

Abstract
Objectives:
The clinical presentation of patients with nonclassic 21-hydroxylase deficiency (N21OHD) is similar with that for other disorders of androgen excess. The diagnosis of N21OHD typically requires cosyntropin stimulation. Additionally, the management of such patients is limited by the lack of reliable biomarkers of androgen excess. Herein, we aimed to: 1) compare the relative contribution of traditional and 11-oxyandrogens in N21OHD patients; and 2) identify steroids that accurately diagnose N21OHD with a single baseline blood draw.
Design:
We prospectively enrolled patients who underwent a cosyntropin stimulation test for suspected N21OHD in two tertiary referral centers between January, 2016 and August, 2019.
Methods:
Baseline sera were used to quantify 15 steroids by liquid chromatography-tandem mass spectrometry. Logistic regression modeling was implemented to select steroids that best discriminate N21OHD from controls.
Results:
Of 86 participants (72 females), median age 26, 32 patients (25 females) had N21OHD. Age, sex distribution, and BMI were similar between patients with N21OHD and controls. Both testosterone and androstenedione were similar in patients with N21OHD and controls, while four 11-oxyandrogens were significantly higher in patients with N21OHD (ratios between medians: 1.7- to 2.2, p < 0.01 for all). 17α-hydroxyprogesterone (6.5-fold), 16α-hydroxyprogesterone (4.1-fold), 21-deoxycortisol (undetectable in 80% of the controls) were higher, while corticosterone was 3.6-fold lower in patients with N21OHD than in controls (p < 0.001). Together, baseline 17α-hydroxyprogesterone, 21-deoxycortisol, and corticosterone showed perfect discrimination between N21OHD and controls.
Conclusions:
Adrenal 11-oxyandrogens are disproportionately elevated compared to conventional androgens in N21OHD. Steroid panels can accurately diagnose N21OHD in unstimulated blood tests.
Key terms: 21-hydroxylase deficiency, congenital adrenal hyperplasia, steroids, androgens
Introduction
Congenital adrenal hyperplasia comprises a series of autosomal recessive defects in enzymes required for cortisol biosynthesis. The most common form of congenital adrenal hyperplasia is 21-hydroxylase deficiency (21OHD) (1). In its most severe forms, also called “classic”, 21OHD typically presents at birth, with virilized external genitalia in girls and life-threatening adrenal insufficiency. Patients with milder enzymatic defects, conventionally termed “nonclassic” or “late-onset” 21OHD, have normal glucocorticoid and mineralocorticoid production, owing to a compensatory increase of corticotropin (ACTH) secretion. The increased ACTH stimulation, however, in conjunction with the 21-hydroxylase defect, leads to excess adrenal androgen production. Patients with nonclassic 21OHD present with clinical manifestations such as premature pubarche, hirsutism, acne, irregular menses, and infertility or miscarriages, features shared with other disorders of androgen excess. In particular, the clinical phenotype of women with nonclassic 21OHD is similar to that for polycystic ovarian syndrome (PCOS) (2–5).
The diagnosis of 21OHD relies on serum 17α-hydroxyprogesterone (17OHP4) measurements (1). Patients with classic 21OHD have marked 17OHP4 elevations in random samples and are typically diagnosed by newborn screening in most developed countries (6). In contrast, the diagnosis of nonclassic 21OHD usually requires dynamic testing with synthetic ACTH1−24 (cosyntropin) stimulation, when 17OHP4 is elevated at screening (> 200 ng/dL, 6 nmol/L) but not above the diagnostic threshold of 1000 ng/dL (30 nmol/L) (7–9). An additional limitation of 17OHP4 screening testing is that false-positive elevations are common, particularly so in premature and sick newborns, in reproductive-age women with irregular menses, and when measured by immunoassays (10–15).
Beyond 17OHP4, 21OHD promotes the accretion of other 21-carbon (C21) steroids, such as 16α-hydroxyprogesterone (16OHP4) (16), which is normally a minor product of 17α-hydroxylase/17,20-lyase (CYP17A1), and 21-deoxycortisol (21dF) (16–19), which results from the 11β-hydroxylation of the abundantly accumulated 17OHP4. Similarly, 11β-hydroxylase (CYP11B1) can also utilize as substrates androstenedione (A4) and testosterone (T), leading to ample amounts of 11β-hydroxyandrostenedione (11OHA4) and 11β-hydroxytestosterone (11OHT), respectively (20, 21). While T and A4 are also frequently elevated in patients with classic 21OHD, these steroids are often normal in patients with nonclassic 21OHD, and do not always account for the clinical manifestations of hyperandrogenism observed in these patients. We and others have shown that the potent androgen 11-ketotestosterone (11KT) and the other 11-oxygenated C19 steroids (11-oxyandrogens) are elevated in patients with classic 21OHD (22, 23). The 11-oxyandrogens correlate well with surrogates of poor disease control in classic 21OHD (24), but data in patients with nonclassic 21OHD are lacking.
Using liquid chromatography-tandem mass spectrometry (LC-MS/MS) assays, multiple steroids can be quantified from a small-volume aliquot of serum. With the current study, we aimed to assess the steroid fingerprints in patients with nonclassic 21OHD of both sexes, with a dual goal: 1) to compare the relative contribution of traditional and adrenal-specific 11-oxyandrogens in these patients; and 2) to identify steroid panels that can diagnose nonclassic 21OHD reliably with a single baseline blood draw.
