

Abstract
Sulfonylureas (SUs) provide an efficacious first-line treatment in patients with hepatocyte nuclear factor 1α (HNF1A) diabetes, but SUs have limitations due to risk of hypoglycemia. Treatment based on the incretin hormones glucose-dependent insulinotropic peptide (GIP) and glucagon-like peptide 1 (GLP-1) is characterized by their glucose-dependent insulinotropic actions without risk of hypoglycemia. The effect of SUs together with GIP or GLP-1, respectively, on insulin and glucagon secretion in patients with HNF1A diabetes is currently unknown. To investigate this, 10 HNF1A mutation carriers and 10 control subjects without diabetes were recruited for a double-blinded, placebo-controlled, crossover study including 6 experimental days in a randomized order involving 2-h euglycemic-hyperglycemic clamps with coadministration of: 1) SU (glimepiride 1 mg) or placebo, combined with 2) infusions of GIP (1.5 pmol/kg/min), GLP-1 (0.5 pmol/kg/min), or saline (NaCl). In HNF1A mutation carriers, we observed: 1) hypoinsulinemia, 2) insulinotropic effects of both GIP and GLP-1, 3) additive to supra-additive effects on insulin secretion when combining SU+GIP and SU+GLP-1, respectively, and 4) increased fasting and arginine-induced glucagon levels compared with control subjects without diabetes. Our study suggests that a combination of SU and incretin-based treatment may be efficacious in patients with HNF1A diabetes via potentiation of glucose-stimulated insulin secretion.
Introduction
Hepatocyte nuclear factor 1α (HNF1A) diabetes is a monogenic subtype of diabetes, also known as maturity-onset diabetes of the young (MODY) type 3 (MODY3 or HNF1A-MODY). HNF1A mutation carriers are characterized by an impaired insulin response to a glucose stimulus (1). A mutation in the transcription factor HNF1A causes impaired insulin secretion due to decreased expression of proteins involved in insulin gene transcription, glucose uptake (GLUT2), and metabolism (glycolysis and citric acid cycle) in β-cells (2). The disrupted glucose uptake and metabolism result in reduced intracellular levels of ATP, which under normal circumstances plays a vital role in glucose-stimulated insulin secretion. ATP binds to and closes KATP channels, which in turn causes membrane depolarization, initiating a cascade of events that results in secretion of insulin (2,3). Sulfonylureas (SUs) stimulate insulin secretion by enhancing ATP-independent closure of the KATP channel (4,5) and thus bypassing the low level of ATP in the pancreatic β-cells. In mechanistic and clinical studies, HNF1A mutation carriers have been demonstrated to be highly sensitive to SUs due to robust increments in insulin secretion (4,5). Clinically, this translates into a potent glucose-lowering effect when using SUs, which is why they are recommended as first-line treatment of HNF1A-diabetes (6,7). The main limitation of SU treatment in patients with HNF1A diabetes is that treatment intensification with additional glucose-lowering drugs is often needed in the long run to provide glycemic control (8). Additional limitations are problems with recurrent hypoglycemia with SUs in some patients (9,10) and that SUs may also induce body weight gain, as observed in patients with type 2 diabetes (11).
Cross-sectional studies indicate that patients with HNF1A-diabetes suffer from both microvascular and macrovascular complications to the same extent as patients with type 1 and type 2 diabetes (12,13). Thus, investigating add-on treatment to SUs is important to prevent diabetic complications. We have previously shown that HNF1A mutation carriers have impaired insulinotropic effects of the incretin hormones glucagon-like peptide 1 (GLP-1) and glucose-dependent insulinotropic peptide (GIP) compared with control subjects without diabetes (14) but, in contrast, that treatment with pharmacological doses of a GLP-1 receptor agonist in patients with HNF1A diabetes has glucose-lowering properties almost similar to SUs, with an 18-fold lower risk of hypoglycemia (9).
In the current study, we hypothesized that administration of a single dose of an SU (glimepiride) and exogenous infusions of GLP-1 and GIP, respectively, have additive effects on insulin secretion in HNF1A mutation carriers and control subjects without diabetes. The infusion rates of GLP-1 and GIP in this study were chosen to imitate postprandial plasma levels of endogenous GLP-1 and GIP during treatment with dipeptidyl peptidase 4 inhibitor (DPP-4i).
Research Design and Methods
Ethical Approval
This study was approved by the Scientific Ethical Committee of the Capital Region (protocol number H-16038140) and the Data Protection Agency (HGH-2017–050, I-Suite 05657) and registered at ClinicalTrials.gov (NCT03081676). The study was conducted in accordance with the Declaration of Helsinki, and all participants gave oral and written consent before inclusion.
Participants
Ten carriers of mutations in HNF1A (7 treated with glucose-lowering therapies) and 10 control subjects without diabetes were individually matched 1:1 according to age, sex, and BMI (Table 1). HNF1A mutations had previously been established with heterozygous loss-of-function mutations verified by Sanger sequencing (Table 2). Mutations were considered pathogenic if they met at least one of the following criteria: 1) previously published reports on disease-causing effect of the specific mutation, 2) the presence of a truncating mutation, and/or 3) cosegregation of the mutation with diabetes within the family. Participants attended a screening visit after an overnight fast (10 h). Medical history, anthropometric data, and blood samples were obtained. HNF1A mutation carriers were recruited either from the diabetic outpatient clinic at Steno Diabetes Center Copenhagen, Gentofte Hospital, University of Copenhagen, or via letter to an HNF1A mutation registry located at the University of Copenhagen. Inclusion criteria for HNF1A mutation carriers were: 1) pathogenic HNF1A mutation verified by genetic testing, 2) treated with diet, SU monotherapy, or noninsulin treatments with or without SU, 3) aged ≥18 years, and 4) informed consent. Inclusion criteria for control subjects without diabetes were: 1) fasting plasma glucose ≤6 mmol/L, 2) glycated hemoglobin A1c (HbA1c) ≤6.1% (43 mmol/mol), 3) no family history of type 1 or type 2 diabetes, 4) aged ≥18 years, and 5) informed consent. Exclusion criteria in both groups were pregnancy, breastfeeding, and abnormal blood or urine biochemistry (hemoglobin, liver enzymes [alanine and aspartate aminotransferases], plasma creatinine, and urine albumin-to-creatinine ratio). Apart from the antidiabetic drugs (Table 2), none of the participants were treated with drugs suspected to influence the plasma/serum levels of glucose, insulin, C-peptide, glucagon, or incretin hormones.
Table 1.
Participant characteristics
| HNF1A mutation carriers | Control subjects | Difference (P value) | |
|---|---|---|---|
| Sex (male/female) | 4/6 | 4/6 | |
| Age (years) | 35.3 (8.1) | 33.9 (8.0) | |
| Weight (kg) | 67.4 (7.0) | 67.9 (14.3) | |
| Height (m) | 1.74 (0.1) | 1.74 (0.1) | |
| BMI (kg/m2) | 22.4 (1.5) | 22.2 (2.4) | |
| Waist circumference (cm) | 76.3 (7.3) | 76.2 (7.2) | 0.9757 |
| Hip circumference (cm) | 93.5 (4.6) | 90.7 (8.0) | 0.3495 |
| Waist-to-hip ratio | 0.82 (0.08) | 0.85 (0.08) | 0.4684 |
| HbA1c (%) | 6.0 (0.6) | 5.1 (0.3) | <0.0001 |
| HbA1c (mmol/mol) | 42.3 (6.1) | 31.9 (2.9) | <0.0001 |
| Fasting plasma glucose (mmol/L) | 6.6 (2.1) | 5.1 (0.4) | 0.0113 |
| HOMA-IR | 1.4 (0.6) | 1.4 (0.6) | 0.7972 |
| Patients with diabetes (%)* | 80 (n = 8) | ||
| Diabetes duration (years)† | 18 (6) | ||
| Complications | |||
| Retinopathy (%) | 40 (n = 4) | ||
| Neuropathy (%) | 10 (n = 1) | ||
| Treatment | |||
| Diet (%) | 30 (n = 3) | ||
| Glimepiride (%) | 30 (n = 3) | ||
| Glimepiride + linagliptin (%) | 10 (n = 1) | ||
| Glimepiride + liraglutide (%) | 10 (n = 1) | ||
| Linagliptin (%) | 10 (n = 1) | ||
| Repaglinide + metformin (%) | 10 (n = 1) | ||
Data are mean (SD) unless otherwise indicated. Participants were matched according to sex, age, and BMI. Participants met fasting (10 h) without morning medication on screening day. Diabetes is defined as HbA1c ≥48 mmol/mol (n = 7) or prior gestational diabetes mellitus (n = 1) diagnosed with an oral glucose tolerance test.
