

Abstract
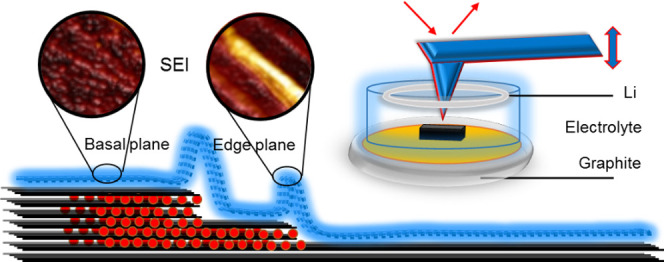
Understanding and ultimately controlling the properties of the solid–electrolyte interphase (SEI) layer at the graphite anode/liquid electrolyte boundary are of great significance for maximizing the performance and lifetime of lithium-ion batteries (LIBs). However, comprehensive in situ monitoring of SEI formation and evolution, alongside measurement of the corresponding mechanical properties, is challenging due to the limitations of the characterization techniques commonly used. This work provides a new insight into SEI formation during the first lithiation and delithiation of graphite battery anodes using operando electrochemical atomic force microscopy (EC-AFM). Highly oriented pyrolytic graphite (HOPG) is investigated first as a model system, exhibiting unique morphological and nanomechanical behavior dependent on the various electrolytes and commercially relevant additives used. Then, to validate these findings with respect to real-world battery electrodes, operando EC-AFM of individual graphite particles like those in commercial systems are studied. Vinylene carbonate (VC) and fluoroethylene carbonate (FEC) are shown to be effective additives to enhance SEI layer stability in 1 M LiPF6/ethylene carbonate/ethyl methyl carbonate (EC/EMC) electrolytes, attributed to their role in improving its structure, density, and mechanical strength. This work therefore presents an unambiguous picture of SEI formation in a real battery environment, contributes a comprehensive insight into SEI formation of electrode materials, and provides a visible understanding of the influence of electrolyte additives on SEI formation.
Keywords: lithium-ion battery, electrochemical AFM, HOPG, graphite, degradation, in situ
1. Introduction
During the electrochemical operation of lithium-ion batteries (LIBs), a solid–electrolyte interphase (SEI) layer is formed on the anode (graphite) surface due to side reactions with the electrolyte solvent and salt.1 This SEI is ionically conductive but electronically insulating, acting as a physicochemical barrier that suppresses both co-intercalation of anion molecules from the edge plane and further solvent decomposition on the electrode surface.2 Ideally, the SEI layer should be a dense and thin film with high ionic (Li+) conductivity and good mechanical strength, uniformly distributed over the anode surface. It should also be fully formed during the first (formation) cycle and remain stable in the following electrochemical cycles and for the remainder of the cell lifetime.3 The formation of the SEI layer causes an initial irreversible capacity loss in the cell, but it is vital for its long-term cyclability, rate capability, self-discharge characteristics, and safety.1 Therefore, the formation of a robust and stable SEI layer on the graphite anode during the formation cycle is essential. According to the chemical composition analysis, the SEI generally contains a lower inorganic layer and an upper organic layer, which are mainly comprised of Li2CO3 and alkyl carbonates, respectively, depending on the type of electrolytes and additives.4,5 Although the formation, composition, morphology, and long-term structural and chemical evolution of the SEI layer have been widely studied, these properties are still poorly understood due to difficulties during sample preparation, transfer or characterization processes such as the sensitivity of samples to the atmosphere, and the limitations of using liquid electrolytes in vacuum conditions.
In situ/operando observation of SEI formation is a direct and efficient method to reveal the electrochemical reaction mechanism of batteries in their initial stages. Various spectroscopy and microscopy techniques have been employed to characterize the chemical and physical properties of battery materials and their degradation during electrochemical processes.6 For instance, the chemical environment of electrodes has been probed via a range of diffraction/spectroscopy techniques including X-ray diffraction (XRD),7 X-ray photoelectron spectroscopy (XPS),8 Raman spectroscopy and Fourier-transform infrared spectroscopy (FTIR);9 the morphology and structural information of electrodes are obtained by imaging techniques such as scanning electron microscopy (SEM),10 transmission electron microscopy (TEM),11 and X-ray computed tomography (X-ray CT).12 However, the above methods cannot efficiently provide accurate structural information of SEI formation on electrodes during electrochemical cell operation due to limitations in either their resolution, operational requirements (e.g., vacuum), or adverse influence on electrode behavior.6
Built on the backbone of “contact” and “tapping” imaging modes, atomic force microscopy (AFM) with a mechanical probe can reveal the three-dimensional (3D) topography of a sample surface with nanoscale resolution in a nondestructive way.13 A form of tapping mode, peak-force tapping AFM, provides pN-level interaction force resolution, while simultaneously probing the morphology of a surface. Thus, by performing peak-force tapping AFM on electrodes under electrochemical operation in liquid electrolytes (i.e., perform electrochemical AFM, EC-AFM) the nanoscale topography and surface mechanical properties can be determined as a function of electrode potential.14,15 Therefore, EC-AFM can deliver operando analysis of battery electrodes during cycling and identify SEI morphology as well as assessing the SEI properties.
Generally, during the first discharge of a graphite anode, SEI layer formation is driven below 1.4 and then 0.9 V, corresponding to the decomposition of additives and electrolyte solvents, respectively, followed by the lithium intercalation below 0.2 V (all potentials vs Li/Li+).5 However, the SEI layer that forms is rarely uniform, with significant variations in the structural and chemical properties across an unevenly distributed and poorly ordered layer being common; this is determined by the solvent, lithium salt, and electrolyte additives used.16 In addition, the properties of the SEI are also significantly affected by temperature, concentration of the electrolyte salt, and reduction current.17 It is well understood that lithium ions only intercalate into graphite layers through the edge plane rather than the basal plane.5 This increased flux drives more interphasial species to accumulate at step edges, causing differences in the SEI layer structure.
