

Abstract
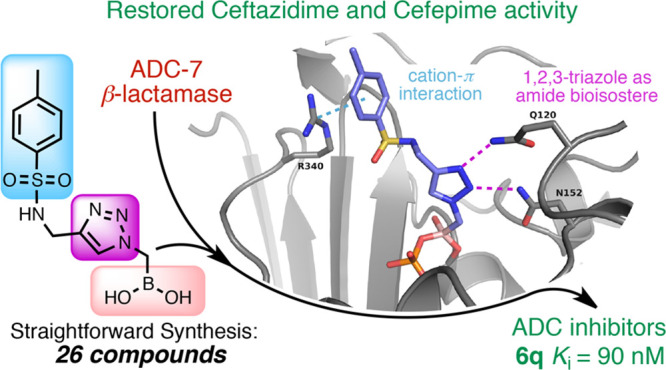
Boronic acid transition state inhibitors (BATSIs) are known reversible covalent inhibitors of serine β-lactamases. The selectivity and high potency of specific BATSIs bearing an amide side chain mimicking the β-lactam’s amide side chain are an established and recognized synthetic strategy. Herein, we describe a new class of BATSIs where the amide group is replaced by a bioisostere triazole; these compounds were designed as molecular probes. To this end, a library of 26 α-triazolylmethaneboronic acids was synthesized and tested against the clinically concerning Acinetobacter-derived cephalosporinase, ADC-7. In steady state analyses, these compounds demonstrated Ki values ranging from 90 nM to 38 μM (±10%). Five compounds were crystallized in complex with ADC-7 β-lactamase, and all the crystal structures reveal the triazole is in the putative amide binding site, thus confirming the triazole–amide bioisosterism. The easy synthetic access of these new inhibitors as prototype scaffolds allows the insertion of a wide range of chemical groups able to explore the enzyme binding site and provides insights on the importance of specific residues in recognition and catalysis. The best inhibitor identified, compound 6q (Ki 90 nM), places a tolyl group near Arg340, making favorable cation−π interactions. Notably, the structure of 6q does not resemble the natural substrate of the β-lactamase yet displays a pronounced inhibition activity, in addition to lowering the minimum inhibitory concentration (MIC) of ceftazidime against three bacterial strains expressing class C β-lactamases. In summary, these observations validate the α-triazolylboronic acids as a promising template for further inhibitor design.
Keywords: boronic acids, Acinetobacter, amide bioisostere, click chemistry, β-lactamase inhibitors
Antimicrobial resistance (AMR) is a major global health threat. Regrettably, this crisis is aggravated by the lack of new therapeutic agents in the current pharmaceutical pipeline. Economic analyses indicate that AMR increases health-care costs, the length of stay in the hospital, morbidity, and mortality.1 For these reasons, the World Health Organization (WHO) has recently designated AMR as one of the three most important problems facing human health.2 The WHO Priority List has recently assigned Acinetobacter baumannii as a critical priority pathogen due to the high prevalence of cephalosporin and carbapenem resistance and its ability to survive in adverse environmental conditions, making it one of the most threatening nosocomial pathogens.3
Common AMR mechanisms found in Acinetobacter spp. include modification of the enzymes that the antibiotic targets, decreased permeability of the outer membrane, efflux pumps, and the production of enzymes that attack and inactivate antibiotics (β-lactamases).3,4 Current antimicrobials used in the clinic to treat infections caused by multidrug resistant (MDR) or extreme drug resistant (XDR) A. baumannii are siderophore containing β-lactams (cefidericol), polymyxins, tigecycline, and aminoglycosides. All these drugs display uncertain clinical efficacy, a high level of toxicity, and mounting resistance.4 The urgent need for new drugs active against this pathogen has recently accelerated drug development, and new therapeutic options are under study.
The attractiveness of identifying β-lactamase inhibitors effective against A. baumannii relies upon the β-lactam’s intrinsic mechanism of action. The use of combination therapy, where a β-lactam antibiotic is combined with a β-lactamase inhibitor, is a time-honored and extremely effective approach to overcome resistance. Three new β-lactam/β-lactamase inhibitor combinations recently entered the market, namely, the diazabicyclooctane avibactam with ceftazidime (Avycaz),5 relebactam with imipenem/cilistatin (Recarbrio), and the boronic acid vaborbactam with Meropenem (Vabomere).6 While these combinations prove to be useful in the treatment of infections caused by carbapenem resistant Enterobacteriacae and multi drug-resistant (MDR) P. aeruginosa, they are not uniformly active against A. baumannii.7A. baumannii possesses many clinically diverse β-lactamases from all four classes; the most significant portion of β-lactam resistance in A. baumannii is expressed by class C Acinetobacter-derived cephalosporinases (ADCs), chromosomally encoded β-lactamases responsible for resistance to advanced generation cephalosporins.
In previous work, we systematically evaluated the activity of a series of boronic acids against ADC-7, a representative class C enzyme found in A. baumannii.8,11 Boronic acid transition state inhibitors (BATSIs) are known reversible covalent inhibitors of β-lactamases, due to the electrophilic character of the boronic moiety, which upon attack of the nucleophilic serine residue, forms a tetrahedral adduct with the enzyme.9 Selectivity and high potency of specific BATSIs toward β-lactamases were identified in several studies, by means of changing the substituents on the carbon atom attached to the boron. The first scaffold (A) that proved active against ADC-7 was a chiral α-acylaminoalkaneboronate (Figure 1),10 where the α-carbon atom was substituted by a canonical R1 amide side chain in position C6/C7, typical of penicillins/cephalosporins, and a R2 group bearing a carboxylate, which is always present in position C3/C4 of the same β-lactam antibiotics. To this scaffold belongs compound SM23, the best inhibitor of this series with a Ki of 21 nM for ADC-7.8
Figure 1.

BATSI scaffolds for ADC-7 inhibitors.
A second scaffold (B) was subsequently designed that replaced the amide group with a sulfonamide.11 With this class of derivatives, the natural substrate mimetics of scaffold A (both the R1 and R2 inspired by the β-lactam structures) were advanced into a series of compounds that could better “fit” into the enzyme active site. Compound CR192 from series B demonstrated a Ki of 0.45 nM, proving one of the most potent inhibitors of ADC-7 ever designed. Finally, in series C, the amide/sulfonamide was replaced by a triazole ring. Triazoles are nonclassical amide bioisosteres12 and share with the amide a wide range of properties such as planarity, size, dipole moment, and hydrogen bonding capabilities. Indeed, even though S06017 is a less potent inhibitor (Ki = 6.1 μM) compared to the achiral sulfonamide CR192, the structural information from the X-ray crystal structure of the enzyme–inhibitor complex suggested that the triazole maintained two of the canonical interactions in the amide binding site, thus behaving as a good amide bioisostere.11
Encouraged by the bioisosterism and the easy synthetic access of α-triazolylboronic acids C, we chose to explore the potential of this particular scaffold in the present analysis, specifically 1,2,3-triazoles 1,4-disubstituted that are easily accessible through 1–3-dipolar Cu-catalyzed azide–alkyne cycloaddition (CuAAC).13 Our goal was to use these compounds as molecular probes to elucidate structure activity relationships, SAR. The Cu-based process employs click chemistry, which proceeds in mild conditions, using inexpensive reagents, with high efficiency and simple product isolation. Furthermore, we have already demonstrated the tolerance of boronic esters with CuAAC.14 In this paper, 26 compounds were synthesized and characterized via kinetic analysis and microbiological assays. The extraordinary inhibitory activity against ADC-7 was determined (Ki values spanning from 90 nM to 33 μM) and compared with vaborbactam binding affinity of 0.72 μM (IC50 14.6 μM). Additionally, the X-ray crystal structures of ADC-7 in complex with 5 of these compounds were determined to resolutions ranging from 1.74 to 2.04 Å. Despite being different from the amide, we hypothesized that the triazole would maintain significant potency and selectivity while allowing for easy and straightforward access to a wide variety of derivatives.
