

Supplemental Digital Content is available in the text
Keywords: Fluid resuscitation, molecular hydrogen, syndecan-1, TNF-α, vascular permeability, xanthine oxidoreductase
ABSTRACT
Background:
Hydrogen gas (H2) inhalation during hemorrhage stabilizes post-resuscitation hemodynamics, improving short-term survival in a rat hemorrhagic shock and resuscitation (HS/R) model. However, the underlying molecular mechanism of H2 in HS/R is unclear. Endothelial glycocalyx (EG) damage causes hemodynamic failure associated with HS/R. In this study, we tested the hypothesis that H2 alleviates oxidative stress by suppressing xanthine oxidoreductase (XOR) and/or preventing tumor necrosis factor-alfa (TNF-α)-mediated syndecan-1 shedding during EG damage.
Methods:
HS/R was induced in rats by reducing mean arterial pressure (MAP) to 35 mm Hg for 60 min followed by resuscitation. Rats inhaled oxygen or H2 + oxygen after achieving shock either in the presence or absence of an XOR inhibitor (XOR-I) for both the groups. In a second test, rats received oxygen alone or antitumor necrosis factor (TNF)-α monoclonal antibody with oxygen or H2. Two hours after resuscitation, XOR activity, purine metabolites, cytokines, syndecan-1 were measured and survival rates were assessed 6 h after resuscitation.
Results:
H2 and XOR-I both suppressed MAP reduction and improved survival rates. H2 did not affect XOR activity and the therapeutic effects of XOR-I and H2 were additive. H2 suppressed plasma TNF-α and syndecan-1 expression; however, no additional H2 therapeutic effect was observed in the presence of anti-TNF-α monoclonal antibody.
Conclusions:
H2 inhalation after shock stabilized hemodynamics and improved survival rates in an HS/R model independent of XOR. The therapeutic action of H2 was partially mediated by inhibition of TNF-α-dependent syndecan-1 shedding.
INTRODUCTION
The dynamic equilibrium of the vascular system is profoundly disturbed by hemorrhagic shock (HS). Even after restoration of lost blood volume by fluid resuscitation, vascular dysfunction continues to progress depending on shock severity and the type of resuscitation fluids used (1). Endothelial dysfunction is known to be a major cause of hemodynamic failure and secondary organ dysfunction, and so is a target for successful resuscitation after HS (2–6).
The endothelial glycocalyx (EG) is a network of membrane-bound proteoglycans and glycoproteins covering the endothelial lumen that maintains vascular homeostasis, including controlling vascular permeability, controlling microvascular tone, preventing microvascular thrombosis, and regulating leukocyte adhesion (7–10). Intravital microscopy has shown that EG thickness was reduced after hemorrhage in skeletal muscle and mesenteric venules (11) and degradation of EG is pertinent to increased HS and fluid resuscitation (HS/R) microvascular permeability (1, 4, 12). Fluid resuscitation further damages the EG via ischemia-reperfusion injury after initial HS-induced degradation (13). Syndecan-1, a transmembrane heparan sulfate proteoglycan and a primary component of the EG, is shed into the blood during HS/R (12, 14). Recent studies revealed that plasma syndecan-1 is a marker for glycocalyx shedding that is strongly correlated with both reduced EG thickness and increased microvascular permeability when different fluids are used for resuscitation in hemorrhaged rats (4). Therapeutics for maintaining vascular endothelial barrier function in the microcirculation by minimizing EG damage are unmet clinical needs.
The EG is degraded by metalloproteinases, heparanase, and hyaluronidase (15, 16). These sheddases are activated by reactive oxygen species (ROS) and pro-inflammatory cytokines including tumor necrosis factor-alpha (TNF-α) and interleukin-1beta (IL-1β) (17, 18). One of the immediate responses in the complex pathophysiology of HS is the activation of the NFκB pathway, primarily through macrophages that detect damage-associated molecular patterns (19). Subsequently, TNF-α is released in the acute phase (20) and plays a pivotal role in the shedding of vascular EG, eventually increasing vascular permeability (21, 22).
Hydrogen (H2) is a versatile medical gas that can be used effectively in various situations of ischemia-reperfusion (I/R) injury (23). We have previously reported that inhaled H2 stabilizes hemodynamics and improves short-term survival after HS/R (24). However, several issues need to be addressed before clinical application. First, inhalation of H2 was initiated at the beginning of hemorrhage, which is inconsistent with clinical settings. Second, mandatory mechanical ventilation at a fixed rate may have impaired the respiratory compensation during shock which left the criticism that effect of H2 may have augmented. Third, resuscitation was performed with a saline infusion that was four times the shed blood volume. This large infusion of saline may provoke acidosis due to chlorine load. Fourth, the mechanisms and targets of H2 have not been clarified. We therefore modified our previous protocol to better mimic clinical conditions to determine the efficacy of H2, with spontaneous breathing after the induction of shock and resuscitation with balanced crystalloids. We hypothesize that H2 stabilizes hemodynamics in rats after HS/R by targeting vascular EG.
H2 has an antioxidative effect, but its mechanism has not been fully elucidated (23, 25). Xanthine oxidoreductase (XOR) is one of the major sources of ROS production after I/R injury (26, 27). XOR catalyzes the oxidation of hypoxanthine to xanthine, and then xanthine to uric acid. XOR exists in two interconvertible forms: xanthine dehydrogenase (XDH) and xanthine oxidase (XO). XDH is dominant in healthy tissue and uses NAD+ as an electron acceptor. It is converted to the XO form either reversibly by the oxidation of sulfhydryl residues or irreversibly by proteolysis (28). Since XO uses oxygen (O2) as a terminal electron acceptor to ROS, conversion from XDH to XO has been proposed to cause I/R injury. XOR is a homodimer with four molybdenum-containing molybdopterin cofactors per monomer. Metallic molybdenum is produced through reduction of molybdenum trioxide by using H2 as a reducing agent. Thus, we hypothesize that H2 suppresses ROS production by reducing the active center molybdenum thereby suppressing the activity of XOR.
