

Abstract
Hypoplasia is defined in the Merriman-Webster dictionary as “a condition of arrested development in which an organ, or part, remains below the normal size, or in an immature state.” The degree of reduced size is not definitional. Renal hypoplasia, however, has historically been defined as a more marked reduction in renal mass such that presentation in childhood is the norm. There are 3 commonly recognized types of renal hypoplasia, simple hypoplasia, oligomeganephronic hypoplasia (oligomeganephronia) and segmental hypoplasia (Ask-Upmark kidney). They have in common a reduction in the number of renal lobes. A fourth type, not widely recognized, is cortical hypoplasia where nephrogenesis is normal but there is a reduction in the number of nephron generations. Recently there has been great interest in milder degrees of reduced nephron mass, known as oligonephronia because of its association with risk of adult-onset hypertension and chronic kidney disease. Since the last pathology review of this topic was published by Jay Bernstein in 1968, an update of the renal pathology findings in renal hypoplasia is provided with a review of 18 new cases. The renal hypoplasias are then framed within the modern concept of oligonephronia, its diverse causes and prognostic implications.
Key Words: renal hypoplasia, oligomeganephronia, segmental hypoplasia, simple hypoplasia, oligonephronia, cortical hypoplasia
INTRODUCTION
Renal hypoplasia may be one of the most misused terms in renal developmental anomalies. Historically, many authorities considered all small kidneys as hypoplastic, thus, the literature is cluttered with a host of diseases, both congenital and acquired, that have in common small overall renal size or limited amount of renal parenchyma, such as pyelonephritic, hydronephrotic, and dysplastic kidneys, as well as, truly hypoplastic kidneys.1–5 Although overly inclusive definitions of renal hypoplasia noted above have contributed to the confusion, basing the diagnosis on clinical findings, especially imaging studies, rather than having a tissue-based histopathologic diagnosis is equally important. For instance, Eskström1 in 1955 published a radiologic study of 179 cases of “renal hypoplasia.” Forty-six were studied histologically. Only 6 of 46 were small with histologically normal nephrons, conforming to the diagnostic criteria for a hypoplasia as defined by Rubenstein et al6 in 1961 as a “congenitally decreased amount of histologically normal renal tissue.” The Rubenstein criteria were seminal because they establish a pathologic mandate for a diagnosis of renal hypoplasia.
There are 3 commonly recognized types of renal hypoplasia: segmental hypoplasia (Ask-Upmark kidney), oligomeganephronic hypoplasia (oligomeganephronia), and simple hypoplasia.6–9 Although there are gross and microscopic differences between the 3 types of hypoplasia, they have in common a reduction in number of renal lobes. There is a fourth type of hypoplasia, cortical hypoplasia, not codified in most hypoplasia classifications but recognized for many years.7,8 It consists of a reduced number of nephron generations resulting in cortical thinning and medullary reduction. Last, there is the concept of reduced nephron number, or reduced nephron endowment, known as oligonephronia (not to be confused with oligomeganephronia), that imparts a predisposition to adult-onset hypertension and chronic kidney disease (CKD).10,11 The anatomic basis for oligonephronia and its possible relationship to the known types of hypoplasia has not been characterized.
The most recent review of the pathologic features of renal hypoplasia was by Bernstein in 1968.8 In this excellent review, he discussed the clinical aspects of the 3 types of hypoplasia and presented details of their gross and microscopic features. He further emphasized that metanephric dysgenesis (renal dysplasia) should not be present, thereby, excluding small dysplastic kidneys, often referred to as hypodysplasia, from renal hypoplasia lexicon. Similarly, the Academy of Pediatrics Committee on Classification, Nomenclature and Terminology in 1989 recognized these same 3 types of hypoplasia, a classification that continues today.9
This manuscript will provide a historical review of the 3 commonly recognized types of renal hypoplasia and cortical hypoplasia, discussing their clinical, gross, and histologic features. The renal hypoplasias will then be framed within the modern concept of oligonephronia and its diverse causes and consequences.10,11
HISTORICAL REVIEW OF RENAL HYPOPLASIAS
Segmental Hypoplasia (Ask-Upmark Kidney)
The first type of renal hypoplasia described was segmental hypoplasia reported by Ask-Upmark in a 1929 German publication.12 It is often referred to as Ask-Upmark kidney. Segmental hypoplasia is rarely diagnosed, or possibly, rarely recognized. Ljungqvist and Lagergren13 in 1962 reported 2 cases in a study of 1000 autopsies and 100 nephrectomies. Segmental hypoplasia is usually identified in children presenting with hypertension (70%) or urinary tract infections.8,13–18 The hypertension is often severe. In Ask-Upmark’s original series, 7 of 8 patients had malignant hypertension.12 Hypertension is also usually present in patients presenting with urinary tract infection. Vesicoureteral reflux is identified in 70% to 80% of all cases of segmental hypoplasia8,15,17 whether presenting with infection or hypertension.
Segmental hypoplasia may be unilateral or bilateral and has a marked female predominance (72%). The hypertension appears renin-mediated. Elevated renal vein renin levels have been measured in veins draining the hypoplastic foci.19–21 In addition, renin granules have been documented by Bowie stain and by immunohistochemistry, and renin protogranules have been demonstrated by electron microscopy within the hypoplastic foci unrelated to a glomerulus or juxtaglomerular apparatus since neither are present.16,22,23 Surgical excision of the hypoplastic foci has resulted in immediate cure of the hypertension in some, but not all cases.15,21,23–27 The prospect for a surgical cure is increased in unilateral disease with a single lesion, and when the contralateral kidney is normal. A normal contralateral kidney is most likely if imaging studies are normal, the kidney is enlarged (compensatory hypertrophy), and proteinuria is absent.
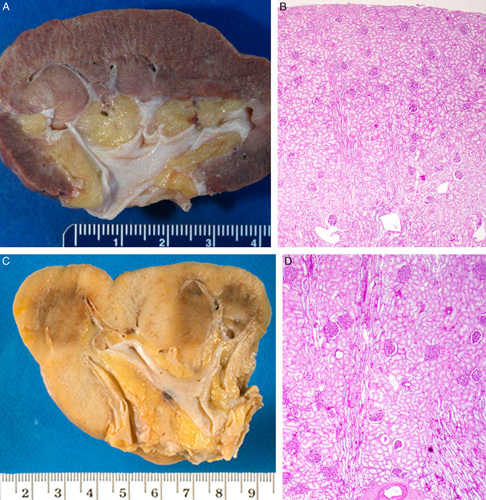
The kidney in segmental hypoplasia is usually small, <2 SD from the mean.8,13–18 Renal weight is often <50 g in adult cases and as small as12 to 25 g in children. The kidney is small because it has a reduced number of renal lobes. It surface is distinctive; having 1 or more transverse grooves that overly a pelvic recess that represent a dilated calyx, or calices (Figs. 1A–D, 2). The renal parenchyma adjacent to the hypoplastic focus is grossly normal but may be hypertrophied making the groove(s) even more prominent. On cut surface there is a thin strip of parenchyma that consists of attenuated cortex with a rudimentary or absent renal pyramid. The groove appears to represent a renal lobe that failed to form. Evidence of failed or arrested lobar development was provided by Ljungqvist and Lagergren’s13 microangiographic studies that showed a corticomedullary arterial pattern in the hypoplastic foci with arcuate and interlobular arteries, and anastomoses between efferent and afferent arterioles.
FIGURE 1.
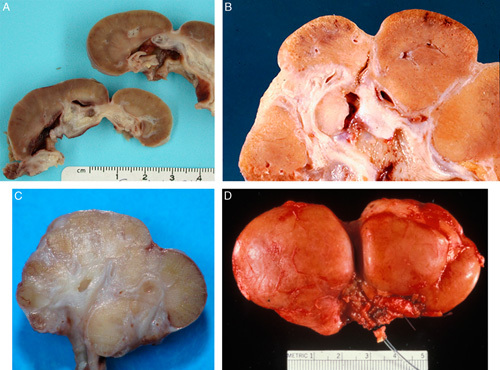
A, This gross photograph of a bivalved kidney from a 19-year-old female with segmental hypoplasia shows a single deep groove characteristic of segmental hypoplasia. The kidney was 7 cm long (normal: 11 to 12 cm). The hypoplastic focus is tan with no evidence of corticomedullary differentiation. The adjacent cortex appears normal. The underlying collecting system shows marked hydronephrosis. B, This gross photograph of a bivalved kidney is from a 33-year-old female with segmental hypoplasia. The kidney was 6 cm long and weighed 38 g (normal: 115 to 125 g). Four hypoplastic foci were preset; 3 are visible in this photograph. C, This gross photograph of a bivalved kidney from a 2-year-old female was 2.5 cm long (normal: 5.5 to 7.8 cm) and weighed 3 g (normal: 35 to 50 g). There are several adjacent hypoplastic foci without intervening normal lobes. Two normal lobes are at the top and on the right, respectively. D, This gross photograph of a child with segmental hypoplasia shows the deep circumferential groove characteristic of this entity. Jay Bernstein Consultative Collection, with permission.
FIGURE 2.
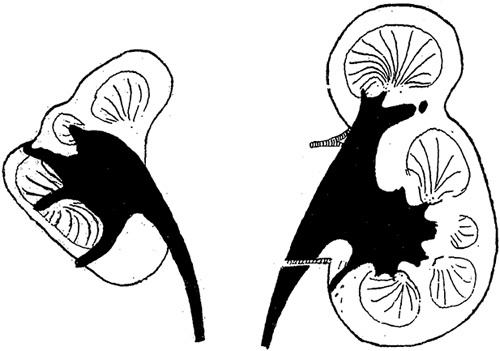
This drawing is from Dr Ask-Upmark’s 1929 article on segmental hypoplasia depicts kidneys with hypoplastic foci. Notice the deep cortical grooves that approximate underlying minor calyces. Reprinted from Ask-Upmark,12 Fig. 1. With permission from John Wiley and Sons. Copyright John Wiley and Sons, Hoboken, NJ. All permission requests for this image should be made to the copyright holder.
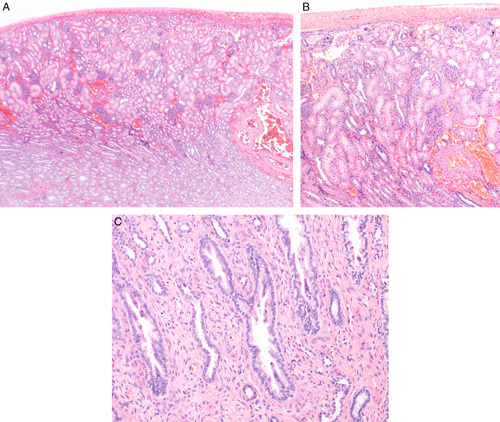
The microscopic features of the hypoplastic focus consist of a sharply delineated lesion bracketed on both sides by normal-appearing cortical parenchyma.8,13–18,21 The hypoplastic focus contains a few tubules, loose interstitial tissue, with no or only rare glomeruli and little or no inflammation (Figs. 3A–D). There are large dilated veins and thick-walled arteries. Cysts and dysplastic features are absent, although occasional subcapsular immature-appearing cell nests or rarely a small island of fetal cartilage may be seen. Sclerotic glomeruli, atrophic tubules with thick replicated basement membranes and interstitial fibrosis are absent supporting a developmental abnormality rather than a sclerosing or atrophic process.
FIGURE 3.
