

Frequent recombination and strong purifying selection facilitate the cross-species transmission of coronaviruses to humans.
Abstract
COVID-19 has become a global pandemic caused by the novel coronavirus SARS-CoV-2. Understanding the origins of SARS-CoV-2 is critical for deterring future zoonosis, discovering new drugs, and developing a vaccine. We show evidence of strong purifying selection around the receptor binding motif (RBM) in the spike and other genes among bat, pangolin, and human coronaviruses, suggesting similar evolutionary constraints in different host species. We also demonstrate that SARS-CoV-2’s entire RBM was introduced through recombination with coronaviruses from pangolins, possibly a critical step in the evolution of SARS-CoV-2’s ability to infect humans. Similar purifying selection in different host species, together with frequent recombination among coronaviruses, suggests a common evolutionary mechanism that could lead to new emerging human coronaviruses.
INTRODUCTION
The severe respiratory disease COVID-19 was first noticed in late December 2019 (1). It rapidly became an epidemic in China, devastating public health and economy. At the beginning of May, COVID-19 had spread to ~150 countries and infected more than 3.3 million people (2). On 11 March 2020, the World Health Organization officially declared it a pandemic.
The etiological agent of COVID-19 (3), severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) (4), was identified as a new member of the genus Betacoronavirus, which includes a diverse reservoir of coronaviruses (CoVs) isolated from bats (5–7). While genetically distinct from the betacoronaviruses that cause SARS and Middle East respiratory syndrome (MERS) in humans (8, 9), SARS-CoV-2 shares the highest level of genetic similarity (96.3%) with CoV RaTG13, sampled from a bat in Yunnan in 2013 (8). Recently, CoV sequences closely related to SARS-CoV-2 were obtained from confiscated Malaya pangolins in two separate studies (10, 11). These pangolin SARS-like CoVs (Pan_SL-CoV) form two distinct clades corresponding to their locations of origin: The first clade, Pan_SL-CoV_GD, sampled from Guangdong (GD) province in China is genetically more similar to SARS-CoV-2 (91.2%) than the second clade, Pan_SL-CoV_GX, sampled from Guangxi (GX) province (85.4%).
Understanding the origin of SARS-CoV-2 may help develop strategies to deter future cross-species transmissions and to establish appropriate animal models. Recombination plays an important role in the evolution of CoVs (12, 13). Viral sequences nearly identical to SARS and MERS viruses were found in civets and dromedary camels, respectively (14, 15), demonstrating that they originated from zoonotic transmissions with intermediate host species between the bat reservoirs and humans, a common pattern leading to CoV zoonosis (5–7). However, nonhuman viruses nearly identical to SARS-CoV-2 have not yet been found. Here, we demonstrate, through localized genomic analysis, a complex pattern of evolutionary recombination and strong purifying selection between CoVs from distinct host species and cross-species infections that likely originated SARS-CoV-2.
RESULTS
Acquisition of receptor binding motif through recombination
Phylogenetic analysis of 43 complete genome sequences from three clades (SARS-CoVs and Bat_SL-CoVs in clade 3; SARS-CoV-2, Bat_SL-CoVs, and Pan_SL-CoVs in clade 2; and two divergent Bat_SL-CoVs in clade 1) within the Sarbecovirus group (9) confirms that RaTG13 is, overall, the closest sequence to SARS-CoV-2 (fig. S1). Pan_SL-CoV_GD is the next closest virus, followed by Pan_SL-CoV_GX. Among the Bat CoV sequences in clade 2 (fig. S1), ZXC21 and ZC45, sampled from bats in 2005 in Zhoushan, Zhejiang, China, are the most divergent, with the exception of the beginning of the ORF1a gene (region 1; Fig. 1A). All other Bat_SL-CoV and SARS-CoV sequences form a separate clade 3, while clade 1 comprises BtKY72 and BM48-31, the two most divergent Bat_SL-CoV sequences in the Sarbecovirus group (fig. S1). Recombination in the first SARS-CoV-2 sequence (Wuhan-Hu-1) with other divergent CoVs has been previously noted (3). Here, to better understand the role of recombination in the origin of SARS-CoV-2 among these genetically similar CoVs, we compare Wuhan-Hu-1 to six representative Bat_SL-CoVs, one SARS-CoV, and the two Pan_SL-CoV_GD sequences using SimPlot analysis (16). RaTG13 has the highest similarity across the genome (8), with two notable exceptions where a switch occurs (Fig. 1A). In phylogenetic reconstructions, SARS-CoV-2 clusters closer to ZXC21, ZC45, and Longquan than RaTG13 at the beginning of the ORF1a gene (region 1; Fig. 1B) and, as previously reported (10, 17), to a Pan_SL-CoV_GD sequence in region 2 (Fig. 1C and fig. S2), which spans the receptor angiotensin-converting enzyme 2 (ACE2) binding site in the spike (S) glycoprotein gene. When comparing Wuhan-Hu-1 to Pan_SL-CoV_GD and RaTG13, as representative of distinct host-species branches in the evolutionary history of SARS-CoV-2, using the recombination detection tool RIP (18), we find considerable recombination breakpoints before and after the ACE2 receptor binding motif (RBM) (fig. S2A) (19, 20). This suggests that SARS-CoV-2 carries a history of cross-species recombination between bat and pangolin CoVs.
Fig. 1. SARS-CoV-2 recombination with Pan_SL-CoV and Bat_SL-CoV.

(A) SimPlot genetic similarity plot between SARS-CoV-2 Wuhan-Hu-1 and representative CoV sequences using a 400–base pair (bp) window at a 50-bp step and the Kimura two-parameter model. Phylogenetic trees of regions of disproportional similarities, showing high similarities between SARS-CoV-2 and ZXC21 (B) or GD/P1La (C), high genetic divergences of all Pan_SL-CoV sequences (D), and high similarities between GD/P1La and divergent Bat_SL-CoV sequences (E). All positions are relative to Wuhan-Hu-1. Red arrows indicate the discordant clustering relationship of SARS-CoV-2 or Pan_SL-CoV sequences with other CoV sequences. In (A), we use the ORF1a and ORF1b nomenclature consistent with the original publication from of the Wuhan virus (3); however, the National Center for Biotechnology Information (NCBI) betacoronavirus reference sequences (see SAR-CoV-2 and NC_045512.2 for an example) designate a single longer stretch called ORF1ab (from 266 to 21,555) that spans both 1a and 1b.