MATERIALS AND METHODS
Animals
Male Sprague-Dawley rats (300 g–350 g) were used in all experiments (Sankyo Labo Service Corporation, Inc, Tokyo, Japan). Animals had food and water ad libitum at all times. Animals were housed at 22 ± 1°C and 55 ± 5% humidity with a 12:12-h light:dark cycle. Rats were allowed to acclimatize for at least 5 days after arrival. The study was approved by the Institutional Animal Care and Use Committee (Keio University, No. 13002-3). All animal experiments are in concordance with ARRIVE guidelines (29).
Anesthesia, analgesia, and surgical procedures
Rats were anesthetized by intraperitoneal injection of medetomidine, midazolam, and butorphanol (0.15 mg/kg, 2.0 mg/kg, and 2.5 mg/kg, respectively). An additional third of the initial dose was injected upon limb extraction response to pain stimulation. Temperature was measured using a rectal probe (BAT-10, Physitemp Instruments Inc, Clifton, NJ) and maintained at 37°C with a heating pad. Polyethylene catheters (PE50, Natsume, Tokyo, Japan) were placed into the left femoral artery and vein for blood withdrawal/sampling and fluid administration, respectively. A Millar transducer catheter (SPR-320; AD Instruments, Bella Vista, NSW, Australia) was placed into the abdominal aorta from the right femoral artery for blood pressure monitoring. Electrocardiograms, heart rate, arterial blood pressure, and body temperature were continuously recorded using a polygraph (Powerlab System, LabChart; AD Instruments, Bella Vista, NSW, Australia).
HS/R protocol
HS was induced using the fixed-pressure model as previously described with minor modifications (24). Briefly, blood was withdrawn over 15 min to achieve a mean arterial pressure (MAP) of 35 ± 2 mm Hg to induce HS (Fig. 1). MAP was maintained at this level until 60 min after initial induction of HS by further blood withdrawal or reinfusion of shed blood. After the shock phase, rats were resuscitated with an infusion of three times the volume of shed blood of lactated-Ringer solution over 30 min. Maintenance fluid of lactated-Ringer at 1 mL/kg/h was administered during the observation phase. Rats were sacrificed by blood withdrawal under anesthesia at various time points. Sham-operated rats received all procedures except blood withdrawal.
Fig. 1.
Experimental protocol for hemorrhagic shock and resuscitation.
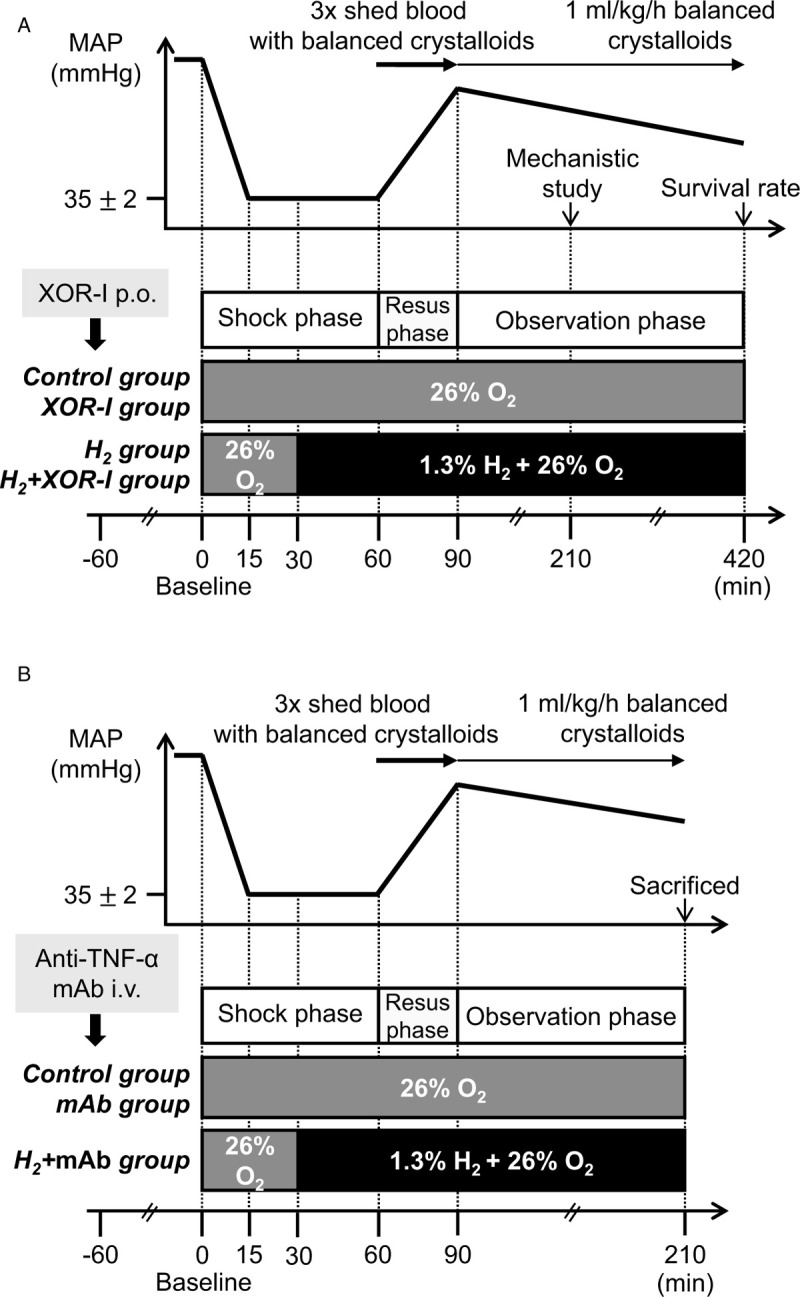
Hemorrhagic shock was induced using the fixed-pressure model. MAP of 35 ± 2 mm Hg was achieved over 15 min by blood withdrawal and maintained until 60 min from the induction of hemorrhagic shock. After the shock phase, rats were resuscitated with an infusion of three times the volume of the shed blood of balanced crystalloids over 30 min. Maintenance fluid was administered during the observation phase. A, Rats inhaled oxygen or H2 + oxygen after achieving shock either in the presence or absence of an XOR-I for both groups. B, Rats received oxygen alone or anti-TNF-α monoclonal antibody with oxygen or H2 + oxygen. H2 indicates hydrogen gas; i.v., intravenous injection; MAP, mean arterial pressure; P.O., per Os; Resus, resuscitation; TNF-α, tumor necrosis factor-alfa; XOR-I, xanthine oxidoreductase inhibitor.
