µM, but moderate antioxidant activity (ORAC= 1.0 and 1.3, respectively). In spite of its lower potency against PDE10 (IC50= 0.64 µM), compound 161c was selected as lead-compound for further investigation, due to its best balanced multi-target action, with good antioxidant activity (ORAC=2.3) and adequate BBB penetration evidenced by the predicted topological polar surface area (TPSA) of 89.06 [161].
Fig. (63).

Rational design of the series of quinazoline-based hybrid compounds 160 and 161 with PDE10 inhibitory and antioxidant activities.
It is well established in the literature that BACE-1 contributes to the accumulation of Aβ peptides in the brain and leads to neuronal death, via the catalytic cleavage of amyloid precursor protein (APP) and plays an important role in the pathogenesis of AD. Due to this fact, Haghighijoo et al. synthesized and evaluated a series of quinazolinone-hydrazone hybrid derivatives (165, Fig. 64) designed as BACE-1 inhibitors, aiming at the modulation of the production of all Aβ forms. The design strategy was based on the combination of key fragments of the 2-aminiquinazolines 162, MK-8931 (163) and the iminopyrimidinone skeleton 164, leading to a novel series of quinazoline-based hydrazone hybrids, represented by compounds 165a and 165b (Fig. 64). Docking studies revealed a strong interaction between compound 165a and the key residues of BACE-1 active site, which was confirmed in vitro with a high inhibitory activity of BACE-1 (IC50= 3.7 μM). On the other hand, compound 165b showed a moderate inhibition of BACE-1 (IC50 = 27.6 μM), but with a significant antioxidant effect with an IC50 value of 8.4 μM [162].
Fig. (64).

Design strategy and chemical structures for the most potent quinazolinone-hydrazone hybrids 165a-b.
Based on SAR analysis of H3R antagonists, Huang et al. used the strategy of MH for combination of the structures of the AChEI BYYT-25 (166) with the BACE-1 inhibitor 167 in the design of a quinoxaline-based hybrid family (168) as potential MTDLs for AD treatment (Fig. 65). The biological screening of seventeen compounds revealed moderate to potent inhibitory/antagonistic activities towards AChE, BACE-1 and H3R. Compound 168a, showed the most effective MTDL profile, acting as an antagonist and inverse agonist of H3R (IC50= 280 and 189.3 nM, respectively), besides inhibition of AChE IC50= 483 nM) and BACE-1 in 46.6% at 20 µM and high selectivity over histamine receptors subtypes H1/H2/H4 [163].
Fig. (65).

Design of quinoxaline-based hybrid compounds (168) based in AChEI BYYT-25 (166) with BACE 1 inhibitor compound 167.
Searching for new dual inhibitors of AChE and β-secretase (BACE-1), Deng co-workers synthetized a series of tetrahydroisoquinoline derivatives (171) designed by MH of (−)-gallocatechin gallate (169) and (+)-(S)-dihydro-ar-tumerone (170, Fig. 66). The biological evaluation led to the identification of compound 171a as the most active dual inhibitor of AChE and BACE-1, with IC50 values of 25.48 nM and 4.45 µM, without significant toxicity. In addition, 171a exhibited Hydrogen superoxide suppression activity which is thought to play a role in the reduction of ROS generation in AD brain [164].
Fig. (66).

MH between (−)-gallocatechin gallate (169), (+)-(S)-dihydro-ar-tumerone (170) in the design of the dual AChE and BACE-1 inhibitor 171.
Hong and co-workers synthesized and evaluated a series of genistein (172)-based hybrid derivatives (174, Fig. 67) as potential multifunctional anti-AD agents. Based on previous results, the goal of this attempt was to optimize de pharmacological potential of derivative 173 by conjugating with pyrrolidine or imidazole subunits in the novel hybrid genistein-O-alkylamine scaffold 174 by using different lengths of carbon chain as a tether. By this approach, compound 174a was discovered as a stronger AChE inhibitor than rivastigmine (IC50= 6.53 µM) with an IC50 value of 0.034 µM. These results were corroborated by computational studies that showed that 174a targeted both CAS and PAS of AChE. In addition to AChE inhibitory activity, this compound was also capable to chelate biometals such as Cu2+ and Zn2+, with potent peroxyl-scavenging activity, without obvious toxic effects on HepG2 and PC12 cell viability at the concentration of 100 μM [165].
Fig. (67).

Design strategy for the genistein-O-alkylamine derivatives 174.
Considering a number of scientific evidences reported in the literature highlighting that the endocannabinoid system could be an interesting molecular target in the AD pathophysiology, Rizzo and co-workers rationalized that molecules capable to modulate CB1 receptors along with inhibition of AChE and Aβ-aggregation could lead to innovative multifunctional ligands of therapeutical interest. Thus, they considered the structures of the CB1 antagonist LY320135 (175a) and its homologue SKF (175), with anti-Aβ aggregating properties, both with a common 2-arylbenzofuran system identified in their earlier studies as an important pharmacophore for anti-amyloid and neuroprotective effects, for MH with AChE inhibitory long-chain N-methyl benzyl-methylene moiety (176) [166].
As a result, the authors obtained a series of 2-arylbenzofuran-based hybrid compounds rationally designed as multi-potent disease-modifying agents (177, Fig. 68). Among them, regioisomers 177a and 177b were identified as the most promising cholinesterase inhibitors, with a capital influence of the position of the O-methylene-amine substituent in their potency and selectivity. Compound 177a, the para-substituted isomer showed inhibition of 47% in the formation of Aβ-fibrils and an antagonist effect on CB1 receptor (EC50= 0.79 µM), but with the low capability of discriminating human AChE and BuChE with IC50 values of 40.7 and 38.1 µM, respectively. Conversely, the meta-regioisomer 177b exhibited significantly increased potency in ChE inhibition, with IC50 values of 0.24 and 2.88 µM for AChE and BuChE, respectively, with almost 10-fold higher selectivity for AChE. However, in spite of the increasing effect on cholinesterase inhibition, the meta-isomer lost the CB1 antagonism and showed less ability (35%) in the inhibition of Aβ-fibrils [166].
Fig. (68).

MH of pharmacophore subunits of compounds SKF-64346 (175), LY320135 (175a) and 2-arylbenzofuran derivatives 176 in the design of the multipotent compounds 177.
In another approach, the benzofuran functionality was exploited by Nadri and co-workers in the design of a series of oxobenzofuranylidene pyridinium derivatives, as new hybrid AChEIs based on the structure of benzofuran analogues 178 and donepezil 179 (Fig. 69). Among twenty-one derivatives, compound 180 showed the best AChE inhibitory activity with IC50= 10 nM [167].
Fig. (69).

Design of the target compound 180.
Aiming to explore the modulation of nicotine acetylcholine receptors (nAChRs) in the pathobiology of AD, Nencini co-workers designed two new hybrid skeletons A and B, rationally planned in the basis of the structure of compounds 181 and 182, respectively (Fig. 70). Both compounds have been previously identified as agonists of α1, α3 and α7 nicotinic receptor subtypes (IC50= 15.1, 7.3 and 0.07 µM, respectively) and of serotonin receptor subtype 5HT3 (IC50 >30 µM). However, in spite of the high potency of compound 181 towards nAChRs, it has no selectivity. Conversely, compound 182 showed a good selectivity, but lacks in potency (IC50= 0.9 µM for α7 and IC50 >30 µM for both α1 and α3). In order to optimize their selectivity and potency for α7 nAChRs and based on previous SAR and predicted drug-like properties, these two molecules were additionally studied by molecular docking, leading to a novel series of sixteen hybrid molecules designed by combination of optimal the features of the urea-derivative 181 and the pyrazole analogue 182. The biological data showed that compound 183 (Fig. 70) was selective and the most potent α7 nAChR agonist (IC50= 0.22 μM), with IC50 values for α1 and α3 nAChR and 5HT3 receptor greater than 30 μM [168].
Fig. (70).

Design strategy for the multipotent hybrid compounds 181 and 182, and the optimized selective α7 nAChR agonist 183.
In another work, Sang and co-workers designed and synthesized a series of 2-acetyl-5-O-(amino-alkyl)phenol derivatives (Fig. 71) as multifunctional AChE and MAOs inhibitors for the treatment of AD. Biological results highlighted compound 186 as a selective AChEI (eeAChE, IC50= 0.69 µM) with a selective index (SI) of 32.7 in relation to BuChE. In silico and kinetic studies suggested that 186 could simultaneously bind to the CAS and PAS of AChE. Compound 186 was also capable to inhibit selectively MAO-B (IC50 = 6.8 µM), exhibiting neuroprotective and antioxidant (ORAC= 1.5 eq) properties, as well as a selective metal-chelating ability agent, with adequate in vitro BBB penetration [169].
Fig. (71).

MH of compounds 184 and 185 in the design of the new 2-acetyl-5-O-(amino-alkyl)phenol scaffold, represented by the multipotent derivative 186.
3. MOLECULAR HYBRIDS DESIGNED AS DRUG CANDIDATE PROTOTYPES FOR PD
Dopaminergic approach represents the major therapeutic strategy to counteract the symptoms associated with PD. At present, despite PD remains incurable, L-dopa is the first-choice drug for symptomatic treatment. It is a metabolic precursor, which is able to cross the BBB and, then be converted into dopamine by the action of dopa-decarboxylase. In parallel, A2A adenosine receptors (ARs) antagonists have been used to amplify the therapeutic effects of L-dopa and reduce motor complications resulting from its long-term use. Dalpiaz and co-workers proposed an innovative molecular scaffold, represented by compound 190 (Fig. 72), that was expected to act as a pro-drug with multipotent activity against PD. The rationale basis for the design of 190 considered the MH between a dopamine (187) moiety coupled to the A2A receptor antagonist 189 through a succinic spacer subunit (188). Preliminary evaluation of plasma stability showed a half-life of 2.73 h for the hybrid 190. Metabolic studies evidenced that 190 was not only subjected to hydrolysis at the diamide spacer subunit, leading to the release of the active metabolites 187 and 189, but also being able to the generation of 191 and 192, with 189 and 192 as the most abundant metabolites. This metabolic profile was particularly interesting because it guarantees a continuous and prolonged release of dopamine, which is the product of a second metabolism step of 192. When assessed integrally, compound 190 was not able to interact with dopaminergic receptors, but showed a good affinity for A2A receptors (Ki= 2.07 and 7.32 nM in rat striatum or hA2ACHO cells, respectively [170].
Fig. (72).

Design of the prodrug 190 by MH between dopamine (187) with a new A2A antagonist 189 linked by a succinic spacer (188).
In 2010, Anzini and co-workers reported the synthesis and biological evaluation of new hybrids planned for acting on NDs, particularly against PD, based on the combination of the structure of riluzole (193) and fragments of 1400W (194), aminoguanidiane (195) and L-NAME (196, Fig. 73). Riluzole is a 2-amino-benzothiazole derivative, with neuroprotective, anticonvulsant, anxiolytic and anti-ischemic properties, approved as the first-choice drug for the treatment of ALS, but with also relevant neuroprotective effects in in vivo models of PD, HD and cerebral ischemia. Its mechanism of action has been described as based on the blockage of tetrodotoxin (TTX)-sensitive sodium channels, with also antagonist effect on kainate and NMDA receptors. Compound 194 is reported for its anti-inflammatory and neuroprotective activities, whereas 195 and 196 showed NOS-inhibition activity. Based on these findings, the authors designed a new family of potential multifunctional agents with anti-inflammatory, neuroprotective and voltage-dependent Ca2+ channels modulatory activities. Preliminary results led to the identification of the ligand 197, with direct ROS scavenging activity, regulatory property on voltage-dependent Ca2+ channels, but with weaker activity than 193. Thus, Chauhan and co-workers synthesized new benzothiazines based on compound 197 (Fig. 73) seeking for optimized new compounds with improved neuroprotective activity. As a result, additional thirteen 197-based analogues were prepared and evaluated for their ability to inhibit the release of glutamate and lactate dehydrogenase (LDH) by human neuronal cells (SH-SY5Y) and in rat cortical brain slices, with compound 198 exhibiting a significant inhibition in the release of glutamate and LDH in rat cortical brain slices with minimal effective concentration (MIC) of 0.1 µM for both studies. Compound 198 also showed neuroprotective and ROS inhibitory activities towards glutamate-induced damage in SH-SY5Y cell line [171].
Fig. (73).

