

SUMMARY
Major histocompatibility complex class I (MHC-I)-restricted immune responses are largely attributed to cytotoxic T lymphocytes (CTLs). However, natural killer (NK) cells, as predicted by the missing-self hypothesis, have opposing requirements for MHC-I, suggesting that they may also demonstrate MHC-I-restricted effects. In mice, the Ly49 inhibitory receptors prevent NK cell killing of missing-self targets in effector responses, and they have a proposed second function in licensing or educating NK cells via self-MHC-I in vivo. Here we show MHC-I-restricted control of murine cytomegalovirus (MCMV) infection in vivo that is NK cell dependent. Using mice lacking specific Ly49 receptors, we show that control of MCMV requires inhibitory Ly49 receptors and an inhibitory signaling motif and the capacity for MCMV to downregulate MHC-I. Taken together, these data provide definitive evidence that the inhibitory receptors are required for missing-self rejection and are relevant to MHC-I-restricted NK cell control of a viral infection in vivo.
Graphical Abstract
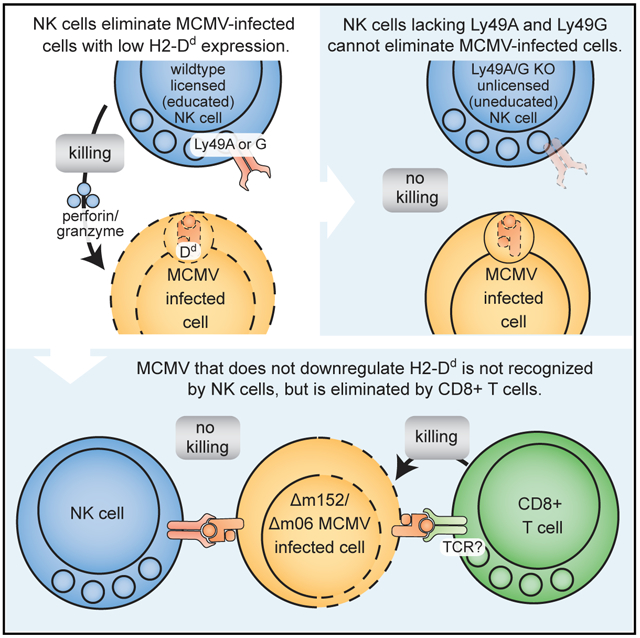
In Brief
Parikh et al. show that major histocompatibility complex class I (MHC-I)-restricted control of viral infection is due to natural killer (NK) cells rather than cytotoxic T cells. Inhibitory NK cell receptors are essential for protection, requiring NK cell licensing (education) by self-MHC-I and missing-self recognition due to virus-induced MHC-I downregulation.
INTRODUCTION
Major histocompatibility complex (MHC) class I (MHC-I) molecules are central to adaptive and innate immune responses. They are required for adaptive CD8+ MHC-I-restricted T cell recognition in antigen priming and responses as well as shaping adaptive T cell receptor repertoires in thymocyte development. In support of this central role for MHC-I in T cell immunity, many viruses have evolved mechanisms to suppress MHC-I expression to thwart T cell effector responses. In innate immunity, MHC-I has opposing effects on natural killer (NK) cells. The missing-self hypothesis predicts that NK cells should kill cells that downregulate MHC-I (Ljunggren and Kärre, 1990), as supported by MHC-I-specific inhibitory NK cell receptors that prevent NK cell killing of cells with normal MHC-I expression in vitro (Karlhofer et al., 1992; Kärre et al., 1986). Although supported by NK cell elimination of adoptively transferred MHC-I-deficient cells, the role of inhibitory NK cell receptors during in vivo responses is still poorly understood, largely because of the large number of polymorphic, germline-encoded receptors with overlapping specificities and apparently opposing functional outcomes. Improved clarity of these processes is required to better understand the basis for association of human MHC-I (HLA-I) alleles with certain diseases, which is generally thought to be due to CD8+ T cells (Trowsdale and Knight, 2013).
In mice, the Ly49 NK cell receptor family is encoded in a gene cluster in the NK gene complex (NKC) on mouse chromosome 6 (Yokoyama and Plougastel, 2003). Although Ly49s display profound allelic polymorphisms with several haplotypes and allelic forms (Rahim and Makrigiannis, 2015), most Ly49s mediate inhibitory function in effector responses through their cytoplasmic immunoreceptor tyrosine-based inhibitory motifs (ITIMs) (Long et al., 2013). Inhibitory Ly49 receptors bind MHC-I alleles, but their specificities have been reportedly identical; for example, Ly49AB6, Ly49CB6, Ly49GB6, and Ly49IB6 all recognize H2Dd (Dam et al., 2003; Hanke et al., 1999). Ly49s are stochastically expressed on overlapping subsets of NK cells with individual NK cells simultaneously expressing two or more Ly49s, limiting the capacity to interpret the effects of antibody depletion studies to a particular receptor specificity or quantitative effects on NK cell numbers.
The Ly49 receptors also display polymorphism at another level. In contrast to inhibitory Ly49s, Ly49 activation receptors lack ITIMs and are coupled to immunoreceptor tyrosine-based activation motif (ITAM)-containing chains, such as DAP12 (Long et al., 2013), even though they may be serologically related to the inhibitory receptors. The Ly49HB6 activation receptor is responsible for genetic resistance of C57BL/6 (B6) mice to murine cytomegalovirus (MCMV) infection, providing vital early viral control, even in mice with intact adaptive immunity (Brown et al., 2001). Ly49HB6 recognizes an MCMV-encoded MHC-I-like molecule, m157 (Arase et al., 2002). The Ly49P1NOD/Ltj, Ly49L-BALB, and Ly49D2PWK/Pas activation receptors serve similar functions, albeit with ligands distinct from m157 (Pyzik et al., 2011). Additionally, the inhibitory Ly49I129 receptor also binds m157 (Arase et al., 2002; Corbett et al., 2011), suggesting that inhibitory Ly49 receptors could play critical roles in viral control, through effects that are not well understood.
Interestingly, the inhibitory Ly49 receptors appear to provide a second function in a process termed licensing (or education) by self-MHC-I. Only inhibitory receptors specific for self-MHC-I in vivo can confer a licensed phenotype whereby licensed NK cells exhibit enhanced responsiveness to stimulation through activation receptors in vitro (Kim et al., 2005). Prior studies suggested that licensed NK cells hamper MCMV control through inhibitory receptors for host MHC-I (Orr et al., 2010). However, other studies suggest that inhibitory Ly49s may enhance MCMV control, but these approaches have used mouse strains with poorly characterized Ly49s and Ly49-depleting antibodies with unclear specificities and that also affect total NK cell number, confounding interpretations (Babić et al., 2010; Pyzik et al., 2011; Pyzik et al., 2014). Additionally, because of the potential roles of other receptors (Guillamón et al., 2018; Rahim et al., 2015) in licensing, the in vivo role of licensed NK cells and inhibitory Ly49s in viral infections remains controversial.
Herein, we found that NK cells mediate MHC-restricted control of MCMV infection. We used CRISPR-Cas9 to simultaneously and cleanly target multiple Ly49 receptors, allowing definitive evaluation of the role of inhibitory Ly49s in MHC-restricted resistance to MCMV infection.
RESULTS
MHC-Restricted, NK Cell-Dependent Protection against MCMV in Mice
To investigate the role of NK cells in MCMV control in different MHC backgrounds, we used C57BL/10 (B10, H2b) and B10.D2 (H2d) MHC-congenic mouse strains that are closely related to B6 (H2b) and share Ly49 haplotypes (Higuchi et al., 2010). Susceptibility at day 4 post-infection (d4 p.i.) to wild-type (WT) MCMV in B10 mice was NK cell dependent, and antibody blocking studies showed that protection against WT MCMV was dependent upon Ly49H (Figure 1A), as shown for B6 mice (Brown et al., 2001). To examine other mechanisms of viral resistance applicable to MCMV isolates from the wild lacking m157 (Corbett et al., 2011), we used Δm157-MCMV that contains a single nucleotide deletion in m157 that prevents full-length expression. Although B10 mice were unable to control Δm157-MCMV infection at d4 p.i., MHC-congenic B10.D2 (H2d) mice were resistant (Figure 1B), demonstrating an MHC-restricted effect.
Figure 1. H2d-Dependent Protection against MCMV Lacking m157 Requires Cytotoxic NK Cells.
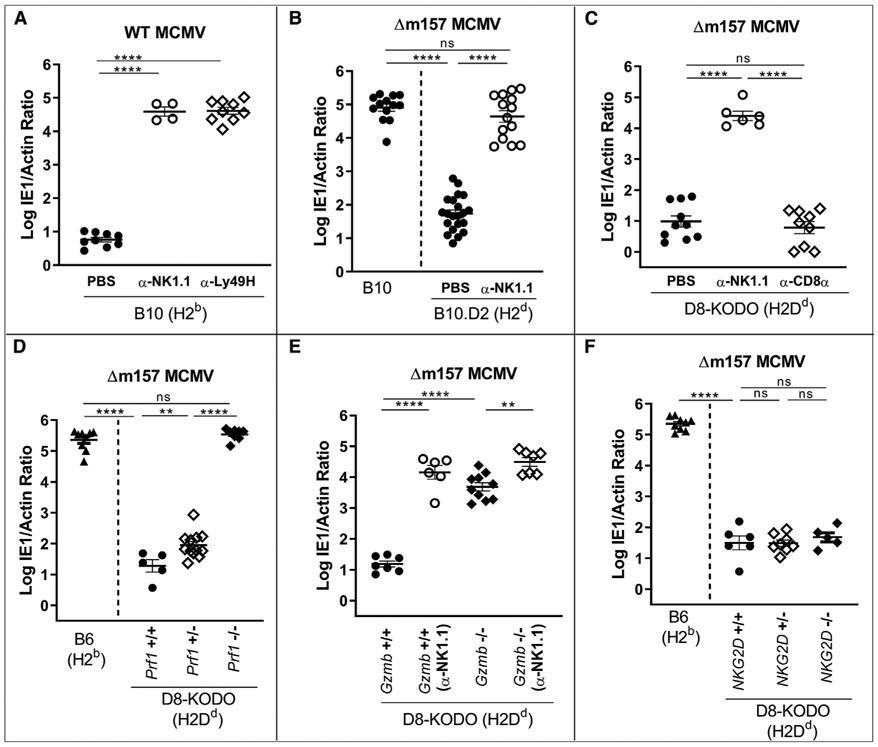
(A–F) Splenic viral titers in mice depleted of total NK cells or CD8+T cells or where Ly49H+ receptors on NK cells were blocked. Data represent a composite of two independent experiments with three to seven mice per group, with individual points representing a single mouse using (A) H2b-expressing B10 mice, (B) B10 and H2d-expressing B10.D2 mice, (C) H2Dd-expressing (D8-KODO) mice, and (D–F) D8-KODO mice with wild-type, heterozygous, or no expression of (D) perforin (Prf1), (E) granzyme B (Gzmb), and (F) NKG2D (Klrk1).