Alongside in situ analysis of the morphological and chemical properties of the SEI as it forms, understanding its mechanical properties is also important in revealing the electrochemical mechanisms and degradation processes.18 The Young’s modulus of the SEI is known to change as the electrochemical processes, resulting in inhomogeneity over the anode surface.19
Although changes in morphology have been demonstrated during lithiation of highly oriented pyrolytic graphite (HOPG) using in situ EC-AFM,20−22 with other studies showing select mechanical data,2,5,16,19,23−26 this pristine carbon is far from representative of the graphite utilized in commercial cells. Commercial graphite particles can be coated with pyrolyzed carbon for better interfacial stability, have a high density of step edges, consist of small or mixed-size particles, and offer less ordered strain behavior. All of these effects will modify behaviors observed in real battery carbons from those seen on HOPG. An operando AFM investigation correlating morphological and mechanical changes during SEI growth at both pristine and battery-industry-relevant graphite is still missing from the literature.27,28
In this work, operando EC-AFM is applied to investigate SEI formation on graphite anode materials during the first discharge/charge in 1 M LiPF6/ethylene carbonate/ethyl methyl carbonate (EC/EMC) electrolytes with and without additives of vinylene carbonate (VC) and fluoroethylene carbonate (FEC). SEI formation and corresponding mechanical property evolution are first observed on the model surface of HOPG. These observations are then, for the first time, corroborated on individual graphite particles that are representative of those used in commercial systems, in the same cell configuration. Similar morphological and mechanical changes are noted during SEI layer formation and evolution process, but important differences are found. Therefore, this work provides a link between the fundamental discoveries at model anode systems and real-world applications, driving battery optimization.
2. Materials and Methods
2.1. Materials
Working electrode HOPG (grade ZYB) with 5 × 5 mm2 area and 1 mm thickness was purchased from Bruker Corp and back-contacted via a copper foil substrate, connected via Ag adhesive. Graphite sheets (15 μm particle size, surface area 120–150 m2 g–1) were purchased from Sigma-Aldrich and supported on HOPG and Si substrates by sonicating in acetonitrile for 10 min, before being drop cast onto the support and dried under Ar. The graphite particles acted as isolated “islands” on the flat substrate surface for a better scanning condition for the AFM probe.
To restrict the reaction to the electrode surface (avoiding Cu or Ag adhesive exposure to solution), the working electrode was covered by an adhesive polyimide film with a round area (3 mm diameter) cut out to define the electrode area. The counter/reference electrode was a Ni wire wrapped with lithium foil, which were placed to surround the working electrode inside the electrolyte. The 1 M LiPF6-EC/EMC (3/7 (v/v)) electrolyte, with 2% VC and 2% VC + 15% FEC additive electrolytes, was supplied by SoulBrain MI.
2.2. Methods
Operando EC-AFM (Bruker Dimension Icon with ScanAsyst) experiments were carried out in an Ar-filled glovebox (Mbraun YKG series) with H2O < 0.1 ppm, O2 < 0.1 ppm combined with CH Instrument electrochemical workstation (Model 700E Series Bipotentiostat). PeakForce tapping mode was adopted in all of the EC-AFM imaging with an RTESPA-300 silicon probe with reflective Al coating (Bruker Corp., k = 40 N m–1, f0 = 300 kHz). Before every experiment, the probe was calibrated by using a standard HOPG (modulus = 18 GPa) sample in the Ar atmosphere for a precise measurement of the mechanical properties. Nanoindentation mechanical measurements were conducted at the same time as the morphology was mapped. By recording the load and displacement of the specialized tips and cantilevers when being pressed into the surface a load–displacement curve could be generated, which was further used to calculate the hardness, elastic modulus, and various viscoelastic properties of the materials. All of the results obtained by EC-AFM were analyzed by Nanoscope Analysis software.
Cyclic voltammetry (CV) measurements and electrochemical impedance spectroscopy (EIS) of the coin cells were conducted using a Gamry Interface 1000. The EIS spectra were conducted with an oscillation amplitude of 10 mV over a frequency range from 1 MHz to 0.1 Hz at open-circuit potential (OCP). Galvanostatic discharge/charge cycling was carried out using a Biologic BCS-800 series at ambient temperature.
The CR-2032 coin cells were constructed from a graphite working electrode (90% graphite, 5% PVDF, and 5% carbon black on copper foil; an active material loading of 6.57 mg cm–2), lithium metal counter electrode, polypropylene separator, and the same electrolytes as in the EC-AFM cells.
3. Results and Discussion
3.1. CV Tests of Electrochemical Cells and Coin Cells
The LiPF6-EC/EMC electrolyte is widely considered to be the best combination of lithium salt and solvent among a variety of candidates, due to merits such as high ionic conductivity, low viscosity, good thermal compatibility, good stability to electrodes, and the relative stability of the as-formed SEI film.29 With additives such as VC and FEC, the SEI layers that form on the surface of the electrode materials (including Li metal anode, Si anode, graphite anode, NMC cathode, sulfur cathode, etc.) during the initial cycle have been demonstrated to be more stable due to their better thermal stability and mechanical properties.23,30,31 Therefore, in this work, 1 M LiPF6-EC/EMC (3/7) is chosen as a standard reference electrolyte and the effects of additives of 2% VC and 2% VC + 15% FEC on the SEI formation are investigated.32
A photo and schematic diagram of the electrochemical cell configuration used for EC-AFM studies is shown in Figure S1. To correlate the electrochemical performance of the HOPG anode in the EC-AFM cell with that of standard graphite LIB anodes (copper current collector with graphite powder, see Materials and Methods) in the coin cells, CVs were measured in the different electrolytes (Figure 1). Figure 1a shows representative first cycle CV curves for the HOPG at 1 mV s–1. In the cathodic scans in EC/EMC and EC/EMC/VC electrolytes, the current shows little change until 1.0 V. By contrast, a constantly increasing current is observed from 2.0 V in the EC/EMC/VC/FEC electrolyte, which may be caused by the early stage decomposition of FEC.1 The current peaks in the potential range of 0.7–0.3 V for EC/EMC, 0.8–0.4 V for EC/EMC/VC, and 1.5–0.5 V for EC/EMC/VC/FEC electrolyte are due to the decomposition of additives and solvents and the formation of the SEI layer on the surface of HOPG. Additionally, sharp peaks at the potential close to 0 V are attributed to the Li ion intercalation in the graphite layers.16 During the anodic scan, all three cells display a positive current at potentials above 1.0 V, indicating a polarization during the deintercalation and decomposition reactions.
Figure 1.

CV curves recorded from the EC-AFM cell (a) 1 mV s–1 and coin cells (b) 1 mV s–1; (c) 0.1 mV s–1 with the three electrolytes of EC/EMC, EC/EMC/VC, and EC/EMC/VC/FEC.