Results
Design
The α-triazolylmethaneboronic acid S06017 (Figure 1), described in a previous publication,11 displayed a lower binding affinity to ADC-7 (Ki = 6.1 μM) compared to α-sulfonamido and α-acylamidomethane boronic acids. However, the crystal structure of the ADC-7/S06017 complex showed that the triazole maintained two of the three canonical interactions in the amide binding site, with two nitrogen atoms interacting with Gln120 and Asn152, suggesting that this heterocycle could behave as a good amide bioisostere. Furthermore, the triazolylmethaneboronic acid scaffold was synthetically accessible and allowed for the introduction of different groups at position 4 of the triazole (Scheme 1). For these reasons, our goals in this study were to validate whether the α-triazolylmethaneboronic acid group (i) is a good scaffold for ADC-7 inhibition; (ii) serves as a template for new ADC-7 BATSIs capable of restoring antibiotic activity.
Scheme 1. Synthesis of α-Triazolylmethaneboronate 6a–q.

(i) NaN3, CH3CN, 85 °C; (ii) (+)-pinanediol, SiO2, H2O2, overnight; (iii) a–q, CuSO4, Na ascorbate, t-BuOH/H2O, 2 h, 60 °C; (iv) isobutylboronic acid, HCl, acetonitrile, n-hexane, r.t.
To these ends, we strategically designed four series of α-triazolylmethaneboronic acids (Table 1). In Series I, five compounds (6a–e) contain a phenyl ring directly bound to the triazole with a substituent on the aromatic moiety. Series II consists of three triazoles (6f–h) bearing electron rich and electron poor heterocyclic rings replacing the phenyl, whereas Series III (compounds 6i–l) and IV (compounds 6m–q) introduce a phenyloxymethyl substituent or a substituted aminomethyl bridge on the triazole in order to confer more flexibility to the structures. The substituents for each series are represented in Table 1.
Table 1.

Synthesis
The synthesis of α-triazolylmethaneboronate was successfully performed as depicted in Scheme 1. The commercially available bromomethanetrifluoroborate 1 was reacted with sodium azide in acetonitrile at 85 °C to afford the azidomethanetrifluoroborate 2 in 90% yield.
Conversion of the organotrifluoroborate 2 into the (+)-pinanediol α-azidomethaneboronate 3 was performed in degassed water in the presence of silica gel (1.5 equiv) and a stoichiometric amount of (+)-pinanediol (90% yield).15−17 Compound 3 is one of the partners of CuAAC; the acetylene counterparts 4a–q were conveniently purchased or synthesized following literature procedures (see the Methods). The cyclization reactions were carried out as described.14 The expected 1,4-disubstituted triazoles 5a–q, differently substituted at the R1 group (see Table 1), were easily isolated by extraction and used as such for the next step. Final deprotection of (+)-pinanediol ester 5a–q was accomplished by transesterification with isobutylboronic acid (0.95 equiv) and HCl 3 M (3 equiv) in a biphasic system of acetonitrile/n-hexane, allowing one to obtain final boronic acids 6a–q.
Inhibition Kinetics and Antibiotic Susceptibility (Minimum Inhibitory Concentrations, MICs)
The binding affinities (Ki) for each of the BATSIs with ADC-7 were determined using competition kinetics with nitrocefin (NCF) used as chromophore substrate. The Ki values (average data from 3 experiments) for all BATSIs, corrected for the NCF affinity (Km 20 μM), are reported in Table 2.
Table 2. Binding Affinities (Ki) of Compounds 6a–q and Their Contribution to Ceftazidime (CAZ) Susceptibility (MIC)a.

In contrast, the vaborbactam affinity for ADC-7 is 0.72 ± 0.1μM.
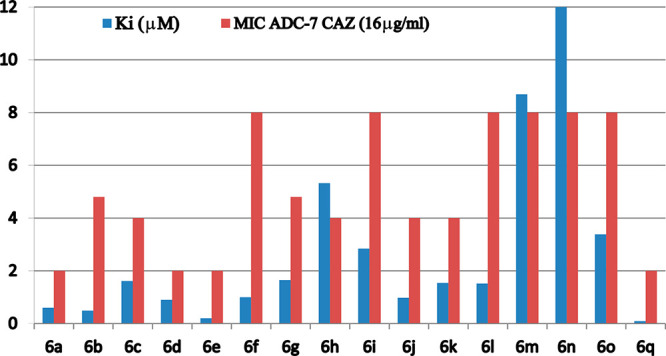
All compounds show inhibition of ADC-7 β-lactamase in the low micromolar range. Compounds from Series I, with an aromatic phenyl ring directly bound to the triazole, exhibit Ki values spanning from 0.2 μM (compound 6e) to 1.6 μM (compound 6c). When the aromatic moiety is a heterocycle (Series II) or a substituted benzyloxy group (Series III), inhibition remains in the low micromolar range, with the thiophene substituent (6f) being the best from Series II (Ki 1.0 μM) and the 3-chlorophenyl (6j), from Series III (Ki 0.98 μM). Compounds from Series IV (6m–q) show the most surprising results, suggesting that the addition of moieties to the triazole to increase flexibility is not always beneficial: activity varies from 33.8 μM for compound 6p having an amide as a bridge between the triazole and the phenyl ring down to as low as 90 nM for 6q, which replaces the amide of 6p with a sulfonamide. This 300-fold difference in activity suggested a possible second round of inhibitor structure refinement. Nevertheless, we wanted to confirm 6q as the best lead compound using a microbiological profile as well.
The inhibition constant (Ki) values and MIC data (Table 2) for compounds 6a–q are plotted in Chart 1. Data show a good agreement between kinetic and antimicrobial activity: the lower the MIC, the higher is the affinity of the compound. Compound 6q proved to be the best compound under both kinetic and microbiological profiles.
Chart 1. Correlation between Synergistic Activity of BATSIs in Combination with CAZ (MICs) against E. coli Expressing blaADC-7 and Their Binding Affinity for Purified ADC-7 Enzyme (Ki).
To further improve the structure of 6q, we designed another series of triazolyl BATSIs (Series V, Figure 2). Nine additional compounds 6r–z were synthesized with the replacement of the tolyl group of 6q with a trifluoroethyl (6r), a thiophene (6s), five different benzyl groups (6t–x), and two p-substituted phenyl groups (6y, 6z).
Figure 2.

General structure and substituents in Series V.
Synthesis of compounds 6r–z followed the same synthetic Scheme 1 (see Methods for detailed description), and their affinity toward ADC-7 (Ki’s) and the enhanced activity with the antibiotic ceftazidime (CAZ) or cefepime (FEP) (MICs) are summarized in Table 3.