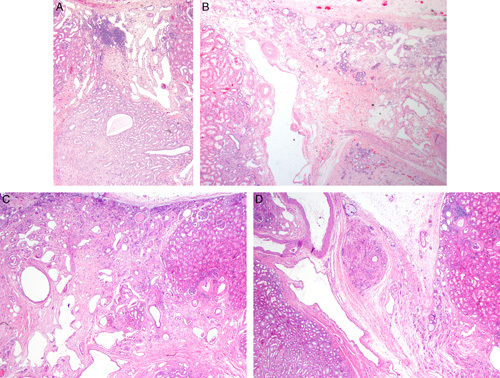
A, This case of segmental hypoplasia from a 2-year-old shows a hypoplastic focus. Notice that the focus is bordered on each side by normal-appearing cortex. The focus is devoid of glomeruli, both intact and sclerotic. It contains large dilated veins, a few tubules, loose connective tissue stroma, and a focus of lymphoid inflammation. Medullary tissue is visible at the bottom of the image. It consists of collecting ducts and cellular stroma. No loops of Henle are present. B, This case of segmental hypoplasia from a 33-year-old shows another hypoplastic focus. The focus is bordered on the left by normal-appearing cortex. The focus itself is devoid of glomeruli, both intact and sclerotic. It contains a large dilated vein, a few tubules, loose connective tissue stroma and several foci of adipocytes. C, This case of segmental hypoplasia from a 22-year-old shows another hypoplastic focus bordered on the right by normal-appearing cortex. The focus itself is devoid of glomeruli, both intact and sclerotic. It contains large dilated veins and loose connective tissue. A few small immature-appearing tubules are noted in the subcapsular region. D, This case of segmental hypoplasia from a 22-year-old shows a distorted appearing hypoplastic focus bordered by normal-appearing cortex on each side. The presumed cortical tissue is on the right side of the hypoplastic focus. On the left side is an oval nodule of rudimentary medullary tissue. To its left is a large artery and large dilated vein.
The underlying medulla is rudimentary and abnormally developed. The limited medullary tissue contains collecting ducts but lacks loops of Henle (Figs. 4A–D). Since loops of Henle are derived from glomeruli their absence is to be expected since glomeruli are absent in the hypoplastic foci. The collecting ducts may be set within a distinctive appearing cellular mesenchymal tissue unlike that of a normal renal pyramid or have collarets of spindle cells, features are often referred to as medullary dysplasia.20,28 The medullary stromal cells and collarets of spindle cells often express estrogen and/or progesterone receptor (Fig. 5). Similar aberrant expression has been described surrounding the immature ducts of dysplastic kidneys.
FIGURE 4.
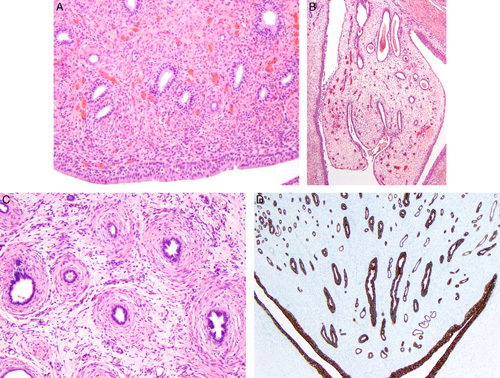
A, The medullary tissue of this hypoplastic focus from a 7-year-old contains collecting ducts widely separated by densely cellular stroma. No endothelial-like loops of Henle are visible. Note urothelium at the bottom of the image. B, This hypoplastic focus from a 33-year-old shows a polyploidy-appearing medullary tissue within a urothelial-lined calyx. The medullary tissue consisting of a few ducts within loose edematous-appearing stroma. No loops of Henle are present. C, The medullary tissue in this hypoplastic focus from a 22-year-old contains collecting ducts that have collarets of spindled cells. No loops of Henle are present. D, This cytokeratin 7 stains highlights the collecting ducts. Notice that no endothelial-like loops of Henle are visible. Immunoperoxidase stain for cytokeratin 7.
FIGURE 5.
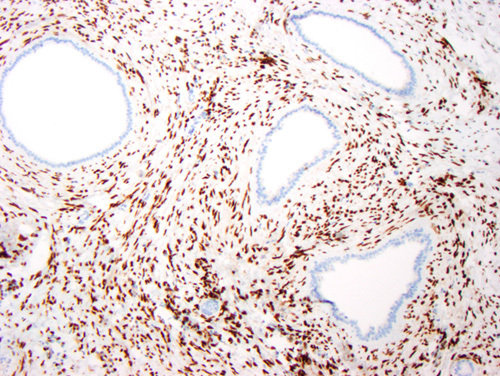
The medullary stromal cells in this case strongly express estrogen receptor. The progesterone receptor stain was similarly positive. The collecting ducts are negative. Immunoperoxidase stain for estrogen receptor.
The radiologic and gross differential diagnosis of segmental hypoplasia includes chronic pyelonephritis and remote infarct. The renal arteries supplying the hypoplastic foci are small, but patent, and otherwise appear normal. This argues against infarction in the pathogenesis of the lesions. Histologically, a remote infarct contains acellular collapsed glomerular tufts and acellular tubular basement membrane remnants, features not seen in segmental hypoplasia. However, since the renal arteries supplying the hypoplastic foci are small and extrarenal vascular anomalies such as fibromuscular dysplasia or renal artery aneurysm occur in 40% of cases, a role for ischemia in the disease process is possible but further supports developmental contributions to the lesions.22,29,30
Chronic pyelonephritis is particularly challenging radiologically and clinically to distinguish from segmental hypoplasia since pyelocalyceal abnormalities and a history of urinary tract infections may be present in both. However, histologic differences between chronic pyelonephritis and segmental hypoplasia are marked. In chronic pyelonephritis there is a prominent inflammatory component, and sclerotic glomeruli or glomeruli with periglomerular fibrosis are present. In addition, clusters of atrophic tubules with thick replicated basement membranes, atrophic tubules with colloid casts, so-called thyroidization of tubules and dense interstitial fibrosis are present in chronic pyelonephritis but are not encountered in segmental hypoplasia. However, since vesicoureteral reflux and urinary tract infections are common in segmental hypoplasia, concomitant pyelonephritic changes can be expected in some patients which can complicate recognition of underlying segmental hypoplasia.
The etiology of segmental hypoplasia was originally regarded as congenital in origin, likely due to congenital reflux that damages a ureteric bud branch impairing development of the lobe. Most reported presentations occur in children and neonatal presentations have been described confirming in utero onset in some cases.15,18,31 However, adult presentations also occur as supported by several of the cases reported herein. Furthermore, some cases with typical gross and microscopic features of segmental hypoplasia appear acquired based upon serial imaging studies. Shindo et al18 in 1983 reported apparent progression from a normal kidney, to kidneys with the typical radiologic, gross, and microscopic features of segmental hypoplasia. They suggested that segmental atrophy is a more appropriate term at least in some cases. However, imaging studies performed in the 1980s are less sensitive compared with current imaging technology. It is conceivable that the development of a lesion with features of segmental hypoplasia in a previously normal kidney may actually represent a lesion that was initially unable to be imaged. Regardless of whether lesions of segmental hypoplasia have onset in utero, are acquired, or both, vesicoureteral reflux appears to be the common pathogenic insult.
Oligomeganephronia (Oligomeganephronic Hypoplasia)
A second type of hypoplasia, oligomeganephronic hypoplasia, commonly referred to as oligomeganephronia, was first described by Röyer and colleagues32,33 in 1962 in the French literature. The name oligomeganephronia reflects its distinctive histologic features, a paucity of nephrons which are markedly enlarged. Although several additional reports appeared in French in the 1960s the first detailed description in the English literature was by Bernstein8 in his 1968 hypoplasia review. The literature on oligomeganephronia is scant and mostly consists of case reports or small series.33–44 The only large series was reported by Broyer et al41 in 1997, the group who first described the disease in the 1960s. They reported the clinical and pathologic features of 67 patients. Twenty-nine were followed for 3 to 17 years. Twelve patients had renal biopsies and 21 nephrectomies were studied. Their report provides the most comprehensive description of this rare renal disease. Broyer and colleagues’ criteria for a diagnosis of oligomeganephronia were rigorous as follows.
Reduction of kidney size with the sum of both kidney lengths ≤80% of 1 kidney of a same-sized child.
Reduction in glomerular filtration rate to 30% normal.
Absence of urinary tract malformations and no evidence of significant vesicoureteral reflux (small amounts of reflux were accepted provided no scars were identified).
As we will see later, many reports of so-called “oligomeganephronia” have employed far more relaxed criteria.
Oligomeganephronia as defined above is by definition a pediatric disease. Broyer et al41 reported that 29 of 50 patients presented at birth or by 1 year of age, and the remaining 21 patients presented by ages 10 to 15 years. Patients are typically born premature or small for gestational age. Most cases are sporadic, but familial forms occur. It has been reported in twins and in siblings.36,39 There is a 3:1 male predominance. The clinical presentation begins with concentrating defects, such as dehydration, polyuria, and polydipsia, in the first few years of life. Hyperfiltration with proteinuria develops later, followed by progressive renal failure, the onset dependent upon the combined renal mass.41 In Broyer and colleagues’ 1997 series end-stage renal disease (ESRD) occurred between 6 months and 17 years. Hypertension, unlike segmental hypoplasia, is rarely present. Genetic causes of oligomeganephronia have been reported but with more lenient diagnostic criteria. The list includes renal-coloboma syndrome (PAX2 mutation), branchiootorenal syndrome (EAY1 mutation), acrorenal syndrome (RET1 mutation), and chromosome 4 deletion syndrome.42–45 These are discussed further below.
Oligomeganephronia is a bilateral disease with kidneys symmetrically affected unless associated with unilateral agenesis, an infrequent association. The kidneys are very small at birth with reduced number of renal lobes, 5 to 6 or fewer, sometimes a few as 1 or 2 lobes. The number of nephrons per lobe is also reduced. Broyer et al41 reported that in 21 nephrectomies performed at the time of renal transplantation cortical hypoplasia was present with radial glomerular counts markedly reduced to 2 to 6 rather than the normal of 9 to 10.
Early in the disease the entire nephron including glomeruli, juxtaglomerular apparatus and tubules are markedly enlarged compared with normal nephrons (Figs. 6A–C). Anecdotally, the number of capillary loops also appear markedly increased. In 1969, Fetterman and Habib46 performed nephron microdissection on kidneys from a 13-year-old male. His kidneys weighed 30 and 35 g (normal: 110 g). The mean glomerular diameter and volume were 2 and 12 times normal, respectively. The mean proximal tubular length and tubular cell volume was 4 and 17 times normal, respectively (Fig. 7A). Griffel et al47 also performed microdissection studies in an 8.5-year-old with oligomeganephronia and unilateral renal agenesis. Although, they did not report the size or weight of the kidney, their dissections demonstrated markedly enlarged glomeruli, and longer and more voluminous tubules compared with an age-matched control (Fig. 7B). Table 1 lists gross findings and glomerular morphometrics for a number of reported cases of oligomeganephronia demonstrating the marked glomerular enlargement.
FIGURE 6.
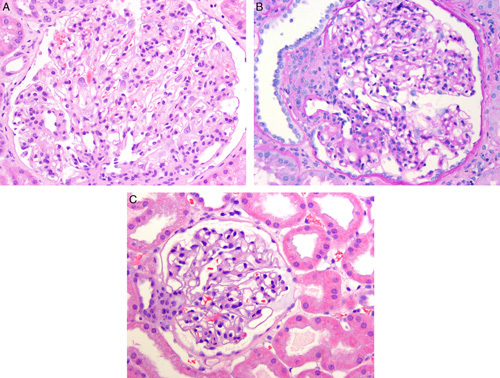
A, This glomerulus from a renal biopsy in a 6-year-old with oligomeganephronia shows marked enlargement. Also note that the number of glomerular capillary loops appears greatly increased compared with a normal glomerulus shown in (C). B, Another glomerulus from a renal biopsy in a 6-year-old with oligomeganephronia shows a glomerulus with an enlarged juxtaglomerular apparatus on the left. Periodic-acid Schiff stain. C, A normal-sized glomerulus from a 6-year-old with minimal change disease shown at similar magnification as (A) and (B) for comparison.