Experimental groups
In the XOR inhibition experiment, rats were randomly sorted into four groups: 26% O2, 74% N2 gas (control); H2 gas (H2); oral XOR-I and 26% O2, 74% N2 gas (XOR-I); and oral XOR-I and H2 gas (H2+XOR-I) (Fig. 1A). In the experiment with anti-TNF-α monoclonal antibody (mAb), rats were randomized into three groups: 26% O2, 74% N2 gas (control); anti-TNF-α and 26% O2, 74% N2 gas (mAb); and anti-TNF-α and H2 gas (H2+mAb) (Fig. 1B). Given the nature of the protocol, it was impossible to blind the investigator to the treatments.
Gas inhalation
Control and H2 (1.3% H2, 26% O2, 72.7% N2) gases were manufactured at Taiyo Nippon Sanso Corporation (Tokyo, Japan). Control gas inhalation was initiated at the beginning of the experiment. H2 gas inhalation began 30 min after induction of HS and continued until the end of the experiment (Fig. 1). Gas was provided through a nose cone to allow spontaneous respiratory compensation during HS/R.
Xanthine oxidase inhibition
The XOR-I Topiroxostat (Sanwa Kagaku Kenkyusho Co, Ltd, Aichi, Japan) was suspended in 0.5% methylcellulose and 10 mg/kg was orally administered 60 min before the HS induction. Control and H2 groups were administered equivalent amounts of methylcellulose.
Anti-TNF-α antibody administration
TNF-α was neutralized by systemic administration of anti-TNF-α mAb as previously described with minor modifications (30). Anti-TNF-α (MAB510-100, R&D Systems, Minneapolis, Minn) was diluted with phosphate-buffered saline and 1 μg/kg was administered intravenously 60 min before the induction of HS. Control rats received an equivalent amount of phosphate-buffered saline as vehicle.
Blood analysis
Arterial blood samples were collected at baseline (0 min; beginning of the shock phase), 60 min (end of the shock phase), and 150 min (1 h from the end of the resuscitation phase). The pH, oxygen tension, carbon dioxide tension, bicarbonate, base excess, hemoglobin, and potassium concentrations were assessed by arterial blood gas analyses (ABG) (iSTAT, Abbott Point Care Inc, Princeton, NJ). Lactate levels were measured using a Lactate Pro2 meter (ARKRAY, Kyoto, Japan). Blood samples were centrifuged at 1,500 × g for 10 min at 4°C, and plasma was stored at −80°C. Plasma concentrations of IL-1β (Invitrogen, Calif), TNF-α (Invitrogen), and syndecan-1 (Signalway Antibody LLC, Md) were measured with ELISA kits according to the manufacturer's instructions.
Bronchoalveolar lavage
A tracheostomy was performed after rats were sacrificed 2 h after resuscitation. Bronchoalveolar lavage was performed twice with 8 mL cold phosphate-buffered saline. Numbers of red blood cells (RBC) in bronchoalveolar lavage fluid (BALF) were manually counted.
XOR activity and purine metabolite measurement
XOR activity and purine metabolite concentrations, hypoxanthine, xanthine, and uric acid were measured in plasma, liver, kidney, and lung as previously described (31, 32).
Statistical analysis
Data are expressed as the means ± standard error mean. Differences between survival rates were tested using the Kaplan–Meier analysis. A log-rank test was used to compare survival rates. One-way analysis of variance followed by a Tukey–Kramer multiple comparison test was used to compare the variables among groups. Hemodynamic data were analyzed using a mixed-effects model for repeated measures as previously described (24). Spearman rank correlation coefficient was used for evaluation of correlations between variables. P values under 0.05 were considered significant. All data were analyzed using SPSS 25.0 statistical software (SPSS Inc, Chicago, Ill).
RESULTS
H2 and XOR-I improve MAP and survival after HS/R
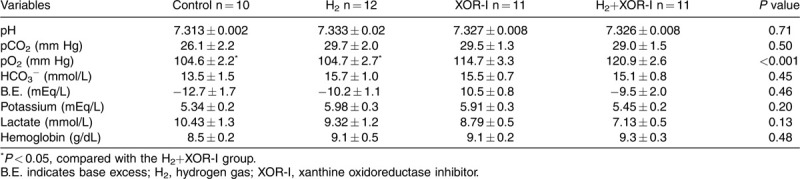
We tested the hypothesis that H2 suppresses enzyme activity of XOR to control I/R injury by comparing the therapeutic effects of H2 and XOR-I alone or in combination after HS/R. ABG variables at baseline were comparable among all groups (See Table, Supplemental Digital Content 1, which demonstrates the ABG at baseline). Shed blood volume was not different between groups (Table 1). At the end of the shock phase, hemorrhaged rats developed severe lactic acidosis with respiratory compensation (Table 2). XOR-I and H2+XOR-I administration appeared to suppress the accumulation of lactate during hypovolemic shock, but the differences in lactate levels between these groups and the control were not statistically significant (Table 2).
Table 1.
Blood withdrawal and resuscitation fluid variables
Table 2.
Arterial gas analysis at 60 min after induction of shock
Lactic acidosis improved after fluid resuscitation; however, lactate accumulation 60 min after completion of resuscitation was reduced in the single-treatment groups and this reduction was additive in the combination treatment group (Table 3).
Table 3.