Pan_SL-CoV sequences are generally more similar to SARS-CoV-2 than other CoV sequences, with the exception of RaTG13 and ZXC21, but are more divergent from SARS-CoV-2 at two regions in particular: the beginning of the ORF1b gene and the highly divergent N terminus of the S gene (regions 3 and 4, respectively; Fig. 1A). Within-region phylogenetic reconstructions show that Pan_SL-CoV sequences become as divergent as BtKY72 and BM48-31 in region 3 (Fig. 1D), while less divergent in region 4, where Pan_SL-CoV_GD clusters with ZXC21 and ZC45 (Fig. 1E). Together, these observations suggest ancestral cross-species recombination between pangolin and bat CoVs in the evolution of SARS-CoV-2 at the ORF1a and S genes. Furthermore, the discordant phylogenetic clustering at various regions of the genome among clade 2 CoVs also supports extensive recombination among these viruses from bats and pangolins.
The SARS-CoV-2 S glycoprotein mediates viral entry into host cells and therefore represents a prime target for drug and vaccine development (12, 19). While SARS-CoV-2 sequences share the greatest overall genetic similarity with RaTG13, this is no longer the case in parts of the S gene. Specifically, amino acid sequences of RBM in the S1 subunit are nearly identical to those in two Pan_SL-CoV_GD viruses, with only one amino acid difference (Q498H)—although the RBM region has not been fully sequenced in one of Guangdong pangolin viruses (Pan_SL-CoV_GD/P2S) (Fig. 2A). Pangolin CoVs from Guangxi are much more divergent. Phylogenetic analysis based on the amino acid sequences of this region shows three distinct clusters of SARS-CoV–, SARS-CoV-2–, and bat CoV–only viruses, respectively (Fig. 2B). While SARS-CoV and SARS-CoV-2 viruses use ACE2 for viral entry, all CoVs in the third cluster have a 5–amino acid deletion and a 13– to 14–amino acid deletion in RBM (Fig. 2A) and do not infect human target cells (5, 21, 22).
Fig. 2. Impact of SARS-CoV-2 recombination on coreceptor binding.
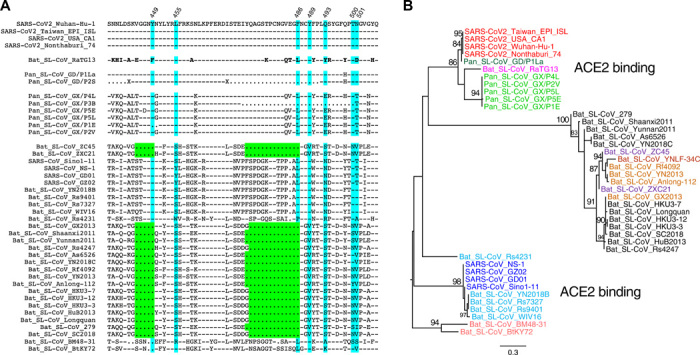
(A) Amino acid sequences of the receptor binding motif (RBM) in the S gene among Sarbecovirus CoVs compared with Wuhan-Hu-1 (top). Dashes indicate identical amino acids, and dots indicate deletions. ACE2 critical contact sites are highlighted in blue, and two large deletions in green. (B) Phylogenetic tree analysis of amino acid sequences of RBM. Viruses with the ability to bind ACE2 form two distinct clusters (one including SARS_CoVs and the other including SARS_CoV-2s). Bat SL-CoVs with large deletions form another distinct cluster.
Although both SARS-CoV and SARS-CoV-2 use the human ACE2 as their receptors (8, 23), they show a high level of genetic divergence (Fig. 1 and fig. S1). However, structures of the S1 unit of the S protein from both viruses are highly similar (20, 24–26), with the exception of a loop that bends differently (Fig. 3A). The root mean square deviation (RMSD) between the two S proteins is 1.2 Å over 174 Cα residues (24). This suggests that conformational similarity of the binding motif enables viral entry through molecular recognition of ACE2. These structural studies also thoroughly analyzed the contact residues between the S protein and human ACE2 (20, 24). Previous structural and mutagenesis studies have identified two hotspots, K31 and K353, at the S/ACE2 interface in SARS-CoV. In SARS-CoV-2, these two hotspots were slightly weakened because of different residues on its S protein, but the loop that takes different conformations from SARS-CoV provides additional interaction that strengthens the interaction (26). Among 17 distinct amino acids between SARS-CoV-2 and RaTG13 in the RBM region (Fig. 2A), five contact sites based on the structural studies (24) are different, likely affecting RaTG13’s binding to ACE2 (Fig. 3B and table S1). The single amino acid difference at position 498 (Q or H) between SARS-CoV-2 and Pan_SL-CoV_GD is at the edge of the ACE2 contact interface; neither Q nor H at this position forms hydrogen bonds with ACE2 residues (Fig. 3C). Thus, a functional RBM nearly identical to the one in SARS-CoV-2 is naturally present in Pan_SL-CoV_GD viruses. The very distinctive RaTG13 RBM suggests that this virus will not likely infect human cells efficiently. Indeed, a recent study showed that the RaTG13 pseudovirus is much less efficient than SARS-CoV-2 pseudoviruses in using ACE2 to infect cells, and this is most likely due to the L486F and Y493Q substitutions, which result in lower ACE2 binding in RaTG13 (26). Therefore, it is likely that the acquisition of a complete functional RBM by a RaTG13-like CoV through a recombination event with a Pan_SL-CoV_GD–like virus enabled it to more efficiently use ACE2 for human infection.
Fig. 3. Structure analysis of the RBM and ACE2 interface.
