

Abstract
Ligand-switch reactions at the heme iron are common in biological systems, but their mechanisms and the features of the polypeptide fold that support dual ligation are not well understood. In cytochrome c (cyt c), two low-stability loops (Ω-loop C and Ω-loop D) are connected by a heme propionate HP6. At alkaline pH, the native Met80 ligand from Ω-loop D switches to a Lys residue from the same loop. Deprotonation of an as yet unknown group triggers the alkaline transition. We have created two cyt c variants T49V/K79G and T78V/K79G with altered connections of these two loops to HP6. Electronic absorption, NMR and EPR studies demonstrate that at pH 7.4 ferric forms of these variants are Lys-ligated, whereas ferrous forms maintain the native Met80 ligation. Measurements of protein stability, cyclic voltammetry, pH-jump and gated electron-transfer kinetics have revealed that these Thr-to-Val substitutions greatly affect the alkaline transition in both ferric and ferrous proteins. The substitutions modify the stability of the Met-ligated species and reduction potentials of the heme iron. Kinetics of ligand-switch processes are also altered and analyses of these effects implicate redox-dependent differences in metal-ligand interactions and the role of the protein dynamics, including cross-talk between the two Ω-loops. With the two destabilized variants, it is possible to map energy levels for the Met- and Lys-ligated species in both ferric and ferrous proteins and assess the role of the protein scaffold in redox-dependent preferences for these two ligands. The estimated shift in the heme iron reduction potential upon deprotonation of the “trigger’ group is consistent with those associated with deprotonation of an HP, suggesting that HP6, on its own or as a part of a hydrogen-bonded cluster, is a likely “trigger” for the Met-to-Lys ligand switch.
Graphical Abstract
Synopsis
The pH-driven Met-to-Lys ligand switch at the heme iron (so-called ‘alkaline transition’) is examined in ferric and ferrous variants of cytochrome c with a variety of spectroscopic techniques and electrochemistry to elucidate the role of heme propionates in defining structure and redox properties of this important metalloprotein.
Introduction
Functional properties of heme proteins are largely governed by the heme iron ligands and the surrounding polypeptide.1,2 An increasing body of evidence suggests that the propionate groups at the periphery of the heme cofactor, even though somewhat distant from the iron center, could be important but less understood contributors to structure and reactivity of these metalloproteins.3–6 Heme propionates (HPs) affect the dynamics of ligand replacement7 and appear to be critical for function of multiple redox enzymes8–10 as well as for electron delivery both in inter- and intramolecular electron transfer (ET).11–14
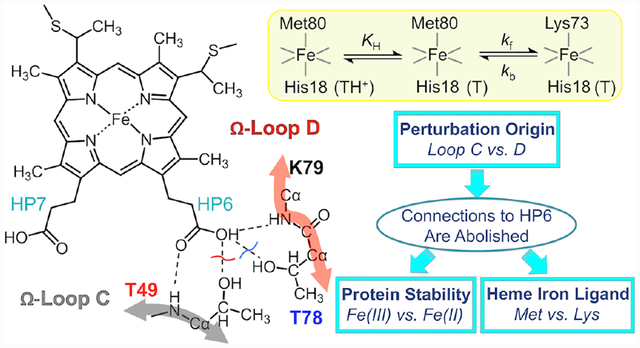
In proteins HPs frequently form hydrogen-bonding interactions with the polypeptide.15 In cytochrome c (cyt c, Figure 1),16 these connectivities contribute to the formation of the compact Met80-ligated native structure, and HPs have been suggested to play a role in cyt c ligand-switch transitions.7,17–19 The outer HP6 in yeast cyt c forms hydrogen-bonding interactions with two Thr residues, Thr49 and Thr78. These threonines are highly conserved in mitochondrial cyt c proteins and when not, they are replaced by Ser or Asn,20 which can also hydrogen bond. Through HP6, the two residues Thr49 and Thr78 connect the two least stable Ω-loops21 (C and D, respectively) in the protein to the heme (Figure 1) and these interactions may be important for the integrity of the tertiary structure. Indeed, we have recently shown that the T49V substitution in horse heart cyt c results in the formation of the Lys-ligated ferric protein at near neutral pH.19
Figure 1.
Structure of yeast iso-1 cyt c (PDB: 2YCC, ref. 16). The highlighted residues, Thr49 (magenta) and Thr78 (cyan), are involved in the intraprotein hydrogen-bonding network, as indicated by the dashed lines. The two Ω-loops are shown in gray (Ω-loop C, residues 40–57, nested-yellow foldon) and red (Ω-loop D, residues 71–85, red foldon).
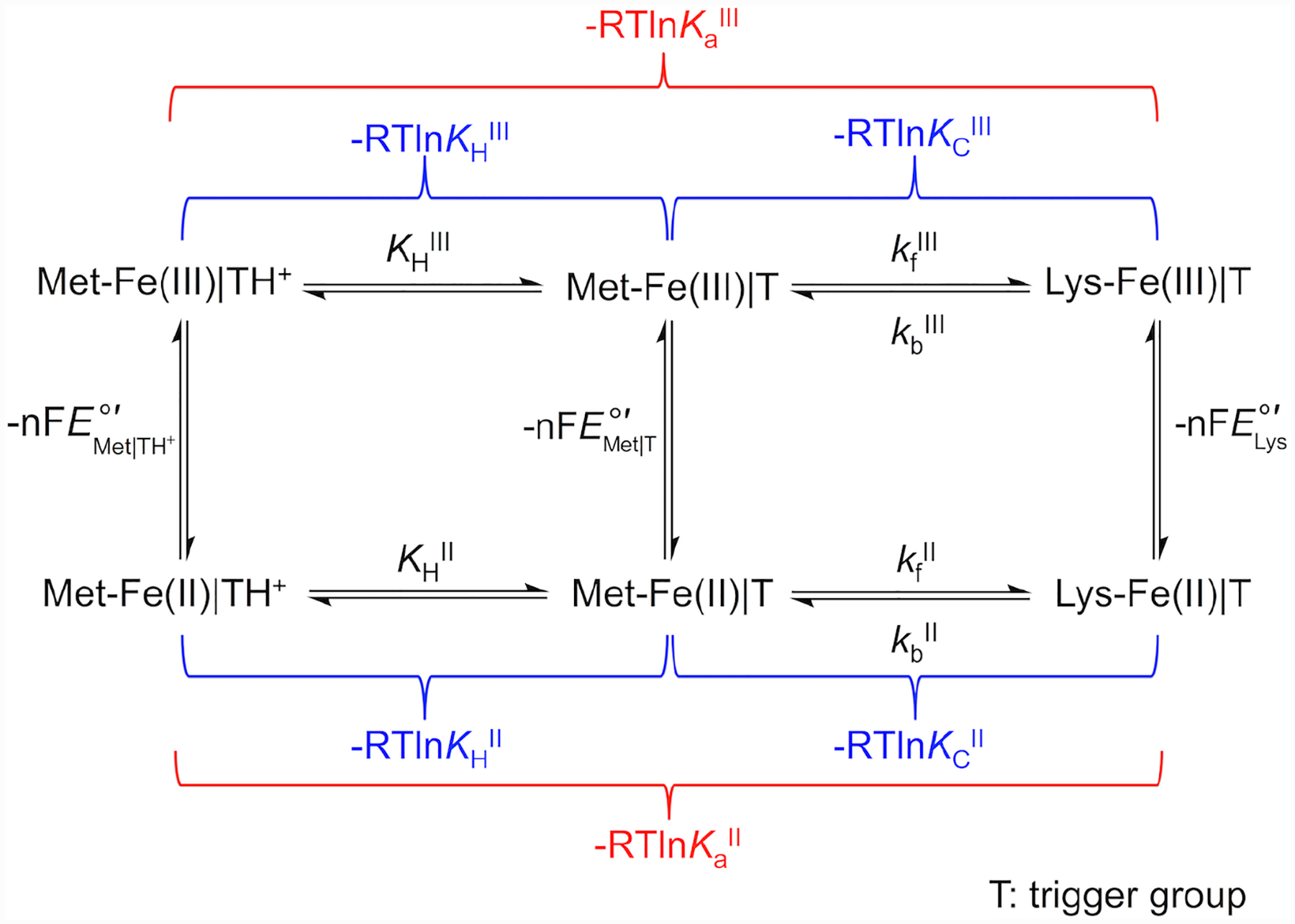
The replacement of the native Met80 ligand to the heme iron in cyt c by a Lys residue within Ω-loop D is termed the alkaline transition because it typically takes place at high solution pH.22,23 First described in the 1940s,24–27 this conformational rearrangement has recently become a subject of increased research interest28–31 owing to potential relevance of non-native cyt c conformers in the mechanism of apoptosis32–34 and the discovery of multiple proteins having Lys as a ligand to the heme iron.35–38 Scheme 1 depicts redox and acid-base equilibria relevant to the alkaline transition, where kf and kb are the forward and backward rate constants associated with Met-to-Lys ligand switch, respectively, and KH is the ionization constant for the “trigger” group whose deprotonation precedes the conformational change.22,39 In this scheme the Met-ligated species with the “trigger” group protonated, Met|TH+, and deprotonated, Met|T, are distinguished.
Scheme 1.
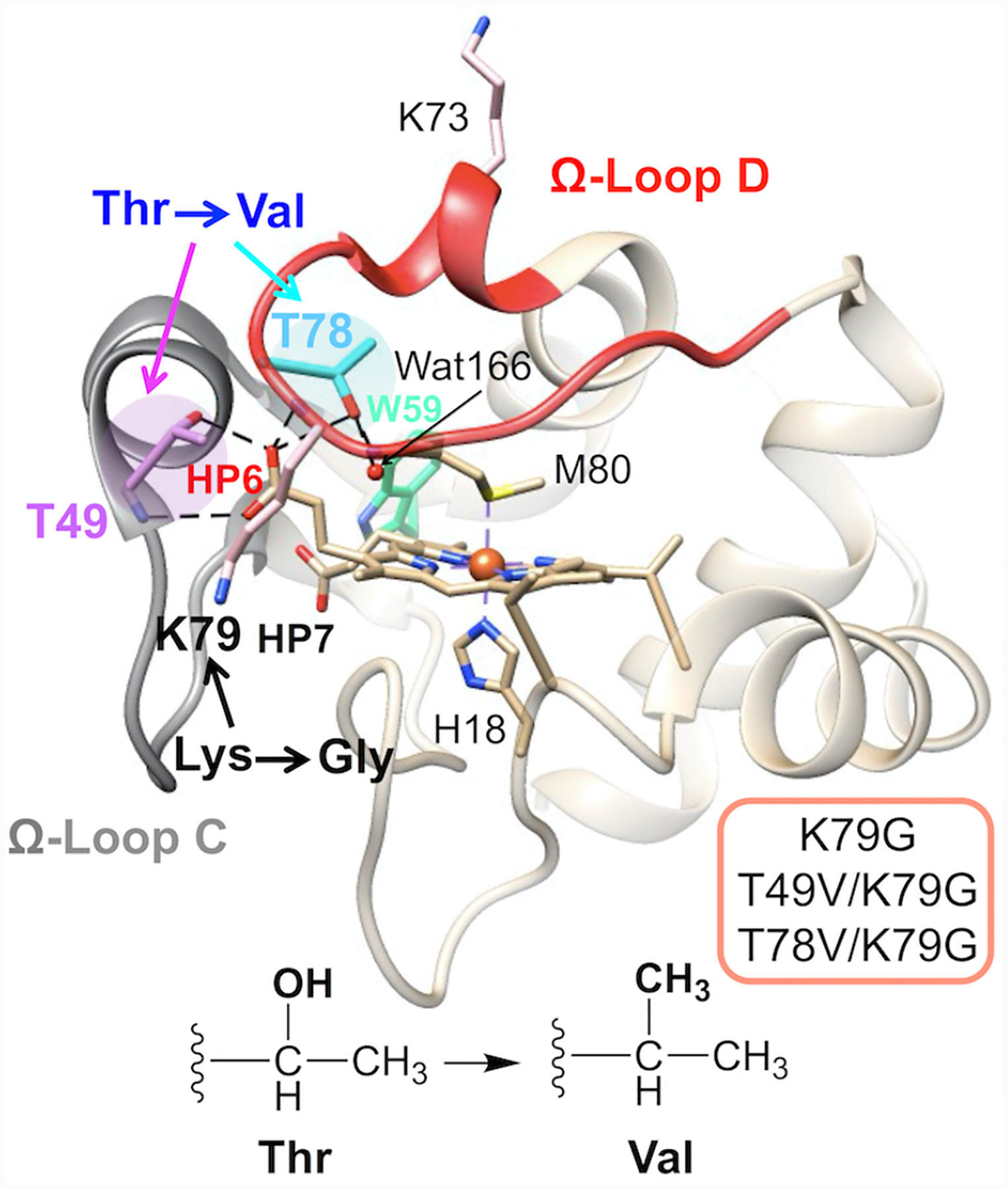
Herein, we systematically compare effects of Thr-to-Val substitutions at residues 49 and 78 on the conformational and redox properties of cyt c. Our recently developed K79G variant of yeast iso-1 cyt c40 simplifies the study of the ligand-switch reactions as only Lys73 can replace Met80 at the heme iron, and we incorporate this background substitution into two cyt c variants, T49V/K79G and T78V/K79G, to target the environment near HP6. Both Thr-to-Val substitutions modify reduction potentials and ligand-switch properties of cyt c but their effects differ. The comprehensive analyses of stability, acid-base equilibria, ligand-switch kinetics in both ferric and ferrous forms of these protein variants offer new insights on the mechanism of the alkaline transition and define the role of the heme-polypeptide interactions, including those that involve HPs, in redox-dependent preferences for a particular axial ligand to the heme iron.
Materials and Methods
Site-Directed Mutagenesis, Protein Expression and Purification.
The two Thr-to-Val variants in this work were created using the plasmid encoding K72A/K79G/C102S iso-1 cyt c40 as a template for mutagenesis. The background substitutions K72A and C102S were used to prevent the misligation of Lys72 to the heme iron and protein dimerization via Cys102, respectively. Mutations were performed using a QuikChange kit (Agilent). The DNA sequences of the prepared plasmids were analyzed at the Molecular Biology Core Facilities of Dartmouth College. The verified plasmids were subsequently transformed into chemically competent BL21 Star DE3 E. coli cells (Invitrogen) for expression. Expression and purification of the variants followed established protocols.41
WT*, K79G, M80A, and M80K* variants were employed for control experiments; their preparation and characterization have been described in our previous work.30,40,42 Similarly to the two Thr-to-Val variants in this work, these control proteins also had K72A and C102S background substitutions; M80K* had an additional K73A substitution.
Spectroscopic Measurements.
Electronic absorption spectra were collected on Agilent 8453 diode-array and JASCO V-630 scanning spectrophotometers. The extinction coefficients of the variants were determined by conducting hemochrome assays according to the published procedures.43 The circular dichroism (CD) spectra of ferric proteins were acquired on a JASCO-J815 CD spectropolarimeter. To measure the far-UV (200–250 nm) CD spectra, proteins with a final concentration of around 15 μM were prepared in a 100 mM sodium phosphate buffer at pH 7.4 or a 100 mM sodium acetate buffer at pH 4.5. For near-UV (250–300 nm) CD spectra measurements, the final concentrations of the proteins were increased to about 100 μM. The path-lengths of the quartz cuvettes for far-UV and near-UV CD measurements were 1 mm and 4 mm, respectively. Fluorescence spectra were recorded on a Horiba Jobin Yvon Spectrolog III fluorimeter and the following parameters were employed for the detection of Trp59 signals: λex = 290 nm and λem = 300–400 nm.
The X-band electron paramagnetic resonance (EPR) spectra of the oxidized variants were acquired on a Bruker EMX 300 spectrometer at 10 K. All the experimental settings were adapted from those for previous measurements.30
The one-dimensional proton nuclear magnetic resonance (1H NMR) spectra were recorded on a 500 MHz Bruker NMR spectrometer. The 1H NMR spectra of ferric proteins were measured at three different pH conditions; protein stocks were exchanged into a 50 mM CAPS (pD 10.5), sodium phosphate (pD 7.4), or acetic acid-d4 (pD 4.5) buffer in D2O and final protein concentrations were between 0.5 and 1 mM. The ferrous protein samples were prepared right before the NMR measurements by exchanging the protein solution into a 50 mM sodium phosphate buffer containing 10% D2O at pH 7.4 and adding 1 mM sodium dithionite in a glove box (COY Laboratory Products) under nitrogen atmosphere. The pulse sequences employed for these measurements were the same as in our previous studies.19
Denaturation Experiments.
The protein global stability and lability of the Met80 ligation were characterized by chemical denaturation experiments using guanidine hydrochloride (GuHCl). The ellipticity at 222 nm is a measure of the extent of secondary structure, whereas the absorbance at 695 nm reflects the amount of protein that is Met-ligated. Stock GuHCl solutions were first made in a 100 mM sodium acetate buffer at pH 4.5 or a 100 mM sodium phosphate buffer at pH 7.4, dilutions with the corresponding buffer yielded desired GuHCl concentrations, and then the final pH of the resulting solutions was adjusted to either 4.5 or 7.4. Final protein concentrations were about 10 μM and 200 μM for CD and electronic absorption measurements, respectively. Changes in ellipticity at 222 nm versus GuHCl concentration were fit to a two-state model by eq 1,44 where mf and bf are the slope and y-intercept of the signal from the folded protein, respectively, mu and bu are the slope and y-intercept of the signal from the unfolded protein, respectively. Cm and mD are the midpoint and the slope of the unfolding transition, respectively. The Gibbs free energy of denaturation, , is the product of mD and Cm.
| (1) |
For experiments with ferrous proteins, GuHCl stock solutions were thoroughly degassed on a vacuum line and all sample preparations were performed in a glove box purged with nitrogen. A small amount of sodium dithionite was added to each sample to keep the protein in the ferrous form during the data collection. Absorption spectra before and after CD measurements verified that the heme iron stayed reduced throughout the experiments.
pH Titrations and Singular-Value Decomposition.
The pH-dependence of the 320–650 nm and 580–750 nm regions of the electronic absorption spectra of ferric proteins were examined separately with the final protein concentrations of about 10 and 100 μM, respectively. The protein samples were first prepared in a 100 mM sodium phosphate buffer at pH 7.4 and subsequently titrated by adding either 1 M NaOH or 1 M HCl. The pH of the protein solution was recorded by an AB15 pH meter (Fisher Scientific) and the absorption spectrum for each pH condition was acquired on an Agilent 8453 diode-array spectrophotometer.
The resulting series of spectra were analyzed by singular-value decomposition (SVD) according to published procedures.45,46 The pH titration profile A(λm, pHn) was deconvoluted into three matrices: the wavelength-dependent matrix U(λ), the pH-dependent matrix V(pH), and the square roots of the eigenvalues S(k) upon the SVD operation (A=USVT) using MATLAB. Multiple criteria were used to determine the number of major components, i.45,46 The most weighted V vectors, Vi(pH), were fit to eq 2 using the global fitting function of SigmaPlot 10.0:
| (2) |
where ti, ni, and pKapp,i are the amplitude, slope, and the apparent pKa value of the ith transition, respectively, and C is the y-intercept for the initial state. The fractional population changes of major components, F(pH), were calculated with the these pKa values and then used to construct the spectra of i components, D(λ), using eq 3:
| (3) |
The spectra for the component corresponding to the Met-ligated species (DMet) in both Soret and charge-transfer regions were calculated using the spectra of the rest of the components (Drest) from the reference set (Table S1). DMet was determined as DMet= (A−DrestFrest)FMet−1, where FMet and Frest are the changes in the fractional populations of the Met-ligated species and the rest of the components, respectively.
Variants WT* (pH 7.4), M80A (pH 4.5), and M80K* (pH 7.4) provided reference spectra for Met/His, H2O/His, and Lys/His cyt c species (Table S1). At pH 2, M80A is unfolded and composed of two-differently ligated species: H2O/H2O (75%) and H2O/His (25%).47 Unfolded M80A in 3 M GuHCl at pH 4.5 was used as a reference for pure (100%) H2O/His unfolded species, and the spectrum of H2O/H2O unfolded species was obtained by linear decomposition of the spectrum for M80A at pH 2 (Figure S1).
Alkaline transitions were also examined in samples of ferrous variants at 10 μM by analyzing absorption changes in the pH range from pH 7.4 to 13.0. An excess of sodium dithionite was added to the protein solutions to keep the heme iron in the ferrous form during titrations.
pH Transitions Monitored by Trp59 Fluorescence and Trp-Heme Distance Calculations.
Samples of ferric proteins at 2 μM were prepared in a 100 mM sodium phosphate buffer at pH 7.4 and their pH were adjusted by adding 1 M HCl or NaOH to create a series of samples in the pH range from 2.0 to 6.0. For each pH condition, the fluorescence spectrum was corrected by subtracting that of the buffer.