Arterial gas analysis at 150 min after induction of shock
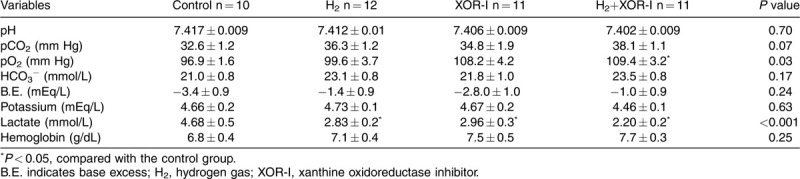
MAP recovered in response to fluid resuscitation; however, in the control group, it decreased to less than 65 mm Hg immediately after the completion of resuscitation. In contrast, MAP was significantly maintained in the H2, XOR-I, and H2+XOR-I groups (Fig. 2A). MAP was higher in the H2 group than in the XOR-I group 60 min after completing fluid resuscitation; however, this difference subsequently resolved over time. With the combination therapy, MAP was maintained at a higher level than either therapy alone. Time course changes in heart rate were comparable among the four groups (Fig. 2B).
Fig. 2.
Mean arterial pressure and short-term survival was attenuated with H2 and XOR-I.
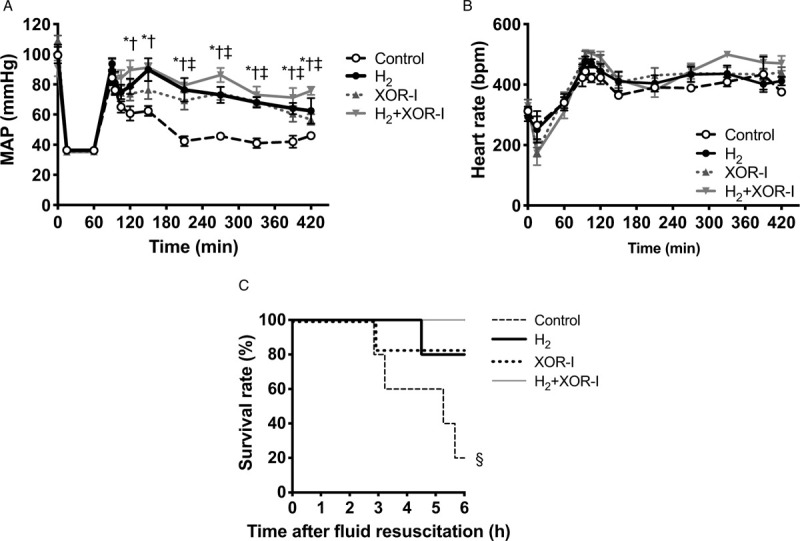
A, MAP was significantly maintained in the H2, XOR-I, and H2+XOR-I groups. With the combination therapy, MAP was maintained at a higher level than either monotherapy. B, Time course changes in heart rate were comparable among the four groups. C, The survival rates 6 h after initiation of fluid resuscitation were 20% (1/5) in the control group, and were 83% (5/6), 80% (4/5), and 100% (5/5) in the XOR-I, H2, and H2+XOR-I treatment groups, respectively. Values are mean ± SEM; n ≥ 5 animals in each group. ∗P < 0.05, H2 compared with the control group; †P < 0.01, H2+XOR-I compared with the control group; ‡P < 0.01, XOR-I compared with the control group; §P = 0.03, compared with the control group. H2 indicates hydrogen gas; MAP, mean arterial pressure; XOR-I, xanthine oxidoreductase inhibitor.
Survival 6 h after initiation of fluid resuscitation was 20% in the control group, and 83%, 80%, and 100% in the XOR-I, H2, and H2+XOR-I groups, respectively (Fig. 2C). These survival rates paralleled the hemodynamic changes after HS/R.
XOR activity and purine metabolite levels are unaffected by H2 treatment
XOR-I suppressed XOR activity as shown by increased hypoxanthine and decreased xanthine and uric acid before HS, during HS, and after resuscitation (see Figures, Supplemental Digital Content 2 and 3, which demonstrate XOR activity and purine metabolites before, during and after HS/R). However, XOR activity and purine metabolite levels in the H2 group were similar to those in the control group.
Attenuation of glycocalyx shedding and vascular hyperpermeability by H2
Inhaled H2 attenuated symptoms including abdominal bloating and oliguria or anuria. The number of RBCs in the BALF, a marker for lung permeability, was significantly reduced by H2 (Fig. 3A). Plasma syndecan-1 was not detected in sham rats but was increased by HS/R. H2 treatment after HS/R reduced this increase in plasma syndecan-1 (Fig. 3B). In contrast, XOR-I did not lower plasma syndecan-1 levels. Moreover, a combination of XOR-I and H2 showed no additive effect on plasma syndecan-1 shedding. Plasma syndecan-1 levels were negatively correlated with MAP measurements (Fig. 3C).
Fig. 3.
Glycocalyx shedding and vascular hyperpermeability was attenuated with H2 inhalation.
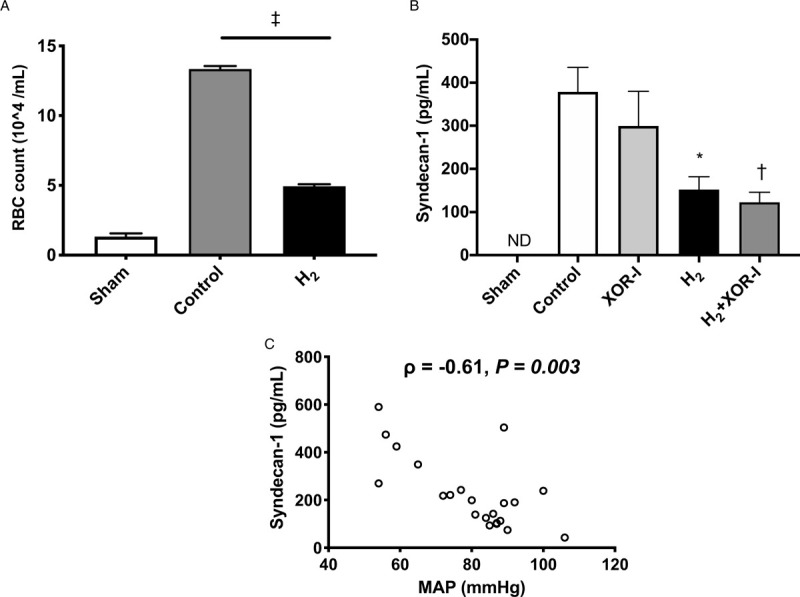
A, RBC counts in bronchoalveolar lavage fluid were measured as a marker for vascular permeability. The RBC counts were elevated in the control group after HS/R. H2 inhalation significantly attenuated RBC leakage into the bronchoalveolar lavage fluid compared with the control group. B, Plasma syndecan-1 was reduced with H2 treatment. However, XOR-I did not lower plasma syndecan-1 level (P = 0.70). Moreover, a combination of XOR-I and H2 did not show additive effect in attenuating plasma syndecan-1 levels. C, MAP and plasma syndecan-1 are negatively correlated. Values are mean ± SEM; N ≥ 10 animals in each group. ∗P = 0.02, H2 versus control group; †P = 0.008, H2+XOR-I versus control group; ‡P < 0.001. H2 indicates hydrogen gas; MAP, mean arterial pressure; ND, not detected; RBC, red blood cell.