HOMA-IR, HOMA of insulin resistance.
Two HNF1A mutation carriers had never presented an HbA1c ≥48 mmol/mol.
HNF1A mutation carriers treated with glucose-lowering agents only.
Table 2.
Overview of HNF1A mutations, treatment, and diabetes status
| Subject | Mutation chromosome 12 | Treatment | Diabetes status |
|---|---|---|---|
| HNF1A-1 | Cys241Gly | Glimepiride | Diabetes |
| HNF1A-2 | IVSnt-2 a->g | Diet | No diabetes |
| HNF1A-3 | c.1623+1 g->t | Glimepiride + linagliptin | Diabetes |
| HNF1A-4 | Pro291fsinsC | Glimepiride + liraglutide | Diabetes |
| HNF1A-5 | Ala559fsinsA | Linagliptin | Diabetes |
| HNF1A-6 | Deletion (exon2–10) | Glimepiride | Diabetes |
| HNF1A-7 | Pro291fsinsC | Repaglinide + metformin | Diabetes |
| HNF1A-8 | Pro291fsinsC | Glimepiride | Diabetes |
| HNF1A-9 | Pro379fsdelCT | Diet | No diabetes |
| HNF1A-10 | Glu234Term | Diet | Diabetes* |
“Diabetes” is defined as one measurement of HbA1c ≥48 mmol/mol at any time.
Patient diagnosed with gestational diabetes mellitus during two pregnancies verified with oral glucose tolerance tests.
Peptides
Synthetic GIP and GLP-1 (Bachem, Bubendorf, Switzerland) were subjected to sterile filtration and microbiological testing and dispensed into vials by the Capital Region Pharmacy (Herlev, Denmark). The peptides were dissolved in sterilized water containing 0.5% human albumin (Statens Serum Institut, Copenhagen, Denmark). All infusions (GIP, GLP-1, and NaCl) had an identical transparent appearance.
Tablets
Tablets with 1 mg glimepiride (1 mg Amaryl) (Sanofi Denmark A/S, Copenhagen, Denmark) and placebo had identical appearance and were provided by the hospital pharmacy of the Capital Region (Herlev, Denmark). Both glimepiride and placebo were gelatin capsules containing trace amounts of lactose monohydrate, potato starch, talc, and magnesium stearate.
Study Design
This study was a double-blinded, crossover study with 6 experimental days (separated by a minimum of 4 days) performed in randomized order over a period of at least 3 months. Employees who were not otherwise involved in the study prepared all interventions to ensure blinding of both investigators and participants. Antidiabetic treatments were discontinued prior to each experimental day (repaglinide 24 h, glimepiride 72 h, and metformin/linagliptin/liraglutide 14 days before). After an experimental day, patients recommenced their antidiabetic treatments only if the time interval before the next experimental day was greater than the treatment-specific washout period. Participants were instructed to continue their usual diet (with at least 250 g carbohydrates the day prior to an experimental day) and avoid strenuous exercise and alcohol consumption 24 h before experimental days. After an overnight fast (10 h), the participants rested in a recumbent position, and a cannula was inserted in a cubital vein of each arm, one for infusions and one for collection of arterialized blood samples. Arterialized venous blood was obtained by a modified heated-hand technique by wrapping the forearm and hand with a heating pad (50°C) throughout the experiment (15). A tablet of 1 mg glimepiride or placebo was administered 90 min before the clamp procedures. The two-step glucose clamp consisted of: step 1 at time 0–60 min with a glucose level targeted at the fasting plasma glucose (determined as mean plasma glucose measured at time −105, −100, and −90 min) and step 2 at time 60–125 min at 1.5 × fasting plasma glucose (mimicking postprandial plasma glucose levels). From time 0 to 125 min, GIP (1.5 pmol/kg/min), GLP-1 (1.5 pmol/kg/min), or saline was infused. Infusion of glucose (200 mg/mL) was given from time 0 to 125 min at a rate adjusted according to bedside measurements of plasma glucose, performed every 5th minute. A bolus of 20% glucose was given at time 60 min for 30 s to increase plasma glucose levels to a target of 1.5 × fasting plasma glucose. At the end of the clamp (time 120 min), a bolus of 5 g l-arginine (given as 10% arginine HCl) was infused for 30 s, and from time 120 to 125 min, the rate of the glucose infusion was not changed.
Data Collection
Plasma glucose was measured at time −115, −100, and −90 min and every 5th minute from time 0 to 120. For bedside measurement of plasma glucose, blood was collected into fluoride tubes and centrifuged immediately for 30 s at room temperature and 7,500g. For the analysis of plasma glucagon, GIP, and GLP-1, blood was collected in chilled tubes (on ice) containing EDTA and a specific DPP-4i (valine pyrrolidine, 0.01 mmol/L) (a gift from Novo Nordisk, Måløv, Denmark). For analyses of serum insulin and C-peptide, blood was sampled in plain tubes for coagulation (20 min at room temperature). EDTA tubes and plain tubes were centrifuged for 15 min at 2,900g and 4°C. Plasma samples for glucagon, GIP, and GLP-1 were stored at −20°C and serum samples for insulin and C-peptide at −80°C until analysis.
Laboratory Methods
Plasma glucose was measured bedside by the glucose oxidase method (Model 2900 Series Biochemistry Analyzers; YSI Incorporated, Yellow Springs, OH). Serum insulin and C-peptide concentrations were measured with a two-sided electrochemiluminescence immunoassay (Siemens Healthcare, Ballerup, Denmark). Plasma concentrations of total GIP (16), total GLP-1 (17), and glucagon (18) were measured by radioimmunoassays as described previously. For the GIP, GLP-1, and glucagon assays, plasma samples (EDTA) were extracted with ethanol (70% v/v) to eliminate unspecific interference.
Statistical Analyses and Calculations
All results in the text and figures are presented as mean ± SEM unless stated otherwise. Differences resulting in P values of <0.05 were considered significant. Area under the curve (AUC) and baseline-subtracted AUC (bsAUC) values were calculated using the trapezoidal rule. Time −105, −100, and −90 min were defined as baseline values for calculations of bsAUC0–60 min, bsAUC60–120 min, and bsAUC0–120 min. Primary end points were differences between interventions in bsAUC0–60 min, bsAUC60–120 min, and bsAUC0–120 min for C-peptide. For calculation of incremental AUC120–125 min (iAUC120–125 min), values were subtracted from the value at time 120 min. Insulin secretion rate (ISR) was calculated based on C-peptide elimination rates and deconvolution as previously described (19,20). To check whether the targeted plasma glucose levels were obtained, the AUC0–120 min for the plasma glucose/fasting plasma glucose ratio was calculated (optimal value: 150 mmol/L/mmol/L/min). Statistical analyses were carried out within each group using linear mixed models with an unrestricted covariance structure and the Kenward-Roger approximation of the df using the algorithm of y = SU × infusion × SU*infusion, in which y is the variant of interest, subject ID is random effect, infusion (GIP, GLP-1, or NaCl) and SU (SU or placebo) are fixed effects, and SU*infusion to test for interaction. To test for differences between groups, we added “group” to the algorithm (y = SU × infusion × group × SU*infusion) and tested the significance level of group. When calculating the total amount of glucose given, we adjusted for fasting plasma glucose levels for HNF1A mutation carriers. To guard against false positives, all comparisons including primary end points were adjusted for multiple testing using the Tukey multiple-comparison test. Extreme outliers were identified according to Tukey fences (21), and extreme outliers are presented explicitly in the results. All analyses were performed in SAS Studio 9.4M5 (SAS Institute, Cary, NC) and graphical presentations in GraphPad Prism 8.0 (GraphPad Software, San Diego, CA).