To isolate the MHC-restricted effect to a single MHC-I allele, we showed that Δm157-MCMV resistance was comparable with B10.D2 mice in B6 mice transgenically expressing only H2Dd, in the genetic absence of H2Kb and H2Db (D8-KODO) (Figure 1C), indicating that resistance was specifically due to H2Dd. Resistance was clearly dependent upon NK cells but not CD8+ T cells, as shown by antibody depletion of NK or CD8+ T cells, respectively (Figures 1B and 1C). Thus, MHC-restricted resistance to Δm157-MCMV is due to H2Dd and is NK cell dependent.
Perforin-deficient (Prf1−/−) D8-KODO mice were as susceptible as WT B6 mice, whereas both perforin WT (Prf1+/+) and perforin heterozygous (Prf1+/−) D8-KODO mice controlled Δm157-MCMV (Figure 1D). Likewise, granzyme-deficient D8-KODO mice could not control Δm157-MCMV (Figure 1E). Although NKG2D enhances NK cell responses to MCMV in B6 mice (Nabekura et al., 2017; Zafirova et al., 2009), we found no change in viral control by NKG2D-deficient D8-KODO mice compared with D8-KODO mice (Figure 1F). Thus, NK cell-dependent control of Δm157-MCMV requires cytotoxicity, implying direct target contact, but NKG2D was not required.
NK Cell Resistance Requires Ly49 Receptor Expression
As Ly49s recognize MHC-I, we assessed their candidacy for being responsible for the MHC-I-restricted, NK-dependent resistance to Δm157-MCMV by using CRISPR-Cas9 to target their deletion directly in B6 zygotes. When we used a guide RNA (gRNA) intentionally chosen for its promiscuity for several Ly49s, we generated ΔLy49-1 mice with two distinct deletions: (1) a 149 kb deletion between Ly49a and Ly49g with an out-of-frame fusion and (2) a 66 kb deletion between two pseudogenes, Ly49n (Klra14-ps) and Ly49k (Klra11-ps), such that Ly49h was deleted (Figure 2A). Flow cytometry confirmed the loss of Ly49A, Ly49C, Ly49G, and Ly49H expression. Ly49D expression was markedly decreased, but its coding sequence was intact, suggesting an as yet unidentified locus control region within one of the deleted segments. We also generated single and compound Ly49-deleted mice, detailed below (Figures S1 and S2). Predicted potential off-target sites were absent by PCR amplification and sequencing (Table S1). To further eliminate any off-target effects and genetic mosaicism, we backcrossed all CRISPR-Cas9 founder mice to WT B6 for two generations, followed by additional crosses to KODO mice, then D8 (H2Dd) transgenic mice, to generate the indicated homozygous Ly49-knockout (KO) mice on the D8-KODO background.
Figure 2. NK Cell Resistance to Δm157-MCMV Requires Specific Ly49 Receptors.

(A) The CRISPR single-guide RNA (sgRNA) used for targeting is shown; PAM site underlined, 10 nt core in yellow. Mismatches between the sgRNA and targeted Ly49s are represented below the guide. B6 Ly49 cluster with the number of predicted mismatches is shown below. Loss of genetic regions is indicated as a red dashed line; flow cytometry histograms indicate wild-type D8-KODO (shaded) and ΔLy49-1 D8-KODO (solid line) NK cell expression.
(B–E) Δm157-MCMV splenic titers; data represent a composite of at least two independent experiments with three to seven mice per group: (B) D8-KODO, ΔLy49-1 D8-KODO and the indicated F1 mice; (C–E) D8-KODO mice (C) lacking DAP10 and DAP12, (D) with Ly49m deletions, and (E) lacking Ly49C and Ly49I.
MHC-restricted, NK cell-dependent resistance to Δm157-MCMV in D8-KODO mice was absent in ΔLy49-1 D8-KODO mice (Figure 2B). Additionally, KODO mice with intact Ly49s complemented ΔLy49-1 D8-KODO mice, as their F1 hybrids showed fully restored Δm157-MCMV resistance (Figure 2B), indicating that heterozygous expression of Ly49A, Ly49C, Ly49G, and Ly49H (Figure S2), deleted in ΔLy49-1, was sufficient for resistance. Similarly, (D8-KODO × KODO) F1 hybrids were also resistant (Figure 2B), indicating that H2Dd heterozygosity is sufficient for antiviral protection when Ly49 genes were intact.
Inhibitory Ly49A and Ly49G Receptors Are Required for H2Dd-Dependent Resistance
To decipher which Ly49 receptor(s) are involved in Δm157-MCMV resistance in D8-KODO mice, we first considered Ly49 activation receptors. However, a cross between resistant D8-KODO mice with susceptible B6 (H2b) mice generating heterozygosity for H2Dd and H2b resulted in an intermediate infection phenotype (Figure 2B), highlighting an apparent role of MHC-I context for antiviral protection and contrasting MCMV resistance explained by Ly49 activation receptors when complete reversal was found in crossing susceptible and resistant mice (Depatie et al., 1997; Pyzik et al., 2011; Xie et al., 2009). Nonetheless, as ΔLy49-1 D8-KODO mice had low levels of Ly49D and lacked Ly49h, and both receptors require DAP12 (and DAP10 to a lesser extent) for expression, including Ly49H-mediated resistance to MCMV, we evaluated the role of Ly49D and Ly49H by studying D8-KODO mice deficient in DAP10 and DAP12. For reasons not immediately clear, Ly49A, Ly49F, and Ly49G expression was markedly decreased (Figure S2). Regardless, these mice were still resistant (Figure 2C), indicating that activation signals through DAP10/DAP12 are not required, consistent with the lack of involvement of NKG2D (Figure 1E), which also requires DAP10 or DAP12 for surface expression, depending on its isoform (Figure S2; Diefenbach et al., 2002). Thus, Ly49 activation receptors are surprisingly not required for resistance to Δm157-MCMV in D8-KODO mice.
Having ruled out Ly49 activation receptors, we assessed Ly49 pseudogenes, which could theoretically contribute to resistance by splicing events, which has been considered for other Ly49 genes (McQueen et al., 1999; Smith et al., 1994). To test Ly49m (Klra13-ps), harboring a premature stop codon (third exon), we generated four independent CRISPR-Cas9 KO strains. When crossed to D8-KODO background, all four lines showed resistance to Δm157-MCMV, indicating no apparent role for Ly49m (Figure 2D). Of the other remaining regions of the Ly49 cluster disrupted in the ΔLy49-1 strain, we did not pursue Ly49j, Ly49k, and Ly49n, because they were predicted to encode severely truncated proteins, not expressed as receptors on NK cells (McQueen et al., 1999).
We then turned our attention to Ly49 inhibitory receptors. As Ly49C and Ly49I reportedly have specificity for H2d (Dam et al., 2003; Hanke et al., 1999), they could be relevant to ΔLy49-1 D8-KODO mice, which express Ly49I but not Ly49C (Figure 2A). However, lack of only these two Ly49s in a new D8-KODO KO strain had no effect on H2Dd resistance to Δm157-MCMV, which remained entirely dependent upon NK cells (Figure 2E).
Finally, we evaluated the potential contribution of Ly49A and Ly49G, as both are deleted in ΔLy49-1 mice, and there are substantial data indicating that they both recognize H2Dd (Jonsson et al., 2010; Silver et al., 2002). In D8-KODO mice, Ly49A depletion had no significant change in viral titers, while Ly49G depletion moderately increased Δm157-MCMV levels (Figure 3A). Interestingly, depletion of both Ly49A and Ly49G led to a major loss of viral control, similar to complete NK cell depletion (Figure 3A), but Ly49G depletion results in approximately 50% decrease of all NK cells (Figure S2). Nonetheless, Ly49D depletion, also affecting 50%–60% of NKcells, did not alter viral resistance, suggesting a potential redundant contribution of Ly49A and Ly49G, independent of quantitative NK cell loss.
Figure 3. Ly49G and Ly49A Are Required for H2Dd-Dependent Δm157-MCMV Resistance.

(A–D and F) Δm157-MCMV splenic titers; data represent a composite of multiple independent experiments, as noted, with three to seven mice per group, with individual points representing a single mouse. Mice used were (A) D8-KODO, five experiments; (B) two lines lacking Ly49A (black versus red), three experiments; (C) Ly49G knockout mice, representative of two experiments; (D) two lines lacking Ly49A and Ly49G, five experiments; and (F) ΔLy49-1 D8-KODO with or without the Ly49A knockin, two experiments.
(E) Composite survival analysis of D8-KODO and Ly49A/G-knockout mice; two experiments.
To definitively determine the role of Ly49A and Ly49G in resistance, we derived new Ly49 KO strains on the D8-KODO background (Figures S1 and S2). Although deletion of Ly49A alone was insufficient to reverse MCMV resistance, Ly49G depletion in these mice allowed MCMV titers higher than levels with Ly49G depletion in Ly49A-sufficient mice and similar to that with NK cell depletion (Figure 3B). Reciprocally, Ly49G-KO mice were resistant and Ly49A depletion reversed resistance, again comparable with NK cell depletion (Figure 3C). Finally, we studied two mouse strains with KOs of both Ly49A and Ly49G (Ly49AG KO) (Figure S1), which displayed relatively unchanged NK cell numbers, repertoire of other Ly49s, and development (Figure S2). Infection of either Ly49AG-KO strain resulted in viral titers similar to NK cell depletion of D8-KODO mice (Figure 3D) and lethality (Figure 3E). To provide additional evidence, we generated Ly49A-knockin (KI) mice (into Ncr1 on chromosome 7), which expressed Ly49A at near normal levels on all NKp46+ NK cells (Figure S3). ΔLy49-1 Ly49A KI D8-KODO mice demonstrated NK cell-dependent resistance to Δm157-MCMV, unlike susceptible parental ΔLy49-1 D8-KODO mice, showing complementation by Ly49A (Figure 3F). Therefore, Ly49A and Ly49G act redundantly in H2Dd mice to promote NK cell-dependent resistance to Δm157-MCMV, unequivocally establishing the protective role of inhibitory Ly49 receptors in NK cell-dependent viral control.
