

Abstract
Monoamine oxidase B (MAO-B) is a potential biomarker for Parkinson’s disease (PD), a neurodegenerative disease associated with the loss of motor activities in human subjects. The disease state is associated with dopamine deprival, and so the inhibitors of MAO-B can serve as therapeutic drugs for PD. Since the expression level of MAO-B directly correlates to the disease progress, the distribution and population of this enzyme can be employed to monitor disease development. One of the approaches available for estimating the population is two-photon imaging. The ligands used for two-photon imaging should have high binding affinity and binding specificity toward MAO-B along with significant two-photon absorption cross sections when they are bound to the target. In this article, we study using a multiscale modeling approach, the binding affinity and spectroscopic properties (one- and two-photon absorption) of three (Flu1, Flu2, Flu3) of the currently available probes for monitoring the MAO-B level. We report that the binding affinity of the probes can be explained using the molecular size and binding cavity volume. The experimentally determined one-photon absorption spectrum is well reproduced by the employed QM/MM approaches, and the most accurate spectral shifts, on passing from one probe to another, are obtained at the coupled-cluster (CC2) level of theory. An important conclusion from this study is also the demonstration that intrinsic molecular two-photon absorption strengths (δ2PA) increase in the order δ2PA (Flu1) > δ2PA (Flu2) > δ2PA (Flu3). This is in contrast with experimental data, which predict similar values of two-photon absorption cross sections for Flu1 and Flu3. We demontrate, based on the results of electronic-structure calculations for Flu1 that this discrepancy cannot be explained by an explicit account for neighboring residues (which could lead to charge transfer between a probe and neighboring aromatic amino acids thus boosting δ2PA). In summary, we show that the employed multiscale approach not only can optimize two-photon absorption properties and verify binding affinity, but it can also help in detailed analyses of experimental data.
Introduction
It is known in disease pathology that aberrant expression of certain biomolecules can be used as a key parameter to diagnose a specific disease. The reason behind this is that the cells follow newer metabolic pathways when compared to normal disease-free condition and these activities naturally involve expression of one or more enzymes which are then indicative of the disease condition and so can be used as disease biomarkers.1 Biomarkers can be identified by doing comparative protein profiling of the samples collected from ill and normal subjects. When it comes to Parkinson’s disease (PD), which is a neurodegenerative disease associated with the loss of motor activities, monamine oxidase B (MAO-B) serves as a potential biomarker. There exist a number of other biomarkers such as proteasome and caspase components or alpha-synuclein fibrils.2 MAO-B is an enzyme made of 500 amino acids and requires FAD cofactor for its functional activities. It is involved in the oxidation of neurotransmitters such as dopamine, phenethylamine, and benzylamine. It is mostly expressed in brain and, to some extent, in other human organs such as liver, kidney, heart, and lungs.3 In brain it is expressed largely in serotonergic neurons as well as in non-neuronal cells such as astrocytes and radial glia cells.3,4 Studies using autopsied brain homogenates showed 83% increase of MAO-B in the case of patients with PD which is again an indication that MAO-B is a potential biomarker for this disease.5,6
In most of the cases the biomarker for a specific disease can as well serve as a drug target for the same disease. Likewise, MAO-B serves as a potential drug target for PD. The inhibition of MAO-B with drugs such as safinamide and rasagiline improves the symptoms of PD. This disease is associated with deprival of dopamine which is connected to motor activities, memory, and motivation. The MAO-B inhibition improves the dopamine deficient state and contributes to improvement in motor function. Considering the paramount importance associated with the therapeutic and diagnostic values of MAO-B, numerous inhibitor molecules and diagnostic agent were developed, and there is also considerable effort to design ligands with high specificity and binding affinity for this target. In particular the diagnostic agents involve the ligands which can be used for optical and PET imaging of MAO-B.
The general mechanism behind the inhibition of MAO-B is through the binding of the ligands to the substrate cavity. In fact, the binding site is divided into a substrate binding site (with cavity volume 490 Å3) and entrance cavity (with a cavity volume of 290 Å3) and these two domains are separated by a loop made of residues Tyr326, Leu171, Ile199, and Phe168. The residue Ile199 serves as a gate for the ligand binding and depending upon the state of binding it exists in an open or closed form. The inhibitors such as rasagiline, clorgyline, and selegyline covalently bind to FAD and occupy the substrate binding site. The irreversible inhibitors such as safinamide also bind to the same site.7,8 Recent studies by an author of this article also demonstrated that the tau PET tracers also target the same binding site as the MAO-B inhibitors.9,10 Moreover, we have found that the PET tracers such as SL25.118811 also bind to the same substrate binding site. It is also speculated that when the substrate site is not available due to the binding of irreversible MAO-B inhibitors or because the ligand molecular volume is larger than the cavity volume then the entrance cavity is the preferred site for ligand binding. Furthermore, there is an ongoing discussion on the availability of a new binding site referred to as the imidazolium binding site in MAO-B which is targeted by certain PET tracers such as BU99008.12
When it comes to diagnosis, one aims to estimate the population and distribution of MAO-B using certain ligands which can specifically bind to these target biomarkers. Due to the relative simplicity of imaging measurements and its cost-effectiveness, there are numerous molecules suitable for optical tracking of MAO-B that have been developed.13,14 Most of the optical probes exhibit turn on fluorescence upon binding to MAO-B. This means the original ligand is not fluorescent but it is converted into fluorescent analog due to the catalytic activity of the MAO-B enzyme.13 Although many probes based on fluorescence phenomenon are available for the MAO-B target only a limited number of two-photon probes have been reported for this target.15 Even though the enzymatic reaction behind the turn-on one- and two-photon properties is known, the binding pathway and mode of binding of such optical tracers are not understood clearly. In addition, it is not clear whether the MAO-B specific fluorescence and two-photon absorption are an intrinsic property of the probe or should be attributed to the bound state of the probe within the MAO-B target. Further it is not known whether the one- and two-photon optical probes bind to the same substrate binding site within MAO-B similar to MAO-B inhibitors.
Motivated by the significance of the subject, in this manuscript, the main focus is put on studying the ligands which light-up MAO-B through the two-photon absorption process. Such ligands should fulfill the following properties: (i) They bind with higher binding affinity and binding specificity to MAO-B target alone and this way only this specific biomarker is lighted up even when there are other biomolecules which can coexist along with this target. (ii) They exhibit significant two-photon absorption cross section in their MAO-B bound state or when they are converted into certain byproducts by the enzyme. Since the byproduct can be formed only in the presence of MAO-B, the two-photon absorption properties are pronounced only in the presence of this enzyme. As two-photon-active compound cannot be formed in the presence of other targets or in water solvent, there is no two-photon-excited fluorescence and the two-photon action cross section is a direct indicator of the presence or population of MAO-B. (iii) It is preferable that the process of two-photon excitation occurs due to the absorption of near-infrared or infrared radiation which has favorable cell penetration property. Even though there are reports of many one-photon probes for MAO-B, there are not many molecules reported in the literature which meet all the above requirements and could be used for two-photon imaging of MAO-B. It was reported for the first time in 2014 where a few fluorogenic molecules, labeled as Flu1, Flu2, and Flu3 (see Figure 1) were proposed for the real-time imaging of MAO-B activities in cells.15 In this contribution we have studied the binding affinity, one-photon absorption properties, and two-photon absorption cross sections for all three molecules in MAO-B. In order to locate the binding site for the three compounds in MAO-B, we have carried out the molecular docking study. Subsequently, the molecular dynamics simulations were carried out for all three fluorophore–MAO-B complexes. The trajectories were used for computing the binding free energies for the fluorophores using the molecular mechanics generalized Born surface area approach (MM-GBSA).16,17 The final snapshots from the molecular dynamics simulations were used as the input for carrying out hybrid QM/MM molecular dynamics simulations. Around 50 independent configurations from the ab initio molecular dynamics simulations were extracted for computing the one- and two-photon properties by employing TD-DFT/MM and CC2/MM levels of theory. In particular, we used an electrostatic embedding scheme which allows for the polarization of the QM region by the protein and solvent environment during the response calculation. In order to shed light on whether the two-photon enhancement is an intrinsic property of the molecule or dependent on the bound nature of the ligand to the enzyme, we have as well computed the two-photon properties of the ligand in the water solvent environment. We will present the details about the computational scheme in the next section and then present the results and discussion in the subsequent section.