Figure 1b,c displays the CV curves of the graphite LIB anodes at scan rates of 1 and 0.1 mV s–1, respectively. At 1 mV s–1, similar processes can be observed as those seen for the HOPG for all three electrolytes, except that anodic reactions occur at a lower potential, indicating an alleviated polarization due to lower electronic resistance in the coin cells. At a slower scan rate, the three cells show very similar discharge and charge processes, with the only difference being the starting point of the SEI formation (Figure 1c). According to previous reports, the lowest unoccupied molecular orbital (LUMO) energy levels for VC (−0.88 eV) and FEC (−0.84 eV) are lower than EC (−0.40 eV), which means that VC and FEC additives have higher reduction capability to accept electrons under reductive environments.33 This accounts for the earlier onset of SEI formation during discharge in the above CV scans. In addition, with VC and FEC additives, a more stable SEI on the graphite anode surface is expected to form, which prevents continuous electrolyte decomposition reactions occurring during further charge and discharge cycles.34
3.2. Operando EC-AFM of HOPG in EC/EMC
Figure 2 presents a series of operando EC-AFM images of the HOPG anode with the EC/EMC electrolyte discharged from 3.0 V (open-circuit potential) to 0.0 V and then charged to 3.0 V. Each image was captured over 1.0 V, which took 512 s for one capture (scan rate = 1 Hz, 512 lines × 512 dots), i.e., the potential was scanned during image capture. Height and Derjaguin–Muller–Toporov (DMT) modulus (reduced Young’s modulus calculated according to the DMT model, explained in the Supporting Information) are presented within a square scan area of 10 × 10 μm2. The images were captured from the top to bottom for 3.0–2.0 V, bottom to top for 2.0–1.0 V, and so forth (as indicated in the figure). The first column of images and Figure S2 (enlarged figures and section line data) displays the morphology of the initial (3.0–2.0 V) HOPG surface. The freshly cleaved HOPG has a very clean and smooth surface after it is submerged into the electrolyte. The long vertical lines are graphite edge planes, which show the presence of carbon terraces with step heights up to 4 nm (a 4 nm step is indicated by the white arrows), corresponding to ∼10 carbon layers. The DMT modulus (20–25 GPa) is uniformly recorded along each line scan and over the whole area, which is close to the theoretical value of graphite (18 GPa).19 From ∼1.5 V (second column) to ∼0.7 V (third column), the measured modulus decreases by ∼5 GPa. This is concurrent with the cathodic current increase, and therefore, this can be attributed to the early stages of electrolyte decomposition, forming an intermediate radical anion near the surface of the graphite.5
Figure 2.

Operando EC-AFM images (1.0 V per capture) captured continuously from the electrochemical cell with the EC/EMC electrolyte in a 10 × 10 μm2 area on the surface of the HOPG anode. The signals of height and DMT modulus are presented, and corresponding scale bars are shown at the right side of the images. All voltages are quoted vs Li/Li+.
Figure 3a,b gives enlarged images of the height and modulus data in Figure 2, column 3 (1.0–0.0 V). Here, it can be seen that prominent morphological changes are initiated at 0.7 V. The height difference of the marked step (white arrows, Figure 2) sharply increases from up to 4 nm to eventually over 200 nm (∼0.25 V, green line in Figure 3a), forming “mountains” along the step edges. An average height increase of about 36 (±5) nm per carbon layer is calculated. In addition, particle-like structures simultaneously form on the basal plane of the graphite, producing a rough surface with a height difference of 40–50 nm. The height increase of the mountains is much larger than that of the basal plane area, which is consistent with previous reports of preferential SEI formation at the step edges of graphite.16,35,36
Figure 3.

Enlarged images of height (a) and modulus (b) of the HOPG surface between 1.0 and 0.0 V (vs Li/Li+) (column 3 in Figure 2, scanned from top to bottom within 512 s) and section line data as marked correspondingly. (c–e) Force–distance curves of the three spots from area (c, d, e) in (b).
At 0.75 V (red line and area c in Figure 3b), the modulus is consistently ∼15 GPa across basal planes and step edges, which is slightly lower than the starting value. Comparatively, the modulus value at 0.25 V (blue line, Figure 3b) shows a significant difference between the basal plane (area d) and step edge (area e), which are ∼14.5 and 0.2–1.5 GPa, respectively. Representative force–distance curves from the three regions indicated in Figure 3b (labeled c, d, and e) are shown in Figure 3c–e. The shapes of the curves at spots c and d, both on the basal plane, are quite alike, leading to similar Young’s modulus. The deformation distance between the start of the repulsive interaction (∼180 nm) to the utmost force between the probe and sample surface (200 nm) can be used to estimate the thickness of the soft SEI layer. At spots d and e, this is calculated at 20 and 80 nm, respectively. From the force–distance curve of areas d and e, the withdrawing of the probe from the mountain region at the step edge has a much larger dissipation energy due to the stronger adhesion. This confirms that the SEI layer at the basal plane has a higher modulus, suggesting greater density and smaller thickness, while the SEI layers at the edge plane are much softer and thicker. These results are consistent with other reports by FTIR and XPS analyses that the SEI layer at step edges is mainly composed of loosely packed organic lithium alkali carbonates and polymeric compounds and the SEI layer at the basal plane contains more inorganic materials like Li2O, LiF, and Li2CO3.20,37,38 It has also been reported that the SEI layer is composed of a soft, polymeric upper layer and a hard, saltlike lower layer covering both edges and basal planes of graphite.4
The fourth column of Figure 2 shows a well-established SEI layer over the whole area after 0.0 V. Beyond 1.0 V on the reverse scan, most of the SEI particles at the basal plane appear to dissolve, while the SEI at the step edges remains. Figure S3 presents the enlarged images of column 6 (2.0–3.0 V, Figure 2), which exhibits partially recovered morphology, including dissolved basal plane SEI particles and slightly reduced mountain height. The modulus is also recovered to a higher level than that of 0.0–1.0 V.
To eliminate the possibility that the moving probe has an influence on the sample during imaging, a second discharge process at 1 mV s–1 was carried out while a larger area of 20 μm2 was scanned, as shown in Figure S4. The probe has an insignificant influence on the observed morphology, peak force error, and modulus, as observed from the inner 10 × 10 μm2 square area and zoomed-out area. In addition, little change is observed during the second discharge due to the stabilized SEI layer on the HOPG surface.