NK Cell Licensing and Missing-Self Rejection Both Require Ly49A and Ly49G Receptors in D8-KODO Mice
To further delineate the potential mechanism for MHC-restricted viral resistance, we investigated whether loss of Ly49A and Ly49G would have an impact on NK cell licensing by stimulating NK cells from D8-KODO, Ly49AG-KO D8-KODO, and TKO (KODO β2 m−/−) mice with plate-bound anti-NK1.1 for interferon gamma production (Kim et al., 2005). Remarkably, the total NK cell pool in Ly49AG-KO D8-KODO mice exhibited significantly reduced IFN-gamma production compared with D8-KODO NK cells and similar to unlicensed NK cells from TKO mice (Figures 4A and 4B). These results strongly suggest that Ly49A and Ly49G are required for NK cell licensing through H2Dd.
Figure 4. NK Cell Licensing and Missing-Self Rejection in D8-KODO Mice Both Require Ly49A and Ly49G.
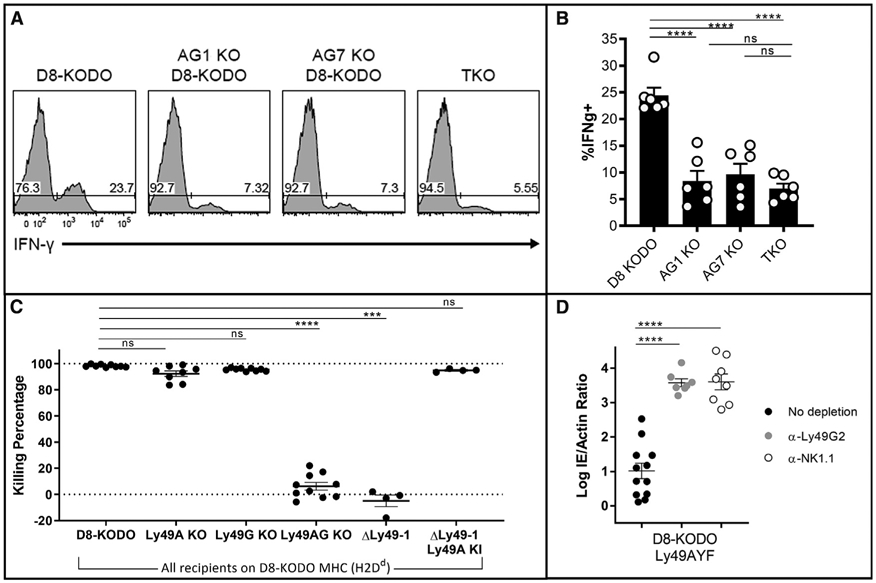
(A) Representative histogram plots depicting the frequency of interferon gamma positive (IFNg+) NK cells following NK1.1 stimulation in D8-KODO, Ly49A/G-KO mice, and TKO (KODO β2 m-KO) mice.
(B) Composite plot of IFNg+ frequencies from (A); two independent experiments with three mice per group.
(C) In vivo cytotoxicity of KODO or D8-KODO splenocytes. Data are cumulative over two independent experiments with four or five recipient mice per group.
(D) Δm157-MCMV infection as in Figure 2B; three independent experiments.
We next tested whether loss of inhibitory Ly49s would affect missing-self recognition in vivo, as suggested by in vitro studies (Bern et al., 2017; Karlhofer et al., 1992) and anti-Ly49 antibody experiments in vivo (Sun et al., 2012). Upon injection, labeled KODO (MHC-I-deficient) splenocytes were effectively cleared in D8-KODO mice that were either WT or lacked only Ly49A or only Ly49G (Figures 4C and S4), in an NK cell-dependent manner as shown by NK cell depletion (Figures S4A and S4B). Importantly, ΔLy49-1 D8-KODO mice and Ly49AG-KO D8-KODO mice were unable to reject KODO targets, while complementation of ΔLy49-1 D8-KODO mice with Ly49A KI restored this capacity. Thus, these results strongly suggest that Ly49A and Ly49G are redundant missing-self receptors in D8-KODO mice, as they enable NK cells to reject missing-self targets through NK cell licensing.
Inhibitory Ly49s Mediate Protection from Δm157-MCMV Is ITIM Dependent
We recently generated KI mice carrying Ly49A with a non-functional ITIM (termed Ly49AYF), demonstrating that effector inhibition and licensing are both mediated by the Ly49A ITIM (Bern et al., 2017). Ly49AYF D8-KODO mice were resistant to Δm157-MCMV (Figure 4D) similar to D8-KODO mice, but in contrast to D8-KODO mice (Figures 3A and 3B), Ly49G depletion led to significantly elevated viral titers, similar to anti-NK1.1 depletion (Figure 4D). These data also mirror Ly49G depletion of Ly49A-KO D8-KODO mice (Figure 3B), suggesting that Ly49A mediates resistance to Δm157-MCMV in an ITIM-dependent manner. We also observed that Ly49G-depleted Ly49AYF D8-KODO mice were unable to reject missing-self targets, indicating that the ITIM is required for Ly49A to mediate missing-self recognition in vivo (Figure S4). These findings strongly suggest that Ly49A mediates resistance to Δm157-MCMV in an ITIM-dependent manner.
MCMV Infection Generates Targets for Missing-Self Rejection
We next investigated the mechanism by which Δm157-MCMV might contribute to the Ly49 effects. We initially focused on two MCMV immunoevasins known to downmodulate MHC-I, m06 and m152 (Pinto et al., 2006), although they have variable effects dependent upon MHC-I alleles (Wagner et al., 2002). To establish their relevance here, we first infected SV40-immortalized D8-KODO mouse embryonic fibroblasts (MEFs) with a GFP-expressing Δm157-MCMV, indicating that H2Dd was downregulated in GFP-positive infected cells (Figures 5A and 5B). Next, we produced mutant Δm157-GFP viruses deficient for m06, m152, or both. Remarkably, H2Dd downmodulation was completely abrogated only when both m06 and m152 were targeted (Figures 5A and 5B), indicating that m04, an open reading frame (ORF) previously shown to interact with MHC-I (Kleijnen et al., 1997), does not play a role in MHC-I regulation for H2Dd. In contrast, WT m152 (Δm06Δm157 MCMV) or WT m06 (Dm152Δm157 MCMV) decreased surface expression of H2Dd in infected cells, consistent with virus-free overexpression systems, demonstrating that both m06 (Reusch et al., 1999) and m152 (Ziegler et al., 1997) redundantly promote MHC downregulation.
Figure 5. Downregulation of H2Dd through m06 and m152 Affects Host MCMV Control.

(A) Histogram plots and (B) bar charts of 24 h in vitro infection and flow cytometric evaluation of D8-KODO MEFs with GFP-expressing MCMV lacking the indicated ORFs; representative of two independent infections.
(C) MFI of H2Dd in mice homozygous and heterozygous for the D8 transgene (left), compared with BALB/c (H2d/d) and BALB/c η B6 F1 (H2b/d) mice (right); representative of two independent experiments.
(D and E) Three h (D) or 24 h (E) in vivo cytotoxicity with KODO or D8-KODO heterozygous (D8-Het; H2Dd+/−) donor cells. Composite data from two independent experiments.
(F and G) Δm157 or Δm157/m06/m152 MCMV infection of B6 or Rag1−/−(F) and D8-KODO or ΔLy49-1 D8-KODO (G) mice. (F) and (G) are representative titers from two independent experiments.
D8-KODO mice heterozygous for H2Dd (Figure 2B) displayed H2Dd expression at an MFI of nearly 50% of homozygous mice (Figure 5C), comparable with the loss seen during in vitro MCMV infection (Figures 5A and 5B). There was a similar level of H2Dd expression in (BALB/c × B6) F1 hybrid mice, confirming that physiologically relevant levels of H2Dd were being assessed, both in vitro and in vivo. Indeed, heterozygous D8 targets in uninfected homozygous D8-KODO mice were protected from clearance following a 3 h (Figure 5D) or 24 h (Figure 5E) in vivo cytotoxicity assay. In contrast, KODO targets were completely eliminated at both 24 h (Figure 5E) and 48 h (Figure 4C) and partially eliminated at 3 h after injection (Figure 5D). During MCMV infection, there was enhanced clearance of both MHC-null and MHC-heterozygous targets at all times post-infection (Figure 5D), consistent with our previous study, in which we identified a role for cytokines modulating missing-self rejection during MCMV infection (Bern et al., 2019). Although heterozygous target cell rejection never reached levels seen with MHC null targets, NK cell-dependent clearance of missing-self targets can discriminate between normal levels and partial downregulation of MHC molecules in the context of MCMV infection.
Deletion of MCMV Genes m06 and m152 Prevents NK-Dependent Clearance while Enhancing CD8+ T Cell-Dependent Protection
To determine if m06 and m152 perturb NK-dependent viral resistance, we generated an independent set of Δm157-MCMV stocks in a virulent MCMV strain with mutations in both m06 and m152, termed Δm157/Δm06/Δm152-MCMV. Δm157-MCMV and Δm157/Δm06/Δm152-MCMV showed similar rates of in vitro growth and viral titers, though Δm157-MCMV had a slight advantage (Figure S6). Regardless, infection of Rag1−/− mice on the B6 background indicated that both viruses had comparable replication in vivo (Figure 5F). By contrast, Δm157/Δm06/Δm152-MCMV infection of B6 and ΔLy49-1 D8-KODO mice, otherwise susceptible to Δm157-MCMV (Figures 1B and 2B), resulted in a resistant phenotype (Figures 5F and 5G). D8-KODO mice were also resistant to Δm157/Δm06/Δm152-MCMV, but resistance was not dependent on NK cells (Figure 5G), contrasting their NK cell-dependent resistance to Δm157-MCMV (Figure 1C). Instead, CD8 depletion significantly reversed resistance to Δm157/Δm06/Δm152-MCMV (Figure 5G), unlike the absence of an effect on resistance to Δm157-MCMV (Figure 1C).
DISCUSSION
Here, we clearly demonstrate that specific NK cell Ly49 inhibitory receptors have a critical MHC-restricted role in controlling viral infection in vivo. Their role is dependent on their specificity for self-MHC-I, which influences their effects on NK cell licensing. Viral modulation of MHC-I was required because when MHC-I was no longer downregulated, early resistance was due to CTLs instead of NK cells. Therefore, both the host (Ly49a and Ly49g) and the virus (m06 and m152) encode multiple molecules involved in this resistance, a redundancy highlighting the ongoing arms race between the host and pathogen and providing definitive support for the missing-self hypothesis.