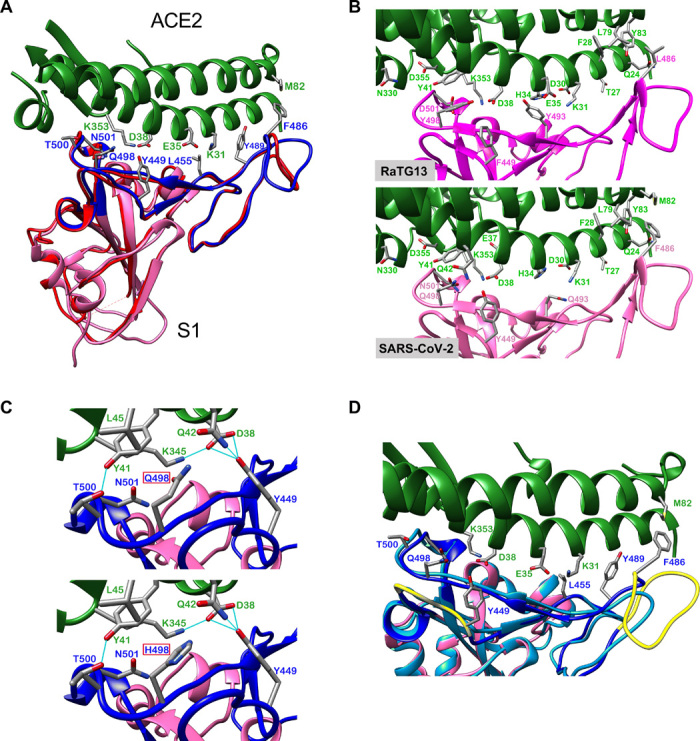
(A) SARS-CoV and SARS-CoV-2 receptor binding domains (RBD). Human ACE2 in green (PDB 6M0J) at the top and the RBD of the S protein at the bottom; SARS-CoV S protein (PDB 2AJF) in red, and SARS-CoV-2 S protein (PDB 6M0J) in magenta with RBM in blue. All structure backbones are shown as ribbons with key residues at the interface shown as stick models, labeled using the same color scheme. (B) Impact of different RBM amino acids between SARS-CoV-2 RaTG13 on ACE2 binding. (C) Impact of an amino acid at position 498 (Q in SARS-CoV-2, top; H in RaTG13, bottom) on ACE2 binding. Same color coding as in (A) with additional hydrogen bonding as light-blue lines. (D) Impact of two deletions on ACE2 binding interface in some bat SL-CoVs; positions indicated in yellow, and modeled structure with long deletion between residues 473 and 486 in light blue.
Three small insertions are identical in SARS-CoV-2 and RaTG13 but not found in other CoVs in the Sarbecovirus group (27, 28). The RaTG13 sequence was sampled in 2013, years before SARS-CoV-2 was first identified. It is unlikely that both SARS-CoV-2 and RaTG13 independently acquired identical insertions at three different locations in the S gene. Thus, it is plausible that an RaTG13-like virus served as a progenitor to generate SARS-CoV-2 by gaining a complete human ACE2 binding RBM from Pan_SL-CoV_GD–like viruses through recombination. Genetic divergence at the nucleic acid level between Wuhan-Hu-1 and Pan_SL-CoV_GD viruses is significantly reduced from 13.9% (Fig. 1E) to 1.4% at the amino acid level (Fig. 2B) in the RBM region, indicating recombination between RaTG13-like CoVs and Pan_SL-CoV_GD–like CoVs. Furthermore, SARS-CoV-2 has a unique furin cleavage site insertion (PRRA) not found in any other CoVs in the Sarbecovirus group (fig. S3) (27), although similar motifs are also found in MERS and more divergent bat CoVs (29). This PRRA motif makes the S1/S2 cleavage in SARS-CoV-2 much more efficient than in SARS-CoV and may expand its tropism and/or enhance its transmissibility (20). A recent study of bat CoVs in Yunnan, China, identified a three–amino acid insertion (PAA) at the same site (30). Although it is not known whether this PAA motif can function similar to the PRRA motif, the presence of a similar insertion at the same site indicates that such insertion may already be present in the wild bat CoVs. The more efficient cleavage of S1 and S2 subunits of the S glycoprotein (29) and efficient binding to ACE2 by SARS-CoV-2 (20, 25) may have allowed SARS-CoV-2 to jump to humans, leading to the rapid spread of SARS-CoV-2 in China and the rest of the world.
Strong purifying selection among SARS-CoV-2 and closely related viruses
Recombination with Pan_SL-CoV_GD at the RBM and the unique furin cleavage site insertion prompted us to examine the SARS-CoV-2 sequences at these regions. Amino acid sequences from SARS-CoV-2, RaTG13, and all Pan_SL-CoV viruses (group A) are identical or nearly identical in the region before and after the RBM and at the region after the furin cleavage site (S2 subunit), while all other CoVs (group B) are very distinctive (Fig. 4A and fig. S4). The average of all pairwise dN/dS ratios, defined as ω, among SARS-CoV-2, RaTG13, and Pan_SL-CoV viruses at the S2 subunit is ω = 0.013, compared with the much higher values ω = 0.053 in the S1 region preceding the furin cleavage site, and ω = 0.042 at the S2 subunit for all other CoVs (Fig. 4B). The much lower ω value at the S2 subunit among the SARS-CoV-2, RaTG13, and Pan_SL-CoV viruses indicates that this region is under strong purifying selection within these sequences. A plot of synonymous and nonsynonymous substitutions relative to Wuhan-Hu-1 highlights the regional differences across the region before and after the furin cleavage site (Fig. 4A): The S2 subunit is highly conserved among the SARS-CoV-2, RaTG13, and Pan_SL-CoV viruses (group A), while far more nonsynonymous mutations are observed in the rest of the CoV sequences (group B). The shift in selective pressure at the S1/S2 cleaveage site among these related viruses versus other CoVs begins near codon 368 (Fig. 4B): The two graphs show the cumulative plots of the average behavior of each codon for all pairwise comparisons in the input data, for synonymous mutations, nonsynonymous mutations, and indels of group A sequences and group B sequences. The nonsynonymous plot shows a marked change in slope (vertical step) in the group A sequences at codon 368 but not in group B sequences. Similarly, when looking at all the dS/dN ratios (ω) for each group A sequence compared with the Wuhan-Hu-1 sequence, we see that these ratios are much lower in the 5′ end of the region, before codon 368 (nucleic acid position 1104), compared with the 3′ end, and no such difference is observed in the group B sequences (Fig. 4C).
Fig. 4. Strong purifying selection after furin cleavage in S gene among SARS-CoV-2 and closely related viruses.