Patients and methods
Study Participants
Patients undergoing clinical evaluation for hyperandrogenism with cosyntropin stimulation testing in two tertiary referral centers (University of Michigan and the National Institutes of Health Clinical Center) for suspected nonclassic 21OHD between January, 2016 and August, 2019 were included in this study. All tests were conducted in the morning, in outpatient setting. The diagnosis of nonclassic 21OHD was determined based on clinical phenotype and stimulated serum 17OHP4 concentrations of > 1,000 ng/dL (30 nmol/L) and < 10,000 ng/dL (300 nmol/L). In all but three 21OHD cases, genetic studies were also performed and were consistent with the diagnosis. The diagnosis of PCOS was based on the Androgen Excess and PCOS Society criteria (25). All studies were conducted with Institutional Board Review (IRB) approval from each of the two participating institutions. Adult patients and parents of participating minors seen at NIH provided informed consent, and all minors at least eight years old gave written assent. For patients seen at the University of Michigan, a waiver of consent was granted by the IRB for using leftover serum collected as part of standard clinical care.
Steroid quantitation by mass spectrometry
Quantitation of 15 unconjugated Δ4 steroids, including cortisol precursors, T4, A4 and four 11-oxyandrogens, from sera obtained at baseline was performed by LC-MS/MS, as previously described (26).
Statistical analyses
Mann-Whitney U test was used for two-group comparisons of continuous variables, and Chi-square test was used to compare sex distributions between groups. Correlations between steroids were assessed using Spearman’s correlation coefficients. Receiver-operating characteristics (ROC) curves were used to characterize the prediction performance. Logistic regression with a lasso penalty, combined with clinical knowledge (relevant due to high correlation between biomarkers) was implemented for the selection of a small set of steroids that best discriminate patients with nonclassic 21OHD from controls.
Results
In total, 86 patients (72 females), with ages between 6–70 years, median age 26, participated in this study. Of these, 32 patients (25 females) met criteria for nonclassic 21OHD (Table 1). In the remaining 54 patients (47 females), 21OHD was excluded based on cosyntropin-stimulated 17OHP4 values < 30 nmol/L. Of the latter, 27 (50%) patients met criteria for PCOS. Overall, age, sex distribution, and BMI were similar between patients with vs. those without 21OHD.
Table 1.
Demographic and clinical characteristics of study participants
| Controls | N21OHD | p | ||
|---|---|---|---|---|
| n | 54 | 32 | ||
| Age (years) | 27 (6–70) | 25 (6–66) | 0.223 | |
| Sex (F/M) | 47/7 | 25/7 | 0.367 | |
| BMI (kg/m2) | 27.9 [23.3–32.9] | 25.1 [20.6–29.7] | 0.085 | |
| Diagnosis (n) | ||||
| PCOS | 27 | |||
| Hirsutism | 9 | |||
| Menstrual dysfunction | 3 | |||
| Precocious pubarche | 5 | |||
| Suspicion for AI | 4 | |||
| Short stature | 1 | |||
| Early onset alopecia | 1 | |||
| Suspicion for CAH* | 4 | |||
Data are expressed as medians and range for age, and interquartile range for BMI.
Patients referred from outside for cosyntropin stimulation with the suspicion of congenital adrenal hyperplasia (CAH), without other clinical details available.
N21OHD, nonclassic 21-hydroxylase deficiency; PCOS, polycystic ovary syndrome; AI, adrenal insufficiency; F, females; M, males.
Comparison of androgens between patients with or without nonclassic 21OHD
Of the C19 steroids measured, both T and A4 were similar in patients with nonclassic 21OHD and those without 21OHD (p = 0.2 and 0.8, respectively). In contrast, all four 11-oxyandrogens were significantly higher in patients with nonclassic 21OHD, from 1.7-fold for 11KA4 to 2.2-fold for 11OHT (p < 0.01 for all, Table 2 and Figure 1). Furthermore, 11KT/T (1.8 [1.2–3.0] vs. 0.6 [0.2–0.9]) and 11OHA4/A4 (2.6 [2.1–4.6] vs. 1.2 [0.7–2.1]) were significantly higher in patients with nonclassic 21OHD than in unaffected individuals (p < 0.001 for both). In contrast, the A4/T ratio was similar between the two groups (p = 0.3).
Table 2.
Comparison of steroid concentrations between patients with nonclassic 21-hydroxylase deficiency (N21OHD) and controls
| Steroid (nmol/L) | N21OHD (N=32) | Controls (N=54) | Ratio of medians N21OHD/Controls | p |
|---|---|---|---|---|
| 17OHP4 | 1.1 [0.5–2.7] | 1.6 [0.8–4.3] | 6.5 | <0.0001 |
| 16OHP4 | 2.4 [1.4–4.9] | 0.6 [0.4–0.9] | 4.1 | <0.0001 |
| 21dF | 1.8 [1.3–5.3] | 0 [0–0.3] | N/A | <0.0001 |
| Progesterone | 0.8 [0.5–1.4] | 0.4 [0.3–0.8] | 1.8 | 0.0018 |
| Cortisol | 202.5 [125.9–442.8] | 249.1 [208.9–373.7] | 0.8 | 0.197 |
| Cortisone | 53.7 [30.5–72.3] | 51.5 [40.3–60.8] | 1.0 | 0.855 |
| Corticosterone | 1.6 [0.8–3.5] | 5.9 [3.5–13.6] | 0.3 | <0.0001 |
| 11dF | 0.8 [0.5–1.4] | 0.9 [0.4–1.6] | 0.8 | 0.626 |
| DOC | 0.3 [0.3–0.4] | 0.5 [0.4–0.9] | 0.6 | 0.113 |
| A4 | 4.3 [2.0–8.1] | 3.9 [2.2–8.5] | 1.1 | 0.798 |
| T | 1.1 [0.5–2.7] | 1.6 [0.8–4.3] | 0.7 | 0.195 |
| 11OHA4 | 9.6 [5.1–25.9] | 4.5 [2.9–7.1] | 2.1 | <0.0001 |
| 11KA4 | 1.1 [0.7–1.7] | 0.6 [0.4–0.8] | 1.7 | 0.0001 |
| 11OHT | 1.0 [0.3–1.7] | 0.4 [0.3–0.6] | 2.2 | 0.0019 |
| 11KT | 1.8 [1.2–3.3] | 0.9 [0.6–1.3] | 2.0 | <0.0001 |
| 11KT/T | 1.8 [1.2–3.0] | 0.6 [0.2–0.9] | 3.3 | <0.0001 |
| 11OHA4/A4 | 2.6 [2.1–4.6] | 1.2 [0.7–2.1] | 2.0 | <0.0001 |
| A4/T | 3.8 [2.8–4.5] | 3.5 [2.4–4.2] | 1.3 | 0.274 |
Data are expressed as median [interquartile range]. 17OHP4, 17α-hydroxyprogesterone; 16OHP4, 16- hydroxyprogesterone; 21dF, 21-deoxycortisol; 11dF, 11-deoxycortisol; DOC, 11-deoxycorticosterone; A4, androstenedione; T, testosterone; 11OHA4, 11β-hydroxyandrostenedione; 11KA4, 11-ketoandrostenedione; 11OHT 11β-hydroxytestosterone; 11KT, 11-ketotestosterone.