FIGURE 7.
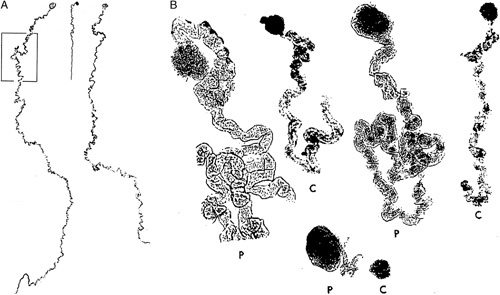
A, This is a mosaic photograph of 2 glomeruli with proximal tubules microdissected from a kidney of a patient with oligomeganephronia, left and right side. These are compared with an age-matched control glomerulus with proximal tubule shown in the center. Reprinted from Fetterman and Habib,35 Fig. 2 with permission from Oxford University Press. Copyright Oxford University Press, New York, NY. All permission requests for this image should be made to the copyright holder. B, This image shows glomeruli and proximal tubules (P) microdissected from a patient with oligomeganephronia compare to an age-matched control (C). Reprinted from Griffel et al,47 Fig. 2 with permission from Springer Nature. Copyright Springer Nature, New York, NY. All permission requests for this image should be made to the copyright holder.
TABLE 1.
Oligomeganephronia—Autopsy and Biopsy Findings

In a renal biopsy the diagnosis of oligomeganephronia can be challenging since knowledge of kidney size is required if the stringent criteria of Broyer et al41 are applied. In addition, information on the number of renal lobes would be particularly helpful. Unfortunately, both data points are usually lacking. Histologically, there is marked enlargement of glomeruli and tubules (Figs. 6A, B). Because of hypertrophy of both glomeruli and tubules, the glomerular density is decreased resulting in fewer glomeruli per millimeter of cortex compared with a normal biopsy in a patient of similar age. In an uncomplicated scenario, the combination of bilateral small kidneys, very large nephrons and absence of nephrosclerosis eliminates acquired atrophy with secondary nephron hypertrophy from consideration. However, with disease progression, segmental, and global glomerulosclerosis with tubulointerstitial scarring develops complicating the histologic recognition of oligomeganephronia. For a diagnosis of oligomeganephronia in this situation a specific underlying disease, glomerulonephritis, pyelonephritis, etc., should be excluded and the renal size should be markedly reduced, beyond that expected from ESRD of other causes, a difficult determination.
The literature on oligomeganephronia is quite limited and muddled with questionable cases. Excluding Broyer and colleagues’ large series, <30 nonsyndromic cases have been reported.33–40,46–49 Many reported cases fall short of fulfilling Broyer and colleagues’ criteria. For instance, cases have been reported in children with renal sizes in the 7.5 to 10 cm range, significantly exceeding Broyer and colleagues’ size criteria.50 Cases have also been reported in adults with advanced CKD despite oligomeganephronia, by definition, representing a pediatric disease with such severe reduction in nephron mass that ESRD develops in childhood or adolescence.51,52 Although the kidneys were smaller than normal in adult cases reported, in the range of 7 to 8.5 cm, CKD was also present.51,52 Therefore, the original kidney size would have been significantly larger before onset of atrophic changes. Acquired atrophy with secondary nephron hypertrophy is more likely in these cases rather congenital oligomeganephronic hypoplasia.
Simple Hypoplasia
The third type of hypoplasia is simple hypoplasia. The affected kidneys have been referred to in the older literature as a doll’s kidney, dwarf kidney, miniature kidney or “trop petit rein.”1–8,53–56 The diagnostic criteria for simple hypoplasia was defined by Rubenstein et al6 in 1961 as 1 kidney weighing 50% or less than expected, or if both kidneys were small, their combined weight was <33% of that expected. Kissane further opined in 1966 that “more important than … mass is enumeration of renal lobules” (Figs. 8, 9A–D). He was referencing renal lobes. “A truly hypoplastic kidney possesses a markedly reduced number of reniculi and calyces, 5 or fewer, in contrast to the normal complement of 10 or more.”56
FIGURE 8.
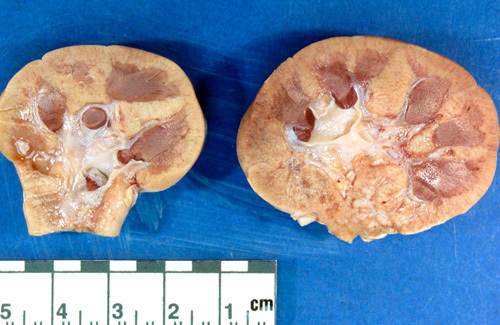
These bivalved kidneys are from a child with bilateral simple hypoplasia. They are markedly reduced in size and have only 3 to 4 and 5 to 6 renal lobes, respectively.
FIGURE 9.
A, This gross photograph from a 50-year-old female shows a simple hypoplastic kidney. The kidney weighed 50 g and was 6 cm in length. There is a marked reduction in number of renal lobes; only 5 renal pyramids are visible in this bivalved specimen. B, Histology from the kidney in (A) is normal. There is no atrophy, cysts or dysplastic features. The number of nephron generations counted in several lobes ranged from 9 to 10. Periodic-acid Schiff stain. C, This gross photograph of a simple hypoplastic kidney from an adult appears somewhat distorted. Only 4 lobes and 3 renal pyramids are noted in this bivalved specimen. D, Histology from the kidney in (C) is normal. There is no atrophy, cysts, or dysplastic features. The number of nephron generations counted in several lobes ranged from 9 to 10. Periodic-acid Schiff stain.
Simple hypoplasia is often associated with hypertension, and if bilateral, progressive renal insufficiency. When unilateral, the contralateral kidney may be hypertrophied and renal function normal. Simple hypoplasia may be an isolated abnormality but is most often associated with other extrarenal nomalies.8,56 In simple hypoplasia the kidney, other than its small size, is normal in all other aspects. Radiologic assessment is not adequate for the diagnosis which can reveal small size but not normal histology.
Histologically, in simple hypoplasia the nephrons are normal (Figs. 9A, B). The issue of nephron size is not addressed in most reports which were all published many years ago, but nephron enlargement should not be present since it is the single definitional criteria in distinguishing simple hypoplasia from oligomeganephronia.1–8,53–56 Furthermore, there should be no cysts or renal dysplasia. Simple hypoplasia is most obvious in the young patient with unilateral disease. With bilateral disease, acquired injury due to low nephron mass and progressive CKD with aging makes it more difficult to histologically distinguish atrophy in a hypoplastic kidney, from atrophy in a developmentally normal kidney.
When strict anatomic criteria are applied, simple renal hypoplasia is very rare. Rubenstein et al6 in a 1961 review of 2153 pediatric autopsies, found only 58 cases (0.27%) of hypoplasia. All were simple hypoplasia. Fifty-six cases were bilateral and 32 of these cases had malformations of other organ systems raising the prospect of a genetic cause. Similarly, in 1975, Risdon et al57 in a radiologic study of 49 small kidneys with pathology correlation identified only 2 cases of hypoplasia, both were examples of simple hypoplasia. No examples of oligomeganephronia or segmental hypoplasia were identified in either study.
Cortical Hypoplasia
A fourth type of hypoplasia, cortical hypoplasia, was introduced in a renal hypoplasia classification by Bonsib58 in 2012 but has been known for many years. Cortical hypoplasia was first described in dogs in 1957.59 Bernstein and Meyer in the 1960s discussed cortical hypoplasia in humans.7,8 In this form of hypoplasia, there are reduced numbers of nephron generations (Figs. 10A–C). Cortical hypoplasia presumably represents premature cessation or arrest of nephrogenesis resulting in fewer nephron generations than normal, and therefore, smaller overall renal size. Medullary rays are smaller as are the renal pyramids.
FIGURE 10.
A, This case of isolated cortical hypoplasia is from a 17-day-old with a 4 cm/9.8 g kidney (normal: 4.5 to 6 cm/13 to 19 g). The cortex contains 3 to 5 nephron generations. The medullary tissue at the bottom is normal. B, This is another lobe from the above case has 3 normal nephron generations and fourth generation of aberrantly formed glomeruli just beneath the renal capsule at the top of the image. C, In several lobes from the above case the medullary tissue contained collecting ducts within a dense cellular stroma. Loops of Henle were not present.
Cortical hypoplasia may, rarely, occur alone as noted in 1 of 18 cases reported above. It is often associated with other forms of renal hypoplasia. In Broyer et al41 large series of oligomeganephronia reduced radial glomerular counts were identified in 21 nephrectomy cases. In the 18 cases of hypoplasia reported herein, 3 of 9 cases of segmental hypoplasia and 5 of 8 cases of simple hypoplasia also showed reduced radial glomerular counts. There are also other clinical associations (Table 2) most related to developmental abnormalities of ampullary buds. In 1980 Henneberry and Stephens60 published an autopsy study of renal hypoplasia and dysplasia in 19 patients with urethral valves. They noted that in the mildest forms of ureteral ectopia near normal renal histology may be observed but associated with reduced radial glomerular counts. In a second study of 32 autopsies and 11 surgical nephrectomies in patients with partial ureteral obstruction and vesicoureteral reflux, Sommers and Stephens61 identified reduced radial glomerular counts to 6 generations, or less. Hinchcliffe et al62 showed that reduced radial glomerular counts are commonly present in nonscarred lobes in patients with vesicoureteral reflux-associated chronic pyelonephritis. They graded cortical hypoplasia based upon radial glomerular counts as follows: none >10, minimal 8 to 10, moderate 4 to 8, severe <4. Severe hypoplasia was identified in 47 of 86 patients.
TABLE 2.
Clinical Contexts Associated With Cortical Hypoplasia/Reduced Radial Glomerular Counts

THE AUTHOR’S PERSONAL EXPERIENCE WITH 18 CASES
Eighteen cases of renal hypoplasia have been collected by the author over many years while practicing at several medical centers. The 18 cases comprise 9 cases of segmental hypoplasia with or without cortical hypoplasia, 8 cases of simple hypoplasia with or without cortical hypoplasia, and 1 case of cortical hypoplasia (normal number of renal lobes). No cases of oligomeganephronia are included. Limited demographic data was available on each case primarily consisting of age and sex. Most cases included renal size and/or weight. The diagnostic criteria employed in this study for each type of hypoplasia are listed in Table 3. The cases satisfy the criteria for hypoplasia as defined by Rubinstein and colleagues.6,8
TABLE 3.