Design and optimization of a new hybrid compound, represented by compounds 197 and 198, with multipotent activities against PD.
Literature data suggest that inhibition of AChE and βA deposits may contribute positively to the treatment not only of AD, but also of PD. One example is 3-substituted coumarin derivatives (199), previously reported as AChEIs with anti-Aβ aggregation and dopamine inducing release activities, and tetracyclic tacrine derivatives, which in addition to AChE inhibitory properties, exhibited neuroprotective activity in different neurodegenerative conditions [172, 173].
Taking these data into account, Sashidhara and co-workers designed a new series of potential neuroprotective, antioxidant and α-synuclein aggregation inhibitors MTDLs, based on the combination of 3-aryl coumarin and tacrine (48) fragments into a single skeleton (Fig. 74). Preliminary evaluation of α-synuclein inhibition highlighted compounds 200a and 200b as that was also able to activate the dopaminergic system with lower cytotoxicity than tacrine against HepG2 and HEK-293 cell lines. Additionally, both compounds were investigated for their ability to activate the DAF-16 transcription factor, an important factor on the antioxidant pathway, with derivative 200 showing a relevant activity [174].
Fig. (74).

Molecular planning to obtain the hybrids 200.
Considering the known ability of carbonyl α,β-unsaturated compounds, such as ethyl cinnamate (201), to activate Nrf2 by inhibition of Keap1 via Michael addition of a cysteine residue present in Keap1, and the ability of the neuro-hormone melatonin (86) to activate antioxidant processes, Buendia and co-workers used MH to obtain a new family of cinnamate-melatonin hybrids, represented by compound 202 (Fig. 75) designed as multipotent candidates against PD. In order to investigate Nrf2 activation ability, AREc32 cells, a HepG2-derived cell line programed to express antioxidant response element (ARE) was used. In this assay, compound 202 was capable to double the rate of Nrf 2 activation (at the concentration of 5.56 µM) in relation to standard untreated cells. This result could explain the observed antioxidant properties, evidenced by the ORAC value of 9.21 Trolox equivalents. In addition, 202 showed 49.7% of neuroprotection on rotenone-induced injury in SH-SY5Y cells, being more effective than melatonin (29.3%). Face to these promising results, the authors investigated the neuroprotective activity of 202 against okadaic acid (OA)-induced cytotoxicity, a model that stablish a close relationship with PD pathophysiology. OA is a toxin able to inhibit protein phosphatases 1 and 2A, inducing tau protein hyperphosphorylation and aggregation, and consequently, induces cell death by oxidative stress damage. In addition, 202 showed 49.7% of neuroprotection on rotenone-induced injury in SH-SY5Y cells, being more effective than melatonin (29.3%). Face to these promising results, the authors investigated the neuroprotective activity of 202 against okadaic acid (OA)-induced cytotoxicity, a model that stablish a close relationship with PD pathophysiology. OA is a toxin able to inhibit protein phosphatases 1 and 2A, inducing tau protein hyperphosphorylation and aggregation, and consequently, induces cell death by oxidative stress damage. Compound 202 showed higher neuroprotective activity than melatonin at concentrations of 1 and 3 µM and, thus, was reported as an innovative MTDL with a promising pharmacological profile for development against PD [175].
Fig. (75).

Molecular design of the melatonin-cinnamate hybrid 202 with neuroprotective, antioxidant and Nrf2-activation activity.
Focusing on the inhibition of MAO-B and H3R receptors as suitable molecular targets for controlling PD, Affini and co-workers sought inspiration in the structures of ciproxifan (203) and UCL2190 (204, Fig. 76), two H3R antagonists with Ki values of 46 and 11 nM, respectively. The other two bioactive prototypes were the indanone derivatives 205 and 206 that showed MAO-B inhibitory activity (IC50= 3.0 and 9.2 nM, respectively), used in the design of new phenoxy-indanone hybrids, represented by compound 207 as multifunctional MAOs inhibitors and H3R antagonists. Biological evaluation led to compound 207 as the most balanced multipotent hybrid, showing a reversible and selective inhibition of MAO-B (IC50= 262 nM) with a SI > 36, and a Ki value of 6.5 nM for H3R interaction. In spite of did not show MAO-B inhibition in comparable potency to approved drugs, such as L-deprenyl (IC50= 3.9 nM), the authors highlighted compound 207 due to its innovative MTDL profile of action, which could of great relevance in the development of new effective drugs for PD [176].
Fig. (76).

MH as strategy for the design of phenoxy-indanone hybrids, with compound 207 with the best MTDL profile as potential anti-PD agent.
In another approach, Elmabruk and co-workers proposed the synthesis of new molecular hybrids, represented by 211 and 212 (Fig. 77), rationally designed by MH of the pharmacophore subunits of compounds 208, 209 and 210 as MTDLs potentially useful in the development of drugs to treat PD. The rational basis for the selection of the parent prototypes took in to account that all three compounds are capable to modulate important molecular targets related to PD pathobiology. Compound P7C3A20 (208) have been described as a potent antioxidant, due to the presence of the aminopropyl carbazole subunit, whereas compound 5-OH-DPAT (209) has shown potent in vivo activity in PD models, and compound D-512 (210) showed to be a potent D2/D3 receptor agonist. In vitro pharmacological screening results led to the identification of hybrids 211 and 212 as the best D2/D3 agonists, with compound 212 showing the higher potency (EC50= 0.87 and 0.23 nM for D2R and D3R subtypes, respectively). Conversely, compound 211 was less potent (EC50= 48.7 and 0.96 nM, for D2R and D3R subtypes, respectively), but around 51-fold more selective for D3R. In the 6-OHDA-induced neurotoxicity model, compound 212 significantly prevents neurotoxicity 5 and 10 µM, and both 211 and 212 showed relevant neuroprotective effects after 30 days of treatment at the concentration of 172.9 µM of cells exposed to α-synuclein [177].
Fig. (77).

MH of the parent compounds 208, 209 and 210 in the design of the hybrids 211 and 212 with remarkable D2/D3 agonist and neuroprotective activities.
In addition to the already discussed importance of MAO-B inhibition in the therapeutics of PD, the inhibition of Ca2+ influx in neurons may play an additional important role in the neuroprotection effect. In this context, selegiline (83), a known potent and selective inhibitor of MAO-B (IC50 <10 nM) and NGP1-01 (213, Fig. 78), capable to modulate the Ca2+ influx in neurons, were taken as structural models by Zindo and co-workers in the design of hybrid compounds that could modulate concomitantly both molecular targets [178].
Fig. (78).

Molecular design of the Ca2+ influx modulator and MAO inhibitor hybrid 214 by combination of the structures of selegiline (83) and NGP1-01 (213).
The resultant synthetic hybrids were tested for their ability to inhibit MAOs, regulation of Ca2+ influx related to the inhibition of L-type voltage-gated calcium channel (VGCC) and their antagonist activity on N-methyl-D-aspartate (NMDA) receptors, a specific receptor inducing the increase of levels of Ca2+ within the neuron. Biological results evidenced compound 214 that showed selective inhibition of MAO-B (IC50= 28.18 µM), 54% neuroprotection against cytotoxicity induced by MPP+ and downregulation of Ca2+ influx through the inhibition in 25.7 and 32.5% of VGCC and NMDA (32.5%) at 10 µM, without further toxic effects at 10 µM [178].
4. MOLECULAR HYBRIDS DESIGNED AS DRUG CANDIDATES PROTOTYPES FOR HD AND ALS
As described earlier, riluzole 193 is neuroprotective, anticonvulsant, anxiolytic and anti-ischemic properties approved drug for the treatment of ALS, but is reported as with low efficacy in clinics. Aiming to improve its pharmacological profile, Calabro and co-workers used the benzothiazole scaffold of 193 and the substituted benzamide subunit 215, reported as responsible for neuroprotective properties, for the design of two series of multifunctional 2-amino-benzoxazole and 2-amino-benzothiazole hybrids (216 and 217, Fig. 79). The rationale behind this approach was based on the evidences that the multi-target mechanism of action of 215 is related to the modulation of excitotoxicity by inhibition of glutamate release and its postsynaptic effects by noncompetitive blockade of N-methyl-D-aspartate (NMDA) receptor. In addition, compound 215 also potentiates the function of postsynaptic GABAA-receptor and affects a voltage-dependent Na+ channels. Considering that excitotoxicity and damage in motor neurons is one of the main hallmarks of ALS, the authors inferred that the riluzole-based structural pattern could be conserved or modified by a bioisosteric exchange of the thiazole ring by an oxazole system, coupled to benzamide functionality 257 to generate novel chemical entities with improved additional voltage-dependent ion channels blockade and neuroprotective activities. As a result, eight synthetic hybrid riluzole-amide (216) and riluzole-urea (217) derivatives were synthetized and evaluated for their abilities of reducing voltage-dependent Na+ current in primary cultures of cerebellar and cortical neurons, as well as their effects on GABA and NMDA receptors. Compounds 216a and 216b were identified as the most active in both series, decreasing in 97 and 98% the voltage-dependent Na+ current in both neuronal cultures. However, no significant affinities at GABA and NMDA receptors were evidenced, suggesting that the lead-compounds could be further investigated as good potential blockers of voltage-dependent Na+ channels, but without multifunctional pharmacological profile [179].
Fig. (79).

Benzoxazole and benzothiazole riluzole-based hybrid derivatives 216 and 217 designed as voltage-dependent Na+-channels blockers and GABA and NMDA receptors antagonists.
Despite dramatic social and individual impacts and intense attention from researchers, there are few approved treatments for amyotrophic lateral sclerosis (ALS), with only riluzole (Rilutek®, 193) and edaravone (Radicava®, 218) currently available to patients. Unfortunately, the mechanistic basis of the activity of these drugs is currently not well-defined, limiting the ability to the design new drugs for ALS. Riluzole-like heterocyclic small molecule frameworks are also known to demonstrate potential neuroprotective properties. Thus, pifithrin-α (219) contains a partially saturated bicyclic thiazolyl core and an aromatic substituent located at the end of a C2- tether attached at the N1 position. A similar substituent pattern is conserved in riluzole analogues 220, with the presence of a functionalized ethyl side-chain endowing neuroprotection activity at similar levels to riluzole in vivo models. As a novel chemical target with potential for neuroprotection and bearing in mind the pyrazinone core of edaravone, Sweeney and co-workers described the synthesis of triazoles 221 (Fig. 80) as new chemical matter with structural resemblance to riluzole and related molecules. Seven substances were identified as having neuroprotective activity, with two compounds having similar activity to riluzole. The pyridyl substituted compounds 221a and 221b are the most promising, with neuroprotective properties greater than riluzole in two independent in vitro assays on primary neurons. These two riluzole derivatives were able to preserve motor neuron morphology in response to kainite-induced damage, and significantly increased the number of neuronal processes extending between 5-10 radii compared to kainate treatment. These findings highlighted compounds 221a and 221b as interesting ligands for further development aiming ALS treatment [180].
Fig. (80).