(A) Phylogenetic tree (left) and highlighter plot (right) of sequences around the RBM and furin cleavage site compared with SARS-CoV-2 Wuhan-Hu-1 [nucleic acid (na) positions 22,541 to 24,391]. ACE2 RBM and furin cleavage site highlighted in light-gray boxes. Mutations compared with Wuahn-Hu-1 are light blue for synonymous and red for nonsynonymous. Dominance of synonymous mutations within group A compared with group B highlighted on the right. Position numbers are counted in number of nucleotides (NA) from the beginning of the region. (B) Cumulative plots of each codon average behavior for all pairwise comparisons for indels, synonymous (light blue), and nonsynonymous (red) mutations, by group. The abrupt slope change of the nonsynonymous curve in group A at around codon 368 (na 1104) is indicative of a shift in localized accumulations of nonsynonymous mutations after the furin cleavage site. Group B instead lacks this abrupt change in slope at the same position. Values of ω denote average ratios of the rate of nonsynonymous substitutions per nonsynonymous site (dN/dS) for each group and region. Position numbers are counted in number of amino acids (AA) from the beginning of the region. (C) Sequence dS/dN ratios compared with Wuhan-Hu-1 within codons 1 to 368 (na 1 to 1104; green) and codons 369 to 620 (na 1105 to 1893; dark blue) in group A and group B sequences.
This strong purifying selection observed in the S2 subunit of the S gene is not unexpected given its role in cell entry by fusing the viral and host cell membranes (5, 19). Following the binding of receptor binding domains (RBD) to the ACE2 receptor, heptad repeat region 1 (HR1) and HR2 within the S2 subunit rearrange to form the fusion core, bringing together the viral and cell membranes for fusion and infection (fig. S5A). Because of the mechanistic constraints for this assembly for fusion, the protein segments that take part in this assembly are well preserved (20, 31). Furthermore, some regions of the S2 subunit are covered by S1 in the trimer conformation of the S protein (fig. S5B). On the basis of the currently available, but incomplete, cryo-EM (cryogenic electron microscopy) structure of the S trimer, we estimate that 60 to 65% of the S2 amino acids are buried. This adds further structural constraints on changing amino acids in S2.
While hundreds of new SARS-CoV-2 sequences are added to the GISAID (Global Initiative on Sharing All Influenza Data) repertoire every day (32), we note that the RBM region currently remains highly conserved. No amino acid within 6 Å of the ACE2 binding site has repeated variations, with the exception of G476S, a very rare mutation found in eight sequences from a local cluster in the Washington state, out of 6400 total sequences from GISAID (13 April 2020). In addition, we observe similar patterns of purifying selection pressure in other parts of the genome, including the E and M genes, as well as the partial ORF1a and ORF1b genes (figs. S6 and S7). The viruses affected by purifying selection pressure vary depending on which genes are analyzed. SARS-CoV-2, RaTG13, all Pan_SL-CoV, and the two bat CoVs (ZXC21 and ZC45) are under the similar purifying selection in both the E and M genes (Fig. 5A and fig. S6). In the S2 subunit, similar purifying selection is only observed for SARS-CoV-2, RaTG13, and all Pan_SL-CoV (Fig. 5B). A few viruses including only SARS-CoV-2, RaTG13, and pangolin CoVs from Guangdong are under similar purifying selection in the partial regions of ORF1a and ORF1b (Fig. 5C and fig. S7). Strong purifying selection pressure on SARS-CoV-2, RaTG13, and Pan_SL-CoV_GD viruses, as indicated by consistently low ω values, suggests that these complete and partial genes are under similar functional/structural constraints among the different host species. In two extreme cases, amino acid sequences of the E gene and the 3′ end of ORF1a are identical among the compared CoV sequences, although genetic distances are quite large among these viruses at the nucleic acid level (Fig. 5, A and C). Such evolutionary constraints in many parts of the viral genome, especially at functional domains in the S gene, which plays an important role in cross-species transmission (5, 12), coupled with frequent recombination, may facilitate cross-species transmissions between RaTG13-like bat and/or Pan_SL-CoV_GD–like viruses.
Fig. 5. Strong purifying selection on complete and partial gene regions among SARS-CoV-2, RaTG13, and Pan_SL-CoV viruses.

Purifying selection pressure on complete and partial genes among different viruses (red boxes), as evident by shorter branches in amino acid sequence trees compared with nucleic acid sequence trees. Distinct purifying selection patterns are observed among different viruses: (A) SARS-CoV-2, RaTG13, all Pan_SL-CoV, and Bat_CoV ZXC21 and ZC45; (B) SARS-CoV-2, RaTG13, and all Pan_SL-CoV sequences; and (C) SARS-CoV-2, RaTG13, and Pan_SL-CoV_GD. Cumulative plots of the average behavior of each codon for all pairwise comparisons for synonymous mutations, nonsynonymous mutations, and indels within each gene region. ω denotes the average ratio of the rate of nonsynonymous substitutions per nonsynonymous site (dN/dS) for each group.
Frequent recombination between SARS-CoVs and Bat_SL-CoVs
Previous studies using limited sequence sets found that SARS-CoVs originated through multiple recombination events between different bat CoVs (10, 12, 21, 33, 34). Our phylogenetic analyses of individual genes confirms this and shows that SARS-CoV sequences tend to cluster with YN2018B, Rs9401, Rs7327, WIV16, and Rs4231 (group A) for some genes and with Rf4092, YN2013, Anlong-112, and GX2013 (group B) for others (fig. S8). SimPlot analysis using both groups of Bat_SL-CoVs and the closely related bat CoV YNLF-34C (34) shows that SARS-CoV GZ02 shifts in similarity among different Bat_SL-CoVs at various regions of the genome (Fig. 6A). In particular, phylogenetic reconstruction of the beginning of ORF1a (region 1) confirms that SARS-CoVs cluster with YNLF-34C (34), and this cluster is distinctive compared with all other CoVs (Fig. 6B). YNLF-34C is more divergent from SARS-CoV than other bat CoV viruses before and after this region, confirming the previously reported complex recombinant nature of YNLF-34C (Fig. 5A) (34). At the end of the S gene (region 2), SARS-CoVs cluster with group A CoVs, forming a highly divergent clade (Fig. 6C). In region 3 (ORF8), SARS-CoVs and group B CoVs, together with YNLF-34C, form a very divergent and distinctive cluster (Fig. 6D). To further explore the recombinant nature of SARS-CoVs, we compared GZ02 to representative bat CoV sequences using the RIP recombination detection tool (18). We identified four significant breakpoints (at 99% confidence) between the two parental lineages (fig. S9A), further supported by phylogenetic analysis (fig. S9, B to D). In addition, the two aforementioned groups of bat CoVs (shown in light brown and light blue in the trees) show similar cluster changes across the five recombinant regions, suggesting multiple events of historic recombination among bat SL-CoVs. These results demonstrate that SARS-CoV shares a recombinant history with at least three different groups of bat CoVs and confirm the major role of recombination in the evolution of these viruses.