In other strains of mice, there are likely differences in the NK cell response to MCMV due to Ly49 polymorphisms, particularly their specificities for MHC-I, and receptor repertoire, including subset distribution. Similarly, hosts may have different MHC-I alleles with varying capacities to license NK cells and susceptibilities to downregulation by viral MHC-I inhibitors. These factors likely account for differences in the MHC-restricted phenotypes described here. Moreover, there appear to be activation receptors, akin to Ly49H in B6 mice, that may dominate NK cell function if their ligands are expressed on infected cells, in which case licensed NK cells may play a secondary role. Finally, MCMV itself has evolved alleles of its ORFs that modulate these processes. All of these processes have confounded previous attempts to clearly elucidate MHC restriction by NK cells.
Our studies suggest that MCMV should encode molecules to specifically inhibit licensed NK cells. MCMV-encoded m157 can engage Ly49 inhibitory receptors in mouse strains that do not have Ly49H activation receptor equivalents, such as Ly49I in 129 mice (Arase et al., 2002) and Ly49C in BALB/c (Pyzik et al., 2014). We predict that m157 inhibition of NK cell function will depend on whether these inhibitory receptors are in hosts with appropriate MHC-I alleles for licensing. Indeed, m157 effects on inhibiting NK cell control appear to be MHC-dependent (Pyzik et al., 2014), suggesting such potential interactions. Moreover, MCMV has 11 ORFs with predicted MHC-I folds (Smith et al., 2002), some of which have been verified by crystallographic studies (Mans et al., 2007), and thus may be similarly involved in modulating NK cells. Prior findings have suggested that in addition to m06 and m152, a third immunoevasin, m04, also interacts with MHC-I. However, instead of downregulating MHC-I, m04 stabilizes certain MHC-I alleles on the surface of infected cells recognized by the Ly49A inhibitory receptor in BALB/c mice (Babić et al., 2010). As an apparent host countermeasure, m04-MHC-I complexes are recognized by Ly49P, an activation receptor in MA/My mice, and correlate with NK cell-dependent resistance to MCMV (Kielczewska et al., 2009). Nevertheless, although we did not address a potential role for m04 in our studies, the MHC-I-restricted, inhibitory NK cell receptor-dependent anti-viral effects described here are fundamentally and mechanistically different from prior reports.
Our findings clearly suggest an important role for inhibitory Ly49 receptors in clearance of MCMV, implying a role for NK cell activation receptors. Although our studies rule out a role for activation receptors that require DAP10/12 signaling adaptors, such as NKG2D, other activation receptors that do not require these adaptors may be involved. For example, the activating receptor DNAM-1 has been described to bind to virus-infected cells via recognition of CD155 and CD112, thereby promoting NK cell-mediated elimination of infected cells (de Andrade et al., 2014). In addition to DNAM-1 in mice, the natural cytotoxicity receptor 1 (NCR1/NKp46), 2B4 that engages CD48, and the lymphocyte function-associated antigen 1 (LFA-1) that binds ICAM-1 and ICAM-2 (Long et al., 2013) may also play important roles in missing-self-dependent killing, whether in the absence or presence of infection. Furthermore, the inflammatory milieu, independent of the MHC-I-modulating abilities of MCMV, could also influence the efficacy of missing-self cytotoxicity. Clarification of the involvement of these or additional uncharacterized activation pathways is required to advance our understanding of these intertwined yet complex pathways.
Beyond viral control, our studies also establish that Ly49 inhibitory receptors play a critical role in missing-self rejection, as previously predicted, on the basis of in vitro observations and mice with global defects in MHC-I expression that have unlicensed NK cells. Here we clearly show that absence of specific inhibitory Ly49 receptors in a mouse expressing MHC-I results in unlicensed NK cells and an inability to perform missing-self rejection. It should be noted that prior studies of Ly49 specificities were often dependent on overexpression systems that may not reflect physiological interactions. Understanding of these ambiguities will be aided by the Ly49-KO mice described here as well as complementation by restoring expression of a single Ly49 receptor. Taken together, these data also provide definitive evidence that the inhibitory receptors are required for missing-self rejection in vivo.
During the preparation of our manuscript, another group published their findings on missing-self recognition using indepenadently derived Ly49-KO mice (Zhang et al., 2019). Their experimental evidence supports our claims of the requisite role of Ly49 receptors in missing-self recognition and confirms the lack of major perturbations in maturation and expression patterns of other NK cell receptors following CRISPR-mediated deletions. However, their work did not address MHC allele specificity, whereas our work additionally demonstrates that a single well-defined H2Dd MHC-I molecule is redundantly recognized by Ly49A and Ly49G in missing-self recognition, while unequivocally excluding the role of additional Ly49 and NK cell receptors in mice expressing only H2Dd. Moreover, our restoration of missing-self killing using a KI approach using only Ly49A and abrogation using an ITIM-defective Ly49A further substantiate the role of inhibitory Ly49s and their ITIMs in missing-self in vivo.
In addition, Gamache et al. (2019) recently studied NK cell control of MCMV in the MA/My mouse strain, which has a different Ly49 haplotype from B6 mice (Higuchi et al., 2010) studied here. Interestingly, recognition of H-2Dk by both the inhibitory Ly49G2 and the Ly49R activation receptors was required in MA/My mice for viral control. Although methodological similarities exist between their findings and ours, some key differences preclude generalization of results. First, we described the viral ORFs essential for missing-self effects in an MCMV strain that lacks an important viral ligand for an NK cell activation receptor. More important, our system clearly delineates that licensing and missing-self killing are coincident with viral control, whereas the work by Gamache et al. (2019) describes a mechanism based on both activation and inhibitory receptors. It is still unclear whether these alleles of NK cell receptors in non-B6 strains function similarly in B6 mice or are significantly influenced by MHC-dependent and independent factors. These considerations underscore the challenges of relating unique but potentially strain-specific NK cell-mediated effects across all mouse strains.
Beyond viral clearance, NK cell effects on tumor control greatly rely on both NK cell activation and inhibitory receptor signaling (Daher and Rezvani, 2018). MHC-I downmodulation during tumor growth to evade CTLs provides an attractive target for oncogenic control, yet the in vivo requirements for this potential critical function are poorly understood. Indeed, we recently showed that missing-self rejection in a mouse with inducible β2 m deletion is markedly enhanced by inflammatory stimuli, such as MCMV infection, otherwise licensed NK cells can lose the licensed phenotype when profound MHC-I deletion occurs (Bern et al., 2019), consistent with results reported here. Thus, our studies on NK cell control of viral infection, demonstrating of the complex in vivo interaction between inhibitory receptors and self-MHC-I, can serve as an analogous framework for considering how to modulate NK cells for controlling cancer.
Finally, our studies highlight a role for NK cells in MHC-restricted immune responses, beyond that of MHC-restricted T cell responses, potentially for human autoimmune diseases that are associated with specific HLA-I alleles, such as HLA-B27 and ankylosing spondylitis. Despite the description of this association several decades ago, its basis is still not understood, even though it is widely thought to be due to CD8+ T cells (Bowness, 2015). Our studies suggest that NK cells may also play an important role in this and other HLA-I disease associations.
STAR ★ METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact author Wayne M. Yokoyama (yokoyama@wustl.edu).
Materials Availability
All viruses and mouse lines developed for this study are available under Material Transfer Agreements from Washington University School of Medicine.
Data and Code Availability
The published article includes all data generated or analyzed during this study. Original source data for Figures in the paper are available upon request to the Lead Contact author. No proprietary software was used in the data analysis.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
C57BL/6Ncr (B6) and BALB/cAnNCr (BALB/c) mice were purchased from Charles River Laboratories. C57BL/10SnJ (B10; 000666), B10.D2/nSnJ (B10.D2; 000463), Prf1−/− (002407) and Rag1−/− (002216) strains were purchased from Jackson Laboratory. DAP10−/−DAP12−/− mice were provided by T. Takai (Inui et al., 2009). NKG2D−/− (Klrk1−/−) mice were obtained from Bojan Polic (University of Rijeka, Croatia) (Zafirova et al., 2009). Granzyme B KO (GzmB−/−) mice were obtained from T. Ley (Washington University, St. Louis) (Heusel et al., 1994). H2Kb−/− H2Db−/− (KODO) mice were purchased from Taconic Farms. Triple knockout (TKO) mice which are H2Kb−/− H2Db−/− and lack β2 m were obtained from T. Hansen (Washington University, St. Louis). D8 transgenic mice expressing an H2Dd genomic construct (Bieberich et al., 1986) were provided by D. Marguiles (National Institute of Allergy and Infectious Diseases, Bethesda, MD). D8-KODO mice were generated by crossing D8-transgenic mice to KODO mice (Choi et al., 2011). Generation and characterization of the ITIM mutant AYF mice on the H2d background have been previously described (Bern et al., 2017). KLRA7em1(IMPC)J (Ly49G KO) mice on the C57BL/6NJ background were purchased from the Jackson Laboratory (027444); this allele was generated at the Jackson Laboratory by CRISPR-Cas9 injection of Cas9 RNA and 3 gRNAs: TCTTGTACTTGTGCATAACC, CAGTCCTCACTAGTTTCTGC and GACATGGACTGACCAAATT resulting in a 241 bp deletion beginning in 5-prime upstream sequence and ending within exon 1. Additional strains of mice generated through CRISPR-Cas9 are described below. All mice, with the exception of the Rag1−/− strain, used in these studies were initially obtained or generated on the B6 genetic background and later backcrossed to the KODO and then D8-KODO background (H2Dd MHC). B6 mice used in experiments were obtained directly from Charles River Laboratories. All other experimental and control mice were bred in-house at Washington University. Mice were 8–14-wk old at the start of experiments unless otherwise stated. Male and/or female mice were used in individual experiments without blinding or randomization. This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The studies were approved by the Animal Studies Committee at Washington University School of Medicine under animal protocol 20180293.
