

Nanostructured mineral coatings delivering chemically modified or unmodified therapeutic mRNA improve diabetic wound healing.
Abstract
Nonviral mRNA delivery is an attractive therapeutic gene delivery strategy, as it achieves efficient protein overexpression in vivo and has a desirable safety profile. However, mRNA’s short cytoplasmic half-life limits its utility to therapeutic applications amenable to repeated dosing or short-term overexpression. Here, we describe a biomaterial that enables a durable in vivo response to a single mRNA dose via an “overexpress and sequester” mechanism, whereby mRNA-transfected cells locally overexpress a growth factor that is then sequestered within the biomaterial to sustain the biologic response over time. In a murine diabetic wound model, this strategy demonstrated improved wound healing compared to delivery of a single mRNA dose alone or recombinant protein. In addition, codelivery of anti-inflammatory proteins using this biomaterial eliminated the need for mRNA chemical modification for in vivo therapeutic efficacy. The results support an approach that may be broadly applicable for single-dose delivery of mRNA without chemical modification.
INTRODUCTION
Drug delivery strategies involving active biologics, such as recombinant proteins, are frequently limited by low biological activity in vivo. This limitation has led to delivery of supraphysiological doses of therapeutic proteins and, in turn, frequent negative side effects in clinical use (1–3). Nucleic acid delivery represents an attractive alternative approach, as targeted cells can produce their own therapeutic proteins in situ with high biologic activity. However, safely delivering the foreign nucleic acid to target cells in vivo remains a substantial challenge (4). Nonviral delivery of mRNA has a number of attractive features that may combine high gene transfer efficacy with a positive safety profile, in contrast with other nucleic acid delivery approaches (5–7). Specifically, mRNA delivery does not require transport of the nucleic acid to the nucleus, as illustrated in Fig. 1A, and therefore, transgenes can be efficiently expressed in nonmitotic cell populations often encountered in vivo (8). In addition, mRNA is not a suitable molecule for homologous recombination or insertional mutagenesis; thus, the safety profile of mRNA delivery is highly desirable.
Fig. 1. Mineral-coated microparticle–mediated mRNA delivery is a highly efficient approach for overexpressing transgenes in nonmitotic primary human cells.
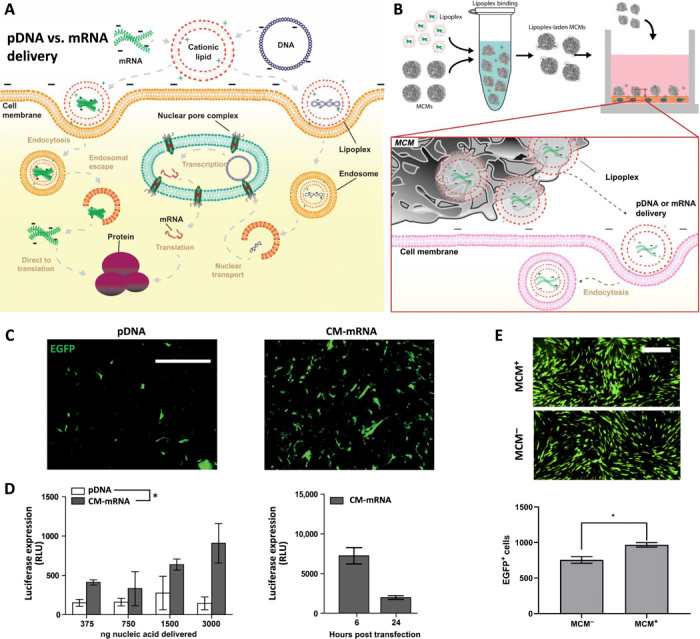
(A) Schematic of intracellular trafficking differences between nonviral delivery of pDNA and mRNA. Chemical transfection of pDNA and chemically modified mRNA (CM-mRNA) follows similar cellular internalization pathways. Upon cytoplasmic delivery, however, mRNA can proceed directly to translation, obviating nuclear transport that prevents efficient nonviral pDNA transfection in nonmitotic cells. (B) Schematic for lipoplex loading onto mineral-coated microparticles (MCMs), delivery to human dermal fibroblast (hDF), and subsequent cellular uptake in vitro. (C) Representative epifluorescence micrographs of mitomycin C–treated hDFs transfected with EGFP-encoding pDNA and CM-mRNA taken 12 hours after transfection with EGFP+ cells in green. Scale bar, 500 μm. (D) Luciferase expression increases (left) in mitomycin C–treated hDFs transfected with FLuc-encoding CM-mRNA, but not pDNA. n = 3; *P ≤ 0.05, one-way analysis of variance (ANOVA). Luciferase expression peaks at 6 hours after transfection (right) in mitotically inactivated hDFs and sharply decreases by 24 hours. RLU, relative luminescence unit. (E) Epifluorescence micrographs (top) of mitomycin C–treated hDFs transfected with EGFP CM-mRNA with (+) and without (−) MCMs. Comparison of number of EGFP+ cells (bottom) transfected with (+) and without (−) MCMs. n = 3; *P ≤ 0.05, Student’s t test.