Data and Resource Availability
The data sets generated during and/or analyzed during the current study are available from the corresponding authors on reasonable request.
Results
Participant Characteristics
HNF1A mutation carriers and control subjects without diabetes were well matched according to sex, age, and BMI, and the two groups had similar HOMA of insulin resistance but differed, as expected, on fasting plasma glucose and HbA1c (Table 1). One study day in a control subject without diabetes (SU+GLP-1) and one study day in an HNF1A mutation carrier (SU+placebo) were excluded from the analysis because the randomization sequence had not been followed, resulting in erroneousness infusions (human error). Regarding glucagon, one participant without diabetes qualified as an extreme outlier and was excluded from glucagon data analyses. This participant had extraordinarily high fasting glucagon concentrations (mean 54 pmol/L, range 30–101 pmol/L) compared with other control subjects without diabetes (mean 9.4 pmol/L) but was kept in other analyses because glucagon concentrations were suppressed and insulin/C-peptide response was comparable to other control subjects without diabetes.
Plasma Levels of GIP and GLP-1
Mean baseline concentrations of GIP (HNF1A mutation carriers, 18 ± 3 pmol/L, and control subjects without diabetes, 18 ± 1 pmol/L; P = 0.9167) and GLP-1 (HNF1A mutation carriers, 9 ± 1 pmol/L, and control subjects without diabetes, 11 ± 1 pmol/L; P = 0.2821) did not differ between groups (Fig. 1A–D). GIP peak concentrations were similar on study days with GIP infusion in HNF1A mutation carriers (131 ± 7 pmol/L [placebo+GIP] and 123 ± 10 pmol/L [SU+GIP]; P = 0.2821) and in the group of control subjects without diabetes (124 ± 6 pmol/L [placebo+GIP] and 117 ± 6 pmol/L [SU+GIP]; P = 0.4438). Likewise, GLP-1 peak concentrations were similar on days with GLP-1 infusion in HNF1A mutation carriers (56 ± 6 pmol/L [placebo+GLP-1] and 62 ± 6 pmol/L [SU+GLP-1]; P = 0.2620) and in control subjects without diabetes (60 ± 5 pmol/L [placebo+GLP-1] and 66 ± 3 pmol/L [SU+GLP-1]; P = 0.5830).
Figure 1.

GIP, GLP-1, plasma glucose, and glucose infused. Plasma GIP (A and B), GLP-1 (C and D), and glucose (E and F) vs. time during the 2-h two-step glucose clamp in 10 HNF1A mutation carriers and 10 control subjects without diabetes. Accumulated glucose infused at time 60 min and 120 min (G and H) (subtracted bolus given at time 60 min) in HNF1A mutation carriers (G) and control subjects without diabetes (H). Data are presented as mean ± SD (A–F) and mean ± SEM (G and H). Continuous infusions of saline (NaCl), GIP, or GLP-1 were started at time 0 min preceded by either single-dose SU 1 mg glimepiride or placebo (PLA) at time −90 min. Symbols show significant differences (P < 0.05) between interventions: *significantly greater than PLA+NaCl; †significantly greater than SU+NaCl; ‡significantly greater than PLA+GIP; §significantly greater than PLA+GLP-1. FPG, fasting plasma glucose.
Plasma Glucose and Glucose Infused
Fasting plasma glucose was 3.7 ± 1.1 mmol/L higher in HNF1A mutation carriers compared with control subjects without diabetes (P = 0.0041), with no difference in fasting plasma glucose between experimental days within each group (Table 3). The targeted glucose concentrations (expressed as the AUC0–120 min for plasma glucose/fasting plasma glucose) during the glucose clamp procedure were achieved without differences between groups (P = 0.6135) or between study days within the two groups (Fig. 1E and F and Table 3). The amount of glucose (grams) needed to maintain the plasma glucose concentrations during the experimental days was greatest with the combination of SU+GIP and SU+GLP-1, respectively, in both HNF1A mutation carriers and control subjects without diabetes (Fig. 1G and H and Table 3).
Table 3.
Glucose, C-peptide, C-peptide/glucose, and glucagon
| Intervention | PLA+NaCl | PLA+GIP | PLA+GLP-1 | SU+NaCl | SU+GIP | SU+GLP-1 | Interaction P value |
|---|---|---|---|---|---|---|---|
| Plasma glucose | |||||||
| HNF1A mutation carriers | |||||||
| Baseline (mmol/L) | 8.6 ± 1.1 | 8.9 ± 1.0 | 8.6 ± 1.0 | 8.9 ± 1.0 | 8.9 ± 1.1 | 8.7 ± 1.1 | |
| AUC0–120 min (mol/L × min) | 1.28 ± 0.15 | 1.33 ± 0.15 | 1.28 ± 0.15 | 1.34 ± 0.16 | 1.31 ± 0.16 | 1.28 ± 0.16 | |
| PG/FPG AUC0–120 (min−1) | 150 ± 1 | 149 ± 1 | 149 ± 1 | 149 ± 1 | 147 ± 1 | 147 ± 1 | |
| Control subjects without diabetes | |||||||
| Baseline (mmol/L) | 4.8 ±0.1 | 5.0 ± 0.1 | 4.9 ± 0.1 | 5.0 ± 0.1 | 5.0 ± 0.1 | 5.0 ± 0.1 | |
| AUC0–120 min (mol/L × min) | 0.72 ± 0.02 | 0.75 ± 0.02 | 0.73 ± 0.02 | 0.75 ± 0.02 | 0.73 ± 0.01 | 0.74 ± 0.01 | |
| PG/FPG AUC0–120 min (min) | 149 ± 1 | 149 ± 1 | 149 ± 1 | 150 ± 1 | 148 ± 1 | 148 ± 1 | |
| Glucose infused | |||||||
| HNF1A mutation carriers | |||||||
| Time 0–60 min (g) | 3.6 ± 1.0 | 5.7 ± 1.4 | 6.2 ± 1.0 | 6.5 ± 1.3 | 9.7 ± 1.8* | 9.3 ± 1.5* | 0.6745 |
| Time 60–120 min (g) | 14 ± 1.0 | 16 ± 2.0 | 18 ± 1.6 | 19 ± 1.3 | 28 ± 2.7*†‡§ | 24 ± 2.2*‡§ | 0.0144 |
| Time 0–120 min (g) | 18 ± 1.7 | 22 ± 3.2 | 24 ± 2.3 | 26 ± 2.1 | 37 ± 4.2*†‡§ | 33 ± 3.3*‡§ | 0.0472 |
| Control subjects without diabetes | |||||||
| Time 0–60 min (g) | 0.8 ± 0.6 | 4.6 ± 0.9* | 4.9 ± 0.8* | 3.7 ± 0.9 | 6.1 ± 1.3* | 9.3 ± 1.2*†‡§‖ | 0.1237 |