Development of CRISPR-modified knockout mice
While NCBI BLAST (https://blast.ncbi.nlm.nih.gov/) was initially used to assess sequence similarity of potential sgRNAs, GT-Scan (https://gt-scan.csiro.au/gt-scan) (O’Brien and Bailey, 2014) and CCTop (https://crispr.cos.uni-heidelberg.de/) (Stemmer et al., 2015) were primarily used to confirm the correct and specific targeting our sgRNA designs. The sequences of synthetic guide RNAs (sgRNA) and the strains of mice generated are shown in Table S1. Cas9 mRNA and sgRNA synthesis, RNA micro-injection into zygotes is identical to what we described earlier (Parikh et al., 2015a). Specifically, we used 20ng of each guide when multiple guides were indicated, and 100ng of Cas9 mRNA. Pups were screened for Ly49 gene disruption at birth by PCR. If multiple Ly49 alleles were present, they were separated by one backcross to B6 and re-screened by PCR. All mice with in-frame insertions or deletions (indels) were excluded from further analysis. Multiple lines with out-of-frame indels were chosen for downstream characterization. These genetic lesions were verified by absence of PCR amplicons for each of the indicated Ly49 genes in homozygous mice, and specific PCR amplicons for the deleted genomic regions, that were sequenced to determine the exact breakpoints (Figures S1 and S2). Finally, Ly49 protein expression was examined, where antibodies were available, at 8 weeks in homozygous mice following at least one additional backcross to parental B6. Since all sgRNAs targeted the second exon of Ly49 genes, we focused on this exon to specifically interrogate on-target and off-target analysis. As shown in Figure S1, all exons and the sgRNA with on-target and off-target potential are depicted. Sanger sequence traces of homozygous mice are shown below the schematic. Flow analysis across genes expressed within the NKC in D8-KODO mice is presented as Figure S2. Specifically, we generated Ly49C/I-double-deficient mice (two lines; one used in experiments), Ly49m-deficient mice (four lines used in experiments), Ly49A-deficient mice (two lines used in experiments), Ly49A/G-double-deficient mice (two lines used in experiments), and the ΔLy49-1 D8-KODO multi-deficient mice (one line used in experiments). The Ly49G-deficient mice were purchased from Jackson Laboratory, however, full on-target, off-target (Figure S1) and flow cytometric (Figure S2) analyses was performed by our laboratory. Importantly, no major impact on maturation was observed in any of these mice.
Development of CRISPR-modified Ly49A knockin mice
The NKp46 (Ncr1) locus was chosen for insertion of the Ly49A cDNA (Figure S3). The donor construct was designed to replace the stop codon of NKp46 (while maintaining the 3-prime untranslated region) with a P2A peptide-cleavage site upstream of the Ly49A cDNA obtained from reverse transcription of Ly49A-expressing B6 NK cells. The result is that Ly49A expression would be restricted to all NK cells. Given that the Ly49A knockin is located on chromosome 7, we were able to cross this mouse with the ΔLy49-1 mouse with Ly49 deletions on chromosome 6 without linkage restrictions. GT-Scan (O’Brien and Bailey, 2014) and CCTop (Stemmer et al., 2015) were primarily used to confirm the correct and specific targeting our sgRNA to the NKp46 locus. The sequences of guide RNAs are shown in Table S1. Cas9 mRNA and sgRNA synthesis, RNA and donor DNA micro-injection is identical to what we described earlier (Parikh et al., 2015a). Specifically, we used 10ng of donor DNA, 20ng of each guide (2 total), and 100ng of Cas9 mRNA. Pups were screened for the donor DNA by PCR soon after birth and maintained as heterozygotes for flow-based confirmation at 8 weeks. After two rounds of B6 backcrossing, the mice were bred to ΔLy49-1 D8-KODO. Mice homozygous for the NKC deletion but heterozygous for the Ly49A knockin were screened for expression of Ly49A (Figure S3). Subsequently, various NKC surface molecules were analyzed by flow cytometry (Figure S2). Again, no major impact on maturation was observed.
Development of CRISPR-Cas9 modified MCMV
Δm157-MCMV and GFP-expressing MCMV were modified to knockout various viral ORFs using CRISPR-Cas9 editing to obviate the need for bacterial artificial chromosome modification. The GFP-expressing MCMV was a generous gift from S.C. Henry and J. Hamilton (Duke University, Durham, NC, USA) (Henry et al., 2000) and previously described by our lab to harbor a non-functional m157 gene (Hsu et al., 2009). The GFP-expressing MCMV and CRISPR-derived progeny were used in in vitro studies while the Δm157-MCMV derived from WT1 (Cheng et al., 2010) and its CRISPR-modified progeny were used in the in vivo analyses. CCTop (Stemmer et al., 2015) was used to identify and confirm specific targeting our sgRNAs to the MCMV ORFs. The stand-alone version of CCTop was loaded with the MCMV genome (GenBank Accession GU305914.1) to effectively eliminate off-target cleavage potential. The table of all possible guide site for MCMV is provided (Table S2). The pX330 vector (Cong et al., 2013; Ran et al., 2013) was obtained from Addgene and modified by replacing the SbfI-Psil site with the Neo marker (SbfI-HincII fragment) from pCDNA3. The oligonucleotide duplexes containing single sgRNAs specific for MCMV m06 or m152 (Table S3) were cloned into this vector that was subsequently transfected into SV40-immortalized B6 MEFs with G418 selection. Parental virus was used to infect these sgRNA and Cas9-expressing MEFs at a low MOI (0.1). Once confluent lysis was observed, viral supernatants were used to reinfect B6 MEFs, individual plaques were picked, and sequence variants were confirmed (Figure S5). For double-deficient MCMV ORF knockouts, a pure stock of virus (e.g., m06 knockout) was used to infect MEFs expressing Cas9 and the alternate sgRNA of interest (e.g., m152). For in vivo analysis, plaque-purified virus was used to generate salivary gland passaged stocks in BALB/c mice as described above. Salivary gland-derived MCMV lacking m06, m152, and m157 was compared to MCMV lacking m157 in a multi-step growth curve on NIH 3T12 (ATCC CCL-164) fibroblasts (MOI = 0.1). Viral genome copies quantified from cell lysates and culture supernatants, as previously described (Parikh et al., 2015b), demonstrated that replication of the triple knockout virus was unimpaired yet delayed slightly in overall growth (Figure S6).
METHOD DETAILS
Viral infection and quantification
The salivary gland propagated MCMV stocks were generated from purified and sequenced clones. Virus was inoculated via the intraperitoneal (IP) route in a total volume of 200 μL PBS at a dose of 40,000 plaque forming units (pfu) per mouse for WT-MCMV and pfu per mouse for m157-deficient MCMV (Parikh et al., 2015b) (Δm157-MCMV) and Δm157/m06/m152-MCMV for determination of splenic titers at day 4. Viral titers from infected spleens were quantified as a ratio of MCMV IE DNA to host beta-actin DNA using real-time PCR and Taqman probes as previously described (Parikh et al., 2015b). When shown, individual data points represent a single mouse. Survival analysis endpoints were determined as either death or >20% weight loss from starting weights, using 300,000 pfu of Δm157-MCMV per mouse. To examine H2Dd MHC-I downmodulation, SV40-immortalized D8-KODO MEFs were infected with GFP-expressing MCMV at an MOI of 1 for 24 h and subsequently released from plates with Versene (Thermo Fisher, Waltham, MA) prior to antibody staining and flow cytometric analysis.
Antibody depletions
Purified 3D10(α-Ly49H) (Smith et al., 2000), PK136 (α-NK1.1), JR9 (α-Ly49A), 4E4(α-Ly49D)(Idris et al., 1999), LGL-1 (α-Ly49G) (Mason et al., 1988) and YTS-169 (α-CD8α) were obtained from hybridomas purified by the Rheumatic Diseases Core Center Protein Purification and Production Facility (Washington University). Antibodies were injected IP at a dose of 200 μg per mouse 48 h prior to infection. >98% depletion was confirmed via flow cytometry in a subset of treated mice. Injection of PBS alone was used as a control where indicated.
Antibodies and Flow Cytometry
The following antibodies and reagents were purchased from eBioscience: anti-CD3e (145-2C11), anti-CD19 (eBio1D3), anti-NK1.1 (PK136), anti-NKp46 (29A1.4), anti-CD27 (LG.7F9), anti-CD11b (M1/70), anti-Ly49D (eBio4E5), anti-Ly49E/F (CM4), anti-Ly49G2 (eBio4D11), anti-Ly49H (3D10), anti-Ly49I (YLI-90), anti-CD94 (18d3), anti-NKG2AB6 (16a11), anti-IFN-γ (XMG1.2), anti-CD122 (5H4), anti-CD127 (SB/199), anti-CD69 (H1.2F3), anti-2B4 (2B4), anti-NKG2D (CX5) and Fixable Viability Dye eFluor 506. The following antibodies and reagents were purchased from BD Biosciences: anti-Ly49F (HBF-719), anti-Ly49G2 (4D11), and streptavidin (SA)-phycoerythrin. The following antibodies and reagents were purchased from BioLegend: anti-NK1.1 (PK136), anti-H2Dd (34-2-12) and SA-allophycocyanin. Anti-Ly49I (YLI-90) was purchased from Abcam. Anti-Ly49A (JR9) was purified in our laboratory from hybridoma supernatants and subsequently conjugated to biotin. The JR9 hybridoma was generously provided by Jacques Roland (Pasteur Institute, Paris, France) (Roland and Cazenave, 1992). Anti-Ly49C (4LO3311) (Brennan et al., 1996) was purified in our laboratory from hybridoma supernatants and subsequently conjugated to biotin. The 4LO3311 hybridoma was generously provided by Suzanne Lemieux (Institut National de la Recherche Scientifique-Institut Armand-Frappier, Laval, Quebec, Canada). Anti-NK1.1 (PK136) was purified in our laboratory from hybridoma supernatants. The PK136 hybridoma was purchased from American Type Culture Collection. Fc receptor blocking was performed with 2.4G2 (anti-FcgRII/III) hybridoma (American Type Culture Collection) culture supernatants. Surface staining was performed on ice in staining buffer (1% BSA and 0.01% NaN3 in PBS). Samples were collected using a FACSCanto (BD Biosciences), and data were analyzed using FlowJo v10.1 for Windows (BD Biosciences).
In vivo cytotoxicity
Mice used for donor splenocytes in in vivo cytotoxicity assays were 8–12 weeks old at the time of transfer. WT-MCMV, where indicated, was inoculated 3 days prior at a dose of 10,000 pfu per mouse (Parikh et al., 2015b). Donor splenocytes were harvested and labeled in vitro with 2.5 μM CFSE (Thermo Fisher) and 5 or 0.2 μM CellTrace violet (CT violet; Thermo Fisher). Recipient mice were injected intravenously with 2 × 106 of each donor. Spleens from recipient mice were harvested 3 (Figure 5B), 24 (Figure 5C) or 48 (Figure 4C; Figure S4) h after transfer of donor cells. NK cell-specific rejection was calculated by gating on transferred CFSE-positive cells and excluding dead cells by forward scatter and side scatter. Rejection was quantified as Killing Percentage = [1 – (Target/Control)/(Target/Control)Average(NK depleted)] × 100, where the target was the MHC-deficient (H2Dd−/−)or heterozygous (H2Dd+/−) donor cell and the control was a H2Dd+/+ donor cell. The ratio of target to control cells was normalized to the average ratio recovered from NK cell-depleted mice to calculate rejection by NK cells.