Chemical structures of riluzole (193) and edavarone (218) and pifithrin-α with neuroprotective properties and riluzole-based hybrids 221, designed as optimized riluzole-based hybrid ligands for ALS.
Conclusion
The development of more effective drugs for NDs remains a big challenge for Medicinal Chemistry, whether in Academia or in Pharma Industry. Due to the characteristic multifactorial pathogenesis of all NDs, involving common biochemical and physiological changes related to oxidative stress, neuroinflammation, mitochondrial damage, depletion in energy supply, synaptic impairment and neuronal loss, the discovering and the development of innovative disease-modifying drugs is aproblem to be solved to overcome current clinical limitations and therapeutical inefficiency. Among NDs, some of them still have no available therapy, and others, such as the most prevalent AD, PD, HD and ALS, have been treated with a very restrict chemotherapeutic arsenal, with serious limitations in efficacy and tolerance. Their treatments are based on symptomatic medications, trying to break the disease progress and severity, offering a better life quality for patients and putting off their vegetative condition and death. In this review article, it became clear that as the most prevalent ND worldwide, AD has centralized the attention of researchers in the discovery of new drug candidates, followed by studies on PD. To our knowledge, ALS and HD remain to call very low attention from medicinal chemists, which are exemplified by the very few works found in the literature focusing the discovery of new ligands specifically designed for such diseases. Regarding this complex multifactorial pathophysiology, polypharmacology, and especially the multifunctional or MTDLs approach, a new perspective is represented and started a new paradigm for the design and the discovery process of novel bioactive chemical entities. By this concept, a single molecule could be capable to act in a simultaneous modulation of diverse molecular targets, aiming to block out compensative mechanisms and alternative biochemical routes, avoiding drug-drug interaction, hepatic stress, higher adverse effects form active metabolites, with probable improvement in therapy effectiveness and, hopefully, the cure. In the last twenty years, it has been witnessed that a strong movement towards the design of new molecules based on the Molecular Hybridization approach, leads to hybrid molecular scaffolds, explores a multitude of chemical diversity in terms of pharmacophore, auxophore and tether subunits, leading to a vast chemical library of potential drug candidates. Consequently, new drug prototype candidates have been routinely identified and, if they do not be able to direct drug development, they represent a number of opportunities and starting points of a revolutionary new way of combat NDs. Once discovered, a new potential drug candidate, balancing the potency and relative selectivity of the different actions and targets involved in the multifunctional profile of a single molecule is an additional challenge to be solved. In spite of none of these, MTDL drug candidates have reached clinical phase of development, the number of promise bioactive molecules that have been described in this review, represent the hope of the Medicinal Chemistry Community, that in the next few years, it is possible to discover innovative drugs capable to modify the course of these devastating pathologies effectively.
Acknowledgements
The authors are grateful to the Brazilian Agencies CNPq (#454088/2014-0, #400271/2014-1, #310082/2016-1), FAPEMIG (#CEX-APQ-00241-15), FINEP, INCT-INOFAR (#465.249/2014-0), PRPPG-UNIFAL for financial support and fellowships.
Consent for Publication
Not applicable.
Funding
This study was also financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior - Brazil (CAPES) - Finance Code 001.
Conflict of Interest
The authors declare no conflict of interest, financial or otherwise.
REFERENCES
- 1.Bolognesi M.L., Matera R., Minarini A., Rosini M., Melchiorre C. Alzheimer’s disease: new approaches to drug discovery. Curr. Opin. Chem. Biol. 2009;13(3):303–308. doi: 10.1016/j.cbpa.2009.04.619. [DOI] [PubMed] [Google Scholar]
- 2.Youdim M.B.H., Buccafusco J.J. Multi-functional drugs for various CNS targets in the treatment of neurodegenerative disorders. Trends Pharmacol. Sci. 2005;26(1):27–35. doi: 10.1016/j.tips.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 3.Ross C.A., Poirier M.A. Protein aggregation and neurodegenerative disease. Nat. Med. 2004;10(S7) Suppl.:S10–S17. doi: 10.1038/nm1066. [DOI] [PubMed] [Google Scholar]
- 4.Association A. Alzheimer’s Association. 2011 Alzheimer’s disease facts and figures. Alzheimers Dement. 2011;7(2):208–244. doi: 10.1016/j.jalz.2011.02.004. [DOI] [PubMed] [Google Scholar]
- 5. https://www.alz.co.uk/research/statistics
- 6.Fraga C.A.M., Barreiro E.J. New Insights for multifactorial disease therapy: The challenge of the symbiotic drugs. Curr. Drug Ther. 2008;3(1):1–13. doi: 10.2174/157488508783331225. [DOI] [Google Scholar]
- 7.Zhang H-Y. One-compound-multiple-targets strategy to combat Alzheimer’s disease. FEBS Lett. 2005;579(24):5260–5264. doi: 10.1016/j.febslet.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 8.Mattson M.P., Magnus T. Ageing and neuronal vulnerability. Nat. Rev. Neurosci. 2006;7(4):278–294. doi: 10.1038/nrn1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Poewe W., Seppi K., Tanner C.M., Halliday G.M., Brundin P., Volkmann J., Schrag A-E., Lang A.E. Parkinson disease. Nat. Rev. Dis. Primers. 2017;3:17013. doi: 10.1038/nrdp.2017.13. [DOI] [PubMed] [Google Scholar]
- 10.Heppner F.L., Ransohoff R.M., Becher B. Immune attack: the role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 2015;16(6):358–372. doi: 10.1038/nrn3880. [DOI] [PubMed] [Google Scholar]
- 11. https://www.alz.org/alzheimers-dementia/facts-figures
- 12. http://www.alzheimermed.com.br/conceitos/aspectos-socioeconomicos
- 13.Gitler A.D., Dhillon P., Shorter J. Neurodegenerative disease: models, mechanisms, and a new hope. Dis. Model. Mech. 2017;10(5):499–502. doi: 10.1242/dmm.030205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pozo Devoto V.M., Falzone T.L. Mitochondrial dynamics in Parkinson’s disease: a role for α-synuclein? Dis. Model. Mech. 2017;10(9):1075–1087. doi: 10.1242/dmm.026294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. https://www.ninds.nih.gov/Disorders/Patient-Caregiver-Education/Fact-Sheets/Amyotrophic-Lateral-Sclerosis-ALS-Fact-Sheet
- 16.Viegas-Junior C., Danuello A., da Silva Bolzani V., Barreiro E.J., Fraga C.A. Molecular hybridization: a useful tool in the design of new drug prototypes. Curr. Med. Chem. 2007;14(17):1829–1852. doi: 10.2174/092986707781058805. [DOI] [PubMed] [Google Scholar]
- 17.Dias K.S.T., Viegas C., Jr, Viegas C. Multi-target directed Drugs: A modern approach for design of new drugs for the treatment of Alzheimer’s disease. Curr. Neuropharmacol. 2014;12(3):239–255. doi: 10.2174/1570159X1203140511153200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de Freitas Silva M., Dias K.S.T., Gontijo V.S., Ortiz C.J.C., Viegas C., Jr Multi-target directed drugs as a modern approach for drug design towards alzheimer’s disease: an update. Curr. Med. Chem. 2018;25(29):3491–3525. doi: 10.2174/0929867325666180111101843. [DOI] [PubMed] [Google Scholar]
- 19.Ivasiv V., Albertini C., Gonçalves A.E., Rossi M., Bolognesi M.L. Molecular hybridization as a tool for designing multitarget drug candidates for complex diseases. Curr. Top. Med. Chem. 2019;19(19):1694–1711. doi: 10.2174/1568026619666190619115735. [DOI] [PubMed] [Google Scholar]
- 20.Francis P.T., Palmer A.M., Snape M., Wilcock G.K. The cholinergic hypothesis of Alzheimer’s disease: a review of progress. J. Neurol. Neurosurg. Psychiatry. 1999;66(2):137–147. doi: 10.1136/jnnp.66.2.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rosini M., Simoni E., Bartolini M., Cavalli A., Ceccarini L., Pascu N., McClymont D.W., Tarozzi A., Bolognesi M.L., Minarini A., Tumiatti V., Andrisano V., Mellor I.R., Melchiorre C. Inhibition of acetylcholinesterase, β-amyloid aggregation, and NMDA receptors in Alzheimer’s disease: a promising direction for the multi-target-directed ligands gold rush. J. Med. Chem. 2008;51(15):4381–4384. doi: 10.1021/jm800577j. [DOI] [PubMed] [Google Scholar]
- 22.Möller H-J., Graeber M.B. The case described by Alois Alzheimer in 1911. Historical and conceptual perspectives based on the clinical record and neurohistological sections. Eur. Arch. Psychiatry Clin. Neurosci. 1998;248(3):111–122. doi: 10.1007/s004060050027. [DOI] [PubMed] [Google Scholar]
- 23.Kumar A., Singh A. Ekavali, A review on Alzheimer’s disease pathophysiology and its management: an update. Pharmacol. Rep. 2015;67(2):195–203. doi: 10.1016/j.pharep.2014.09.004. [DOI] [PubMed] [Google Scholar]
- 24.Grøntvedt G.R., Schröder T.N., Sando S.B., White L., Bråthen G., Doeller C.F. Alzheimer’s disease. Curr. Biol. 2018;28(11):R645–R649. doi: 10.1016/j.cub.2018.04.080. [DOI] [PubMed] [Google Scholar]
- 25.Chong F.P., Ng K.Y., Koh R.Y., Chye S.M. Tau proteins and tauopathies in alzheimer’s disease. Cell. Mol. Neurobiol. 2018;38(5):965–980. doi: 10.1007/s10571-017-0574-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Viegas F.P.D., Simões M.C.R. Rocha; Castelli, M. R.; Moreira, V.; Junior, C. Doença de Alzheimer: Caracterização, evolução e implicações do processo neuroinflamatório. Rev. Virtual Quim. 2011;3(4):286–306. [Google Scholar]