H2 inhalation reduces inflammatory cytokine production
Two hours after the completion of fluid resuscitation, plasma TNF-α and IL-1β levels were elevated in hemorrhaged rats. H2 reduced plasma TNF-α levels (Fig. 4A), as well as IL-1β, but this reduction was not significant (Fig. 4B). Plasma TNF-α levels were negatively correlated with MAP (Fig. 4C) but positively correlated with plasma syndecan-1 (Fig. 4D).
Fig. 4.
Inflammatory cytokines were attenuated with H2 inhalation and negatively correlated with syndecan-1.
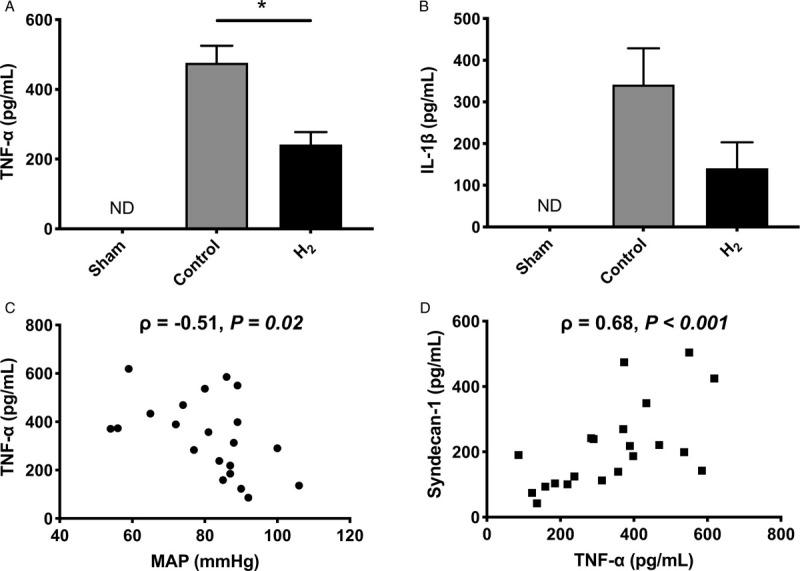
A, H2 inhalation reduced plasma TNF-α. B, H2 inhalation reduced IL-1β but this reduction was not significant (P = 0.09). C, MAP and plasma TNF-α are negatively correlated after HS/R. D, TNF-α and syndecan-1 are positively correlated after HS/R. ∗P < 0.001. Values are mean ± SEM; n ≥10 animals in each group. H2 indicates hydrogen gas; IL-1β, interleukin-1 beta; MAP, mean arterial pressure; ND, not detected; TNF-α, tumor necrosis factor-alfa.
H2 maintains hemodynamic stability after HS by suppressing TNF-α-mediated endothelial glycocalyx shedding
We investigated the effect of TNF-α on endothelial glycocalyx degradation, vascular hyperpermeability, and hemodynamic instability after hemorrhagic shock (See Tables, Supplemental Digital Content 4–7, which demonstrates HS/R variables and ABG). After treatment with anti-TNF-α antibody, we observed a reduction in plasma TNF-α (Fig. 5A), syndecan-1 shedding (Fig. 5B), and RBC leakage in the BALF (Fig. 5C), as well as stabilization of hemodynamics (Fig. 5D) after HS. The change in heart rate over time was comparable among all groups.
Fig. 5.
Systemic administration of anti-TNF-α antibody attenuated elevation of plasma TNF-α, improved MAP and endothelial glycocalyx degradation.
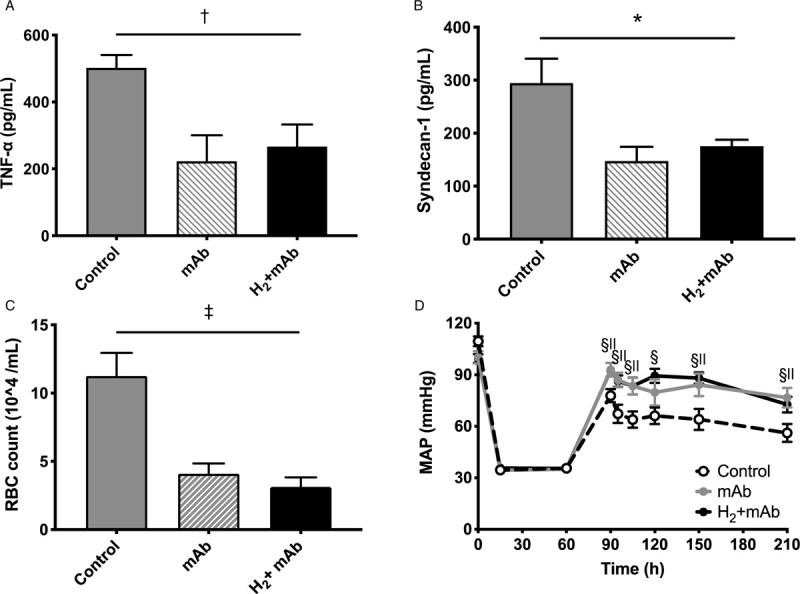
Systemic administration of an anti-TNF-α monoclonal antibody: (A) attenuated plasma TNF-α concentration and addition of H2 did not confer an additive effect on plasma TNF-α concentration; (B) attenuated plasma syndecan-1concentration, but there was no observable additive effect of H2; (C) significantly reduced the number of leaked RBCs in the bronchoalveolar lavage fluid, but did not have an additive effect in combination with H2 inhalation; (D) attenuated MAP, but an additive effect was not observed with the concomitant treatment of H2. ∗P = 0.04, ANOVA; †P = 0.004, ANOVA; ‡P < 0.001, ANOVA; §P < 0.01 mAb versus control group; ||P < 0.01, H2+mAb versus control group. Values are mean ± SEM; n ≥ 8 animals in each group. H2 indicates hydrogen gas; mAb, anti-TNF-α monoclonal antibody; MAP, mean arterial pressure; RBC, red blood cell; TNF-α, tumor necrosis factor-alfa.