Figure 1. Comparison of traditional and 11-oxyandrogens between patients with nonclassic 21-hydroxylase deficiency (N21OHD) and controls.
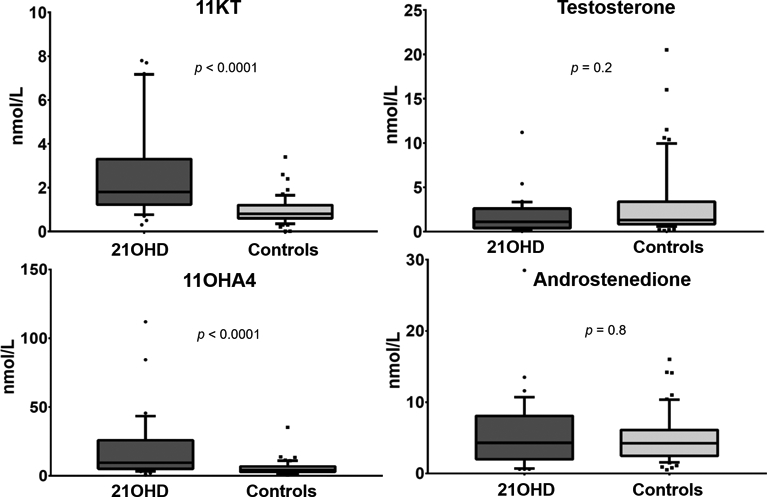
11KT, 11-ketotestosterone; 11OHA4, 11β-hydroxyandrostenedione.
In a subgroup analysis by sex, age and BMI were similar between female patients with and without nonclassic 21OHD, while male patients with nonclassic 21OHD were younger than their unaffected counterparts (Table 3). All four 11-oxyandrogens were significantly higher in female patients with nonclassic 21OHD than in those without 21OHD (from 2.1-fold for 11KA4 to 2.9-fold for 11KT, p < 0.0001 for all, Table 3). Similar results were observed when comparing adult women with nonclassic 21OHD with PCOS women (Table 4). In addition, the correlations between 11KT and 11OHA4 with their precursors, T and A4, respectively were tighter in female patients with nonclassic 21OHD than those unaffected, suggesting a predominant adrenal origin for all these androgens in female patients with nonclassic 21OHD and a predominant ovarian source of T and A4 in other disorders of androgen excess (Figure 2). In males, hormonal differences could not be rigorously compared, due to the small number of patients and differences in age distribution between the groups (Table 3).
Table 3:
Comparison between patients with nonclassic 21-hydroxylase deficiency (N21OHD) and controls of the same sex
| Females | Males | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| N21OHD (n=25) | Controls (n=47) | Ratio* | P | N21OHD (n=7) | Controls (n=7) | Ratio* | P | ||
| Age (years) | 27 [18–34] | 28 [21–36] | 0.696 | 9 [8–13] | 18 [12–52] | 0.036 | |||
| BMI (kg/m2) | 26 [22–32] | 28 [23–33] | 0.224 | 21 [19–26] | 26 [15–31] | 0.62 | |||
| 17OHP4 (nmol/L) | 16.8 [8.1–55.7] | 2.3[1–4] | 7.4 | <0.0001 | 24.9 [13.6–55.7] | 2.8 [2.1–3.8] | 9.0 | 0.0006 | |
| 16OHP4 (nmol/L) | 2.3 [1–7.7] | 0.6 [0.3–0.9] | 4.1 | <0.0001 | 2.8 [1.5–7.7] | 0.6 [0.5–0.9] | 5.0 | 0.002 | |
| 21dF (nmol/L) | 1.8 [1.2–5.1] | 0.0 [0.0–0.0] | N/A | <0.0001 | 1.5 [1.3–5.1] | 0.0 [0.0–0.5] | N/A | 0.0006 | |
| Progesterone (nmol/L) | 0.8 [0.4–1.5] | 0.4 [0.3–0.8] | 1.8 | 0.014 | 1.0 [0.5–1.5] | 0.4 [0.3–0.8] | 2.2 | 0.052 | |
| Cortisol (nmol/L) | 233.4 [128.1–555] | 256.0 [206.6–423.6] | 0.9 | 0.429 | 189.7 [81.4–222.1] | 219.0 [129.4–245.4] | 0.9 | 0.318 | |
| Cortisone (nmol/L) | 57 [31.2–64.2] | 52.5 [44.6–63.7] | 1.1 | 0.823 | 50.4 [23.4–64.2] | 39.4 [20.6–52.9] | 1.3 | 0.710 | |
| Corticosterone (nmol/L) | 2.0 [0.8–1.7] | 5.4 [3.3–15.3] | 0.4 | <0.0001 | 1.4 [0.5–1.7] | 5.0 [3.3–14.7] | 0.3 | 0.0006 | |