- 27.Liu Q., Xie F.R.R., Moreira P.I., Nunomura A., Zhu X., Smith M.A.G.P. Prevention and treatment of alzheimer disease and aging: antioxidants mini-reviews in medicinal chemistry mini-reviews. Med. Chem. 2007;7(2):171–180. doi: 10.2174/138955707779802552. [DOI] [PubMed] [Google Scholar]
- 28.Azzi A. Oxidative stress: A dead end or a laboratory hypothesis? Biochem. Biophys. Res. Commun. 2007;362(2):230–232. doi: 10.1016/j.bbrc.2007.07.124. [DOI] [PubMed] [Google Scholar]
- 29.Legg K. Neurodegenerative diseases: An alternative path to reduce neuroinflammation. Nat. Rev. Drug Discov. 2011;10(12):901. doi: 10.1038/nrd3607. [DOI] [PubMed] [Google Scholar]
- 30.Cummings J., Lee G., Ritter A., Sabbagh M., Zhong K. Alzheimer’s disease drug development pipeline: 2019. Alzheimers Dement. (N. Y.) 2019;5(1):272–293. doi: 10.1016/j.trci.2019.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sanabria-Castro A., Alvarado-Echeverría I., Monge-Bonilla C. Molecular pathogenesis of alzheimer’s disease: an update. Ann. Neurosci. 2017;24(1):46–54. doi: 10.1159/000464422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Das T.K., Mas W.R., Kaneez F.S., Fatima S.K. Oxidative stress gated by fenton and haber weiss reactions and its association with Alzheimer’s disease. Arch. Neurosci. 2014;2(3):20078. [Google Scholar]
- 33.Starkov A.A., Beal F.M. Portal to Alzheimer’s disease. Nat. Med. 2008;14(10):1020. doi: 10.1038/nm1008-1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reddy P.H., Beal M.F. Amyloid beta, mitochondrial dysfunction and synaptic damage: implications for cognitive decline in aging and Alzheimer’s disease. Trends Mol. Med. 2008;14(2):45–53. doi: 10.1016/j.molmed.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nesi G., Sestito S., Digiacomo M., Rapposelli S. Oxidative stress, mitochondrial abnormalities and proteins deposition: multitarget approaches in Alzheimer’s disease. Curr. Top. Med. Chem. 2017;17(27):3062–3079. doi: 10.2174/1568026617666170607114232. [DOI] [PubMed] [Google Scholar]
- 36.Bolós M., Perea J.R., Avila J. Alzheimer’s disease as an inflammatory disease. Biomol. Concepts. 2017;8(1):37–43. doi: 10.1515/bmc-2016-0029. [DOI] [PubMed] [Google Scholar]
- 37.Akiyama H., Barger S., Barnum S., Bradt B., Bauer J., Cole G.M., Cooper N.R., Eikelenboom P., Emmerling M., Fiebich B.L., Finch C.E., Frautschy S., Griffin W.S., Hampel H., Hull M., Landreth G., Lue L., Mrak R., Mackenzie I.R., McGeer P.L., O’Banion M.K., Pachter J., Pasinetti G., Plata-Salaman C., Rogers J., Rydel R., Shen Y., Streit W., Strohmeyer R., Tooyoma I., Van Muiswinkel F.L., Veerhuis R., Walker D., Webster S., Wegrzyniak B., Wenk G., Wyss-Coray T. Inflammation and Alzheimer’s disease. Neurobiol. Aging. 2000;21(3):383–421. doi: 10.1016/S0197-4580(00)00124-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Irwin M.R., Vitiello M.V. Implications of sleep disturbance and inflammation for Alzheimer’s disease dementia. Lancet Neurol. 2019;18(3):296–306. doi: 10.1016/S1474-4422(18)30450-2. [DOI] [PubMed] [Google Scholar]
- 39.Giunta B., Fernandez F., Nikolic W.V., Obregon D., Rrapo E., Town T., Tan J. Inflammaging as a prodrome to Alzheimer’s disease. J. Neuroinflammation. 2008;5(1):51. doi: 10.1186/1742-2094-5-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kamal M., Greig N., Reale M. Anti-Inflammatory properties of acetylcholinesterase inhibitors administred in Alzheimers disease. Antiinflamm. Antiallergy Agents Med. Chem. 2009;8(1):85–100. doi: 10.2174/187152309787580810. [DOI] [Google Scholar]
- 41.Heneka M.T., O’Banion M.K. Inflammatory processes in Alzheimer’s disease. J. Neuroimmunol. 2007;184(1-2):69–91. doi: 10.1016/j.jneuroim.2006.11.017. [DOI] [PubMed] [Google Scholar]
- 42.Parkinson’s Foundation Statistics of Parkinson’s disease. https://parkinson.org/Understanding-Parkinsons/Statistics
- 43.Przedborski S. The two-century journey of Parkinson disease research. Nat. Rev. Neurosci. 2017;18(4):251–259. doi: 10.1038/nrn.2017.25. [DOI] [PubMed] [Google Scholar]
- 44.Obeso J.A., Rodriguez-Oroz M.C., Goetz C.G., Marin C., Kordower J.H., Rodriguez M., Hirsch E.C., Farrer M., Schapira A.H.V., Halliday G. Missing pieces in the Parkinson’s disease puzzle. Nat. Med. 2010;16(6):653–661. doi: 10.1038/nm.2165. [DOI] [PubMed] [Google Scholar]
- 45.Wang Q., Liu Y., Zhou J. Neuroinflammation in Parkinson’s disease and its potential as therapeutic target. Transl. Neurodegener. 2015;4(1):19. doi: 10.1186/s40035-015-0042-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Valdinocci D., Simões R.F., Kovarova J., Cunha-Oliveira T., Neuzil J., Pountney D.L. Intracellular and intercellular mitochondrial dynamics in parkinson’s disease. Front. Neurosci. 2019;13:930. doi: 10.3389/fnins.2019.00930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang W., Wang T., Pei Z., Miller D. S., Wu X., Block M. L., Wilson B., Zhang W., Zhou Y., Hong J.-S. Aggregated-synuclein activates microglia: a process leading to disease progresion in Parkinson’s disease. 2005. [DOI] [PubMed]
- 48.Zarei S., Carr K., Reiley L., Diaz K., Guerra O., Altamirano P.F., Pagani W., Lodin D., Orozco G., Chinea A. A comprehensive review of amyotrophic lateral sclerosis. Surg. Neurol. Int. 2015;6:171. doi: 10.4103/2152-7806.169561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. https://www.ninds.nih.gov/Disorders/Patient-Caregiver-Education/Fact-Sheets/Amyotrophic-Lateral-Sclerosis-ALS-Fact-Sheet
- 50.Kiernan M.C., Vucic S., Cheah B.C., Turner M.R., Eisen A., Hardiman O., Burrell J.R., Zoing M.C. Amyotrophic lateral sclerosis. Lancet. 2011;377(9769):942–955. doi: 10.1016/S0140-6736(10)61156-7. [DOI] [PubMed] [Google Scholar]
- 51.Ghosh R., Tabrizi S.J. Huntington disease. Handb. Clin. Neurol. 2018;147:255–278. doi: 10.1016/B978-0-444-63233-3.00017-8. [DOI] [PubMed] [Google Scholar]
- 52.Dayalu P., Albin R.L. Huntington disease: Pathogenesis and treatment. Neurol. Clin. 2015;33(1):101–114. doi: 10.1016/j.ncl.2014.09.003. [DOI] [PubMed] [Google Scholar]
- 53.Bates G.P., Dorsey R., Gusella J.F., Hayden M.R., Kay C., Leavitt B.R., Nance M., Ross C.A., Scahill R.I., Wetzel R., Wild E.J., Tabrizi S.J. Huntington disease. Nat. Rev. Dis. Primers. 2015;1:15005. doi: 10.1038/nrdp.2015.5. [DOI] [PubMed] [Google Scholar]
- 54.Bolognesi M.L., Cavalli A. Multitarget drug discovery and polypharmacology. ChemMedChem. 2016;11(12):1190–1192. doi: 10.1002/cmdc.201600161. [DOI] [PubMed] [Google Scholar]
- 55.Lansbury P.T., Jr Back to the future: the ‘old-fashioned’ way to new medications for neurodegeneration. Nat. Med. 2004;10(S7) Suppl.:S51–S57. doi: 10.1038/nrn1435. [DOI] [PubMed] [Google Scholar]
- 56.Bolognesi M.L. Polypharmacology in a single drug: Multitarget drugs. Curr. Med. Chem. 2013;20(13):1639–1645. doi: 10.2174/0929867311320130004. [DOI] [PubMed] [Google Scholar]
- 57.Dias K S T, de Paula C. T., Riquiel M. M., Lago S. T., Costa K. C. M., Vaz S. M., Machado R. P., Lima L. M. S., Viegas C. 2015.
- 58.Wang N., Qiu P., Cui W., Yan X., Zhang B., He S. Recent advances in multi-target anti-Alzheimer disease compounds (2013 up to the present). Curr. Med. Chem. 2019;26(30):5684–5710. doi: 10.2174/0929867326666181203124102. [DOI] [PubMed] [Google Scholar]
- 59.Lazar C., Alicja K., Taira K., Konishi Y. Drug evolution concept in drug design: 1.h ybridization method. J. Med. Chem. 2004;47(27):6973–6982. doi: 10.1021/jm049637+. [DOI] [PubMed] [Google Scholar]
- 60.Schmitt B., Bernhardt T., Moeller H-J., Heuser I. Frölich, L. Combination therapy in Alzheimer’s disease. CNS Drugs. 2004;18(13):827–844. doi: 10.2165/00023210-200418130-00001. [DOI] [PubMed] [Google Scholar]
- 61.Piau A., Nourhashémi F., Hein C., Caillaud C., Vellas B. Progress in the development of new drugs in Alzheimer’s disease. J. Nutr. Health Aging. 2011;15(1):45–57. doi: 10.1007/s12603-011-0012-x. [DOI] [PubMed] [Google Scholar]
- 62.Iraji A., Khoshneviszadeh M., Firuzi O., Khoshneviszadeh M., Edraki N. Novel small molecule therapeutic agents for Alzheimer disease: Focusing on BACE1 and multi-target directed ligands. Bioorg. Chem. 2020;97:103649. doi: 10.1016/j.bioorg.2020.103649. [DOI] [PubMed] [Google Scholar]
- 63.Li Q., He S., Chen Y., Feng F., Qu W., Sun H. Donepezil-based multi-functional cholinesterase inhibitors for treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2018;158:463–477. doi: 10.1016/j.ejmech.2018.09.031. [DOI] [PubMed] [Google Scholar]
- 64.Knowles J. Donepezil in alzheimer’s disease: An evidence-based review of its impact on clinical and economic outcomes. Core Evid. 2006;1(3):195–219. [PMC free article] [PubMed] [Google Scholar]
- 65.Dias Viegas F.P., de Freitas Silva M., Divino da Rocha M., Castelli M.R., Riquiel M.M., Machado R.P., Vaz S.M., Simões de Lima L.M., Mancini K.C., Marques de Oliveira P.C., Morais É.P., Gontijo V.S., da Silva F.M.R., D’Alincourt da Fonseca Peçanha D., Castro N.G., Neves G.A., Giusti-Paiva A., Vilela F.C., Orlandi L., Camps I., Veloso M.P., Leomil Coelho L.F., Ionta M., Ferreira-Silva G.Á., Pereira R.M., Dardenne L.E., Guedes I.A., de Oliveira Carneiro W., Junior, Quaglio Bellozi P.M., Pinheiro de Oliveira A.C., Ferreira F.F., Pruccoli L., Tarozzi A., Viegas C., Jr Design, synthesis and pharmacological evaluation of N-benzyl-piperidinyl-aryl-acylhydrazone derivatives as donepezil hybrids: Discovery of novel multi-target anti-alzheimer prototype drug candidates. Eur. J. Med. Chem. 2018;147:48–65. doi: 10.1016/j.ejmech.2018.01.066. [DOI] [PubMed] [Google Scholar]
- 66.Li T., Pan W., Wang K., Liu W., Ma Q., Sang Z. Novel ferulic acid-donepezil hybrids as multifunctional agents for th e treatment of alzheimer’s disease with butyrylcholinesterase, amyloid- β, antioxidant and neuroprotective properties. Lett. Drug Des. Discov. 2017;14(8):918–929. doi: 10.2174/1570180814666170421181517. [DOI] [Google Scholar]