The fluorescence spectrum of the fully unfolded K79G (in 4 M GuHCl) has provided a reference FDA(λ) for Trp59 fluorescence in the unfolded protein. By taking into the account the previously reported Trp-to-heme FRET efficiency in unfolded cyt c (E = 0.55),48 the fluorescent intensity of the donor in the absence of the acceptor FD(λ) was calculated using eq 4. This value was then used with the experimental fluorescence intensity, FDA(λ), to calculate the E value for each protein sample in the studied pH range (eq 4). The E values and the known Föster distance (R0 = 34 Å)48 were used to find Trp59-to-heme distances, r, (eq 5). These distances have been referenced to that in folded K79G to characterize the relative increase in this parameter upon Thr-to-Val substitutions and protein unfolding.
| (4) |
| (5) |
pH-Jump Kinetics.
The pH-jump experiments were performed with ferric proteins using a BioLogic SFM-300 stopped-flow instrument. Immediately before the measurements, proteins were exchanged into unbuffered solutions containing 100 mM NaCl. For upward pH-jump measurements, the initial pH conditions of ferric T49V/K79G and T78V/K79G were adjusted to pH 5.0 and 5.8, respectively, by adding 1 M HCl or 1 M NaOH. A series of 12 mM jump buffers (sodium phosphate, pH 6.0–7.4; Tris, pH 7.8–8.6; boric acid, pH 9.0–10.0) containing 100 mM NaCl were made to achieve the final pH range of 6.0–10.0 upon mixing with the protein solution. For downward pH-jump experiments, the initial pH of both T49V/K79G and T78V/K79G were adjusted to pH 8.0. The jump buffers, all at a concentration of 12 mM (sodium phosphate, pH 6.0–7.0) containing 100 mM NaCl, were used to achieve the final pH range of 6.0–7.0. The mixing ratio for protein and jump buffer was 5:1 (vol:vol) to yield the final protein concentration of about 10 μM.
Absorption changes at 405 nm, the wavelength of maximum change in absorbance, were monitored following the rapid mixing of the jump buffer with the protein solution. The traces from the upward (or downward) pH-jump experiments were fit to monoexponential rise (or decay) functions yielding rate constants . The dependences of on pH were fit to eq 6 according to the mechanism in Scheme 122:
| (6) |
The kfIII and KHIII values were determined by the fitting the pH-dependence of to eq 6, whereas kbIII values were obtained by averaging the first three values in the low-pH region of the dependence. The conformational equilibrium constant, KCIII, was calculated as kfIII/kbIII.
Cyclic Voltammetry Measurements.
Gold electrodes were cleaned and prepared with a self-assembled monolayer of 3-mercapto-1-propanol before cyclic voltammetry was carried out in a three-electrode cell as previously described.30 The reference cell side-arm was filled with the same buffer used for the working cell and sparged with argon before final assembly of the reference electrode. Protein samples were dialyzed into buffer solutions of 0.1 M sodium phosphate at pH 7.4 or pH 6.0 or 0.1 M sodium acetate at pH 4.5. The protein solution (≈ 100 μM) in the working cell was allowed to incubate for over 30 minutes under argon flow to avoid the electrocatalytic reduction of oxygen at the low potential limit (≈ −300 mV in these experiments). All potentials were corrected to the standard hydrogen electrode (SHE) using the relationship SHE = SCE + 243 mV at 22 °C.49
The 3-mercapto-1-propanol surface modification and buffer conditions used here are resistant to protein adsorption.50 Nevertheless, to limit the possible influence of protein adsorption over time, scans were collected as quickly as possible, starting with the faster scan rates (those over 1 V/s) before performing slower ones. A fresh electrode was also used to repeat measurements at slower scan rates. As an additional control, scans in the absence of protein were used to find the lower potential limit available at each scan rate and pH.
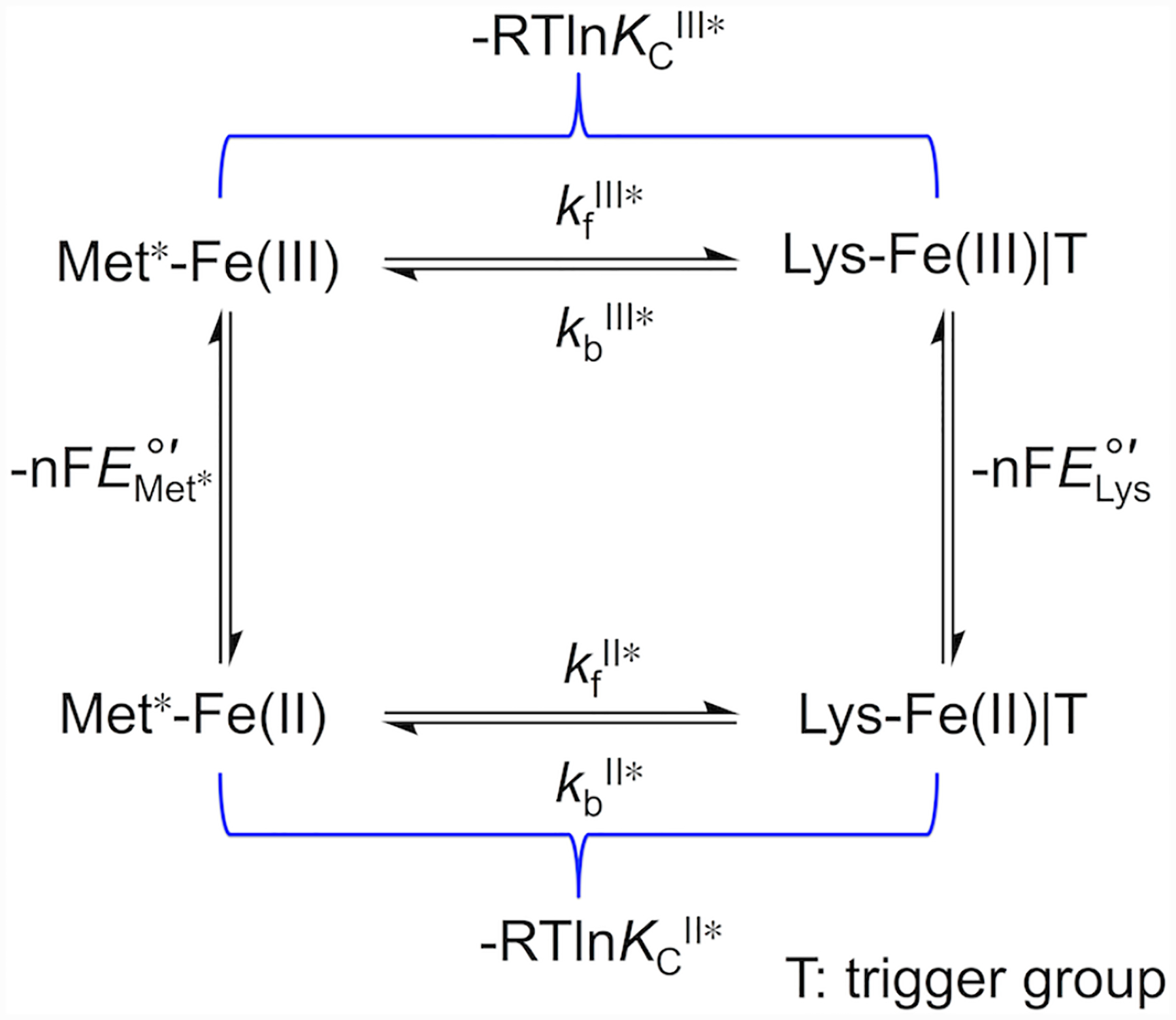
Voltammograms were both analyzed and simulated with software included with the potentiostat (CH Instruments) and DigiElch (Gamry). The modeling software is limited in the complexity of systems amenable to modeling, therefore we have reduced the full mechanistic Scheme 1 to a scheme consisting of only one square (Scheme 2). Instead of differentiating between the Met-ligated species with protonated (Met|TH+) and deprotonated (Met|T) “trigger” group, the effects of the protonation equilibrium of the “trigger” group were folded into the composite conformational exchange with an equilibrium constant KC*, as has been previously done in spectroscopic studies of the alkaline transition.51 The relationship between the equilibrium constant for this modeled ligand switch, KC*, and the true conformational equilibrium constant, KC, is determined by eq 7 and rate constants kf* and kb* are related to kf and kb as follows: kf* = kf × (KH/(KH + [H+])) and kb* = kb. At high scan rates (10 – 50 V/s), positions of signals in voltammograms were not affected by coupling to ligand-switch processes and peak positions were used to find reduction potentials for the Met-ligated (, an equilibrium mixture of Met|TH+ and Met|T species) and Lys-ligated () forms of the protein. These values, together with KCIII* values calculated from experimental pKCIII and pKHIII values, were used to find KCII* values using Scheme 2.
Scheme 2.
| (7) |
To model the voltammetry according to Scheme 2, simulations were carried out based on the rate constants kfIII and kbIII from spectroscopic measurements and reduction potentials and from fast scan voltammetry. The separation of peak positions increased as progressively faster scan rates were used49,52 and heterogeneous interfacial electron exchange constants were adjusted until the simulation matched the experiment. With all the parameters optimized for modeling the voltammetry at the highest and slowest scan rates, simulations were done at scan rates that would correspond to the onset of reversibility for the signal of the Lys-ligated form. The input value for ferrous kbII was systematically varied in 0.5 s−1 increments, and the narrowest range of values was found for each variant that replicated the scan rates where reversibility was reached in the experiment.
Comparison of voltammetry to digital simulation was also used to measure the background capacitance (Cd).49 The Cd value was then multiplied by the scan rate to find the corresponding contribution to the current response. When necessary, this background current was subtracted from the observed voltammetry using SOAS.53
Kinetics of Heme Iron Reduction.
The reduction reactions of ferric yeast iso-1 cyt c variants were carried out with a BioLogic SFM-300 stopped-flow instrument under anaerobic conditions. The reductant, hexaammineruthenium(II) complex (a6Ru2+), was synthesized according to the published method54 from commercial-grade [Ru(NH3)6]Cl3 (Strem Chemicals). The successful production of the a6Ru2+ complex was confirmed by IR spectroscopy and the concentration of a6Ru2+ solution was determined based on the extinction coefficient (ε400 = 30 M−1cm−1) by electronic absorption spectroscopy.55 The protein and a6Ru2+ solutions in a 10 mM sodium phosphate buffer containing 100 mM NaCl at pH 7.0 were made in a glove box right before the experiments. The protein concentration was around 5 μM and the concentration of a6Ru2+ was at least ten-fold greater than that of the protein in each of the studied reactions to achieve pseudo-first-order reaction conditions. For each concentration of a6Ru2+, at least three kinetic traces corresponding to the changes in the absorption at 550 nm as a function of time were collected and fit to a biexponential rise function.
Structural Modeling.
Structural models of ferric and ferrous Met-ligated variants were created using VMD 1.8.6 (Visual Molecular Dynamics)56 and NAMD (Not just A Molecular Dynamics)57 simulations following previously published procedures.40 Force-field parameters for heme c have been published elsewhere.58 Solvation of the protein was achieved by building a water box with a thickness of 3 Å from the protein surface. The timestep was 2 fs and a snapshot structure was saved every 500 steps, resulting in a total of 130 snapshots by the end of each simulation. Analyses of root-mean-square deviations (RMSDs) of the protein backbone atoms in these snapshots suggested a lack of large conformational fluctuations after 0.04 ns of simulations (Figure S2). The last thirty snapshots were analyzed to obtain the statistics in Tables S2 and S3 and averaged to generate structural models in Figure 2. Analyses of the hydrogen-bonding interactions were performed in Chimera 1.10.1.59 The Lys-ligated conformers were modeled using the crystal structure of the Lys73-ligated T78C/K79G variant of cyt c (PDB: 4Q5P)30 with the Rotamers60 tool in Chimera. The solvent accessible surface area of the heme group and the volume of the heme pocket were calculated with POPS (Parameter Optimized Surfaces)61 and POCASA (Pocket-Cavity Search Application)62, respectively.
Figure 2.
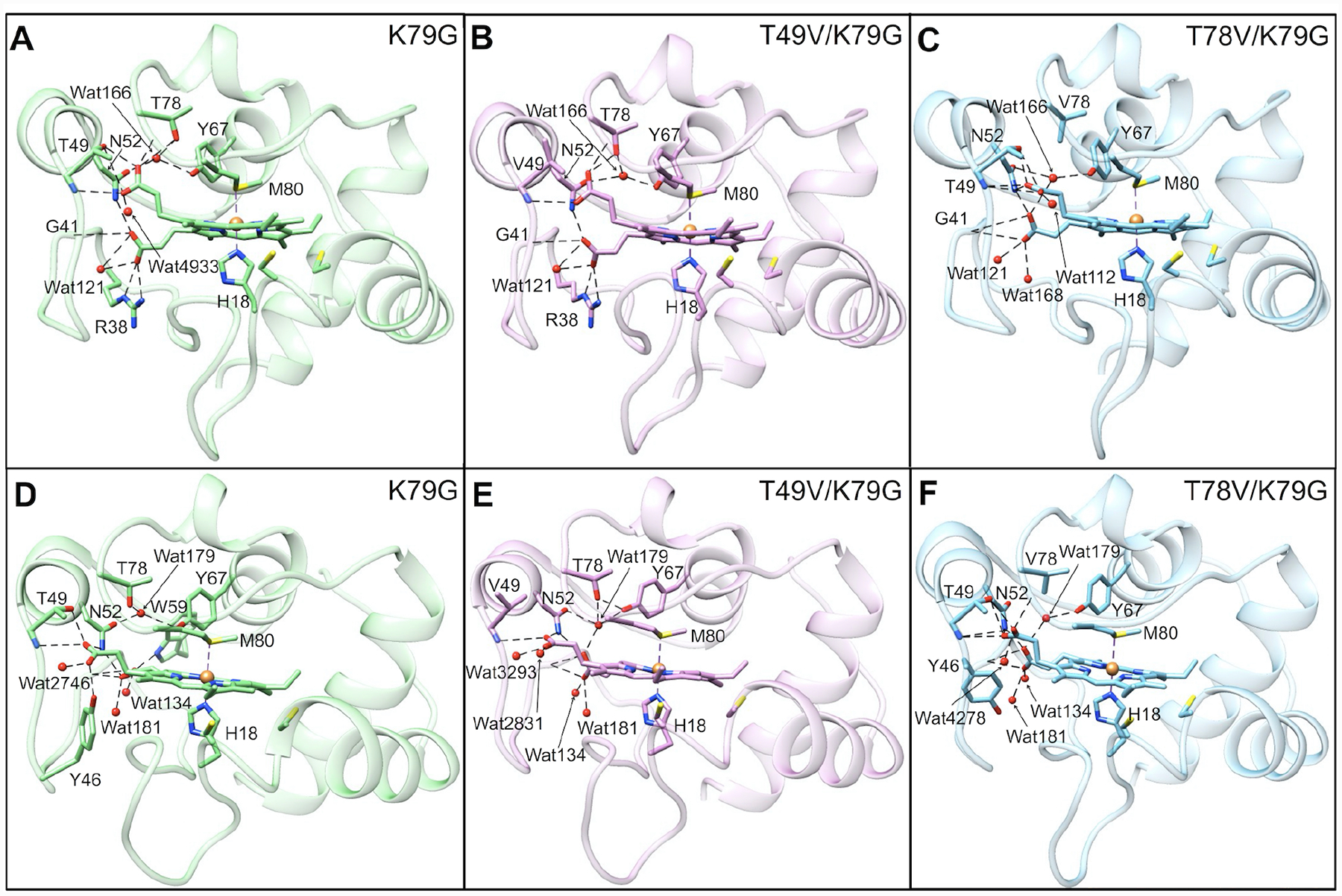
Averaged structural models of Met80-ligated (A–C) ferric and (D–F) ferrous (A, D) K79G, (B, E) T49V/K79G, and (C, F) T78V/K79G. Water molecules are represented by red spheres and the heme iron is represented by an orange sphere. All variants contain K72A/C102S background mutations. Hydrogen-bonding networks in the proximity of the two propionate groups are displayed by dashed lines in all the models.
Results
Characterization of Heme Iron Ligation, Structural Properties, and Stability of Cyt c Variants at pH 7.4.
Spectroscopic features of T49V/K79G and T78V/K79G were compared to their parent variant, K79G, to investigate the effect of the introduced Thr-to-Val substitutions on the axial ligation of yeast iso-1 cyt c. Electronic absorption spectra (Figure 3A) of ferric T49V/K79G and T78V/K79G variants at pH 7.4 have revealed a blue shift for the Soret band (λmax = 406 nm) compared to that for K79G (λmax = 409 nm), suggesting changes in the coordination environment of the heme iron.40 Further, the charge-transfer band at 695 nm characteristic of Met ligation to the ferric heme iron decreases in intensity. The g values from EPR spectra of the two ferric variants are very similar and distinct from those of the Met80-ligated K79G (Figure 3B), but resemble those of Lys-ligated cyt c proteins.63 In addition, in the 1H NMR spectra of ferric T49V/K79G and T78V/K79G resonances at 35 to 30 ppm, which are associated with the Met-ligated heme iron, decrease in intensity and new resonances appear between 25 and 10 ppm, which are characteristic of the alkaline conformers (Figure 3C).64 Collectively, these spectroscopic results suggest that in T49V/K79G and T78V/K79G a Lys residue is able to take the place of Met80 as an axial ligand to the ferric heme iron at pH 7.4, significantly lower than the harsher alkaline conditions required for such ligand replacement in WT and K79G cyt c.22,40
Figure 3.
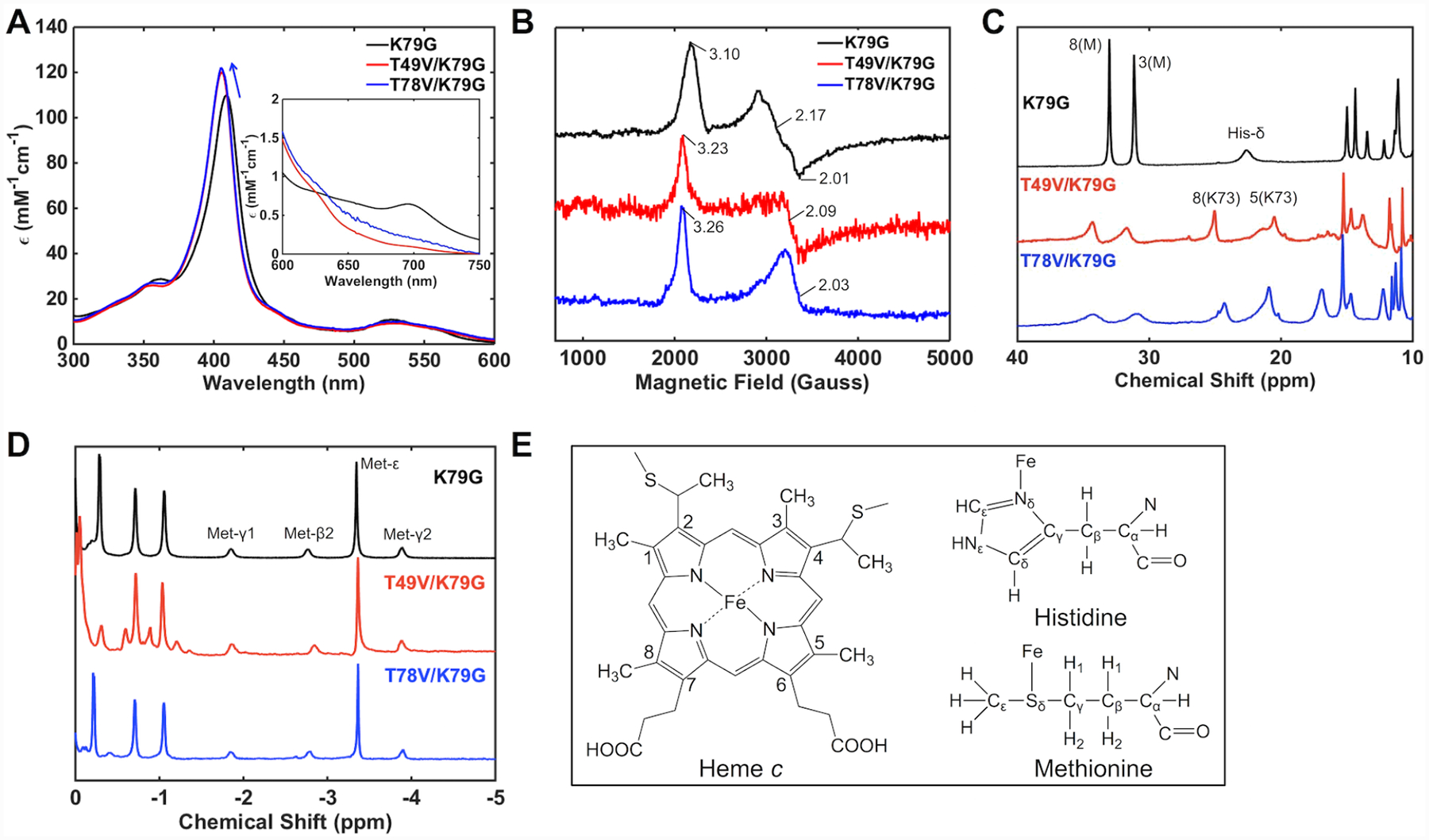
Spectra of K79G (black), T49V/K79G (red), and T78V/K79G (blue) at pH 7.4. (A) Electronic absorption spectra of ferric proteins at 22 ± 2 °C. The arrow indicates a shift in the position of the Soret band. The inset is the comparison of charge-transfer bands at 695 nm. (B) EPR spectra of ferric proteins at 10 K. 1H NMR spectra of (C) ferric and (D) ferrous proteins at pD (pH) 7.4 and 25 °C. (E) Structures of heme, Met, and His showing the labeling nomenclature. Numbered peaks correspond to protons of heme methyl groups, with the identity of the axial ligand (M = Met, K = Lys) in parentheses.