Renal Hypoplasia Diagnostic Criteria
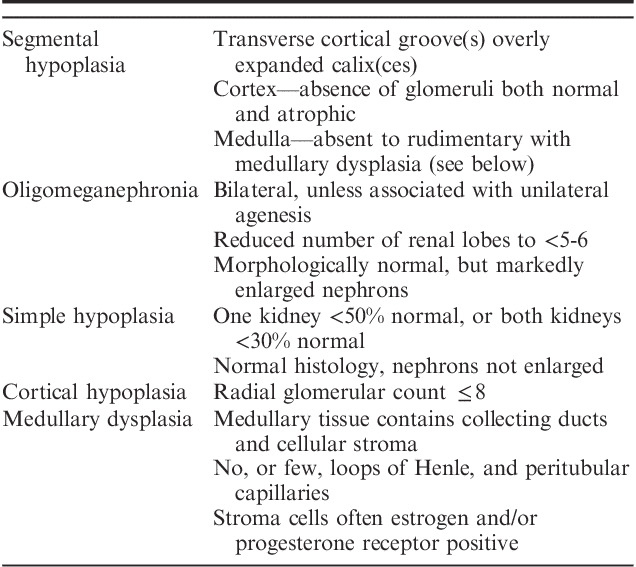
The number of nephron generations was estimated by the Radial Glomerular Count Method which employed sections oriented along medullary rays.63 The glomerular layers on each side of the medullary ray were counted. The normal number of nephron generation ranges from 12 to 14, although usually no >10 generations can be observed in optimally oriented sections. Cortical hypoplasia is defined by radial glomerular counts of ≤8 nephron generations.63 The age-related normal ranges for renal weights and sizes utilized published data from several sources.64–66
Nine cases of segmental hypoplasia were reviewed (Table 4). The patient ages ranged from 11 months to 44 years. Five were 19 years of age, or less. All 9 cases were unilateral. One patient presented with malignant hypertension, blood pressure 220/120, which was cured by nephrectomy. Two patients had ureteral ectopia. Two other kidneys were hydronephrotic. One other patient presented with a urinary tract infection.
TABLE 4.
Renal Hypoplasia—Clinical and Pathologic Features of 18 Cases

All 9 cases of segmental hypoplasia were diagnosed on a nephrectomy specimen. The kidneys in 8 cases were much smaller in size and/or weighed much less than that expected for the patient’s age (Table 4). In 1 case the kidney size and weight were not available. Grossly, from 2 to 5 hypoplastic foci were identified by external examination. The hypoplastic foci were deeply indented and often sharply delineated on each side by normal-appearing cortex (Figs. 1A, B). In cases with multiple adjacent hypoplastic foci, the foci merged resulting in a very small kidney with an irregular profile (Fig. 1C). In a bivalved kidney the hypoplastic foci grossly appeared gray-white in contrast to the adjacent red-brown normal cortex. No grossly discernable corticomedullary differentiation was present in the hypoplastic foci.
Microscopically the “cortical” tissue contained small foci of small tubules (Figs. 3A–D). There were multiple large dilated veins and thick-walled arteries which permitted identification of the tissue as cortical since large veins and arteries are not present in the medulla. The cortical stroma was loose without dense fibrosis. Focal lymphocytic inflammation and foci of adipocytes (not present in the normal cortex) were present in several cases. No intact or sclerotic glomeruli were noted in the hypoplastic foci in 8 cases. One hypoplastic focus in 1kidney contained several normal-appearing glomeruli without tubular segments. One kidney contained a few immature-appearing or embryonal-like cell nests and tubules in the immediate subcapsular area. No cortical cysts, cartilage, or immature/dysplastic ducts were present as seen in dysplastic kidneys.
All hypoplastic foci histologically contained medullary tissue (Figs. 3A–D, 4A–D). The medullary tissue was much smaller in quantity compared with the adjacent normal renal pyramids. It was usually deep to the cortex as expected but appeared isolated from the cortical tissue and not connected to the scattered cortical tubules. Specifically, no medullary rays were present, the usual route of corticomedullary tubular connection. Occasionally, medullary tissue was off-set, adjacent to, lateral to, the cortex or (Fig. 3D). The medullary tissue contained collecting ducts. However, loops of Henle were absent, as expected since they would be derived from glomeruli which were not present in the hypoplastic cortex (Figs. 4A–D). The absence of loops of Henle was readily demonstrated by cytokeratin 7 staining in which variably-sized collecting duct cuboidal to columnar cells are seen with no intervening endothelial-like thin loops of Henle cells (Fig. 4D). Most medullary tissue had a cellular immature-appearing stroma unlike the stroma of normal renal pyramids. In 2 cases the medullary collecting ducts had collarets of spindled cells (Fig. 4C). The stromal cells and collecting duct collarets strongly expressed estrogen receptor in 5 of 5 cases stained and progesterone receptor in 2 of 5 cases stained (Fig. 5). This combination of findings is often referred to as medullary dysplasia (Table 2).
The cortex adjacent to the hypoplastic foci contained histologically normal nephrons in 7 cases. In 2 cases, segmental glomerulosclerosis (31 and 35 y old) was noted affecting some glomeruli. Although histologically normal nephrons were present in nonhypoplastic lobes in all 9 cases, in 4 cases multiple lobes showed cortical hypoplasia with reduced radial glomerular counts (Figs. 11A, B). The radial glomerular counts in 3 cases were severely reduced to 2 to 3 nephron generations and in 1 case the radial glomerular counts were moderately reduced to 6 to 7 generations, compared with 9 to 10 nephron generations in the other cases (Table 4). In addition, in 1 case the medulla underlying normal-appearing cortex (2 y old) had cellular immature-appearing stroma similar to the medullary changes in the hypoplastic foci. Estrogen and progesterone receptor stains were negative in that case.
FIGURE 11.
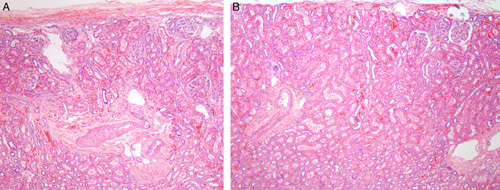
A, This image in a case of segmental hypoplasia from an 11-year-old shows cortical hypoplasia in a lobe adjacent to a hypoplastic focus. Only 2 nephron generations are present. Normal-appearing outer medulla is visible below the arteries in each image. B, This is a second image in a case of segmental hypoplasia from the same 11-year-old that showed cortical hypoplasia in a lobe adjacent to another hypoplastic focus. Only 2 nephron generations are present. Normal-appearing outer medulla is visible below the arteries in each image.
Nine cases of simple and/or cortical hypoplasias were studied (Table 4). They consisted of 1 case of cortical hypoplasia, 3 cases of simple hypoplasia and 5 cases of combined simple and cortical hypoplasia. The patients ages ranged from 17 days to 50 years old. Seven were pediatric cases. All cases were unilateral. The tissue reviewed consisted of 4 autopsies and 5 nephrectomies.
The single case of isolated cortical hypoplasia occurred in a 17-day-old female (Table 4). The kidney was grossly reniform and modestly reduced in weight at 9.8 g (normal: 13 to 19 g). The contralateral kidney was of normal size, 16.5 g. On cut surface corticomedullary differentiation was present in the hypoplastic lobes but the cortex appeared thin and the renal pyramids small. The cortex in the hypoplastic foci contained predominately histologically normal nephrons without glomerulosclerosis, tubulointerstitial scarring, or arterial sclerosis (Figs. 10A, B). However, immediately beneath the capsule a few scattered small dysmorphic glomeruli and occasional small immature-appearing tubules were present. The normal nephrons in the hypoplastic focus were not enlarged. Medullary rays were absent or only minimally developed since the number of nephron generations, and therefore, descending and ascending tubules that populate the medullary rays, were few. Radial glomerular counts ranged from 3 to 5 (Table 4). The underlying renal medulla appeared normal in some lobes (Fig. 10A) while in other lobes medullary dysplasia was present consisting of collecting ducts widely separated by cellular stroma with few loops of Henle (Fig. 10C).
Three cases of simple hypoplasia were reviewed (Table 4). Two cases were from adults and 1 was from a 17-year-old. All were unilateral. In 2 cases, the kidney was one third smaller in size and weight compared with a normal kidney (Table 4). The kidney in 1 case was 50% or less, smaller or lighter, than normal (Figs. 9A, C). The number of renal lobes in 2 cases where lobes were counted were 6 and 7 lobes, respectively. The nephrons were histologically normal (Figs. 9B, D). Specifically, no glomerulosclerosis, tubulointerstitial scarring, vascular sclerosis, cysts, or dysplastic development were noted in any case. The glomerular diameter in 1 adult case was normal. The glomerular diameters in 2 cases were enlarged. The largest glomeruli were 300 and 350 µm compared with a normal adult glomerulus of 200 to 250 µm. Nephron generation ranged from 9 to 10. The medullary tissue was histologically normal.
Combined simple and cortical hypoplasia was identified in 5 cases (Table 4). The patient ages ranged from 3 weeks to 11 years. All cases were unilateral. The hypoplastic kidney sizes were markedly decreased to one third of normal in 1 case, and to less than one fourth of normal in 4 cases (Fig. 12A, Table 4). The contralateral kidney was reported as normal in size in all cases. The nephrons were histologically normal and not enlarged (Figs. 12B, 13A, D). No glomerulosclerosis, tubulointerstitial scarring or vascular sclerosis were present. In addition, no cysts or dysplastic development was present. However, a single island of cartilage that was noted among normal nephrons in 1 case (Fig. 13B). The medullary rays were inconspicuous and poorly developed (Fig. 13C). The nephron generations ranged from as low as 2 to 3, to a maximum of 5 to 6. The medulla was dysplastic in 2 cases containing collecting ducts within cellular immature stroma with only a few loops of Henle identified (Fig. 13D). No medullary abnormalities were noted in the other 3 cases.
FIGURE 12.
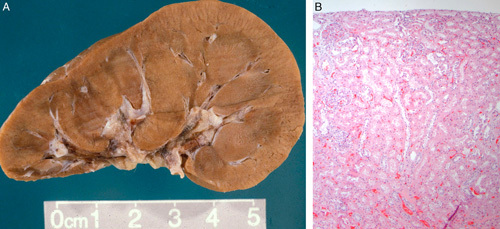
A, This gross photograph shows a bivalved kidney from 11-year-old with both simple hypoplasia and focal cortical hypoplasia. The kidney weighed 40 g and was 7.5 cm long. In the center left of the specimen is focal cortical hypoplasia. The cortex is thin and 3 small renal pyramids are visible. B, The hypoplastic cortex from the case in (A) has histologically normal nephrons but only 3 nephron generations are present in this lobe.
FIGURE 13.
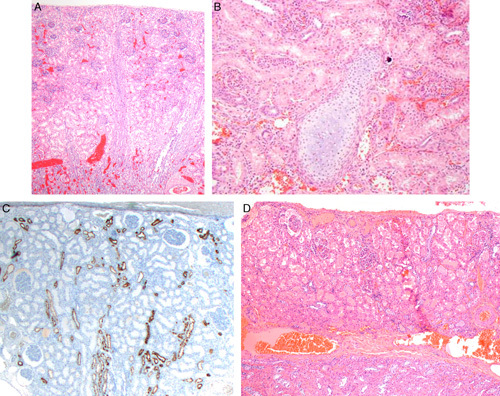
A, This example of simple and cortical hypoplasia is from a 3-week-old. The kidney weighed 8 g. The opposite kidney weighed 34 g. The histology of both cortex and medulla is normal. However, only 5 to 6 nephron generations are present. B, Although no cysts or dysplastic developmental abnormalities were noted in the above case a single focus of cartilage was noted among otherwise normal nephrons. C, Immunoperoxidase staining for epithelial membrane antigen on the case in Figures 11A and B shows poorly developed medullary rays in the center with only a few distal tubules and collecting ducts. D, This is a case of mixed simple and cortical hypoplasia from a 7-year-old. The kidney weighed 15.6 g (normal for age: 65 to 85 g). The histology of the cortex is normal but only 2 nephron generations are present in this lobe. The underlying medullary tissue at the bottom of the photograph shows collecting ducts in a densely cellular stoma. A few loops of Henle were present but not readily seen in this image.