NK cell stimulation and intracellular staining
Splenocytes were stimulated with anti-NK1.1 (PK136) as previously described (Kim et al., 2005). Briefly, 24-well culture plates were coated with 500 μL of purified PK136 (1 μg/mL). Plates were washed with PBS, and 5 × 106 splenocytes were then added to each well in 500 μL of R10 (RPM1 1640 supplemented with 10% fetal bovine serum) media. Splenocytes were stimulated in parallel with 0.5 μg/mL PMA (Sigma-Aldrich) and 4 μg/mL ionomycin (Sigma-Aldrich) and incubated at 37°C and in 5% CO2 for a total of 7 h. Brefeldin A (GolgiPlug; BD Biosciences) was added to the cells after 1 h. After staining surface antigens, cells were fixed and permeabilized (Cytofix/Cytoperm; BD Biosciences) followed by staining for IFN-γ. NK cells were gated as viable CD3− CD19− NKp46+ lymphocytes.
Sequence analysis
PCR amplicons were amplified with the Phusion high-fidelity DNA polymerase (NEB, Ipswich, MA) using manufacturer recommended cycling conditions, column purified (Macherey-Nagel, Bethlehem, PA) and were sequenced on an ABI 3730 at Genewiz, Inc (South Plainfield, NJ, USA). The resulting chromatograms were aligned using SnapGene software (GSL Biotech, Chicago, IL) and the relevant reference sequences for MCMV (GenBank Accession GU305914.1) or the C57BL/6 (GRCm38/mm10). All oligonucleotides were synthesized by IDT (Coralville, IA).
QUANTIFICATION AND STATISTICAL ANALYSIS
Significance for all statistical tests was determined at p values < 0.05 and is shown as ****p < 0.0001, ***p < 0.001, **p < 0.01, *p < 0.05, ns = not significant in all figures. All data were assessed for normality using probability plots and the Kolmogorov-Smirnov test for normality (GraphPad Prism v8.2.1). Normal distributions of viral load, flow cytometry frequencies, and survival analysis were compared using either two-tailed Student’s t test (2 analyzed groups) or one-way ANOVA (> 2 analyzed groups) and corrected for multiple comparison using the Bonferroni method (GraphPad Prism v8.2.1). Killing percentages were treated as non-normal data and compared using Mann-Whitney U (2 analyzed groups) or Kruskal-Wallis (> 2 analyzed groups) tests and corrected for multiple comparison using Dunn’s method (GraphPad Prism v8.2.1). Error bars in all figures represent the standard error of the mean (SEM).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD19 Monoclonal Antibody (eBio1D3 (1D3)), FITC | eBioscience/Thermo Fisher Scientific | Cat# 11-0193-82; RRID: AB_657666 |
| CD3e Monoclonal Antibody (145-2C11), FITC | eBioscience/Thermo Fisher Scientific | Cat# 11-0031-82; RRID: AB_464882 |
| CD122 Monoclonal Antibody (TM-b1 (TM-beta1)), eFluor 450 | eBioscience/Thermo Fisher Scientific | Cat# 48-1222-82; RRID: AB_2016697 |
| CD314 (NKG2D) Monoclonal Antibody (CX5), PE | eBioscience/Thermo Fisher Scientific | Cat# 12-5882-82; RRID: AB_465996 |
| CD335 (NKp46) Monoclonal Antibody (29A1.4), PerCP-eFluor 710 | eBioscience/Thermo Fisher Scientific | Cat# 46-3351-80; RRID: AB_1834442 |
| Anti-Mouse Ly49H (3D10) APC antibody | eBioscience/Thermo Fisher Scientific | Cat# 17-5886-80; RRID: AB_10596369 |
| Rat Anti-Mouse CD127 (SB/199) Monoclonal Antibody, PE-Cy7 | BD Biosciences | Cat# 560733; RRID: AB_1727424 |
| Anti-mouse NK-1.1 (PK136) antibody, Brilliant Violet 650 | Biolegend | Cat# 108736; RRID: AB_2563159 |
| Anti-mouse NK-1.1 (PK136) antibody, PerCP-Cy5.5 | eBioscience/Thermo Fisher Scientific | Cat# 45-5941-82; RRID:AB_914361 |
| Anti-mouse NKG2A B6, Biotin | eBioscience/Thermo Fisher Scientific | Cat# 13-5897-85; RRID: AB_466781 |
| Anti-mouse NKG2D (CD314, CX5), PE | eBioscience/Thermo Fisher Scientific | Cat# 12-5882-82; RRID: AB_465996 |
| Anti-mouse Ly49F (HBF-719), PE | Thermo Fisher Scientific | Cat# MLY49F04; RRID: AB_2539785 |
| Anti-Mouse Ly49E/F PerCP-eFluor 710 | eBioscience/Thermo Fisher Scientific | Cat# 46-5848-80; RRID: AB_2016642 |
| Anti-Mouse Ly49A (JR9), Biotin | Hybridoma provided by Jacques Roland (Roland and Cazenave, 1992) | Biotin labeled in lab |
| Anti-Mouse Ly49C (4LO3311), Biotin | Hybridoma provided by Suzanne Lemieux (Brennan et al., 1996) | Biotin labeled in lab |
| Purified anti-mouse NK1.1 antibody (PK136) | Purified from PK136 Hybridoma, American Type Culture Collection, ATCC | Cat# HB-191; RRID: CVCL_7695 |
| Fc Receptor blocking antibody, 2.4G2 | Purified from 2.4G2 Hybridoma, American Type Culture Collection, ATCC | Cat# HB-197; RRID: CVCL_9148 |
| Anti-mouse CD27 Antibody (LG.7F9), APC | eBioscience/Thermo Fisher Scientific | Cat# 17-0271-82; RRID: AB_469370 |
| Anti-mouse CD11b Antibody (M1/70), eFluor 450 | eBioscience/Thermo Fisher Scientific | Cat# 48-0112-82; RRID: AB_1582236 |
| Anti-mouse CD69 Antibody (H1.2F3), eFluor 450 | eBioscience/Thermo Fisher Scientific | Cat# 48-0691-82; RRID: AB_10719430 |
| Anti-mouse Ly49G2 Antibody (4D11), APC | BD Biosciences | Cat# 555316; RRID: AB_398590 |
| Anti-mouse Ly49D Antibody (4E5), APC | eBioscience/Thermo Fisher Scientific | Cat# 17-5782-82; RRID: AB_10717290 |
| Anti-mouse Ly49I Antibody (YLI-90), Biotin | Abcam | Cat# ab25203; RRID: AB_448709 |
| Anti-mouse CD94 Antibody (18d3), eFluor 450 | eBioscience/Thermo Fisher Scientific | Cat# 48-0941-82; RRID: AB_11218905 |
| Anti-mouse CD244.2 (2B4) Antibody (eBio244F4), PE-Cy7 | eBioscience/Thermo Fisher Scientific | Cat# 25-2441-82; RRID: AB_2573432 |
| Anti-mouse IFN gamma Antibody (XMG1.2), eFluor 450 | eBioscience/Thermo Fisher Scientific | Cat# 48-7311-82; RRID: AB_1834366 |
| Anti-mouse H-2Dd antibody (34-2-12), PE | Biolegend | Cat# 110608; RRID: AB_313489 |
| Anti-mouse H-2Dd antibody (34-2-12), Alexa Fluor 647 | Biolegend | Cat# 110612; RRID: AB_492913 |
| Anti-mouse CD8a antibody (53-6.7), FITC | eBioscience/Thermo Fisher Scientific | Cat# 11-0081-86; RRID: AB_464917 |
| Streptavidin-Phycoerythrin | BD Biosciences | Cat# 554061; RRID: AB_10053328 |
| Streptavidin-Allophycocyanin | Biolegend | Cat# 405207 |
| Purified Ly49G (LGL-1) antibody | Purified from hybridoma Rheumatic Diseases Core Center Protein Purification and Production Facility (Washington University) | (Mason et al., 1988) |
| Purified Ly49D (4E4) antibody | Purified from hybridoma Rheumatic Diseases Core Center Protein Purification and Production Facility (Washington University) | (Idris et al., 1999) |
| Purified Ly49H (3D10) antibody | Purified from hybridoma Rheumatic Diseases Core Center Protein Purification and Production Facility (Washington University) | (Smith et al., 2000) |
| Purified CD8α (YTS-169) antibody | BioXCell | Cat# BE0117, RRID:AB_10950145 |
| Bacterial and Virus Strains | ||
| Δm157 MCMV | (Parikh et al., 2015b) | N/A |
| Δm157 MCMV, green fluorescent protein (GFP) reporter | (Henry et al., 2000) | N/A |
| Δm157/Δm06 MCMV (GFP) | This study | N/A |
| Δm157/Δm0152 MCMV (GFP) | This study | N/A |
| Δm157/Δm06/Δm0152 MCMV (GFP) | This study | N/A |
| Δm157/Δm06/Δm0152 MCMV | This study | N/A |
| Wild-type murine cytomegalovirus (MCMV) | (Cheng et al., 2010) | Genebank Accession GU305914 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Fixable Viability Dye eFluor 506 | eBioscience | Cat# 65-0866-14 |
| Phorbol 12-myristate 13-acetate (PMA) | Sigma-Aldrich | Cat# P8139 |
| Ionomycin, calcium salt | Sigma-Aldrich | Cat# I0634 |
| Cytofix/perm kit with Brefeldin A (GolgiPlug) | BD Biosciences | Cat# 555028 |
| Critical Commercial Assays | ||
| mMESSAGE mMACHINE T7 Ultra Transcription Kit | Thermo Fisher Scientific | Cat# AM1345 |
| MEGAclear Transcription Clean-Up Kit | Thermo Fisher Scientific | Cat# AM1908 |
| CellTrace CFSE Cell Proliferation Kit | Thermo Fisher Scientific | Cat# C34554 |
| CellTrace Violet Cell Proliferation Kit | Thermo Fisher Scientific | Cat# C34557 |
| MEGAshortscript T7 Transcription Kit | Thermo Fisher Scientific | Cat# AM1354 |
| Experimental Models: Cell Lines | ||
| Cells: BALB/3T12-3 fibroblasts | American Type Culture Collection, ATCC | Cat# CCL-164, RRID:CVCL_0637 |
| Cells: SV40-immortalized B6 murine embryonic fibroblasts (MEFs) | This study | E12.5 MEFs from C57BL/6NCrl mice |
| Cells: SV40-immortalized D8-KODO MEFs | This study | E12.5 MEFs from D8-KODO mice |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6NCrl | Charles River Laboratories | Cat#027, RRID:IMSR_CRL:027 |
| Mouse: BALB/cAnNCrl | Charles River Laboratories | Cat# CRL:028, RRID:IMSR_CRL:028 |
| Mouse: Klra7em1(IMPC)J (Ly49G KO) | The Jackson Laboratory (now at MMRRC) | Cat# MMRRC:048353-UCD |
| Mouse: C57BL/10SnJ (B10) | The Jackson Laboratory | IMSR Cat# JAX:000666, RRID:IMSR_JAX:000666 |
| Mouse: B10.D2/nSnJ (B10.D2) | The Jackson Laboratory | IMSR Cat# JAX:000463, RRID:IMSR_JAX:000463 |
| Mouse: H2Kb−/− H2Db−/− (KODO) | Taconic Farms | IMSR Cat# TAC:4215, RRID:IMSR_TAC:4215 |
| Mouse: Prf1−/− | The Jackson Laboratory | IMSR Cat# JAX:002407, RRID:IMSR_JAX:002407 |
| Mouse: Rag1−/− | The Jackson Laboratory | IMSR Cat# JAX:002216, RRID:IMSR_JAX:002216 |
| Mouse: DAP10−/−, DAP12−/− | Provided by T. Takai | (Inui et al., 2009) |
| Mouse: NKG2D−/− (KLRK1 KO) | Provided by Bojan Polic (University of Rijeka, Croatia) | (Zafirova et al., 2009) |
| Mouse: H2Kb−/− H2Db−/−, β2 m−/− | Obtained from Ted Hansen (Washington University, St. Louis) | N/A |