Figure 1.
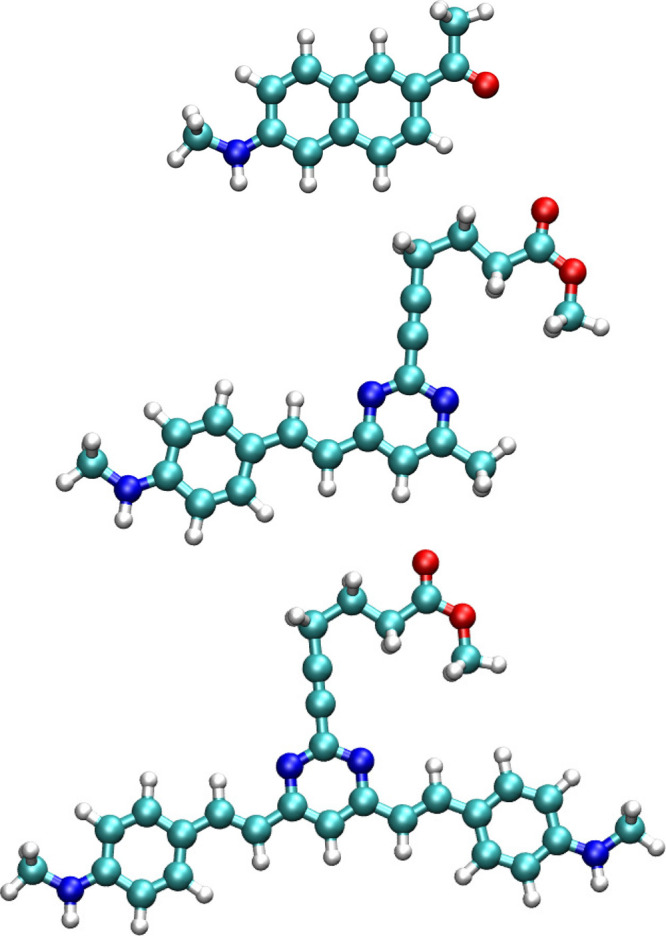
Molecular structure of Flu1 (top), Flu2 (middle), and Flu3 (bottom) compounds.
Computational Details
Various computational approaches were employed to estimate the binding affinity and one- and two-photon absorption properties of three molecules, Flu1, Flu2, and Flu3, in monoamine oxidase B. The initial structure for MAO-B is based on the PDB structure, 2V5Z, where the MAO-B structure is reported with cofactor FAD and ligand safinamide (see Figure 2).18 In the current study, safinamide was removed, but the cofactor FAD was kept as it is in the original structure. The molecular docking studies were carried out using Autodock 4.0 software19 to find the binding site for the compounds in MAO-B. The docking studies were carried out by including the FAD cofactor. A blind docking is carried out as we do not know a priori the binding site. The grid box dimensions were chosen as 120 × 120 × 120 (with default grid spacing of 0.375 Å) so that it can locate both surface and core binding sites within the enzyme. A Lamarckian-algorithm-based search was employed to find the high affinity binding modes for the fluorophores within the enzyme. The most stable complex structure obtained from molecular docking has been used as the input configuration for subsequent molecular dynamics calculations. The simulation box contained MAO-B, cofactor-FAD, and the ligands solvated with approximately 19000 water molecules. The MAO-B and FAD are negatively charged (total charge of MAO-B and FAD complex is −5), and so the whole complex was neutralized with 5 Na+ ions. The adopted simulation box was orthorhombic, and the dimensions are approximately 76, 97, and 92 Å in the case of Flu1 ligand. The electrostatic potential fitted charges obtained using the B3LYP/6-31G(d) level of theory and the GAFF force field20 were adopted for the fluorophores for carrying out molecular dynamics simulations. The FF99SB and TIP3P21 force-fields were used for describing the MAO-B enzyme and water solvents, respectively. The minimization and simulation in NVT and NPT ensembles were carried out in that order. Followed by the equilibration simulation of length scale 5 ns, a final production run in isothermal–isobaric ensemble for a total time scale of 50 ns has been carried out. To show that the 50 ns molecular dynamics simulation was sufficient, we have computed various structural and dynamical properties. In the Supporting Information in Figures S1(a) and (b) we show the RMSD and RMSF computed for three MAO-B:Flu1-Flu3 complexes. The former plot shows convergence in RMSD with time which is an indication that the systems are fairly equilibrated. Further, RMSF shows that except the tail ends the backbone atoms of all residues only show normal thermal fluctuations which is again an indication that the protein structure is fairly stable in all three cases investigated and it should not affect the optical properties. All the simulations were carried out using Amber software.22 The time step used for the integration of the equation of motion was 2 fs. Subsequently, the free energy calculations were carried out for all three MAO-B:fluorophore complexes to estimate the comparative binding affinity of the three compounds to the target. The 2500 configurations picked up from the trajectory of the last 10 ns were used for the free energy calculations. The binding free energies were estimated using a molecular mechanics–generalized Born surface area approach as provided by the MMPBSA.py toolbox.23 In order to estimate the one- and two-photon properties, we employed Car–Parrinello hybrid quantum mechanics/molecular mechanics (QM/MM) molecular dynamics and hybrid QM/MM response calculations at the coupled cluster-molecular mechanics (CC2/MM) level of theory. In the latter two sets of calculations, the fluorophores were treated at the QM level while the enzyme or solvent environment was treated using the molecular mechanics force-field. For Car–Parrinello molecular dynamics simulations, we have employed the BLYP functional which is computationally less demanding so that a sufficient length scale of ab initio MD can be achieved (up to few tens of ps). We have also shown in our previous works that geometries for various probes obtained using such a functional during ab initio MD successfully reproduced the one- and two-photon absorption spectra when compared to experiments.24−27 The time step used for solving the equation of motion was 5 au, and we have used a fictitious mass of 600 amu. These values are chosen based on our previous experience in doing hybrid QM/MM molecular dynamics simulation. Overall, molecular docking has been used to locate various binding sites for fluorophores in MAO-B. The stability of the fluorophore:enzyme complex has been addressed using molecular dynamics and MM-GBSA based free energy calculations. Finally the photophysical properties were studied using the hybrid CC2/MM level of theory.
Figure 2.