The sensitivity of NMR experiments allows for detection of a small population of Met-ligated species at pH 7.4 for both variants (Figure 3C) but these conformers fully disappear at pH 10.5 where we find only Lys-ligated species with all the above spectroscopic techniques (Figure S3). Monitoring of the charge-transfer band at 695 nm as a function of pH has yielded pKa values for the Met-to-Lys ligand switch (Table 1). These pKa values are the same for the two variants with Thr-to-Val substitutions and almost two pH units lower than that of the parent variant K79G.40 Since both Lys72 and Lys79 within Ω-loop D are substituted by a non-coordinating residue in T49V/K79G and T78V/K79G, Lys73 is the ligand to the heme iron.
Table 1.
Thermodynamic and Kinetic Parameters Associated with Met-to-Lys Ligand Switch in Ferric Yeast iso-1 Cytochrome c Variants
The 1H NMR spectra of the two ferrous variants display identical features to that of K79G (Figure 3D), including a characteristic pattern in the upfield region that arises from Met ligation to the heme iron, suggesting that Met80 persists as an axial ligand at pH 7.4. The electronic absorption spectra of ferrous K79G, T49V/K79G, and T78V/K79G are indistinguishable from each other (Figure S4A). Increasing the pH has revealed two pH-driven transitions in the ferrous variants (Figure S4B and C and Table 2) but neither was associated with a Met-to-Lys ligand switch (Figure S4D). It appears that Met80 persists as a ligand to the ferrous heme iron and, as revealed by CD studies (Figure S4E), its loss is concomitant with protein denaturation at alkaline pH. The switch in ligation from Lys73 to Met80 upon reduction of the heme iron implies that ligand-switch reactions accompany redox reactions in T49V/K79G and T78V/K79G and suggests redox-dependent differences in stabilization of the native Met80 ligation to the heme iron in cyt c.
Table 2.
Thermodynamic and Kinetic Parameters Associated with Met-to-Lys Ligand Switch in Ferrous Yeast iso-1 Cytochrome c Variants
| Variant | Measured pKaII | KfII (s−1)a | KbII (s−1)b | pKCIIc | pKHII | Calculated pKaII | ||
|---|---|---|---|---|---|---|---|---|
| pKa,1II | pKa,2II | using pKHIId | using pKaIIe | |||||
| K79G | n.a. | 11.4±0.1 | (2.8±2.0)×10−3 | 3.5±0.5 | 3.1±0.6 | 12.5±0.3f | 15.6±0.7 | 15.5±0.3 |
| T49V/K79G | 10.8±0.2 | 11.4±0.1 | (1.4±1.0)×10−1 | 7.0±0.5 | 1.7±0.4 | 10.8±0.2g | 12.5±0.4 | 12.5±0.3 |
| T78V/K79G | 10.0±0.4 | 11.5±0.1 | (2.3±1.7)×10−3 | 11.5±0.5 | 3.7±0.6 | 10.0±0.4g | 13.7±0.7 | 13.8±0.4 |
Determined as kfII = kbII × KCII.
Determined from cyclic voltammetry and simulations based on Scheme 2.
Determined according to eq 7 based on KCII* and pKHII.
Determined as pKaII = pKHII + pKCII.
Calculated according to Scheme 1 using , and pKaIII, and the experimentally determined as an estimate for .
Based on the assumption pKHII = pKHIII + 2.
Based on the assumption that pKHII = pKa,1II for the two Thr-to-Val variants.
CD spectra of T49V/K79G and T78V/K79G variants were acquired to evaluate conformational effects of the Thr-to-Val substitutions. In the far-UV region of the CD spectra (Figure 4A), the signals for ferric T49V/K79G and T78V/K79G variants are indistinguishable from each other but differ from that for the parent variant K79G. Similar trends are observed in the near-UV region (Figure 4B). Decreased signals at 222 nm and in the region between 280 and 290 nm suggest a loss of some α-helical content and rearrangement of the tertiary structure upon the Thr-to-Val substitutions. In contrast, in ferrous T49V/K79G and T78V/K79G neither the secondary structure nor the tertiary structure at pH 7.4 changes compared to that in K79G (Figure S5).
Figure 4.
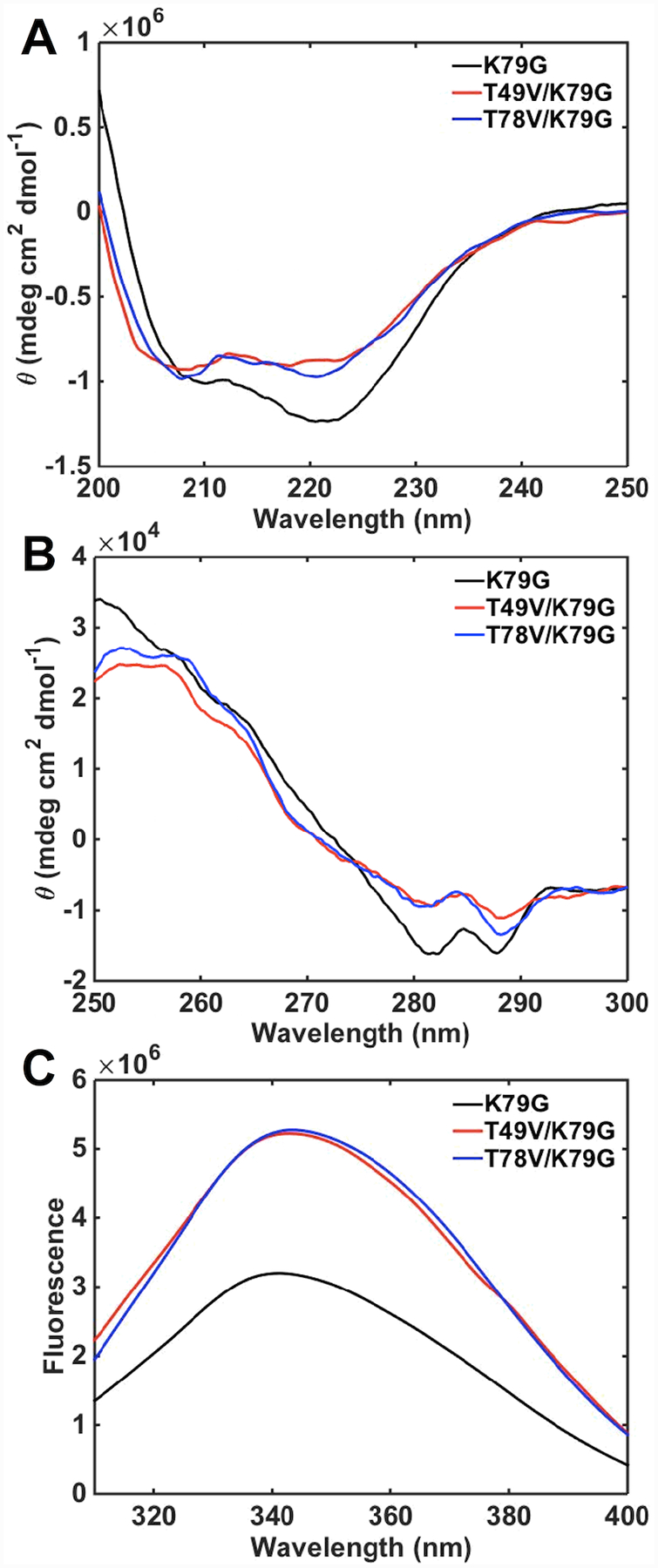
(A) Far- and (B) near-UV CD spectra and (C) Trp59 fluorescence spectra of ferric K79G (black), T49V/K79G (red), and T78V/K79G (blue) in a 100 mM sodium phosphate buffer at pH 7.4 and 22 ± 2 °C.
Trp59 is an intrinsic fluorophore in cyt c and its signal is largely quenched in the folded protein owing to the proximity of this residue to the heme group.48 The higher intensities of Trp59 fluorescence signals for T49V/K79G and T78V/K79G compared to that for K79G (Figure 4C) reveal that the average Trp59-to-heme distance increases as a result of these Thr-to-Val substitutions. However, the magnitude of this change in intramolecular distance (about 10%) is less than that upon transition of native cyt c to the unfolded or even molten-globular state,65,66 suggesting that many protein tertiary contacts are likely preserved in the two variants. Based on these spectroscopic findings, we conclude that, as Ω-loop D reshapes to accommodate Lys73 coordination to the heme iron in ferric T49V/K79G and T78V/K79G, some structural changes take place but these are not major perturbations in the polypeptide fold.
Changes in the CD spectra associated with the loss of the protein α-helical structure in GuHCl denaturation experiments were used to assess global stability of ferric and ferrous variants at pH 7.4 (Figure S6). Considering the error bars of thermodynamic parameters in Table 3, the global stabilities of Lys73-ligated ferric T49V/K79G and T78V/K79G are comparable to that of the Met80-ligated K79G. The midpoints of the unfolding transitions show variation among the variants but the differences are small. The T49V substitution appears to influence the stability parameters the most. The same is true for the ferrous variants: the effects on the value are minimal (T49V/K79G) or none (T78V/K79G), but values do pick up the differences among the variants and suggest again that the Thr-to-Val substitution at residue 49 may be more destabilizing than the one at residue 78. By taking into account the reduction potential of GuHCl-unfolded cyt c (Scheme S1),67 redox-linked changes in protein stability are calculated (Table 3). For T49V/K79G and T78V/K79G, these include changes in both the oxidation state and ligation of the heme iron.
Table 3.
Thermodynamic Parameters for the Unfolding Transitions of Ferric and Ferrous Yeast iso-1 Cyt c Variants at pH 7.4
| Variant | Ferric | Ferrous | ΔΔGb |
|||||
|---|---|---|---|---|---|---|---|---|
| Ligand Xa | ||||||||
| K79G | Met80 (100%) | 1.31±0.03 | 16.9±2.8 | 22.1±3.0 | 3.74±0.03 | 14.3±1.8 | 53.5±6.7 | −17.9±7.5 |
| T49V/K79G | Lys73 (78–85%) | 1.10±0.06 | 14.9±3.0 | 16.4±3.5 | 3.08±0.07 | 11.7±2.9 | 36.0±9.1 | −6.1±9.9 |
| T78V/K79G | Lys73 (78–85%) | 1.23±0.04 | 15.5±2.8 | 19.1±3.4 | 3.18±0.04 | 13.3±2.0 | 42.3±6.5 | −9.7±7.5 |
Recovery of Met80 Ligation to the Ferric Heme Iron at pH 4.5.
At pH 4.5, features of Met-ligated cyt c are apparent in spectra of T49V/K79G and T78V/K79G (Figure 5). The charge-transfer band at 695 nm, albeit at lower intensity compared to that for K79G, is clearly evident in the electronic absorption spectra and resonances associated with heme methyls of Met-ligated cyt c show up in the downfield region of the 1H NMR spectra of the variants. The g values from EPR for K79G and T49V/K79G are also similar. In the case of T78V/K79G, however, EPR spectra resemble more those of the H2O-ligated K73A/K79G/M80K (at pH 4.5)30 and microperoxidase AcMP8.68 A high-spin ferric heme iron species is present at pH 4.5 in both T49V/K79G and T78V/K79G as evident from the charge-transfer band at 624 nm in the electronic absorption spectra, the broad 1H resonances at around 60 ppm, and low-field EPR signals. However, the differences in the spectra of the two variants suggest a higher population of the high-spin species in T78V/K79G. The Soret band of this variant is blue-shifted (λmax = 401 nm) and higher in intensity compared to that of T49V/K79G (λmax = 403 nm). Whereas the high-spin species is only a minor component in the EPR spectrum of T49V/K79G, the spectrum of T78V/K79G is dominated by this species.
Figure 5.
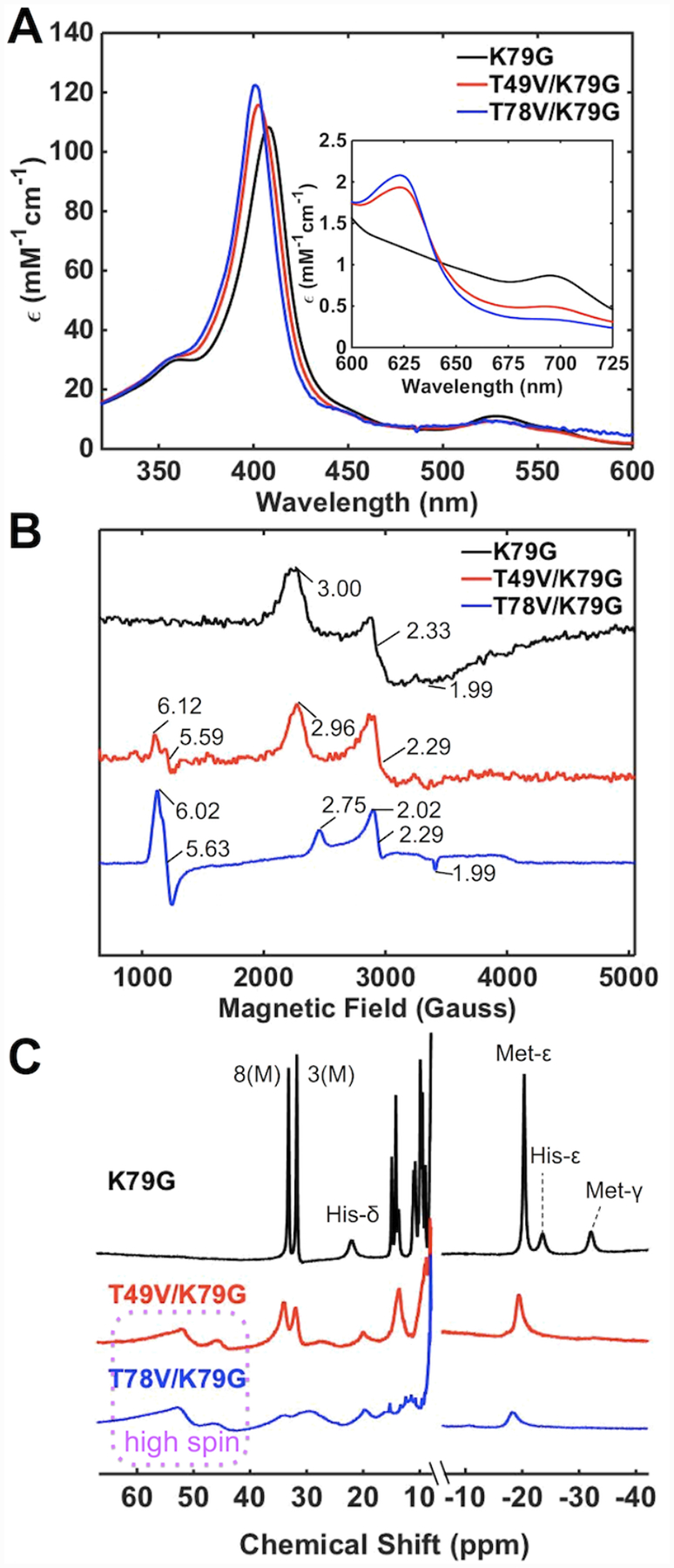
Spectra of ferric K79G (black), T49V/K79G (red), and T78V/K79G (blue) at pH 4.5. (A) Electronic absorption spectra of ferric proteins at 22 ± 2 °C. The inset is the comparison of charge-transfer bands at 695 nm. (B) EPR spectra of ferric proteins at 10 K (C) 1H NMR spectra of ferric proteins at pD 4.5. The labeling nomenclature is the same as in Figure 3.
Thermodynamic parameters from GuHCl denaturation studies (Figure S7A) indicate that the variants become less stable at lower pH, which is typical of most cyt c variants (Table S4).42,69 Given the error bars for these parameters and the presence of at least two types of heme iron species at pH 4.5, quantitative comparisons of parameters for global denaturation of the variants are difficult. The trends in these dependencies in Figure S7A, however, do suggest that both variants are less stable than their parent K79G variant.
We also examined each variant at varying GuHCl concentrations by monitoring the 695 nm charge-transfer band, which allowed us to exclusively focus on the Met-ligated species and properties of Ω-loop D under these experimental conditions. The unfolding transition for T78V/K79G is not well defined (Figure S7B) and lacks cooperativity. The classical two-state model is not adequate for the analysis of denaturation of this variant. The unfolding curves for K79G and T49V/K79G do show a sigmoidal character but their pre- and post-transition regions are steep, suggesting that the two-state model may not be appropriate for the analyses of these two variants either. We have conservatively chosen not to fit these results but instead focus on qualitative comparisons. The curves in Figure S7B suggest that Ω-loop D becomes less stable when tertiary contacts near HP6 are altered and illustrate the particular ease of unfolding of this loop for T78V/K79G.
Insights from Structural Modeling.
We have recently discussed a comparison of ferric K79G and WT* structures.40 Analyses of structural snapshots of K79G and WT* have revealed that, while the hydrogen bond between Thr49 and HP6 persisted over the time course of simulations, the one between Thr78 and HP6 fluctuated between the “on” and “off” states (Figure S8).
Herein, we employ simulations to construct structural models of Met-ligated ferric and ferrous T49V/K79G and T78V/K79G variants (Figure 2). The average volume of the heme pocket and the solvent accessible surface area (SASA) of the heme do not change upon the Thr-to-Val substitutions, in either ferric or ferrous proteins (Tables S2 and S3). These results highlight the preservation of the tight polypeptide fold that surrounds the heme. However, distinct changes in the hydrogen-bonding network and hydration are apparent.
The model of Met-ligated ferric T49V/K79G (Figure 2B) highly resembles that of K79G (Table S2). The number of buried water molecules remains the same as that in K79G, while the conserved water molecule, Wat166, moves closer to the ferric heme iron atom. Notably, hydrogen bonds between the side chain of residue 49 and HP6 as well as between HP6 and Wat166 are gone upon the T49V substitution. An external water molecule Wat4933 next to HP6 in K79G disappears in T49V/K79G.
The T78V substitution (Figure 2C) does not change the number of buried water molecules on the side proximal to Met80, but introduces an additional water molecule (Wat168) on the side proximal to His18. The distance between Wat166 and the ferric heme iron is shortened as well but not as much as in T49V/K79G. As a consequence of the T78V substitution, the original Thr78(OH)-to-HP6 hydrogen bond in K79G is no longer possible. HP6 is able to rotate toward Thr49, allowing its oxygen atoms to hydrogen bond to the backbone amide of Thr49 while maintaining the hydrogen bonds to the side-chain hydroxyl of Thr49. The second propionate, HP7, moves to a position where it could engage in hydrogen-bonding interactions with three nearby water molecules (Wat121, Wat166, and Wat168). In contrast, only one water molecule is within a hydrogen-bonding distance to HP7 in the structural models of ferric K79G and T49V/K79G.
In the model of ferrous K79G (Figure 2D), the Thr78(OH)-to-HP6 hydrogen bond found for ferric K79G disappears, whereas the Thr49(OH)-to-HP6 hydrogen bond remains and a new hydrogen bond is formed between HP6 and the side chain of Tyr46. In ferrous T49V/K79G (Figure 2E), the hydrogen bond partners of HP6 are limited to amides of residues 49 and 52. The hydration of propionate groups is improved, with two water molecules (Wat2831 and Wat3293) next to HP6 and three (Wat134, Wat179 and Wat181) next to HP7. The distance between the ferrous heme iron and water molecule Wat179 in the side proximal to Met80 slightly decreases. In ferrous T78V/K79G (Figure 2F), the number of buried water molecules and hydrogen-bonding interactions with HPs are the same as in ferric T78V/K79G (Figure 2C).
Force-field parameters for Lys-ligated hemes are currently not available. Instead, models of Lys-ligated variants were constructed based on the structure of the Lys73-ligated variant of cyt c T78C/K79G (Figure S9).30 Despite these limitations, some comments about our variants can be made. Since residues 49 and 78 are distant from the heme pocket (Figure S9A) in both the Lys73-ligated T78C/K79G and the Lys73-ligated alkaline form of cyt c,70 it is unlikely that substitutions at these sites influence structural integrity or heme exposure of the Lys-ligated species.