Genetic Causes of Hypoplasia
Normal nephrogenesis is dependent upon proper ampullary bud formation and arborization, and subsequent nephron induction. This is a complex process that requires the intricate coordination, expression, and interaction of many genes. Some of the most important currently known genes are PAX2 (paired box 2). RET (receptor tyrosine kinase), GDNF (glial-derived neurotrophic factor), WT1 (Wilms tumor 1), and wnt-4, to name a few.67,68 Mutation of 1 or more of these genes can affect ampullary bud function leading to “congenital anomalies of the kidney and urinary tract”, a family of mutations of which renal hypoplasia is one of its members. Table 5 lists many of the other malformations.
TABLE 5.
Congenital Abnormalities of the Kidney and Urinary Tract Malformations

Genetic causes of renal hypoplasia are discussed separately, herein, because most reports of simple and oligomeganephronic hypoplasia appeared before the availability of genetic testing. Many would likely have a genetic basis if tested today. Many genetic causes of small kidneys cannot be placed within the known anatomic types of hypoplasia since they lack documentation of renal size, renal weights, and/or histologic evaluation (Table 6). Even when such documentation has been provided, cases often fall short of the stringent criteria established for simple and oligomeganephronic hypoplasia by Rubenstein and colleagues.6,8
TABLE 6.
Genetic Studies of Renal Hypoplasia

One of the most important and widely studied genes in renal hypoplasia is PAX2. PAX2 is a nuclear transcription factor critical to formation of the nervous system and genitourinary tract.67,68 It is a master organizer and one of the first genes activated during fetal development. It is expressed in the nephric duct, ampullary bud and metanephric mesenchyme. Small kidneys have been reported with PAX2 mutations in a sporadic setting, as well as syndromic, specifically the renal-coloboma syndrome (papilledema-renal syndrome).
Porteus et al69 in 2000, studied 29 patients (5 children and 24 adults) with renal-coloboma syndrome. Radiologic evidence of small but otherwise normally formed kidneys was the most common renal abnormality found in 17 patients. Salomon et al42 also studied 9 patients with small but radiologically normal-appearing kidneys. Three of the 9 patients were found to have PAX2 mutations. In retrospect, minor ocular abnormalities were detected in 2 of 3 patients suggesting that they may have a mild form of renal-coloboma syndrome. Kidney sizes were not provided in either study. Salomon and colleagues also reviewed the literature on PAX2 mutations and found 32 cases;18 cases were labeled as having renal hypoplasia. Subtle forms of renal disease may also be caused by PAX2 mutations. Quinlan et al70 in a study of 168 newborns measured kidney volumes by ultrasound. They identified a PAX2 haplotype (AAA) present in 18.5% of newborns that was associated with a 10% reduction in renal volume compared with the most common haplotype (GGG/GGG). Unfortunately, histology was not part of these 3 studies.
Histology was part of 2 PAX2 mutation and renal hypoplasia studies. Nishimoto et al43 tested for PAX2 mutations in 20 patients with radiologically small kidneys. Only 2 patients harbored PAX2 mutations, ages 4 and 8 years. Their kidneys were 67% to 78% of normal size, much larger than the original definitions for both simple and oligomeganephronic hypoplasia. Renal biopsies were performed in 11 patients. Ten had “oligomeganephronia” based upon having the combination of small kidneys and normal but greatly enlarged nephrons. One patient was classified as having simple hypoplasia. The size of the kidneys and patient ages in 18 of 20 cases with small kidneys but lacking PAX2 mutations were not provided. The latter is important since oligomeganephronia as classically defined is a pediatric disease with marked reduction in renal size. Martinovic-Bouriel et al74 reported autopsy findings in 2 fetuses with renal-coloboma syndrome. One fetus, 24 weeks of gestational age, had unilateral renal hypoplasia with kidney weight 10% of normal and a contralateral cystic dysplastic kidney. The other fetus, 18 weeks of gestational age, had kidneys with a combined weight 50% normal for gestational age. Histology showed a scant nephrogenic zone. Collectively, these 5 studies indicate that PAX2 mutation can cause small kidneys, widely variable in size, but PAX2 mutation accounts for only a small percentage of small but radiologically normal kidneys.
Several other mutations have been identified associated with smaller than normal kidneys. Zhang et al71 studied 136 normal newborns and identified a RET single nucleotide polymorphism (RET 1476A allele) in Caucasian newborns. Affected patients had 10% smaller renal volumes and 9% less cystatin C levels (a measure of renal function). So-called oligomeganephronia was identified in some patients with branchio-oto-renal syndrome due to EYA1 gene mutation and in Townes-Brocks syndrome due to SALL1 mutation.72,73 Finally, Anderson et al45 identified small kidneys, 6.8 and 7.7 cm, and large glomeruli on biopsy, in patients with Seckel syndrome and chromosomal 4 abnormalities.
Oligonephronia
All types of hypoplastic kidneys have in common reduced nephron number. However, nephron quantifications have demonstrated that even patients with putatively normal kidneys may have too few nephrons, referred to as oligonephronia. Nephron quantifications were first performed almost 100 years ago.75,76 It was discovered from the beginning that there is broad range of nephron number in the “normal” kidney, that is, a kidney that is grossly and histologically normal, in a patient with normal renal function. The broad range in nephron number in otherwise normal individuals has received attention over the past few decades because of the association of congenitally low nephron number with adult-onset hypertension and CKD.10,11,77–82 Although methodologies have varied, modern glomerular quantifications consistently show a Gaussian distribution in nephron numbers in the normal population. The “average” kidney contains ∼900,000 to 1,000,000 nephrons.77–87 However, nephron numbers range from as low as 210,000, to as high as 2,700,000, almost a 12.9-fold range (Table 7).
TABLE 7.
Glomerular Numbers—Range and Average in Adults

Glomerular number is known to be influenced by many factors (Table 8).42–45,67–105 Gestational causes of oligonephronia, maternal, and intrauterine, are particularly important because of their prevalence. Maternal factors include smoking, gestational hyperglycemia, nutritional deficiencies, drug and alcohol abuse, and chronic infections.88–90 Although potentially preventable, maternal factors represent enormous social and economic issues not easily addressed in the United States but especially challenging in Third World Countries. The World Health Organization estimates that worldwide there are 1.22 billion smokers and 422 million adults living with diabetes, while the United Nation’s Food and Agriculture Organization estimated that the incidence of malnutrition worldwide in 2014 was 795 million.88
TABLE 8.
Causes of Oligonephronia

Intrauterine factors such as prematurity and growth retardation/small for gestational age are major sources of oligonephronia.11,78–81,86–102 It has been demonstrated that there is a direct linear correlation between birth weight and nephron number80,81,87 (Fig. 14A). For each kilogram increase in birth weight there is an increase in 185,000 to 300,000 nephrons per kidney.81 Like maternal factors, intrauterine factors are extremely prevalent worldwide. The World Health Organization (WHO) estimated that in 2010 that 14.9 million, or 11.1% of all births worldwide, were preterm defined as birth occurring at 37 weeks, or less.88 Furthermore, a daunting 36% of live births are either preterm, low birth weight defined as <2500 g, small for gestational age, or a combination, underscoring the massive global potential for CKD due to oligonephronia.88
FIGURE 14.
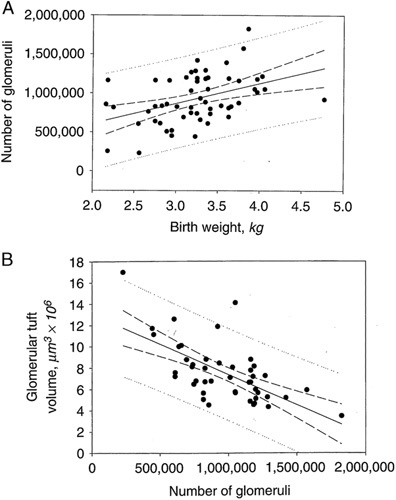
A, The relationship between birth weight and total glomerular number among all cases that includes infants, children, and adults. Number of glomeruli versus birth weight (●); number of glomeruli versus birth weight regression (____); 95% regression confidence interval (--); regression prediction interval (……). The regression coefficient predicts a gain of 257,426 glomeruli/kg increase in birth weight (r=0.423, P=0.0012, N=56). Reprinted from Hughson et al,86 Fig. 4 with permission from Elsevier. B, Relationship between glomerular number and glomerular volume. Reprinted from Hughson et al,86 Fig. 6 with permission from Elsevier. Copyright Elsevier, Amsterdam, Netherlands. All permission requests for this image should be made to the copyright holder.
The developmental defect underlying prematurity-associated oligonephronia is premature cessation of nephrogenesis leading to cortical hypoplasia.89,94,103–105 Glomerular number is a biological variable fixed at birth in the normal individual; no new nephrons are formed after 34 to 36 weeks gestational age. Most nephron are formed in the third trimester; 60% are formed after the 28th week gestation a period when many preterm fetuses are delivered. Rodriguez et al103 studied 56 very preterm and extremely preterm infants between 23 and 30 weeks estimated gestational age and found a marked reduction in radial glomerular counts when corrected for gestational age compared with term infants. Similarly, Sutherland et al105 in an autopsy study of preterm infants that survived up to 68 days noted a decreased thickness of the nephrogenic zone. Furthermore, they noted an increased number of abnormal glomeruli (up to 18%) and enlarged glomeruli, evidence that even formed nephrons experience developmental injury due to prematurity.
Although, in the preterm fetus a limited degree of nephrogenesis occurs in the neonatal period it is insufficient to make up the nephron deficit. Rodriguez et al103 found that glomerulogenesis can continue up to 40 days of life postterm in premature babies but only 1 nephron generation is formed per month rather than the 3 to 4 generations per month in the later stages of a normal gestation. Very premature infants also have reduced renal function. Kandassamy et al95 found that babies born <32 weeks when studied at term corrected age (preterm age plus extrauterine age) compared with term babies have a lower glomerular filtration rate. They also have a smaller kidney volume, a surrogate for nephron number.