We then examined whether H2 exerts additional therapeutic effects when TNF-α-mediated endothelial glycocalyx shedding was blocked in hemorrhaged rats. When hemorrhaged rats were pretreated with an anti-TNF-α antibody, no further decrease in plasma TNF-α was observed by the administration of H2 (Fig. 5A). Under these conditions, no additive effect on plasma syndecan-1 shedding (Fig. 5B), RBC leakage in BALF (Fig. 5C), and hemodynamic improvement (Fig. 5D) was observed, indicating that the therapeutic effect of H2 is exerted in part through the suppression of TNF-α-mediated EG shedding.
DISCUSSION
We have demonstrated that H2 mitigated the harmful effects of protein-free fluid resuscitation therapy after shock. This effect of H2 was at least in part mediated by the suppression of TNF-α-mediated EG degradation.
Previously, we reported that H2 inhalation upon induction of HS enhanced tolerance to blood loss and hemodynamic stability after fluid resuscitation with four times the shed blood volume of normal saline (24). However, that experimental protocol was inconsistent with typical clinical scenarios and the high infusion volume was harmful (33, 34). Therefore, in this study H2 inhalation was started after shock, mimicking H2 inhalation in an ambulance. Rats maintained spontaneous respiration such that respiratory compensation for metabolic acidosis was not impaired. Furthermore, fluid resuscitation was performed with three times the amount of the shed blood with balanced crystalloids.
We asked if H2 exerts its therapeutic effect by suppressing the activity of XOR and reducing molybdenum localized at the active center of this enzyme. H2 inhalation did not affect XOR activity, nor were hypoxanthine, xanthine, and urate concentrations changed during HS or 120 min after resuscitation. From these results, we conclude that H2 exerts a therapeutic effect independent of XOR activity.
XOR-I had a hemodynamic stabilization effect equivalent to H2 in the rat HS model after fluid resuscitation. However, as additive an effect of XOR-I and H2 was observed, and XOR-I alone had no effect on syndecan-1 shedding, the mechanisms by which XOR-I and H2 provided stabilization were different. XOR-I increased the pool of salvageable purines and increased ATP levels (35), while the inhibition of XOR increased the NAD/NADH ratio. The conversion of pyruvate to lactate is suppressed under conditions where the NAD/NADH ratio was increased (36, 37). Indeed, accumulation of lactate during shock was suppressed by the pre-administration of XOR-I, which may have beneficial effects related to reinforcement of ATP synthesis due to increased hypoxanthine levels.
From the subjective improvements observed in symptoms including abdominal bloating and anuria, we hypothesized that H2 targets vascular endothelial cells and suppresses vascular hyperpermeability, unregulated vasodilation, microvascular thrombosis, and augmented leukocyte adhesion. Normally, the EG provides a barrier against vascular permeability in part by serving as a negatively charged molecular sieve. This barrier limits transvascular movement of negatively charged and/or > 70-kDa molecules (38). Moreover, the EG senses fluid-shear forces and transmits them to endothelial cells, initiating nitric oxide-mediated vasorelaxation. The EG has anticoagulant and anti-adhesive effects on the surface of endothelial cells, whose functions are significantly altered following hypovolemic shock by both ischemia and reperfusion. Activation of endothelial cells leads to release of vasoactive substances and inflammatory mediators, including TNF-α, which induces the production of several matrix metalloproteinases and heparanase in endothelial cells (15, 39). As a result, the EG is degraded and its function impaired, leading to enhanced microvascular permeability, edema caused by fluid leakage into surrounding tissue, augmented leukocyte adhesion, platelet aggregation, and dysregulated vasodilation.
Several studies indicate that TNF-α plays a pivotal role in vascular failure and end-organ damage during acute HS and subsequent resuscitation (20–22). In this study, we showed that systemic administration of anti-TNF-α antibody reduced circulating levels of syndecan-1 and improved hemodynamic stability, suggesting a substantial role for TNF-α in glycocalyx degradation. We found that H2 suppresses the release of TNF-α and therefore suppresses degradation of the EG, preventing vascular endothelial dysfunction after HS/R.
While our results provide new information relevant to the treatment of HS, we did not directly observe a change in microvascular glycocalyx thickness. We used lactated-Ringer solution for fluid resuscitation and therefore cannot comment on how the use of H2 in HS/R may work in conjunction with the use of plasma in clinical resuscitation, which is reported to cause less shedding of endothelial glycocalyx than protein-free solutions (14, 40, 41). Therefore, further studies are needed to evaluate the therapeutic effects of H2 during plasma resuscitation. The mechanism of action for H2 in HS/R is not only the suppression of systemic release of TNF-α; it is also necessary to understand the effects of H2 on the nervous, endocrine, and metabolic systems. Although we focused on hyperpermeability after HS/R in this study, TNF-α also decreases vascular reactivity through decreased endothelial nitric oxide synthase (NOS) expression and elevated expression of inducible NOS. Future ex-vivo studies will clarify the positive effects of H2 on reduced vascular reactivity after HS/R. Despite these questions, H2 is a promising treatment option for HS/R that can protect EG and be used in addition to traditional treatments before definitive hemostasis is attained in both field and clinical settings.