Despite the potential advantages of mRNA delivery, several challenges limit its use as a biologic therapy. One major challenge is the short cytoplasmic half-life of mRNA, which limits the duration of in situ protein production to just hours. Repeated dosing could potentially address the issue of poor mRNA half-life, but reagents used for nonviral mRNA delivery have their own issues of safety and local toxicity, rendering repeated dosing potentially dangerous (9). An additional limitation is the antigenicity of in vitro–transcribed mRNA. This limitation has been addressed via the use of chemically modified ribonucleobases, which improve efficacy of mRNA delivery (10), but may reduce translation efficiency (10, 11), decrease or result in unintended interactions with RNA binding proteins that endogenously recognize and regulate mRNA dynamics through these chemical modifications (12); it may also increase production cost. Thus, there is a need for new strategies that capitalize on the potentially transformative benefits of nonviral mRNA delivery by amplifying therapeutic effects and minimizing the doses required for therapeutic efficacy.
Previously, we have demonstrated that mineral-coated microparticles (MCMs) can improve plasmid DNA (pDNA) transfection efficiencies via a localized delivery mechanism illustrated in Fig. 1B, while simultaneously reducing the reagent-associated cytotoxicity (13). Similarly, delivery of mRNA via MCMs here increased the number of enhanced green fluorescent protein–positive (EGFP+) cells relative to standard mRNA transfection. In addition, we have previously found that nanostructured mineral coatings can sequester and stabilize labile growth factors against denaturation, allowing for sustained protein release and prolonged stimulation of the intended biological response (14, 15). On the basis of these previous observations, we hypothesized that MCMs locally delivering mRNA could drive efficient production of a therapeutic protein and that the MCMs could sequester and stabilize the protein, thereby extending the duration of the intended biological response.
Here, we demonstrate that mRNA delivered via MCMs is highly effective in promoting transient overexpression in vitro and in vivo. The MCMs also locally sequester and stabilize the overexpressed secreted proteins, resulting in increased biological effects of the proteins. We demonstrate the utility of this approach as an in vivo therapy using a murine model of diabetic wound healing. We also provide a first demonstration that localized codelivery of wild-type mRNA (WT-mRNA) with anti-inflammatory proteins results in transfection efficiency that is equivalent to chemically modified mRNA (CM-mRNA) delivery, providing an alternative, materials-based approach for effective mRNA delivery. Together, these results provide a new approach for an effective single-dose mRNA treatment, which may be applicable to a number of regenerative medicine applications.