- 67.Sang Z., Pan W., Wang K., Ma Q., Yu L., Yang Y., Bai P., Leng C., Xu Q., Li X., Tan Z., Liu W. Design, synthesis and evaluation of novel ferulic acid-O-alkylamine derivatives as potential multifunctional agents for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2017;130:379–392. doi: 10.1016/j.ejmech.2017.02.039. [DOI] [PubMed] [Google Scholar]
- 68.Sang Z., Pan W., Wang K., Ma Q., Yu L., Liu W. Design, synthesis and biological evaluation of 3,4-dihydro-2(1H)-quinoline-O-alkylamine derivatives as new multipotent cholinesterase/monoamine oxidase inhibitors for the treatment of Alzheimer’s disease. Bioorg. Med. Chem. 2017;25(12):3006–3017. doi: 10.1016/j.bmc.2017.03.070. [DOI] [PubMed] [Google Scholar]
- 69.Dias K.S.T., de Paula C.T., Dos Santos T., Souza I.N.O., Boni M.S., Guimarães M.J.R., da Silva F.M.R., Castro N.G., Neves G.A., Veloso C.C., Coelho M.M., de Melo I.S., Giusti F.C., Giusti-Paiva A., da Silva M.L., Dardenne L.E., Guedes I.A., Pruccoli L., Morroni F., Tarozzi A., Viegas C. Jr Design, synthesis and evaluation of novel feruloyl-donepezil hybrids as potential multitarget drugs for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2017;130:440–457. doi: 10.1016/j.ejmech.2017.02.043. [DOI] [PubMed] [Google Scholar]
- 70.Mishra C.B., Kumari S., Manral A., Prakash A., Saini V., Lynn A.M., Tiwari M. Design, synthesis, in-silico and biological evaluation of novel donepezil derivatives as multi-target-directed ligands for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2017;125:736–750. doi: 10.1016/j.ejmech.2016.09.057. [DOI] [PubMed] [Google Scholar]
- 71.García-Osta A., Cuadrado-Tejedor M., García-Barroso C., Oyarzábal J., Franco R. Phosphodiesterases as therapeutic targets for Alzheimer’s disease. ACS Chem. Neurosci. 2012;3(11):832–844. doi: 10.1021/cn3000907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hu J., Huang Y.D., Pan T., Zhang T., Su T., Li X., Luo H. Bin; Huang, L. Design, synthesis, and biological evaluation of dual-target inhibitors of acetylcholinesterase (ache) and phosphodiesterase 9a (pde9a) for the treatment of Alzheimer’s disease. ACS Chem. Neurosci. 2019;10(1):537–551. doi: 10.1021/acschemneuro.8b00376. [DOI] [PubMed] [Google Scholar]
- 73.Hiremathad A., Chand K., Tolayan L., Rajeshwari K.R.S., Esteves A.R., Cardoso S.M., Chaves S., Santos M.A. Hydroxypyridinone-benzofuran hybrids with potential protective roles for Alzheimer’s disease therapy. J. Inorg. Biochem. 2018;179:82–96. doi: 10.1016/j.jinorgbio.2017.11.015. [DOI] [PubMed] [Google Scholar]
- 74.Huang L., Miao H., Sun Y., Meng F., Li X. Discovery of indanone derivatives as multi-target-directed ligands against Alzheimer’s disease. Eur. J. Med. Chem. 2014;87:429–439. doi: 10.1016/j.ejmech.2014.09.081. [DOI] [PubMed] [Google Scholar]
- 75.Yan J., Hu J., Liu A., He L., Li X., Wei H. Design, synthesis, and evaluation of multitarget-directed ligands against Alzheimer’s disease based on the fusion of donepezil and curcumin. Bioorg. Med. Chem. 2017;25(12):2946–2955. doi: 10.1016/j.bmc.2017.02.048. [DOI] [PubMed] [Google Scholar]
- 76.Wang L., Esteban G., Ojima M., Bautista-Aguilera O.M., Inokuchi T., Moraleda I., Iriepa I., Samadi A., Youdim M.B., Romero A., Soriano E., Herrero R., Fernández Fernández A.P. Ricardo-Martínez-Murillo.; Marco-Contelles, J.; Unzeta, M. Donepezil + propargylamine + 8-hydroxyquinoline hybrids as new multifunctional metal-chelators, ChE and MAO inhibitors for the potential treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2014;80:543–561. doi: 10.1016/j.ejmech.2014.04.078. [DOI] [PubMed] [Google Scholar]
- 77.Wu M.Y., Esteban G., Brogi S., Shionoya M., Wang L., Campiani G., Unzeta M., Inokuchi T., Butini S., Marco-Contelles J. Donepezil-like multifunctional agents: Design, synthesis, molecular modeling and biological evaluation. Eur. J. Med. Chem. 2016;121:864–879. doi: 10.1016/j.ejmech.2015.10.001. [DOI] [PubMed] [Google Scholar]
- 78.Fernandes T.B., Cunha M.R., Sakata R.P., Candido T.M., Baby A.R., Tavares M.T., Barbosa E.G., Almeida W.P., Parise-Filho R. Synthesis, molecular modeling, and evaluation of novel sulfonylhydrazones as acetylcholinesterase inhibitors for Alzheimer’s disease. Arch. Pharm. (Weinheim) 2017;350(11):1–16. doi: 10.1002/ardp.201700163. [DOI] [PubMed] [Google Scholar]
- 79.Moradi A., Faraji L., Nadri H., Hasanpour Z., Moghadam F.H., Pakseresht B., Golshani M., Moghimi S., Ramazani A., Firoozpour L. Synthesis, docking study, and biological evaluation of novel umbellipherone/hymecromone derivatives as acetylcholinesterase/butyrylcholinesterase inhibitors. Med. Chem. Res. 2018;27(7):1741–1747. doi: 10.1007/s00044-018-2187-8. [DOI] [Google Scholar]
- 80.Estrada Valencia M., Herrera-Arozamena C., de Andrés L., Pérez C., Morales-García J.A., Pérez-Castillo A., Ramos E., Romero A., Viña D., Yáñez M., Laurini E., Pricl S., Rodríguez-Franco M.I. Neurogenic and neuroprotective donepezil-flavonoid hybrids with sigma-1 affinity and inhibition of key enzymes in Alzheimer’s disease. Eur. J. Med. Chem. 2018;156:534–553. doi: 10.1016/j.ejmech.2018.07.026. [DOI] [PubMed] [Google Scholar]
- 81.Xie S.S., Wang X., Jiang N., Yu W., Wang K.D., Lan J.S., Li Z.R., Kong L.Y. Multi-target tacrine-coumarin hybrids: cholinesterase and monoamine oxidase B inhibition properties against Alzheimer’s disease. Eur. J. Med. Chem. 2015;95:153–165. doi: 10.1016/j.ejmech.2015.03.040. [DOI] [PubMed] [Google Scholar]
- 82.Xie S.S., Lan J.S., Wang X., Wang Z.M., Jiang N., Li F., Wu J.J., Wang J., Kong L.Y. Design, synthesis and biological evaluation of novel donepezil-coumarin hybrids as multi-target agents for the treatment of Alzheimer’s disease. Bioorg. Med. Chem. 2016;24(7):1528–1539. doi: 10.1016/j.bmc.2016.02.023. [DOI] [PubMed] [Google Scholar]
- 83.Cai P., Fang S.Q., Yang H.L., Yang X.L., Liu Q.H., Kong L.Y., Wang X.B. Donepezil-butylated hydroxytoluene (BHT) hybrids as Anti-Alzheimer’s disease agents with cholinergic, antioxidant, and neuroprotective properties. Eur. J. Med. Chem. 2018;157:161–176. doi: 10.1016/j.ejmech.2018.08.005. [DOI] [PubMed] [Google Scholar]
- 84.Berwaldt G.A., Gouvêa D.P., da Silva D.S. das Neves, A.M.; Soares, M.S.P.; Azambuja, J.H.; Siqueira, G.M.; Spanevello, R.M.; Cunico, W. Synthesis and biological evaluation of benzothiazin-4-ones: a possible new class of acetylcholinesterase inhibitors. J. Enzyme Inhib. Med. Chem. 2019;34(1):197–203. doi: 10.1080/14756366.2018.1543286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Huang W., Wang Y., Li J., Zhang Y., Ma X., Zhu P., Zhang Y. Design, synthesis, and evaluation of genipin derivatives for the treatment of Alzheimer’s Disease. Chem. Biol. Drug Des. 2019;93(2):110–122. doi: 10.1111/cbdd.13194. [DOI] [PubMed] [Google Scholar]
- 86.Samadi A., de la Fuente Revenga M., Pérez C., Iriepa I., Moraleda I., Rodríguez-Franco M.I., Marco-Contelles J. Synthesis, pharmacological assessment, and molecular modeling of 6-chloro-pyridonepezils: new dual AChE inhibitors as potential drugs for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2013;67:64–74. doi: 10.1016/j.ejmech.2013.06.021. [DOI] [PubMed] [Google Scholar]
- 87.Jin H. Acetylcholinesterase and butyrylcholinesterase inhibitory properties of functionalized tetrahydroacridines and related analogs. Med. Chem. (Los Angeles) 2014;4(10):688–696. doi: 10.4172/2161-0444.1000213. [DOI] [Google Scholar]
- 88.Hussein W., Sağlık B.N., Levent S., Korkut B., Ilgın S., Özkay Y., Kaplancıklı Z.A. Synthesis and biological evaluation of new cholinesterase inhibitors for Alzheimer’s disease. Molecules. 2018;23(8):2033. doi: 10.3390/molecules23082033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Camps P., Formosa X., Galdeano C., Gómez T., Muñoz-Torrero D., Scarpellini M., Viayna E., Badia A., Clos M.V., Camins A., Pallàs M., Bartolini M., Mancini F., Andrisano V., Estelrich J., Lizondo M., Bidon-Chanal A., Luque F.J. Novel donepezil-based inhibitors of acetyl- and butyrylcholinesterase and acetylcholinesterase-induced β-amyloid aggregation. J. Med. Chem. 2008;51(12):3588–3598. doi: 10.1021/jm8001313. [DOI] [PubMed] [Google Scholar]
- 90.Viayna E., Gómez T., Galdeano C., Ramírez L., Ratia M., Badia A., Clos M.V., Verdaguer E., Junyent F., Camins A., Pallàs M., Bartolini M., Mancini F., Andrisano V., Arce M.P., Rodríguez-Franco M.I., Bidon-Chanal A., Luque F.J., Camps P., Muñoz-Torrero D. Novel huprine derivatives with inhibitory activity toward β-amyloid aggregation and formation as disease-modifying anti-Alzheimer drug candidates. ChemMedChem. 2010;5(11):1855–1870. doi: 10.1002/cmdc.201000322. [DOI] [PubMed] [Google Scholar]
- 91.Sola I., Aso E., Frattini D., López-González I., Espargaró A., Sabaté R., Di Pietro O., Luque F.J., Clos M.V., Ferrer I., Muñoz-Torrero D. Novel levetiracetam derivatives that are effective against the Alzheimer-like phenotype in mice: synthesis, in vitro, ex vivo, and in vivo efficacy studies. J. Med. Chem. 2015;58(15):6018–6032. doi: 10.1021/acs.jmedchem.5b00624. [DOI] [PubMed] [Google Scholar]
- 92.Li G., Hong G., Li X., Zhang Y., Xu Z., Mao L., Feng X., Liu T. Synthesis and activity towards Alzheimer’s disease in vitro: Tacrine, phenolic acid and ligustrazine hybrids. Eur. J. Med. Chem. 2018;148:238–254. doi: 10.1016/j.ejmech.2018.01.028. [DOI] [PubMed] [Google Scholar]
- 93.Hiremathad A., Keri R.S., Esteves A.R., Cardoso S.M., Chaves S., Santos M.A. Novel Tacrine-Hydroxyphenylbenzimidazole hybrids as potential multitarget drug candidates for Alzheimer’s disease. Eur. J. Med. Chem. 2018;148:255–267. doi: 10.1016/j.ejmech.2018.02.023. [DOI] [PubMed] [Google Scholar]