| Mouse: D8-B6 (transgenic mice expressing H2Dd) | Provided by D. Marguiles (National Institute of Allergy and Infectious Diseases, Bethesda, MD) | N/A |
| Mouse: D8-KODO (H2Kb−/− H2Db−/− mice expressing H2Dd transgene) | Previously generated in lab | (Choi et al., 2011) |
| Mouse: GzmB−/− (Granzyme B KO) | Obtained from T. Ley (Washington University, St. Louis) | (Heusel et al., 1994) |
| Mouse: D8-KODO AYF (ITIM mutant of Ly49A) | Previously generated in lab | (Bern et al., 2017) |
| Mouse: Ly49A−/− (strain A15) | This study | N/A |
| Mouse: Ly49A−/− (strain A20) | This study | N/A |
| Mouse: Ly49C−/−, Ly49I−/− (strain C14B) | This study | N/A |
| Mouse: Ly49M−/− (strains M1, M4, M7, M8) | This study | N/A |
| Mouse: Ly49A−/−, Ly49G−/− (strain AG1) | This study | N/A |
| Mouse: Ly49A−/−, Ly49G−/− (strain AG7) | This study | N/A |
| Mouse: Ly49A−/−, Ly49C−/− Ly49H−/−, Ly49G−/− (strain ΔLy49-1) | This study | N/A |
| Mouse: Ly49A knockin into NKp46 locus (Ly49A-KI) | This study | N/A |
| Oligonucleotides | ||
| Guide RNA for MCMV disruption | See Table S3 | All oligonucleotides were synthesized by IDT (Coralville, IA) |
| Primers for Ly49 sequence analysis | See Table S4 | All oligonucleotides were synthesized by IDT (Coralville, IA) |
| Primers for MCMV sequence analysis | See Table S5 | All oligonucleotides were synthesized by IDT (Coralville, IA) |
| Guide RNA for Ly49 disruption | See Table S1 | All oligonucleotides were synthesized by IDT (Coralville, IA) |
| Primers for MCMV quantification | Generated previously in lab | (Parikh et al., 2015b) |
| Recombinant DNA | ||
| p2A-Ly49A cDNA cassette | This study | GeneBlock synthesized at IDT and inserted into NKp46 genomic DNA in pACYC177 |
| pCDNA3.1 Mammalian Expression Vector | Thermo Fisher Scientific | Cat# V79020 |
| Plasmid pX330-U6-Chimeric_BB-CBh-hSpCas9 | (Ran et al., 2013) | RRID:Addgene_42230 |
| SV401: pBSSVD2005 | Addgene | RRID:Addgene_21826 |
| Plasmid pACYC177 | New England Biolabs | Cat # E4151 |
| Software and Algorithms | ||
| SnapGene® software | GSL Biotech | Version 4.3 |
| FlowJo Software | BD Biosciences | v10.1 for Windows |
| CCTop | https://crispr.cos.uni-heidelberg.de/ | (Stemmer et al., 2015) |
| ABI StepOnePlus qPCR software | Applied Biosystems | Version 2.3 |
| GraphPad Prism | GraphPad Software | Version 8.2.1 for Windows |
| GT-Scan | https://gt-scan.csiro.au/gt-scan | (O’Brien and Bailey, 2014) |
Highlights.
MHC-I-restricted control of MCMV infection requires specific Ly49 receptors
The Ly49 receptors mediate NK cell licensing (education) and missing-self rejection
Ly49-mediated control requires viral open reading frames that downregulate MHC-I
ACKNOWLEDGMENTS
We thank J. Michael White (Transgenic, Knockout, and Micro-Injection Core at Washington University) for CRISPR-Cas9 injections and Andrew Cao, Trenton J. (T.J.) Dawson, and Darryl Higuchi for technical assistance. Experimental support was provided by the Protein Production and Purification Core Facility. This work was supported by National Institutes of Health grants R01AI129545 and R01AI131680 (to W.M.Y.) and K08-AI104991 (to B.A.P.)
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j. celrep.2020.107969.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Arase H, Mocarski ES, Campbell AE, Hill AB, and Lanier LL (2002). Direct recognition of cytomegalovirus by activating and inhibitory NK cell receptors. Science 296, 1323–1326. [DOI] [PubMed] [Google Scholar]
- Babić M, Pyzik M, Zafirova B, Mitrović M, Butorac V, Lanier LL, Krmpotić A, Vidal SM, and Jonjić S (2010). Cytomegalovirus immunoevasin reveals the physiological role of “missing self” recognition in natural killer cell dependent virus control in vivo. J. Exp. Med 207, 2663–2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bern MD, Beckman DL, Ebihara T, Taffner SM, Poursine-Laurent J, White JM, and Yokoyama WM (2017). Immunoreceptor tyrosine-based inhibitory motif-dependent functions of an MHC class I-specific NK cell receptor. Proc. Natl. Acad. Sci. USA 114, E8440–E8447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bern MD, Parikh BA, Yang L, Beckman DL, Poursine-Laurent J, and Yokoyama WM (2019). Inducible down-regulation of MHC class I results in natural killer cell tolerance. J. Exp. Med 216, 99–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieberich C, Scangos G, Tanaka K, and Jay G (1986). Regulated expression of a murine class I gene in transgenic mice. Mol. Cell. Biol 6, 1339–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowness P (2015). HLA-B27. Annu. Rev. Immunol 33, 29–48. [DOI] [PubMed] [Google Scholar]
- Brennan J, Lemieux S, Freeman JD, Mager DL, and Takei F (1996). Heterogeneity among Ly-49C natural killer (NK) cells: characterization of highly related receptors with differing functions and expression patterns. J. Exp. Med 184, 2085–2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MG, Dokun AO, Heusel JW, Smith HRC, Beckman DL, Blattenberger EA, Dubbelde CE, Stone LR, Scalzo AA, and Yokoyama WM (2001). Vital involvement of a natural killer cell activation receptor in resistance to viral infection. Science 292, 934–937. [DOI] [PubMed] [Google Scholar]
- Cheng TP, Valentine MC, Gao J, Pingel JT, and Yokoyama WM (2010). Stability of murine cytomegalovirus genome after in vitro and in vivo passage. J. Virol 84, 2623–2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi T, Ferris ST, Matsumoto N, Poursine-Laurent J, and Yokoyama WM (2011). Ly49-dependent NK cell licensing and effector inhibition involve the same interaction site on MHC ligands. J. Immunol 186, 3911–3917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, and Zhang F (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbett AJ, Coudert JD, Forbes CA, and Scalzo AA (2011). Functional consequences of natural sequence variation of murine cytomegalovirus m157 for Ly49 receptor specificity and NK cell activation. J. Immunol 186, 1713–1722. [DOI] [PubMed] [Google Scholar]
- Daher M, and Rezvani K (2018). Next generation natural killer cells for cancer immunotherapy: the promise of genetic engineering. Curr. Opin. Immunol 51, 146–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dam J, Guan R, Natarajan K, Dimasi N, Chlewicki LK, Kranz DM, Schuck P, Margulies DH, and Mariuzza RA (2003). Variable MHC class I engagement by Ly49 natural killer cell receptors demonstrated by the crystal structure of Ly49C bound to H-2K(b). Nat. Immunol 4, 1213–1222. [DOI] [PubMed] [Google Scholar]
- de Andrade LF, Smyth MJ, and Martinet L (2014). DNAM-1 control of natural killer cells functions through nectin and nectin-like proteins. Immunol. Cell Biol 92, 237–244. [DOI] [PubMed] [Google Scholar]
- Depatie C, Muise E, Lepage P, Gros P, and Vidal SM (1997). High-resolution linkage map in the proximity of the host resistance locus Cmv1. Genomics 39, 154–163. [DOI] [PubMed] [Google Scholar]
- Diefenbach A, Tomasello E, Lucas M, Jamieson AM, Hsia JK, Vivier E, and Raulet DH (2002). Selective associations with signaling proteins determine stimulatory versus costimulatory activity of NKG2D. Nat. Immunol 3, 1142–1149. [DOI] [PubMed] [Google Scholar]
- Gamache A, Cronk JM, Nash WT, Puchalski P, Gillespie A, Wei H, Gray L, Hammarskjold ML, Xu W, and Brown MG (2019). Ly49R activation receptor drives self-MHC-educated NK cell immunity against cytomegalovirus infection. Proc. Natl. Acad. Sci. USA 116, 26768–26778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillamón CF, Martínez-Sánchez MV, Gimeno L, Mrowiec A, Martínez-García J, Server-Pastor G, Martínez-Escribano J, Torroba A, Ferri B, Abellán D, et al. (2018). NK cell education in tumor immune surveillance: DNAM-1/KIR receptor ratios as predictive biomarkers for solid tumor outcome. Cancer Immunol. Res 6, 1537–1547. [DOI] [PubMed] [Google Scholar]
- Hanke T, Takizawa H, McMahon CW, Busch DH, Pamer EG, Miller JD, Altman JD, Liu Y, Cado D, Lemonnier FA, et al. (1999). Direct assessment of MHC class I binding by seven Ly49 inhibitory NK cell receptors. Immunity 11, 67–77. [DOI] [PubMed] [Google Scholar]
- Henry SC, Schmader K, Brown TT, Miller SE, Howell DN, Daley GG, and Hamilton JD (2000). Enhanced green fluorescent protein as a marker for localizing murine cytomegalovirus in acute and latent infection. J. Virol. Methods 89, 61–73. [DOI] [PubMed] [Google Scholar]
- Heusel JW, Wesselschmidt RL, Shresta S, Russell JH, and Ley TJ (1994). Cytotoxic lymphocytes require granzyme B for the rapid induction of DNA fragmentation and apoptosis in allogeneic target cells. Cell 76, 977–987. [DOI] [PubMed] [Google Scholar]
- Higuchi DA, Cahan P, Gao J, Ferris ST, Poursine-Laurent J, Graubert TA, and Yokoyama WM (2010). Structural variation of the mouse natural killer gene complex. Genes Immun. 11, 637–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu KM, Pratt JR, Akers WJ, Achilefu SI, and Yokoyama WM (2009). Murine cytomegalovirus displays selective infection of cells within hours after systemic administration. J. Gen. Virol 90, 33–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Idris AH, Smith HR, Mason LH, Ortaldo JR, Scalzo AA, and Yokoyama WM (1999). The natural killer gene complex genetic locus Chok encodes Ly-49D, a target recognition receptor that activates natural killing. Proc. Natl. Acad. Sci. USA 96, 6330–6335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inui M, Kikuchi Y, Aoki N, Endo S, Maeda T, Sugahara-Tobinai A, Fujimura S, Nakamura A, Kumanogoh A, Colonna M, and Takai T (2009). Signal adaptor DAP10 associates with MDL-1 and triggers osteoclastogenesis in cooperation with DAP12. Proc. Natl. Acad. Sci. USA 106, 4816–4821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson AH, Yang L, Kim S, Taffner SM, and Yokoyama WM (2010). Effects of MHC class I alleles on licensing of Ly49A+ NK cells. J. Immunol 184, 3424–3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlhofer FM, Ribaudo RK, and Yokoyama WM (1992). MHC class I alloantigen specificity of Ly-49+ IL-2-activated natural killer cells. Nature 358, 66–70. [DOI] [PubMed] [Google Scholar]