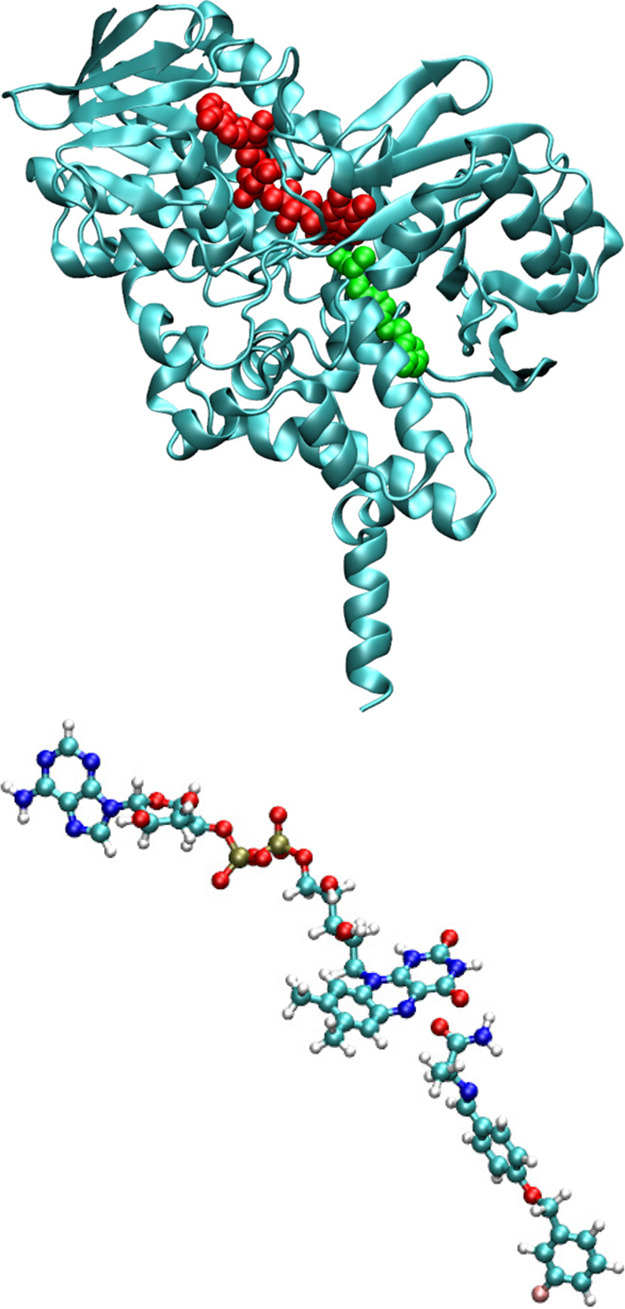
Crystal structure for MAO-B bound to a reversible inhibitor safinamide (top, PDB ID is 2v5z). The safinamide is shown in green while the cofactor FAD is shown in red. FAD–safinamide complex in a ball and stick model as it is in the crystal structure (bottom).
The two-photon strengths of Flu1, Flu2, and Flu3 were computed by employing quadratic response theory28−30 combined with TD-DFT and CC2 methods and the TZVP basis set of Ahlrichs et al.31 B3LYP32 and CAM-B3LYP33 functionals were employed to assess these widely used density functional approximations in simulations of spectroscopic properties (absorption maximum, two-photon absorption cross section, and shift in absorption wavelength on passing from Flu1 to Flu3). RI-CC2 calculations were performed using the TURBOMOLE program29,34 while TD-DFT calculations were performed using DALTON software.35
The rotationally averaged two-photon strength (δ2PA) is given by
| 1 |
The second-order transition moment matrix elements (Sαβ) appearing in the above expression are defined within the DFT framework as36,37
| 2 |
where 0, i, and f stand for the ground, intermediate, and final excited state and ℏω is the excitation energy to a given excited state. The two-photon absorption cross section was calculated based on the following formula:
| 3 |
where g(2ω) is the line shape function, a0 is the Bohr radius, α is the fine structure constant, and c is the speed of light. In this work we used the Lorentzian shape and the broadening parameter set 0.1 eV and estimated the two-photon cross sections in GM units.38 The two-photon transition probabilities for Flu1–Flu3 were computed for all snapshots, and given their similar values they were used to determine average two-photon absorption cross sections.
Results and Discussion
Binding Affinity of Fluorophores
The binding modes for all three fluorophores are shown in Figure 3. The cofactor FAD is shown in blue while the fluorophores are shown in red. As can be seen, there is a striking difference in their binding modes within the MAO-B, and in particular the Flu1 and Flu2 bind to the substrate binding site while Flu3 binds to both the substrate and the entrance cavity. The Flu1 and Flu2 are located next to the cofactor while the Flu3 is placed relatively at a far distance which, likewise, indicates that the Flu3 binds to the entrance cavity. The binding modes of the ligands are dictated by their molecular volume, and those ligands which can fit into the cavity volume of the substrate binding site can bind to this site. In addition to the molecular volume, there should also be complementary groups in the ligands to further stabilize the formed complex. In order to understand the reason behind the binding behavior, we have computed the molecular volume of all three ligands and they are respectively 144.3, 276.5, and 392.4 cm3/mol. Since the substrate binding site can only accommodate the ligands with certain molecular volume, the ligands Flu1 and Flu2 bind to this site while Flu3 binds to the entrance site. In order to maximize the interaction with the MAO-B, the ligand should possess favorable molecular volume and complementary functional groups. Table 1 presents the free energy of binding for all three ligands and, as can be seen, there is an increase in binding affinity on passing from Flu1 to Flu2 while there is no significant increase in the binding affinity on going from Flu2 to Flu3. The rationale behind these trends is that the Flu3 does not fit well into the substrate binding site which is reported to be the high affinity binding site in MAO-B. Judging by the individual contributions to the total free energy of binding it can be suggested that the binding is mainly driven by hydrophobic interactions between the ligand and MAO-B. The van der Waals contributions to the total binding free energy increase following the order −36, −59, and −65 kcal/mol for Flu1, Flu2, and Flu3 ligands, respectively. As expected, the van der Waals interactions increase with molecular size, and in all three cases these are dominantly contributing to the stabilization of the complexes. The electrostatic interactions (the sum of protein–ligand Coulombic and polar solvation free energy) do not favor the complex formation. We have also analyzed the residue wise contributions to total binding free energy for all three cases, and the results are shown in Figure 4. The interaction patterns for Flu1 and Flu2 are comparable suggesting that the similar residues are involved in the stabilization of the formed complex. In the case of Flu1, the residues GLY58, VAL169, LEU171, CYS172, LEU186, GLY204, VAL324, GLY396. TRP432, and SER433 contribute to the binding free energy amounting to more than 0.5 kcal/mol (in terms of magnitude). In the case of Flu2, the dominantly contributing residues are TYR97, ARG100, THR166, VAL169, ASN170, ARG197, ILE198, ILE199, SER200, GLY204, THR314, VAL324, MET341, GLY396, and SER433. As can be seen, many of the residues are common and are involved in the stabilization of the complex of both compounds. This again confirms that Flu1 and Flu2 bind to the same binding site. Further it is worth mentioning that the cofactor is also involved in the stabilization of the complex and it contributes by −2.4 and −0.7 kcal/mol in the case of Flu1 and Flu2, respectively. Subsequently, we will analyze the stabilizing contacts in the case of Flu3. It can be seen clearly that the substrate binding site residues (LEU88, HIS90, TYR97, GLY101, PRO102, PRO104, ILE110, LEU113, ASN117, THR158, LYS162, ALA165, ARG197, and THR314) do not contribute to the binding free energy suggesting that Flu3 has a unique binding site. Furthermore, there is no contribution from the cofactor to the binding energy of Flu3. Let us now discuss the binding site preference of the fluorophores in comparison to other binders of MAO-B. Experimental crystallographic studies report that the MAO-B inhibitors such as safinamide, clorgyline, and paragyline bind to the substrate binding site.7 We have seen in our previous study that MAO-B inhibitors e.g. safinamide and tau tracers such as PBB3, FDDNP, THK5351, and T807 bind to the substrate binding site.9 Similar to these reports Flu1 and Flu2 bind to the substrate binding site. However, due to the relatively larger molecular size, Flu3 binds to the entrance site surrounded by the residues around ARG100. The binding preference is dictated by the complementarity of residues in the binding site as well as the molecular volume and size of the ligands. For the fluorophore to be used for bioimaging it should exhibit high binding affinity and specificity toward MAO-B and the free energy of binding should be the most negative. All three compounds fulfill this criteria with the Flu2 and Flu3 being the binders with relatively larger affinity.
Figure 3.