3.3. Operando EC-AFM of HOPG in Different Electrolytes
To investigate the influence of electrolyte additives on the morphology and properties of the SEI layer, operando EC-AFM was conducted via the same processes as above in different electrolytes with VC and FEC additives. Figure 4 shows three sets of 3D images of height and modulus obtained in the different electrolytes (a) EC/EMC, (b) EC/EMC/VC, and (c) EC/EMC/VC/FEC at the stages of 3.0–2.0 V (column 1) 1.0–0.0 V (column 2), and 2.0–3.0 V (column 3). Figure S5 shows this data in more detail. As with the EC/EMC, during a potential sweep of 3.0–2.0 V, no changes to the HOPG surface are observed in the electrolytes with VC and FEC additives. The surface remains flat (step height up to 4 nm) and uniform modulus value (∼20 GPa) is maintained over the 10 × 10 μm2 area. Subsequently, within the voltage range of 1.0–0.0 V, thick SEI mountains appear along the edge of the graphite and particle SEI forms at the basal plane area in all samples. However, different from the EC/EMC electrolyte (Figure 4a, 1.0–0.0 V), obvious morphology change occurs at a slightly higher voltage (about 0.8 V) in the two other electrolytes (Figure 4b,c, 1.0–0.0 V), which agrees well with the CV curves (Figure 1). This is attributed to a lower reductive activation energy (13 kcal mol–1) and higher reduction potential (1.05–1.4 V vs Li/Li+) for VC than that of EC (24.9 kcal mol–1 and 0.65–0.9 V vs Li/Li+, respectively). SEI forms at higher potential (0.1–0.2 V higher) in VC-containing electrolytes, which is reported to possess improved stability due to higher mechanical strength.34 It is observed that with VC or VC/FEC additives, the height increase of the mountain region is much smaller than without any additive. As calculated, the height increment is 22 (±5) nm per carbon layer in EC/EMC/VC and 25 (±5) nm in EC/EMC/VC/FEC, both notably smaller than 36 (±5) nm per layer in EC/EMC.
Figure 4.

3D images of height (upper row) and modulus (lower row) obtained in the different electrolytes of (a) EC/EMC, (b) EC/EMC/VC, and (c) EC/EMC/VC/FEC at the stages of 3.0–2.0 V (column 1), 1.0–0.0 V (column 2), and 2.0–3.0 V (column 3). All of the images have a scan area of 10 × 10 μm2. Height and modulus scale bars are shown at the right side of each image. All voltages quoted vs Li/Li+.
The height of the SEI formed at the basal plane in the three electrolytes is all in the same range (all have a peak/trough hight variation of ∼50 nm), meaning which means that the thicknesses of basal plane SEI layers are all approximately equal to 40–50 nm (Figure S6). However, compared to that in EC/EMC, the basal plane SEI height profiles in EC/EMC/VC and EC/EMC/VC/FEC show a smoother shape, indicating a more consistent and homogeneous SEI structure. Both of them also have a higher overall SEI modulus than that in EC/EMC, indicating a greater density or different layer structure. Also different from the EC/EMC electrolyte, the basal plane SEI in the latter two electrolytes is maintained after the discharge/charge cycle (2.0–3.0 V), suggesting greater SEI stability. This could reduce additional SEI formation on subsequent cycles, leading to lower irreversible capacity losses. Although the EC/EMC/VC electrolyte leads to a higher modulus at the basal plane than that of EC/EMC/VC/FEC, the latter exhibits a more uniform modulus between the basal plane and edge plane areas than the former.
To study the structure and mechanical properties of SEI more precisely, terraces in the three samples were selected as marked (white double arrows) in the first column in Figure 4. For a fair comparison, all three terraces have the same original step thickness of 1 nm, which is ∼3 carbon layers (section line height in Figure S7). The height, width, and modulus of the “mountain” SEI formed along the three terraces (one spot selected every 1 μm over the ∼10 μm edge) after the first CV scan (data obtained from 2.0–3.0 V images) are compared in Figure 5a–c, respectively. In Figure 5a, a larger mountain SEI height in EC/EMC is demonstrated, compared to its counterparts with additives. Although the EC/EMC/VC produced a similar SEI height as that of EC/EMC/VC/FEC, the mountain width is much larger and more dispersed (Figure 5b). As can be seen from Figure 4b column 3, the mountain region is spread widely, while the other two samples have relatively slim and straight mountain morphology. After the discharge/charge process, the mountain area SEI modulus in the EC/EMC electrolyte is much lower than that with additives, as shown in Figure 5c. The mountain area SEI in the EC/EMC/VC/FEC electrolyte has the highest and most consistent modulus values, indicating a uniformly robust SEI. In fact, the calculated standard deviation (SD) based on the height, width, and modulus data in EC/EMC/VC/FEC are the smallest, revealing better overall uniformity of the edge-plane SEI layer than others.
Figure 5.

Height (a), width (b), and modulus (c) data obtained from Figure 4, column 3 (2.0–3.0 V), at the same graphite terrace as the double white arrows marked in Figure 4, column 1. Each set of data contains 10 different spots from the marked terraces, which has the same original thickness. (d–f) Modulus evolution in the complete CV process at the three regions, as marked in Figure 4 column 2 (1.0–0.0 V), in the three electrolytes. All voltages quoted vs Li/Li+.
For a more comprehensive comparison of modulus values, three representative regions (i, ii, and iii) were selected as marked in Figure 4, column 2 (1.0–0.0 V). Figure 5d–f shows the evolution of the modulus (average value of each marked region) during the CV test. On the basal plane areas (i and ii), the modulus values decrease slightly upon SEI build-up. The SEI modulus in EC/EMC recovered to ∼10 GPa after the discharge/charge cycle. By contrast, within EC/EMC/VC and EC/EMC/VC/FEC electrolytes, the modulus recovered to 15–20 GPa. At step edges (region iii), the SEI modulus in all electrolytes drops significantly as the cell approaches 0 V. However, the EC/EMC/VC/FEC system maintains a higher value of ∼10 GPa after the CV test, while the others showed much lower modulus, <1.0 GPa for EC/EMC and <3.0 GPa for EC/EMC/VC. This means that the mixed VC/FEC additive system has a denser SEI, enabling greater stability and better protection of the graphite anode.