| 11dF (nmol/L) | 0.8 [0.5–1.4] | 0.6 [0.4–1.4] | 1.2 | 0.482 | 1.1 [0.6–1.4] | 1.1 [0.6–1.6] | 1.0 | 0.477 | |
| DOC (nmol/L) | 0.3 [0.3–0.4] | 0.4 [0.2–0.7] | 0.8 | 0.322 | 0.3 [0.3–0.4] | 0.6 [0.5–0.8] | 0.5 | 0.001 | |
| A4 (nmol/L) | 4.7 [2.6–3.4] | 4.8 [3.1–7.1] | 1.0 | 0.890 | 1.9 [0.6–3.4] | 2.1 [1.0–2.4] | 0.9 | 0.901 | |
| T (nmol/L) | 1.2 [0.8–11.2] | 1.2 [0.9–2.8] | 1.0 | 0.696 | 0.4 [0.2–11.2] | 9.4 [0.6–16] | 0.04 | 0.137 | |
| 11OHA4 (nmol/L) | 10.1 [5.8–28.4] | 4.4 [2.9–7.3] | 2.3 | <0.0001 | 5.4 [3.1–28.4] | 4.2 [1.6–5.3] | 1.3 | 0.318 | |
| 11KA4 (nmol/L) | 1.3 [0.7–1.4] | 0.6 [0.5–0.8] | 2.1 | 0.0003 | 0.9 [0.4–1.4] | 0.6 [0.3–0.8] | 1.4 | 0.105 | |
| 11OHT (nmol/L) | 1.1 [0.5–0.5] | 0.4 [0.3–0.6] | 2.6 | <0.0001 | 0.3 [0.2–0.5] | 0.3 [0.1–0.4] | 1.1 | 0.685 | |
| 11KT (nmol/L) | 2.4 [1.4–2.3] | 0.8 [0.6–1.3] | 2.9 | <0.0001 | 1.4 [0.9–2.3] | 0.4 [0.3–1.0] | 3.2 | 0.024 | |
Data are expressed as median [interquartile range]. 17OHP4, 17α-hydroxyprogesterone; 16OHP4, 16α-hydroxyprogesterone; 21dF, 21-deoxycortisol; 11dF, 11-deoxycortisol; DOC, 11-deoxycorticosterone; A4, androstenedione; T, testosterone; 11OHA4, 11β-hydroxyandrostenedione; 11KA4, 11-ketoandrostenedione; 11OHT 11β-hydroxytestosterone; 11KT, 11-ketotestosterone; F, females; M, males.
Ratio of medians N21OHD/Controls
Table 4.
Comparison between women with nonclassic 21-hydroxylase deficiency (N21OHD) and women with PCOS
| F N21OHD* (N=22) | PCOS (N=27) | Ratio of medians N21OHD/Control | p | |
|---|---|---|---|---|
| Age (years) | 30 [23–36] | 28 [22–35] | 0.73 | |
| BMI (kg/m2) | 27 [23–33] | 31 [27–36] | 0.12 | |
| A4 (nmol/L) | 5.4 [3.8–8.3] | 5.9 [4–9.9] | 0.9 | 0.37 |
| T (nmol/L) | 1.4 [0.9–2.7] | 2.1 [1–4.2] | 0.7 | 0.24 |
| 11OHA4 (nmol/L) | 13.2 [7.6–33.4] | 4.4 [3.1–8.6] | 3.0 | <0.0001 |
| 11KA4 (nmol/L) | 1.3 [0.7–1.8] | 0.7 [0.6–0.9] | 1.9 | 0.0054 |
| 11OHT (nmol/L) | 1.3 [0.8–2] | 0.5 [0.4–0.7] | 2.8 | <0.0001 |
| 11KT (nmol/L) | 2.7 [1.2–3.8] | 1.0 [0.6–1.3] | 2.6 | <0.0001 |
3 pre-pubertal girls were excluded from this group, to preserve comparable ages between the two groups.
Data are expressed as median [interquartile range]. A4, androstenedione; T, testosterone; 11OHA4, 11β-hydroxyandrostenedione; 11KA4, 11-ketoandrostenedione; 11OHT 11β-hydroxytestosterone; 11KT, 11-ketotestosterone; F, females.
Figure 2. Correlations between key androgens in female patients with nonclassic 21-hydroxylase deficiency (N21OHD) and controls.
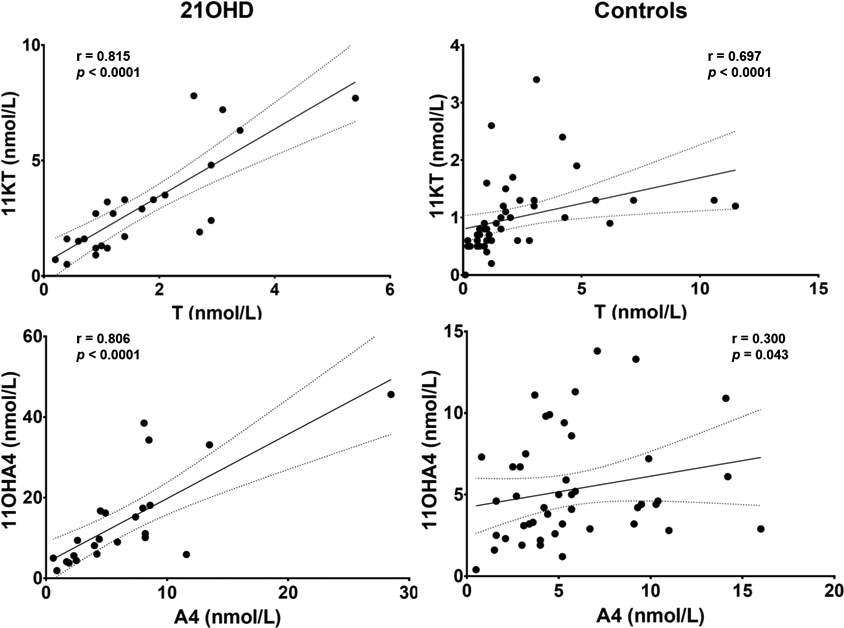
T, testosterone; 11KT, 11-ketotestosterone; A4, androstenedione; 11OHA4, 11β-hydroxyandrostenedione.