In conclusion, we report for the first time that inhalation of H2 after HS stabilized hemodynamics and short-term survival after fluid resuscitation with balanced crystalloids. This effect was independent of XOR activity and reduced EG degradation in part through the attenuation of TNF-α production. Thus, we propose that H2 may be useful for reducing inflammatory cytokines and vascular endothelial dysfunction that further exacerbate organ dysfunction associated with HS/R.
Supplementary Material
Supplementary Material
Supplementary Material
Acknowledgments
The authors thank S. Suzuki, Y. Miyake (Keio University School of Medicine), and S. Kotoda (Bioresearch Center) for excellent technical assistance.
Footnotes
Reprints will not be ordered.
The research was supported by a research grant from Taiyo Nippon Sanso Corporation to MSa, and xanthine oxidoreductase inhibitor (Topiroxat) was provided by Sanwa Kagaku Kenkyusho Co, Ltd. TT, MSa, and MSu received a travel grant from the Taiyo Nippon Sanso Corporation.
The authors report no conflicts of interest.
REFERENCES
- 1.Torres Filho IP, Torres LN, Salgado C, Dubick MA. Plasma syndecan-1 and heparan sulfate correlate with microvascular glycocalyx degradation in hemorrhaged rats after different resuscitation fluids. Am J Physiol Heart Circ Physiol 310 (11):H1468–H1478, 2016. [DOI] [PubMed] [Google Scholar]
- 2.Lipowsky HH. The endothelial glycocalyx as a barrier to leukocyte adhesion and its mediation by extracellular proteases. Ann Biomed Eng 40 (4):840–848, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Becker BF, Chappell D, Bruegger D, Annecke T, Jacob M. Therapeutic strategies targeting the endothelial glycocalyx: acute deficits, but great potential. Cardiovasc Res 87 (2):300–310, 2010. [DOI] [PubMed] [Google Scholar]
- 4.Chignalia AZ, Yetimakman F, Christiaans SC, Unal S, Bayrakci B, Wagener BM, Russell RT, Kerby JD, Pittet JF, Dull RO. The glycocalyx and trauma: a review. Shock 45 (4):338–348, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tuma M, Canestrini S, Alwahab Z, Marshall J. Trauma and endothelial glycocalyx: the microcirculation helmet? Shock 46 (4):352–357, 2016. [DOI] [PubMed] [Google Scholar]
- 6.Johansson PI, Stensballe J, Ostrowski SR. Shock induced endotheliopathy (SHINE) in acute critical illness—a unifying pathophysiologic mechanism. Crit Care 21 (1):25, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Curry FE, Adamson RH. Endothelial glycocalyx: permeability barrier and mechanosensor. Ann Biomed Eng 40 (4):828–839, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nieuwdorp M, Meuwese MC, Vink H, Hoekstra JB, Kastelein JJ, Stroes ES. The endothelial glycocalyx: a potential barrier between health and vascular disease. Curr Opin Lipidol 16 (5):507–511, 2005. [DOI] [PubMed] [Google Scholar]
- 9.Reitsma S, Slaaf DW, Vink H, van Zandvoort MA, oude Egbrink MG. The endothelial glycocalyx: composition, functions, and visualization. Pflugers Arch 454 (3):345–359, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pries AR, Secomb TW, Gaehtgens P. The endothelial surface layer. Pflugers Arch 440 (5):653–666, 2000. [DOI] [PubMed] [Google Scholar]
- 11.Torres Filho I, Torres LN, Sondeen JL, Polykratis IA, Dubick MA. In vivo evaluation of venular glycocalyx during hemorrhagic shock in rats using intravital microscopy. Microvasc Res 85:128–133, 2013. [DOI] [PubMed] [Google Scholar]
- 12.Johansson PI, Stensballe J, Rasmussen LS, Ostrowski SR. A high admission syndecan-1 level, a marker of endothelial glycocalyx degradation, is associated with inflammation, protein C depletion, fibrinolysis, and increased mortality in trauma patients. Ann Surg 254 (2):194–200, 2011. [DOI] [PubMed] [Google Scholar]
- 13.Rubio-Gayosso I, Platts SH, Duling BR. Reactive oxygen species mediate modification of glycocalyx during ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol 290 (6):H2247–H2256, 2006. [DOI] [PubMed] [Google Scholar]
- 14.Kozar RA, Peng Z, Zhang R, Holcomb JB, Pati S, Park P, Ko TC, Paredes A. Plasma restoration of endothelial glycocalyx in a rodent model of hemorrhagic shock. Anesth Analg 112 (6):1289–1295, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schmidt EP, Yang Y, Janssen WJ, Gandjeva A, Perez MJ, Barthel L, Zemans RL, Bowman JC, Koyanagi DE, Yunt ZX, et al. The pulmonary endothelial glycocalyx regulates neutrophil adhesion and lung injury during experimental sepsis. Nat Med 18 (8):1217–1223, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Manon-Jensen T, Multhaupt HA, Couchman JR. Mapping of matrix metalloproteinase cleavage sites on syndecan-1 and syndecan-4 ectodomains. FEBS J 280 (10):2320–2331, 2013. [DOI] [PubMed] [Google Scholar]
- 17.Becker BF, Jacob M, Leipert S, Salmon AH, Chappell D. Degradation of the endothelial glycocalyx in clinical settings: searching for the sheddases. Br J Clin Pharmacol 80 (3):389–402, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lipowsky HH, Lescanic A. The effect of doxycycline on shedding of the glycocalyx due to reactive oxygen species. Microvasc Res 90:80–85, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Altavilla D, Saitta A, Squadrito G, Galeano M, Venuti SF, Guarini S, Bazzani C, Bertolini A, Caputi AP, Squadrito F. Evidence for a role of nuclear factor-kappaB in acute hypovolemic hemorrhagic shock. Surgery 131 (1):50–58, 2002. [DOI] [PubMed] [Google Scholar]