- 94.Najafi Z., Mahdavi M., Saeedi M., Karimpour-Razkenari E., Asatouri R., Vafadarnejad F., Moghadam F.H., Khanavi M., Sharifzadeh M., Akbarzadeh T. Novel tacrine-1,2,3-triazole hybrids: In vitro, in vivo biological evaluation and docking study of cholinesterase inhibitors. Eur. J. Med. Chem. 2017;125:1200–1212. doi: 10.1016/j.ejmech.2016.11.008. [DOI] [PubMed] [Google Scholar]
- 95.Jiang X.Y., Chen T.K., Zhou J.T., He S.Y., Yang H.Y., Chen Y., Qu W., Feng F., Sun H.P. Dual GSK-3β/AChE inhibitors as a new strategy for multitargeting anti-alzheimer’s disease drug discovery. ACS Med. Chem. Lett. 2018;9(3):171–176. doi: 10.1021/acsmedchemlett.7b00463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hepnarova V., Korabecny J., Matouskova L., Jost P., Muckova L., Hrabinova M., Vykoukalova N., Kerhartova M., Kucera T., Dolezal R., Nepovimova E., Spilovska K., Mezeiova E., Pham N.L., Jun D., Staud F., Kaping D., Kuca K., Soukup O. The concept of hybrid molecules of tacrine and benzyl quinolone carboxylic acid (BQCA) as multifunctional agents for Alzheimer’s disease. Eur. J. Med. Chem. 2018;150:292–306. doi: 10.1016/j.ejmech.2018.02.083. [DOI] [PubMed] [Google Scholar]
- 97.Girek M., Szymański P. Tacrine hybrids as multi-target-directed ligands in Alzheimer’s disease: influence of chemical structures on biological activities. Chem. Pap. 2019;73:269–289. doi: 10.1007/s11696-018-0590-8. [DOI] [Google Scholar]
- 98.Sameem B., Saeedi M., Mahdavi M., Shafiee A. A review on tacrine-based scaffolds as multi-target drugs (MTDLs) for Alzheimer’s disease. Eur. J. Med. Chem. 2017;128:332–345. doi: 10.1016/j.ejmech.2016.10.060. [DOI] [PubMed] [Google Scholar]
- 99.Spilovska K., Korabecny J., Nepovimova E., Dolezal R., Mezeiova E., Soukup O., Kuca K. Multitarget tacrine hybrids with neuroprotective properties to confront Alzheimer’s disease. Curr. Top. Med. Chem. 2017;17(9):1006–1026. doi: 10.2174/1568026605666160927152728. [DOI] [PubMed] [Google Scholar]
- 100.Camps P., Formosa X., Galdeano C., Muñoz-Torrero D., Ramírez L., Gómez E., Isambert N., Lavilla R., Badia A., Clos M.V., Bartolini M., Mancini F., Andrisano V., Arce M.P., Rodríguez-Franco M.I., Huertas O., Dafni T., Luque F.J. Pyrano[3,2-c]quinoline-6-chlorotacrine hybrids as a novel family of acetylcholinesterase- and β-amyloid-directed anti-Alzheimer compounds. J. Med. Chem. 2009;52(17):5365–5379. doi: 10.1021/jm900859q. [DOI] [PubMed] [Google Scholar]
- 101.Di Pietro O., Viayna E., Vicente-García E., Bartolini M., Ramón R., Juárez-Jiménez J., Clos M.V., Pérez B., Andrisano V., Luque F.J., Lavilla R., Muñoz-Torrero D. 1,2,3,4-Tetrahydrobenzo[h][1,6]naphthyridines as a new family of potent peripheral-to-midgorge-site inhibitors of acetylcholinesterase: synthesis, pharmacological evaluation and mechanistic studies. Eur. J. Med. Chem. 2014;73:141–152. doi: 10.1016/j.ejmech.2013.12.008. [DOI] [PubMed] [Google Scholar]
- 102.Di Pietro O., Pérez-Areales F.J., Juárez-Jiménez J., Espargaró A., Clos M.V., Pérez B., Lavilla R., Sabaté R., Luque F.J., Muñoz-Torrero D. Tetrahydrobenzo[h][1,6]naphthyridine-6-chlorotacrine hybrids as a new family of anti-Alzheimer agents targeting β-amyloid, tau, and cholinesterase pathologies. Eur. J. Med. Chem. 2014;84:107–117. doi: 10.1016/j.ejmech.2014.07.021. [DOI] [PubMed] [Google Scholar]
- 103.Spilovska K., Korabecny J., Sepsova V., Jun D., Hrabinova M., Jost P., Muckova L., Soukup O., Janockova J., Kucera T., Dolezal R., Mezeiova E., Kaping D., Kuca K. Novel tacrine-scutellarin hybrids as multipotent anti-Alzheimer’s agents: design, synthesis and biological evaluation. Molecules. 2017;22(6):1006. doi: 10.3390/molecules22061006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Liao S., Deng H., Huang S., Yang J., Wang S., Yin B., Zheng T., Zhang D., Liu J., Gao G., Ma J., Deng Z. Design, synthesis and evaluation of novel 5,6,7-trimethoxyflavone-6-chlorotacrine hybrids as potential multifunctional agents for the treatment of Alzheimer’s disease. Bioorg. Med. Chem. Lett. 2015;25(7):1541–1545. doi: 10.1016/j.bmcl.2015.02.015. [DOI] [PubMed] [Google Scholar]
- 105.Wang M., Qin H-L., Leng J. Ameeduzzafar; Amjad, M.W.; Raja, M.A.G.; Hussain, M.A.; Bukhari, S.N.A. Synthesis and biological evaluation of new tetramethylpyrazine-based chalcone derivatives as potential anti-Alzheimer agents. Chem. Biol. Drug Des. 2018;92(5):1859–1866. doi: 10.1111/cbdd.13355. [DOI] [PubMed] [Google Scholar]
- 106.Fu Y., Mu Y., Lei H., Wang P., Li X., Leng Q., Han L., Qu X., Wang Z., Huang X. Design, synthesis and evaluation of novel tacrine-ferulic acid hybrids as multifunctional drug candidates against Alzheimer’s disease. Molecules. 2016;21(10):1338. doi: 10.3390/molecules21101338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Dgachi Y., Martin H., Malek R., Jun D., Janockova J., Sepsova V., Soukup O., Iriepa I., Moraleda I., Maalej E., Carreiras M.C., Refouvelet B., Chabchoub F., Marco-Contelles J., Ismaili L. Synthesis and biological assessment of KojoTacrines as new agents for Alzheimer’s disease therapy. J. Enzyme Inhib. Med. Chem. 2019;34(1):163–170. doi: 10.1080/14756366.2018.1538136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hamulakova S., Poprac P., Jomova K., Brezova V., Lauro P., Drostinova L., Jun D., Sepsova V., Hrabinova M., Soukup O., Kristian P., Gazova Z., Bednarikova Z., Kuca K., Valko M. Targeting copper(II)-induced oxidative stress and the acetylcholinesterase system in Alzheimer’s disease using multifunctional tacrine-coumarin hybrid molecules. J. Inorg. Biochem. 2016;161:52–62. doi: 10.1016/j.jinorgbio.2016.05.001. [DOI] [PubMed] [Google Scholar]
- 109.Ceschi M.A., da Costa J.S., Lopes J.P.B., Câmara V.S., Campo L.F., Borges A.C.A., Gonçalves C.A.S., de Souza D.F., Konrath E.L., Karl A.L.M., Guedes I.A., Dardenne L.E. Novel series of tacrine-tianeptine hybrids: Synthesis, cholinesterase inhibitory activity, S100B secretion and a molecular modeling approach. Eur. J. Med. Chem. 2016;121:758–772. doi: 10.1016/j.ejmech.2016.06.025. [DOI] [PubMed] [Google Scholar]
- 110.Makhaeva G.F., Grigoriev V.V., Proshin A.N., Kovaleva N.V., Rudakova E.V., Boltneva N.P., Serkov I.V., Bachurin S.O. Novel conjugates of tacrine with 1,2,4,-thiadiazole as highly effective cholinesterase inhibitors, blockers of NMDA receptors, and antioxidants. Dokl. Biochem. Biophys. 2017;477(1):405–409. doi: 10.1134/S1607672917060163. [DOI] [PubMed] [Google Scholar]
- 111.Luo W., Li Y.P., He Y., Huang S.L., Tan J.H., Ou T.M., Li D., Gu L.Q., Huang Z.S. Design, synthesis and evaluation of novel tacrine-multialkoxybenzene hybrids as dual inhibitors for cholinesterases and amyloid beta aggregation. Bioorg. Med. Chem. 2011;19(2):763–770. doi: 10.1016/j.bmc.2010.12.022. [DOI] [PubMed] [Google Scholar]
- 112.Bareggi S.R., Cornelli U. Clioquinol: Review of its mechanisms of action and clinical uses in neurodegenerative disorders. CNS Neurosci. Ther. 2012;18(1):41–46. doi: 10.1111/j.1755-5949.2010.00231.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Di Vaira M., Bazzicalupi C., Orioli P., Messori L., Bruni B., Zatta P. Clioquinol, a drug for Alzheimer’s disease specifically interfering with brain metal metabolism: structural characterization of its zinc(II) and copper(II) complexes. Inorg. Chem. 2004;43(13):3795–3797. doi: 10.1021/ic0494051. [DOI] [PubMed] [Google Scholar]
- 114.Hu J., Pan T., An B., Li Z., Li X., Huang L. Synthesis and evaluation of clioquinol-rolipram/roflumilast hybrids as multitarget-directed ligands for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2019;163:512–526. doi: 10.1016/j.ejmech.2018.12.013. [DOI] [PubMed] [Google Scholar]
- 115.Song Q., Li Y., Cao Z., Liu H., Tian C., Yang Z., Qiang X., Tan Z., Deng Y. Discovery of novel 2,5-dihydroxyterephthalamide derivatives as multifunctional agents for the treatment of Alzheimer’s disease. Bioorg. Med. Chem. 2018;26(23-24):6115–6127. doi: 10.1016/j.bmc.2018.11.015. [DOI] [PubMed] [Google Scholar]
- 116.Li F., Wu J.J., Wang J., Yang X.L., Cai P., Liu Q.H., Kong L.Y., Wang X.B. Synthesis and pharmacological evaluation of novel chromone derivatives as balanced multifunctional agents against Alzheimer’s disease. Bioorg. Med. Chem. 2017;25(14):3815–3826. doi: 10.1016/j.bmc.2017.05.027. [DOI] [PubMed] [Google Scholar]
- 117.Xie S., Chen J., Li X., Su T., Wang Y., Wang Z., Huang L., Li X. Synthesis and evaluation of selegiline derivatives as monoamine oxidase inhibitor, antioxidant and metal chelator against Alzheimer’s disease. Bioorg. Med. Chem. 2015;23(13):3722–3729. doi: 10.1016/j.bmc.2015.04.009. [DOI] [PubMed] [Google Scholar]
- 118.Grandy K., Melatonin J. Therapeutic Intervention in Mild Cognitive Impairment and Alzheimer Disease. J. Neurol. Neurophysiol. 2013;04(02):2–7. doi: 10.4172/2155-9562.1000148. [DOI] [Google Scholar]
- 119.Cheng S., Zheng W., Gong P., Zhou Q., Xie Q., Yu L., Zhang P., Chen L., Li J., Chen J., Chen H., Chen H. (-)-Meptazinol-melatonin hybrids as novel dual inhibitors of cholinesterases and amyloid-β aggregation with high antioxidant potency for Alzheimer’s therapy. Bioorg. Med. Chem. 2015;23(13):3110–3118. doi: 10.1016/j.bmc.2015.04.084. [DOI] [PubMed] [Google Scholar]
- 120.López-Iglesias B., Pérez C., Morales-García J.A., Alonso-Gil S., Pérez-Castillo A., Romero A., López M.G., Villarroya M., Conde S., Rodríguez-Franco M.I. New melatonin-N,N-dibenzyl(N-methyl)amine hybrids: potent neurogenic agents with antioxidant, cholinergic, and neuroprotective properties as innovative drugs for Alzheimer’s disease. J. Med. Chem. 2014;57(9):3773–3785. doi: 10.1021/jm5000613. [DOI] [PubMed] [Google Scholar]