Heme Iron Reduction Potentials.
At pH 7.4, cyclic voltammograms at 0.1 V/s for both T78V/K79G and T49V/K79G exhibit a quasireversible signal at high potential from the Met-ligated species and a peak at lower potential from irreversible reduction of the Lys-ligated species (Figures 6A and 6B). Similar signals are also observed for K79G at pH 10 (Figure S10), and this absence of electrochemical reversibility is suggestive of a redox-linked ligand switch (Scheme 1).71,72 Apparent reduction potentials for the Met-ligated species determined from positions of the high-potential signals are distinctly lower at slower scan rates (<0.5 V/s, Table S5). Such shifts are expected when a reduction is coupled to another chemical process involving species with distinct reduction potentials,73,74 and thus this result also points to interconversion between the Met- and Lys-ligated species.
Figure 6.
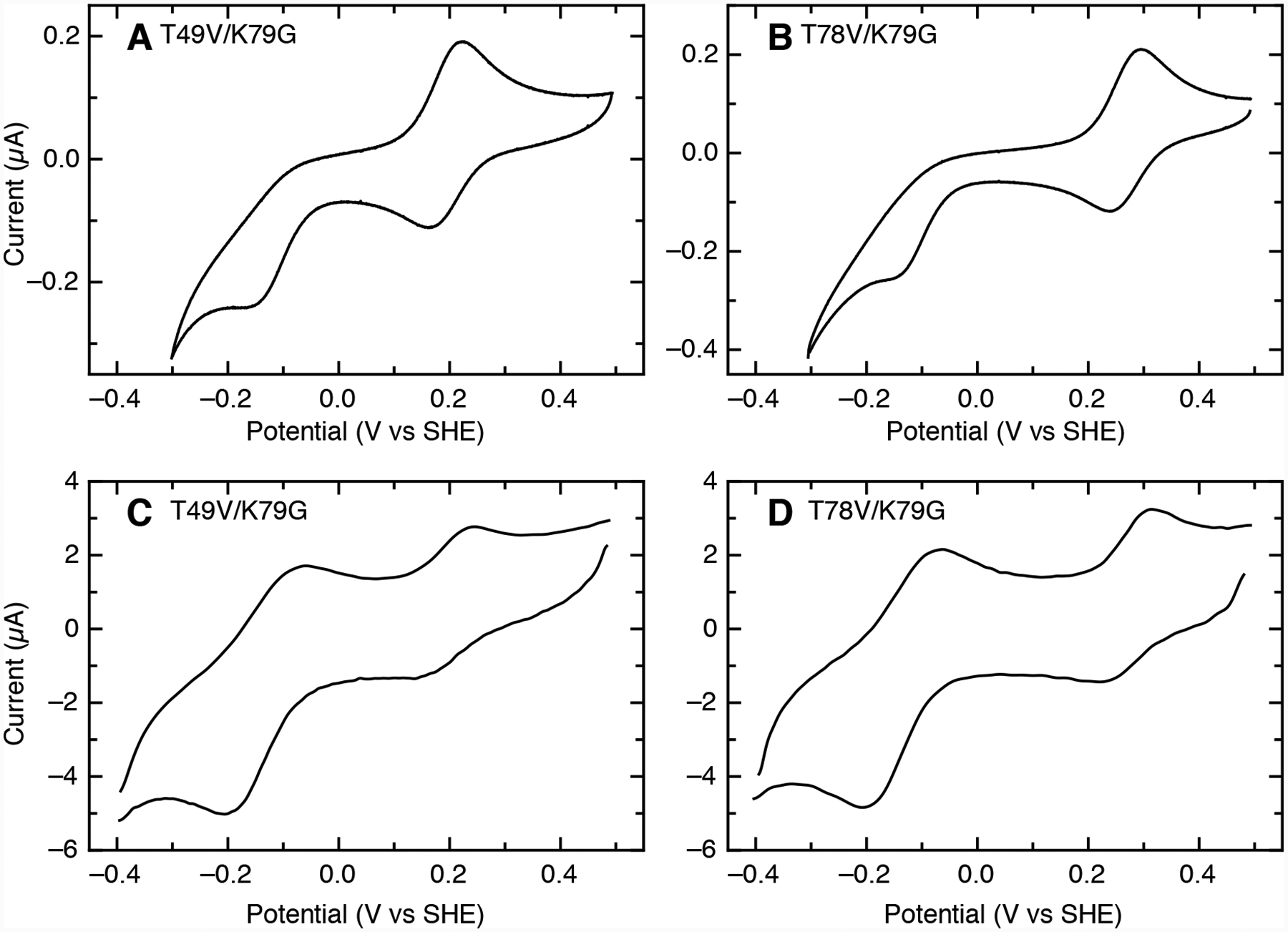
Cyclic voltammograms at (A, B) 0.1 V/s and (C, D) 20 V/s for (A, C) T49V/K79G and (B, D) T78V/K79G at pH 7.4 (100 μM samples in 0.1 M sodium phosphate). Background capacitative currents were subtracted in each direction, and voltammograms at 20 V/s were treated with a fast Fourier transform noise filter.
At fast scan rates (>10 V/s), the voltammetric experiment readily outruns the Met-to-Lys ligand-switch process and the positions of the two signals correspond more closely to the thermodynamic potentials of Met- and Lys-ligated species. At pH 7.4 for both T49V/K79G and T78V/K79G, a nearly reversible signal for the Met-ligated species () is observed at a high potential along with a separate reversible signal for the Lys-ligated species () at a much lower potential (Figures 6C and 6D). Compared to K79G, the value for T78V/K79G is slightly higher, while that for T49V/K79G is about 60 mV lower (Table 4).
Table 4.
Reduction Potentials and Calculated Energy Differences at pH 7.4 for Yeast iso-1 Cytochrome c Variants
| Variant | Reduction potential (mV vs SHE) | Calculated ΔΔG (kJ/mol) | |||||
|---|---|---|---|---|---|---|---|
| a | b | b | a | c | c | d | |
| K79G | 262±3 | 265±38 | 148±36 | −145±10e | −25.6±3.7 | 14.0±1.0 | −32.4±3.9 |
| T49V/K79G | 197±4 | 201±28 | 48±25 | −141±10 | −19.4±2.7 | 13.6± 1.0 | −15.4±2.9 |
| T78V/K79G | 275±3 | 277±45 | 184±38 | −140±4 | −26.7±4.3 | 13.5±0.4 | −22.7±4.5 |
Experimental values measured at scan rates 20 V/s.
Calculated values according to Scheme 1.
Calculated from the reduction potentials of the Met-Fe|TH+ or Lys-Fe|T species by or .
Calculated according to Scheme S2, where the diagonal . KeqIII is a pH-adjusted equilibrium constant between the Lys-Fe(III)|T and Met-Fe(III)|TH+ species and is determined by .
The reduction potential of Lys-ligated K79G was measured at pH 10, while the other potentials and all of the potentials were measured at pH 7.4.
The potentials (−140 to −145 mV) are very similar in all three variants. These values are also similar to previously reported reduction potentials obtained for the alkaline form of cyt c using modified-gold electrodes,75,76 but differ from those using pyrolytic graphite (−190 mV and less).72 As previously noted,71 such a disparity may arise from differences in buffer composition and other experimental factors. It is also known that polished graphite edges have negative surface charges that can affect heterogeneous ET.77
The three variants were also examined by electrochemistry at pH 4.5 and at pH 6.0. At pH 6.0, the voltammetry is reversible for all three variants and the reduction potentials observed are consistent with Met ligation (Table S6). As in previous studies, there is a small increase in potential as pH is lowered from 7.4 to 6.0.40,78 At pH 4.5, both K79G and T49V/K79G exhibit reversible voltammetry over a wide range of scan rates, and both have a higher reduction potential than that at pH 6.0, though some part of this upward shift may result from the change from phosphate buffer to acetate.40,79 However, T78V/K79G has a potential that is 20 mV lower at pH 4.5 than at pH 6.0. At slower scan rates (≤ 0.1 V/s), T78V/K79G exhibits a single reversible signal in voltammetry at pH 4.5. As the scan rate increased to >1 V/s, an additional peak at a lower potential becomes apparent in the reductive sweep (Figure S11), but no additional peak for reoxidation is observed, even at 50 V/s. These results suggest the presence of a new species with a lower potential (120–130 mV) in fast exchange with the Met-ligated species (270 mV). The lower potential of this additional species may arise from an increase in solvent accessibility of the heme or a change in the heme iron ligation.
Matrix Analyses of pH-Dependent Changes in Ligation in Ferric Variants.
Our spectroscopic experiments have revealed the presence of at least three different heme iron species for ferric T49V/K79G and T78V/K79G within the pH range from 4.5 to 7.4: the high-spin (five-coordinate or six-coordianate H2O-ligated) and low-spin Met- and Lys-ligated species. To understand the properties of the protein conformers involved, we have performed SVD analyses of electronic absorption spectra over a wide pH range, from 2 to 11. Owing to the large difference in extinction coefficients in the Soret and charge-transfer regions, separate titration experiments, at different protein concentrations, were employed to monitor changes in these two regions of the electronic absorption spectra. For both ferric T49V/K79G and T78V/K79G, the consistent description of the two spectral series has required the inclusion of four components (states) accounting for two acidic and one alkaline transitions. Global fitting of the selected V vectors (Figures S12 and S13) yielded pKa and n values for the proposed transitions (Table S7).
We refer to the two acid protein states as the lower-pH acid state A1 and the higher-pH acid state A2, respectively, based on the pH conditions at which they dominate. Previous studies of variants of yeast iso-1 cyt c have identified the presence of H2O/H2O and H2O/His heme iron species at pH 2.47 The positons and intensities of spectral bands in spectra of our variants at the low-pH range where the A1 and A2 states are populated are consistent with these species (Table S1). Our spectroscopic studies at pH 4.5 (Figure 5) and 7.4 (Figure 3) suggest that the alkaline transition (pKa,3 = 6.7 ± 0.1) involves the replacement of the Met80 ligand at the ferric heme iron by a Lys residue and therefore we refer to the two states associated with this transition as M and K states, respectively.
The sum of n values associated with the two acidic transitions (A1→A2 and A2→M) was close to three for both variants, in good agreement with the typical number of protons found for the acidic transition of cyt c.80 The two-stage acidic transition is reminiscent of the multi-stage acid unfolding process of several other variants of yeast iso-1 cyt c that have substitutions affecting the stability of Ω-loops and/or their connectivities.81,82 The pKa value for the first transition (A1→A2) is very similar for the two variants. However, the pKa value for the second transition (A2→M) is almost two pH units higher for T78V/K79G than that for T49V/K79G suggesting distinct differences in the nature of the A2 state for the two variants.
We have employed far-UV CD and Trp59 fluorescence spectroscopies, which are sensitive to the properties of the polypeptide fold, to assess conformational properties of the A2 state. With CD and Trp59 signals for A1 and M states known from low- and high-pH regions of the pH dependence data (Figure 7), the corresponding signals for A2 were calculated. The polypeptide ensemble of T78V/K79G in the pH range from 3.5 to 6.0 is comprised of conformers with a degree of α-helical structure and the average Trp-to-heme distance very similar to that in native K79G (Figures 4 and 7) suggesting that the conformers comprising the A2 state in this variant are folded. In contrast, the A2 state in T78V/K79G shows a 70% decrease in α-helicity and its Trp59-to-heme distance (26.0 Å) is similar to that in the unfolded A1 state. We conclude that, in contrast to T78V/K79G, acid denaturation of T49V/K79G proceeds through a largely unfolded state A2 suggesting that increased polypeptide dynamics may be necessary for acid-induced Met80 loss from the heme iron in this variant. The low pKa,2 = 3.4 ± 0.1 for T49V/K79G suggests that the bis-His species previously reported to be present during acid unfolding of horse heart cyt c83 is an unlikely component of the A2 state for this variant. High concentrations of phosphate84 could have affected the makeup of the conformational ensemble in this earlier work and subsequent reports no longer invoke this species.47,85 We do not believe that bis-His coordination at the heme is particularly relevant for the A2 state of T78V/K79G (pKa,2 = 5.0 ± 0.1) either since under the pH conditions when this state dominates the conformational properties of this variant are largely unperturbed, preventing such coordination, and the high-spin species is the primary component of the protein ensemble (Figure 5).
Figure 7.
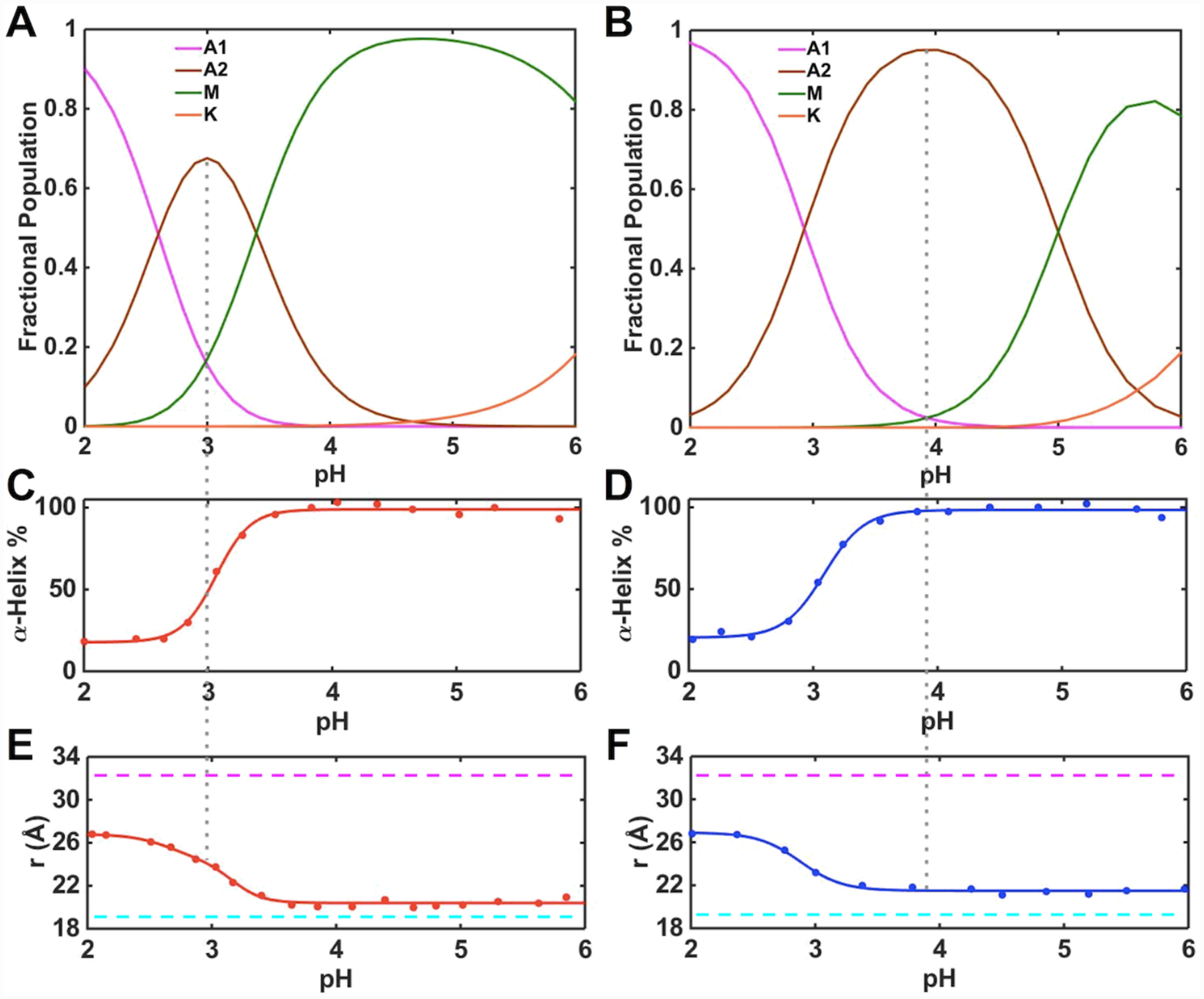
Changes in the heme iron ligation and properties of the polypeptide fold in ferric (A, C, E) T49V/K79G and (B, D, F) T78V/K79G in the pH range from 2 to 6. (A, B) Fractional populations of differently-ligated heme iron species. (C, D) Percentage of the α-helical content relative to that at pH 6.0. (E, F) Distances between Trp59 and heme calculated from the fluorescence data. The magenta and cyan dashed lines indicate the distance between Trp59 and heme in fully unfolded (33 Å) and fully folded (18.8 Å) K79G, respectively. The gray dashed line represents the pH condition under which the A2 state is most populated.
The pKa values for the alkaline transition (M→K) are the same in ferric T49V/K79G and T78V/K79G. The n values are determined to be about one, suggesting that the transition involves one proton. The fractional population maps (Figures 7A and 7B) were computed based on the sequential model (A1→A2→M→K). These results suggest that at pH 4.5, for which we have extensive characterization by multiple spectroscopic methods (Figure 5), the conformers in the M state are a major component (>95%) of the protein ensemble for T49V/K79G. In contrast, the A2 state dominates (>80%) the protein ensemble at pH 4.5 for T78V/K79G. The intensities of the 624-nm and 695-nm bands and broad downfield signals in the 1H NMR spectra of this latter variant are consistent with the coexistence and exchange between the high-spin H2O-ligated and the low-spin Met-ligated species. In contrast, the population of the A2 species should be very low at this pH for T49V/K79G, yet the 624-nm band associated with the high-spin heme iron species is clearly visible and the signals in the NMR spectra are still broad. Further, the 695-nm band is much weaker than that of K79G. Although the decreased intensity of the 695-nm band is not a definitive sign of loss of Met coordination to the heme iron and the origin of this band is still open to debate,23 these collective observations of spectral discrepancies suggest that additional factors may be at play here. We have found the same intensity ratio of the 624- and 695-nm bands in proteins from multiple preparations, and this profile has persisted after repeated purification of the protein samples and at varying (0.05–0.70 mM) protein concentrations. These findings suggest that the presence of the high-spin species does not come from protein degradation and/or aggregation but rather is an intrinsic property of the protein ensemble at this pH.
The high-spin species is present within a broad pH range, even at pH values much higher than pKa,2 when the steep rise in the population of the H2O-ligated A2 species occurs. Further, as pH decreased from 7.4 to 5 for T49V/K79G and from 7.4 to 6 for T78V/K79G, the intensity of both 624-nm and 695-nm bands increased. These findings argue against a branched model involving the direct exchange between the A2 and K states and instead suggest that the high-spin species in equilibrium with the Met-ligated species must be distinct from the H2O-ligated A2 species. Since this high-spin species coexists with the Met-ligated species before the M→A2 transition, we include both these species as components of the M state. Because only three distinct pH transitions (and thus four states) are apparent in pH titrations, such composite description of the M state does not alter insights about pH-dependent equilibria within the four-state sequential model (A1→A2→M→K) but still allows for the comparison of the two variants in their propensity for Met coordination to the heme iron. Figure 8 summarizes heme iron ligation states accessible to ferric T49V/K79G and T78V/K79G and the corresponding pKa values.
Figure 8.