- Kärre K, Ljunggren HG, Piontek G, and Kiessling R (1986). Selective rejection of H-2-deficient lymphoma variants suggests alternative immune defence strategy. Nature 319, 675–678. [DOI] [PubMed] [Google Scholar]
- Kielczewska A, Pyzik M, Sun T, Krmpotic A, Lodoen MB, Munks MW, Babic M, Hill AB, Koszinowski UH, Jonjic S, et al. (2009). Ly49P recognition of cytomegalovirus-infected cells expressing H2-Dk and CMV-encoded m04 correlates with the NK cell antiviral response. J. Exp. Med 206, 515–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Poursine-Laurent J, Truscott SM, Lybarger L, Song YJ, Yang L, French AR, Sunwoo JB, Lemieux S, Hansen TH, and Yokoyama WM (2005). Licensing of natural killer cells by host major histocompatibility complex class I molecules. Nature 436, 709–713. [DOI] [PubMed] [Google Scholar]
- Kleijnen MF, Huppa JB, Lucin P, Mukherjee S, Farrell H, Campbell AE, Koszinowski UH, Hill AB, and Ploegh HL (1997). A mouse cytomegalovirus glycoprotein, gp34, forms a complex with folded class I MHC molecules in the ER which is not retained but is transported to the cell surface. EMBO J. 16, 685–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ljunggren HG, and Kärre K (1990). In search of the ‘missing self’: MHC molecules and NK cell recognition. Immunol. Today 11, 237–244. [DOI] [PubMed] [Google Scholar]
- Long EO, Kim HS, Liu D, Peterson ME, and Rajagopalan S (2013). Controlling natural killer cell responses: integration of signals for activation and inhibition. Annu. Rev. Immunol 31, 227–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mans J, Natarajan K, Balbo A, Schuck P, Eikel D, Hess S, Robinson H, Simic H, Jonjic S, Tiemessen CT, and Margulies DH (2007). Cellular expression and crystal structure of the murine cytomegalovirus major histocompatibility complex class I-like glycoprotein, m153. J. Biol. Chem 282, 35247–35258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason L, Giardina SL, Hecht T, Ortaldo J, and Mathieson BJ (1988). LGL-1: a non-polymorphic antigen expressed on a major population of mouse natural killer cells. J. Immunol 140, 4403–4412. [PubMed] [Google Scholar]
- McQueen KL, Lohwasser S, Takei F, and Mager DL (1999). Expression analysis of new Ly49 genes: most transcripts of Ly49j lack the transmembrane domain. Immunogenetics 49, 685–691. [DOI] [PubMed] [Google Scholar]
- Nabekura T, Gotthardt D, Niizuma K, Trsan T, Jenus T, Jonjic S, and Lanier LL (2017). Cutting edge: NKG2D signaling enhances NK cell responses but alone is insufficient to drive expansion during mouse cytomegalovirus infection. J. Immunol 199, 1567–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien A, and Bailey TL (2014). GT-scan: identifying unique genomic targets. Bioinformatics 30, 2673–2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr MT, Murphy WJ, and Lanier LL (2010). ‘Unlicensed’ natural killer cells dominate the response to cytomegalovirus infection. Nat. Immunol 11, 321–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh BA, Beckman DL, Patel SJ, White JM, and Yokoyama WM (2015a). Detailed phenotypic and molecular analyses of genetically modified mice generated by CRISPR-Cas9-mediated editing. PLoS ONE 10, e0116484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh BA, Piersma SJ, Pak-Wittel MA, Yang L, Schreiber RD, and Yokoyama WM (2015b). Dual requirement of cytokine and activation receptor triggering for cytotoxic control of murine cytomegalovirus by NK cells. PLoS Pathog. 11, e1005323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto AK, Munks MW, Koszinowski UH, and Hill AB (2006). Coordinated function of murine cytomegalovirus genes completely inhibits CTL lysis. J. Immunol 177, 3225–3234. [DOI] [PubMed] [Google Scholar]
- Pyzik M, Charbonneau B, Gendron-Pontbriand EM, Babić M, Krmpotić A, Jonjić S, and Vidal SM (2011). Distinct MHC class I-dependent NK cell-activating receptors control cytomegalovirus infection in different mouse strains. J. Exp. Med 208, 1105–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyzik M, Dumaine A, Charbonneau B, Fodil-Cornu N, Jonjic S, and Vidal SM (2014). Viral MHC class I-like molecule allows evasion of NK cell effector responses in vivo. J. Immunol 193, 6061–6069. [DOI] [PubMed] [Google Scholar]
- Rahim MMA, and Makrigiannis AP (2015). Ly49 receptors: evolution, genetic diversity, and impact on immunity. Immunol. Rev 267, 137–147. [DOI] [PubMed] [Google Scholar]
- Rahim MMA, Chen P, Mottashed AN, Mahmoud AB, Thomas MJ, Zhu Q, Brooks CG, Kartsogiannis V, Gillespie MT, Carlyle JR, and Makrigiannis AP (2015). The mouse NKR-P1B:Clr-b recognition system is a negative regulator of innate immune responses. Blood 125, 2217–2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat. Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reusch U, Muranyi W, Lucin P, Burgert HG, Hengel H, and Koszinowski UH (1999). A cytomegalovirus glycoprotein re-routes MHC class I complexes to lysosomes for degradation. EMBO J. 18, 1081–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roland J, and Cazenave PA (1992). Ly-49 antigen defines an alpha beta TCR population in i-IEL with an extrathymic maturation. Int. Immunol 4, 699–706. [DOI] [PubMed] [Google Scholar]
- Silver ET, Lavender KJ, Gong D-E, Hazes B, and Kane KP (2002). Allelic variation in the ectodomain of the inhibitory Ly-49G2 receptor alters its specificity for allogeneic and xenogeneic ligands. J. Immunol 169, 4752–4760. [DOI] [PubMed] [Google Scholar]
- Smith HR, Karlhofer FM, and Yokoyama WM (1994). Ly-49 multigene family expressed by IL-2-activated NK cells. J. Immunol 153, 1068–1079. [PubMed] [Google Scholar]
- Smith HR, Chuang HH, Wang LL, Salcedo M, Heusel JW, and Yokoyama WM (2000). Nonstochastic coexpression of activation receptors on murine natural killer cells. J. Exp. Med 191, 1341–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith HRC, Heusel JW, Mehta IK, Kim S, Dorner BG, Naidenko OV, Iizuka K, Furukawa H, Beckman DL, Pingel JT, et al. (2002). Recognition of a virus-encoded ligand by a natural killer cell activation receptor. Proc. Natl. Acad. Sci. U S A 99, 8826–8831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stemmer M, Thumberger T, Del Sol Keyer M, Wittbrodt J, and Mateo JL (2015). CCTop: an intuitive, flexible and reliable CRISPR/Cas9 target prediction tool. PLoS ONE 10, e0124633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun K, Alvarez M, Ames E, Barao I, Chen M, Longo DL, Redelman D, and Murphy WJ (2012). Mouse NK cell-mediated rejection of bone marrow allografts exhibits patterns consistent with Ly49 subset licensing. Blood 119, 1590–1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trowsdale J, and Knight JC (2013). Major histocompatibility complex genomics and human disease. Annu. Rev. Genomics Hum. Genet 14, 301–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner M, Gutermann A, Podlech J, Reddehase MJ, and Koszinowski UH (2002). Major histocompatibility complex class I allele-specific cooperative and competitive interactions between immune evasion proteins of cytomegalovirus. J. Exp. Med 196, 805–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X, Stadnisky MD, and Brown MG (2009). MHC class I Dk locus and Ly49G2+ NK cells confer H-2k resistance to murine cytomegalovirus. J. Immunol 182, 7163–7171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama WM, and Plougastel BFM (2003). Immune functions encoded by the natural killer gene complex. Nat. Rev. Immunol 3, 304–316. [DOI] [PubMed] [Google Scholar]
- Zafirova B, Mandarić S, Antulov R, Krmpotić A, Jonsson H, Yokoyama WM, Jonjić S, and Polić B (2009). Altered NK cell development and enhanced NK cell-mediated resistance to mouse cytomegalovirus in NKG2D-deficient mice. Immunity 31, 270–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Feng J, Chen S, Yang H, and Dong Z (2019). Synergized regulation of NK cell education by NKG2A and specific Ly49 family members. Nat. Commun 10, 5010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler H, Thale R, Lucin P, Muranyi W, Flohr T, Hengel H, Farrell H, Rawlinson W, and Koszinowski UH (1997). A mouse cytomegalovirus glycoprotein retains MHC class I complexes in the ERGIC/cis-Golgi compartments. Immunity 6, 57–66. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The published article includes all data generated or analyzed during this study. Original source data for Figures in the paper are available upon request to the Lead Contact author. No proprietary software was used in the data analysis.