Fig. 6. Multiple recombination of SARS-CoVs with different Bat_SL-CoVs.

(A) SimPlot genetic similarity plot between SARS-CoV GZ02 and SARS_SL-CoVs, using a 400-bp window at a 50-bp step and the Kimura two-parameter model. Group A CoVs (YN2018B, Rs9401, Rs7327, WIV16, and Rs4231) are shown in blue, group B CoVs (Rf4092, YN2013, Anlong-112, and GX2013) in orange, YNLF-34C in green, and outlier control HKU3-12 in red. Phylogenetic trees for high-similarity regions between GZ02 and YNLF-34C (B), group A (C), and group B (D). All positions are relative to Wuhan-Hu-1. Red arrows indicate the distinct clustering relationship of SARS-CoV sequences with other different CoV sequences.
Of the bat SL-CoVs that contributed to the recombinant origin of SARS-CoV, only group A viruses bind to ACE2. Group B bat SL-CoVs do not infect human cells (5, 21, 22) and have two deletions in the RBM (Figs. 1E and 2A). The short deletion between residues 445 and 449, and in particular the loss of Y449, which forms three hydrogen bonds with ACE2, will significantly affect the overall structure of the RBM (Fig. 3, C and D). The region encompassing the large deletion between residues 473 and 486 contains the loop structure that accounts for the major differences between the S protein of SARS-CoV and SARS-CoV-2 (Fig. 3A) and strengthens the interaction of the latter to ACE2 (26). This deletion causes the loss of contact site F486 and affects the conserved residue F498’s hydrophobic interaction with residue M82 on ACE2 (Fig. 3D). These two deletions will render RBM in those CoVs incapable of binding human ACE2. Therefore, recombination may play a role in enabling cross-species transmission in SARS-CoVs through the acquisition of an S gene type that can efficiently bind to the human ACE2 receptor.
ORF8 is one of the highly variable genes in CoVs, and its function has not yet been well elucidated (5, 12, 35). Recombination breakpoints within this region show that recombination occurred at the beginning and at the end of ORF8 (fig. S10), where nucleic acid sequences are nearly identical among both SARS-CoVs and group B bat CoVs. Moreover, all compared viruses form three highly distinct clusters (Fig. 6D), suggesting that the ORF8 gene may be biologically constrained and evolves through modular recombination. The third recombination region at the beginning of ORF1a is near where SARS-CoV-2 also recombined with other bat CoVs (region 1 in Fig. 1A). This region is highly variable (5, 12), and recombination within this part of the genome was also found in other CoVs, suggesting that it may be a recombination hotspot and may factor into cross-species transmission.
DISCUSSION
There are three important aspects to betacoronavirus evolution that should be carefully considered in phylogenetic reconstructions among more distant CoVs. First, there is extensive recombination among all of these viruses (Figs. 1 and 5) (10, 12, 21, 33, 34), making standard phylogenetic reconstructions based on full genomes problematic, as different regions of the genome have distinct ancestral relationships. Second, between more distant sequences, synonymous substitutions are often fully saturated, which can confound analyses of selective pressure and add noise to phylogenetic analysis. Last, there are different selective pressures at work in different lineages, which is worth considering when interpreting trees.
The currently sampled pangolin CoVs are too divergent from SARS-CoV-2 to be its recent progenitors, but it is noteworthy that these sequences contain an RBM that can most likely bind to human ACE2. While RaTG13 is the most closely related CoV sequence to SARS-CoV-2, it has a distinctive RBM. In addition, a recent study showed that the RaTG13 pseudovirus is much less efficient than the SARS-CoV-2 pseudovirus in using ACE2 to infect cells (26). SARS-CoV-2 has a nearly identical RBM to the one found in the pangolin CoVs from Guangdong. Thus, it is plausible that RaTG13-like bat CoV viruses may have obtained the RBM sequence binding to human ACE2 through recombination with Pan_SL-CoV_GD–like viruses. We hypothesize that this, and/or other ancestral recombination events between viruses infecting bats and pangolins, may have played a key role in the evolution of the strain that led to the introduction of SARS-CoV-2 into humans. It is also possible that other not yet identified hosts infected with CoVs that can jump to human populations through cross-species transmission if they can successfully infect human cells through ACE2 or other receptors. An analysis of 6400 SARS-CoV-2 sequences from GISAID (36, 37) identifies only one very rare mutation, G476S that is a direct ACE2 contact residue. It was found in a local cluster of sequences from the Washington state. However, it is at the periphery of the receptor contact surface and so may not significantly affect the virus’ receptor binding affinity.
All three human CoVs (SARS, MERS, and SARS-2) are the result of recombination among CoVs. Recombination in all three viruses involved the S gene, likely a precondition to zoonosis that enabled efficient binding to human receptors (5, 12). Extensive recombination among bat CoVs and strong purifying selection pressure among viruses from humans, bats, and pangolins may allow such closely related viruses to readily jump between species and adapt to new hosts. Many bat CoVs have been found able to bind to human ACE2 and replicate in human cells (10, 21, 22, 38–40). Serological evidence has revealed that additional, otherwise undetected, spillovers have occurred in people in China living in proximity to wild bat populations (41). Continuous surveillance of CoVs in their natural hosts and in humans will be the key to rapidly control new CoV outbreaks.