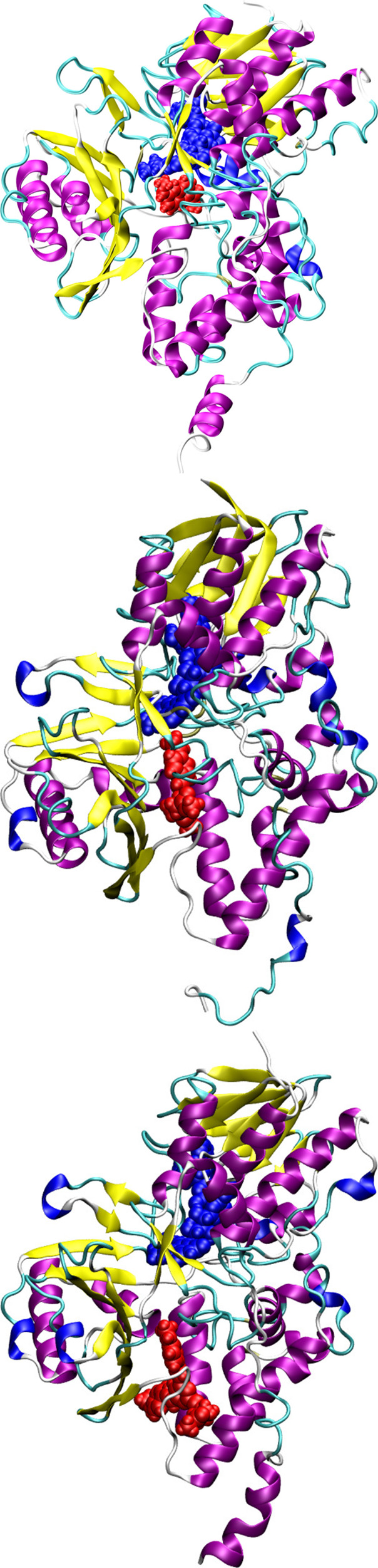
Binding mode of Flu1 (top), Flu2 (middle), and Flu3 (bottom) in MAO-B. The cofactor FAD (in blue) is also shown. The fluorophore is shown in red.
Table 1. Binding Free Energies of the Studied Compounds (in kcal/mol) in MAO-B Using an MM-GBSA Approach.
| Site | ΔEvdw | ΔEelec | ΔGGB | ΔGSA | ΔGbinding |
|---|---|---|---|---|---|
| Flu1 | –36.1 | –5.9 | 15.1 | –4.2 | –31.2 |
| Flu2 | –58.5 | –14.3 | 31.6 | –7.7 | –48.9 |
| Flu3 | –64.6 | –10.3 | 35.6 | –8.9 | –48.2 |
Figure 4.
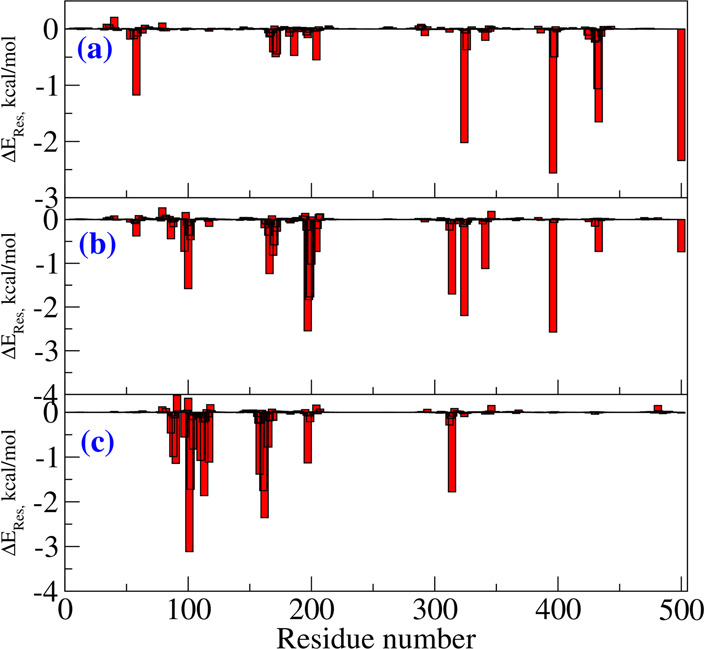
Residue-wise contributions for the total binding free energy of Flu1, Flu2, and Flu3 compounds with MAO-B target.
One-Photon Absorption of Fluorophores in MAO-B
The absorption spectra have been experimentally measured for Flu1, Flu2, and Flu3,15 and the authors reported that the long-wavelength absorption band maxima were located at 352, 384, and 441 nm, respectively. As we see, on passing from Flu1 to Flu3, the position of the absorption band maximum is found to be red-shifted. The shifts are 32 nm (Flu1 → Flu2) and 89 nm (Flu1 → Flu3). The increase in hyperconjugation is usually associated with the red shift in the absorption spectra, and here also it appears to be the plausible explanation. On passing from Flu1 to Flu2, the addition of a CH=CH spacer group contributes to an increase in hyperconjugation, while on passing from Flu2 to Flu3 the addition of a vinyl-benzene aromatic moiety contributes to increased conjugation. We have computed the one-photon absorption spectra for all three ligands using two different density functional approximations (B3LYP and CAM-B3LYP) as well as with the more reliable coupled-cluster RI-CC2 model,29 which is much more computationally demanding than density functional theory. Electronic-structure calculations using the CC2 method were not feasible for relatively bigger molecular systems (composed of dozens of atoms) until only recently. The implementation of resolution-of-identity CC2 (RI-CC2)29 made it feasible to use this level of theory for computing one- and two-photon properties of medium-sized molecules.34 However, due to computational demands associated with the coupled-cluster methods, the DFT-based approaches will still be the preferable choice for studying the energetics and properties of relatively large-sized molecules. With this in mind, in this study we have also compared the performance of the two exchange-correlation functionals (B3LYP, CAM-B3LYP) against the results obtained using the RI-CC2 method. Moreover, as will be demonstrated below, we will also employ the fragmentation approach to study not only fluorophores but also the neighboring bioenvironment, which is only feasible at the DFT level. There is vast literature demonstrating the failure of B3LYP in the description of electronic charge-transfer excitations (see e.g. ref (39) and references therein). In order to alleviate these deficiencies, the CAM-B3LYP functional was proposed, and it is now widely applied for charge-transfer excitations.33,40 All these calculations were performed using an electrostatic embedding scheme which, together with the more advanced polarizable embedding scheme, was developed to model the optical properties of molecules in an environment described with various electric moments both at DFT as well as RI-CC2 levels.41Table 2 contains the average absorption wavelengths (over all considered snapshots) computed for the two lowest electronic excitations for the three compounds in the bioenvironment (MAO-B). Orbitals involved in electronic excitations are shown in the Supporting Information. The two density functional approximations correctly reproduce the red shift in wavelength for S0 → S1 excitation on passing from Flu1 to Flu3; however, the level of accuracy is very system-dependent. The predicted vertical excitation energies by the B3LYP functional for Flu1 and Flu2 are 349 and 396 nm, respectively, which are in good agreement with the experimental absorption band maxima located at 352 and 384 nm. In the case of Flu3, B3LYP reveals its well recognized deficiency with a description of charge-transfer excitations, i.e. it overestimates the wavelength for the S0 → S1 transition by 105 nm. The CAM-B3LYP underestimates the wavelength for the two compounds by −37 nm (Flu1) and −29 nm (Flu2). For Flu3, the calculated absorption maximum deviates from the experimental value (441 nm) to a larger extent (by −65 nm). However, the predicted value is superior than that computed using the B3LYP functional. In summary, CAM-B3LYP deviates more from experimental reference for Flu1 and Flu2 in comparison with B3LYP, but the results for Flu3 demonstrate that the CAM-B3LYP functional does not suffer from erroneous description of charge-transfer excitations to the same extent as B3LYP. Now, we will discuss the one-photon absorption spectra as predicted by the RI-CC2 method. The results from Table 2 demonstrate that RI-CC2 systematically underestimates the wavelength corresponding to the S0 → S1 transition in comparison with the experimental value by −27 nm (Flu1), −17 nm (Flu2), and −45 nm (Flu3). The average absolute deviations from experiment follow the order: CAM-B3LYP (44 nm) > B3LYP (40 nm) > CC2 (30 nm). Although B3LYP, on average, seems slightly more accurate in predicting excitation energies than CAM-B3LYP, it should not be overlooked that the former functional delivers less systematic (i.e., more unpredictable) errors reaching up to roughly 100 nm. Moreover, it should be highlighted that the spectral shifts for the S0 → S1 absorption band on passing from Flu1 → Flu2 and Flu1 → Flu3 are well reproduced by the RI-CC2 and CAM-B3LYP approaches. The experimentally predicted shfits are 32 and 89 nm while the former method predicts 42 and 71 nm and the latter method predicts 40 and 61.