There are many other causes of oligonephronia which includes genetic factors, ethnicity, possibly sex, and even height.42–45,67–72,74,78,80 Previously discussed are mutation of PAX2, RET1, EAY1, and chromosome 4 that can result in variable reductions in nephron number from mild decreases in renal size to severely hypoplastic kidneys42–45,67–74 (Table 6). Ethnicity is another determinate of nephron numbers although maternal and intrauterine factors mentioned above contribute in some ethnic groups.78–81 Hoy et al78 found that nephron numbers in United States Caucasians and African American are nearly equivalent, but nephron numbers in African Americans span a much larger range than Caucasians. Both groups have average nephron numbers that are much greater than Australian Aborigines.78,81,99 Japanese have been found to have smaller kidneys than United States Caucasians at autopsy, presumably due to low nephron number which may partially explain their higher incidence of hypertension.96 Hoy et al78 found that males have 9% to 12% more nephrons than females.80 Curiously, taller individuals have been shown to have more nephrons than shorter individuals with an increase of >28,000 glomeruli per centimeter increase in height.78
Oligonephronia and the Fetal Origin of Essential Hypertension
Twenty-three percent of American adults develop essential hypertension which places them at risk for CKD.10,11 Although the pathogenesis of hypertension is heterogeneous, the kidneys appears to be the primary culprit in many cases. Studies of transplant patients have shown that when a normotensive recipient receives a transplant from a hypertensive donor he/she is more likely to develop hypertension than a recipient who receives a kidney from a normotensive patient.106 Conversely, a hypertensive recipient who receives a kidney from a normotensive patient is more likely to become normotensive than if he/she receives a kidney form a hypertensive donor.107
In 1988 Brenner et al10 postulated that essential hypertension in many patients may result from reduced nephron endowment. They argued that low nephron number results in a decrease in total glomerular filtration surface area, that leads to elevated glomerular capillary pressure and hyperfusion injury, that culminates in glomerulosclerosis. The Brenner hypothesis of an inverse relationship between nephron number and hypertension has been confirmed in multiple studies.10,11,76–82,86,88,103 Hayman Jr et al75 in 1929 were the first to establish an inverse relationship between nephron number and hypertension. They counted glomeruli in suspensions and correlated glomerular numbers with blood pressure. Hypertension was present in 15% of people with >1,000,000 glomeruli, 33% of patients with 700,000 to 1,000,000 glomeruli, and 100% of patients with <700,000 glomeruli. Similar to Hayman and colleagues’ findings, it has been shown that United States Caucasians and Australian Aborigines do not develop hypertension if they have >1,000,000 nephrons.81 Finally, Keller et al77 studied 10 normotensive and 10 hypertensive adult patients with primary hypertension. Patients with hypertension had 46% fewer nephrons, average 702,379 versus 1,429,200, compared with age-matched controls. In addition to hypertension and CKD, the diminished renal reserve of low nephron numbers may negatively affect the renal outcome when other primary renal diseases are superimposed. This includes not only potentially progressive diseases such as IgA nephropathy, focal segmental glomerulosclerosis, and autosomal dominant polycystic kidney disease, but diseases generally regarded as not progressive such as minimal change disease.108–113
Glomerular Hypertrophy and Chronic Kidney Disease
The hypertensive diathesis of low nephron number and risk of CKD is associated with nephron hypertrophy. Nephron hypertrophy is regarded as an indication of adverse glomerular hemodynamics and maladaptation due to the stress of low nephron number. The resultant decrease in total glomerular filtration area leads to glomerular hypertension, hyperfiltration, and nephron enlargement that culminates in nephron sclerosis. Numerous studies have demonstrated an inverse correlation between nephron number and glomerular volume in normal adults, premature infants and small for gestational age/growth retarded infants, and in ethnic groups with high incidences of hypertension and CKD such as Native Americans, African Americans, and Australian Aborigines (76 to 83, 85 to 88, 92, 93) (Fig. 14B). Large glomerular volume is also seen in other conditions associated with elevated risk of CKD such as diabetes mellitus, smoking, gout, and obesity114–117 (Table 9).
TABLE 9.
Causes of Nephron Hypertrophy

RENAL HYPOPLASIA: AN CONTEMPORARY PERSPECTIVE BASED UPON HISTORICAL REVIEW AND THE AUTHORS’ PERSONAL EXPERIENCE
Our understanding of renal hypoplasias has evolved considerably since the definitions were first proposed many decades ago. Simple hypoplasia and oligomeganephronia as described by Rubenstein and colleagues,6–8 restricted the diagnosis to the most extreme degrees of nephron deficit such that presentation in childhood was inevitable in those with bilateral disease. Although, the current definitions of hypoplasia remain as originally proposed, in practice, more relaxed definitions are now often employed that accommodate the anatomic continuum in number of renal lobes, number of nephron generations and absolute nephron numbers. This is in accord with dictionary definitions of the general term, hypoplasia, which are not restrictive. For instance, in the Merriman-Webster dictionary hypoplasia is defined as “a condition of arrested development in which an organ or part, remains below the normal size, or in an immature state.118” The degree of reduced size is not definitional.
Even with the strict original definitions of renal simple and oligomeganephronic hypoplasia there is a spectrum of lobar reduction from 1 to 5 lobes, and a broad range of the resultant nephron deficit. Interposed between the extreme degrees of nephron deficit of classic simple and oligomeganephronic hypoplasia and “normal” nephron numbers of 900,000 to 1,000,000, are milder degrees of clinically important nephron deficiency. For instance, nephropathologists on occasion encounter an adolescent whose kidneys are significantly smaller than normal (for instance 7 to 9 cm) presenting with proteinuria and/or renal insufficiency. A biopsy may show marked nephron enlargement with normal histology excluding atrophy as a cause for small renal size and renal impairment. Although the kidneys are larger than classically defined for hypoplastic kidneys, the patient has clinical (proteinuria and/or renal insufficiency) and has biopsy (nephron hypertrophy) features associated with reduced nephron mass. The anticipated renal prognosis is progressive renal disease. Patients with this scenario merge with the lower end of the oligonephronia spectrum, such as those with <700,000 nephrons, in which hypertension and CKDs are inevitable, but delayed until adulthood unless additional stress or renal injury is imposed. This anatomic continuum of kidney size and nephron number defy specification of a diagnostic threshold for renal hypoplasia since the consequences of a nephron deficit may present in a neonate, infant, adolescence, or be delayed until adulthood dependent upon nephron endowment.
The 3 classic forms of hypoplasia, segmental, oligomeganephronic and simple, have in common a reduced number of renal lobes that implicates early ampullary bud abnormalities in development. The finding that several genes important in ampullary bud arborization are associated with renal hypoplasia in humans and experimental demonstration in animals, and that mutation of these same genes enhance ampullary bud apoptosis and reduces ampullary bud branching leading to low nephron number, supports this postulate.67–74,119–121 Reduced ampullary bud branching may also be associated with cortical hypoplasia in otherwise normal-appearing lobes although premature cessation of nephrogenesis could also be primary, especially in the intrauterine causes.
Segmental hypoplasia differs from simple hypoplasia and oligomeganephronia in that there is nearly complete lobar failure of 1, or more lobes, with normal, or near normal, development of most other lobes. Although nephrogenesis is lacking in the hypoplastic lobes rudimentary medullary differentiation is present indicative of limited ampullary bud activity. The seemingly normal-appearing lobes, however, may not completely escape ampullary bud injury since some lobes demonstrate varying degrees of cortical hypoplasia with reduced nephron generations. Interestingly, in several closely related ampullary bud-associated developmental defects such as vesicoureteral reflux, ureteral ectopia, and urethral valves, cortical hypoplasia may also occur.60–62
The main anatomic difference between simple hypoplasia and oligomeganephronic hypoplasia is the presence or absence of nephron hypertrophy. However, this definitional difference may be a matter of circumstance, or timing, rather than a fundamental difference in the disease process. The scattered reports of bilateral simple hypoplasia are often in a neonate or very young infant at autopsy.1–6,53–56 Should these patients have survived longer; nephron hypertrophy due to maladaptation to low nephron number may have developed, thereby, qualifying as oligomeganephronia. In addition, nephron hypertrophy was usually not addressed these reports since most cases occurred before the description of oligomeganephronia in the late 1960s and 1970s.
The most important type of renal hypoplasia because of its prevalence is oligonephronia although it is not formally included in hypoplasia classifications. The anatomic basis for many causes of oligonephronia are not known, that is, how kidneys in the lower quartile of nephron numbers relate to simple, oligomeganephronic, and cortical hypoplasia. A reduced number of renal lobes, a reduced number of nephron generations, or a combination, must be present since these are the only mechanisms in which low nephron numbers can occur. Unfortunately, the number of renal lobes or nephron generations in oligonephronic kidneys were not addressed in studies of nephron quantification.75–86 These features may not have been of interest to the investigators, or the quantification techniques may not have allowed determination of renal lobe number or performance of radial glomerular counts.
Maternal and intrauterine causes of oligonephronia are particularly of concern on a global scale as previously discussed. The magnitude of genetic contribution to oligonephronia has yet to be defined. Although several genes have been associated with small kidneys and renal hypoplasia, many other genes may ultimately be described. Many patients with a genetic cause would not qualify for the classic definition of simple hypoplasia or oligomeganephronic hypoplasia, but they have kidneys smaller than normal and many have nephron hypertrophy, features know to predispose to hypertension and CKD.
In summary, the initial descriptions of a disease entity invariably identify the most severe cases. Establishing a diagnostic threshold for a disease is often problematic, somewhat arbitrary and may become a moving target. Such is true for renal hypoplasias. The original definitions targeted onset in neonates and young children. With the advent of renal biopsy onset in childhood are seen, or may be presumed. If oligonephronia is wrapped into renal hypoplasia classification, then adult presentations occur. In the final analysis, renal hypoplasia and oligonephronia are common and clinically important conditions worldwide, but are deceptive, often clinically silent, lurking in the background until renal consequences develop. Unfortunately, they are difficult to identify both clinically and on renal biopsy, even if diagnostically considered.
Footnotes
The author has no funding or conflicts of interest to disclose.
All figures can be viewed online in color at www.anatomicpathology.com.