RESULTS
Nonviral mRNA delivery via MCMs improved transfection in nonmitotic primary human cells when compared to pDNA delivery or standard mRNA lipofection
We used mitotically inactivated primary cells to demonstrate the potential utility of MCM-mediated delivery of mRNA over pDNA delivery in therapeutic applications. Primary cells were selected, as many transformed cell lines are quite easy to transfect, while primary cells are often less so. We mitotically inactivated these cells to inhibit passive nuclear transport of pDNA during mitosis, as primary cells in vivo are largely nonmitotic. After mitotic inactivation via mitomycin C treatment, we observed higher efficiency transfection of EGFP mRNA chemically modified (CM-mRNA) with ribonucleobases pseudouridine (ψU) and 5-methylcytidine (5-meC) when compared to EGFP pDNA in primary human dermal fibroblasts (hDFs; Fig. 1C). These two base modifications have been previously established to improve protein expression from delivery of in vitro transcribed mRNA in multiple cell types including hDF (16–18). In addition, CM-mRNA provided a dose-dependent transgene response, while pDNA did not, as evidenced by the increase in bioluminescence after transfection with CM-mRNA encoding for firefly luciferase (FLuc; Fig. 1D). Delivery of EGFP CM-mRNA lipoplexes from MCMs resulted in a 1.28-fold increase in transfection efficiency when compared to standard CM-mRNA lipofection (Fig. 1E). However, the CM-mRNA–based transfection resulted in only short-term transgene expression, as bioluminescence decreased to 21.8% of peak expression after 24 hours (Fig. 1D), and delivery via MCMs did not cause an increase in the duration of transgene expression (fig. S1).
MCMs improved the biological effects of basic fibroblast growth factor CM-mRNA delivery in primary human cells via a protein sequestering mechanism
MCM-mediated transfection of CM-mRNA encoding for basic fibroblast growth factor (bFGF; fig. S2, A and B) increased hDF proliferation relative to transfection without MCMs (Fig. 2, A and B). Epifluorescence micrographs illustrated this increase using a click-chemistry compatible thymine analog [Click-iT EdU (5-ethynyl-2’-deoxyuridine)] that labels cells which have progressed through S phase (Fig. 2A). CM-mRNA delivery via MCMs resulted in a 3.5-fold increase in S phase+ hDFs at 2 days after transfection (Fig. 2B). While the bFGF expression level in the CM-mRNA delivery conditions were equivalent with or without MCMs (Fig. 2B), most of the overexpressed bFGF in the MCM+ condition (58.1 ± 4.5%) was not freely soluble in the media (free) but instead sequestered in the MCMs (Fig. 2B). This result indicated that MCMs could both deliver the mRNA cargo and subsequently sequester the cell-secreted gene product, in this case, bFGF.
Fig. 2. MCMs extend biological effects of mRNA delivery via an “overexpress and sequester” mechanism.
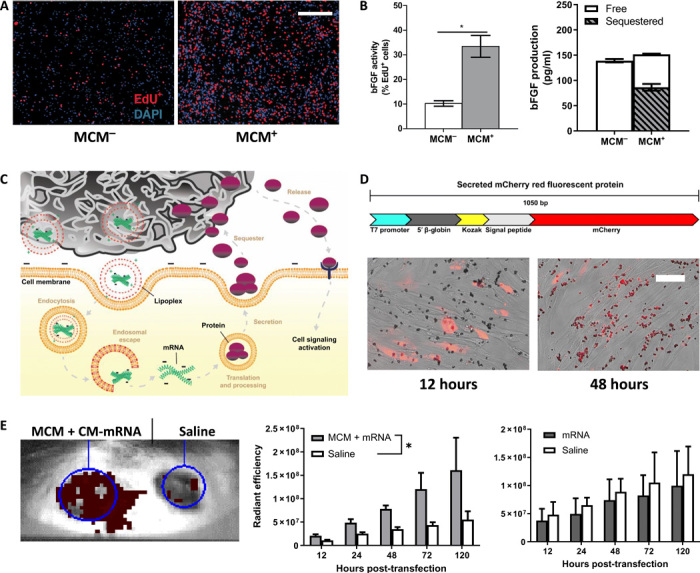
(A) Representative epifluorescence micrographs of hDFs transfected with 150-ng bFGF CM-mRNA with (+) and without (−) MCMs, 48 hours after transfection. Scale bar, 500 μm. (B) Left: S phase+ cells (red) quantified as the percentage of total 4′,6-diamidino-2-phenylindole (DAPI; blue) nuclei. n = 3; *P ≤ 0.05, one-way Student’s t test. Right: bFGF production and sequestering after bFGF CM-mRNA transfection. “Free” represents bFGF in the media, and “sequestered” is the measured bFGF after mineral coating dissolution via EDTA. n = 3. (C) Schematic of overexpress and sequester mechanism. MCMs deliver mRNA lipoplexes and then bind and sequester the secreted protein, sustaining growth factor release over time and prolonging the biological response. (D) Schematic (top) for model secreted mCherry protein and (bottom) merged phase and epifluorescence micrographs of transfected hDFs 12 (left) and 48 (right) hours after transfection. Scale bar, 50 μm. (E) Representative IVIS image of the secreted mCherry CM-mRNA + MCM–treated dermal wound (left wound) and saline control (right wound) 48 hours after delivery. (F) Red fluorescence radiant efficiency within each transfected or saline wound site over five with MCMs (left) and without (right). n = 3; *P < 0.05 by two-way ANOVA.