Table 2. Average Absorption Wavelength (λ, nm), Oscillator Strength (f), and Two-Photon Absorption Cross Section (σ2PA, GM) for the Two Lowest Excitations for Three Fluorophores Computed Using the TD-DFT/MM and RI-CC/MM Levels of Theory.
| System | Flu1 | Flu2 | Flu3 | |
|---|---|---|---|---|
| B3LYP/TZVP | ||||
| S0 → S1 | λ | 349 | 396 | 546 |
| f | 0.269 | 0.258 | 0.157 | |
| σ2PA | 31 | 193 | 154 | |
| S0 → S2 | λ | 332 | 370 | 431 |
| f | 0.039 | 0.033 | 0.231 | |
| σ2PA | 6 | 7 | 176 | |
| CAM-B3LYP/TZVP | ||||
| S0 → S1 | λ | 315 | 355 | 376 |
| f | 0.205 | 0.288 | 0.353 | |
| σ2PA | 16 | 176 | 227 | |
| S0 → S2 | λ | 306 | 293 | 337 |
| f | 0.061 | 0.100 | 0.137 | |
| σ2PA | 7 | 14 | 190 | |
| CC2/TZVP | ||||
| S0 → S1 | λ | 325 | 367 | 396 |
| f | 0.165 | 1.175 | 0.789 | |
| σ2PA | 30 | 507 | 541 | |
| S0 → S2 | λ | 313 | 305 | 347 |
| f | 0.110 | 0.016 | 0.492 | |
| σ2PA | 31 | 22 | 216 | |
| Experiment | ||||
| S0 → S1 | λ | 352 | 384 | 441 |
| σ2PA | 674 | 293 | 619 | |
Two-Photon Absorption of Fluorophores in MAO-B
Two-photon action cross sections (product of two-photon absorption cross section and fluorescence quantum yield) were reported for all three compounds in the recent experimental study.15 The corresponding values are 128, 44, and 192 GM for Flu1, Flu2, and Flu3 ligands, respectively. Moreover, the quantum yields have been reported as well and the values are respectively 0.19, 0.15, and 0.31. Since quantum yields cannot be computed reliably for large molecules (and require computing the radiative and nonradiation decay rates), we only compare the calculated two-photon absorption cross section values with experimentally estimated values. The experimentally measured two-photon absorption cross section values for three compounds are 674, 293, and 619 GM, respectively. Interestingly, Flu2 appears to have much lower two-photon absorption cross section value than Flu1 while Flu3 has comparable value to that of Flu1. Even though the hyperconjugation appears to increase on passing from Flu1 to Flu3, the trend in experimental values of two-photon absorption cross sections does not reflect this. In order to shed more light on this striking and counterintuitive result, we have performed electronic structure calculations, and the results are reported in Table 2. The results of calculations show that all three compounds exhibit significant two-photon absorption cross sections for the S0 → S1 transition, i.e. 31–193 GM (B3LYP) and 16–227 GM (CAM-B3LYP) and 30–541 GM (RI-CC2). These results demonstrate that all compounds studied here are suitable for two-photon imaging of MAO-B with Flu3 being the superior two-photon probe (as it is associated with the largest two photon-cross sections). However, the experimental trend is not satisfactorily reproduced by electronic-structure calculations. The most striking experimental result is that Flu1, the least extended π-conjugated system, shows the largest value of two-photon absorption cross section. What is even more striking is that the experimental trends are not in line with the results of electronic-structure calculations, i.e. all three employed methods predict the two-photon absorption cross section of Flu1 to be rather moderate (in the range 13–30 GM). Table 2 contains also the results for the S0 → S2 excitation. The RI-CC2 results demonstrate than on passing from Flu1 to Flu2/Flu3 the separation between S1 and S2 increases from 12 nm to 62/49 nm, respectively. In the case of Flu1, the separation between the S1 and S2 is insignificant but the two-photon absorption cross sections are comparable. Hence, the two-photon properties of S2 are not sufficient to explain the discrepancies between the results of electronic-structure calculations and experimental measurements for Flu1. One of the plausible explanations of this discrepancy is that some of the neighboring residues might be involved in intermolecular charge-transfer excitations between fluorophore and a residue, thus contributing to two-photon absorption cross section. In order to shed light on this notion, in what follows we will discuss the effect of possible charge-transfer between fluorophores and residues and its influence on the two-photon absorption cross sections. As demonstrated in the recent two decades the QM/MM approach is very robust in the description of the interaction between the QM and MM subsystems and it effectively describes the changes in electron density due to heterogeneous or bioenvironments. However, the success of the QM/MM approach heavily relies on the division of the system into QM and MM regions, and for example, processes involving electron transfers between subsystems present in QM and MM regions cannot be properly described. However, when the environment is more polar and has functional units with electron-accepting/donating groups with the ability to strongly attract the electron density from the solute, this description is usually not sufficient. In such cases it is necessary to describe all the molecular fragments involved in the charge transfer in the QM part. So, on those occasions where there is a possible charge transfer between solute and bioenvironment upon excitation, the important fragment has to be included in the QM part. It is worth recalling that in the case of yellow fluorescent protein the charge transfer between the chromophore and neighboring tyrosine residue (TYR203) has been reported to contribute to enhancement of two-photon cross sections.42 A similar intermolecular charge-transfer-induced enhancement in 2PA cross sections has also been reported in supermolecular π-stacked systems.43,44 So, we have also analyzed the neigboring residues located closer to Flu1 when it is bound to MAO-B. The reason for investigating this system, as discussed above, is that experimentally it was shown to have the largest 2PA cross sections (order of magnitude larger than predicted by electronic-structure calculations). It should be highlighted that there are at least five tyrosine residues (TYR396, TYR433, TYR58, TYR324, and TYR186) within a distance range of 10 Å from the center of mass of Flu1. Interestingly, the two tyrosine residues TYR58 and TYR396 located closer to Flu1 and have favorable π-stacking geometry suitable for charge transfer (see Figure 5 where these two residues are shown in green along with Flu1 shown in yellow) and may potentially contribute to the enhancement in 2PA cross sections if this is a universal feature. Interestingly, in the case of Flu2, there are again two tyrosine residues but their relative orientation is not favorable for any charge transfer between the solute and these residues (see Figure 6). So, we have carried out one- and two-photon absorption calculations for ligand–residue intermolecular complex systems. The calculations were carried out for all the residues within 10 Å. The residues were capped with hydrogen atoms to fulfill the valencies created due to fragmentation along peptide bonds. Note that in these QM calculations we neglected the effect of environment beyond the indicated region. The two-photon absorption cross section values computed for 50 such ligand–residue intermolecular complexes are shown in Figure 7. As can be seen the two-photon absorption cross section can be altered by a maximum of 30% when using the B3LYP level of theory. In the case of the CAM-B3LYP level of theory the changes in σ2PA due to the presence of amino acid fragments is also significant. Overall, the charge transfer character yields some changes in σ2PA, and the computed values are still much smaller than the experimental values reported for the Flu1 probe. Given the accuracy of coupled-cluster theory, which predicts that intrinsic two-photon properties are much larger for Flu3 than Flu1, we put forward a notion that the experimental results for Flu1 either are burdened by inaccuracies in determination of fluorescence quantum yields or they reveal a two-photon excitation of a complex that is very different from the one considered here.