While the SARS- and MERS-originating strains have been found in civets and dromedary camels, respectively (14, 15), so far, efforts to identify a similarly close link in the original pathway of SARS-CoV-2 into humans have failed. If the new SARS-CoV-2 strain did not cause widespread infections in its natural or intermediate hosts, then such a strain may never be identified. The close proximity of animals of different species in a wet market setting may increase the potential for cross-species spillover infections by enabling recombination between more distant CoVs and the emergence of recombinants with novel phenotypes. While the direct reservoir of SARS-CoV-2 is still being sought, one thing is clear: Reducing or eliminating direct human contact with wild animals is critical to preventing new CoV zoonosis in the future.
MATERIALS AND METHODS
Sequences analysis
All 43 CoV complete genome sequences were obtained from GenBank and GISAID (36, 37) and were selected to be representative of the diversity (tables S2 and S3). Pan_SL-CoV_GD/P1La sequence was generated by combining Pan_SL-CoV_GD/P1L (10) with some additional sequences from the National Center for Biotechnology Information (NCBI) BioProject database PRJNA5732983 (11, 42) to have a maximal coverage of the complete genome sequence for analysis. A new CoV sequence from pangolin (EPI_ISL_410721) (43) was not included here, as it became available after we had already completed the analyses in this study. Once it became available, we observed that it was as close to SARS-CoV-2 as the sequences we had already used and, hence, did not change the interpretation of our results. Whole-genome sequences were first aligned using Clustal X2 (44). The alignments for all coding regions were manually optimized on the basis of the amino acid sequence alignment using SeaView 5.0.1.
Recombination analyses
SimPlot 3.5.15 (16) was used to determine the percent identity of the query sequence to reference sequences. Potential recombinant regions among analyzed sequences were identified by sliding a 400–base pair (bp) window at a 50-bp step across the alignment using the Kimura two-parameter model. Phylogenetic trees were constructed by the maximum likelihood method using the Generalized Time Reversible (GTR) model (45), and their reliability was estimated from 1000 bootstrap replicates. The positions of the analyzed sequence regions were based on those in the reference SARS-CoV-2 Wuhan-Hu-1 (MN908947). Recombination regions and breakpoints were also analyzed using the LANL (Los Alamos National Laboratory) database (46) tool RIP (18) with a 400-bp window. Regions between breakpoints were identified using a 99% confidence threshold.
Selection analyses
Cumulative plots of the average behavior of each codon for all pairwise comparisons in the input data, for insertions and deletions (indels), synonymous (syn), and nonsynonymous (nonsyn) mutations and values of the ratios of the rate of synonymous nucleotide substitutions per synonymous site and nonsynonymous substitutions per nonsynonymous site (dN/dS or ω), were obtained using the LANL database tool SNAP (47). To avoid counting instances where synonymous mutations were saturated, averages of all pairwise dN/dS ratios were calculated excluding pairs that yielded dS values greater than 1.
Structure modeling of receptor binding
To investigate the single mutation Q498H in RBM between SARS-CoV-2 and Pan_SL-CoV_GD, Q498 in the crystal structure of S/ACE2 complex was mutated to H498 using Chimera (48). Local energy minimization (only H498 was allowed to move) was computed using Chimera’s built-in functions. To investigate the impact of the deletion between residue 473 to 486 to the binding interface between SARS-CoV-2 and human ACE2, a homology model with the deletion was generated using I-TASSER (49). The top five best models provided by the server have confidence scores (C-score) of 0.86, −2.33, −4.01, −4.17, and −4.49. The C-score was used to estimate the quality of the models, which should be between −5.0 and 2; the higher the value, the higher the confidence in the model (49). On the basis of the C-score, model 1 was used in Fig. 3D. The interaction of the RBD of RaTG13 and ACE2 was modeled on PDB 6M0J, a structure of RBD of SARS-CoV-2 in complex with human ACE2 (24) using the ICM software package (50), and the mutational differences of the Gibbs free energy (table S1) were calculated with the built-in algorithm.
Supplementary Material
Acknowledgments
We thank all those who have contributed SARS-CoV-2 genome sequences to the GISAID database (https://gisaid.org). We also thank X. Wang from Tsinghua University for sharing the PDB 6M0J structure with us before its official release date. Funding: E.E.G., B.K., S.G., and B.F. acknowledge support by the Laboratory Directed Research and Development program of Los Alamos National Laboratory under project number 20200554ECR. Author contributions: Project conceptualization: F.G., B.K., and E.E.G. Structure analysis: C.X., X.-P.K., and S.G. Sequence analysis: F.G., B.K., X.L., E.E.G., M.H.M., Y.C., and B.F. Phylogenetic analysis: F.G., B.K., X.L., E.E.G., M.H.M., and Y.C. Recombination analysis: F.G., E.E.G., B.K., X.L., M.H.M., and B.F. Manuscript writing: F.G., B.K., and E.E.G. Manuscript editing: F.G., B.K., E.E.G., X.L., C.X., and X.-P.K. F.G. and B.F. supervised the project. Competing interests: All authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/27/eabb9153/DC1
REFERENCES AND NOTES
- 1.Zhu N., Zhang D., Wang W., Li X., Yang B., Song J., Zhao X., Huang B., Shi W., Lu R., Niu P., Zhan F., Ma X., Wang D., Xu W., Wu G., Gao G. F., Tan W.; for the China Novel Coronavirus Investigating and Research Team , A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 382, 727–733 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.W. H. Organization, Novel Coronavirus (COVID-19) Situation (2020); https://experience.arcgis.com/experience/685d0ace521648f8a5beeeee1b9125cd.