- 20.Altavilla D, Cainazzo MM, Squadrito F, Guarini S, Bertolini A, Bazzani C. Tumour necrosis factor-alpha as a target of melanocortins in haemorrhagic shock, in the anaesthetized rat. Br J Pharmacol 124 (8):1587–1590, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Henry CB, Duling BR. TNF-alpha increases entry of macromolecules into luminal endothelial cell glycocalyx. Am J Physiol Heart Circ Physiol 279 (6):H2815–H2823, 2000. [DOI] [PubMed] [Google Scholar]
- 22.Morita Y, Clemens MG, Miller LS, Rangan U, Kondo S, Miyasaka M, Yoshikawa T, Bulkley GB. Reactive oxidants mediate TNF-alpha-induced leukocyte adhesion to rat mesenteric venular endothelium. Am J Physiol 269 (6):H1833–H1842, 1995. [DOI] [PubMed] [Google Scholar]
- 23.Sano M, Suzuki M, Homma K, Hayashida K, Tamura T, Matsuoka T, Katsumata Y, Onuki S, Sasaki J. Promising novel therapy with hydrogen gas for emergency and critical care medicine. Acute Med Surg 5 (2):113–118, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matsuoka T, Suzuki M, Sano M, Hayashida K, Tamura T, Homma K, Fukuda K, Sasaki J. Hydrogen gas inhalation inhibits progression to the “irreversible” stage of shock after severe hemorrhage in rats. J Trauma Acute Care Surg 83 (3):469–475, 2017. [DOI] [PubMed] [Google Scholar]
- 25.Iuchi K, Imoto A, Kamimura N, Nishimaki K, Ichimiya H, Yokota T, Ohta S. Molecular hydrogen regulates gene expression by modifying the free radical chain reaction-dependent generation of oxidized phospholipid mediators. Sci Rep 6:18971, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Battelli MG, Bolognesi A, Polito L. Pathophysiology of circulating xanthine oxidoreductase: new emerging roles for a multi-tasking enzyme. Biochim Biophys Acta 1842 (9):1502–1517, 2014. [DOI] [PubMed] [Google Scholar]
- 27.Cantu-Medellin N, Kelley EE. Xanthine oxidoreductase-catalyzed reactive species generation: a process in critical need of reevaluation. Redox Biol 1:353–358, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nishino T. The conversion of xanthine dehydrogenase to xanthine oxidase and the role of the enzyme in reperfusion injury. J Biochem 116 (1):1–6, 1994. [DOI] [PubMed] [Google Scholar]
- 29.Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol 8 (6):e1000412, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zingarelli B, Squadrito F, Altavilla D, Calapai G, Di Rosa M, Caputi AP. Role of tumor necrosis factor-alpha in acute hypovolemic hemorrhagic shock in rats. Am J Physiol 266 (4 pt 2):H1512–H1515, 1994. [DOI] [PubMed] [Google Scholar]
- 31.Murase T, Nampei M, Oka M, Ashizawa N, Matsumoto K, Miyachi A, Nakamura T. Xanthine oxidoreductase activity assay in tissues using stable isotope-labeled substrate and liquid chromatography high-resolution mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 1008:189–197, 2016. [DOI] [PubMed] [Google Scholar]
- 32.Nakamura T, Murase T, Nampei M, Morimoto N, Ashizawa N, Iwanaga T, Sakamoto R. Effects of topiroxostat and febuxostat on urinary albumin excretion and plasma xanthine oxidoreductase activity in db/db mice. Eur J Pharmacol 780:224–231, 2016. [DOI] [PubMed] [Google Scholar]
- 33.Torres LN, Chung KK, Salgado CL, Dubick MA, Torres Filho IP. Low-volume resuscitation with normal saline is associated with microvascular endothelial dysfunction after hemorrhage in rats, compared to colloids and balanced crystalloids. Crit Care 21 (1):160, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cannon JW, Khan MA, Raja AS, Cohen MJ, Como JJ, Cotton BA, Dubose JJ, Fox EE, Inaba K, Rodriguez CJ, et al. Damage control resuscitation in patients with severe traumatic hemorrhage: a practice management guideline from the Eastern Association for the Surgery of Trauma. J Trauma Acute Care Surg 82 (3):605–617, 2017. [DOI] [PubMed] [Google Scholar]
- 35.Johnson TA, Jinnah HA, Kamatani N. Shortage of cellular ATP as a cause of diseases and strategies to enhance ATP. Front Pharmacol 10:98, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ido Y, Chang K, Williamson JR. NADH augments blood flow in physiologically activated retina and visual cortex. Proc Natl Acad Sci U S A 101 (2):653–658, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee CF, Chavez JD, Garcia-Menendez L, Choi Y, Roe ND, Chiao YA, Edgar JS, Goo YA, Goodlett DR, Bruce JE, et al. Normalization of NAD+ redox balance as a therapy for heart failure. Circulation 134 (12):883–894, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gulati A. Vascular endothelium and hypovolemic Shock. Curr Vasc Pharmacol 14 (2):187–195, 2016. [DOI] [PubMed] [Google Scholar]
- 39.Hanemaaijer R, Koolwijk P, le Clercq L, de Vree WJ, van Hinsbergh VW. Regulation of matrix metalloproteinase expression in human vein and microvascular endothelial cells. Effects of tumour necrosis factor alpha, interleukin 1 and phorbol ester. Biochem J 296 (pt 3):803–809, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu F, Peng Z, Park PW, Kozar RA. Loss of syndecan-1 abrogates the pulmonary protective phenotype induced by plasma after hemorrhagic shock. Shock 48 (3):340–345, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nelson A, Statkevicius S, Schott U, Johansson PI, Bentzer P. Effects of fresh frozen plasma, Ringer's acetate and albumin on plasma volume and on circulating glycocalyx components following haemorrhagic shock in rats. Intensive Care Med Exp 4 (1):6, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.