REFERENCES
- 1.Eskström T. Renal hypoplasia: a clinical study of 179 cases. Acta Chir Scand. 1955;79(suppl 203):1–74. [PubMed] [Google Scholar]
- 2.Boissonnat P. What to call hypoplastic kidney? Arch Dis Child. 1962;37:142–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kanasawa M, Moller J, Good RA, et al. Dwarfed kidneys in children. Am J Dis Child. 1959;109:130–140. [PubMed] [Google Scholar]
- 4.Emmett JL, Alvarez-Ierena JJ, McDonald JR. Atrophic pyelonephritis versus congenital renal hypoplasia. J Am Med Assoc. 1952;148:1470–1478. [DOI] [PubMed] [Google Scholar]
- 5.Slungaard RK, Jaeck JL. Bilateral dwarfed kidneys. Am J Dis Child. 1959;97:575–582. [DOI] [PubMed] [Google Scholar]
- 6.Rubenstein M, Meyer R, Bernstein J. Congenital abnormalities of the urinary system I: a postmortem study of developmental anomalies and acquired congenital lesions in a children’s hospital. J Pediatr. 1961;58:356–366. [DOI] [PubMed] [Google Scholar]
- 7.Bernstein J, Meyer R. Some speculations on the nature and significance of developmentally small kidneys (renal hypoplasia). Nephron. 1964;1:137–143. [DOI] [PubMed] [Google Scholar]
- 8.Bernstein J. Developmental abnormalities of the renal parenchyma—renal hypoplasia and dysplasia. Pathol Annu. 1968;2:213–2247. [Google Scholar]
- 9.Glassberg KI, Stephens FD, Lebowitz RL, et al. Renal dysgenesis and cystic disease of the kidney: a report of the Committee on Terminology, Nomenclature and Classification, Section on Urology, American Academy of Pediatrics. J Urol. 1989;138:1085–1092. [DOI] [PubMed] [Google Scholar]
- 10.Brenner BM, Garcia DL, Anderson S. Glomeruli and blood pressure. Less of one, more of the other? Am J Hypertens. 1988;1:335–347. [DOI] [PubMed] [Google Scholar]
- 11.Brenner BM, Chertow GM. Congenital oligonephronia and the etiology of adult hypertension and progressive renal disease. Am J Kidney Dis. 1994;23:171–175. [PubMed] [Google Scholar]
- 12.Ask-Upmark E. Über juvenile maligne nephrosklerose und ihr verhältnis zu störungen in der nierenentwicklung. Acta Path et Microbiol Scand. 1929;6:383–442. [Google Scholar]
- 13.Ljungqvist A, Lagergren C. The Ask-Upmark kidney: a congenital renal anomaly studied by micro-angiography and histology. Acta Pathol Microbiol Scand. 1962;56:277–283. [PubMed] [Google Scholar]
- 14.Habib R, Courtecuisse V, Ehrensperger J, et al. Hypoplasie segmentaire du rein avec hypertension arteriélle chez l’enfant. Ann Pediatr (Paris). 1965;12:262–278. [PubMed] [Google Scholar]
- 15.Royer P, Habib R, Broyer M, et al. Segmental hypoplasia of the kidney in children. Adv Nephrol Necker Hosp. 1971;1:145–159. [PubMed] [Google Scholar]
- 16.Habib R.Hudson J, Kincaide-Smith P. Pathology of renal segmental corticopapillary scarring in children with hypertension: the concept of segmental hypoplasia. Reflux Nephropathy. New York, NY: Masson Publishing USA Inc.; 1979:220–239. [Google Scholar]
- 17.Arant BS, Jr, Sotelo-Avila C, Bernstein J. Segmental “hypoplasia” of the kidney (Ask Upmark). J Pediatr. 1979;95:931–939. [DOI] [PubMed] [Google Scholar]
- 18.Shindo S, Bernstein J, Arant BS., Jr Evolution of renal segmental atrophy (Ask-Upmark kidney) in children with vesicoureteral reflux: radiographic and morphologic studies. J Pediatr. 1983;102:847–854. [DOI] [PubMed] [Google Scholar]
- 19.Godard C, Vallotton MB, Broyer M. Plasma renin activity in segmental hypoplasia of the kidneys with hypertension. Nephron. 1973;10:528–534. [DOI] [PubMed] [Google Scholar]
- 20.Zezulka AV, Arkell DG, Beevers DG. The association of hypertension, the Ask-Upmark kidney and other congenital abnormalities. J Urol. 1986;135:1000–1001. [DOI] [PubMed] [Google Scholar]
- 21.Bonsib SM, Meng RL, Johnson FP., Jr Ask-Upmark kidney with contralateral renal artery fibromuscular dysplasia. Am J Nephrol. 1985;5:450–456. [DOI] [PubMed] [Google Scholar]
- 22.Amat D, Camilleri JP, Phat VN, et al. Renin localization in segmental renal hypoplasia: immunohistochemical determination in two cases. Virchows Arch A Pathol Anat Histol. 1981;390:193–204. [DOI] [PubMed] [Google Scholar]
- 23.Barajas L, Marks LS, Trygstad CW. Unilateral renal hypoplasia with associated venous anomaly and hypertension. Virchows Arch A Pathol Anat Histol. 1977;374:169–182. [DOI] [PubMed] [Google Scholar]
- 24.Meares EM, Jr., Gross DM. Hypertension owing to unilateral renal hypoplasia. J Urol. 1972;108:197–201. [DOI] [PubMed] [Google Scholar]
- 25.Johnson JH, Mix LW. The Ask-upmark kidney: a form of ascending pyelonephritis? Br J Urol. 1976;48:393–398. [DOI] [PubMed] [Google Scholar]
- 26.Babin J, Sackett M, Delage C, et al. The Ask-Upmark kidney: a curable cause of hypertension in young patients. J Hum Hypertens. 2005;19:315–316. [DOI] [PubMed] [Google Scholar]
- 27.Sobel JD, Hampel N, Kursbaum A. Hypertension due to Ask-Upmark kidney. Br J Urol. 1977;49:477–480. [DOI] [PubMed] [Google Scholar]
- 28.Dein RW, Walker D, Hackett RL. The Ask-Upmark kidney. A case report. Arch Pathol. 1973;96:10–13. [PubMed] [Google Scholar]
- 29.Marwali MR, Rossi NF. Ask-Upmark kidney associated with renal and extrarenal arterial aneurysms. Am J Kidney Dis. 1999;33:1–5. [DOI] [PubMed] [Google Scholar]
- 30.Sugimoto T, Tanaka Y, Niita N, et al. Renal segmental hypoplasia, Ask-Upmark kidney, in a patient with adult-onset hypertension. Intern Med. 2006;45:1101–1102. [DOI] [PubMed] [Google Scholar]
- 31.Valderrama E, Berkman JI. Ask-Upmark kidney in a premature patient. Clin Nephrol. 1979;11:313–317. [PubMed] [Google Scholar]
- 32.Röyer P, Habib R, Mathieu H, et al. L’hypoplasie rénale bilatérale congénitale avec réduction du nombre et hypertrophie des néphrons chez l’enfant. Ann Pediatr (Paris). 1962;38:753–766. [Google Scholar]
- 33.Habib R, Courtecuisse V, Mathieu H, et al. Un type anatomoclinique particulier d’insuffisance rénale chronique de l’enfant. L’hypoplasie oligonéphronique congénitale bilatérale. J Urol Nephrol (Paris). 1962;68:139–143. [PubMed] [Google Scholar]
- 34.Morita T, Wenzl J, McCoy J, et al. Bilateral renal hypoplasia with oligomeganephronia: a quantitative and electron microscopic study. Am J Clin Pathol. 1973;59:104–112. [DOI] [PubMed] [Google Scholar]
- 35.Wing LNg, Cheung MF, Chan CW, et al. Oligomeganephronic renal hypoplasia. Pathology. 1980;12:639–645. [DOI] [PubMed] [Google Scholar]
- 36.Moerman Ph, van Damme B, Proesmans W, et al. Oligomeganephronic renal hypoplasia in two siblings. J Pediatr. 1984;105:75–77. [DOI] [PubMed] [Google Scholar]
- 37.Miltényi M, Balogh L, Schmidt K, et al. A new variant of the acrorenal syndrome associated with bilateral oligmeganephronic hypoplasia. Eur J Pediatr. 1984;142:40–43. [DOI] [PubMed] [Google Scholar]
- 38.Kusuyama Y, Tsukino R, Oomori H, et al. Familial occurrence of oligomeganephronia. Acta Pathol Jpn. 1985;35:449–457. [DOI] [PubMed] [Google Scholar]
- 39.Weir MR, Salinas JA, Rawling PC. Intrauterine twin demise with oligomeganephronia. Nephron. 1985;40:482–484. [DOI] [PubMed] [Google Scholar]
- 40.Hopkins K, Mowery J, Houghton D. Congenital oligomeganephronia: computed tomography appearance. Clin Pract. 2013;3:e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Broyer M, Soto B, Gagnadoux M-F, et al. Oligomeganephronic renal hypoplasia. Adv Nephrol Necker Hosp. 1997;26:47–63. [PubMed] [Google Scholar]
- 42.Salomon R, Tellier A-L, Attie-Bitach T, et al. PAX2 mutations in oligomeganephronia. Kidney Int. 2001;59:457–462. [DOI] [PubMed] [Google Scholar]
- 43.Nishimoto K, Iijima K, Shirakawa T, et al. PAX2 gene mutation in a family with isolated renal hypoplasia. J Am Soc Nephrol. 2001;12:1769–1772. [DOI] [PubMed] [Google Scholar]
- 44.Park SH, Chi JG. Oligomeganephronia associated with 4p deletions type chromosomal anomaly. Pediatr Pathol. 1984;13:731–740. [DOI] [PubMed] [Google Scholar]
- 45.Anderson CE, Wallerstein R, Zamerowski ST, et al. Ring chromosome 4 mosaicism coincidence of oligomeganephronia and signs of Seckel syndrome. Am J Med Genet. 1997;72:281–285. [PubMed] [Google Scholar]
- 46.Fetterman GH, Habib R. Congenital bilateral oligomeganephronic renal hypoplasia with hypertrophy of nephrons (oligoméganéphronie), Studies by microdissection. Am J Clin Pathol. 1969;52:199–207. [Google Scholar]
- 47.Griffel B, Pewzner S, Berandt M. Unilateral “oligoméganéphronie” with agenesis of the contralateral kidney, studied by microdissection. Virchows Arch A Pathol Anat Histol. 1977;357:179–186. [DOI] [PubMed] [Google Scholar]
- 48.Van Acker KJ, Vincke VJ, Quatacker J, et al. Congenital oligomeganephronic renal hypoplasia with hypertrophy of nephrons (oligomeganephronia). Arch Dis Child. 1971;46:321–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Scheinman JI, Abelson HT. Bilateral renal hypoplasia with oligonephronia. J Pediatr. 1970;76:369–376. [DOI] [PubMed] [Google Scholar]
- 50.Carter JE, Lirenman DS. Bilateral renal hypoplasia with oligomeganephronia: oligomeganephronic renal hypoplasia. Am J Dis Child. 1970;120:537–542. [DOI] [PubMed] [Google Scholar]
- 51.Fuke Y, Hemmi S, Kajiwara M, et al. Oligomeganephronia in an adult without end stage renal failure. Clin Exp Nephrol. 2012;16:325–328. [DOI] [PubMed] [Google Scholar]
- 52.Kawanishi K, Takai T, Kojima C, et al. Three cases of late-onset oligomeganephronia. Nephrol Dial Transplant. 2011;4:14–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gutierrez R. Surgical aspects of renal agenesis with special reference to hypoplastic kidney, renal aplasia and congenital absence of one kidney. Arch Surg. 1933;27:686–732. [Google Scholar]
- 54.Kansawa M, Moller J, Good RA, et al. Dwarfed kidneys in children: the classification, etiology, and significance of bilateral small kidneys in 11 children. Am J Dis Child. 1965;109:130–140. [PubMed] [Google Scholar]
- 55.Graham AP. Hypoplastic kidney, bilateral: case report. J Urol. 1948;60:581–585. [DOI] [PubMed] [Google Scholar]
- 56.Kissane J.Heptinstall RH. Congenital malformations. Pathology of the Kidney. Boston, MA: Little Brown and Company; 1966:68. [Google Scholar]
- 57.Risdon RA, Young LW, Chrispan AR. Renal hypoplasia and dysplasia: a radiological and pathological correlation. Pediatr Radiol. 1977;3:213–225. [DOI] [PubMed] [Google Scholar]
- 58.Bonsib SM. Atlas of Medical Renal Pathology. New York, NY: Springer; 2013:22–28. [Google Scholar]
- 59.Krook L. The pathology of cortical hypoplasia in the dog. Nord Vet Med. 1957;9:161–176. [Google Scholar]
- 60.Henneberry MO, Stephens FD. Renal hypoplasia and dysplasia in infants with posterior urethral valves. J Urol. 1980;123:912–915. [DOI] [PubMed] [Google Scholar]
- 61.Sommers JT, Stephens FD. Morphogenesis of nephropathy with partial ureteral obstruction and vesicoureteral reflux. J Urol. 1981;125:67–72. [DOI] [PubMed] [Google Scholar]
- 62.Hinchcliffe SA, Chan Y-F, Jones H, et al. Renal hypoplasia and postnatally acquired cortical loss in children with vesicoureteral reflex. Pediatr Nephrol. 1992;6:439–444. [DOI] [PubMed] [Google Scholar]
- 63.Hinchliffe SA, Sargent PH, Chan YF, et al. “Medullary ray glomerular counting” as a method of assessment of human nephrogenesis. Pathol Res Pract. 1992;188:775–782. [DOI] [PubMed] [Google Scholar]