| Time 60–120 min (g) | 9.7 ± 1.9 | 20 ± 3.6 | 20 ± 4.4 | 17 ± 2.6 | 30 ± 5.3*† | 39 ± 5.8*†‡§ | 0.0180 |
| Time 0–120 min (g) | 11 ± 2.3 | 25 ± 4.3 | 25 ± 5.0 | 21 ± 3.2 | 36 ± 6.3*† | 48 ± 6.7*†‡§ | 0.0205 |
| C-peptide | |||||||
| HNF1A mutation carriers | |||||||
| Baseline (pmol/L) | 302 ± 14 | 299 ± 21 | 314 ± 23 | 299 ± 24 | 314 ± 18 | 310 ± 19 | |
| bsAUC0–60 min (nmol/L × min) | 0.7 ± 0.9 | 7.5 ± 1.7 | 5.8 ± 1.4 | 8.0 ± 2.4 | 19 ± 3.8*†‡§ | 18 ± 3.4*†‡§ | 0.1617 |
| bsAUC60–120 min (nmol/L × min) | 9.6 ± 1.6 | 20 ± 3.3 | 26 ± 3.1* | 21 ± 4.3 | 45 ± 6.5*†‡ | 46 ± 5.6*†‡§ | 0.0190 |
| bsAUC0–120 min (nmol/L × min) | 10 ± 2.1 | 28 ± 4.7* | 32 ± 4.0* | 29 ± 6.5 | 64 ± 10*†‡§ | 63 ± 8.6*†‡§ | 0.0294 |
| Control subjects without diabetes | |||||||
| Baseline (pmol/L) | 358 ± 42 | 415 ± 35 | 370 ± 27 | 394 ± 50 | 372 ± 34 | 426 ± 39 | |
| bsAUC0–60 min (nmol/L × min) | −2.7 ± 2.4 | 5.5 ± 2.2 | 5.1 ± 2.4 | 3.9 ± 1.8 | 12 ± 2.9* | 24 ± 3.9*†‡§‖ | 0.0233 |
| bsAUC60–120 min (nmol/L × min) | 17 ± 5.8 | 48 ± 6.7 | 60 ± 11* | 40 ± 7.7 | 80 ± 14*† | 131 ± 17*†‡§‖ | 0.0097 |
| bsAUC0–120 min (nmol/L × min) | 14 ± 7.8 | 53 ± 8.2 | 65 ± 13 | 43 ± 8.9 | 93 ± 16*† | 156 ± 20*†‡§‖ | 0.0078 |
| C-peptide/glucose | |||||||
| HNF1A mutation carriers | |||||||
| Baseline (pmol/L) | 41 ± 6.4 | 40 ± 7.3 | 42 ± 6.3 | 36 ± 5.1 | 39 ± 6.1 | 42 ± 6.5 | |
| bsAUC0–60 min (nmol/L × min) | −0.0 ± 0.12 | 0.8 ± 0.2* | 0.7 ± 0.2* | 0.9 ± 0.3 | 2.5 ± 0.4*†‡§ | 2.2 ± 0.4*†‡§ | 0.0398 |
| bsAUC60–120 min (nmol/L × min) | 0.1 ± 0.1 | 1.0 ± 0.3 | 1.6 ± 0.3* | 1.0 ± 0.3 | 3.1 ± 0.5*†‡ | 3.3 ± 0.5*†‡§ | 0.0042 |
| bsAUC0–120 min (nmol/L × min) | 0.1 ± 0.2 | 1.8 ± 0.4* | 2.3 ± 0.5* | 2.0 ± 0.5 | 5.6 ± 0.9*†‡§ | 5.5 ± 0.8*†‡§ | 0.0077 |
| Control subjects without diabetes | |||||||
| Baseline (pmol/L) | 75 ± 9.2 | 83 ± 6.8 | 75 ± 5.9 | 78 ± 8.5 | 75 ± 7.3 | 75 ± 7.0 | |
| bsAUC0–60 min (nmol/L × min) | −0.7 ± 0.52 | 1.2 ± 0.5 | 1.1 ± 0.5 | 0.8 ± 0.4 | 2.6 ± 0.6* | 5.0 ± 0.9*†‡§‖ | 0.0260 |
| bsAUC60–120 min (nmol/L × min) | 0.8 ± 0.9 | 4.7 ± 0.9 | 6.6 ± 1.6 | 3.5 ± 1.0 | 9.3 ± 1.9*† | 16 ± 2.4*†‡§‖ | 0.0166 |
| bsAUC0–120 min (nmol/L × min) | 0.2 ± 0.1 | 5.8 ± 1.3 | 7.7 ± 2.1 | 4.4 ± 1.3 | 12 ± 2.4*† | 21 ± 3.2*†‡§‖ | 0.0148 |
| Glucagon | |||||||
| HNF1A mutation carriers | |||||||
| Baseline (pmol/L) | 13 ± 1 | 12 ± 1 | 11 ± 1 | 11 ± 2 | 13 ± 1 | 12 ± 2 | |
| bsAUC0–60 min (pmol/L × min) | −306 ± 59 | −182 ± 59 | −381 ± 59 | −186 ± 59 | −152 ± 59 | −255 ± 59 | 0.5167 |
| bsAUC60–120 min (pmol/L × min) | −463 ± 57 | −435 ± 57 | −488 ± 57 | −351 ± 60 | −353 ± 57 | −420 ± 57 | 0.9094 |
| bsAUC0–120 min (pmol/L × min) | −770 ± 106 | −618 ± 106 | −869 ± 106 | −560 ± 111 | −505 ± 106 | −675 ± 106 | 0.8320 |
| Control subjects without diabetes | |||||||
| Baseline (pmol/L) | 8 ± 2 | 9 ± 2 | 9 ± 2 | 10 ± 2 | 8 ± 2 | 13 ± 2 | |
| bsAUC0–60 min (nmol/L × min) | −93 ± 76 | −163 ± 76 | −248 ± 76 | −148 ± 76 | −52.7 ± 76 | −321 ± 81 | 0.2472 |
| bsAUC60–120 min (nmol/L × min) | −325 ± 100 | −388 ± 100 | −408 ± 100 | −374 ± 100 | −274 ± 100 | −538 ± 105 | 0.2707 |
| bsAUC0–120 min (nmol/L × min) | −419 ± 171 | −605 ± 177 | −655 ± 171 | −523 ± 171 | −325 ± 171 | −857 ± 179 | 0.1979 |
Data are mean ± SEM. A significant interaction describes a supra-additive of combining SU+GIP and/or SU+GLP-1.
FPG, fasting plasma glucose; PG, plasma glucose; PLA, placebo.
Symbols show significant differences (P < 0.05) between interventions:
significantly greater than PLA+NaCl;
significantly greater SU+NaCl;
significantly greater than PLA+GIP;
significantly greater than PLA+GLP-1;
significantly greater than SU+GIP.
C-Peptide, Insulin, and ISR
Mean fasting C-peptide concentrations in HNF1A mutation carriers were significantly lower than in control subjects without diabetes (308 ± 16.8 vs. 387 ± 31.7 pmol/L; P = 0.0442), even though their plasma glucose was higher (Table 3). When looking across all indices of insulin secretion (C-peptide, insulin, ISR, C-peptide/glucose, insulin/glucose, and ISR/glucose), the overall trends were the same (Figs. 2 and 3, Table 3, and Supplementary Table 1); below, detailed results for C-peptide are presented. In HNF1A mutation carriers, combinations of SU+GIP and SU+GLP-1, respectively, were significantly more insulinotropic (based on C-peptide bsAUC0–60 min, bsAUC60–120 min, and bsAUC0–120 min) compared with administration of placebo+GIP, placebo+GLP-1, placebo+NaCl, and SU+NaCl (Table 3). In HNF1A mutation carriers, both placebo+GIP and placebo+GLP-1, respectively, compared with placebo+NaCl resulted in significantly greater C-peptide bsAUC0–120 min values. Other analyses (insulin, insulin/glucose, ISR, and ISR/glucose) demonstrated an insignificant insulinotropic trend. SU+NaCl was not significantly more insulinotropic compared with placebo+NaCl (in all insulin secretion parameters). In control subjects without diabetes, SU+GLP-1 was more insulinotropic (C-peptide bsAUC0–120 min) compared with all other interventions, while SU+GIP was the second most insulinotropic intervention (Table 3). In control subjects without diabetes, placebo+GLP-1 and placebo+GIP alone were more insulinotropic (C-peptide bsAUC0–120 min) compared with placebo+NaCl, while SU+NaCl was not significantly different from placebo+NaCl.
Figure 2.