Sequestering of secreted mCherry via MCMs also visually illustrated the protein sequestering mechanism (Fig. 2, C and D). Specifically, we designed a DNA template for mRNA synthesis that combined gene sequences for the secretion signal peptide from mouse matrix metalloproteinase 9 with mCherry fluorescent protein (Fig. 2D). Transfection of CM-mRNA generated from this template resulted in a red fluorescent protein that could be observed 12 hours after transfection in the cytoplasm but lost by 48 hours (Fig. 2D). After 48 hours, MCMs began to fluoresce red, indicating that they had sequestered the secreted mCherry protein (Fig. 2D). At the 12 hours post-transfection time point, where the observed red fluorescence was still cytoplasmic, and in control conditions without secreted mCherry encoding CM-mRNA, we observed no colocalized red fluorescence with the MCMs (Fig. 2D). In addition, the cell culture media increased in red fluorescence after 48 hours for cells transfected without MCMs, but not with MCMs, providing further evidence that the mCherry protein was being efficiently sequestered into the MCMs (fig. S3A).
Protein sequestering also extended the retention of an overexpressed secreted protein in a murine dermal wound (Fig. 2E). We created two contralateral wounds in each animal and delivered mCherry CM-mRNA to one wound, with or without MCMs, and saline to the contralateral wound within the same animal. We examined each wound at 12, 24, 48, 72, and 120 hours after transfection and measured mCherry red fluorescence within the wound borders using an in vivo imaging system (IVIS). Over this 5-day time course, MCM-based delivery resulted in higher red fluorescence within the treated wound, relative to the contralateral saline control, while soluble delivery did not (Fig. 2E). The higher red fluorescence in the treated wound relative to the saline control indicated an enrichment and retention of the overexpressed mCherry protein over this 5-day time course (Fig. 2E). To quantify this local mCherry enrichment within the intended wound site, we normalized the red fluorescence in the treated wound to the saline control within each animal (fig. S3B). After 12 hours, MCM-mediated delivery resulted in a 1.9-fold increase in mCherry enrichment in the treated wound and, by day 5, mCherry fluorescence was 2.9-fold higher in the treated wound (fig. S3B). In comparison, soluble delivery resulted in 0.9- and 1.2-fold mCherry enrichment in the treated wound relative to the saline control for soluble delivery after 12 hours and 5 days, respectively (fig. S3B). Together, these results demonstrated that MCMs could locally sequester cell-secreted, mRNA-encoded proteins both in vitro and in vivo. We next endeavored to determine whether this protein sequestering mechanism could result in greater activity of the secreted proteins.
bFGF produced in situ via MCM-mediated CM-mRNA delivery had greater bioactivity than recombinant bFGF
Previous studies (19) have shown that in vitro treatment with recombinant human bFGF (rhbFGF) stimulates proliferation in hDFs. Thus, to evaluate biological activity of our mRNA delivery approach, we evaluated hDF proliferation in response to rhbFGF delivered via MCMs versus bFGF produced in situ via MCM-mediated CM-mRNA delivery (Fig. 3A). MCM delivery of rhbFGF (1 or 10 ng/ml) led to a 1.9 ± 0.8% or 38.3 ± 2.7% increase in hDF proliferation, respectively (Fig. 3, B and C). MCM-based delivery of CM-mRNA resulted in proliferation rates equivalent to MCM delivery of rhbFGF (10 ng/ml; Fig. 3, B and C), as each caused ~20-fold higher hDF proliferation than soluble rhbFGF (1 ng/ml; Fig. 3C). However, the cell-secreted bFGF concentration was 53-fold lower than the rhbFGF condition (10 ng/ml), indicating that the cell-secreted bFGF had substantially higher bioactivity (Fig. 3C).