- 3.Wu F., Zhao S., Yu B., Chen Y.-M., Wang W., Song Z.-G., Hu Y., Tao Z.-W., Tian J.-H., Pei Y.-Y., Yuan M.-L., Zhang Y.-L., Dai F.-H., Liu Y., Wang Q.-M., Zheng J.-J., Xu L., Holmes E. C., Zhang Y.-Z., A new coronavirus associated with human respiratory disease in China. Nature 579, 265–269 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Coronaviridae Study Group of the International Committee on Taxonomy of Viruses , The species severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 5, 536–544 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cui J., Li F., Shi Z. L., Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 17, 181–192 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin X.-D., Wang W., Hao Z.-Y., Wang Z.-X., Guo W.-P., Guan X.-Q., Wang M.-R., Wang H.-W., Zhou R.-H., Li M.-H., Tang G.-P., Wu J., Holmes E. C., Zhang Y.-Z., Extensive diversity of coronaviruses in bats from China. Virology 507, 1–10 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Banerjee A., Kulcsar K., Misra V., Frieman M., Mossman K., Bats and coronaviruses. Viruses 11, 41 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou P., Yang X.-L., Wang X.-G., Hu B., Zhang L., Zhang W., Si H.-R., Zhu Y., Li B., Huang C.-L., Chen H.-D., Chen J., Luo Y., Guo H., Jiang R.-D., Liu M.-Q., Chen Y., Shen X.-R., Wang X., Zheng X.-S., Zhao K., Chen Q.-J., Deng F., Liu L.-L., Yan B., Zhan F.-X., Wang Y.-Y., Xiao G.-F., Shi Z.-L., A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579, 270–273 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lu R., Zhao X., Li J., Niu P., Yang B., Wu H., Wang W., Song H., Huang B., Zhu N., Bi Y., Ma X., Zhan F., Wang L., Hu T., Zhou H., Hu Z., Zhou W., Zhao L., Chen J., Meng Y., Wang J., Lin Y., Yuan J., Xie Z., Ma J., Liu W. J., Wang D., Xu W., Holmes E. C., Gao G. F., Wu G., Chen W., Shi W., Tan W., Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 395, 565–574 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lam T. T.-Y., Shum M. H.-H., Zhu H.-C., Tong Y.-G., Ni X.-B., Liao Y.-S., Wei W., Cheung W. Y.-M., Li W.-J., Li L.-F., Leung G. M., Holmes E. C., Hu Y.-L., Guan Y., Identifying SARS-CoV-2 related coronaviruses in Malayan pangolins. Nature, (2020). [DOI] [PubMed] [Google Scholar]
- 11.Liu P., Chen W., Chen J.-P., Viral metagenomics revealed Sendai virus and coronavirus infection of Malayan pangolins (Manis javanica). Viruses 11, 979 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Graham R. L., Baric R. S., Recombination, reservoirs, and the modular spike: Mechanisms of coronavirus cross-species transmission. J. Virol. 84, 3134–3146 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rehman S. U., Shafique L., Ihsan A., Liu Q., Evolutionary trajectory for the emergence of novel coronavirus SARS-CoV-2. Pathogens 9, 240 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guan Y., Zheng B. J., He Y. Q., Liu X. L., Zhuang Z. X., Cheung C. L., Luo S. W., Li P. H., Zhang L. J., Guan Y. J., Butt K. M., Wong K. L., Chan K. W., Lim W., Shortridge K. F., Yuen K. Y., Peiris J. S. M., Poon L. L. M., Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science 302, 276–278 (2003). [DOI] [PubMed] [Google Scholar]
- 15.Azhar E. I., El-Kafrawy S. A., Farraj S. A., Hassan A. M., Al-Saeed M. S., Hashem A. M., Madani T. A., Evidence for camel-to-human transmission of MERS coronavirus. N. Engl. J. Med. 370, 2499–2505 (2014). [DOI] [PubMed] [Google Scholar]
- 16.Lole K. S., Bollinger R. C., Paranjape R. S., Gadkari D., Kulkarni S. S., Novak N. G., Ingersoll R., Sheppard H. W., Ray S. C., Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 73, 152–160 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wong M. C., Javornik Cregeen S. J., Ajami N. J., Petrosino J. F., Evidence of recombination in coronaviruses implicating pangolin origins of nCoV-2019. bioRxiv, 2020.2002.2007.939207 (2020). [Google Scholar]
- 18.Siepel A. C., Halpern A. L., Macken C., Korber B. T., A computer program designed to screen rapidly for HIV type 1 intersubtype recombinant sequences. AIDS Res. Hum. Retrovir. 11, 1413–1416 (1995). [DOI] [PubMed] [Google Scholar]
- 19.Li F., Structure, function, and evolution of coronavirus spike proteins. Annu. Rev. Virol. 3, 237–261 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walls A. C., Park Y.-J., Tortorici M. A., Wall A., McGuire A. T., Veesler D., Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 180, 281–292.e6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hu B., Zeng L.-P., Yang X.-L., Ge X.-Y., Zhang W., Li B., Xie J.-Z., Shen X.-R., Zhang Y.-Z., Wang N., Luo D.-S., Zheng X.-S., Wang M.-N., Daszak P., Wang L.-F., Cui J., Shi Z.-L., Discovery of a rich gene pool of bat SARS-related coronaviruses provides new insights into the origin of SARS coronavirus. PLOS Pathog. 13, e1006698 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Letko M., Marzi A., Munster V., Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat. Microbiol. 5, 562–569 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li W., Moore M. J., Vasilieva N., Sui J., Wong S. K., Berne M. A., Somasundaran M., Sullivan J. L., Luzuriaga K., Greenough T. C., Choe H., Farzan M., Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 426, 450–454 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lan J., Ge J., Yu J., Shan S., Zhou H., Fan S., Zhang Q., Shi X., Wang Q., Zhang L., Wang X., Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 581, 215–220 (2020). [DOI] [PubMed] [Google Scholar]