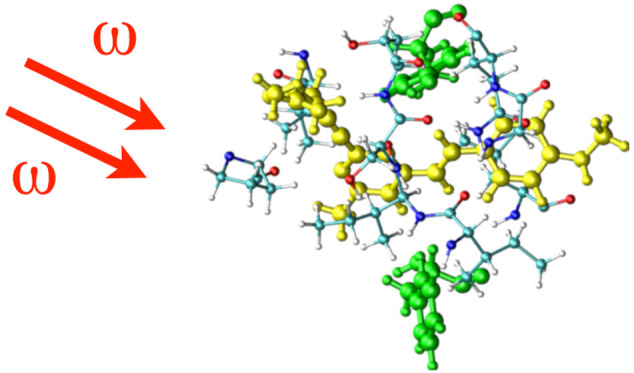
Figure 5.
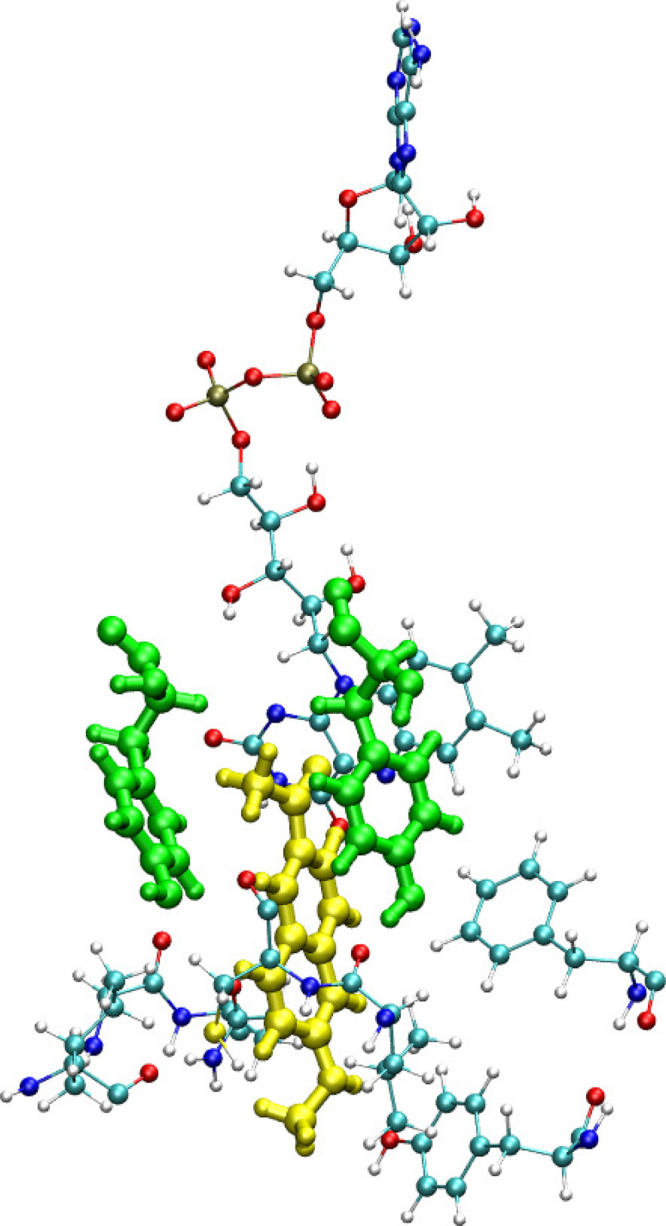
Residues with potential to participate in charge transfer with ligand, Flu1. Two tyrosine residues are located closer to Flu1 ligand. Also the cofactor is shown.
Figure 6.
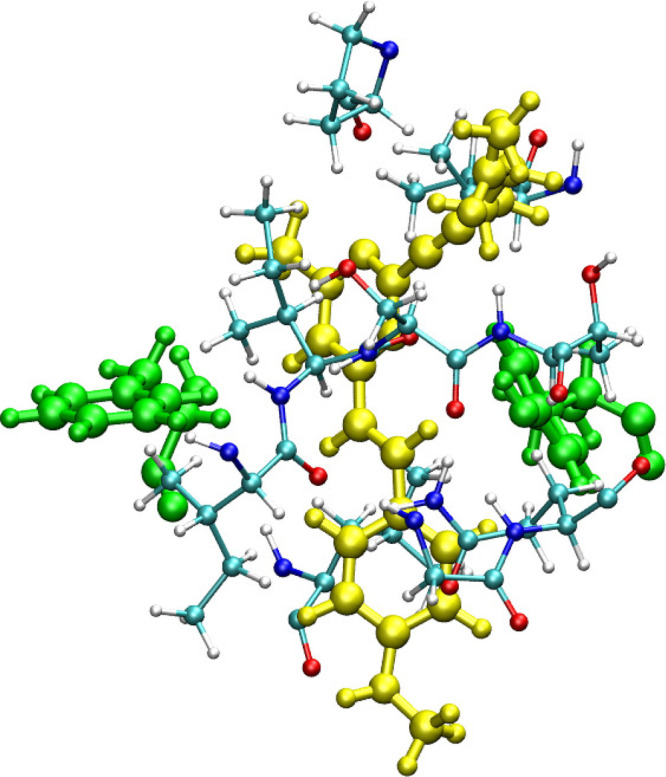
Residues with potential to participate in charge transfer with ligand, Flu2. Two tyrosine residues are located closer to Flu2 ligand, but the relative orientation is suitable for charge transfer. The cofactor is not shown.
Figure 7.
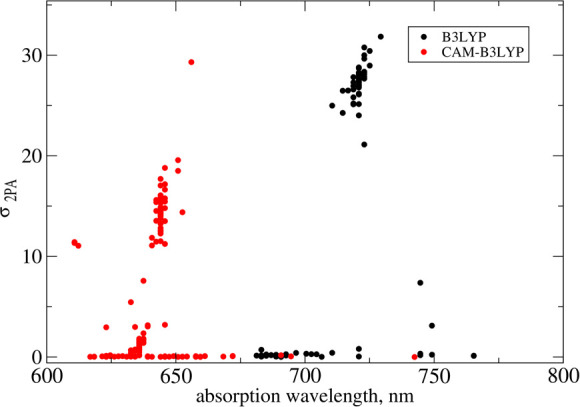
Two-photon absorption cross section computed for the Flu1–amino acids molecular complex. The amino acids within 10 Å are included in the model.