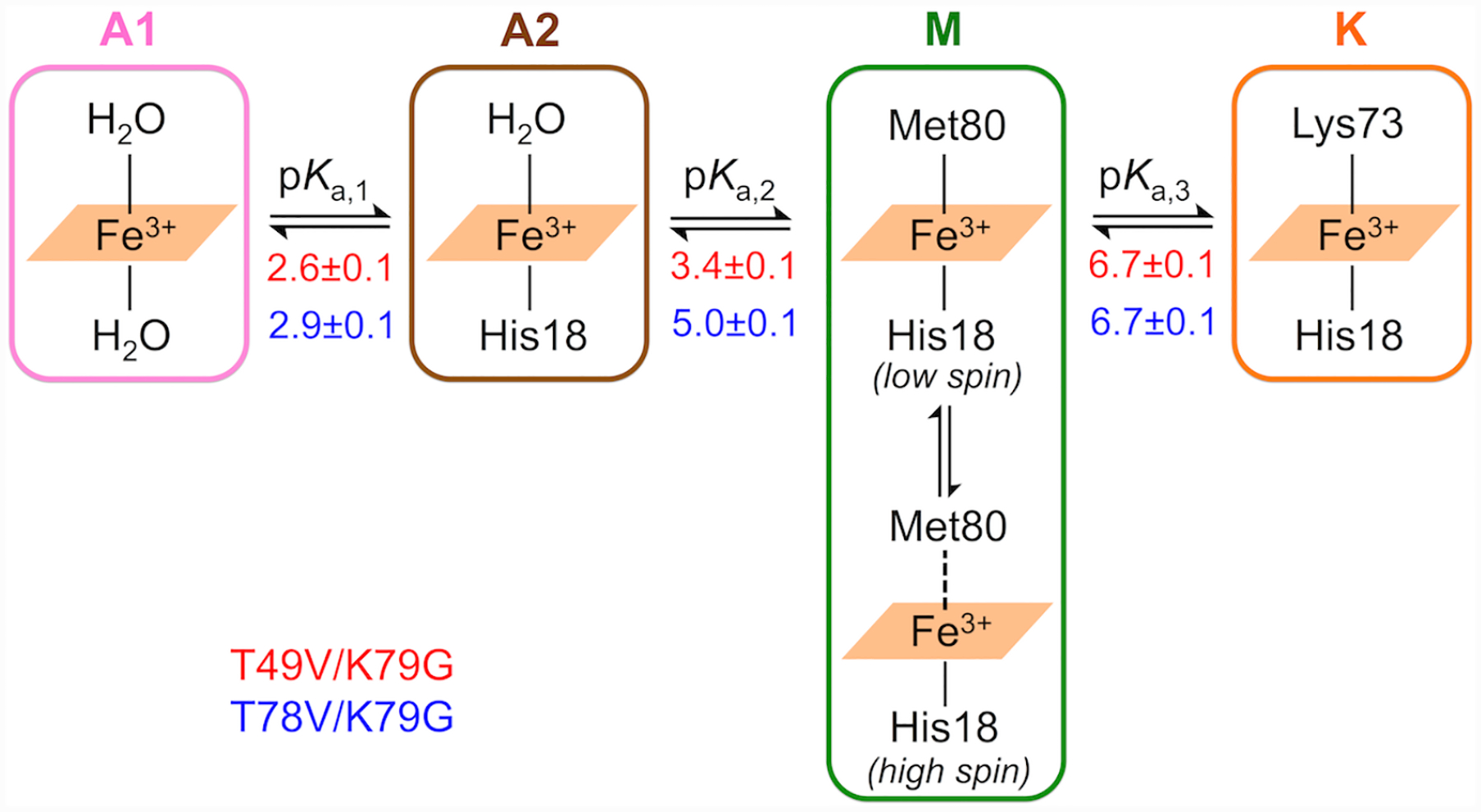
pH-dependent changes in the ligation to ferric heme iron based on the SVD analyses. The apparent pKa values for each transition (Tables 1 and S7) in T49V/K79G (in red) and T78V/K79G (in blue) are listed under the equilibrium arrows.
An equilibrium between high-spin and low-spin species has previously been described in Met-ligated cyt c from Wolinella succinogenes86 and several cyt c4 proteins.87,88 The identity of the high-spin species has not been firmly established but given the weak nature of the Met-iron interaction in cyt c89 it may be envisioned as a species lacking the Met bond to the heme iron. Mechanistic studies of peroxidase activity suggest that an equilibrium between the low-spin six-coordinate (Met-on) and high-spin five-coordinate species (Met-off) is relevant to native cyt c and to its destabilized variants, in particular.90–92 We adapt this notation in our description of the M state.
For ferric T49V/K79G, the spectra of H2O/H2O unfolded, H2O/His unfolded, and Lys/His folded species (Table S1) were employed as the input basis spectra for A1, A2, and K states, respectively. The same input spectra for the A1 and K states were used for ferric T78V/K79G, while the spectrum of H2O/H2O folded was employed as the input basis spectrum for the A2 state. For both T49V/K79G and T78V/K79G variants, the calculated spectra for the M state (Figure S12 and S13) have Soret bands blue-shifted relative to their typical positions for the Met-ligated species (409 nm) as well as absorption bands at both 624 nm and 695 nm. These spectroscopic features are consistent with the mixture of the Met-on and Met-off components in the M state (Figure 8). Linear decomposition of these spectra has revealed that the Met-on species constitute about 50% and 70% of the M state for T49V/K79G and T78V/K79G, respectively. The lower population of the Met-on species for the T49V/K79G variant suggests that the Thr-to-Val substitution at residue 49 might have weakened the Fe-S bond more than the analogous substitution at residue 78.
Kinetics of pH-Induced Met-to-Lys Ligand Switch at the Heme Iron in Ferric Variants.
Time-dependent changes in absorbance of ferric variants at 405 nm were monitored in pH-jump experiments (Figure S14). All kinetic progress curves from both upward and downward pH-jump experiments were monoexponential, yielding an observed rate constant . Kinetic parameters obtained from the dependence of on proton concentration (Figure 9) are listed in Table 1. For both T49V/K79G and T78V/K79G, the rate constant for the forward alkaline transition corresponding to the Met80-to-Lys73 ligand switch (kfIII) is smaller than the rate constant for Met80 dissociation,40 suggesting that Lys73 coordination, rather than Met80 dissociation, is the rate-controlling step for the forward ligand switch. The kfIII value for T78V/K79G is about two-fold greater than that for T49V/K79G but both variants have kfIII values smaller than that for the parent K79G.40
Figure 9.
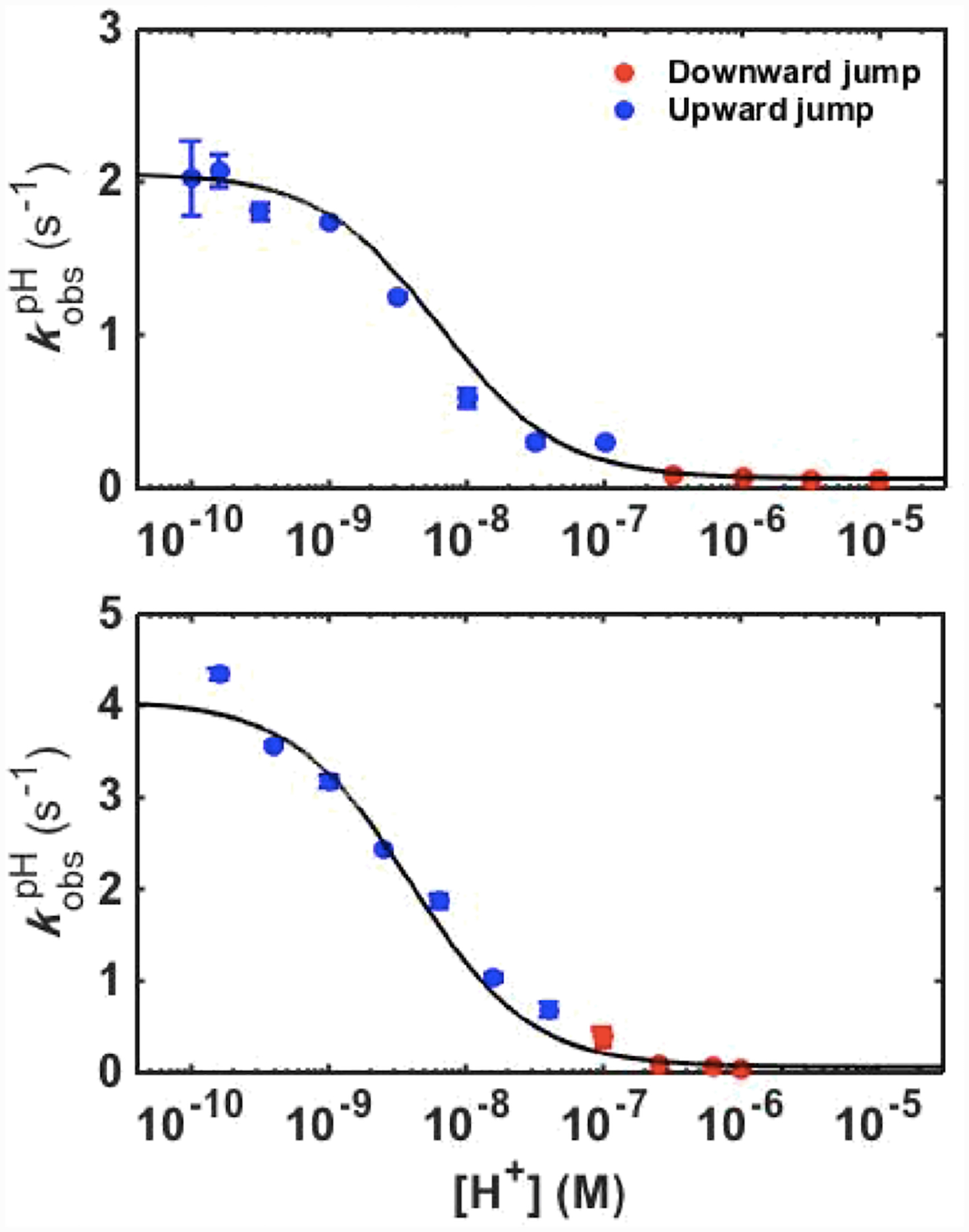
Plots of versus proton concentration for ferric T49V/K79G (top) and T78V/K79G (bottom) variants. The curves are fits of the data to eq 6 with parameters listed in Table 1.
The rate constants for the Lys73-to-Met80 back conversion, kbIII, are comparable for all three variants (Table 1) suggesting that the studied Thr-to-Val substitutions do not appear to influence the energy barrier for the Lys-to-Met ligand switch. The similarity of these values reinforces our previous conclusion that slow Lys dissociation from the ferric heme iron is the rate-controlling step for the backward ligand switch.40
The conformational equilibrium constant KCIII and the acid dissociation constant of the “trigger” group KHIII from kinetic data in Figure 9 (Table 1) allow for further analysis of the ligand-switch mechanism. The KCIII value for T78V/K79G is the same as the one for K79G, whereas the KCIII value for T49V/K79G is slightly lower. Both Thr-to-Val substitutions result in the same low apparent pKaIII but evidently contributions to this effect differ for the two variants. For T78V/K79G, the effect solely arises from a decrease in the pKHIII value. For T49V/K79G, pKHIII decreases even more but at the same time pKCIII increases, resulting in the same apparent pKaIII.
ET Reactions of Ferric Variants with a6Ru2+.
Kinetics of reduction of ferric T49V/K79G and T78V/K79G by a6Ru2+ were examined at pH 7.0, at which both variants exist as mixtures of the Met- and Lys-ligated species. At this pH, Met80 is the ligand to the ferrous heme iron in these variants, therefore, simultaneous studies of both ET and ligand-switch processes are possible.93 Since the Met-ligated species in both variants have potentials higher than that of a6Ru2+ (E°′ = 51 mV)94 (Table 4), these protein species undergo a direct bimolecular ET reaction with this reducing agent. The negative reduction potentials (Table 4) make direct ET of the Lys-ligated species with a6Ru2+ unfavorable and a Lys73-to-Met80 ligand switch must take place before ET occurs (Scheme 3).
Scheme 3.
For both variants, kinetics progress curves show two phases (Figure S15): a fast one with a rate constant, , linearly dependent on the concentration of a6Ru2+ ([a6Ru2+]), and a slow phase for which the rate constant, , is [a6Ru2+]-independent (Figure 10). Based on these dependencies, the fast phase is assigned to the reduction of the Met-ligated conformers and the slow one to the Lys73-to-Met80 ligand switch, a step that gates reduction of the Lys-ligated conformers. The bimolecular ET rate constants, kET,M, obtained from analyses of , and ligand-switch rate constants, , are summarized in Tables S8 and 1, respectively. Since the Lys73-to-Met80 ligand switch at the ferric heme iron is the same process in both pH-jump and ET experiments, values provide an independent measure of the rate constant for the backward alkaline transition kbIII. Indeed, the values for both variants are in excellent agreement with the respective kbIII values from the pH-jump studies (Table 1).
Figure 10.
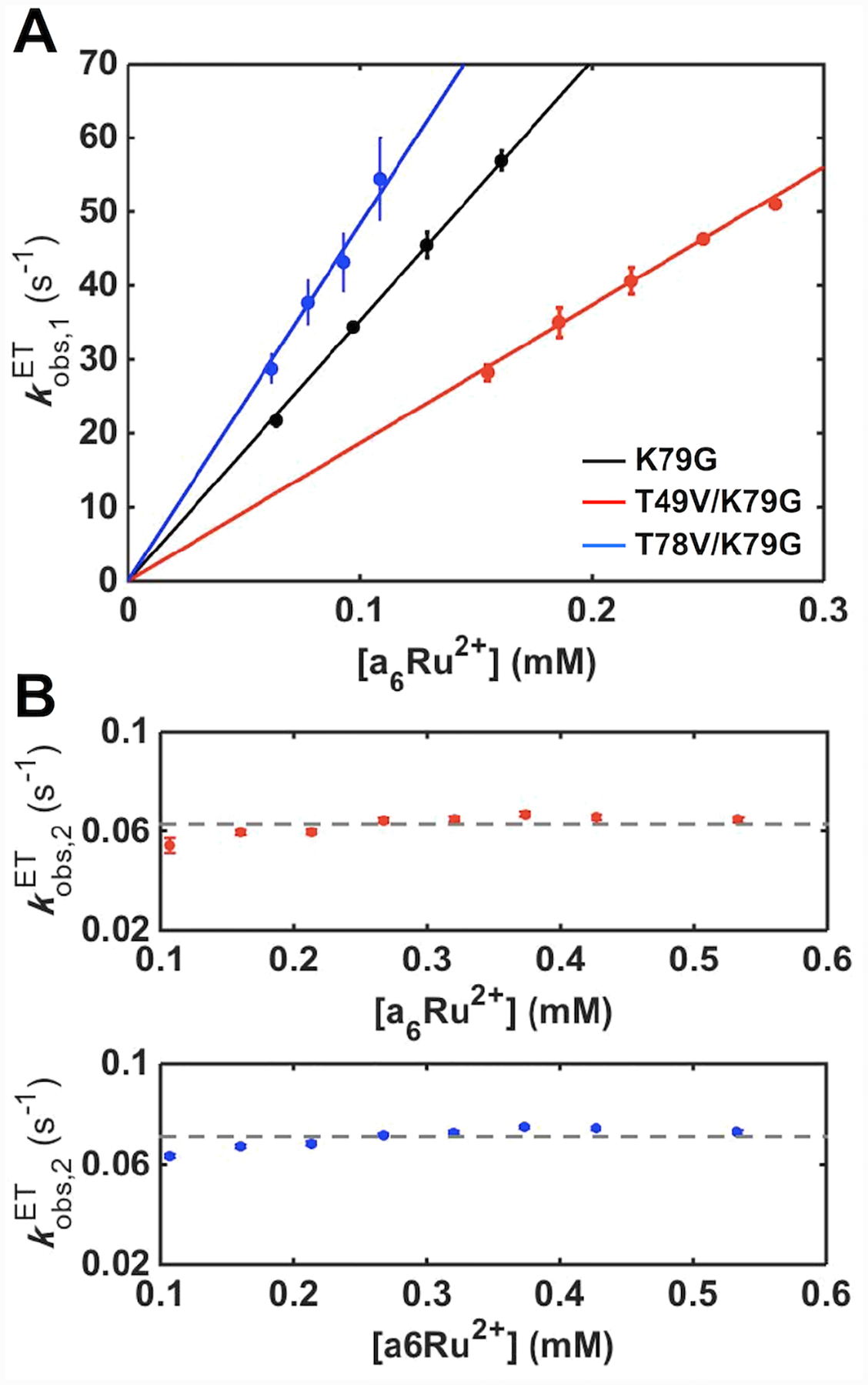
The dependence of (A) and (B) on a6Ru2+ concentration for ET reactions of K79G (black), T49V/K79G (red) and T78V/K79G (blue) in a 10 mM sodium phosphate buffer at pH 7.0 containing 0.1 M NaCl. The solid curves are fits of the dependencies to . The dashed lines display the average of all the values for each variant.
Thermodynamics and Kinetics of the Met-to-Lys Ligand Switch at the Heme Iron in Ferrous Variants.
Absorption spectra of the Lys- and Met-ligated low-spin ferrous hemes are very similar, which complicates spectroscopic analyses of ligand-switch processes in these species. These challenges are further exacerbated by the lack of a discernible population of the Lys-ligated species in ferrous cyt c under non-denaturing conditions (Figure S4D). However, analyses of cyclic voltammetry data were able to provide characterization of these species.
With KCII* values readily obtained from experimentally available , , and KCIII* values according to the relationships in Scheme 2, KCII can be subsequently found according to eq 7, but only if pKHII or pKaII values are independently known. The pKHIII values for all the variants were available from our pH-jump experiments (Table 1) and it has been previously shown that, when an ionizable group resides near the heme in cyt c, pKHII is greater than pKHIII by 0.3–2.1 pH units.23,95 Accordingly, we looked for acid-base equilibria affecting spectroscopic properties of the heme in ferrous variants within this pH range. For K79G, the high pH required for the deprotonation of the “trigger” group is associated with the Met-to-hydroxide ligand switch and concomitant denaturation of the protein.96 However, for T49V/K79G and T78V/K79G, there are clear pH-dependent changes affecting electronic absorption spectra of the heme group (Figure S4) with pKa,1II values of 10.8 ± 0.2 and 10.0 ± 0.4, respectively. We propose that these transitions, which retain Met80 coordination to the heme iron, correspond to structural perturbations upon deprotonation of the “trigger” group i. e. pKHII = pKa,1II. Because pKHIII values are dramatically lowered by Thr-to-Val substitutions in ferric T49V/K79G and T78V/K79G, we now can observe the “trigger” group deprotonation processes experimentally in ferrous forms of these variants as well. Since this deprotonation process cannot be experimentally observed for K79G, an additional assumption of pKHII = pKHIII + 2 was made, to give a similar increase in the pKH value upon the heme iron reduction as that in the other two variants. With this information, pKCII and then pKaII values were calculated (Table 2).
It is also possible to find pKaII values from pKaIII values and and through the relationships in Scheme 1, but the latter reduction potential is not readily measured under conditions of fast pre-equilibrium. However, using the experimentally observed reduction potentials at high scan rates as the values for , as was done elsewhere,72 gave pKaII values which agree within errors with the values found by our method above (Table 2).
Once both KCIII and KCII were known, the rightmost square in Scheme 1 was used to determine reduction potentials of the Met-Fe|T species () (Table 4). Reduction potentials of the Met-Fe|TH+ species, , were found from the complete cycle using , pKaIII, and pKaII. The potentials for the Met-Fe|TH+ species agree quite closely with those obtained for each variant at lower pH values where ligand switching is not a complicating factor and the “trigger” group is protonated (Table S6). This finding suggests that the observed decrease in the reduction potential of Met-ligated cyt c as pH is increased is largely due to the effect of the “trigger” group deprotonation and the ensuing alkaline transition.
At slower scan rates in voltammetry experiments, reduction of the Lys-ligated species is followed by a ligand switch at the heme iron that results in the Met-ligated species. This conformational exchange gives an apparently irreversible reduction, with no peak observed for reoxidation of the Lys-ligated species. However, as the scan rate is increased, the ligand switch is outpaced and the reduction and oxidation of the Lys-ligated species become reversible in the voltammetry. The onset of this behavior is seen by the appearance of a small peak corresponding to the reoxidation of the Lys-ligated species (Figure S10). This peak grows in size as scan rate is further increased (Figure 6C and 6D). The scan rates necessary to attain and achieve reversibility of redox reactions for the Lys-ligated species differed for the three variants (Figure S10). For K79G scan rates of 0.15 to 0.2 V/s were sufficient to detect the appearance of a small peak corresponding to the reoxidation of the protein. Faster scan rates were necessary to observe a comparable signal in T49V/K79G (0.4 to 0.6 V/s) and T78V/K79G (0.6 to 0.9 V/s). Digital simulations of the voltammetry signals were used to find kbII values (Table 2) that best matched the observed onset of reversibility for each variant. While robust kbII values could be obtained from the voltammetry, the kfII values were not directly measurable because of the extremely slow time scale for the forward ligand switch and were calculated from KCII values instead.
Energy Diagram for the Redox-Linked Met-to-Lys Ligand Switch in Cyt c.
The knowledge of protein stability (Table 3), reduction potentials (Table 4), pKa values and ligand-switch kinetics (Tables 1 and 2) from this work allowed us to map out energy levels for the Met- and Lys-ligated ferric and ferrous variants (Figure 11A). Since the derivation of this energy diagram consists of multiple steps, we explicitly list them below and emphasize in this description energy relationships among the species. The Gibbs free energy G°′ of the Met-Fe(III)|TH+ species for K79G was set to zero to serve as a reference for all other energy levels.
Figure 11.