- 121.Borges F., Roleira F., Milhazes N., Santana L., Uriarte E. Simple coumarins and analogues in medicinal chemistry: occurrence, synthesis and biological activity. Curr. Med. Chem. 2005;12(8):887–916. doi: 10.2174/0929867053507315. [DOI] [PubMed] [Google Scholar]
- 122.Stefanachi A., Leonetti F., Pisani L., Catto M., Carotti A. Coumarin: a natural; privileged and versatile scaffold for bioactive compounds. Molecules. 2018;23(2):E250. doi: 10.3390/molecules23020250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Jiang N., Huang Q., Liu J., Liang N., Li Q., Li Q., Xie S.S. Design, synthesis and biological evaluation of new coumarin-dithiocarbamate hybrids as multifunctional agents for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2018;146:287–298. doi: 10.1016/j.ejmech.2018.01.055. [DOI] [PubMed] [Google Scholar]
- 124.He Q., Liu J., Lan J.S., Ding J., Sun Y., Fang Y., Jiang N., Yang Z., Sun L., Jin Y., Xie S.S. Coumarin-dithiocarbamate hybrids as novel multitarget AChE and MAO-B inhibitors against Alzheimer’s disease: Design, synthesis and biological evaluation. Bioorg. Chem. 2018;81(September):512–528. doi: 10.1016/j.bioorg.2018.09.010. [DOI] [PubMed] [Google Scholar]
- 125.Vafadarnejad F., Mahdavi M., Karimpour-Razkenari E., Edraki N., Sameem B., Khanavi M., Saeedi M., Akbarzadeh T. Design and synthesis of novel coumarin-pyridinium hybrids: In vitro cholinesterase inhibitory activity. Bioorg. Chem. 2018;77:311–319. doi: 10.1016/j.bioorg.2018.01.013. [DOI] [PubMed] [Google Scholar]
- 126.Jalili-Baleh L., Nadri H., Forootanfar H., Samzadeh-Kermani A., Küçükkılınç T.T., Ayazgok B., Rahimifard M., Baeeri M., Doostmohammadi M., Firoozpour L., Bukhari S.N.A., Abdollahi M., Ganjali M.R., Emami S., Khoobi M., Foroumadi A. Novel 3-phenylcoumarin-lipoic acid conjugates as multi-functional agents for potential treatment of Alzheimer’s disease. Bioorg. Chem. 2018;79:223–234. doi: 10.1016/j.bioorg.2018.04.030. [DOI] [PubMed] [Google Scholar]
- 127.Jalili-Baleh L., Forootanfar H., Küçükkılınç T.T., Nadri H., Abdolahi Z., Ameri A., Jafari M., Ayazgok B., Baeeri M., Rahimifard M., Abbas Bukhari S.N., Abdollahi M., Ganjali M.R., Emami S., Khoobi M., Foroumadi A. Design, synthesis and evaluation of novel multi-target-directed ligands for treatment of Alzheimer’s disease based on coumarin and lipoic acid scaffolds. Eur. J. Med. Chem. 2018;152:600–614. doi: 10.1016/j.ejmech.2018.04.058. [DOI] [PubMed] [Google Scholar]
- 128.Yang H.L., Cai P., Liu Q.H., Yang X.L., Li F., Wang J., Wu J.J., Wang X.B., Kong L.Y. Design, synthesis and evaluation of coumarin-pargyline hybrids as novel dual inhibitors of monoamine oxidases and amyloid-β aggregation for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2017;138:715–728. doi: 10.1016/j.ejmech.2017.07.008. [DOI] [PubMed] [Google Scholar]
- 129.Lan J.S., Ding Y., Liu Y., Kang P., Hou J.W., Zhang X.Y., Xie S.S., Zhang T. Design, synthesis and biological evaluation of novel coumarin-N-benzyl pyridinium hybrids as multi-target agents for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2017;139:48–59. doi: 10.1016/j.ejmech.2017.07.055. [DOI] [PubMed] [Google Scholar]
- 130.Shishodia S. Molecular mechanisms of curcumin action: Gene expression. Biofactors. 2013;39(1):37–55. doi: 10.1002/biof.1041. [DOI] [PubMed] [Google Scholar]
- 131.Orteca G., Tavanti F., Bednarikova Z., Gazova Z., Rigillo G., Imbriano C., Basile V., Asti M., Rigamonti L., Saladini M., Ferrari E., Menziani M.C. Curcumin derivatives and Aβ-fibrillar aggregates: An interactions’ study for diagnostic/therapeutic purposes in neurodegenerative diseases. Bioorg. Med. Chem. 2018;26(14):4288–4300. doi: 10.1016/j.bmc.2018.07.027. [DOI] [PubMed] [Google Scholar]
- 132.Lan J.S., Hou J.W., Liu Y., Ding Y., Zhang Y., Li L., Zhang T. Design, synthesis and evaluation of novel cinnamic acid derivatives bearing N-benzyl pyridinium moiety as multifunctional cholinesterase inhibitors for Alzheimer’s disease. J. Enzyme Inhib. Med. Chem. 2017;32(1):776–788. doi: 10.1080/14756366.2016.1256883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Fang L., Chen M., Liu Z., Fang X., Gou S., Chen L. Ferulic acid-carbazole hybrid compounds: Combination of cholinesterase inhibition, antioxidant and neuroprotection as multifunctional anti-Alzheimer agents. Bioorg. Med. Chem. 2016;24(4):886–893. doi: 10.1016/j.bmc.2016.01.010. [DOI] [PubMed] [Google Scholar]
- 134.Tang Y-W., Shi C-J., Yang H-L., Cai P., Liu Q-H., Yang X-L., Kong L-Y., Wang X-B. Synthesis and evaluation of isoprenylation-resveratrol dimer derivatives against Alzheimer’s disease. Eur. J. Med. Chem. 2019;163:307–319. doi: 10.1016/j.ejmech.2018.11.040. [DOI] [PubMed] [Google Scholar]
- 135.Bastianetto S., Ménard C., Quirion R. Neuroprotective action of resveratrol. Biochim. Biophys. Acta. 2015;1852(6):1195–1201. doi: 10.1016/j.bbadis.2014.09.011. [DOI] [PubMed] [Google Scholar]
- 136.Regitz C., Fitzenberger E., Mahn F.L., Dußling L.M., Wenzel U. Resveratrol reduces amyloid-beta (Aβ1-42)-induced paralysis through targeting proteostasis in an Alzheimer model of Caenorhabditis elegans. Eur. J. Nutr. 2016;55(2):741–747. doi: 10.1007/s00394-015-0894-1. [DOI] [PubMed] [Google Scholar]
- 137.Schweiger S., Matthes F., Posey K., Kickstein E., Weber S., Hettich M.M., Pfurtscheller S., Ehninger D., Schneider R., Krauß S. Resveratrol induces dephosphorylation of Tau by interfering with the MID1-PP2A complex. Sci. Rep. 2017;7(1):13753. doi: 10.1038/s41598-017-12974-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Cheng G., Xu P., Zhang M., Chen J., Sheng R., Ma Y. Resveratrol-maltol hybrids as multi-target-directed agents for Alzheimer’s disease. Bioorg. Med. Chem. 2018;26(22):5759–5765. doi: 10.1016/j.bmc.2018.08.011. [DOI] [PubMed] [Google Scholar]
- 139.Yang X., Qiang X., Li Y., Luo L., Xu R., Zheng Y., Cao Z., Tan Z., Deng Y. Pyridoxine-resveratrol hybrids Mannich base derivatives as novel dual inhibitors of AChE and MAO-B with antioxidant and metal-chelating properties for the treatment of Alzheimer’s disease. Bioorg. Chem. 2017;71:305–314. doi: 10.1016/j.bioorg.2017.02.016. [DOI] [PubMed] [Google Scholar]
- 140.Xu P., Zhang M., Sheng R., Ma Y. Synthesis and biological evaluation of deferiprone-resveratrol hybrids as antioxidants, Aβ1-42 aggregation inhibitors and metal-chelating agents for Alzheimer’s disease. Eur. J. Med. Chem. 2017;127:174–186. doi: 10.1016/j.ejmech.2016.12.045. [DOI] [PubMed] [Google Scholar]
- 141.Chojnacki J.E., Liu K., Yan X., Toldo S., Selden T., Estrada M., Rodríguez-Franco M.I., Halquist M.S., Ye D., Zhang S. Discovery of 5-(4-hydroxyphenyl)-3-oxo-pentanoic acid [2-(5-methoxy-1H-indol-3-yl)-ethyl]-amide as a neuroprotectant for Alzheimer’s disease by hybridization of curcumin and melatonin. ACS Chem. Neurosci. 2014;5(8):690–699. doi: 10.1021/cn500081s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Lan J.S., Liu Y., Hou J.W., Yang J., Zhang X.Y., Zhao Y., Xie S.S., Ding Y., Zhang T. Design, synthesis and evaluation of resveratrol-indazole hybrids as novel monoamine oxidases inhibitors with amyloid-β aggregation inhibition. Bioorg. Chem. 2018;76:130–139. doi: 10.1016/j.bioorg.2017.11.009. [DOI] [PubMed] [Google Scholar]
- 143.Cao Z., Yang J., Xu R., Song Q., Zhang X., Liu H., Qiang X., Li Y., Tan Z., Deng Y. Design, synthesis and evaluation of 4′-OH-flurbiprofen-chalcone hybrids as potential multifunctional agents for Alzheimer’s disease treatment. Bioorg. Med. Chem. 2018;26(5):1102–1115. doi: 10.1016/j.bmc.2018.01.030. [DOI] [PubMed] [Google Scholar]
- 144.Carreras I., McKee A.C., Choi J.K., Aytan N., Kowall N.W., Jenkins B.G., Dedeoglu A. R-flurbiprofen improves tau, but not Aß pathology in a triple transgenic model of Alzheimer’s disease. Brain Res. 2013;1541:115–127. doi: 10.1016/j.brainres.2013.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Cheng X., Shen Y., Li R. Targeting TNF: A therapeutic strategy for Alzheimer’s disease. Drug Discov. Today. 2014;19(11):1822–1827. doi: 10.1016/j.drudis.2014.06.029. [DOI] [PubMed] [Google Scholar]
- 146.de Freitas Silva M., Pruccoli L., Morroni F., Sita G., Seghetti F., Viegas C., Tarozzi A. The Keap1/Nrf2-ARE Pathway as a Pharmacological Target for Chalcones. Molecules. 2018;23(7):1–22. doi: 10.3390/molecules23071803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Telpoukhovskaia M.A., Patrick B.O., Rodríguez-Rodríguez C., Orvig C. Exploring the multifunctionality of thioflavin- and deferiprone-based molecules as acetylcholinesterase inhibitors for potential application in Alzheimer’s disease. Mol. Biosyst. 2013;9(4):792–805. doi: 10.1039/c3mb25600f. [DOI] [PubMed] [Google Scholar]
- 148.Song Q., Li Y., Cao Z., Qiang X., Tan Z., Deng Y. Novel salicylamide derivatives as potent multifunctional agents for the treatment of Alzheimer’s disease: Design, synthesis and biological evaluation. Bioorg. Chem. 2019;84(84):137–149. doi: 10.1016/j.bioorg.2018.11.022. [DOI] [PubMed] [Google Scholar]
- 149.Bolognesi M.L., Cavalli A., Melchiorre C. Memoquin: a multi-target-directed ligand as an innovative therapeutic opportunity for Alzheimer’s disease. Neurotherapeutics. 2009;6(1):152–162. doi: 10.1016/j.nurt.2008.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Pan W., Hu K., Bai P., Yu L., Ma Q., Li T., Zhang X., Chen C., Peng K., Liu W., Sang Z. Design, synthesis and evaluation of novel ferulic acid-memoquin hybrids as potential multifunctional agents for the treatment of Alzheimer’s disease. Bioorg. Med. Chem. Lett. 2016;26(10):2539–2543. doi: 10.1016/j.bmcl.2016.03.086. [DOI] [PubMed] [Google Scholar]
- 151.Wang J., Cai P., Yang X-L., Li F., Wu J-J., Kong L-Y., Wang X-B. Novel cinnamamide-dibenzylamine hybrids: Potent neurogenic agents with antioxidant, cholinergic, and neuroprotective properties as innovative drugs for Alzheimer’s disease. Eur. J. Med. Chem. 2017;139(9):68–83. doi: 10.1016/j.ejmech.2017.07.077. [DOI] [PubMed] [Google Scholar]