The above results directly confirm the previous first-principles computational studies that VC and FEC additives lead to more compact and stable SEI layers than those formed in EC electrolytes without additives.39 The VC and FEC additives in the electrolyte induce a controlled growth of the SEI layer with more inorganic and stable polymer species. In contrast, in the electrolyte without such additives, the SEI layer growth is uncontrolled due to unstable external layers that are easily attacked by radical species promoting sustained growth, which exhibits a much larger thickness of the SEI layer at the edge plane.33 In addition, VC and FEC are suggested to promote the polymerization of VC with cross-linking and formation of the LiF nanoparticles in the SEI layer, respectively, both of which lead to a high SEI stiffness.40 In summary, with VC and FEC addition, a thinner and stronger SEI layer formed on the HOPG surface leading to a higher stability, which impedes the decomposition, evolution, and thickening of the SEI during the following discharge/charge cycles.
3.4. Electrochemical Study of the Coin Cells
To compare the EC-AFM data with actual battery performance, coin cells containing graphite vs Li were tested with the three electrolyte systems. Electrochemical impedance spectroscopy (EIS) and galvanostatic discharge/charge cycling performance tests were conducted.
Figure S8a presents the EIS Nyquist plots of the three coin cells with different electrolytes before and after the first CV scan at 0.1 mV s–1. For the freshly assembled cells, all spectra consist of a depressed semicircle in the higher frequency range and a sloping line in the lower frequency range, corresponding to the charge transfer resistance (Rct) and diffusion resistance (Wwarburg), respectively. All three cells show a similar charge transfer resistance (∼120–130 Ω), indicating a similar basic bulk resistance (Rb) (including current collector, electrolyte, and separator) and Rct before SEI formation. After the CV scan, all three cells show decreased overall resistance due to the established SEI layer, which is favorable for ion transportation.41 The simplified equivalent circuit and corresponding resistance components are shown in Figure S8b, and the fitted resistance data are given in Table S1. The higher frequency area could be subdivided into two depressed semicircles, corresponding to the resistance of SEI (RSEI) layer and Rct.42 The cells with EC/EMC and EC/EMC/VC exhibit the lowest and highest sum of RSEI and Rct, respectively. Cell impedance is influenced by factors including SEI thickness43 and chemical composition.44 It is also reported that ion transport is sluggish in the rigid polymer chains of the VC-induced SEI layer,33 thus the sum value of RSEI and Rct in EC/EMC electrolytes is the lowest due to better ionic mobility. However, there is a sacrifice of the SEI layer stiffness in contrast to the other two electrolytes with additives. In addition, compared with EC/EMC/VC, the FEC-added electrolyte shows a lower sum resistance of RSEI and Rct, suggesting that ion transportation is improved by FEC due to the inorganic LiF SEI component.45
A thinner and stronger SEI layer, with better ionic mobility, is expected to improve battery cycling performance.33 Galvanostatic cycling operation of the three coin cells was carried out at 0.2C after the first discharge–charge cycle. As shown in Figure S8c, despite a lower capacity than in EC/EMC electrolytes, the coin cell with EC/EMC/VC/FEC electrolytes exhibits a more stable capacity and Coulombic efficiency during the 20 cycles. The slight capacity increase in the initial cycles of the cell with EC/EMC is presumed to be due to the unstable SEI formation. The unstable SEI layer significantly weakens the battery cycling stability, attributed to the repeated formation of new SEI and co-intercalation of Li+-solvent clusters (which can result in graphite exfoliation).46 The capacity drops quickly for the cell with EC/EMC/VC electrolytes, which might be attributed to the sluggish ion transportation in the VC-induced SEI layer. Figure S8d–f gives the voltage profiles in the first five cycles of the three cells. The overlapped curves of second to fifth cycles indicate that the stable SEI layer formed after the second cycle in the electrolyte of EC/EMC/VC/FEC. These results are in alignment with previous reports.33
3.5. Operando EC-AFM of Individual, Industry Relevant, Graphite Oarticles
Operando EC-AFM monitoring of an individual graphite sheet, more representative of those utilized in commercial graphite anodes, supported on an HOPG substrate, was carried out in the EC/EMC electrolyte by discharging/charging at a scan rate of 1 mV s–1. A 4 × 4 μm2 area is presented in Figure 6 (the series of images for the whole process is provided in Figure S9). At OCP (∼3.0 V), the graphite sheet has a ∼5 nm thickness and slightly lower modulus (10–16 GPa) than HOPG (∼18 GPa). In the potential range of 0.75–0.5 V, a height increase is detected at the edge planes both of the background HOPG and graphite particle. The corresponding height profile (Figure 6c) shows the edge has a much larger thickness increase than the inner area, which agrees well with the observations on HOPG. The height of SEI at the particle edge further increases to 50–65 nm during 0.25–0.0 V. In the central area of the graphite particle, another sharp height increase occurs, suggesting a step edge in the center of the sheet. The edge of the graphite sheet shows a slightly lower modulus than that in the center at OCP and 0.75–0.5 V. Being constantly lower than the HOPG substrate, the modulus of the graphite sheet gradually reduced to 1.0–3.0 GPa and shows a uniform distribution across the graphite sheet. After being charged to 0.75–1.0 V, the graphite sheet with an uneven height profile is no longer intact, consistent with the disruption of the anode material due to volume change.1 The modulus over the whole graphite sheet maintains at a lower level (0.5–2.0 GPa) than HOPG, which is likely due to the significantly higher edge/basal ratio of the small graphite sheet than HOPG. For the graphite anode in an actual battery environment, SEI formation is known to be reliant on the particle size, basal/edge ratio, pore size, degree of crystallinity, and surface chemical composition.47 Here, it is confirmed that for a small graphite particle with a higher edge/basal ratio, excess SEI is formed compared with that of HOPG with a lower edge/basal ratio. This effect is corroborated for the in situ EC-AFM measurements of a graphite sheet on a silicon substrate, showing that it is not driven by the underlying substrate (Figure S10).
Figure 6.

Operando EC-AFM of an individual graphite sheet on the HOPG substrate in the electrolyte of EC/EMC. Height (a) and modulus (b) images of four potential steps of OCP ∼3.0, 0.75–0.5, 0.25–0.0, and 0.75–1.0 V are presented. All of the captured images have a 4 × 4 μm2 area and same scale bars for each row. (c) Height and modulus information of the corresponding section line marked in (a) and (b). All voltages are quoted vs Li/Li+.