To illustrate the correlation trends for the majority of the patients, three outliers were excluded for the 11OH4 and A4 panels (two from the N21OHD group and one from the control group), all with disproportionately higher 11OHA4 than A4.
Comparison of C21 steroids between patients with or without nonclassic 21OHD
The highest relative difference between patients with nonclassic 21OHD vs. controls was observed for: 17OHP4 (6.5-fold), 16OHP4 (4.1-fold), and 21-deoxycortisol (21dF, undetectable in 80% of the controls, p < 0.001 for all) (Table 2). Conversely, corticosterone was significantly lower in patients with nonclassic 21OHD (p < 0.001), while cortisol, 11-deoxycortisol and 11-deoxycorticosterone showed no statistical difference between the two groups (Table 2).
Of 32 patients with nonclassic 21OHD, 23 (72%) had a 17OHP4 serum concentration < 30 nmol/L at baseline, and 19 of these were female. Logistic regression modeling showed that combined, 17OHP4 (β1 = 0.05), 21dF (β2 = 1.45), and corticosterone (β3 = −0.43) had an AUC of 1 (Table 5). Of all steroids measured, 21dF showed the highest area under the curve (AUC) when used alone (0.997, p < 0.001), and 21dF values > 0.64 nmol/L had a sensitivity of 96.30 (95% confidence interval, CI: 87.25% - 99.55%) and a specificity of 96.88 (95% CI: 83.78% - 99.92%). 17OHP4 also demonstrated excellent discriminatory power between the two groups when used in isolation, with an AUC of 0.986 (p < 0.001). A 17OHP4 of 5.68 nmol/L showed a sensitivity of 96.30 (95% CI: 87.25% - 99.55%) and a specificity of 93.75 (95% CI: 79.19% - 99.23%).
Table 5.
Logistic regression model utilizing three steroids to discriminate between patients with nonclassic 21-hydroxylase deficiency and unaffected individuals.
| Steroid | Coefficient | OR | 95% CI | p |
|---|---|---|---|---|
| 17OHP4 | 0.05 | 1.05 | 1.05–1.05 | <0.001 |
| 21dF | 1.46 | 4.28 | 4.13–4.45 | <0.001 |
| Corticosterone | −0.43 | 0.65 | 0.65–0.66 | <0.001 |
17OHP4, 17α-hydroxyprogesterone; 21dF, 21-deoxycortisol; OR, odds ratio; CI, confidence interval.
Discussion
Since the 1960s, steroids have been measured by immunoassays, which are limited by cross-reactivity and can yield unreliable results, particularly near the limits of sensitivity and in the presence of high concentrations of similar steroids (27, 28). Many laboratories have transitioned to steroid quantitation by mass spectrometry assays, which are highly sensitive and specific (29–31). In addition, LC-MS/MS also affords quantitation of multiple steroids in a single assay, using small-volume biospecimens. Multiple-steroid assays could assist clinical decision-making and circumvent the need for additional blood draws or dynamic testing. In this first study of 11-oxyandrogens in patients with nonclassic 21OHD, we found that 11-oxyandrogens are disproportionately higher than T and A4 in patients with nonclassic 21OHD as compared to patients with other disorders of androgen excess. Secondly, using a 15-steroid LC-MS/MS assay, we found that the combination of 17OHP4, 21dF and corticosterone measured in unstimulated serum samples showed the highest accuracy in diagnosing nonclassic 21OHD among children and adults undergoing differential diagnostic testing.
Compared to conventional newborn screening using immunoassay-based 17OHP4 alone, LC-MS/MS testing for simultaneous elevations of 21dF and 17OHP4 plus low cortisol was shown to reduce false-positive results (30). Similarly, the ratio [17OHP4 + A4]/cortisol was found to be superior to 17OHP4 for newborn screening for classic 21OHD. In contrast to the marked 17OHP4 elevations seen in patients with classic 21OHD, most hormonal abnormalities, including 17OHP4, are subtle in random samples from patients with nonclassic 21OHD (32). In menstruating girls and women, baseline blood samples should ideally be obtained in the early follicular phase (32), due to the dual adrenal and gonadal origin of 17OHP4 and its rise during the luteal phase. Accurate timing, however, often becomes impractical in patients with irregular menses and/or oligomenorrhea, in whom biochemical testing to distinguish PCOS from nonclassic 21OHD is commonly pursued. Furthermore, serum cortisol and A4 concentrations are typically normal in patients with nonclassic 21OHD. Using LC-MS/MS, we found that baseline 17OHP4 performed well in distinguishing patients with nonclassic 21OHD from unaffected individuals. In addition to 17OHP4, we found that 16OHP4 and 21dF are also significantly higher in patients with nonclassic 21OHD than in unaffected individuals, albeit lower than previously shown in patients with classic 21OHD (16). Conversely, corticosterone was on average almost 4-fold lower in patients with nonclassic 21OHD than in controls, while cortisol, 11-deoxycorticosterone, 11-deoxycortisol, and other intermediates were similar in both groups. Notably, an unstimulated 21dF value of 0.64 nmol/L demonstrated nearly perfect sensitivity and specificity for nonclassic 21OHD. In 21OHD, 17OHP4 is converted to 21dF by fully functional enzyme 11β-hydroxylase (CYP11B1). In unaffected individuals, the unhindered glucocorticoid pathway takes 17OHP4 to 11-deoxycortisol, and subsequently to cortisol. Thus, 21dF does not track with physiologic 17OHP4 elevations, and it has been proposed as a better alternative to 17OHP4 for the diagnosis of 21OHD (33). To date, however, data regarding the utility of 21dF as a single baseline test for the diagnosis of nonclassic 21OHD have been lacking. Together, 17OHP4, 21dF, and corticosterone measured at baseline showed the highest accuracy for distinguishing the patients with nonclassic 21OHD from controls. The added value of such panels could be particularly relevant in the few cases with ambiguous classification based on 17OHP4 or 21dF alone.