Conclusions
With the use of a multiscale computational approach, we have studied the one- and two-photon absorption properties of three two-photon probes reported for MAO-B imaging. Along with the photophysical properties, we have also estimated their relative free energy of binding. Flu2 and Flu3 are found to exhibit relatively larger binding affinity which should be attributed to their larger molecular volumes. As a part of this study, we have also assessed the accuracy of electronic-structure methods in predicting one-photon absorption spectra. The location of the absorption maximum for the two fluorophores is reproduced most accurately by the TD-B3LYP/MM level of theory. However, in the case of the most extended probe, the B3LYP functional fails to correctly predict the location of the band corresponding to charge-transfer excitation and the error exceeds 100 nm. It is thus more reliable to use the RI-CC2 method (or CAM-B3LYP functional) which delivers more systematic errors. It has also been observed that the spectral shifts on passing from Flu1 to Flu2 and Flu2 to Flu3 are better reproduced by the RI-CC2/MM level of theory. Electronic-structure calculations at all levels of theory revealed that among three studied probes Flu2 and Flu3 are the molecules with the largest σ2PA values and higher binding affinity (than Flu1). This demonstrates their potential as two-photon probes. An important conclusion from this study is also the demonstration that intrinsic molecular two-photon absorption properties are increasing in the order δ2PA (Flu1) < δ2PA (Flu2) < δ2PA (Flu3). This is in contrast with experimental data, which predict similar values of two-photon absorption cross section for Flu1 and Flu3. We demonstrate, based on QM calculations for Flu1 that this discrepancy cannot be explained by an explicit account for neighboring residues (which could lead to charge transfer between a probe and neighboring aromatic amino acids). In summary, we show that the employed multiscale approach not only can optimize σ2PA and verify binding affinity but it can also help in detailed analyses of experimental data.
Acknowledgments
The authors acknowledge support from the Swedish Foundation for Strategic Research (SSF) through the project “New imaging biomarkers in early diagnosis and treatment of Alzheimer’s disease” and the support from SLL through the project “Biomolecular profiling for early diagnosis of Alzheimer’s disease”. This work was supported by grants from the Swedish Infrastructure Committee (SNIC) for the projects and “In-silico Diagnostic Probes Design” (SNIC2020-5-2). R.Z. thanks National Science Centre (Poland) for financial support (grant no. 2018/30/E/ST4/00457).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jcim.0c00423.
Analysis of molecular dynamics data, electronic one- and two-photon absorption spectra, and excitation character analysis (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Michell A.; Lewis S.; Foltynie T.; Barker R. Biomarkers and Parkinson’s Disease. Brain 2004, 127, 1693–1705. 10.1093/brain/awh198. [DOI] [PubMed] [Google Scholar]
- Hong Z.; Shi M.; Chung K. A.; Quinn J. F.; Peskind E. R.; Galasko D.; Jankovic J.; Zabetian C. P.; Leverenz J. B.; Baird G. e. DJ-1 and α-Synuclein in Human Cerebrospinal Fluid as Biomarkers of Parkinson’s Disease. Brain 2010, 133, 713–726. 10.1093/brain/awq008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Q. S.; Grimsby J.; Chen K.; Shih J. C. Promoter Organization and Activity of Human Monoamine Oxidase (MAO) A and B Genes. J. Neurosci. 1992, 12, 4437–4446. 10.1523/JNEUROSCI.12-11-04437.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicotra A.; Pierucci F.; Parvez H.; Senatori O. Monoamine Oxidase Expression During Development and Aging. NeuroToxicology 2004, 25, 155–165. 10.1016/S0161-813X(03)00095-0. [DOI] [PubMed] [Google Scholar]
- Tong J.; Rathitharan G.; Meyer J. H.; Furukawa Y.; Ang L.-C.; Boileau I.; Guttman M.; Hornykiewicz O.; Kish S. J. Brain Monoamine Oxidase B and A in Human Parkinsonian Dopamine Deficiency Disorders. Brain 2017, 140, 2460–2474. 10.1093/brain/awx172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He R.; Yan X.; Tang B.; Sun Q. Recent Advances in Biomarkers for Parkinson’s Disease. Front. Aging Neurosci. 2018, 10, 305. 10.3389/fnagi.2018.00305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finberg J. P.; Rabey J. M. Inhibitors of MAO-A and MAO-B in Psychiatry and Neurology. Front. Pharmacol. 2016, 7, 340. 10.3389/fphar.2016.00340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas R. J.; Edmondson D. E.; Almos T.; Scott R.; Massari M. E. Reversible and Irreversible Small Molecule Inhibitors of Monoamine Oxidase B (MAO-B) Investigated by Biophysical Techniques. Bioorg. Med. Chem. 2015, 23, 770–778. 10.1016/j.bmc.2014.12.063. [DOI] [PubMed] [Google Scholar]
- Murugan N. A.; Chiotis K.; Rodriguez-Vieitez E.; Lemoine L.; Ågren H.; Nordberg A. Cross-Interaction of Tau PET Tracers with Monoamine Oxidase B: Evidence From in Silico Modelling and In Vivo Imaging. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 1369–1382. 10.1007/s00259-019-04305-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake L.; Pham J.; Brooks A.; Koeppe R.; Scott P. MAO-B Inhibitor Deprenyl Displaces [18F] AV1451 Binding on Human Brain Tissue Sections and Alters Uptake in Nonhuman Primate Brain. J. Nucl. Med. 2019, 60, 487–487. [Google Scholar]
- Narayanaswami V.; Drake L. R.; Brooks A. F.; Meyer J. H.; Houle S.; Kilbourn M. R.; Scott P. J.; Vasdev N. Classics in Neuroimaging: Development of PET Tracers for Imaging Monoamine Oxidases. ACS Chem. Neurosci. 2019, 10, 1867–1871. 10.1021/acschemneuro.9b00081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonivento D.; Milczek E. M.; McDonald G. R.; Binda C.; Holt A.; Edmondson D. E.; Mattevi A. Potentiation of Ligand Binding Through Cooperative Effects in Monoamine Oxidase B. J. Biol. Chem. 2010, 285, 36849–36856. 10.1074/jbc.M110.169482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G.; Yee D. J.; Gubernator N. G.; Sames D. Design of Optical Switches as Metabolic Indicators: New Fluorogenic Probes for Monoamine Oxidases (MAO A and B). J. Am. Chem. Soc. 2005, 127, 4544–4545. 10.1021/ja0428457. [DOI] [PubMed] [Google Scholar]
- Wu X.; Shi W.; Li X.; Ma H. A Strategy for Specific Fluorescence Imaging of Monoamine Oxidase A in Living Cells. Angew. Chem., Int. Ed. 2017, 56, 15319–15323. 10.1002/anie.201708428. [DOI] [PubMed] [Google Scholar]
- Li L.; Zhang C.-W.; Chen G. Y.; Zhu B.; Chai C.; Xu Q.-H.; Tan E.-K.; Zhu Q.; Lim K.-L.; Yao S. Q. A Sensitive Two-Photon Probe to Selectively Detect Monoamine Oxidase B Activity in Parkinson’s Disease Models. Nat. Commun. 2014, 5, 3276. 10.1038/ncomms4276. [DOI] [PubMed] [Google Scholar]