4. Conclusions
Operando EC-AFM characterization was carried out to investigate the SEI formation and evolution on HOPG and graphite sheet anodes in different electrolytes. The continuous imaging of SEI formation on HOPG shows that obvious morphology change starts at ∼0.7 V, which simultaneously forms two different types of SEI at the edge and basal planes of graphite. Correlated Young’s modulus mapping and morphology imaging of SEI formation is obtained for the first time. It is demonstrated that SEI at edge sites is much thicker and softer than that at the basal plane, but by adding VC and FEC additives an enhanced SEI layer (thinner and stronger) is developed at both edge and basal areas of the graphite. When combined with the electrochemical measurements of battery performance, the enhanced SEI layer is demonstrated to improve the overall battery performance.
By correlating the fundamental discoveries made on the model HOPG substrate with those of the individual industrially relevant graphite particles, it has been shown that although useful and important comparisons can be made, this must be done with care. It is clear that the same processes occur at HOPG and graphite particles, but the significantly different material structure leads to different behavior and device performance.
Overall, this work provides a comprehensive and direct observation of SEI on graphite anode materials. The improved understanding of the SEI formation and degradation mechanism offered, alongside the highlighting of the benefits of different electrolyte additives, can ultimately help drive improvements in LIB technologies.
Acknowledgments
The present research has been supported by the Faraday Institution (EP/S003053/1), degradation project (FIRG001), LiSTAR project (FIRG014), and EPSRC (EP/R023581/1, EP/P009050/1, and EP/N032888/1). T.S.M. thanks the U.K. EPSRC for support via his Fellowship EP/P023851/1. P.R.S. acknowledges The Royal Academy of Engineering (CiET1718/59).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsami.0c11190.
Explanation of how the DMT modulus data was obtained; photo and schematic diagram of the electrochemical cell configuration; additional AFM images and data; electrochemical measurements including EIS and cycling performance of coin cells; the operando EC-AFM images of a graphite-on-silicon substrate, conducted with the same procedures as discussed for HOPG (PDF)
Author Contributions
The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- An S. J.; Li J.; Daniel C.; Mohanty D.; Nagpure S.; Wood D. L. III The State of Understanding of the Lithium-ion-battery Graphite Solid Electrolyte Interphase (SEI) and its Relationship to Formation Cycling. Carbon 2016, 105, 52–76. 10.1016/j.carbon.2016.04.008. [DOI] [Google Scholar]
- Jeong S. K.; Inaba M.; Mogi R.; Iriyama Y.; Abe T.; Ogumi Z. Surface Film Formation on a Graphite Negative Electrode in Lithium-ion Batteries: Atomic Force Microscopy Study on the Effect of Film-forming Additives in Propylene Carbonate Solutions. Langmuir 2001, 17, 8281–8286. 10.1021/la015553h. [DOI] [Google Scholar]
- Xu K. Nonaqueous Liquid Electrolyte for Lithium-based Rechargeable Batteries. Chem. Rev. 2004, 104, 4303–4417. 10.1021/cr030203g. [DOI] [PubMed] [Google Scholar]
- Nie M.; Demeaux J.; Young B. T.; Heskett D. R.; Chen Y.; Bose A.; Woicik J. C.; Lucht B. L. Effect of Vinylene Carbonate and Fluoroethylene Carbonate on SEI Formation on Graphitic Anodes in Li-ion Batteries. J. Electrochem. Soc. 2015, 162, A7008–A7014. 10.1149/2.0021513jes. [DOI] [Google Scholar]
- Cresce A. V.; Russell S. M.; Baker D. R.; Gaskell K. J.; Xu K. In Situ and Quantitative Characterization of Solid Electrolyte Interphases. Nano Lett. 2014, 14, 1405–1412. 10.1021/nl404471v. [DOI] [PubMed] [Google Scholar]
- Tripathi A. M.; Su W. N.; Hwang B. J. In Situ Analytical Techniques for Battery Interface Analysis. Chem. Soc. Rev. 2018, 47, 736–851. 10.1039/C7CS00180K. [DOI] [PubMed] [Google Scholar]
- Shen X.; Li Y.; Qian T.; Liu J.; Zhou J.; Yan C.; Goodenough J. B. Lithium Anode Stable in Air for Low-cost Fabrication of a Dendrite-free Lithium Battery. Nat. Commun. 2019, 10, 900 10.1038/s41467-019-08767-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y.; Jie Y.; Huang F.; Chen Y.; Lei Z.; Zhang G.; Ren X.; Qin L.; Cao R.; Jiao S. Enabling Stable Lithium Metal Anode through Electrochemical Kinetics Manipulation. Adv. Funct. Mater. 2019, 29, 1904629 10.1002/adfm.201904629. [DOI] [Google Scholar]
- Ni K.; Wang X.; Tao Z.; Yang J.; Shu N.; Ye J.; Pan F.; Xie J.; Tan Z.; Sun X.; Liu J.; Qi Z.; Chen Y.; Wu X.; Zhu Y. In Operando Probing of Lithium-ion Storage on Single-layer Graphene. Adv. Mater. 2019, 31, 1808091 10.1002/adma.201808091. [DOI] [PubMed] [Google Scholar]
- Biswal P.; Stalin S.; Kludze A.; Choudhry S.; Archer L. A. Nucleation and Early Stage Growth of Li Electrodeposits. Nano Lett. 2019, 19, 8191–8200. 10.1021/acs.nanolett.9b03548. [DOI] [PubMed] [Google Scholar]
- Yuan Y. F.; Amine K.; Lu J.; Shahbazian-Yassar R. Understanding Materials Challenges for Rechargeable Ion Batteries with in Situ Transmission Electron Microscopy. Nat. Commun. 2017, 8, 15806 10.1038/ncomms15806. [DOI] [Google Scholar]