A second clinical challenge with nonclassic 21OHD is the monitoring and management of androgen excess. Treatment is typically reserved for symptomatic patients with nonclassic 21OHD (32). Symptoms of hyperandrogenism in women with nonclassic 21OHD are indistinguishable from those seen in PCOS, and include hirsutism, acne, irregular menses, and infertility or miscarriages (2–4). To further complicate the clinical assessment, polycystic ovarian morphology is common in women with nonclassic 21OHD (5). Conventional steroids have been unreliable in distinguishing the ovarian vs. adrenal components of androgen excess. Although T and A4 elevations have been reported in children with nonclassic 21OHD (34), these androgens have been more inconsistent in adults with nonclassic 21OHD. In our study, A4 and T were similar in patients with nonclassic 21OHD and those without 21OHD, including when the data analysis was restricted to women. Importantly, however, 11-oxyandrogens were roughly twice as high in patients with nonclassic 21OHD than in their counterparts. We have previously shown that 11-oxyandrogens were 3–4 fold higher in patients with classic 21OHD during treatment with standard hormonal therapy than in sex- and age-matched controls (22). 11KT is recognized as a potent androgen of adrenal origin in human beings (35–37), which mediates clinical manifestations of normal and premature adrenarche (35) and correlates well with clinical surrogates of poor disease control in patients with classic 21OHD (24). Interestingly, 11-oxyandrogens were also found to be higher in women with PCOS than in controls of similar ages (38). In our study, half of the female patients in which 21OHD was excluded were diagnosed with PCOS, and most others had isolated hirsutism or menstrual dysfunction. Our findings suggest that overall, the adrenal 11-oxyandrogens are disproportionately higher in patients with nonclassic 21OHD as compared to other disorders of androgen excess with shared clinical features. Moreover, 11KT displayed a tight positive correlation with T in female patients with nonclassic 21OHD, suggesting tandem elevations of adrenal origin in these patients. In contrast, the weaker correlation between 11KT and T in female patients without 21OHD points toward an ovarian component of T excess.
In summary, we have found that accurate steroid quantitation by mass spectrometry facilitates the diagnosis of nonlcassic 21OHD with a single baseline blood test. While 17OHP4 and 21dF display excellent discriminatory power even individually when measured by mass spectrometry, the added value of multi-steroid panels could circumvent the need for cosyntropin-stimulation testing in equivocal cases. Extended to larger populations, similar steroid panels could potentially differentiate between classic, nonclassic, carriers of 21OHD and healthy individuals in single baseline samples, as was previously proposed with cosyntropin stimulation (17, 18). Second, we now show that 11-oxyandrogens are relatively higher in patients with nonclassic 21OHD as compared to individuals with other disorders of androgen excess. Limitations of our study include its moderate sample size with few males and incomplete clinical phenotyping in some patients. Additionally, patients without 21OHD comprised a heterogeneous spectrum of ages and pathologies. Further studies focusing directly on the comparison of biomarkers between women with nonclassic 21OHD and PCOS, along with careful correlation with stigmata of hyperandrogenism in these women, will further elucidate the mechanisms responsible for 11-oxyandrogen biosynthesis, their clinical significance, and their utility in clinical practice.
Acknowledgements:
We thank Jianwei Ren and Patrick O’Day for their assistance with LC-MS/MS assays, the University of Michigan adrenal research coordinators for their assistance with regulatory processes, and all study participants.
Funding: This work was supported by grants 1K08DK109116 (to AFT) and R01GM086596 (to RJA) from the National Institutes of Health; by the Michigan Institute for Clinical and Health Research (MICHR) Translational Science Award/U046500 to AFT; by the Rare Disease Foundation and the BC Children’s Hospital Foundation Microgrant to AFT; and by the Intramural Research Program of the National Institutes of Health.
Disclosure Statement: DPM received unrelated research funds from Diurnal Limited and Millendo Therapeutics through the National Institutes of Health Cooperative Research and Development Agreement. RJA received unrelated research funds from Millendo Therapeutics, Neurocrine Biosciences, and Spruce Biosciences.