Table 3. Binding Affinities (Ki Values) of Series V BATSI Compounds against ADC-7 Enzyme and MIC Values (μg/mL) of CAZ or FEP in Combination with 4 μg/mL of Series V BATSIs.
| comp | Ki [μM] ADC-7 | E. coli, DH10B blaADC-7 | E. coli, DH10B blaCMY-2 | P. aer. 18SH blaPDC | E. coli, DH10B blaCTX-M-9 |
|---|---|---|---|---|---|
| CAZ | 16 | 128 | 64 | ||
| FEP | 8 | ||||
| 6q | 0.09 ± 0.01 | 2 | 16 | 32 | 1 |
| 6r | 1.46 ± 0.2 | 4 | 32 | 32 | 4 |
| 6s | 0.31 ± 0.04 | 2 | 8 | 64 | 2 |
| 6t | 1.02 ± 0.1 | 2 | 32 | 64 | 4 |
| 6u | 0.39 ± 0.3 | 2 | 64 | 64 | 8 |
| 6v | 0.77 ± 0.08 | 2 | 32 | 64 | 4 |
| 6x | 0.31 ± 0.02 | 2 | 32 | 64 | 2 |
| 6w | 0.70 ± 0.08 | 2 | 32 | 32 | 4 |
| 6y | 0.84 ± 0.07 | 2 | 32 | 64 | 4 |
| 6z | 0.21 ± 0.03 | 2 | 64 | 64 | 4 |
All compounds from Series V present nanomolar (Ki6q 90 nM) to low micromolar (Ki6r 1.46 μM) inhibitory activity against ADC-7 β-lactamase. To assess the capability of these compounds to restore β-lactam susceptibility, broth microdilution MICs were performed against three bacterial strains expressing class C β-lactamases and one expressing a class A β-lactamase. The microdilution MICs were performed in 200 μL wells, with BATSI concentrations maintained at 4 μg/mL. The antibiotic partner for class C β-lactamase strains (E. coli DH10B carrying blaCMY-2 and blaADC-7 and P. aeruginosa 18SH strain, blaPDC) was ceftazidime (CAZ) with increasing concentrations from 0.12 to 128 μg/mL. The antibiotic partner for the class A blaCTX-M-9 strain was chosen to be cefepime (FEP) with variable concentration from 0.12 to 128 μg/mL. The addition of boronic acid inhibitors decreased the CAZ MIC for E. coli DH10B blaADC-7 from 16 to 2 μg/mL. When used against E. coli DH10B blaCMY-2, the most potent inhibitors were 6s and 6q, decreasing the MICs for CAZ from 128 to 8 and 16 μg/mL, respectively. All of the other BATSIs decreased the CAZ MICs by 1- or 2-fold. The effect against P. aeruginosa clinical strain 18SH was minimal (only a 1-fold decrease with the addition of 6q, 6r, or 6w). When FEP was paired with 6q, 6s, or 6x, the E. coli DH10B blaCTX-M-9 became susceptible to cefepime (FEP MICs decrease from 8 to 1 or 2 μg/mL). The other compounds lowered the FEP MICs by 1-fold.
Crystallographic Structures of ADC-7/Novel BATSI Complexes
To identify the structural basis for the observed inhibition of ADC-7 by these novel triazole boronic acids as well as to confirm the triazole functionality as a bioisostere for the R1 amide group found in the natural β-lactam substrates, X-ray crystal structures of five ADC-7/BATSI complexes were determined. Two compounds from Series I (Table 1, 6d and 6e), one compound from Series II (6f), the most effective inhibitor (6q), and one from Series V (6r) were selected for crystallographic analysis.
The ADC-7/BATSI complexes were determined to resolutions ranging from 1.80 to 2.04 Å (Table 4). In summary, all complexes crystallized in the P21 space group with four molecules in the asymmetric unit, as previously observed for ADC-7/BATSI complexes.8,10,11 The quality of the final models was evaluated with the wwPDB validation service18 and showed that 96–98% of all residues were in the favorable region, with 2–4% in the allowed region, of the Ramachandran plots. The complexes with 6f, 6q, and 6r were obtained by soaking ADC-7 crystals in inhibitor solutions, and the complexes with 6d and 6e were obtained through cocrystallization.
Table 4. Crystallographic Summary for ADC-7/Boronic Acid Complexes.
| ADC-7/6d | ADC-7/6e | ADC-7/6f | ADC-7/6q | ADC-7/6r | |
|---|---|---|---|---|---|
| cell constants (Å; deg) | a = 89.62 | a = 88.77 | a = 88.55 | a = 88.48 | a = 88.93 |
| b = 80.78 | b = 81.25 | b = 81.46 | b = 80.62 | b = 81.10 | |
| c = 107.00 | c = 105.92 | c = 105.67 | c = 105.11 | c = 105.94 | |
| β = 112.47 | β = 112.93 | β = 113.10 | β = 113.46 | β = 113.06 | |
| space group | P21 | P21 | P21 | P21 | P21 |
| resolution (Å) | 98.88–1.96 (1.964–1.957)a | 97.55–1.82 (1.822–1.816) | 97.20–2.04 (2.042–2.035) | 50.00–1.80 (1.86–1.80) | 81.82–1.74 (1.837–1.746) |
| unique reflections | 96293 (988) | 123054 (1239) | 86979 (870) | 125707 (12526) | 116473 (5826) |
| Rmerge (%) | 5.0 (41.5) | 5.4 (46.6) | 7.2 (64.7) | 9.0 (71.4) | 8.6 (57.1) |
| Rpim (%) | 2.9 (23.5) | 3.1 (26.5) | 4.3 (37.8) | 4.9 (39.2) | 5.6 (39.3) |
| CC(1/2) | 0.999 (0.929) | 0.998 (0.905) | 0.995 (0.750) | 0.940 (0.760) | 0.995 (0.647) |
| completeness (%) | 94.6 (98.9) | 98.4 (98.8) | 98.0 (98.6) | 100.0 (100.0) | 89.1 (50.0) |
| ⟨I/σI⟩ | 13.3 (2.0) | 11.4 (1.9) | 11.1 (2.1) | 8.48 (2.6) | 8.0 (1.5) |
| resolution range for refinement (Å) | 98.88–1.96 | 97.55–1.82 | 97.19–2.04 | 44.26–1.80 | 81.95–1.74 |
| number of protein residues | 1424 | 1422 | 1422 | 1425 | 1423 |
| number of water molecules | 330 | 488 | 236 | 694 | 871 |
| RMSD bond lengths (Å) | 0.005 | 0.005 | 0.006 | 0.008 | 0.007 |
| RMSD bond angles (deg) | 1.38 | 1.30 | 1.53 | 1.51 | 1.52 |
| R-factor (%) | 21.9 | 22.0 | 22.1 | 19.4 | 21.6 |
| Rfree (%)b | 25.1 | 25.6 | 25.4 | 23.8 | 27.3 |
| average B-factor, protein atoms (Å2) | 44.77 | 47.41 | 46.86 | 35.54 | 40.68 |
| average B-factor, inhibitor atom (Å2) | 58.36 | 66.93 | 48.98 | 58.3 | 54.82 |
Values in parentheses are for the highest resolution shell.
Rfree was calculated with 5% of reflections set aside randomly.
In each case, the initial Fo – Fc electron density maps (contoured at 3σ) revealed unambiguous density that accounted for the presence of the inhibitor bound in the active site as well as covalent attachment to the catalytic Ser64. Inhibitors were built into the observed difference density, and the models were refined with Refmac5.19 PDB-REDO was used to analyze and improve models between rounds of manual rebuilding in Coot.20Fo – Fc omit maps were calculated for the final models (Figure 3) and confirmed the conformation of the inhibitor in the active site. In each complex, the four monomers were superposed, with RMSDs of all common Cα atoms ranging from 0.18 to 0.46 Å. The inhibitors within each complex bound in consistent conformations. For simplicity, the B monomer is used in figures and is representative of all monomers, although some differences are described in more detail below.
Figure 3.