- 64.Geelhoed JJM, Taal RH, Steegers EAP, et al. Kidney growth curves in healthy children from the third trimester of pregnancy until 2 years of age. Pediatr Nephrol. 2010;25:289–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Emery JL, Mithra A. The weights of kidneys in late intra-uterine life and childhood. J Clin Pathol. 1960;13:490–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hansen K, Sung CJ, Huang C, et al. Reference values of second trimester fetal and neonatal organ weights and measurements. Pediatr Dev Pathol. 2003;6:160–167. [DOI] [PubMed] [Google Scholar]
- 67.Harshman LA, Brophy PD. PAX2 in human kidney malformations and disease. Pediatr Nephrol. 2012;27:1265–1275. [DOI] [PubMed] [Google Scholar]
- 68.Dziarmaga A, Quilan J, Goodyer P. Renal hypoplasia: lessons from PAX2. Pediatr Nephrol. 2006;21:26–31. [DOI] [PubMed] [Google Scholar]
- 69.Porteous S, Torban E, Cho N-P, et al. Primary renal hypoplasia in humans and mice with PAX2 mutations: evidence of increased apoptosis in fetal kidneys of PAX21neu +/− mutant mice. Hum Mol Genet. 2000;9:1–11. [DOI] [PubMed] [Google Scholar]
- 70.Quinlan J, Lemire M, Hudson T, et al. A common variant of the PAX2 gene is associated with reduced newborn kidney size. Kidney Int. 2007;18:1915–1921. [DOI] [PubMed] [Google Scholar]
- 71.Zhang Z, Quinlan J, Hoy W, et al. A common RET variant is associated with reduced newborn kidney size and function. J Am Soc Nephrol. 2008;19:2027–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Abdelhak S, Kalatzis V, Heilrig R, et al. A human homologue of the Drosophilia eyes absent gene underlies branchio-oto-renal (BOR) syndrome and identifies a novel gene family. Nat Genet. 1997;15:2247–2255. [DOI] [PubMed] [Google Scholar]
- 73.Keifer SM, Ohlemiller KK, Yang J, et al. Expression of Sall1 transcriptional repressor is responsible for Townes-Brock syndrome birth defects. Hum Mol Genet. 2003;12:2221–2227. [DOI] [PubMed] [Google Scholar]
- 74.Martinovic-Bouriel J, Benachi A, Bonnière M, et al. PAX2 mutations in fetal hypodysplasia. Am J Med Genet A. 2010;152A:830–835. [DOI] [PubMed] [Google Scholar]
- 75.Hayman JM, Jr, Martin JW, Jr, Miller M. Renal function and the number of glomeruli in chronic kidney disease. Arch Intern Med. 1929;64:69–83. [Google Scholar]
- 76.Moore RA. The total number of glomeruli in the normal human kidney. Anat Rec. 1931;48:153–168. [Google Scholar]
- 77.Keller G, Zimmer G, Mall G, et al. Nephron number in patients with primary hypertension. N Engl J Med. 2003;348:101–108. [DOI] [PubMed] [Google Scholar]
- 78.Hoy WE, Bertram JF, Denton RD, et al. Nephron number, glomerular volume, renal disease and hypertension. Curr Opin Nephrol Hypertens. 2008;17:258–265. [DOI] [PubMed] [Google Scholar]
- 79.Hoy WE, Hughson MD, Bertram JF, et al. Nephron number, hypertension, renal disease and renal failure. J Am Soc Nephrol. 2005;16:2557–2564. [DOI] [PubMed] [Google Scholar]
- 80.Hughson M, Douglas-Denton R, Hoy WE. Hypertension, glomerular number and birth weight in African-American and white subjects in the southeastern United States. Kidney Int. 2006;69:671–678. [DOI] [PubMed] [Google Scholar]
- 81.Bertram JF, Douglas-Denton RN, Diouf B, et al. Human nephron number: implications for health and disease. Pediatr Nephrol. 2011;26:1529–1533. [DOI] [PubMed] [Google Scholar]
- 82.Puelles VG, Hoy WE, Hughson MD, et al. Glomerular number and size variability and risk for kidney disease. Curr Opin Nephrol Hypertens. 2011;20:7–15. [DOI] [PubMed] [Google Scholar]
- 83.Nyengaard JR, Brentsen TE. Glomerular number and size in relationship to age, kidney weight, and body surface in normal man. Anat Rec. 1992;232:194–201. [DOI] [PubMed] [Google Scholar]
- 84.Merlet-Bénichou C, Gilbert T, Vilar J, et al. Nephron number: variability is the rule causes and consequences. Lab Invest. 1999;9:515–527. [PubMed] [Google Scholar]
- 85.Hoy WE, Douglas-Denton RN, Hughson MD, et al. A stereological study of glomerular number and volume: preliminary findings in a multiracial study of kidneys at autopsy. Kidney Int Suppl. 2003;83:S31–S37. [DOI] [PubMed] [Google Scholar]
- 86.Hughson M, Farris AB, III, Douglas-Denton R, et al. Glomerular number and size in autopsy kidneys: the relationship to birth weight. Kidney Int. 2003;63:2113–2122. [DOI] [PubMed] [Google Scholar]
- 87.Mañalich R, Reyers L, Herrera M, et al. Relationship between weight at birth and the number and size of renal glomeruli in humans: a histomorphometric study. Kidney Int. 2000;58:770–773. [DOI] [PubMed] [Google Scholar]
- 88.Remuzzi G, Luycka V. The Low Birth Weight and Nephron Number Working Group. The impact of kidney development on the life course: a consensus document for action. Nephron. 2017;136:3–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hinchliffe SA, Lynch MR, Sargent PH, et al. The effect of intrauterine growth retardation on the development of renal nephrons. Br J Obstet Gynaecol. 1992;99:296–301. [DOI] [PubMed] [Google Scholar]
- 90.Luyckz VA, Brenner BM. Birth weight, malnutrition and kidney associated outcomes—a global concern. Nat Rev Nephrol. 2015;11:135–149. [DOI] [PubMed] [Google Scholar]
- 91.Luyckx VA, Bertram JF, Brenner BM, et al. Effect of fetal and child health on kidney development and long-term risk of hypertension and kidney disease. Lancet. 2013;382:273–283. [DOI] [PubMed] [Google Scholar]
- 92.Lackland DT, Egan BM, Fan ZJ, et al. Low birth weight contributes to the excess prevalence of end-stage renal disease in African-Americans. J Clin Hypertens. 2001;3:29–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Luyckx VA, Brenner BM. The clinical importance of nephron mass. J Am Soc Nephrol. 2010;21:898–910. [DOI] [PubMed] [Google Scholar]
- 94.Rodriguez MM, Gómez A, Abithol C, et al. Comparative histomorphometry: a case study of oligonephropathy of prematurity. Pediatr Nephrol. 2005;20:946–949. [DOI] [PubMed] [Google Scholar]
- 95.Kandasamy Y, Smith R, Wright IMR, et al. Extra-uterine renal growth in premature infants: oligonephropathy and prematurity. Pediatr Nephrol. 2013;28:1791–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mackenzie HS, Lawler EV, Brenner BM. Congenital oligonephropathy: the fetal flaw in essential hypertension. Kidney Int. 1996;49(suppl 55):S30–S34. [PubMed] [Google Scholar]
- 97.Lackland DR, Bendall HE, Osmomd C, et al. Low birth weight potentuates to high risk of early-onset chronic renal failure in the Southeastern United States. Arch Intern Med. 2000;22:1472–1476. [DOI] [PubMed] [Google Scholar]
- 98.Barker DJP, Martyn CN. The fetal origins of hypertension. Adv Nephrol Necker Hosp. 1997;26:65–72. [PubMed] [Google Scholar]
- 99.Spencer J, Wang Z, Hoy W. Low birth weight and reduced renal volume in Aboriginal children. Amer J Kidney Dis. 2001;37:915–920. [DOI] [PubMed] [Google Scholar]
- 100.Hughson MD. Low birth weight and kidney function: is there a relationship and is it determined by intrauterine environment? Am J Kidney Dis. 2007;50:531–534. [DOI] [PubMed] [Google Scholar]
- 101.Merllet-Benichou C, Vilar J, Lelievre-Pegerier M, et al. Fetal nephron mass: its control and deficit. Adv Nephrol Necker Hosp. 1997;10:19–45. [PubMed] [Google Scholar]
- 102.White SL, Perkovic V, Cass A, et al. Is low birth weight sand antecendent of CKD in later life? A systematic review of observational studies. Am J Kidney Dis. 2009;54:248–261. [DOI] [PubMed] [Google Scholar]
- 103.Rodríguez MM, Gómez AH, Abitbol CL, et al. Histomorphometric analysis of postnatal glomerulogenesis in extremely preterm infants. Pediatr Dev Pathol. 2004;7:17–25. [DOI] [PubMed] [Google Scholar]
- 104.Faa G, Gerosa C, Fanni D, et al. Marked interindividual variability in renal maturation of preterm infants: lessons from autopsy. J Matern Fetal Neonatal Med. 2010;23(suppl 3):129–133. [DOI] [PubMed] [Google Scholar]
- 105.Sutherland MR, Gubhaju L, Moore L, et al. Accelerated maturation and abnormal morphology in the preterm neonatal kidney. J Am Soc Nephrol. 2011;22:1365–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Strandgaard S, Hansen U. Hypertension in renal allograft recipients may be conveyed by cadaveric kidneys from donors with subarachnoid hemorrhage. Br Med J. 1986;292:1041–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Curtius JJ, Luke RG, Dustan HE, et al. Remission of essential hypertension after renal transplantation. N Engl J Med. 1983;309:1009–1015. [DOI] [PubMed] [Google Scholar]
- 108.Ruggajo P, Svarstad E, Leh S, et al. Low birth weight and risk of progression to end stage renal disease in IgA nephropathy—a retrospective registry-based cohort study. PLoS One. 2016;11:e015319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zidar N, Avgustin Cavic M, Kenda RB, et al. Unfavorable course of minimal change nephrotic syndrome in children with intrauterine growth retardation. Kidney Int. 1998;54:1320–1323. [DOI] [PubMed] [Google Scholar]
- 110.Orskov B, Christensen KB, Feldt-Rasmussen B, et al. Low birth weight is associated with earlier onset of end-stage renal disease in Danish patients with autosomal dominant polycystic kidney disease. Kidney Int. 2012;81:919–924. [DOI] [PubMed] [Google Scholar]
- 111.Hodgin JB, Rasoulpour M, Markowitz GS, et al. Very low birth weight is a risk factor for secondary focal segmental glomerulosclerosis. Clin J Am Soc Nephrol. 2009;4:71–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Duncan RC, Bass PS, Garrett PJ, et al. Weight at birth and other factors influencing progression of idiopathic membranous nephropathy. Nephrol Dial Transplant. 1994;9:875–881. [PubMed] [Google Scholar]
- 113.Rajan T, Barbour SJ, White CT, et al. Low birth weight and nephron mass and their role in the progression of chronic kidney disease: a case report on identical twins with Alport disease. Nephrol Dial Transplant. 2011;26:4136–4139. [DOI] [PubMed] [Google Scholar]
- 114.Rea DJ, Heimbach JK, Grande JP, et al. Glomerular volume and renal histology in obese and non-obese living kidney donors. Kidney Int. 2006;70:1636–1641. [DOI] [PubMed] [Google Scholar]
- 115.Bauer WC, Rosenberg BF. A quantitative study of glomerular enlargement in children with Tetrology of Fallot. A condition of glomerular enlargement without an increase in renal mass. Am J Pathol. 1960;39:695–712. [PMC free article] [PubMed] [Google Scholar]
- 116.Choung H-Y, Bomback AS, Stokes MB, et al. The spectrum of kidney biopsy findings in patients with morbid obesity. Kidney Int. 2019;95:647–654. [DOI] [PubMed] [Google Scholar]
- 117.Denic A, Alexander MP, Kaushik V, et al. Detection and clinical patterns of nephron hypertrophy and nephrosclerosis among apparently healthy adults. Am J Kidney Dis. 2016;68:58–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Merriman-Webster Inc. Merriam-Webster Dictionary. Springfield, MA: Merriman-Webster Inc.; 2018. [Google Scholar]
- 119.Clark AT, Bertram JF. Molecular regulation of nephron endowment. Am J Physiol. 1999;276:F485–F497. [DOI] [PubMed] [Google Scholar]
- 120.Cullen-McEwen LA, Kett MM, Dowling J, et al. Nephron number, renal function, and arterial hypertension in aged Gdnf heterozygous mice. Hypertension. 2003;41:335–340. [DOI] [PubMed] [Google Scholar]
- 121.Dziarmaga A, Clark P, Stayner C, et al. Ureteric bud apoptosis and renal hypoplasia in transgenic PAX2-Bax fetal mice mimics the renal-coloboma syndrome. J Am Soc Nephrol. 2003;14:2767–2774. [DOI] [PubMed] [Google Scholar]