References
- 1.Speiser PW, White PC. Congenital adrenal hyperplasia. The New England journal of medicine. 2003;349(8):776–88. [DOI] [PubMed] [Google Scholar]
- 2.Casteras A, De Silva P, Rumsby G, Conway GS. Reassessing fecundity in women with classical congenital adrenal hyperplasia (CAH): normal pregnancy rate but reduced fertility rate. Clinical endocrinology. 2009;70(6):833–7. [DOI] [PubMed] [Google Scholar]
- 3.Moran C, Azziz R, Carmina E, Dewailly D, Fruzzetti F, Ibanez L, Knochenhauer ES, Marcondes JA, Mendonca BB, Pignatelli D, et al. 21-Hydroxylase-deficient nonclassic adrenal hyperplasia is a progressive disorder: a multicenter study. Am J Obstet Gynecol. 2000;183(6):1468–74. [DOI] [PubMed] [Google Scholar]
- 4.Bidet M, Bellanne-Chantelot C, Galand-Portier MB, Tardy V, Billaud L, Laborde K, Coussieu C, Morel Y, Vaury C, Golmard JL, et al. Clinical and molecular characterization of a cohort of 161 unrelated women with nonclassical congenital adrenal hyperplasia due to 21-hydroxylase deficiency and 330 family members. The Journal of clinical endocrinology and metabolism. 2009;94(5):1570–8. [DOI] [PubMed] [Google Scholar]
- 5.Carmina E, Dewailly D, Escobar-Morreale HF, Kelestimur F, Moran C, Oberfield S, Witchel SF, Azziz R. Non-classic congenital adrenal hyperplasia due to 21-hydroxylase deficiency revisited: an update with a special focus on adolescent and adult women. Hum Reprod Update. 2017;23(5):580–99. [DOI] [PubMed] [Google Scholar]
- 6.Therrell BL, Jr., Berenbaum SA, Manter-Kapanke V, Simmank J, Korman K, Prentice L, Gonzalez J, Gunn S. Results of screening 1.9 million Texas newborns for 21-hydroxylase-deficient congenital adrenal hyperplasia. Pediatrics. 1998;101(4 Pt 1):583–90. [DOI] [PubMed] [Google Scholar]
- 7.New MI, Lorenzen F, Lerner AJ, Kohn B, Oberfield SE, Pollack MS, Dupont B, Stoner E, Levy DJ, Pang S, et al. Genotyping steroid 21-hydroxylase deficiency: hormonal reference data. The Journal of clinical endocrinology and metabolism. 1983;57(2):320–6. [DOI] [PubMed] [Google Scholar]
- 8.Wilson RC, Mercado AB, Cheng KC, New MI. Steroid 21-hydroxylase deficiency: genotype may not predict phenotype. The Journal of clinical endocrinology and metabolism. 1995;80(8):2322–9. [DOI] [PubMed] [Google Scholar]
- 9.Speiser PW, Azziz R, Baskin LS, Ghizzoni L, Hensle TW, Merke DP, Meyer-Bahlburg HF, Miller WL, Montori VM, Oberfield SE, et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an Endocrine Society clinical practice guideline. The Journal of clinical endocrinology and metabolism. 2010;95(9):4133–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pauwels G, Allegaert K, Regal L, Meulemans A. Risk factors for elevated levels of 17-hydroxyprogesterone during neonatal intensive care unit admission. Acta Clin Belg. 2012;67(2):88–93. [DOI] [PubMed] [Google Scholar]
- 11.Ryckman KK, Cook DE, Berberich SL, Shchelochkov OA, Berends SK, Busch T, Dagle JM, Murray JC. Replication of clinical associations with 17-hydroxyprogesterone in preterm newborns. Journal of pediatric endocrinology & metabolism : JPEM. 2012;25(3–4):301–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nordenstrom A, Wedell A, Hagenfeldt L, Marcus C, Larsson A. Neonatal screening for congenital adrenal hyperplasia: 17-hydroxyprogesterone levels and CYP21 genotypes in preterm infants. Pediatrics. 2001;108(4):E68. [DOI] [PubMed] [Google Scholar]
- 13.Olgemoller B, Roscher AA, Liebl B, Fingerhut R. Screening for congenital adrenal hyperplasia: adjustment of 17-hydroxyprogesterone cut-off values to both age and birth weight markedly improves the predictive value. The Journal of clinical endocrinology and metabolism. 2003;88(12):5790–4. [DOI] [PubMed] [Google Scholar]
- 14.Azziz R, Hincapie LA, Knochenhauer ES, Dewailly D, Fox L, Boots LR. Screening for 21-hydroxylase-deficient nonclassic adrenal hyperplasia among hyperandrogenic women: a prospective study. Fertility and sterility. 1999;72(5):915–25. [DOI] [PubMed] [Google Scholar]
- 15.Cavarzere P, Samara-Boustani D, Flechtner I, Dechaux M, Elie C, Tardy V, Morel Y, Polak M. Transient hyper-17-hydroxyprogesteronemia: a clinical subgroup of patients diagnosed at neonatal screening for congenital adrenal hyperplasia. European journal of endocrinology / European Federation of Endocrine Societies. 2009;161(2):285–92. [DOI] [PubMed] [Google Scholar]
- 16.Turcu AF, Rege J, Chomic R, Liu J, Nishimoto HK, Else T, Moraitis AG, Palapattu GS, Rainey WE, Auchus RJ. Profiles of 21-Carbon Steroids in 21-hydroxylase Deficiency. The Journal of clinical endocrinology and metabolism. 2015;100(6):2283–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Costa-Barbosa FA, Carvalho VM, Nakamura OH, Bachega TA, Vieira JG, Kater CE. Zona fasciculata 21-hydroxysteroids and precursor-to-product ratios in 21-hydroxylase deficiency: further characterization of classic and non-classic patients and heterozygote carriers. Journal of endocrinological investigation. 2011;34(8):587–92. [DOI] [PubMed] [Google Scholar]
- 18.Costa-Barbosa FA, Tonetto-Fernandes VF, Carvalho VM, Nakamura OH, Moura V, Bachega TA, Vieira JG, Kater CE. Superior discriminating value of ACTH-stimulated serum 21-deoxycortisol in identifying heterozygote carriers for 21-hydroxylase deficiency. Clinical endocrinology. 2010;73(6):700–6. [DOI] [PubMed] [Google Scholar]
- 19.Milewicz A, Vecsei P, Gruszka S, Szymczak J, Bednarek-Tupikowska G, Grabinski M. Diagnosis of congenital adrenal hyperplasia based on plasma 21-deoxycortisol level determined with a specific radioimmunoassay. Materia medica Polona Polish journal of medicine and pharmacy. 1984;16(2–4):95–8. [PubMed] [Google Scholar]