C-peptide and C-peptide/glucose. Serum C-peptide (A and B) and C-peptide/glucose (E and F) vs. time during the 2-h two-step glucose clamp in 10 HNF1A mutation carriers and 10 control subjects without diabetes. Corresponding bsAUC0–60 min and bsAUC60–120 min are presented in C, D, G, and H. Continuous infusions of saline (NaCl), GIP, or GLP-1 were started at time 0 min preceded by either single-dose SU 1 mg glimepiride or placebo (PLA) at time −90 min. Data from time 120 min to 125 min is magnified in insets in A, B, E, and F. Data are presented as mean ± SEM. Symbols show significant differences (P < 0.05) between interventions: *significantly greater than PLA+NaCl; †significantly greater than SU+NaCl; ‡significantly greater than PLA+GIP; §significantly greater than PLA+GLP-1; ‖significantly greater than SU+GIP.
Figure 3.

Insulin and glucagon. Serum insulin (A and B) and plasma glucagon (E and F) vs. time in 10 HNF1A mutation carriers and 10 control subjects without diabetes. Corresponding bsAUC0–60 min and bsAUC60–120 min are presented in C, D, G, and H. Continuous infusions of saline (NaCl), GIP, or GLP-1 were started at time 0 min preceded by either a single-dose SU 1 mg glimepiride or placebo (PLA) at time −90 min. Data from time 120 min to 125 min is magnified in insets in A, B, E, and F. Data are presented as mean ± SEM. Symbols show significant differences (P < 0.05) between interventions: *significantly greater than PLA+NaCl; †significantly greater than SU+NaCl; ‡significantly greater than PLA+GIP; §significantly greater than PLA+GLP-1.
Supra-additive Effect of Combining SU With an Incretin Hormone
We observed a significant interaction between SU (SU or placebo) and infusions (GIP, GLP-1, or NaCl) for C-peptide (bsAUC60–120 min and bsAUC0–120 min) in both HNF1A mutation carriers (P = 0.0190 and P = 0.0294, respectively) and control subjects without diabetes (P = 0.0097 and P = 0.0078, respectively), which is indicative of a supra-additive effect of combining SU and GIP and/or GLP-1, respectively (Table 3). When looking at bsAUC0–60 min for C-peptide, no interaction was observed in HNF1A mutation carriers (P = 0.1617), while an interaction was observed in control subjects without diabetes (P = 0.0233). Regarding C-peptide/glucose, an interaction was present across all time periods (bsAUC0–60 min, bsAUC60–120 min, and bsAUC0–120 min) in both groups (Table 3). The magnitude of the interaction for C-peptide bsAUC0–120 min and C-peptide/glucose is depicted in Fig. 4. In HNF1A mutation carriers, the supra-additive effect on C-peptide was rather small (∼5–10%); however, it was substantially higher when adjusted for glucose concentrations and C-peptide/glucose (∼25–45%).
Figure 4.
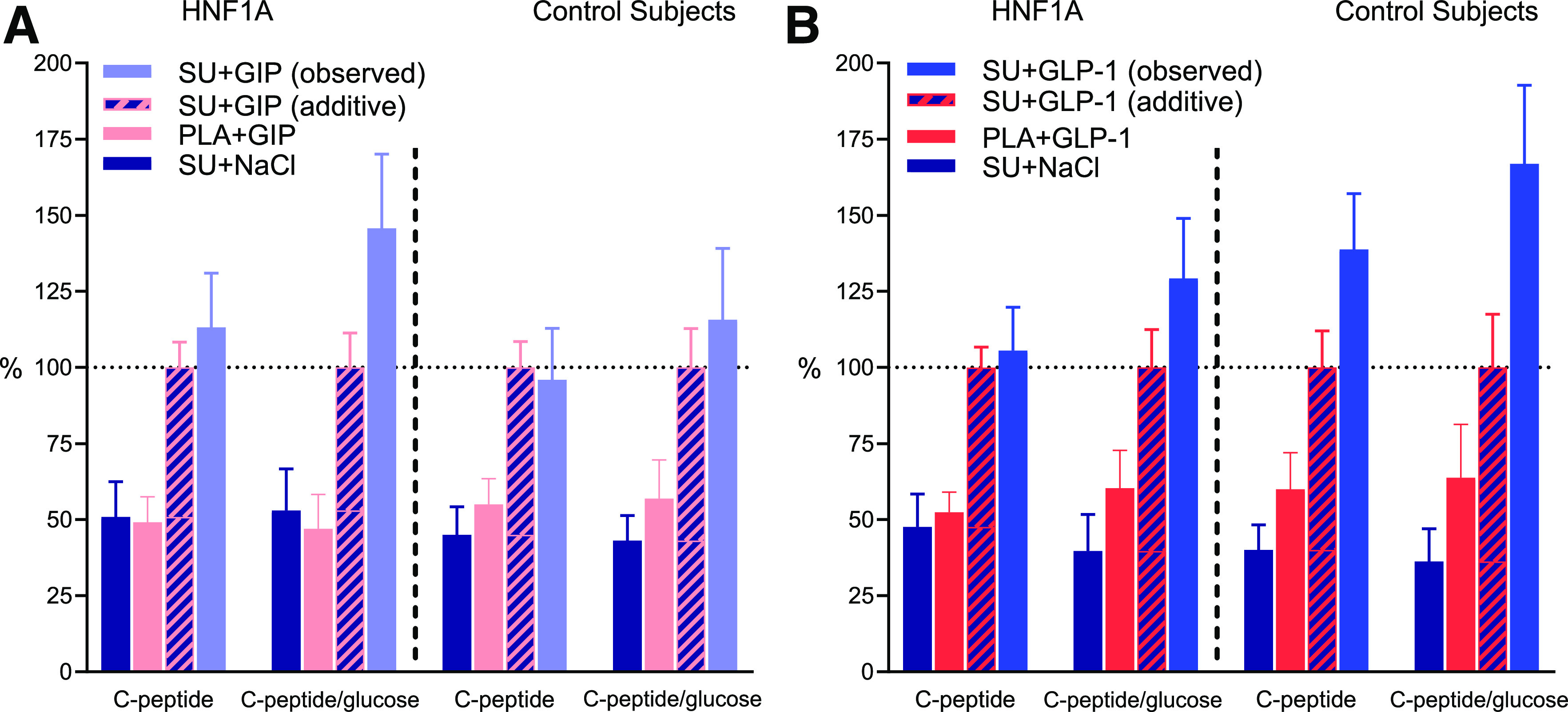
Interaction between SU and the incretin hormones. bsAUC0–120 min for C-peptide and C-peptide/glucose for 10 HNF1A mutation carriers and 10 control subjects without diabetes during the 2-h two-step glucose clamp. Continuous infusions of saline (NaCl), GIP, or GLP-1 were started at time 0 min preceded by either single-dose SU 1 mg glimepiride or placebo (PLA) at time −90 min. Data are shown as mean percentage ± SEM. A: The reference value (100%) is the sum of bsAUC0–120 min, SU+NaCl + bsAUC0–120 min, PLA+GIP, which on the graphs is shown as SU+GIP (additive). SU+GIP (observed) is the observed bsAUC0–120 min, SU+GIP during our study. If SU+GIP (observed) is greater than SU+GIP (additive), this is indicative of an interaction and thus a supra-additive effect. B: The calculations are identical for A, but with GLP-1 instead of GIP.
Arginine-Induced C-Peptide Secretion
The arginine-induced maximal secretion test (Table 4 and insets in Fig. 2A and B) displayed a significantly attenuated C-peptide response in HNF1A mutation carriers compared with control subjects without diabetes. In HNF1A mutation carriers, the greatest peak and AUC120–125 min for C-peptide were observed after administration of arginine on experimental days with SU+GIP and SU+GLP-1, while in control subjects without diabetes, SU+GLP-1 was the most potent stimuli. The concentration of C-peptide at the time of the arginine administration was the most important determinant of the C-peptide response given that the difference of the iAUC120–125 min (subtracted the C-peptide level at time 120 min) is small in both groups across all study days.