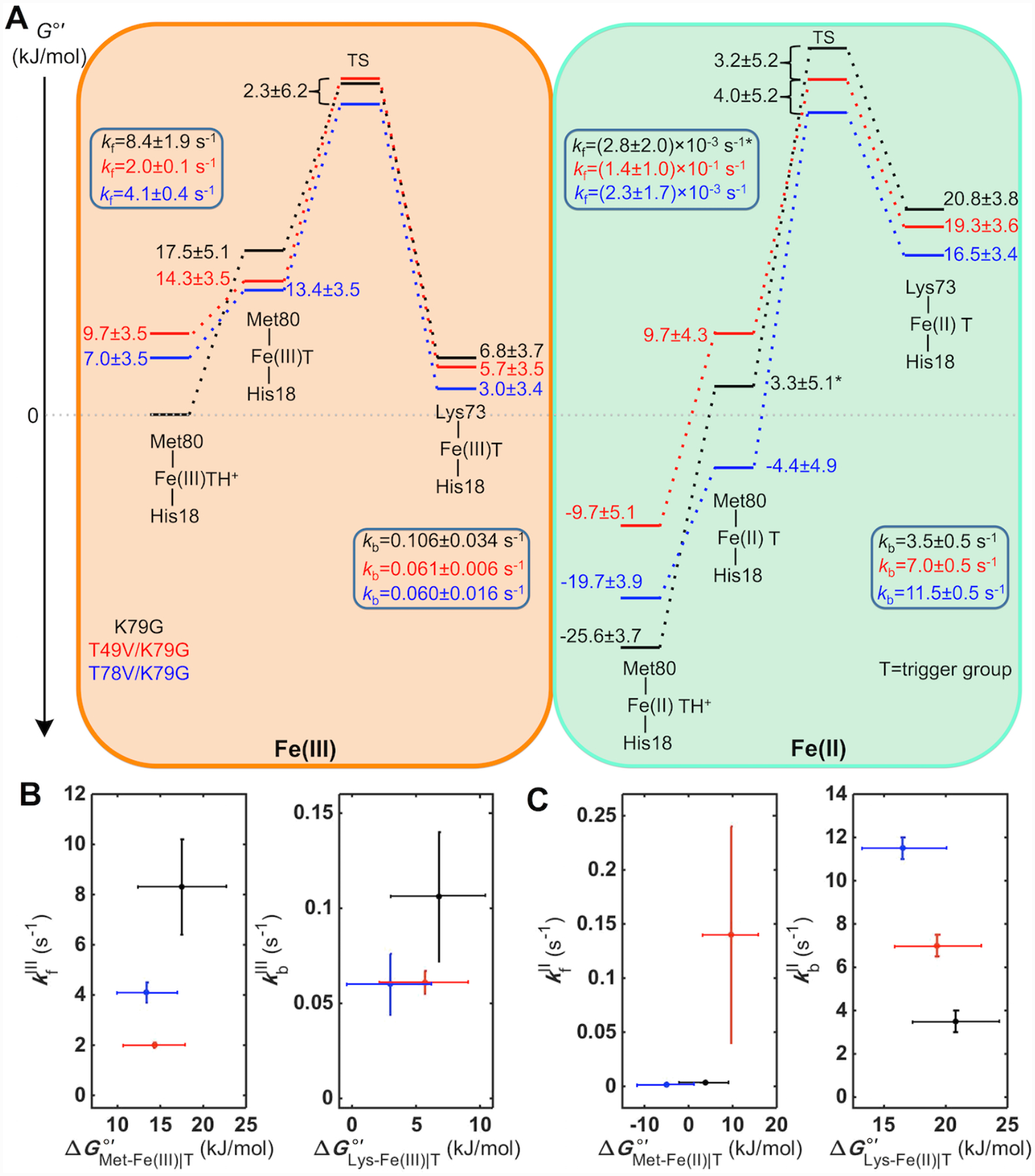
(A) Energy diagram for the alkaline transition in ferric and ferrous K79G, T49V/K79G, and T78V/K79G at pH 7.4. The asterisk-labeled energy level and kfII value for K79G are calculated based on the assumption pKHII = pKHIII + 2. (B, C) The dependencies of the rate constants kf and kb on energies of the corresponding ground states in (B) ferric and (C) ferrous K79G, T49V/K79G, and T78V/K79G.
At pH 7.4, the Lys-ligated species constitute the majority of the protein ensemble for T49V/K79G and T78V/K79G (Table 3). For K79G at this pH, the ferric heme iron species are Met-ligated. Thus, the energy difference between the Lys-Fe(III)|T species for T49V/K79G (or T78V/K79G) and the Met-Fe(III)|TH+ species for K79G was obtained by calculating the difference of their values (Table 3). This approach incorporates an assumption that the free energies of the denatured states do not differ from each other as much as uncertainties in other parameters. The equilibrium constant, KCIII, between Lys-Fe(III)|T and the Met-Fe(III)|T species was obtained by calculating the ratio , using the rate constants in Table 1. To find relative energy levels at pH 7.4 for the acid-base equilibria involved, , a pH-dependent equilibrium constant for the exchange between Met-Fe(III)|TH+ and Lys-Fe(III)|T states, was calculated as . These values were used to calculate the difference in energy of the Lys-Fe(III)|T and the Met-Fe(III)|TH+ species for each of the variants using . The relative energies for the Met-Fe(III)|T species were calculated using . Providing support for our approach, the calculated differences between the Met-Fe(III)|T and Lys-Fe(III)|T species of 11 ± 3 kJ/mol for variants in our study are consistent with the energy difference of 14 kJ/mol between the Gibbs free energies of activation for the forward (ΔGf‡) and backward (ΔGb‡) reactions in horse heart cyt c.97
With the ground-state energy levels of the Met-Fe(III)|T and Lys-Fe(III)|T species mapped out, the changes upon T49V and T78V substitutions in the energy of the transition state (TS) (ΔΔG‡) were then determined by adding to the difference of the energies of the Lys-Fe(III)|T species for K79G and T49V/K79G (or for K79G and T78V/K79G). These calculations have allowed us to differentiate the energies of the Met-Fe(III)|TH+ and Met-Fe(III)|T species and provided a quantitative basis for the analyses of the effects of Thr-to-Val substitutions on the triggering process. Results in Figure 11A suggest that deprotonation of the “trigger” group destabilizes ferric Met-ligated cyt c, priming it for reaching the TS for the Met-to-Lys ligand switch. Without knowledge of the preexponential factors for our variants, it was not possible to define the absolute energy levels for the TS, but the available temperature-dependent kinetics for the alkaline transition in ferric horse heart cyt c put the TS in that protein around 68 kJ/mol higher in energy than the Met-Fe(III)|T species.97
After obtaining the energy levels of the ferric Met-Fe(III)|TH+ and Lys-Fe(III)|T species for each variant, energies of the corresponding ferrous species were calculated based on the reduction potentials (Table 4): or . With the KCII values determined from the analyses of the voltammetry and pH dependence of electronic absorption spectra, energy levels for the Met-Fe(II)|T species relative to Lys-Fe(II)|T were calculated (Figure 11A). As in the case of ferric variants, the changes in the energy of TS (ΔΔG‡) upon the Thr-to-Val substitutions were determined by adding to the difference of the energies of the Lys-Fe(II)|T species for K79G and T49V/K79G (or for K79G and T78V/K79G).
Discussion
Studies of the alkaline transition in cyt c inform about mechanisms of biologically relevant ligand-switch reactions98–101 and the features of the polypeptide fold that support dual ligation in heme proteins. The transition in cyt c is coupled to acid-base equilibria.22,102 First, the Met-to-Lys ligand switch requires deprotonation of a Lys residue. Second, deprotonation of an as yet unknown group, distinct from Lys, triggers the conformational transition.103 The identity of the “trigger” group has been extensively debated;23,28,104 and HP6 has been a prominent target of these discussions,7,19,29,30,105 both as a “trigger” group itself and a linker that ties together the hydrogen-bonding network near the heme. The Thr-to-Val substitutions we have studied here break up hydrogen-bond connections to HP6 (Figure 1) and allow for examination of the importance of these connectivities in the Met-to-Lys ligand-switch mechanism.
Hydrogen-exchange experiments have determined that Ω-loop C and Ω-loop D are the least and the second-to-least stable regions of cyt c structure, respectively.21 The two Ω-loops are stapled together by HP6 through Thr49 and Thr78 in the native Met-ligated cyt c.16 By comparing effects of Thr-to-Val substitutions at these sites on structure, stability, and ligand-switch mechanism, we identify distinct roles of these loops in conformational dynamics of cyt c. With data on both ferric and ferrous cyt c variants, we rationalize effects of these substitutions (and HP6 environment) on heme iron reduction potentials and on redox-dependent preferences in axial ligands.
Effects of Breaking Connections to HP6 on Structure, Stability, and Redox Properties of Cyt c.
Stabilities of ferric and ferrous variants, together with structural models, provide a framework for understanding effects of Thr-to-Val substitutions on redox properties of cyt c. The substitutions modify denaturation curves of both ferric and ferrous cyt c. Although error bars for GuHCl denaturation experiments prevent direct comparison of values for the variants (Table 3), calculations using reduction potentials for Met|TH+ species (Scheme S2 and Table 4) reveal that the stabilities of these species in both ferric and ferrous forms of the Thr-to-Val variants are lower than those of K79G (Figure 11A). These findings suggest that connections to HP6 play a role in stabilization of the native protein fold. The extent of destabilization for ferrous T49V/K79G is greater than that for ferrous T78V/K79G.
Figure 11A incorporates an assumption that the energy levels for the unfolded variants are very similar. T49V/K79G and T78V/K79G have similar composition, so comparisons of these two variants to each other are consistent with such an assumption. However, K79G has two native Thr residues. As reported elsewhere,106,107 Thr-to-Val substitutions can stabilize the protein unfolded state. Thus, the Thr-to-Val substitutions could serve to increase all of the energy levels for the Thr-to-Val variants in Figure 11A relative to unfolded K79G. As a consequence, our finding that the Met|TH+ state in the two Thr-to-Val variants is destabilized relative to that for K79G remains supported, and if anything, is perhaps underestimated.
In ferric T49V/K79G, the move of Wat166 closer to the iron atom would help to stabilize the positively-charged ferric heme moiety. Water molecules next to the heme have been suggested to screen the charge of an anionic HP and draw electron density from HP through hydrogen bonding.108 The loss of a water molecule (Wat4933) found near HP6 in ferric K79G would help then to stabilize ferric T49V/K79G. In contrast, the improvement in hydration of HPs in ferrous T49V/K79G would help to stabilize the neutral ferrous porphyrin moiety. However, these arguments are inconsistent with experimentally derived energy levels in Figure 11A suggesting that one should also consider destabilizing interactions. The removal of hydrogen-bonding interactions between Thr49 and HP6 in the T49V/K79G variant would destabilize both ferric and ferrous forms of the protein. The favorable repositioning of Wat166 and the loss of Wat4933 in ferric T49V/K79G likely partially counteract destabilizing effects of losing what appears to be a critical hydrogen-bonding connection between Ω-loop C and the heme.
For T78V/K79G, the loss of hydrogen bonds from Thr78 upon the T78V substitution is compensated by new interactions HP6 forms with Thr49. The change in the stability of the ferrous species upon the T78V substitution, however, appears to be small, if any (Figure 11A), compared to a clear destabilization of the ferrous species upon the T49V substitution. The increase in the reduction potential for T78V/K79G is noteworthy as most reported substitutions near HPs in cyt c led to decreases in the heme iron reduction potential.39,78,109–111
These analyses, together with the lack of significant changes in SASA or heme pocket volume, illustrate that the effects of our Thr-to-Val substitutions largely come from alterations in the hydrogen-bonding network and hydration around the still largely-buried heme. Studies of crystallographically-characterized N52I, Y67F, and N52I/Y67F variants have found that changes of this kind could modify the stability of cyt c.109,112,113 For that set of cyt c variants, the stabilities of both ferric and ferrous species increase, but the heme iron reduction potentials decrease. For T78V/K79G and T78V/K79G studied here, the stabilities of ferric and ferrous species decrease, but the directions of the potential change are opposite for the two variants. The earlier findings with N52I, Y67F, and N52I/Y67F and outcomes of our work with Thr-to-Val variants suggest that substitutions near the heme could both stabilize or destabilize cyt c, but the extent of these effects may differ for the two oxidation states of the heme iron. Therefore, in order to understand the origin of changes in heme iron reduction potentials, it is important to assess structural features and stabilities of both ferric and ferrous forms of protein variants.
Alkaline Transition in Ferric Cyt c.
Structural changes in the heme pocket upon Thr-to-Val substitutions result in destabilization of the Met-ligated ferric cyt c (Figure 11A). These findings add support to the importance of entatic forces in stabilization of Met80 coordination to the ferric heme iron in cyt c, as suggested by Solomon et al.,114,115 and illustrate that HP6 is an important component of this entatic frame. Interestingly, the decrease in stability of Met-ligated cyt c is coupled with the decrease in rate constants kf as well. These findings, together with faster rates for Met80 dissociation from the ferric heme iron (koffMet, III = 50 ± 7 s−1 in K79G),40 rule out Met80 dissociation as the rate-controlling step. Instead, the Met-to-Lys ligand switch appears to depend on structural reorganization of Ω-loop D. The slower rate constants in T49V/K79G and T78V/K79G variants compared to K79G likely arise from nonproductive contacts formed by a hydrophobic Val residue that impede the conformational search during the formation of the Lys73-ligated species.
There are no differences in kbIII values for the Lys-to-Met ligand switch, nor in the energy levels for the ferric TS and Lys-ligated species for the variants studied here (Figure 11A), suggesting that the Thr-to-Val substitutions modify the alkaline transition in ferric cyt c by predominantly acting on the Met- and not the Lys-ligated species. The rate constants kbIII are of the same order of magnitude as kbIII values for other cyt c variants with a largely intact polypeptide fold116 and also consistent with Lys dissociation rate constants from imidazole binding experiments.63 These similarities of kbIII values as well as similar spectroscopic properties and stabilities of Lys-ligated T49V/K79G and T78V/K79G agree with the lack of specific interactions that residues 49 and 78, respectively, form with the rest of the Lys-ligated protein (Figure S9).
At pH 7.4, the energy gap between the Met-Fe(III)|TH+ and the Met-Fe(III)|T species for K79G is very large (Figure 11A), on the order of values (Table S4), in accord with the low likelihood of the alkaline transition under these pH conditions. This gap gets smaller at higher pH (Figure S16). Because the energy gap between the Met-Fe(III)|TH+ and the Met-Fe(III)|T species for T49V/K79G and T78V/K79G is already small at pH 7.4, the alkaline transition can proceed at lower pH in these two variants.
Since deprotonation of the “trigger” group affects the heme absorption spectrum, this group must reside in the vicinity of the heme. The pKHIII values in T49V/K79G and T78V/K79G are now unequivocally too low to represent the pKa of a Lys residue, providing a strong argument against such an assignment. The two Ω-loops C and D contribute to the enclosure of the heme and because the two loops are interconnected, even remote perturbations within these loops may potentially influence the acid-base properties of the “trigger” group. However, the magnitude of effects of such perturbations is informative. The decrease in pKHIII values in our Thr-to-Val variants is greater than that seen for cyt c variants with substitutions at residue 82,116 suggesting that the deprotonation site is closer to residues 49 and 78. Adding support to this argument, substitutions at residues 81 and 83 in recent studies of human cyt c did not change much the pKHIII value of the “trigger” group.29,117 Similar to our Thr-to-Val variants, substitutions G41S and Y48H in human cyt c, which are closer to HP7 than to HP6, also modify pKHIII,29 but their effects are not as dramatic as that of T49V/K79G and T78V/K79G.
HP6 hydrogen bonds to both Thr49 and Thr78 and thus is a logical candidate to be the “trigger” of the alkaline transition, especially since substitutions at both of these sites dramatically affect the properties of the “trigger” group. The proposed high pKa of this group in the wild-type cyt c (⩾9)7,105 is in accord with the pKHIII value for the alkaline transition. The Thr-to-Val substitutions could lower the pKHIII value of HP6 but not all the way to the range seen in solvent-exposed HPs (between 5.0 and 6.1 for ferric heme peptides).68 The continued hydrogen-bonding interactions involving this group and screening by the surrounding polypeptide could still keep the pKa value of this group elevated in T49V/K79G and T78V/K79G variants.
A cluster of hydrogen-bonded titratable groups could work cooperatively to trigger the transition. Bowler et al. have identified multiple acid-base equilibria important for triggering the alkaline transition in their studies of Met-to-His switchable variants of cyt c.102 Wilson, Worrall, et al. have recently argued that the deprotonation of the hydrogen-bonded unit that links Ω-loops C and D is what triggers the transition.29 HP6 is a central element of this hydrogen-bonding unit and our observations here of prominent effects on the alkaline transition for substitutions near this group are consistent with this interesting proposal. There is clearly cross-talk between the groups within this hydrogen-bonding unit as changes near both HP6 and HP7, albeit to a different extent, influence the properties of the “trigger”.
Alkaline Transition in Ferrous Cyt c.
So far only a few studies have attempted to examine the alkaline transition in ferrous cyt c.72,118 With destabilization of the Met-ligated species in T49V/K79G and T78V/K79G and combined use of spectroscopic and voltammetry measurements, characterization of this elusive process becomes possible.
The kfIII values in ferric proteins (Table 1) reflect the kinetics of conformational search prior to Lys coordination because Met80 dissociation from the ferric heme iron is fast. The calculated kfII values in ferrous proteins (Table 2) are at least an order of magnitude smaller than the kfIII values. Such a large decrease is inconsistent with Lys73 coordination continuing to be the rate-controlling step but agrees with Met80 dissociation from the ferrous heme now being this slow step. The observed decrease in upon the heme iron reduction is consistent with expectations from comparison of Kd values for Met binding to ferric and ferrous forms of AcMP8.89 As the rate of the forward Met-to-Lys ligand switch is controlled by Met80 dissociation, it reports on the bond strength and properties of the surrounding heme pocket needed to accommodate ligand dissociation. Therefore, a faster rate of Met80 dissociation from the ferrous heme in T49V/K79G compared to that in T78V/K79G may be linked to the decreased stability of the Met-ligated ferrous species and a weaker Fe-S bond in the former variant.
The magnitude of kbII values reported here is in accord with those for the disappearance of the transient ferrous alkaline cyt c in kinetics experiments118 and voltammetry results with other cyt c variants.72 The kbII values are greater than the corresponding kbIII values. We hypothesize that the reduced strength of the Fe-N bond upon heme iron reduction is the main reason for the observed enhancement of Lys dissociation rates. While binding affinities of Lys (or His) to ferric and ferrous AcMP8 appear to be similar,63,89 differences in the Fe-N bond strengths have been observed in X-ray spectroscopic studies of bis-imidazole ferric and ferrous porphyrins.114 Moreover, the much lower stability of the Lys-Fe(II)|T species compared to that of the Lys-Fe(III)|T (Table 4) reflects the importance of the protein structure in influencing redox-dependent metal-ligand interactions.
The trends in kbII suggest that both Thr-to-Val substitutions facilitate Lys dissociation from the ferrous heme iron. The increase in kbII, however, is more prominent for T78V/K79G. We have previously observed correlation between rates of Lys dissociation from the ferric heme iron and flexibility of the surrounding polypeptide.19,63 If Lys dissociation from the ferrous heme iron is influenced by these effects as well, Ω-loop D may be more structured in the Lys-ligated T49V/K79G. Further, this result suggests that there may be differences in conformational properties of ferrous and ferric Lys-ligated proteins as the kbIII values of ferric T49V/K79G and T78V/K79G are very similar.
The KCII values for T49V/K79G and T78V/K79G calculated assuming pKa,1II = pKHII are (2.0±1.5)×10−2 and (2.0±1.7)×10−4, respectively. These values are at least three orders of magnitude smaller than the corresponding KCIII values, highlighting the unfavorable nature of the alkaline transition in ferrous proteins. While in ferric proteins the KCIII equilibrium brings down the apparent pKaIII of the alkaline transition, which then can be easily observed experimentally, in ferrous proteins the apparent pKaII for the transition goes up, to inaccessible pH values since the protein denatures. Not only do the Met-ligated species become more stable upon reduction of the heme iron, but Lys-ligated species get destabilized as well.
A recent study by Lecomte et al. of the His-to-Lys ligand switch in ferrous hemoglobin G1bN provides an interesting comparison.36 The alkaline transition is observed in ferrous G1bN but not in the ferric protein. Compared to a Met-ligated species, a His-ligated species has a much lower reduction potential.119 The smaller difference in potential relative to the Lys-ligated species would lead to a smaller disparity in stability for the differently ligated ferrous conformers than we see for cyt c. Therefore, the contribution from the conformational exchange pKCII to the apparent pKaII is likely small and the transition can be easily observed in the experimentally accessible pH range (pKaII = 9.3). The authors have argued that greater flexibility of ferrous GlbN relative to the ferric protein may be responsible for the ease of the ligand switch in that system. We note that cyt c, in contrast, experiences an increase in backbone dynamics upon oxidation of the heme iron.120
Our analyses have revealed that deprotonation of the “trigger” group decreases reduction potentials of the Met-ligated species by about 50–180 mV (Table 4). The energy levels of ferrous species are more sensitive to this deprotonation process than those of ferric species (Figure 11A). These findings suggest that destabilizing effects of the deprotonation may come from introduction of a negative charge in the proximity of the neutral porphyrin moiety in Met-Fe(II)|T. These arguments are consistent with deprotonation of HP6 or a cluster of groups that this HP contributes to but not with a Lys residue since its deprotonation will result in a neutral species. In bacterial cytochromes, the measured shift upon HP7 ionization is 50–70 mV and calculations based on electrostatic models predict shifts over 100 mV.121,122 The site-specific effects of the two Thr-to-Val substitutions studied here implicate HP6 as the region playing a critical role in the alkaline transition. The similarity of our shifts in reduction potentials upon the deprotonation of the “trigger” group to these numbers offers further support to the involvement of this propionate group.