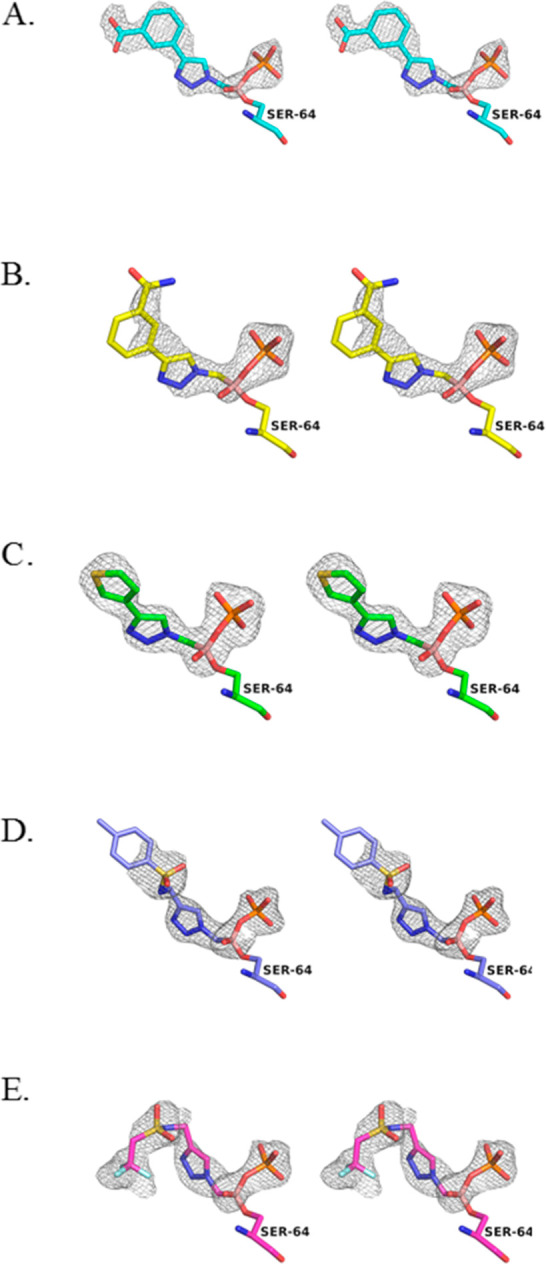
Stereoview of the Fo – Fc omit maps for the ADC-7/BATSI complexes. (A) 6d. (B) 6e. (C) 6f. (D) 6q. (E) 6r. This and all subsequent figures were made with PyMOL.21 Omit maps are contoured at 3.0 σ and displayed as a gray cage surrounding the inhibitor. Carbon atoms are colored cyan for 6d, yellow for 6e, green for 6f, purple for 6q, and magenta for 6r. Oxygen atoms, red; nitrogen, blue; boron atoms, pale pink; fluorine, pale cyan; phosphorus atoms, orange; sulfur, yellow.
The boronic acid moiety interacts as expected with the enzyme in most of the complexes (Figure 4). The O1 hydroxyl group is observed to hydrogen bond with residues that comprise the oxyanion hole (main chain nitrogens of Ser64 and Ser315 and the main chain carbonyl oxygen of Ser315). However, in the complexes with 6e and 6f (Figure 4B,C), only the interactions with the main chain nitrogen of Ser64 and the main chain oxygen of Ser315 are observed. The O2 atom of the boronic acid is modified with a covalently bound phosphate ion, as has been observed in several other ADC-7/BATSI complexes where the BATSI lacks an R2 group.11 The triazole ring of each of the inhibitors is also observed to interact in a similar fashion in each of the complexes. All complexes exhibited the expected hydrogen bonds between atoms N6 and N7 of the triazole ring and the side chain nitrogens of Gln120 and Asn152, albeit with some variations in the distances. Most were between 2.6 and 3.2 Å, although several were slightly longer (3.4–3.6 Å). Overall, these five structures confirm that, in ADC-7, the triazole is an effective amide bioisostere.
Figure 4.
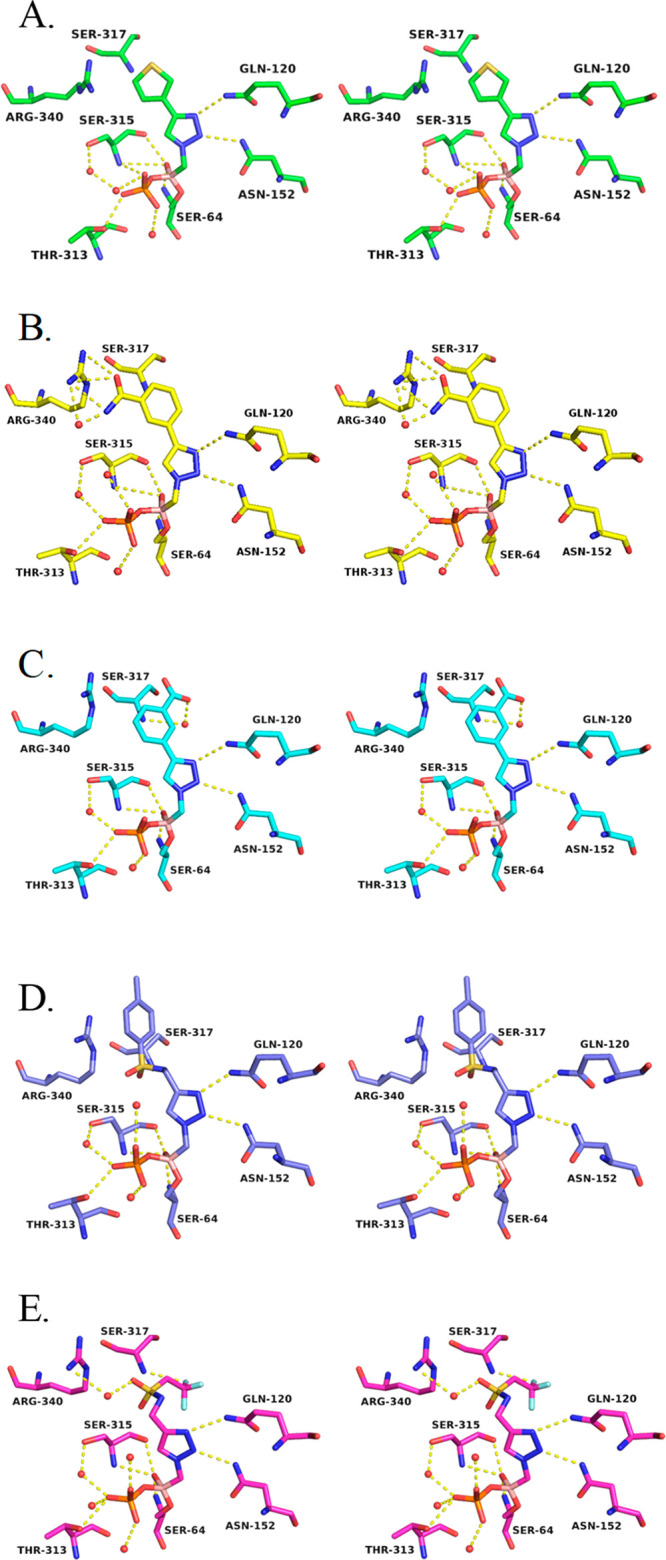
Stereoview of the hydrogen bonding interactions in the ADC-7/BATSI complexes. (A) 6f. (B) 6e. (C) 6d. (D) 6q. (E) 6r. Hydrogen bonding interactions are shown as dashed yellow lines and represent distances from 2.6 to 3.2 Å. Water molecules are shown as red spheres.
Specific Characteristics of the ADC-7/Series I Complexes
In each of the monomers of the ADC-7/6d and ADC-7/6e complexes (Figure 4C,B), the inhibitor is bound in the active site in similar conformations and follows a similar trajectory. The most variability is observed at the distal end of the inhibitors. In ADC-7/6d (Figure 4C), the linear trajectory of the inhibitor orients the benzoate group toward the lip of the active site, with the carboxylate group making a hydrogen bond with the main chain nitrogen of Ser317, via a water molecule, whereas in the ADC-7/6e complex (Figure 4B), the placement of the benzamide group is seen in two distinct conformations. In one (B and C monomers), the benzamide group is oriented toward Arg340, with the benzamide oxygen making a long hydrogen bond with this residue (3.2 Å). In the other (A and D monomers), the benzyl group is rotated ∼180° with the benzamide oriented away from Arg340.