Table 4.
Arginine-induced maximal secretion test
| Intervention | PLA+NaCl | PLA+GIP | PLA+GLP-1 | SU+NaCl | SU+GIP | SU+GLP-1 | Interaction P value |
|---|---|---|---|---|---|---|---|
| C-peptide | |||||||
| HNF1A mutation carriers | |||||||
| Peak (nmol/L) | 1.2 ± 0.1 | 1.4 ± 0.2 | 1.8 ± 0.2*‡ | 1.8 ± 0.2 | 2.2 ± 0.3*†‡ | 2.5 ± 0.3*†‡§ | 0.1963 |
| AUC120–125 min (nmol/L × min) | 4.9 ± 0.4 | 5.4 ± 0.7 | 7.0 ± 0.7* | 6.8 ± 0.8 | 9.3 ± 1.1*†‡ | 10.0 ± 1.0*†‡§ | 0.1193 |
| iAUC120–125 min (nmol/L × min) | 2.2 ± 0.3 | 2.1 ± 0.4 | 2.8 ± 0.4 | 3.1 ± 0.4 | 3.2 ± 0.5 | 3.9 ± 0.6‡ | 0.9680 |
| Control subjects without diabetes | |||||||
| Peak (nmol/L) | 2.2 ± 0.2 | 3.0 ± 0.3 | 3.4 ± 0.4* | 2.9 ± 0.4 | 3.7 ± 0.5 | 5.0 ± 0.6*†‡§ | 0.1801 |
| AUC120–125 min (nmol/L × min) | 8.3 ± 0.9 | 12.3 ± 1.1 | 14.1 ± 1.7* | 11.8 ± 1.7 | 16.1 ± 2.1* | 22.0 ± 2.6*†‡§ | 0.1385 |
| iAUC120–125 min (nmol/L × min) | 4.8 ± 0.6 | 5.3 ± 0.7 | 5.7 ± 0.6 | 5.8 ± 0.9 | 5.9 ± 0.9 | 6.9 ± 1.0 | 0.8340 |
| Glucagon | |||||||
| HNF1A mutation carriers | |||||||
| Peak (nmol/L) | 41 ± 5 | 44 ± 4 | 36 ± 4 | 32 ± 4 | 35 ± 3 | 34 ± 4 | 0.1797 |
| AUC120–125 min (nmol/L × min) | 508 ± 71 | 428 ± 88 | 293 ± 52 | 414 ± 69 | 467 ± 89 | 356 ± 55 | 0.2300 |
| iAUC120–125 min (nmol/L × min) | 128 ± 16 | 129 ± 13 | 104 ± 15 | 95 ± 17 | 112 ± 11 | 109 ± 17 | 0.2671 |
| Control subjects without diabetes | |||||||
| Peak (nmol/L) | 30 ± 4 | 31 ± 3 | 25 ± 3 | 24 ± 3 | 26 ± 3 | 20 ± 3† | 0.9993 |
| AUC120–125 min (nmol/L × min) | 287 ± 50 | 256 ± 41 | 160 ± 39* | 237 ± 41 | 217 ± 32 | 212 ± 54 | 0.2801 |
| iAUC120–125 min (nmol/L × min) | 99 ± 14 | 97 ± 13 | 73 ± 16 | 78 ± 15 | 79 ± 12 | 79 ± 15 | 0.5468 |
Data are mean ± SEM. iAUC120–125 min is the incremental values from time 120 min, when 5 g arginine was given as a bolus.
PLA, placebo.
Symbols show significant differences (P < 0.05) between interventions:
significantly greater than PLA+NaCl;
significantly greater SU+NaCl;
significantly greater than PLA+GIP;
significantly greater than PLA+GLP-1.
Glucagon
Fasting glucagon concentrations were higher in HNF1A mutation carriers compared with control subjects without diabetes (11.8 ± 0.5 vs. 9.5 ± 0.8 pmol/L; P = 0.0163) (Table 3 and Fig. 3E and F). Glucagon concentrations decreased from baseline (time −100 min and −90 min) to time 0 min regardless of SU or placebo administration, and the difference between groups was abolished at time 0 min (HNF1A mutation carriers, 8.9 ± 0.9 pmol/L, vs. control subjects without diabetes, 7.4 ± 1.0 pmol/L; P = 0.2777). The glucagon concentrations decreased with increasing glucose concentrations in both HNF1A mutation carriers and control subjects without diabetes. There were no significant differences in bsAUC0–120 min for glucagon between interventions in any of the groups. We observed an insignificant trend toward a greater decrease of glucagon concentrations on days with placebo+GLP-1 and SU+GLP-1, while the smallest decrements in glucagon levels were observed with SU+GIP in both groups (Fig. 3G and H and Table 3).
Arginine-Induced Glucagon Secretion
The arginine-induced glucagon levels were significantly higher in HNF1A mutation carriers compared with control subjects without diabetes evaluated as peak (P = 0.0215), AUC120–125 min (P = 0.0093), and iAUC120–125 min (P = 0.0332) for glucagon (Table 4 and insets in Fig. 3E and F). In both groups, there was no difference between experimental days.
Discussion
This study investigates the insulinotropic properties of a combination of SU with infusions of either GIP or GLP-1 in HNF1A mutation carriers. The primary finding is that SU combined with GIP or GLP-1 increases C-peptide concentrations in an additive to supra-additive fashion in HNF1A mutation carriers, indicating that a combination of SU and incretin-based therapy may have synergistic effects in the treatment of patients with HNF1A diabetes.
Despite the fact that most patients with HNF1A-diabetes eventually need additional treatment on top of SU, no study has evaluated potential second-line glucose-lowering agents (4,10,14). In the current study, infusion rates of exogenous GIP and GLP-1 were chosen to result in plasma levels seen during treatment with a DPP-4i. A considerable strength of this study is the design, in which we isolate the effects of GIP, GLP-1, and SU on the endocrine pancreas from that of glucose using a two-step glucose clamp. Another strength is the placebo-controlled crossover design, which reduces the intraindividual differences. Considering the rarity of HNF1A mutations, it is also a strength that none of the HNF1A mutation carriers were related. A limitation to our study is the heterogeneity of the HNF1A mutation carriers regarding their diabetes status, fasting plasma glucose, and oral glucose-lowering treatment, which included incretin-based treatment. Our study was powered to detect changes in C-peptide levels but may not be powered adequately to detect changes in glucagon.
We demonstrate a significant insulinotropic effect evaluated as C-peptide bsAUC0–120 min in the current study when using supraphysiological doses of both GIP (1.5 pmol/kg/min) and GLP-1 (0.5 pmol/kg/min). This is in line with a previous study by Vilsbøll et al. (22) that found a significant insulinotropic effect of exogenous infusions of GIP (4 pmol/kg/min) and GLP-1 (1 pmol/kg/min), respectively, compared with saline during a 2-h hyperglycemic clamp (15 mmol/L) in patients with HNF1A diabetes. Together, GIP and GLP-1 are responsible for the incretin effect (i.e., the amplification of insulin secretion with an oral glucose challenge compared with isoglycemic intravenous glucose infusion). Østoft et al. (9,14) described impaired incretin effect in HNF1A mutation carriers and that a GLP-1 receptor agonist has glucose-lowering actions with low risk of hypoglycemia in patients with HNF1A diabetes. Taken together, studies investigating the effect of incretins in HNF1A mutation carriers indicate that a diminished activation of both GIP and GLP-1 receptors contribute to impaired insulin responses and thus hyperglycemia, but that GIP and GLP-1 receptors may constitute viable treatment targets during elevated plasma levels of the peptides seen with incretin-based therapies such as DPP-4i (GIP and GLP-1) and GLP-1 receptor agonist (GLP-1).