Two Dynamic Ω-Loops Stapled by HP6 Direct Metal-Ligand Interactions at the Heme Iron.
It has been previously suggested that the hydrogen-bonding network around HPs in cyt c is in a dynamic state of disruption and reformation,23 and, indeed, we observe these processes in our simulations. The connection HP6-Thr49 linking Ω-loop C to the heme appears to be relatively static but the connection HP6-Thr78 linking Ω-loop D to the heme is dynamic (Figure S8), priming this coordination loop for changes in the heme iron ligation. As our results show, both of these HP6 connections favor the native Met80 coordination to the heme iron and stability of the protein.
Ligand dissociation and polypeptide rearrangement associated with the alkaline transition depend on the plasticity of Ω-loop D.28 Destabilization of Ω-loop D and general destabilization of the cyt c structure have been correlated with the faster local dynamics around the heme crevice,123 but exceptions to the latter correlation have been observed as well.124 While some pairwise correlations can be visualized, the kf and kb values for variants in this work do not show a clear dependence on the global stability of the Met-Fe|T and Lys-Fe|T species within the entire series (Figures 11B and 11C). When effects of local destabilization of Ω-loop D in the Met-ligated ferric species (Figure S7 and Table S4) on kf values are considered, there are no clear trends either. For T78V/K79G, destabilization of Ω-loop D is dramatic: the majority of the Met-ligated species disappear at low GuHCl concentrations, at which the bulk of the α-helical structure is still preserved. Yet, the kfIII value for this variant decreases compared to that in K79G, but not as much as kfIII in T49V/K79G does. Our analyses of the equilibria between the Met-on and Met-off species in folded proteins (Figure 8) imply a further weakening of the Fe-S bond in T49V/K79G compared to that in T78V/K79G, but this result does not provide an explanation for this effect since the rate of the Met-to-Lys ligand switch is not controlled by Met80 dissociation from the ferric heme iron. As discussed earlier, we have attributed these trends to effects of Thr-to-Val substitutions on the conformational search associated with Lys73 coordination. This process, rather than Met80 dissociation, controls the rate of the Met-to-Lys ligand switch in ferric proteins.
The distinct differences in local destabilization of Ω-loop D in T49V/K79G and T78V/K79G relate to the mechanism of the Met-to-Lys ligand switch. As pH decreases, Met80 in T78V/K79G is readily replaced by H2O in the frame of a largely folded protein in the A2 state. This species is distinct from the Met-off species in equilibrium with the Met-on species we find in the M state. In the A2 state, H2O presumably enters the heme pocket and the disruption of the HP6-Thr78 interaction allows for the displacement of Met80 from the heme pocket to promote structural change associated with this entry. In contrast, the same process requires major unfolding in T49V/K79G. Yet, the two variants do not differ much in the stability of the Met-Fe(III)|T or Met-Fe(III)|TH+ species and undergo the alkaline transition at a similar pH.
These results suggest that, while no large structural changes in the heme environment are necessary to dissociate Met80 from the heme iron, the connections of Ω-loop D to HP6 may need to be disrupted to facilitate the ligand displacement. The HP6-Thr78 interaction is already dynamic and it is likely that the deprotonation of the “trigger” group finalizes the break-up of these contacts in T49V/K79G and K79G (and WT) cyt c. Thus, the role for the deprotonation of the “trigger” group could be not just to further destabilize the Met-ligated species and bring them closer in energy to TS, but to specifically disrupt some intraprotein interactions involving Ω-loop D. Because of the great sensitivity of the alkaline transition to substitutions near HP6, it is the likely location of the disrupted contacts and of the “trigger” group. In accord with this proposal, recent mutational studies of human cyt c have revealed that substitutions at residues 81 and 83 do not affect greatly the properties of the “trigger” group.29,117 On the other hand, substitutions at residue 82 modify the pKH value of the “trigger” group as well as kinetics of the alkaline transition in yeast iso-1 cyt c.116 Yeast cyt c is less stable than human cyt c31,80 and we suggest that substitutions of the largely buried Phe82 residue may have been particularly destabilizing to Ω-loop D in yeast cyt c and disrupted interactions of this loop with HP6. The crystal structure of hydroxide-ligated WT* cyt c with Met80 dissociated from the heme iron did not show disruption of hydrogen-bonding interaction of Thr78 with HP6,125 but the break-up of this interaction may have been transient, particularly since this hydrogen-bonding contact appears to be dynamic.
Given the structural perturbations with the T49V substitution, it is not surprising that the stabilities of ferric and ferrous forms of T49V/K79G differ from those of K79G. What is intriguing, however, is the effect of this substitution in Ω-loop C on the alkaline transition that largely concerns rearrangements of Ω-loop D. We have already discussed the more trivial effects of the T49V substitution on the alkaline transition due to global destabilization of the Met-ligated species. Further, the higher population of the Met-off species in the M state suggests this substitution weakens the Fe-S bond more so than with T78V. These results are in accord with the ease of accessing the pentacoordinate heme iron state recently reported for several variants of human cyt c that also have substitutions in Ω-loop C.90,91 Now we focus on its effects on the associated dynamics and properties of the “trigger” group.
NMR hydrogen exchange studies have suggested that unfolding of Ω-loop C gates internal deprotonation in the mechanism of the alkaline transition.103 Unfolding of this low-stability loop provides a pathway to HP6 that links the two loops to facilitate deprotonation of the “trigger” group and induce structural changes in Ω-loop D for Met80 displacement. Unfolding of Ω-loop C will be favored in T49V/K79G, which lacks the Thr49-to-HP6 connection linking this loop to the protein core. The comparison of kfIII values for T49V/K79G and T78V/K79G variants suggests that structural perturbations in Ω-loop C are also linked to the rearrangement of Ω-loop D. With increased conformational heterogeneity of the protein ensemble from destabilization of Ω-loop C, conformational search in T49V/K79G could be slower and a larger fraction of the protein may need to rearrange. Interestingly, the kfIII value of T49V/K79G is comparable to that of N52G/K72A,81 which has also been found to have destabilizing effects on Ω-loop C.
The cross-talk between the two Ω-loops is clearly involved in both thermodynamic and kinetic aspects of the alkaline transition, with HP6 being a critical component of this communication. We further argue that, mediated by a hydrogen-bonding network, propagation of structural perturbations from Ω-loop C to other parts of the protein structure may tune redox properties of cyt c as we have seen in these studies (Table S8) and in our photoinduced ET work.126 This intrinsic dynamic property of the cyt c scaffold may also be at the core of functional effects of disease mutants of human cyt c carrying substitutions in Ω-loop C.29,127
Conclusions
Two Thr-to-Val substitutions near HP6 have dramatic effects on properties of the heme iron within the cyt c fold. The connections to HP6 at these sites are critical for maintaining the native Met80 ligation to the heme iron. Without them, the stabilities of the Met-ligated species decrease in both ferric and ferrous proteins. Our map of energy levels highlights the importance of thorough characterization of both oxidation states to explain shifts in reduction potentials in protein variants; such an approach is rarely taken.
Destabilization of the protein scaffold and a decrease in pKH of the “trigger” group yield the Lys-ligated heme iron in ferric proteins already at neutral pH, confirming a marginal role of the Met80-iron bond in stability of ferric cyt c. The sensitivity of pKH value to both Thr-to-Val substitutions suggests that the trigger group is located near Thr49 and Thr78. This observation, together with the estimates of shifts in the heme iron potentials, strengthens support for HP6 being the “trigger”, either on its own or as part of the hydrogen-bonded cluster. The substitutions also affect kinetics of ligand-switch reactions by altering dynamics of the conformational search and flexibility of the heme pocket. The communication between Ω-loop C and Ω-loop D seen in stability and kinetics effects of the T49V substitution further highlights the importance of HP6 connections linking the two loops.
Supplementary Material
Acknowledgements.
This work was supported by the NSF CHE-1708592 (E.V.P.) and NIH R01-GM098502 (E.V.P.) grants. We also thank the Berry College Faculty Development Grant program for additional support (K.R.H.). We are grateful to Bruce E. Bowler (University of Montana) for the Rbs (WT*) cyt c plasmid. Y.D. was partially supported by China Scholarship Council.
Footnotes
Supporting Information. The Supporting Information is available free of charge on the ACS Publications website at DOI: tbd. Two schemes showing relationships between reduction potentials and protein stability; sixteen figures showing absorption spectra of M80A to determine reference spectra for A1 and A2; RMSDs of MD snapshots of K79G, T49V/K79G, and T78V/K79G; EPR and 1H NMR spectra of ferric K79G, T49V/K79G, and T78V/K79G at pH (or pD) 10.5; spectroscopic characterizations of ferrous K79G, T49V/K79G, and T78V/K79G under alkaline conditions; CD spectra of ferrous K79G, T49V/K79G, and T78V/K79G at pH 7.4; GuHCl titrations of ferric and ferrous K79G, T49V/K79G, and T78V/K79G at pH 7.4; GuHCl titrations of ferric K79G, T49V/K79G, and T78V/K79G at pH 4.5; hydrogen-bond analyses of the structural models of ferric Met-ligated WT* and K79G; structural models of ferric Lys73-ligated K79G, T49V/K79G, and T78V/K79G; cyclic voltammograms at varying scan rates for T49V/K79G and T78V/K79G at pH 7.4 and for K79G at pH 10; cyclic voltammograms at varying scan rates for T78V/K79G at pH 4.5; SVD analysis for the pH titration data of ferric T49V/K79G; SVD analysis for the pH titration data of ferric T78V/K79G; absorption spectra and kinetic curves from pH-jump experiments; representative progress curve for the bimolecular reduction of ferric T49V/K79G by a6Ru2+ at pH 7; changes in the energy gaps versus pH for the alkaline transition in ferric K79G; and eight tables listing species used as reference spectra for the SVD analysis of pH titrations; structural analysis statistics of ferric K79G, T49V/K79G, and T78V/K79G; structural analysis statistics of ferrous K79G, T49V/K79G, and T78V/K79G; thermodynamic parameters for the unfolding transitions of ferric K79G variants at pH 4.5; reduction potentials for K79G, T49V/K79G, and T78V/K79G at varying scan rates; reduction potentials for K79G, T49V/K79G, and T78V/K79G at varying pH; parameters from global fits for the pH-dependence of heme iron ligation in ferric Thr-to-Val variants; and bimolecular ET rate constants.
References
- (1).Biological Inorganic Chemistry: Structure and Reactivity; Bertini I; Gray HB; Stiefel EI; Valentine JS, Eds.; University Science Books: Sausalito, CA, 2007. [Google Scholar]
- (2).Mao J; Hauser K; Gunner MR How cytochromes with different folds control heme redox potentials. Biochemistry 2003, 42, 9829–9840. [DOI] [PubMed] [Google Scholar]
- (3).Hunter CL; Lloyd E; Eltis LD; Rafferty SP; Lee H; Smith M; Mauk AG Role of the heme propionates in the interaction of heme with apomyoglobin and apocytochrome b5. Biochemistry 1997, 36, 1010–1017. [DOI] [PubMed] [Google Scholar]
- (4).Xu C; Ibrahim M; Spiro TG DFT analysis of axial and equatorial effects on heme-CO vibrational modes: applications to CooA and H-NOX heme sensor proteins. Biochemistry 2008, 47, 2379–2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Louro RO; Bento I; Matias PM; Catarino T; Baptista AM; Soares CM; Carrondo MA; Turner DL; Xavier AV Conformational component in the coupled transfer of multiple electrons and protons in a monomeric tetraheme cytochrome. J. Biol. Chem 2001, 276, 44044–44051. [DOI] [PubMed] [Google Scholar]
- (6).Warren JJ; Mayer JM Proton-coupled electron transfer reactions at a heme-propionate in an iron-protoporphyrin-IX model compound. J. Am. Chem. Soc 2011, 133, 8544–8551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Tonge P; Moore GR; Wharton CW Fourier-transform infra-red studies of the alkaline isomerization of mitochondrial cytochrome c and the ionization of carboxylic acids. Biochem. J 1989, 258, 599–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Guallar V Heme electron transfer in peroxidases: the propionate e-pathway. J. Phys. Chem. B 2008, 112, 13460–13464. [DOI] [PubMed] [Google Scholar]
- (9).Brändén G; M. B; Schmidt B; A. MD; Ferguson-Miller S; Brzezinski P The protonation state of a heme propionate controls electron transfer in cytochrome c oxidase. Biochemistry 2005, 44, 10466–10475. [DOI] [PubMed] [Google Scholar]
- (10).Roitberg AE; Holden MJ; Mayhew MP; Kurnikov IV; Beratan DN; Vilker VL Binding and electron transfer between putidaredoxin and cytochrome P450cam. Theory and experiments. J. Am. Chem. Soc 1998, 120, 8927–8932. [Google Scholar]
- (11).Macdonald IK; Badyal SK; Ghamsari L; Moody PC; Raven EL Interaction of ascorbate peroxidase with substrates: a mechanistic and structural analysis. Biochemistry 2006, 45, 7808–7817. [DOI] [PubMed] [Google Scholar]
- (12).Reid LS; Mauk MR; Mauk AG Role of heme propionate groups in cytochrome b5 electron transfer. J. Am. Chem. Soc 1984, 106, 2182–2185. [Google Scholar]
- (13).Dias JM; Alves T; Bonifácio C; Pereira AS; Trincão J; Bourgeois D; Moura I; Romão MJ Structural basis for the mechanism of Ca2+ activation of the di-heme cytochrome c peroxidase from Pseudomonas nautica 617. Structure 2004, 12, 961–973. [DOI] [PubMed] [Google Scholar]
- (14).Bren KL Going with the electron flow: heme electronic structure and electron transfer in cytochrome c. Isr. J. Chem 2016, 56, 693–704. [Google Scholar]
- (15).Smith LJ; Kahraman A; Thornton JM Heme proteins—Diversity in structural characteristics, function, and folding. Proteins 2010, 78, 2349–2368. [DOI] [PubMed] [Google Scholar]
- (16).Berghuis AM; Brayer GD Oxidation state-dependent conformational changes in cytochrome c. J. Mol. Biol 1992, 223, 959–976. [DOI] [PubMed] [Google Scholar]
- (17).Redzic JS; Bowler BE Role of hydrogen bond networks and dynamics in positive and negative cooperative stabilization of a protein. Biochemistry 2005, 44, 2900–2908. [DOI] [PubMed] [Google Scholar]
- (18).Bandi S; Baddam S; Bowler BE Alkaline conformational transition and gated electron transfer with a Lys 79 → His variant of iso-1-cytochrome c. Biochemisry 2007, 46, 10643–10654. [DOI] [PubMed] [Google Scholar]
- (19).Gu J; Shin DW; Pletneva EV Remote perturbations in tertiary contacts trigger ligation of lysine to the heme iron in cytochrome c. Biochemistry 2017, 56, 2950–2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Cytochrome c - A Multidisciplinary Approach; Scott RA; Mauk AG, Eds.; University Science Books: Sausalito, CA, 1996. [Google Scholar]
- (21).Maity H; Maity M; Englander SW How cytochrome c folds, and why: submolecular foldon units and their stepwise sequential stabilization. J. Mol. Biol 2004, 343, 223–233. [DOI] [PubMed] [Google Scholar]
- (22).Rosell FI; Ferrer JC; Mauk AG Proton-linked protein conformational switching: Definition of the alkaline conformational transition of yeast iso-1-ferricytochrome c. J. Am. Chem. Soc 1998, 120, 11234–11245. [Google Scholar]
- (23).Moore GR; Pettigrew GW Cytochromes c: Evolutionary, structural, and physicochemical aspects; Springer-Verlag: New York, 1990. [Google Scholar]
- (24).Theorell H; Åkesson Å Studies on cytochrome c. I. Electrophoretic purification of cytochrome c and its amino acid composition. J. Am. Chem. Soc 1941, 63, 1804–1811. [Google Scholar]
- (25).Theorell H; Åkesson Å Studies on cytochrome c. II. The optical properties of pure cytochrome c and some of its derivatives. J. Am. Chem. Soc 1941, 63, 1812–1818. [Google Scholar]
- (26).Theorell H; Åkesson Å Studies on cytochrome c. III. Titration curves. J. Am. Chem. Soc 1941, 63, 1818–1820. [Google Scholar]
- (27).Theorell H Studies on cytochrome c. IV. The magnetic properties of ferric and ferrous cytochrome c. J. Am. Chem. Soc 1941, 63, 1820–1827. [Google Scholar]
- (28).Cherney MM; Bowler BE Protein dynamics and function: Making new strides with an old warhorse, the alkaline conformational transition of cytochrome c. Coord. Chem. Rev 2011, 255, 664–677. [Google Scholar]
- (29).Deacon OM; Svistunenko DA; Moore GR; Wilson MT; Worrall JAR Naturally occurring disease-related mutations in the 40–57 Ω-Loop of human cytochrome c control triggering of the alkaline isomerization. Biochemistry 2018, 57, 4276–4288. [DOI] [PubMed] [Google Scholar]
- (30).Amacher JF; Zhong F; Lisi GP; Zhu MQ; Alden SL; Hoke KR; Madden DR; Pletneva EV A compact structure of cytochrome c trapped in a lysine-ligated state: loop refolding and functional implications of a conformational switch. J. Am. Chem. Soc 2015, 137, 8435–8449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Nold SM; Lei H; Mou TC; Bowler BE Effect of a K72A mutation on the structure, stability, dynamics, and peroxidase activity of human cytochrome c. Biochemistry 2017, 56, 3358–3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Bradley JM; Silkstone G; Wilson MT; Cheesman MR; Butt JN Probing a complex of cytochrome c and cardiolipin by magnetic circular dichroism spectroscopy: Implications for the initial events in apoptosis. J. Am. Chem. Soc 2011, 133, 19676–19679. [DOI] [PubMed] [Google Scholar]
- (33).Kagan VE; Tyurin VA; Jiang J; Tyurina YY; Ritov VB; Amoscato AA; Osipov AN; Belikova NA; Kapralov AA; Kini V; Vlasova II; Zhao Q; Zou M; Di P; Svistunenko DA; Kurnikov IV; Borisenko GG Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat. Chem. Biol 2005, 1, 223–232. [DOI] [PubMed] [Google Scholar]
- (34).Schweitzer-Stenner R Relating the multi-functionality of cytochrome c to membrane binding and structural conversion. Biophys. Rev 2018, 10, 1151–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Johnson EA; Russo MM; Nye DB; Schlessman JL; Lecomte JTJ Lysine as a heme iron ligand: A property common to three truncated hemoglobins from Chlamydomonas reinhardtii. Biochim. Biophys. Acta Gen. Subj 2018, 1862, 2660–2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Nye DB; Preimesberger MR; Majumdar A; Lecomte JTJ Histidine-lysine axial ligand switching in a hemoglobin: A role for heme propionates. Biochemistry 2018, 57, 631–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Rodrigues ML; Oliveira TF; Pereira IAC; Archer M X-ray structure of the membrane-bound cytochrome c quinol dehydrogenase NrfH reveals novel haem coordination. EMBO J. 2006, 25, 5951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Mowat CG; Rothery E; Miles CS; McIver L; Doherty MK; Drewette K; Taylor P; Walkinshaw MD; Chapman SK; Reid GA Octaheme tetrathionate reductase is a respiratory enzyme with novel heme ligation. Nat. Struct. Mol. Biol 2004, 11, 1023–1024. [DOI] [PubMed] [Google Scholar]