Specific Characteristics of the ADC-7/Series II Complex
In the ADC-7/6f complex (Figure 4A), the inhibitor binds in the same conformation in all active sites of the four monomers. A thiophene replaces the aryl ring of Series I compounds at the distal end but does not make favorable interactions with the enzyme. Additionally, the shorter length of this inhibitor does not extend to the lip of the active site, where interactions with Ser317 were observed in the Series I complexes, and Arg340 is oriented toward the active site, likely due to the smaller sized inhibitor.
Specific Characteristics of the ADC-7/Series IV Complex
Series IV explored more flexible groups that extend from the triazole ring. The sulfonamide linker (Figure 4D) displayed the best inhibition of all molecules tested from any of the series (Ki6q 90 nM). Interestingly, the sulfonamide group itself does not make any favorable interactions with the enzyme. Arg340 is positioned out of the active site, with the distal tolyl group making favorable cation−π interactions with this residue. Distances from Arg340 to the centroid of the aryl ring range from 3.8 to 4.4 Å.
Specific Characteristics of the ADC-7/Series V Complex
To improve the binding affinity of 6q, Series V molecules were designed. The structure of ADC-7 in complex with 6r (Figure 4E), which replaces the tolyl group with a trifluoromethyl, was determined. The sulfonamide is oriented near Arg340 but is not within hydrogen bonding distance in monomers A and B, where Arg340 is swung out away from the active site. However, in monomers C and D, Arg340 adopts a conformation that positions it into the active site, and in these instances, the sulfone interacts with this residue (2.5–2.9 Å). The distal trifluoromethyl substituent is bent away from Arg340 and does not favorably interact with any residues in the active site.
Discussion and Conclusion
This study explores the α-triazolylmethaneboronic acid scaffold as a good template for ADC β-lactamase inhibition. Whereas boronic acids have been identified as protease inhibitors since the 1970s, only in the past decade has this class of compounds been recognized as effective “bullets” in the antimicrobial resistance arsenal.
α-Acylaminoboronic acids (Figure 1A) have been designed as a good starting point to gain the proper interactions with the enzyme. Indeed, several crystal structures of different β-lactamase/α-amidomethaneboronic acid complexes point to the presence of an amide binding site with specific enzyme residues always interacting with the amide. In previous work on ADC-7,11 the triazole-containing compound S06017 (Figure 1) was synthesized, tested, and cocrystallized with the enzyme.
From the crystal structure, we observed that the triazole could behave as a good amide bioisostere, with two lone pair nitrogens able to interact with the canonical R1 amide recognition residues Asn152 and Gln120 that hydrogen bond with the two lone pairs of the amide oxygen. Given the easy and mild access to the triazole ring, with wide functional group tolerance, we wanted to prove triazole is a good amide bioisostere and to improve the activity of S06017 (Ki 6.11 μM). Therefore, in this work, we designed and synthesized 26 α-triazolylmethaneboronic acids, differing the substituent at position 4 of the triazole. The Ki values of these compounds vary from 90 nM to 38 μM, thus confirming a good general affinity for the enzyme and a consistent difference in activity due to insertion of varying functional groups.
Compounds with a substituted phenyl ring directly attached to the triazole (Series I, compounds 6a–e) proved to be very active with Ki values spanning from 200 nM to 1.61 μM. Two compounds (6d and 6e) from this series were crystallized in complex with ADC-7: these complexes confirmed that the triazole makes two of the three canonical interactions of the β-lactam side chain, thus behaving as a good amide bioisostere. Furthermore, from the crystal structure, the benzamide carbonyl oxygen of the best inhibitor from this series, 6e (Ki 200 nM), makes a hydrogen bond with Arg340 (3.2 Å), suggesting the role that interactions with Arg340 may play in increasing binding affinity for these BATSIs. With 6d, the carboxylate group of the benzoate is flipped ∼180° from the benzamide, positioning the negatively charged group away from Arg340. A favorable ionic interaction might be expected between these groups in the other conformation, but rotation of the benzoate results in a steric clash between the two. Therefore, the carboxylate group is instead oriented toward the solvent.
The replacement of the substituted phenyl ring with an electron rich (i.e., the thiophene in 6f) or electron poor (the pyridine and pyrazine in 6g and 6h, respectively) heterocycle maintain a similar level of activity (Ki’s from 1 to 5.3 μM). From this Series II, the structure of the enzyme in complex with compound 6f was superposed with the ADC-7/6e complex (Figure 5). The two compounds have a 5-fold difference in activity (Ki of 1 μM for 6f vs 200 nM for 6e): indeed, the thiophene ring is placed in the same position as the phenyl ring from Series I and does not take advantage of any specific interaction with the enzyme. The most distinctive difference between the two structures is the positioning of Arg340, a residue which exhibits flexibility: ADC-7/6f shows Arg340 oriented toward the active site in the presence of the smaller thiophene inhibitor. In contrast, the ADC-7/6e complex (yellow) shows Arg340 oriented away from the active site to accommodate the binding of a larger inhibitor and to be positioned at a proper distance for hydrogen bonding.
Figure 5.

Flexibility of Arg340 in inhibitor recognition. Superposition of ADC-7 in complex with 6f (green) and 6e (yellow).
In an attempt to gain interactions with Arg340, Series III and IV were synthesized to elongate the substituent on the triazole. The addition of a substituted phenyloxymethyl linker as in Series III (compounds 6i–l) did not significantly improve activity (Ki’s from 0.98 to 2.84 μM), whereas the substituted aminomethyl bridge exploited the most significant differences. In Series IV, activity in fact dramatically dropped when a protonated aminomethyl (compound 6m) or acylamino side chain (compounds 6n and 6p) was introduced (Ki’s from 8.7 to 33.8 μM). In contrast, compound 6q with a p-tolylsulfonylamino substituent displayed the best activity among the α-triazolyl BATSIs (Ki 90 nM), pointing to 6q as one of the best achiral inhibitors of class C β-lactamases. The analysis of the ADC-7/6q complex revealed how the tetrahedral geometry of the sulfonamide, as in 6q, allows for cation−π interactions with Arg340 (Figure 6), which is probably not reached when a planar geometry is introduced through an amide linker as in 6p.
Figure 6.
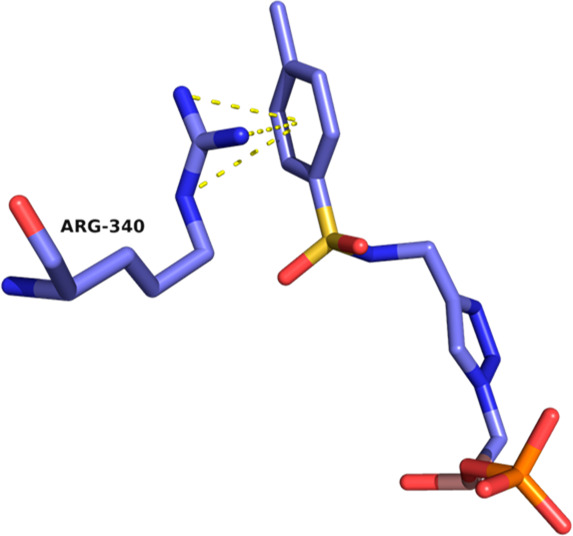
Cation−π interactions between Arg340 and the aryl ring of 6q, the BATSI with the highest affinity to ADC-7. Interactions, indicated by dashed yellow lines, are drawn from Arg340 to the centroid of the aryl ring, with distances ranging from 3.8 to 4.4 Å.
Notably, the structure of 6q does not resemble the natural substrate of the β-lactamase but displays a pronounced inhibition activity. In fact, when compared to α-acylaminomethaneboronic acids previously synthesized22 (Figure 7), compound 6q (Ki 90 nM) is 3 times more active than the boronic acid bearing the ceftazidime side chain (Ki 310 nM) and almost 9 times more active than the cephalothin analog (Ki 780 nM). The activity of the α-triazolylboronic acid is significantly less compared to the one of the α-sulfonylaminomethaneboronic acids bearing a distal tetrazole in the side chain (compound CR192). From the structural analysis of such derivatives, it became evident how the increase in activity was due to the interaction of the negatively charged tetrazole with a distal binding site formed by Asn213 and Ser317.