- Kollman P. A.; Massova I.; Reyes C.; Kuhn B.; Huo S.; Chong L.; Lee M.; Lee T.; Duan Y.; Wang W.; Donini O.; Cieplak P.; Srinivasan J.; Case D. A.; Cheetham T. E. Calculating Structures and Free Energies of Complex Molecules: Combining Molecular Mechanics and Continuum Models. Acc. Chem. Res. 2000, 33, 889–897. 10.1021/ar000033j. [DOI] [PubMed] [Google Scholar]
- Rastelli G.; Rio A. D.; Degliesposti G.; Sgobba M. Fast and Accurate Predictions of Binding Free Energies Using MM-PBSA and MM-GBSA. J. Comput. Chem. 2010, 31, 797–810. 10.1002/jcc.21372. [DOI] [PubMed] [Google Scholar]
- Binda C.; Wang J.; Pisani L.; Caccia C.; Carotti A.; Salvati P.; Edmondson D. E.; Mattevi A. Structures of Human Monoamine Oxidase B Complexes with Selective Noncovalent Inhibitors: Safinamide and Coumarin Analogs. J. Med. Chem. 2007, 50, 5848–5852. 10.1021/jm070677y. [DOI] [PubMed] [Google Scholar]
- Morris G. M.; Huey R.; Lindstrom W.; Sanner M. F.; Belew R. K.; Goodsell D. S.; Olson A. J. Autodock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexiblity. J. Comput. Chem. 2009, 30, 2785–91. 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Wolf R.; Caldwell J.; Kollman P.; Case D. Development and Testing of a General AMBER Force Field. J. Comput. Chem. 2004, 25, 1157–1174. 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- Jorgensen W. L.; Madura J. D. Quantum and Statistical Mechanical Studies of Liquids. 25. Solvation and Conformation of Methanol in Water. J. Am. Chem. Soc. 1983, 105, 1407–1413. 10.1021/ja00344a001. [DOI] [Google Scholar]
- Case D.; Betz R.; Cerutti D. S.; Cheatham T. E. III; Darden T. A.; Duke and Giese T. J.; Gohlke H.; Goetz A.; Homeyer N.; Izadi S.; Janowski P.; Kaus J.; Kovalenko A.; Lee T. S.; LeGrand S.; Li P.; Lin C.; Luchko T.; Luo R.; Madej B.; Mermelstein D.; Merz K. M.; Monard G.; Nguyen H.; Nguyen H. T.; Omelyan I.; Onufriev A.; Roe D. R.; Roitberg A.; Sagui C.; Simmerling C. L.; Botello-Smith W. M.; Swails J.; Walker R. C.; Wang J.; Wolf R. M.; Wu X.; Xiao L.; Kollman P. A.. Amber 16; University of California: San Francisco, 2016. [Google Scholar]
- Miller B. R. III; McGee T. D. Jr; Swails J. M.; Homeyer N.; Gohlke H.; Roitberg A. E. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. 10.1021/ct300418h. [DOI] [PubMed] [Google Scholar]
- Murugan N. A.; Zalesny R.; Ågren H. Unusual Binding-Site-Specific Photophysical Properties of the Benzothiazole-Based Optical Probe in Amyloid Beta Fibrils. Phys. Chem. Chem. Phys. 2018, 20, 20334–20339. 10.1039/C8CP03274B. [DOI] [PubMed] [Google Scholar]
- Kuang G.; Murugan N. A.; Ågren H. Mechanistic Insight into the Binding Profile of DCVJ and α-Synuclein Fibril Revealed by Multiscale Simulations. ACS Chem. Neurosci. 2019, 10, 610–617. 10.1021/acschemneuro.8b00465. [DOI] [PubMed] [Google Scholar]
- Murugan N. A.; Zalesny R.; Kongsted J.; Ågren H. Chelation-Induced Quenching of Two-Photon Absorption of Azacrown Ether Substituted Distyryl Benzene for Metal Ion Sensing. J. Chem. Theory Comput. 2014, 10, 778–788. 10.1021/ct400924u. [DOI] [PubMed] [Google Scholar]
- Murugan N. A.; Rinkevicius Z.; Ågren H. Solvent Dependence on Bond Length Alternation and Charge Distribution in Phenol Blue: A Car–Parrinello Molecular Dynamics Investigation. J. Phys. Chem. A 2009, 113, 4833–4839. 10.1021/jp900190b. [DOI] [PubMed] [Google Scholar]
- Sałek P.; Vahtras O.; Guo J.; Luo Y.; Helgaker T.; Ågren H. Calculations of Two-Photon Absorption Cross Sections by Means of Density-Functional Theory. Chem. Phys. Lett. 2003, 374, 446–452. 10.1016/S0009-2614(03)00681-X. [DOI] [Google Scholar]
- Friese D. H.; Hättig C.; Ruud K. Calculation of Two-Photon Absorption Strengths with the Approximate Coupled Cluster Singles and Doubles Model CC2 Using the Resolution-of-Identity Approximation. Phys. Chem. Chem. Phys. 2012, 14, 1175–1184. 10.1039/C1CP23045J. [DOI] [PubMed] [Google Scholar]
- Hättig C.; Christiansen O.; Jørgensen P. Multiphoton Transition Moments and Absorption Cross Sections in Coupled Cluster Response Theory Employing Variational Transition Moment Functionals. J. Chem. Phys. 1998, 108, 8331–8354. 10.1063/1.476261. [DOI] [Google Scholar]
- Schäfer A.; Huber C.; Ahlrichs R. Fully Optimized Contracted Gaussian Basis Sets of Triple Zeta Valence Quality For Atoms Li to Kr. J. Chem. Phys. 1994, 100, 5829–5835. 10.1063/1.467146. [DOI] [Google Scholar]
- Becke A. D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. 10.1063/1.464913. [DOI] [Google Scholar]
- Yanai T.; Tew D. P.; Handy N. C. A New Hybrid Exchange-Correlation Functional Using the Coulomb-Attenuating Method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. 10.1016/j.cplett.2004.06.011. [DOI] [Google Scholar]
- TURBOMOLE V7.2 2017, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, since 2007; available from http://www.turbomole.com.
- Aidas K.; et al. The Dalton Quantum Chemistry Program System. Wiley Interdisciplinary Reviews: Computational Molecular Science 2014, 4, 269. 10.1002/wcms.1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monson P.; McClain W. Polarization Dependence of the Two-Photon Absorption of Tumbling Molecules with Application to Liquid 1-Chloronaphtalene and Benzene. J. Chem. Phys. 1970, 53, 29–37. 10.1063/1.1673778. [DOI] [Google Scholar]
- McClain W. M. Two-photon molecular spectroscopy. Acc. Chem. Res. 1974, 7, 129–135. 10.1021/ar50077a001. [DOI] [Google Scholar]
- Beerepoot M. T.; Friese D. H.; List N. H.; Kongsted J.; Ruud K. Benchmarking Two-Photon Absorption Cross Sections: Performance of CC2 and CAM-B3LYP. Phys. Chem. Chem. Phys. 2015, 17, 19306–19314. 10.1039/C5CP03241E. [DOI] [PubMed] [Google Scholar]
- Peach M. J. G.; Benfield P.; Helgaker T.; Tozer D. J. Excitation Energies in Density Functional Theory: An Evaluation and a Diagnostic Test. J. Chem. Phys. 2008, 128, 044118. 10.1063/1.2831900. [DOI] [PubMed] [Google Scholar]
- Peach M. J. G.; Helgaker T.; Sałek P.; Keal T. W.; Lutnæs O. B.; Tozer D. J.; Handy N. C. Assessment of a Coulomb-Attenuated Exchange-Correlation Energy Functional. Phys. Chem. Chem. Phys. 2006, 8, 558–562. 10.1039/B511865D. [DOI] [PubMed] [Google Scholar]
- Hrsak D.; Marefat Khah A.; Christiansen O.; Hättig C. Polarizable Embedded RI-CC2Method for Two-Photon Absorption Calculations. J. Chem. Theory Comput. 2015, 11, 3669–3678. 10.1021/acs.jctc.5b00496. [DOI] [PubMed] [Google Scholar]
- Beerepoot M. T.; Friese D. H.; Ruud K. Intermolecular Charge Transfer Enhances Two-Photon Absorption in Yellow Fluorescent Protein. Phys. Chem. Chem. Phys. 2014, 16, 5958–5964. 10.1039/c3cp55205e. [DOI] [PubMed] [Google Scholar]
- Chakrabarti S.; Ruud K. Intermolecular Interaction-Controlled Tuning of the Two-Photon Absorption of Fullerene Bound in a Buckycatcher. J. Phys. Chem. A 2009, 113, 5485–5488. 10.1021/jp902071j. [DOI] [PubMed] [Google Scholar]
- Chakrabarti S.; Ruud K. Large Two-Photon Absorption Cross Section: Molecular Tweezer as a New Promising Class of Compounds for Nonlinear Optics. Phys. Chem. Chem. Phys. 2009, 11, 2592–2596. 10.1039/b822395e. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.