- 20.Bloem LM, Storbeck KH, Schloms L, Swart AC. 11beta-hydroxyandrostenedione returns to the steroid arena: biosynthesis, metabolism and function. Molecules. 2013;18(11):13228–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Strushkevich N, Gilep AA, Shen L, Arrowsmith CH, Edwards AM, Usanov SA, Park HW. Structural insights into aldosterone synthase substrate specificity and targeted inhibition. Molecular endocrinology. 2013;27(2):315–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Turcu AF, Nanba AT, Chomic R, Upadhyay SK, Giordano TJ, Shields JJ, Merke DP, Rainey WE, Auchus RJ. Adrenal-derived 11-oxygenated 19-carbon steroids are the dominant androgens in classic 21-hydroxylase deficiency. European journal of endocrinology / European Federation of Endocrine Societies. 2016;174(5):601–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kamrath C, Wettstaedt L, Boettcher C, Hartmann MF, Wudy SA. Androgen excess is due to elevated 11-oxygenated androgens in treated children with congenital adrenal hyperplasia. The Journal of steroid biochemistry and molecular biology. 2018;178:221–8. [DOI] [PubMed] [Google Scholar]
- 24.Turcu AF, Mallappa A, Elman MS, Avila NA, Marko J, Rao H, Tsodikov A, Auchus RJ, Merke DP. 11-Oxygenated Androgens Are Biomarkers of Adrenal Volume and Testicular Adrenal Rest Tumors in 21-Hydroxylase Deficiency. The Journal of clinical endocrinology and metabolism. 2017;102(8):2701–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Azziz R, Carmina E, Dewailly D, Diamanti-Kandarakis E, Escobar-Morreale HF, Futterweit W, Janssen OE, Legro RS, Norman RJ, Taylor AE, et al. The Androgen Excess and PCOS Society criteria for the polycystic ovary syndrome: the complete task force report. Fertility and sterility. 2009;91(2):456–88. [DOI] [PubMed] [Google Scholar]
- 26.Nanba AT, Rege J, Ren J, Auchus RJ, Rainey WE, Turcu AF. 11-Oxygenated C19 Steroids Do Not Decline With Age in Women. The Journal of clinical endocrinology and metabolism. 2019;104(7):2615–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wong T, Shackleton CH, Covey TR, Ellis G. Identification of the steroids in neonatal plasma that interfere with 17 alpha-hydroxyprogesterone radioimmunoassays. Clinical chemistry. 1992;38(9):1830–7. [PubMed] [Google Scholar]
- 28.Minutti CZ, Lacey JM, Magera MJ, Hahn SH, McCann M, Schulze A, Cheillan D, Dorche C, Chace DH, Lymp JF, et al. Steroid profiling by tandem mass spectrometry improves the positive predictive value of newborn screening for congenital adrenal hyperplasia. The Journal of clinical endocrinology and metabolism. 2004;89(8):3687–93. [DOI] [PubMed] [Google Scholar]
- 29.Schwarz E, Liu A, Randall H, Haslip C, Keune F, Murray M, Longo N, Pasquali M. Use of steroid profiling by UPLC-MS/MS as a second tier test in newborn screening for congenital adrenal hyperplasia: the Utah experience. Pediatric research. 2009;66(2):230–5. [DOI] [PubMed] [Google Scholar]
- 30.Janzen N, Peter M, Sander S, Steuerwald U, Terhardt M, Holtkamp U, Sander J. Newborn screening for congenital adrenal hyperplasia: additional steroid profile using liquid chromatography-tandem mass spectrometry. The Journal of clinical endocrinology and metabolism. 2007;92(7):2581–9. [DOI] [PubMed] [Google Scholar]
- 31.Janzen N, Sander S, Terhardt M, Peter M, Sander J. Fast and direct quantification of adrenal steroids by tandem mass spectrometry in serum and dried blood spots. Journal of chromatography B, Analytical technologies in the biomedical and life sciences. 2008;861(1):117–22. [DOI] [PubMed] [Google Scholar]
- 32.Speiser PW, Arlt W, Auchus RJ, Baskin LS, Conway GS, Merke DP, Meyer-Bahlburg HFL, Miller WL, Murad MH, Oberfield SE, et al. Congenital Adrenal Hyperplasia Due to Steroid 21-Hydroxylase Deficiency: An Endocrine Society Clinical Practice Guideline. The Journal of clinical endocrinology and metabolism. 2018;103(11):4043–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miller WL. Congenital Adrenal Hyperplasia: Time to Replace 17OHP with 21-Deoxycortisol. Hormone research in paediatrics. 2019;91(6):416–20. [DOI] [PubMed] [Google Scholar]
- 34.Armengaud JB, Charkaluk ML, Trivin C, Tardy V, Breart G, Brauner R, Chalumeau M. Precocious pubarche: distinguishing late-onset congenital adrenal hyperplasia from premature adrenarche. The Journal of clinical endocrinology and metabolism. 2009;94(8):2835–40. [DOI] [PubMed] [Google Scholar]
- 35.Rege J, Turcu AF, Kasa-Vubu JZ, Lerario AM, Auchus GC, Auchus RJ, Smith JM, White PC, Rainey WE. 11-Ketotestosterone Is the Dominant Circulating Bioactive Androgen During Normal and Premature Adrenarche. The Journal of clinical endocrinology and metabolism. 2018;103(12):4589–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Storbeck KH, Bloem LM, Africander D, Schloms L, Swart P, Swart AC. 11beta-Hydroxydihydrotestosterone and 11-ketodihydrotestosterone, novel C19 steroids with androgenic activity: a putative role in castration resistant prostate cancer? Molecular and cellular endocrinology. 2013;377(1–2):135–46. [DOI] [PubMed] [Google Scholar]
- 37.Pretorius E, Africander DJ, Vlok M, Perkins MS, Quanson J, Storbeck KH. 11-Ketotestosterone and 11-Ketodihydrotestosterone in Castration Resistant Prostate Cancer: Potent Androgens Which Can No Longer Be Ignored. PloS one. 2016;11(7):e0159867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.O’Reilly MW, Kempegowda P, Jenkinson C, Taylor AE, Quanson JL, Storbeck KH, Arlt W. 11-oxygenated C19 steroids are the predominant androgens in polycystic ovary syndrome. The Journal of clinical endocrinology and metabolism. 2016:jc20163285. [DOI] [PMC free article] [PubMed] [Google Scholar]