Figure 7.
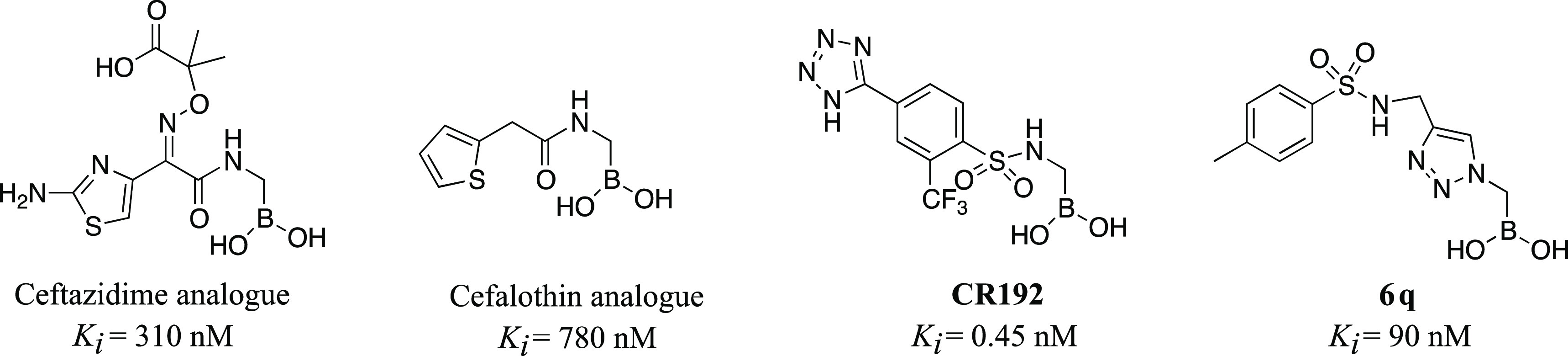
Structures and Ki values of previously synthesized α-acylaminoboronic acids.
Given the length and trajectory of 6q, the distal functional groups of this molecule do not extend to the outer edge of the active site where Asn213 is located. However, the Cβ atom of Ser317 is within the van der Waals distance of the aryl ring of 6q (∼4.3–4.5 Å), thus giving the opportunity for further optimizing the molecule. A validation of the α-triazolylmethaneboronic structure of 6q as a template for further derivatization was obtained through microbiological assays in E. coli expressing ADC-7 of compounds 6a–p. All compounds lowered the MIC (16 μg/mL) of CAZ from 1- to 4-fold, and the MIC values were in good agreement with Ki’s (Chart 1), thus confirming a good permeability of these compounds.
In an attempt to improve 6q activity and eventually reach the distal binding site of ADC-7, we obtained an additional nine compounds (Figure 2, Series V, compounds 6r–z). Unfortunately, none of the compounds of Series V improve activity toward ADC-7 with Ki’s spanning from 0.21 to 1.46 μM (Table 3). Compound 6r was crystallized in complex with ADC-7. In 6r, the tolyl group of 6q is replaced with a trifluoroethyl group, which is unable to make the cation−π interaction seen in the ADC-7/6q complex: Arg340 in fact points away, likely resulting in lower binding affinity of the compound. Compounds 6t–x all contain a methylene linker that may extend the distal group away from Arg340 and prevent this interaction, resulting in lower binding affinities as well. Compounds 6y and 6z more closely resemble 6q and 6r as they lack the flexible methylene linker. Compound 6y contains a cyano group as compared to the tetrazole of 6z, which might impact the ability of the aryl rings to form cation−π interactions with Arg340. Overall, the lower binding affinities of Series V might point to the inability of reaching the distal binding site (Arg213 and Ser317) and at the same time to the loss of interaction with Arg340, which is a residue that is unique to the ADC enzymes as compared to other class C β-lactamases. Known to be a contributor to protein–protein and protein–ligand interactions, the cation−π interaction observed in these ADC-7/inhibitor complexes suggests that it is important for the design of future series. Arg340 may be a key residue to target as it is unique to this class of enzymes and has shown the ability to interact with a variety of different functional groups (amide, carboxylate, trifluoromethyl, phenyl) in a variety of different interactions, such as Coulombic, ionic, hydrogen bond, and cation−π. In addition, the flexibility shown by Arg340 allows ADC-7 to accommodate BATSIs with larger R1 groups that are able to reach the residues at the lip of the active site (such as Asn213 and Ser317).
In summary, when the highly efficient and versatile synthetic method known as click chemistry is employed, a new class of β-lactamases inhibitors has been synthesized, starting from the easily accessible pinanediol azidomethaneboronate. All 26 BATSIs displayed Ki values spanning from low micromolar to nanomolar values, with compound 6q being among the best achiral inhibitors of the class C β-lactamases. Five of these inhibitors were crystallized in complex with ADC-7 revealing that, besides the interaction of the boronic moiety with the catalytic serine residue, the triazole is able to maintain the typical interactions of the extensively explored and parent amidomethaneboronic inhibitors, thus acting as a good amide bioisostere. Finally, this new class of inhibitors proved to be able to restore CAZ and FEP activity against class C and A β-lactamase strains.
Methods
Synthesis
Reactions were monitored by thin layer chromatography (TLC), which were visualized by UV fluorescence and by Hanessian’s cerium molybdate stain. Deoxygenated water was obtained through sonication. Chromatographic purification of the compounds was performed on silica gel (particle size 0.05–0.20 mm). Melting points were measured in open capillary tubes on a Stuart SMP30 Melting Point apparatus. Optical rotations were determined at 20 °C on a PerkinElmer 241 polarimeter and are expressed in 10–1 deg cm2 g–1. 1H and 13C NMR spectra were recorded on a Bruker Avance-400 MHz spectrometer. Chemical shifts (δ) are reported in ppm and were calibrated to the residual signals of the deuterated solvent.21 Multiplicity is given as s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, and br = broad signal; coupling constants (J) are given in Hz. Two-dimensional NMR techniques (COSY, HMBC, HSQC) were used to aid in the assignment of signals in 1H and 13C spectra. Particularly, in the 13C spectra, the signal of the boron-bearing carbon atom, which tends to be broadened, and the signal of the quaternary triazole carbon are often beyond the detection limit, but their resonances were unambiguously determined by HSQC and HMBC; melting points of free boronic acids 6b–z were not reproducible due to dehydration.23 Mass spectra were determined on an Agilent Technologies LC-MS (n) Ion Trap 6310A (ESI, 70 eV). High-resolution mass spectra were recorded on an Agilent Technologies 6520 Accurate-Mass Q-TOF LC/MS.
The purity of all tested compounds was above 95%, determined by analytical HPLC-MS (see the Supporting Information for a detailed description). Synthesis and characterization of compounds 2, 3, 4b–e, 4g–h, 4j–w, 4y, 5b–e, 5g–h, 5j–w, 5y, 5z, 6b–e, 6g–h, and 6j–z are reported in the Supporting Information.
Microbiology
MICs were performed as previously described7 and according to Clinical and Laboratory Standards Institute (CLSI) guidelines,25 using a 6 × 104 cfu/mL inoculum. Bacterial cultures were grown overnight in Mueller-Hinton (MH) broth supplemented with 20 μg/mL chloramphenicol. We employed the E. coli construct that was previously validated as a representative of ADC-7 in a uniform genetic background (blaADC-7 was directionally cloned in pBC SK (−) phagemid vector). Bacterial liquid culture was diluted using MH broth to a 6 × 104 cfu/mL final concentration, and the antibiotic partner, CAZ or FEP, was added at concentrations from 128 to 0.06 μg/mL. BATSIs were constant at 4 μg/mL. The plates were incubated at 37 °C overnight, and the results were recorded the next day.