Interestingly, during the hyperglycemic part of our clamp study, we found a supra-additive effect on insulin secretion with the combination of SU+GIP and SU+GLP-1 in both HNF1A mutation carriers and control subjects without diabetes. This observation could be explained by a combined effect on the KATP channel by SU, GIP, and GLP-1. SU binds directly to the KATP channel, while GIP and GLP-1 receptor activation increase levels of cAMP, in turn activating protein kinase A, which increases the sensitivity of the KATP channel to ATP (23). The combined actions increase the likelihood of KATP channel closure and depolarization and subsequently increased insulin release. In addition, acute GIP and GLP-1 receptor activation also increase insulin secretion via several other mechanisms than the KATP channel (23), and chronic GLP-1 receptor stimulation has been shown to increase glycolysis and ATP production in β-cells through transcriptional activation and expression of glycolytic genes (24). Whether this is the case in HNF1A mutation carriers is unknown; however, case series have indicated remarkable HbA1c reduction and increase peak insulin levels during an intravenous glucose tolerance test in patients with HNF1A-diabetes when adding a DPP-4i to SU (25). In patients with type 2 diabetes, the glucose-lowering effect of DPP-4i is mainly attributed to the increase of GLP-1 levels because the insulinotropic effect of GIP is severely diminished (26); however, our study indicates that GIP could mediate the glucose-lowering effects of DPP-4i to a greater extent in patients with HNF1A diabetes.
Unexpectedly, we observed a greater mean fasting and arginine-induced glucagon concentration in HNF1A mutation carriers compared with control subjects without diabetes. Our study is the first to indicate increased fasting glucagon concentrations in HNF1A mutation carriers compared with control subjects without diabetes, to our knowledge. In addition, postprandial hyperglucagonemic responses during meal tests have also been observed in HNF1A mutation carriers (9,10,14). The observation of hyperglucagonemia in fasting, postprandial, and after arginine infusion could indicate an altered secretion pattern of glucagon in HNF1A mutation carriers.
To our knowledge, the effect of an HNF1A mutation on α-cell functions has not been investigated in either animals or cell lines. The nature of postprandial hyperglucagonemia could theoretically be due to insufficient glucose sensing in the glucagon-producing α-cells (like that observed in β-cells); however, the normal suppression of glucagon during intravenous glucose infusion in our and other studies contradicts this (14,22). Insulin is also known as an inhibitor of glucagon secretion (27), which is why hypoinsulinemia could disrupt the normal paracrine signaling between β- and α-cells, resulting in increased glucagon concentrations. Finally, an explanation could be that the total α-cell mass or α- to β-cell ratio could be increased. A study of a single pancreatic human islet from a 33-year-old diseased donor with HNF1A diabetes displayed elevated α-cell mass and increased α/β-cell ratio compared with seven control subjects without diabetes (28). Arginine induces maximal glucagon release from α-cells via a mechanism independent of both glucose metabolism and KATP channel (29,30) and is thought to be correlated with total α-cell mass (31). Thus, our study could potentially be in line with an increased α-cell mass; however, our data could just as well indicate increased glucagon secretory capacity of α-cells.
In patients with type 2 diabetes, both exogenous GLP-1 infusions and GLP-1 receptor agonists show glucagonostatic properties alleviating postprandial hyperglucagonemia (32). On the contrary, the hyperglucagonemia of HNF1A mutation carriers does not seem to respond to GLP-1, as we did not see a decrease in glucagon with GLP-1 infusion, nor did 6 weeks of GLP-1 receptor agonism change fasting or postprandial glucagon concentrations (9). The elevated plasma glucagon concentrations in HNF1A mutation carriers most likely add to their state of diabetes, and future studies should investigate the role of glucagon in more detail.
Conclusions
We investigated the insulinotropic properties of SU in combination with incretin hormones in HNF1A mutation carriers and report additive to supra-additive effects on insulin secretion with the combination of a low-dose SU with either GIP or GLP-1. We also report increased fasting and arginine-induced levels of glucagon in HNF1A mutation carriers. Our results and previous work indicate that targeting the GIP and/or GLP-1 receptors in combination with SU therapy may constitute a viable strategy for the management of hyperglycemia in patients with HNF1A diabetes. An ongoing clinical trial (EudraCT no. 2017-000204-15) is investigating the efficacy and safety of combined SU and DPP-4i therapy (33).
Article Information
Acknowledgments. The authors thank the study participants for commitment and loyalty, Sisse Marie Schmidt and Inass Al Nachar (Steno Diabetes Center Copenhagen, Gentofte Hospital, Hellerup, Denmark) for laboratory assistance, and coworkers in our department for valuable advice and support.
Funding. The clinical study was performed at Steno Diabetes Center Copenhagen (Gentofte Hospital, Hellerup, Denmark) and supported by private foundations: Aase og Ejnar Danielsens Fond, Fonden til Lægevidenskabens Fremme, Aage og Johanne Louis-Hansens Fond, and Axel Muusfeldts Fond. The Novo Nordisk Foundation Center for Basic Metabolic Research is an independent research center at the University of Copenhagen partially funded by an unrestricted donation from the Novo Nordisk Foundation. J.J.H. was supported by the Novo Nordisk Foundation.
Duality of Interest. H.S. has served on scientific advisory panels for Boehringer Ingelheim and Novo Nordisk. J.J.H. has served on advisory boards for Novo Nordisk. F.K.K. has served on scientific advisory panels and/or been part of speakers bureaus for, served as a consultant to, and/or received research support from Amgen, AstraZeneca, Bayer, Boehringer Ingelheim, Carmot Therapeutics, Inc., Eli Lilly and Company, Gubra, Lupin Limited, MedImmune, LLC, Merck Sharp & Dohme/Merck, Mundipharma, Norgine, Novo Nordisk, Sanofi, and Zealand Pharma. T.V. has served on scientific advisory panels and/or speakers bureaus for, served as a consultant to, and/or received research support from Amgen, AstraZeneca, Boehringer Ingelheim, Bristol-Myers Squibb, Eli Lilly and Company, Merck Sharp & Dohme, Mundipharma, Novo Nordisk, Sanofi, and Sun Pharmaceutical Industries Ltd. No other potential conflicts of interest relevant to this article were reported.
Author Contributions. A.S.C., S.H., H.S., T.H., F.K.K., and T.V. designed the study. A.S.C. wrote the study protocol. A.S.C., K.R., and N.L.H. performed the study. J.J.H. generated data. A.S.C. and T.V. performed the data analysis and wrote the manuscript. All authors critically edited the manuscript and approved the final version. A.S.C. and T.V. are the guarantors of this work and, as such, had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Prior Presentation. Parts of the results from this study were presented at the Annual Meeting of the Danish Endocrine Society, Nyborg, Denmark, 18–19 January 2019; the 79th Scientific Sessions of the American Diabetes Association, San Francisco, CA, 7–11 June 2019; the 3rd EASD Incretin Study Group of Diabetes, Bochum, Germany, 24–26 January 2019; and the 7th Meeting of the EASD Study Group on Genetics of Diabetes, Prague, Czech Republic, 16–18 May 2019.
Footnotes
Clinical trial reg. no. NCT03081676, clinicaltrials.gov
This article contains supplementary material online at https://doi.org/10.2337/figshare.12410447.