- (39).Davies AM; Guillemette JG; Smith M; Greenwood C; Thurgood AG; Mauk AG; Moore GR Redesign of the interior hydrophilic region of mitochondrial cytochrome c by site-directed mutagenesis. Biochemistry 1993, 32, 5431–5435. [DOI] [PubMed] [Google Scholar]
- (40).Deng Y; Zhong F; Alden SL; Hoke KR; Pletneva EV The K79G mutation reshapes the heme crevice and alters redox properties of cytochrome c. Biochemistry 2018, 57, 5827–5840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Hanske J; Toffey JR; Morenz AM; Bonilla AJ; Schiavoni KH; Pletneva EV Conformational properties of cardiolipin-bound cytochrome c. Proc. Natl. Acad. Sci. USA 2012, 109, 125–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Zhong F; Lisi GP; Collins DP; Dawson JH; Pletneva EV Redox-dependent stability, protonation, and reactivity of cysteine-bound heme proteins. Proc. Natl. Acad. of Sci. USA 2014, 111, E306–E315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Berry EA; Trumpower BL Simultaneous determination of hemes-a, hemes-b, and hemes-c from pyridine hemochrome spectra. Anal. Biochem 1987, 161, 1–15. [DOI] [PubMed] [Google Scholar]
- (44).Pace CN; Sholtz JM In Protein structure: a practical approach; Creighton TF, Ed.; Oxford University Press: New York, 1997, p 299–321. [Google Scholar]
- (45).Galo AL; Colombo MF Singular value decomposition and ligand binding analysis. J. Spectr 2013, 2013, 7. [Google Scholar]
- (46).Hendler RW; Shrager RI Deconvolutions based on singular value decomposition and the pseudoinverse: a guide for beginners. Journal of Biochemical and Biophysical Methods 1994, 28, 1–33. [DOI] [PubMed] [Google Scholar]
- (47).Battistuzzi G; Bortolotti CA; Bellei M; Di Rocco G; Salewski J; Hildebrandt P; Sola M Role of Met80 and Tyr67 in the low-pH conformational equilibria of cytochrome c. Biochemistry 2012, 51, 5967–5978. [DOI] [PubMed] [Google Scholar]
- (48).Sanchez KM; Schlamadinger DE; Gable JE; Kim JE Förster resonance energy transfer and conformational stability of proteins. An advanced biophysical module for physical chemistry students. J. Chem. Educ 2008, 85, 1253–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Bard AJ; Faulkner LR Electrochemical Methods: fundamentals and applications; 2nd ed.; John Wiley & Sons; New York, 2001. [Google Scholar]
- (50).Haas AS; Pilloud DL; Reddy KS; Babcock GT; Moser CC; Blasie JK; Dutton PL Cytochrome c and cytochrome c oxidase: Monolayer assemblies and catalysis. J. Phys. Chem. B 2001, 105, 11351–11362. [Google Scholar]
- (51).Nelson CJ; Bowler BE pH dependence of formation of a partially unfolded state of a Lys 73→His variant of iso-1-cytochrome c: Implications for the alkaline conformational transition of cytochrome c. Biochemistry 2000, 39, 13584–13594. [DOI] [PubMed] [Google Scholar]
- (52).Nicholson R Theory and application of cyclic voltammetry for measurement of electrode reaction kinetics. Anal. Chem 1965, 37, 1351–1355. [Google Scholar]
- (53).Fourmond V; Hoke KR; Heering HA; Baffert C; Leroux F; Bertrand P; Léger C SOAS: a free program to analyze electrochemical data and other one-dimensional signals. Bioelectrochemistry 2009, 76, 141–147. [DOI] [PubMed] [Google Scholar]
- (54).Fergusson JE; Love JL; Armor JN Ruthenium ammines In Inorganic Syntheses; John Wiley & Sons, Inc.: 1972, p 208–213. [Google Scholar]
- (55).Meyer TJ; Taube H Electron-transfer reactions of ruthenium ammines. Inorg. Chem 1968, 7, 2369–2379. [Google Scholar]
- (56).Humphrey W; Dalke A; Schulten K VMD - Visual Molecular Dynamics. J. Mol. Graphics 1996, 14, 33–38. [DOI] [PubMed] [Google Scholar]
- (57).Phillips JC; Braun R; Wang W; Gumbart J; Tajkhorshid E; Villa E; Chipot C; Skeel RD; Kalé L; Schulten K Scalable molecular dynamics with NAMD. J. Comput. Chem 2005, 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Autenrieth F; Tajkhorshid E; Baudry J; Luthey-Schulten Z Classical force field parameters for the heme prosthetic group of cytochrome c. J. Comput. Chem 2004, 25, 1613–1622. [DOI] [PubMed] [Google Scholar]
- (59).Pettersen EF; Goddard TD; Huang CC; Couch GS; Greenblatt DM; Meng EC; Ferrin TE UCSF Chimera--a visualization system for exploratory research and analysis. J. Comp. Chem 2004, 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- (60).Dunbrack RL Rotamer libraries in the 21st century. Curr. Opin. Struct. Biol 2002, 12, 431–440. [DOI] [PubMed] [Google Scholar]
- (61).Fraternali F; Cavallo L Parameter optimized surfaces (POPS): analysis of key interactions and conformational changes in the ribosome. Nucleic Acids Research 2002, 30, 2950–2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Yu J; Zhou Y; Tanaka I; Yao M Roll: a new algorithm for the detection of protein pockets and cavities with a rolling probe sphere. Bioinformatics 2010, 26, 46–52. [DOI] [PubMed] [Google Scholar]
- (63).Zhong F; Pletneva EV Ligation and reactivity of methionine-oxidized cytochrome c. Inorg. Chem 2018, 57, 5754–5766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Russell BS; Melenkivitz R; Bren KL NMR investigation of ferricytochrome c unfolding: Detection of an equilibrium unfolding intermediate and residual structure in the denatured state. Proc. Natl. Acad. Sci. USA 2000, 97, 8312–8317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Kataoka M; Hagihara Y; Mihara K; Goto Y Molten globule of cytochrome c studied by small angle X-ray scattering. J. Mol. Biol 1993, 229, 591–596. [DOI] [PubMed] [Google Scholar]
- (66).Pletneva EV; Gray HB; Winkler JR Nature of the cytochrome c molten globule. J. Am. Chem. Soc 2005, 127, 15370–15371. [DOI] [PubMed] [Google Scholar]
- (67).Hilgen-Willis S; Bowden EF; Pielak GJ Dramatic stabilization of ferricytochrome c upon reduction. J. Inorg. Biochem 1993, 51, 649–653. [DOI] [PubMed] [Google Scholar]
- (68).Marques HM Insights into porphyrin chemistry provided by the microperoxidases, the haempeptides derived from cytochrome c Dalton Trans. 2007, 4371–4385. [DOI] [PubMed] [Google Scholar]
- (69).Cohen DS; Pielak GJ Entropic stabilization of cytochrome c upon reduction. J. Am. Chem. Soc 1995, 117, 1675–1677. [Google Scholar]
- (70).Assfalg M; Bertini I; Dolfi A; Turano P; Mauk AG; Rosell FI; Gray HB Structural model for an alkaline form of ferricytochrome c. J. Am. Chem. Soc 2003, 125, 2913–2922. [DOI] [PubMed] [Google Scholar]
- (71).Battistuzzi G; Borsari M; Sola M; Francia F Redox thermodynamics of the native and alkaline forms of eukaryotic and bacterial class I cytochromes c. Biochemistry 1997, 36, 16247–16258. [DOI] [PubMed] [Google Scholar]
- (72).Barker PD; Mauk AG pH-Linked conformational regulation of a metalloprotein oxidation-reduction equilibrium: electrochemical analysis of the alkaline form of cytochrome c. J. Am. Chem. Soc 1992, 114, 3619–3624. [Google Scholar]
- (73).Laviron E; Roullier LJ The square scheme with the electrochemical reactions at equilibrium A study by diffusion or thin layer cyclic voltammetry and by scanning potential coulometry. Electroanalytical Chemistry and Interfacial Electrochemistry 1985, 186, 1–15. [Google Scholar]
- (74).Nicholson RS; Shain I Theory of stationary electrode polarography. Single scan and cyclic methods applied to reversible, irreversible, and kinetic systems. Anal. Chem 1964, 36, 706–723. [Google Scholar]
- (75).Haladjian J; Pilard R; Bianco P; Serre P-A Effect of pH on the electroactivity of horse heart cytochrome c. Bioelectrochem. Bioenerg 1982, 9, 91–101. [Google Scholar]
- (76).Rodrigues CG; Farchione F; Wedd AG Relationship of two electroactive forms of horse heart cytochrome c at gold and glassy carbon electrodes in water and methanol. J. Electroanal. Chem 1987, 218, 251–264. [Google Scholar]
- (77).McCreery RL Advanced carbon electrode materials for molecular electrochemistry. Chem. Rev 2008, 108, 2646–2687. [DOI] [PubMed] [Google Scholar]
- (78).Cutler RL; Davies AM; Creighton S; Warshel A; Moore GR; Smith M; Mauk AG Role of arginine-38 in regulation of the cytochrome c oxidation-reduction equilibrium. Biochemistry 1989, 28, 3188–3197. [DOI] [PubMed] [Google Scholar]
- (79).Battistuzzi G; Borsari M; Dallari D; Lancellotti I; Sola M Anion binding to mitochondrial cytochromes c studied through electrochemistry. Effects of the neutralization of surface charges on the redox potential. Eur. J. Biochem 1996, 241, 208–214. [DOI] [PubMed] [Google Scholar]
- (80).Godbole S; Bowler BE Effect of pH on formation of a nativelike intermediate on the unfolding pathway of a Lys 73 → His variant of yeast iso-1-cytochrome c. Biochemistry 1999, 38, 487–495. [DOI] [PubMed] [Google Scholar]
- (81).Baddam S; Bowler BE Mutation of asparagine 52 to glycine promotes the alkaline form of iso-1-cytochrome c and causes loss of cooperativity in acid unfolding. Biochemistry 2006, 45, 4611–4619. [DOI] [PubMed] [Google Scholar]
- (82).Baddam S; Bowler BE Thermodynamics and kinetics of formation of the alkaline state of a Lys79→Ala/Lys73→His variant of iso-1-cytochrome c. Biochemistry 2005, 44, 14956–14968. [DOI] [PubMed] [Google Scholar]
- (83).Indiani C; de Sanctis G; Neri F; Santos H; Smulevich G; Coletta M Effect of pH on axial ligand coordination of cytochrome c″ from Methylophilus methylotrophus and horse heart cytochrome c. Biochemistry 2000, 39, 8234–8242. [DOI] [PubMed] [Google Scholar]
- (84).Sinibaldi F; Piro MC; Coletta M; Santucci R Salt-induced formation of the A-state of ferricytochrome c - effect of the anion charge on protein structure. FEBS J. 2006, 273, 5347–5357. [DOI] [PubMed] [Google Scholar]
- (85).Milazzo L; Tognaccini L; Howes BD; Smulevich G Probing the non-native states of cytochrome c with resonance Raman spectroscopy: A tool for investigating the structure-function relationship. J. Raman Spect 2018, 49, 1041–1055. [Google Scholar]
- (86).Moura I; Liu MY; Costa C; Liu MC; Pai G; Xavier AV; LeGall J; Payne WJ; Moura JJ Spectroscopic characterization of a high-potential monohaem cytochrome from Wolinella succinogenes, a nitrate-respiring organism. Redox and spin equilibria studies. Eur. J. Biochem 1988. 177, 673–682. [DOI] [PubMed] [Google Scholar]
- (87).Akazaki H; Futami Y; Shibayama N; Shirasaki I; Nakade H; Chida H; Hakamata W; Park S-Y; Nishio T; Oku T Physicochemical properties of diheme cytochrome c4 of unknown function from Vibrio parahaemolyticus strain RIMD2210633. Biosci. Biotechnol. Biochem 2008, 72, 2791–2794. [DOI] [PubMed] [Google Scholar]
- (88).Conrad LS; Karlsson JJ; Ulstrup J Electron transfer and spectral alpha-band properties of the di-heme protein cytochrome c4 from Pseudomonas stutzeri. Eur. J. Biochem 1995, 231, 133–141. [DOI] [PubMed] [Google Scholar]
- (89).Tezcan FA; Winkler JR; Gray HB Effects of ligation and folding on reduction potentials of heme proteins. J. Am. Chem. Soc 1998, 120, 13383–13388. [Google Scholar]
- (90).Deacon OM; Karsisiotis AI; Moreno-Chicano T; Hough MA; Macdonald C; Blumenschein TMA; Wilson MT; Moore GR; Worrall JAR Heightened dynamics of the oxidized Y48H variant of human cytochrome c increases its peroxidatic activity. Biochemistry 2017, 56, 6111–6124. [DOI] [PubMed] [Google Scholar]
- (91).Karsisiotis AI; Deacon OM; Wilson MT; Macdonald C; Blumenschein TM; Moore GR; Worrall JA Increased dynamics in the 40–57 Omega-loop of the G41S variant of human cytochrome c promote its pro-apoptotic conformation. Sci. Rep 2016, 6, 30447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Diederix RE; Ubbink M; Canters GW Peroxidase activity as a tool for studying the folding of c-type cytochromes. Biochemistry 2002, 41, 13067–13077. [DOI] [PubMed] [Google Scholar]
- (93).Baddam S; Bowler BE Conformationally gated electron transfer in iso-1-cytochrome c: Engineering the rate of a conformational switch. J. Am. Chem. Soc 2005, 127, 9702–9703. [DOI] [PubMed] [Google Scholar]
- (94).Lim HS; Barclay DJ; Anson FC Formal potentials and cyclic voltammetry of some ruthenium-ammine complexes. Inorg. Chem 1972, 11, 1460–1466. [Google Scholar]
- (95).Zhong F; Pletneva EV Mechanistic studies of protion-coupled electron transfer in a calorimetry cell. J. Am. Chem. Soc 2019, 141, 9773–9777. [DOI] [PubMed] [Google Scholar]
- (96).Silkstone GG; Cooper CE; Svistunenko D; Wilson MT EPR and optical spectroscopic studies of Met80X mutants of yeast ferricytochrome c. Models for intermediates in the alkaline transition. J. Am. Chem. Soc 2005, 127, 92–99. [DOI] [PubMed] [Google Scholar]
- (97).Davis LA; Schejter A; Hess GP Alkaline isomerization of oxidized cytochrome c. Equilibrium and kinetic measurements. J. Biol. Chem 1974, 249, 2624–2632. [PubMed] [Google Scholar]
- (98).Gilles-Gonzalez MA; Gonzalez G Heme-based sensors: defining characteristics, recent developments, and regulatory hypotheses. J. Inorg. Biochem 2005, 99, 1–22 [DOI] [PubMed] [Google Scholar]
- (99).Shimizu T Binding of cysteine thiolate to the Fe(III) heme complex is critical for the function of heme sensor proteins. J. Inorg. Biochem 2012, 108, 171–177. [DOI] [PubMed] [Google Scholar]
- (100).Marvin KA; Reinking JL; Lee AJ; Pardee KM; Krause HM; Burstyn JN Nuclear receptors Homo sapiens Rev-erb and Drosophila melanogaster E75 are thiolate-ligated heme proteins, which undergo redox-mediated ligand switching and bind CO and NO. Biochemistry 2009, 48, 7056–7071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (101).Gupta N; Ragsdale SW Thiol-disulfide redox dependence of heme binding and heme ligand switching in nuclear hormone receptor Rev-erbβ. J. Biol. Chem 2011, 286, 4392–4403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Martinez RE; Bowler BE Proton-mediated dynamics of the alkaline conformational transition of yeast iso-1-cytochrome c. J. Am. Chem. Soc 2004, 126, 6751–6758. [DOI] [PubMed] [Google Scholar]
- (103).Hoang L; Maity H; Krishna MM; Lin Y; Englander SW Folding units govern the cytochrome c alkaline transition. J. Mol. Biol 2003, 331, 37–43. [DOI] [PubMed] [Google Scholar]
- (104).Weinkam P; Zimmermann J; Sagle LB; Matsuda S; Dawson PE; Wolynes PG; Romesberg FE Characterization of alkaline transitions in ferricytochrome c using carbon-deuterium infrared probes. Biochemistry 2008, 47, 13470–13480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (105).Hartshorn RT; Moore GR A denaturation-induced proton-uptake study of horse ferricytochrome c. Biochem. J 1989, 258, 599–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (106).Fernández-Escamilla AM; Cheung MS; Vega MC; Wilmanns M; Onuchic JN; Serrano L Solvation in protein folding analysis: combination of theoretical and experimental approaches. Proc. Natl. Acad. Sci. U. S. A 2004, 101, 2834–2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (107).König G; Bruckner S; Boresch S Absolute hydration free energies of blocked amino acids: implications for protein solvation and stability. Biophys. J 2013, 104, 453–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (108).Bortolotti CA; Amadei A; Aschi M; Borsari M; Corni S; Sola M; Daidone I The reversible opening of water channels in cytochrome c modulates the heme iron reduction potential. J. Am. Chem. Soc 2012, 134, 13670–13678. [DOI] [PubMed] [Google Scholar]
- (109).Lett CM; Berghuis AM; Frey HE; Lepock JR; Guillemette JG The role of a conserved water molecule in the redox-dependent thermal stability of iso-1-cytochrome c. J. Biol. Chem 1996, 271, 29088–29093. [DOI] [PubMed] [Google Scholar]
- (110).Lett CM; Guillemette JG Increasing the redox potential of isoform 1 of yeast cytochrome c through the modification of select haem interactions. Biochem. J 2002, 362, 281–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).Moore GR; Harris DE; Leitch FA; Pettigrew GW Characterisation of ionisations that influence the redox potential of mitochondrial cytochrome c and photosynthetic bacterial cytochromes c2. Biochim. Biophys. Acta 1984, 764, 331–342. [Google Scholar]
- (112).Berghuis AM; Guillemette JG; McLendon G; Sherman F; Smith M; Brayer GD The role of a conserved internal water molecule and its associated hydrogen bond network in cytochrome c. J. Mol. Biol 1994, 236, 786–799. [DOI] [PubMed] [Google Scholar]
- (113).Berghuis AM; Guillemette JG; Smith M; Brayer GD Mutation of tyrosine-67 to phenylalanine in cytochrome c significantly alters the local heme environment. J. Mol. Biol 1994, 235, 1326–1341. [DOI] [PubMed] [Google Scholar]
- (114).Kroll T; Hadt RG; Wilson SA; Lundberg M; Yan JJ; Weng T-C; Sokaras D; Alonso-Mori R; Casa D; Upton MH; Hedman B; Hodgson KO; Solomon EI Resonant inelastic X-ray scattering on ferrous and ferric bis-imidazole porphyrin and cytochrome c: Nature and role of the axial methionine-Fe bond. J. Am. Chem. Soc 2014, 136, 18087–18099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (115).Mara MW; Hadt RG; Reinhard ME; Kroll T; Lim H; Hartsock RW; Alonso-Mori R; Chollet M; Glownia JM; Nelson S; Sokaras D; Kunnus K; Hodgson KO; Hedman B; Bergmann U; Gaffney KJ; Solomon EI Metalloprotein entatic control of ligand-metal bonds quantified by ultrafast x-ray spectroscopy. Science 2017, 356, 1276–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (116).Pearce LL; Gartner AL; Smith M; Mauk AG Mutation-induced perturbation of the cytochrome c alkaline transition. Biochemistry 1989, 28, 3152–3156. [DOI] [PubMed] [Google Scholar]
- (117).Lei H; Nold SM; Motta LJ; Bowler BE Effect of V83G and I81A substitutions to human cytochrome c on acid unfolding and peroxidase activity below a neutral pH. Biochemistry 2019, 58, 2921–2933. [DOI] [PubMed] [Google Scholar]
- (118).Lambeth DO; Campbell KL; Zand R; Palmer G The appearance of transient species of cytochrome c upon rapid oxidation or reduction at alkaline pH. J. Biol. Chem 1973, 248, 8130–8136. [PubMed] [Google Scholar]
- (119).Raphael AL; Gray HB Semisynthesis of axial-ligand (position 80) mutants of cytochrome c. J. Am. Chem. Soc 1991, 113, 1038–1040. [Google Scholar]
- (120).Fetrow JS; Baxter SM Assignment of 15N chemical shifts and 15N relaxation measurements for oxidized and reduced iso-1-cytochrome c. Biochemistry 1999, 38, 4480–4492. [DOI] [PubMed] [Google Scholar]
- (121).Churg AK; Warshel A Control of the redox potential of cytochrome c and microscopic dielectric effects in proteins. Biochemistry 1986, 25, 1675–1681. [DOI] [PubMed] [Google Scholar]
- (122).Rogers NK; Moore GR On the energetics of conformational changes and pH dependent redox behaviour of electron transfer proteins. FEBS Lett. 1988, 228, 69–73. [DOI] [PubMed] [Google Scholar]
- (123).Bandi S; Bowler BE Probing the dynamics of a His73-heme alkaline transition in a destabilized variant of yeast iso-1-cytochrome c with conformationally gated electron transfer methods. Biochemistry 2011, 50, 10027–10040. [DOI] [PubMed] [Google Scholar]
- (124).McClelland LJ; Bowler BE Lower protein stability does not necessarily increase local dynamics. Biochemistry 2016, 55, 2681–2693. [DOI] [PubMed] [Google Scholar]
- (125).McClelland LJ; Mou TC; Jeakins-Cooley ME; Sprang SR; Bowler BE Structure of a mitochondrial cytochrome c conformer competent for peroxidase activity. Proc. Natl. Acad. of Sci. USA 2014, 111, 6648–6653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (126).Gu J; Yang S; Rajic AJ; Kurnikov IV; Prytkova TR; Pletneva EV Control of cytochrome c redox reactivity through off-pathway modifications in the protein hydrogen-bonding network. Chem. Commun 2014, 50, 5355–5357. [DOI] [PubMed] [Google Scholar]
- (127).Josephs TM; Liptak MD; Hughes G; Lo A; Smith RM; Wilbanks SM; Bren KL; Ledgerwood EC Conformational change and human cytochrome c function: mutation of residue 41 modulates caspase activation and destabilizes Met-80 coordination. J. Biol. Inorg. Chem 2013, 18, 289–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.