Purification and Kinetics
ADC-7 β-lactamase was expressed as previously described8 and purified using cation exchange chromatography. For the purification of ADC-7, cell pellets were suspended in 25 mM 3-(N-morpholino) propanesulfonic acid (MOPS buffer), pH 6.5, with 1× HALT protease inhibitor cocktail (Sigma) and DNase I (50 Units). The solution was sonicated for 4 × 30 s intervals on ice. The lysate was centrifuged at 15 000 rpm at 4 °C for 20 min. The cell-free extract was then loaded onto a carboxymethyl-cellulose column by gravity flow at 4 °C (5 mL resin per gram of cell pellet). The column was washed with 100 mL of 25 mM MOPS, pH 6.5, at a flow rate of 0.3 mL/min followed by elution with a linear gradient of 0–0.5 M NaCl in 25 mM MOPS, pH 6.5. The fractions containing ADC-7 were collected, pooled, and then dialyzed in 2 × 5 L of 25 mM MOPS, pH 6.5 at 4 °C. The dialyzed ADC-7 was concentrated to at least 10 mg/mL using an Amicon Ultra centrifugal filter unit with Ultra-10 membrane (Millipore). The concentration of ADC-7 was determined using the A280 with an extinction coefficient of 46 300 M–1cm–1, as calculated for the expressed residues D24-K383 of ADC-7 by the ProtParam tool on the ExPASy bioinformatics portal.24
The inhibition constants (Ki) for each of the BATSIs with ADC-7 were determined using competition kinetics. When nitrocefin (NCF) was utilized as a colorimetric substrate of ADC-7, boronic acids 7a–q were tested as inhibitors of ADC-7 β-lactamase as previously described.8,10,11 The measurements of the initial velocities were performed with the addition of 100 μM NCF after a 5 min preincubation of the enzyme (2 nM) with increasing concentration of the inhibitor. To determine the average velocities (v0), data from three experiments were fit to the equation:
where vu represents the NCF uninhibited velocity and IC50 represents the inhibitor concentration that results in a 50% reduction of vu. The Ki values for all 26 BATSIs were corrected for the NCF affinity (Km = 20 μM) with the Cheng-Prusoff30 equation:
The data analysis was performed using EnzFitter and Origin 2019b.
Crystallization and Structure Determination
Structures of ADC-7 in complexes with the inhibitors were obtained via both soaking and cocrystallization methods. For soaks, ADC-7 crystals were grown via hanging drop vapor diffusion at room temperature as previously described.11 Preformed crystals were harvested using a nylon loop and soaked in crystallization buffer containing the BATSI at concentrations ranging from 2 to 16 mM for between 5 and 25 min. Co-crystals were grown in 0.1 M succinate/phosphate/glycine (SPG buffer), pH 5.0, 25% w/v PEG-1500, with 3.5–3.75 mg/mL ADC-7 and 1 mM BATSIs in the initial crystallization buffer.
Data for each of the complexes were measured from single crystals at the Advanced Photon Source at Argonne National Laboratory (LS-CAT sector). All diffraction images were processed with XDS25 with the exception of the ADC-7/6q data set, where HKL200026 was used. For the ADC-7/6r data set, additional processing of the structure factors was performed using STARANISO.27 Structures were determined by molecular replacement with Phaser,28 using the ADC-7/S02030 complex (PDB 4U0X), with water, ion, and inhibitor atoms removed, as the starting model. Refinement of the models was done with Refmac5 in the CCP4 suite,29 and model building was done with Coot.20b The coordinates and structure factors for the ADC-7/BATSI complexes were deposited in the Protein Data Bank with the following codes: 6TZF (6d), 6TZG (6e), 6TZH (6f), 6TZI (6r), and 6TZJ (6q).
Acknowledgments
Research reported in this publication was supported in part by facilities and funds provided by Department of Life Sciences, University of Modena and Reggio Emilia, to E.C. (FAR 2016) and by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health (NIH) to R.A.B. under Award Numbers R01AI100560, R01AI063517, and R01AI072219. This study was also supported in part by funds and/or facilities provided by the Cleveland Department of Veterans Affairs, Award Number 1I01BX001974, to R.A.B. from the Biomedical Laboratory Research & Development Service of the VA Office of Research and Development and the Geriatric Research Education and Clinical Center VISN 10. E.R.F. was supported as a Beckman Scholar, provided by the Arnold and Mabel Beckman Foundation and administered by the Office of Undergraduate Research and Scholarship at GVSU. X-ray data was measured at the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility, operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH or the Department of Veterans Affairs. E.C., F.F., M.L.I., M.S., and F.P. thank Diego Pinetti, Maria Cecilia Rossi, and Cinzia Restani of the Centro Interdipartimentale Grandi Strumenti (CIGS) for supervision in high resolution mass spectra and NMR.
Glossary
Abbreviations
- BATSI
boronic acid transition state inhibitors
- ADC
Acinetobacter derived cephalosporinase
- MICs
minimum inhibitory concentrations
- CAZ
ceftazidime
- FEP
cefepime
- HSQC
heteronuclear single-quantum coherence
- HMBC
heteronuclear multiple-bond correlation
- LC/MS
liquid chromatography/mass spectrometry
- CuAAC
copper-catalyzed alkyne azide cycloaddition
- t-BuOH
tert-butanol
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsinfecdis.0c00254.
Synthesis and characterization of compounds 2, 3, 4a–z, 5b–z, and 6b–z; a statement of purity of compounds 6b–z; copies of 1H and 13C NMR spectra of compounds 6b–z (PDF)
Author Contributions
E.C., M.S., M.L.I., F.F., and F.P. synthesized and characterized all of the BATSI compounds. M.A.T. and R.A.B. performed microbiological assays and kinetics. R.A.P., B.J.W., E.R.F., and K.A.S. determined all of the crystal structures. P.N.R. contributed to the analysis of the data. E.C. wrote the first draft of the manuscript; all authors have contributed and have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Zhen X.; Stalsby Lundborg C.; Sun X.; Hu X.; Dong H. (2019) Economic burden of antibiotic resistance in ESKAPE organisms: a systematic review. Antimicrobial Resistance and Infection Control 8, 137. 10.1186/s13756-019-0590-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tacconelli E.; Carrara E.; Savoldi A.; Harbarth S.; Mendelson M.; Monnet D. L.; Pulcini C.; Kahlmeter G.; Kluytmans J.; Carmeli Y.; Ouellette M.; Outterson K.; Patel J.; Cavaleri M.; Cox E. M.; Houchens C. R.; Grayson M. L.; Hansen P.; Singh N.; Theuretzbacher U.; Magrini N.; et al. (2018) Discovery, research, and development of new antibiotics: the WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 18, 318–327. 10.1016/S1473-3099(17)30753-3. [DOI] [PubMed] [Google Scholar]
- Wong D.; Nielsen T. B.; Bonomo R. A.; Pantapalangkoor P.; Luna B.; Spellberg B. (2017) Clinical and Pathophysiological Overview of Acinetobacter Infections: a Century of Challenges. Clin. Microbiol. Rev. 30, 409–447. 10.1128/CMR.00058-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isler B.; Doi Y.; Bonomo R. A.; Peterson D. L. (2019) New Treatment Options against Carbapenem-Resistant Acinetobacter baumannii Infections. Antimicrob. Agents Chemother. 63, e01110–18. 10.1128/AAC.01110-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosley J. F.; Smith L. L.; Parke C. K.; Brown J. A.; Wilson A. L.; Gibbs L. V. (2016) Ceftazidime-Avibactam (Avycaz): For the Treatment of Complicated Intra-Abdominal and Urinary Tract Infections. Pharmacy and Therapeutics 41, 479–483. [PMC free article] [PubMed] [Google Scholar]