- 25.Wrapp D., Wang N., Corbett K. S., Goldsmith J. A., Hsieh C.-L., Abiona O., Graham B. S., McLellan J. S., Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science (New York, N.Y.) 367, 1260–1263 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shang J., Ye G., Shi K., Wan Y., Luo C., Aihara H., Geng Q., Auerbach A., Li F., Structural basis of receptor recognition by SARS-CoV-2. Nature 581, 221–224 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xiao C., Li X., Liu S., Sang Y., Gao S.-J., Gao F., HIV-1 did not contribute to the 2019-nCoV genome. Emerg. Microbes Infect. 9, 378–381 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Andersen K. G., Rambaut A., Lipkin W. I., Holmes E. C., Garry R. F., The proximal origin of SARS-CoV-2. Nat. Med. 26, 450–452 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Coutard B., Valle C., de Lamballerie X., Canard B., Seidah N. G., Decroly E., The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antivir. Res. 176, 104742 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou H., Chen X., Hu T., Li J., Song H., Liu Y., Wang P., Liu D., Yang J., Holmes E. C., Hughes A. C., Bi Y., Shi W., A novel bat coronavirus reveals natural insertions at the S1/S2 cleavage site of the Spike protein and a possible recombinant origin of HCoV-19. bioRxiv, 2020.2003.2002.974139 (2020). [Google Scholar]
- 31.Xia S., Liu M., Wang C., Xu W., Lan Q., Feng S., Qi F., Bao L., Du L., Liu S., Qin C., Sun F., Shi Z., Zhu Y., Jiang S., Lu L., Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 30, 343–355 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.A. Brufsky, Distinct viral clades of SARS-CoV-2: Implications for modeling of viral spread. J. Med. Virol. [Online ahead of print], (2020); 10.1002/jmv.25902. [DOI] [PMC free article] [PubMed]
- 33.Hon C.-C., Lam T.-Y., Shi Z.-L., Drummond A. J., Yip C.-W., Zeng F., Lam P.-Y., Leung F. C.-C., Evidence of the recombinant origin of a bat severe acute respiratory syndrome (SARS)-like coronavirus and its implications on the direct ancestor of SARS coronavirus. J. Virol. 82, 1819–1826 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lau S. K. P., Feng Y., Chen H., Luk H. K. H., Yang W.-H., Li K. S. M., Zhang Y.-Z., Huang Y., Song Z.-Z., Chow W.-N., Fan R. Y. Y., Ahmed S. S., Yeung H. C., Lam C. S. F., Cai J.-P., Wong S. S. Y., Chan J. F. W., Yuen K.-Y., Zhang H.-L., Woo P. C. Y., Severe acute respiratory syndrome (SARS) coronavirus ORF8 protein is acquired from SARS-related coronavirus from greater horseshoe bats through recombination. J. Virol. 89, 10532–10547 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chan J. F.-W., Kok K.-H., Zhu Z., Chu H., To K. K.-W., Yuan S., Yuen K.-Y., Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes Infect. 9, 221–236 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shu Y., McCauley J., GISAID: Global initiative on sharing all influenza data - from vision to reality. Euro Surveill. 22, 30494 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Elbe S., Buckland-Merrett G., Data, disease and diplomacy: GISAID's innovative contribution to global health. Global Chall. 1, 33–46 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Menachery V. D., Yount B. L. Jr., Debbink K., Agnihothram S., Gralinski L. E., Plante J. A., Graham R. L., Scobey T., Ge X.-Y., Donaldson E. F., Randell S. H., Lanzavecchia A., Marasco W. A., Shi Z.-L., Baric R. S., A SARS-like cluster of circulating bat coronaviruses shows potential for human emergence. Nat. Med. 21, 1508–1513 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Menachery V. D., Yount B. L. Jr., Sims A. C., Debbink K., Agnihothram S. S., Gralinski L. E., Graham R. L., Scobey T., Plante J. A., Royal S. R., Swanstrom J., Sheahan T. P., Pickles R. J., Corti D., Randell S. H., Lanzavecchia A., Marasco W. A., Baric R. S., SARS-like WIV1-CoV poised for human emergence. Proc. Natl. Acad. Sci. U.S.A. 113, 3048–3053 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ge X.-Y., Li J.-L., Yang X.-L., Chmura A. A., Zhu G., Epstein J. H., Mazet J. K., Hu B., Zhang W., Peng C., Zhang Y.-J., Luo C.-M., Tan B., Wang N., Zhu Y., Crameri G., Zhang S.-Y., Wang L.-F., Daszak P., Shi Z.-L., Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature 503, 535–538 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang N., Li S.-Y., Yang X.-L., Huang H.-M., Zhang Y.-J., Guo H., Luo C.-M., Miller M., Zhu G., Chmura A. A., Hagan E., Zhou J.-H., Zhang Y.-Z., Wang L.-F., Daszak P., Shi Z.-L., Serological evidence of bat SARS-related coronavirus infection in humans, China. Virol. Sin. 33, 104–107 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.P. Liu, J.-Z. Jiang, X.-F. Wan, Y. Hua, X. Wang, F. Hou, J. Chen, J. Zou, J. Chen, Are pangolins the intermediate host of the 2019 novel coronavirus (2019-nCoV)? bioRxiv, 2020.2002.2018.954628 (2020); 10.1101/2020.02.18.954628. [DOI]
- 43.Wang N., Li S.-Y., Yang X.-L., Huang H.-M., Zhang Y.-J., Guo H., Luo C.-M., Miller M., Zhu G., Chmura A. A., Hagan E., Zhou J.-H., Zhang Y.-Z., Wang L.-F., Daszak P., Shi Z.-L., Isolation and characterization of 2019-nCoV-like coronavirus from Malayan pangolins. bioRxiv, 2020.2002.2017.951335 (2020). [Google Scholar]
- 44.Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Valentin F., Wallace I. M., Wilm A., Lopez R., Thompson J. D., Gibson T. J., Higgins D. G., Clustal W and clustal X version 2.0. Bioinformatics 23, 2947–2948 (2007). [DOI] [PubMed] [Google Scholar]
- 45.Guindon S., Gascuel O., A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52, 696–704 (2003). [DOI] [PubMed] [Google Scholar]
- 46.B. Foley et al., HIV sequence compendium 2018 (Theoretical Biology and Biophysics Group, Los Alamos National Laboratory, NM, LA-UR 18–25673, Los Alamos, New Mexico, 2018).
- 47.B. B. T. Korber, in Computational Analysis of HIV Molecular Sequences, A. G. Rodrigo, G. H. Learn, Eds. (Kluwer Academic Publishers, Dordrecht, Netherlands, 2000), chap. 4, pp. 55–72. [Google Scholar]
- 48.Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E., UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004). [DOI] [PubMed] [Google Scholar]
- 49.Yang J., Zhang Y., Protein structure and function prediction using I-TASSER. Curr. Protoc. Bioinformatics 52, 5.8.1–5.8.15 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Abagyan R., Totrov M., Kuznetsov D., ICM—A new method for protein modeling and design: Applications to docking and structure prediction from the distorted native conformation. J. Comput. Chem. 15, 488–506 (1994). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/27/eabb9153/DC1