- Finegan D. P.; Scheel M.; Robinson J. B.; Tjaden B.; Hunt I.; Mason T. J.; Millichamp J.; Michiel M. D.; Offer G. J.; Hinds G.; Brett D. J. L.; Shearing P. R. In-operando High-speed Tomography of Lithium-ion Batteries during Thermal Runaway. Nat. Commun. 2015, 6, 6924 10.1038/ncomms7924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker C. R.; Strawhecker K. E.; McAllister Q. P.; Lundgren C. A. In Situ Atomic Force Microscopy of Lithiation and Delithiation of Silicon Nanostructures for Lithium Ion Batteries. ACS Nano 2013, 7, 9173–9182. 10.1021/nn4037909. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Wang R.; Yang X.; Lu W.; Wu X.; Wang X.; Li H.; Chen L. Direct Observation of Inhomogeneous Solid Electrolyte Interphase on MnO Anode with Atomic Force Microscopy and Spectroscopy. Nano Lett. 2012, 12, 2153–2157. 10.1021/nl300570d. [DOI] [PubMed] [Google Scholar]
- Benning S.; Chen C.; Eichel R. A.; Notten P. H. L.; Hausen F. Direct Observation of SEI Formation and Lithiation in Thin-Film Silicon Electrodes via in Situ Electrochemcial Atomic Force Microscopy. ACS Appl. Energy Mater. 2019, 2, 6761–6767. 10.1021/acsaem.9b01222. [DOI] [Google Scholar]
- Shi Y.; Yan H. J.; Wen R.; Wan L. J. Direct Visualization of Nucleation and Growth Processes of Solid Electrolyte Interphase Film using in Situ Atomic Force Microscopy. ACS Appl. Mater. Interfaces 2017, 9, 22063–22067. 10.1021/acsami.7b05613. [DOI] [PubMed] [Google Scholar]
- Bhattacharya S.; Riahi A. R.; Alpas A. T. Thermal Cycling Induced Capacity Enhancement of Graphite Anodes in Lithium-ion Cells. Carbon 2014, 67, 592–606. 10.1016/j.carbon.2013.10.032. [DOI] [Google Scholar]
- Moeremans B.; Cheng H. W.; Merola C.; Hu Q.; Oezaslan M.; Safari M.; Bael M. K. C.; Hardy A.; Valtiner M.; Renner F. U. In Situ Mechanical Analysis of the Nanoscopic Solid Electrolyte Interphase on Anodes of Li-ion Batteries. Adv. Sci. 2019, 6, 1900190 10.1002/advs.201900190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng X.; Liu X. R.; Yan H. J.; Wang D.; Wan L. J. Morphology and Modulus Evolution of Graphite Anode in Lithium Ion Battery: an in Situ AFM Investigation. Sci. China: Chem. 2014, 57, 178–183. 10.1007/s11426-013-4988-4. [DOI] [Google Scholar]
- Tsubouchi S.; Domi Y.; Doi T.; Ochida M.; Nakagawa H.; Abe T.; Ogumi Z.; Yamanaka T. Spectroscopic Characterization of Surface Films Formed on Edge Plane Graphite in Ethylene Carbonate-based Electrolytes Containing Film-forming Additives. J. Electrochem. Soc. 2012, 159, A1786–A1790. 10.1149/2.028211jes. [DOI] [Google Scholar]
- Gossage Z. T.; Hui J.; Zeng Y.; Flores-Zuleta H.; Rodriguez-Lopez J. Probing the Reversibility and Kinetics of Li+ during SEI Formation and (De)intercalation on Edge Plane Graphite using Ion-sensitive Scanning Electrochemical Microscopy. Chem. Sci. 2019, 10, 10749–10754. 10.1039/C9SC03569A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campana F. P.; Kotz R.; Vetter J.; Novak P.; Siegenthaler H. In Situ Atomic Force Microscopy Study of Dimensional Changes during Li+ Ion Intercalation/De-intercalation in Highly Oriented Pyrolytic Graphite. Electrochem. Commun. 2005, 7, 107–122. 10.1016/j.elecom.2004.11.015. [DOI] [Google Scholar]
- Shen C.; Hu G.; Gheong L. Z.; Huang S.; Zhang J. G.; Wang D. Direct Observation of the Growth of Lithium Dendrites on Graphite Anodes by Operando EC-AFM. Small Methods 2018, 2, 1700298 10.1002/smtd.201700298. [DOI] [Google Scholar]
- Kim Y. S.; Jeong S. K. Atomic Force Microscopy for Understanding Solvent Cointercalation into Graphite Electrode in Lithium Second Batteries. J. Spectrosc. 2015, 2015, 462140 10.1155/2015/462140. [DOI] [Google Scholar]
- Yamaguchi S.; Asahina H.; Hirasawa K. A.; Sato T.; Mori S. SEI Film Formation on Graphite Anode Surface in Lithium Ion Battery. Mol. Cryst. Liq. Cryst. Sci. Technol., Sect. A 1998, 322, 239–244. 10.1080/10587259808030230. [DOI] [Google Scholar]
- Liu T.; Lin L.; Bi X.; Tian L.; Yang K.; Liu J.; Li M.; Chen Z.; Lu J.; Anime K.; Xu K.; Pan F. In Situ Quantification of Interphasial Chemistry in Li-Ion Battery. Nat. Nanotechnol. 2019, 14, 50–56. 10.1038/s41565-018-0284-y. [DOI] [PubMed] [Google Scholar]
- Koltypin M.; Cohen Y. S.; Markovsky B.; Cohen Y.; Aurbach D. The Study of Lithium Insertion-deinsertion Processes into Composite Graphite Electrodes by In Situ Atomic Force Microscopy (AFM). Electrochem. Commun. 2002, 4, 17–23. 10.1016/S1388-2481(01)00264-8. [DOI] [Google Scholar]
- Aurbach D.; Koltypin M.; Teller H. In Situ AFM Imaging of Surface Phenomena on Composite Graphite Electrodes during Lithium Insertion. Langmuir 2002, 18, 9000–9009. 10.1021/la020306e. [DOI] [Google Scholar]
- Zhang S. S.; Jow T. R.; Amine K.; Henriksen G. L. LiPF6-EC-EMC Electrolyte for Li-ion Battery. J. Power Sources 2002, 107, 18–23. 10.1016/S0378-7753(01)00968-5. [DOI] [Google Scholar]
- Markevich E.; Salitra G.; Aurbach D. Fluoroethylene Carbonate as an Important Component for the Formation of an Effective Solid Electrolyte Interphase on Anodes and Cathodes for Advanced Li-ion Batteries. ACS Energy Lett. 2017, 2, 1337–1345. 10.1021/acsenergylett.7b00163. [DOI] [Google Scholar]