References
- 1.Pearson ER, Velho G, Clark P, et al. . Beta-cell genes and diabetes: quantitative and qualitative differences in the pathophysiology of hepatic nuclear factor-1alpha and glucokinase mutations. Diabetes 2001;50(Suppl. 1):S101–S107 [DOI] [PubMed] [Google Scholar]
- 2.Wang H, Maechler P, Hagenfeldt KA, Wollheim CB. Dominant-negative suppression of HNF-1alpha function results in defective insulin gene transcription and impaired metabolism-secretion coupling in a pancreatic beta-cell line. EMBO J 1998;17:6701–6713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wollheim CB. Beta-cell mitochondria in the regulation of insulin secretion: a new culprit in type II diabetes. Diabetologia 2000;43:265–277 [DOI] [PubMed] [Google Scholar]
- 4.Pearson ER, Starkey BJ, Powell RJ, Gribble FM, Clark PM, Hattersley AT. Genetic cause of hyperglycaemia and response to treatment in diabetes. Lancet 2003;362:1275–1281 [DOI] [PubMed] [Google Scholar]
- 5.Hansen T, Eiberg H, Rouard M, et al. . Novel MODY3 mutations in the hepatocyte nuclear factor-1α gene: evidence for a hyperexcitability of pancreatic β-cells to intravenous secretagogues in a glucose-tolerant carrier of a P447L mutation. Diabetes 1997;46:726–730 [DOI] [PubMed] [Google Scholar]
- 6.International Society for Pediatric and Adolescent Diabetes ISPAD Clinical Practice Consensus Guidelines 2018. Accessed 13 December 2019. Available from https://www.ispad.org/page/ISPADGuidelines2018
- 7.American Diabetes Association 2. Classification and diagnosis of diabetes: Standards of Medical Care in Diabetes--2019. Diabetes Care 2019;42(Suppl. 1):S13–S28 [DOI] [PubMed] [Google Scholar]
- 8.Shepherd MH, Shields BM, Hudson M, et al.; UNITED study . A UK nationwide prospective study of treatment change in MODY: genetic subtype and clinical characteristics predict optimal glycaemic control after discontinuing insulin and metformin. Diabetologia 2018;61:2520–2527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Østoft SH, Bagger JI, Hansen T, et al. . Glucose-lowering effects and low risk of hypoglycemia in patients with maturity-onset diabetes of the young when treated with a GLP-1 receptor agonist: a double-blind, randomized, crossover trial. Diabetes Care 2014;37:1797–1805 [DOI] [PubMed] [Google Scholar]
- 10.Tuomi T, Honkanen EH, Isomaa B, Sarelin L, Groop LC. Improved prandial glucose control with lower risk of hypoglycemia with nateglinide than with glibenclamide in patients with maturity-onset diabetes of the young type 3. Diabetes Care 2006;29:189–194 [DOI] [PubMed] [Google Scholar]
- 11.UK Prospective Diabetes Study (UKPDS) Group Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33) [published correction appears in Lancet 1999;354:602]. Lancet 1998;352:837–853 [PubMed] [Google Scholar]
- 12.Isomaa B, Henricsson M, Lehto M, et al. . Chronic diabetic complications in patients with MODY3 diabetes. Diabetologia 1998;41:467–473 [DOI] [PubMed] [Google Scholar]
- 13.Steele AM, Shields BM, Shepherd M, Ellard S, Hattersley AT, Pearson ER. Increased all-cause and cardiovascular mortality in monogenic diabetes as a result of mutations in the HNF1A gene. Diabet Med 2010;27:157–161 [DOI] [PubMed] [Google Scholar]
- 14.Østoft SH, Bagger JI, Hansen T, et al. . Incretin effect and glucagon responses to oral and intravenous glucose in patients with maturity-onset diabetes of the young--type 2 and type 3. Diabetes 2014;63:2838–2844 [DOI] [PubMed] [Google Scholar]
- 15.Liu D, Moberg E, Kollind M, Lins PE, Adamson U, Macdonald IA. Arterial, arterialized venous, venous and capillary blood glucose measurements in normal man during hyperinsulinaemic euglycaemia and hypoglycaemia. Diabetologia 1992;35:287–290 [DOI] [PubMed] [Google Scholar]
- 16.Lindgren O, Carr RD, Deacon CF, et al. . Incretin hormone and insulin responses to oral versus intravenous lipid administration in humans. J Clin Endocrinol Metab 2011;96:2519–2524 [DOI] [PubMed] [Google Scholar]
- 17.Orskov C, Rabenhøj L, Wettergren A, Kofod H, Holst JJ. Tissue and plasma concentrations of amidated and glycine-extended glucagon-like peptide I in humans. Diabetes 1994;43:535–539 [DOI] [PubMed] [Google Scholar]
- 18.Orskov C, Jeppesen J, Madsbad S, Holst JJ. Proglucagon products in plasma of noninsulin-dependent diabetics and nondiabetic controls in the fasting state and after oral glucose and intravenous arginine. J Clin Invest 1991;87:415–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kjems LL, Christiansen E, Vølund A, Bergman RN, Madsbad S. Validation of methods for measurement of insulin secretion in humans in vivo. Diabetes 2000;49:580–588 [DOI] [PubMed] [Google Scholar]
- 20.Hovorka R, Soons PA, Young MA. ISEC: a program to calculate insulin secretion. Comput Methods Programs Biomed 1996;50:253–264 [DOI] [PubMed] [Google Scholar]
- 21.Tukey JW. Schematic summaries (pictures and numbers). In Exploratory Data Analysis. Reading, MA, Addison-Wesley, 1977, p. 27–56 [Google Scholar]
- 22.Vilsbøll T, Knop FK, Krarup T, et al. . The pathophysiology of diabetes involves a defective amplification of the late-phase insulin response to glucose by glucose-dependent insulinotropic polypeptide-regardless of etiology and phenotype. J Clin Endocrinol Metab 2003;88:4897–4903 [DOI] [PubMed] [Google Scholar]
- 23.Gromada J, Bokvist K, Ding W-G, Holst JJ, Nielsen JH, Rorsman P. Glucagon-like peptide 1 (7-36) amide stimulates exocytosis in human pancreatic β-cells by both proximal and distal regulatory steps in stimulus-secretion coupling. Diabetes 1998;47:57–65 [DOI] [PubMed] [Google Scholar]
- 24.Carlessi R, Chen Y, Rowlands J, et al. . GLP-1 receptor signalling promotes β-cell glucose metabolism via mTOR-dependent HIF-1α activation. Sci Rep 2017;7:2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Katra B, Klupa T, Skupien J, et al. . Dipeptidyl peptidase-IV inhibitors are efficient adjunct therapy in HNF1A maturity-onset diabetes of the young patients--report of two cases. Diabetes Technol Ther 2010;12:313–316 [DOI] [PubMed] [Google Scholar]
- 26.Mentis N, Vardarli I, Köthe LD, et al. . GIP does not potentiate the antidiabetic effects of GLP-1 in hyperglycemic patients with type 2 diabetes. Diabetes 2011;60:1270–1276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hædersdal S, Lund A, Knop FK, Vilsbøll T. The role of glucagon in the pathophysiology and treatment of type 2 diabetes. Mayo Clin Proc 2018;93:217–239 [DOI] [PubMed] [Google Scholar]
- 28.Haliyur R, Tong X, Sanyoura M, et al. . Human islets expressing HNF1A variant have defective β cell transcriptional regulatory networks. J Clin Invest 2019;129:246–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marcelli-Tourvieille S, Hubert T, Pattou F, Vantyghem MC. Acute insulin response (AIR): review of protocols and clinical interest in islet transplantation. Diabetes Metab 2006;32:295–303 [DOI] [PubMed] [Google Scholar]
- 30.Fajans SS, Floyd JC Jr, Knopf RF, et al. . A difference in mechanism by which leucine and other amino acids induce insulin release. J Clin Endocrinol Metab 1967;27:1600–1606 [DOI] [PubMed] [Google Scholar]
- 31.Ryan EA, Lakey JRT, Paty BW, et al. . Successful islet transplantation: continued insulin reserve provides long-term glycemic control. Diabetes 2002;51:2148–2157 [DOI] [PubMed] [Google Scholar]
- 32.Nauck M. Incretin therapies: highlighting common features and differences in the modes of action of glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors. Diabetes Obes Metab 2016;18:203–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sidelmann Christensen A, Storgaard H, Hædersdal S, Hansen T, Krag Knop F, Vilsbøll T. Glimepiride monotherapy versus combination of glimepiride and linagliptin therapy in patients with HNF1A-diabetes: a protocol for a randomised, double-blinded, placebo-controlled trial. BMJ Open 2018;8:e022517. [DOI] [PMC free article] [PubMed] [Google Scholar]