- 152.Hu J., An B., Pan T., Li Z., Huang L., Li X. Design, synthesis, and biological evaluation of histone deacetylase inhibitors possessing glutathione peroxidase-like and antioxidant activities against Alzheimer’s disease. Bioorg. Med. Chem. 2018;26(21):5718–5729. doi: 10.1016/j.bmc.2018.10.022. [DOI] [PubMed] [Google Scholar]
- 153.Oliveira C., Cagide F., Teixeira J., Amorim R., Sequeira L., Mesiti F., Silva T., Garrido J., Remião F., Vilar S., Uriarte E., Oliveira P.J., Borges F. Hydroxybenzoic acid derivatives as dual-target ligands: mitochondriotropic antioxidants and cholinesterase inhibitors. Front Chem. 2018;6:126. doi: 10.3389/fchem.2018.00126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Sang Z., Wang K., Han X., Cao M., Tan Z., Liu W. Design, synthesis, and evaluation of novel ferulic acid derivatives as multi-target-directed ligands for the treatment of alzheimer’s disease. ACS Chem. Neurosci. 2019;10(2):1008–1024. doi: 10.1021/acschemneuro.8b00530. [DOI] [PubMed] [Google Scholar]
- 155.Panek D., Więckowska A., Pasieka A., Godyń J., Jończyk J., Bajda M., Knez D., Gobec S., Malawska B. Design, synthesis, and biological evaluation of 2-(benzylamino-2-hydroxyalkyl)isoindoline-1,3-diones derivatives as potential disease-modifying multifunctional anti-alzheimer agents. Molecules. 2018;23(2):1–15. doi: 10.3390/molecules23020347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Sang Z., Li Y., Qiang X., Xiao G., Liu Q., Tan Z., Deng Y. Multifunctional scutellarin-rivastigmine hybrids with cholinergic, antioxidant, biometal chelating and neuroprotective properties for the treatment of Alzheimer’s disease. Bioorg. Med. Chem. 2015;23(4):668–680. doi: 10.1016/j.bmc.2015.01.005. [DOI] [PubMed] [Google Scholar]
- 157.Xiao G., Li Y., Qiang X., Xu R., Zheng Y., Cao Z., Luo L., Yang X., Sang Z., Su F., Deng Y. Design, synthesis and biological evaluation of 4′-aminochalcone-rivastigmine hybrids as multifunctional agents for the treatment of Alzheimer’s disease. Bioorg. Med. Chem. 2017;25(3):1030–1041. doi: 10.1016/j.bmc.2016.12.013. [DOI] [PubMed] [Google Scholar]
- 158.Bender A.T., Beavo J.A. Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use. Pharmacol. Rev. 2006;58(3):488–520. doi: 10.1124/pr.58.3.5. [DOI] [PubMed] [Google Scholar]
- 159.Castro A., Jerez M.J., Gil C., Martinez A. Cyclic nucleotide phosphodiesterases and their role in immunomodulatory responses: advances in the development of specific phosphodiesterase inhibitors. Med. Res. Rev. 2005;25(2):229–244. doi: 10.1002/med.20020. [DOI] [PubMed] [Google Scholar]
- 160.Francis S.H., Blount M.A., Corbin J.D. Mammalian cyclic nucleotide phosphodiesterases: molecular mechanisms and physiological functions. Physiol. Rev. 2011;91(2):651–690. doi: 10.1152/physrev.00030.2010. [DOI] [PubMed] [Google Scholar]
- 161.Li J., Chen J-Y., Deng Y-L., Zhou Q., Wu Y., Wu D., Luo H-B. Structure-based design, synthesis, biological evaluation, and molecular docking of novel pde10 inhibitors with antioxidant activities. Front Chem. 2018;6(May):167. doi: 10.3389/fchem.2018.00167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Haghighijoo Z., Firuzi O., Hemmateenejad B., Emami S., Edraki N., Miri R. Synthesis and biological evaluation of quinazolinone-based hydrazones with potential use in Alzheimer’s disease. Bioorg. Chem. 2017;74:126–133. doi: 10.1016/j.bioorg.2017.07.014. [DOI] [PubMed] [Google Scholar]
- 163.Huang W., Tang L., Shi Y., Huang S., Xu L., Sheng R., Wu P., Li J., Zhou N., Hu Y. Searching for the multi-target-directed ligands against alzheimer’s disease: discovery of quinoxaline-based hybrid compounds with ache, h3r and bace 1 inhibitory activities. Bioorg. Med. Chem. 2011;19(23):7158–7167. doi: 10.1016/j.bmc.2011.09.061. [DOI] [PubMed] [Google Scholar]
- 164.Deng Y., Jiang Y., Zhao X., Wang J. Design synthesize and bio-evaluate 1,2-dihydroisoquinolin-3(4H)-one derivates as acetylcholinesterase and β-secretase dual inhibitors in treatment with alzheimer’s disease. J. Biosci. Med. (Irvine) 2016;•••:112–123. doi: 10.4236/jbm.2016.41014. [DOI] [Google Scholar]
- 165.Hong C., Guo H.Y., Chen S., Lv J.W., Zhang X., Yang Y.C., Huang K., Zhang Y.J., Tian Z.Y., Luo W., Chen Y.P. Synthesis and biological evaluation of genistein-O-alkylamine derivatives as potential multifunctional anti-Alzheimer agents. Chem. Biol. Drug Des. 2019;93(2):188–200. doi: 10.1111/cbdd.13414. [DOI] [PubMed] [Google Scholar]
- 166.Rizzo S., Tarozzi A., Bartolini M., Da Costa G., Bisi A., Gobbi S., Belluti F., Ligresti A., Allarà M., Monti J.P., Andrisano V., Di Marzo V., Hrelia P., Rampa A. 2-Arylbenzofuran-based molecules as multipotent Alzheimer’s disease modifying agents. Eur. J. Med. Chem. 2012;58:519–532. doi: 10.1016/j.ejmech.2012.10.045. [DOI] [PubMed] [Google Scholar]
- 167.Nadri H., Pirali-Hamedani M., Shekarchi M., Abdollahi M., Sheibani V., Amanlou M., Shafiee A., Foroumadi A. Design, synthesis and anticholinesterase activity of a novel series of 1-benzyl-4-((6-alkoxy-3-oxobenzofuran-2(3H)-ylidene) methyl) pyridinium derivatives. Bioorg. Med. Chem. 2010;18(17):6360–6366. doi: 10.1016/j.bmc.2010.07.012. [DOI] [PubMed] [Google Scholar]
- 168.Nencini A., Castaldo C., Comery T.A., Dunlop J., Genesio E., Ghiron C., Haydar S., Maccari L., Micco I., Turlizzi E., Zanaletti R., Zhang J. Design and synthesis of a hybrid series of potent and selective agonists of α7 nicotinic acetylcholine receptor. Eur. J. Med. Chem. 2014;78:401–418. doi: 10.1016/j.ejmech.2014.03.031. [DOI] [PubMed] [Google Scholar]
- 169.Sang Z., Wang K., Wang H., Wang H., Ma Q., Han X., Ye M., Yu L., Liu W. Design, synthesis and biological evaluation of 2-acetyl-5-O-(amino-alkyl)phenol derivatives as multifunctional agents for the treatment of Alzheimer’s disease. Bioorg. Med. Chem. Lett. 2017;27(22):5046–5052. doi: 10.1016/j.bmcl.2017.09.057. [DOI] [PubMed] [Google Scholar]
- 170.Dalpiaz A., Cacciari B., Vicentini C.B., Bortolotti F., Spalluto G., Federico S., Pavan B., Vincenzi F., Borea P.A., Varani K. A novel conjugated agent between dopamine and an A2A adenosine receptor antagonist as a potential anti-Parkinson multitarget approach. Mol. Pharm. 2012;9(3):591–604. doi: 10.1021/mp200489d. [DOI] [PubMed] [Google Scholar]
- 171.Chauhan K., Sharma M., Saxena J., Singh S.V., Trivedi P., Srivastava K., Puri S.K., Saxena J.K., Chaturvedi V., Chauhan P.M.S. Synthesis and biological evaluation of a new class of 4-aminoquinoline-rhodanine hybrid as potent anti-infective agents. Eur. J. Med. Chem. 2013;62:693–704. doi: 10.1016/j.ejmech.2013.01.017. [DOI] [PubMed] [Google Scholar]
- 172.Piazzi L., Rampa A., Bisi A., Gobbi S., Belluti F., Cavalli A., Bartolini M., Andrisano V., Valenti P., Recanatini M. 3-(4-[[Benzyl(methyl)amino]methyl]phenyl)-6,7-dimethoxy-2H-2-chromenone (AP2238) inhibits both acetylcholinesterase and acetylcholinesterase-induced β-amyloid aggregation: a dual function lead for Alzheimer’s disease therapy. J. Med. Chem. 2003;46(12):2279–2282. doi: 10.1021/jm0340602. [DOI] [PubMed] [Google Scholar]
- 173.Abdelhafez O.M., Amin K.M., Ali H.I., Maher T.J., Batran R.Z. Dopamine release and molecular modeling study of some coumarin derivatives. Neurochem. Int. 2011;59(6):906–912. doi: 10.1016/j.neuint.2011.08.004. [DOI] [PubMed] [Google Scholar]
- 174.Sashidhara K.V., Modukuri R.K., Jadiya P., Rao K.B., Sharma T., Haque R., Singh D.K., Banerjee D., Siddiqi M.I., Nazir A. discovery of 3-arylcoumarin-tetracyclic tacrine hybrids as multifunctional agents against parkinson’s disease. ACS Med. Chem. Lett. 2014;5(10):1099–1103. doi: 10.1021/ml500222g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Buendia I., Navarro E., Michalska P., Gameiro I., Egea J., Abril S., López A., González-Lafuente L., López M.G., León R. New melatonin-cinnamate hybrids as multi-target drugs for neurodegenerative diseases: Nrf2-induction, antioxidant effect and neuroprotection. Future Med. Chem. 2015;7(15):1961–1969. doi: 10.4155/fmc.15.99. [DOI] [PubMed] [Google Scholar]
- 176.Affini A., Hagenow S., Zivkovic A., Marco-Contelles J., Stark H. Novel indanone derivatives as MAO B/H3R dual-targeting ligands for treatment of Parkinson’s disease. Eur. J. Med. Chem. 2018;148:487–497. doi: 10.1016/j.ejmech.2018.02.015. [DOI] [PubMed] [Google Scholar]
- 177.Elmabruk A., Das B., Yedlapudi D., Xu L., Antonio T., Reith M.E.A., Dutta A.K. Design, synthesis, and pharmacological characterization of carbazole based dopamine agonists as potential symptomatic and neuroprotective therapeutic agents for parkinson’s disease. ACS Chem. Neurosci. 2019;10(1):396–411. doi: 10.1021/acschemneuro.8b00291. [DOI] [PubMed] [Google Scholar]
- 178.Zindo F.T., Malan S.F., Omoruyi S.I., Enogieru A.B., Ekpo O.E., Joubert J. Design, synthesis and evaluation of pentacycloundecane and hexacycloundecane propargylamine derivatives as multifunctional neuroprotective agents. Eur. J. Med. Chem. 2019;163:83–94. doi: 10.1016/j.ejmech.2018.11.051. [DOI] [PubMed] [Google Scholar]
- 179.Calabrò M.L., Caputo R., Ettari R., Puia G., Ravazzini F., Zappalà M., Micale N. Synthesis and biological evaluation of new 2-amino-6-(trifluoromethoxy) benzoxazole derivatives, analogues of riluzole. Med. Chem. Res. 2013;22(12):6089–6095. doi: 10.1007/s00044-013-0594-4. [DOI] [Google Scholar]
- 180.Sweeney J.B., Rattray M., Pugh V., Powell L.A. Riluzole-triazole hybrids as novel chemical probes for neuroprotection in amyotrophic lateral sclerosis. ACS Med. Chem. Lett. 2018;9(6):552–556. doi: 10.1021/acsmedchemlett.8b00103. [DOI] [PMC free article] [PubMed] [Google Scholar]