- Hou T.; Yang G.; Rajput N. N.; Self J.; Park S. W.; Nanda J.; Persson K. A. The Influence of FEC on the Solvation Structure and Reduction Reaction of LiPF6/EC Electrolytes and Its Implication for Solid Electrolyte Interphase Formation. Nano Energy 2019, 64, 103881 10.1016/j.nanoen.2019.103881. [DOI] [Google Scholar]
- Ma L.; Xia J.; Xia X.; Dahn J. R. The Impact of Vinylene Carbonate, Fluoroethylene Carbonate and Vinyl Ethylene Carbonate Electrolyte Additives on Electrode/Electrolyte Reactivity Studied Using Accelerating Rate Calorimetry. J. Electrochem. Soc. 2014, 161, A1495–A1498. 10.1149/2.0091410jes. [DOI] [Google Scholar]
- Son H. B.; Jeong M. Y.; Han J. G.; Kim K.; Kim K. H.; Jeong K. M.; Choi N. S. Effect of Reductive Cyclic Carbonate Additives and Linear Carbonate Co-solvents on Fast Chargeability of LiNi0.6Co0.2Mn0.2O2/Graphite Cells. J. Power Sources 2018, 400, 147–156. 10.1016/j.jpowsour.2018.08.022. [DOI] [Google Scholar]
- Tasaki K.; Kanda K.; Kobayashi T.; Nakamura S.; Ue M. Theoretical Studies on the Reductive Decompositions of Solvent and Additives for Lithium-ion Batteries near Lithium Anodes. J. Electrochem. Soc. 2006, 153, A2192–A2197. 10.1149/1.2354460. [DOI] [Google Scholar]
- Domi Y.; Ochida M.; Tsubouchi S.; Nakagawa H.; Yamanaka T.; Doi T.; Abe T.; Ogumi Z. In Situ AFM Study of Surface Film Formation on the Edge Plane of HOPG for Lithium-ion Batteries. J. Phys. Chem. C 2011, 115, 25484–25489. 10.1021/jp2064672. [DOI] [Google Scholar]
- Seidl L.; Martens S.; Ma J.; Stimming U.; Schneider O. In Situ Scanning Tunnelling Microscopy Studies of the SEI Formation on Graphite Electrodes for Li+-ion Batteries. Nanoscale 2016, 8, 14004–14014. 10.1039/C6NR00825A. [DOI] [PubMed] [Google Scholar]
- Zhou Y.; Su M.; Yu X.; Zhang Y.; Wang J. G.; Ren X.; Cao R.; Xu W.; Baer D. R.; Du Y.; Borodin O.; Wang Y.; Wang X. L.; Xu K.; Xu Z.; Wang C.; Zhu Z. Real-time Mass Spectrometric Characterization of the Solid-electrolyte Interphase of a Lithium-ion Battery. Nat. Nanotechnol. 2020, 15, 224–230. 10.1038/s41565-019-0618-4. [DOI] [PubMed] [Google Scholar]
- Shen C.; Wang S.; Jin Y.; Han W. Q. In Situ AFM Imaging of Solid Electrolyte Interfaces on HOPG with Ethylene Carbonate and Fluoroethylene Carbonate-Based Electrolytes. ACS Appl. Mater. Interfaces 2015, 7, 25441–25447. 10.1021/acsami.5b08238. [DOI] [PubMed] [Google Scholar]
- Soto F. A.; Ma Y.; Hoz J. M. M.; Seminario J. M.; Balbuena P. B. Formation and Growth Mechanisms of Solid-electrolyte Interphase Layers in Rechargeable Batteries. Chem. Mater. 2015, 27, 7990–8000. 10.1021/acs.chemmater.5b03358. [DOI] [Google Scholar]
- Heiskanen S. K.; Kim J.; Lucht B. L. Generation and Evolution of the Solid Electrolyte Interphase of Lithium-ion Batteries. Joule 2019, 3, 2322–2333. 10.1016/j.joule.2019.08.018. [DOI] [Google Scholar]
- Tokranov A.; Kumar R.; Li C.; Minne S.; Xiao X.; Sheldon B. W. Control and Optimization of the Electrochemical and Mechanical Properties of the Solid Electrolyte Interphase on Silicon Electrodes in Lithium Ion Batteries. Adv. Energy Mater. 2016, 6, 1502302 10.1002/aenm.201502302. [DOI] [Google Scholar]
- Morales-Ugarte J. E.; Bolimowska E.; Rouault H.; Santos-Pena J.; Santini C. C.; Benayad A. EIS and XPS Investigation on SEI Layer Formation during First Discharge on Graphite Electrode with a Vinylene Carbonate Doped Imidazolium Based Ionic Liquid Electrolyte. J. Phys. Chem. C 2018, 122, 18223–18230. 10.1021/acs.jpcc.8b03636. [DOI] [Google Scholar]
- Dinkelacker F.; Marzak P.; Yun J.; Liang Y.; Bandarenka A. S. Multistage Mechanism of Lithium Intercalation into Graphite Anodes in the Presence of the Solid Electrolyte Interface. ACS Appl. Mater. Interfaces 2018, 10, 14063–14069. 10.1021/acsami.7b18738. [DOI] [PubMed] [Google Scholar]
- Lu P.; Li C.; Schneider E. W.; Harris S. J. Chemistry, Impedance, and Morphology Evolution in Solid Electrolyte Interphase Films during Formation in Lithium Batteries. J. Phys. Chem. C 2014, 118, 896–903. 10.1021/jp4111019. [DOI] [Google Scholar]
- Schwenke K. U.; Solchenbach S.; Demeaux J.; Lucht B. L.; Gasteiger H. A. The Impact of CO2 Evolved from VC and FEC during Formation of Graphite Anodes in Lithium-ion Batteries. J. Electrochem. Soc. 2019, 166, A2035–A2047. 10.1149/2.0821910jes. [DOI] [Google Scholar]
- Ming J.; Cao Z.; Wahyudi W.; Li M.; Kumar P.; Wu Y.; Hwang J. Y.; Hedhili M. N.; Cavallo L.; Sun Y. K.; Li L. J. New Insights on Graphite Anode Stability in Rechargeable Batteries: Li Ion Coordination Structures Prevail over Solid Electrolyte Interphase. ACS Energy Lett. 2018, 3, 335–340. 10.1021/acsenergylett.7b01177. [DOI] [Google Scholar]
- Joho F.; Rykart B.; Blome A.; Novak P.; Wilhelm H.; Spahr M. E. Relation between Surface Properties, Pore Stucture and First-cycle Charge Loss of Graphite as Negative Electrode in Lithium-ion Batteries. J. Power Sources 2001, 97–98, 78–82. 10.1016/S0378-7753(01)00595-X. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.