- Griffith D. C.; Sabet M.; Tarazi Z.; Lomovskaya O.; Dudley M. N. (2019) Pharmacokinetics/Pharmacodynamics of Vaborbactam, a Novel Beta-Lactamase Inhibitor, in Combination with Meropenem. Antimicrob. Agents Chemother. 63, e01659-18. 10.1128/AAC.01659-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong D.; van Duin D. (2017) Novel Beta-Lactamase inhibitors: Unlocking Their Potential in Therapy. Drugs 77, 615–628. 10.1007/s40265-017-0725-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caselli E.; Romagnoli C.; Powers R. A.; Taracila M. A.; Bouza A. A.; Swanson H. C.; Smolen K. A.; Fini F.; Wallar B. J.; Bonomo R. A.; Prati F. (2018) Inhibition of Acinetobacter-Derived Cephalosporinase: Exploring the Carboxylate Recognition Site Using Novel β-Lactamase Inhibitors. ACS Infect. Dis. 4, 337–348. 10.1021/acsinfecdis.7b00153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crompton I. E.; Cuthbert B. K.; Lowe G.; Waley S. G. (1988) β-lactamase inhibitors. The inhibition of serine β-lactamases by specific boronic acids. Biochem. J. 251, 453–459. 10.1042/bj2510453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers R. A.; Swanson H. C.; Taracila M. A.; Florek N. W.; Romagnoli C.; Caselli E.; Prati F.; Bonomo R. A.; Wallar B. J. (2014) Biochemical and Structural Analysis of Inhibitors Targeting the ADC-7 Cephaloporinases of Acinetobacter Baumanii. Biochemistry 53, 7670–7679. 10.1021/bi500887n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouza A. A.; Swanson H. C.; Smolen K. A.; VanDine A. L.; Taracila M. A.; Romagnoli C.; Caselli E.; Prati F.; Bonomo R. A.; Powers R. A.; Wallar B. J. (2018) Structure-Based Analysis of Boronic Acids as Inhibitors of Acinetobacter-Derived Cephalosporinase-7, a Unique Class C β-Lactamase. ACS Infect. Dis. 4, 325–336. 10.1021/acsinfecdis.7b00152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Li H.; Aneja R.; Chaiken I. (2013) Molecules 18, 9797–9817. 10.3390/molecules18089797. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Tron G. C.; Pirali T.; Billington R. A.; Canonico P. L.; Sorba G.; Genazzani A. A. (2008) Med. Res. Rev. 28, 278–308. 10.1002/med.20107. [DOI] [PubMed] [Google Scholar]; c Hou J.; Liu X.; Shen J.; Zhao G.; Wang P. G. (2012) Expert Opin. Drug Discovery 7, 489–501. 10.1517/17460441.2012.682725. [DOI] [PubMed] [Google Scholar]; d Lauria A.; Delisi R.; Mingoia F.; Terenzi A.; Martorana A.; Barone G.; Almerico A. M. (2014) Eur. J. Org. Chem. 2014, 3289–3306. 10.1002/ejoc.201301695. [DOI] [Google Scholar]; e Bonandi E.; Christodoulou M. S.; Fumagalli G.; Perdicchia D.; Rastelli G.; Passarella D. (2017) The 1,2,3-triazole ring as bioisostere in medicinal chemistry. Drug Discovery Today 22, 1572–1581. 10.1016/j.drudis.2017.05.014. [DOI] [PubMed] [Google Scholar]
- a Meldal M.; Tornøe C. W. (2008) Cu-catalyzed azide-alkyne cycloaddition. Chem. Rev. 108, 2952–3015. 10.1021/cr0783479. [DOI] [PubMed] [Google Scholar]; b Rostovtsev V. V.; Green L. G.; Fokin V. V.; Sharpless K. B. (2002) A stepwise Huisgen cycloaddition process: copper(I)-catalyzed regioselective ligation of azides and terminal alkynes. Angew. Chem., Int. Ed. 41, 2596–2599. . [DOI] [PubMed] [Google Scholar]
- a Caselli E.; Romagnoli C.; Vahabi R.; Taracila M. A.; Bonomo R. A.; Prati F. (2015) Click Chemistry in Lead Optimization of Boronic Acids as β-Lactamase Inhibitors. J. Med. Chem. 58, 5445–58. 10.1021/acs.jmedchem.5b00341. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Romagnoli C.; Caselli E.; Prati F. (2015) Synthesis of 1,2,3-triazol-1-yl-methaneboronic acids via click chemistry: an easy access to a new potential scaffold for protease inhibitors. Eur. J. Org. Chem. 2015, 1075–1083. 10.1002/ejoc.201403408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molander G. A.; Cavalcanti L. N.; Canturk B.; Pan P.-S.; Kennedy L. E. (2009) Efficient Hydrolysis of Organotrifluoroborates via Silica Gel and Water. J. Org. Chem. 74, 7364–7369. 10.1021/jo901441u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabalka G. W.; Coltuclu V. (2009) Thermal and Microwave Hydrolysis of Organotrifluoroborates Mediated by Alumina. Tetrahedron Lett. 50, 6271–6272. 10.1016/j.tetlet.2009.09.008. [DOI] [Google Scholar]
- Yuen A. K. L.; Hutton C. A. (2005) Deprotection of Pinacolyl Boronate Esters via Hydrolysis of Intermediate Potassium Trifluoroborates. Tetrahedron Lett. 46, 7899–7903. 10.1016/j.tetlet.2005.09.101. [DOI] [Google Scholar]
- Berman H.; Henrick K.; Nakamura H. (2003) Announcing the worldwide Protein Data Bank. Nat. Struct. Mol. Biol. 10, 980. 10.1038/nsb1203-980. [DOI] [PubMed] [Google Scholar]
- Murshudov G. N.; Vagin A. A.; Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr., Sect. D: Biol. Crystallogr. 53, 240–255. 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- a Joosten R. P.; Long F.; Murshudov G. N.; Perrakis A. (2014) The PDB_REDO server for macromolecular structure model optimization. IUCrJ 1 (Pt 4), 213–220. 10.1107/S2052252514009324. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Emsley P.; Cowtan K. (2004) Coot: Model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 60, 2126–2132. 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- (2010) The PyMOL Molecular Graphics System, Version 1.3, Schrodinger, LLC, New York.
- Drawz S. M.; Babic M.; Bethel C. R.; Taracila M.; Distler A. M.; Ori C.; Caselli E.; Prati F.; Bonomo R. A. (2010) Inhibition of the class C beta-lactamase from Acinetobacter spp.: insights into effective inhibitor design. Biochemistry 49, 329–40. 10.1021/bi9015988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall D. G., Ed. (2005) Boronic Acids, Wiley-VCH. [Google Scholar]
- Gasteiger E., Hoogland C., Gattiker A., Duvaud S., Wilkins M. R., Appel R. D., and Bairoch A. (2005) Protein Identification and Analysis Tools on the ExPASy Server. In The Proteomics Protocols Handbook (Walker J. M., Ed.), pp 571–607, Humana Press. [Google Scholar]
- Kabsch W. (2010) Acta Crystallogr., Sect. D: Biol. Crystallogr. 66, 125–132. 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z.; Minor W. (1997) Methods Enzymol. 276, 307–326. 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Tickle I. J., Flensburg C., Keller P., Paciorek W., Sharff A., Vonrhein C., and Bricogne G. (2018) STARANISO, Global Phasing Ltd., Cambridge, United Kingdom: (http://staraniso.globalphasing.org/cgi-bin/staraniso.cgi).
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674. 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov G. N.; Vagin A. A.; Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr., Sect. D: Biol. Crystallogr. 53, 240–255. 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Cheng Y.-C.; Prusoff W. H. (1973) Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50% inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 22 (23), 3099–3108. 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.