Fig. 3. In situ–produced bFGF has greater bioactivity than recombinantly produced bFGF.
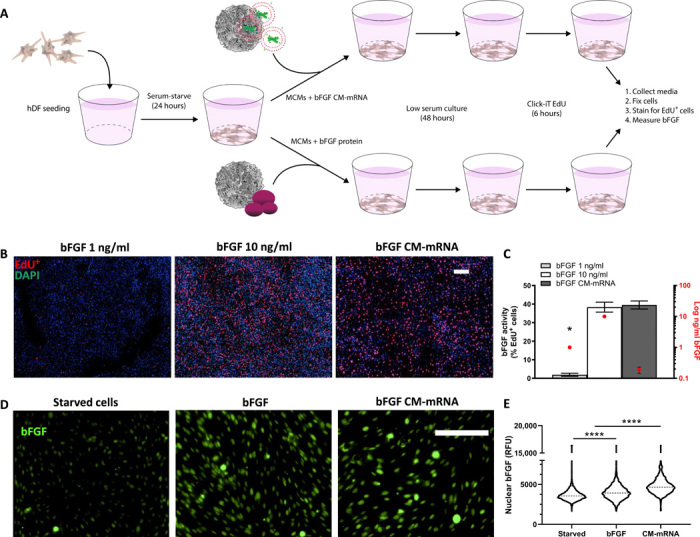
(A) Schematic of in situ recombinantly produced bFGF bioactivity comparison. Serum-starved hDFs were treated with recombinant bFGF (1 or 10 ng/ml) or bFGF CM-mRNA, all via MCMs. Forty-eight hours after treatment, the media was collected for bFGF quantification via enzyme-linked immunosorbent assay and cells analyzed for proliferation. (B) Representative epifluorescent micrographs of 1 (left) and 10 ng/ml (middle) recombinant bFGF and 300-ng CM-mRNA (right) all delivered with MCMs. DAPI+ nuclei (blue) and S phase+ nuclei (red). Scale bar, 100 μm. (C) Number of S phase+ nuclei as a percentage of total nuclei (left axis) and amount of total bFGF delivered or produced (right axis, log scale). n = 3; *P < 0.0001 by one-way ANOVA with Tukey’s post hoc analysis. (D) Right: Representative epifluorescence micrographs for nuclear bFGF immunofluorescence in serum-starved hDF treated with bFGF (10 ng/ml), bFGF CM-mRNA, or no treatment. Scale bar, 100 μm. (E) Image cytometry of nuclear immunofluorescence for bFGF in serum-starved hDF treated with bFGF (10 ng/ml), bFGF CM-mRNA, or no treatment. Nuclei (1500 to 2500) measured, pooled from two experimental replicates. ****P < 0.0001 by one-way ANOVA with Tukey’s post hoc analysis for multiple comparisons. RFU, relative fluorescence units.
One possible reason for the marked increase in bioactivity of cell-secreted, mRNA-encoded bFGF when compared to rhbFGF could involve cell production of other bioactive bFGF isoforms. Thus, we next explored the potential effects of high–molecular weight (HMW) bFGF isoforms, which are known to translocate to the nucleus and promote fibroblast proliferation in low-serum conditions (20), as a potential explanation for the observed higher bFGF bioactivity. Immunofluorescence for bFGF (Fig. 3D) and confocal microscopy (fig. S4) revealed that mRNA delivery had more prominent presence of nuclear bFGF when compared to rhbFGF delivery. In particular, image cytometry–based quantification of the mRNA delivery condition showed a 30.1% relative increase in nuclear bFGF immunofluorescence relative to untreated serum-starved hDFs, while rhbFGF (10 ng/ml) only increased nuclear bFGF by 19.8% (Fig. 3E).
MCM-mediated CM-mRNA delivery improved dermal wound healing in diabetic mice
Next, to evaluate the potential therapeutic effects of bFGF mRNA delivery, we measured the wound perimeters of full-thickness dermal wounds with wound edges fixed to prevent contraction in db+/db+ mice over 19 days (Fig. 4, A and B). We additionally evaluated the final wound resolution quality after 19 days via blinded assessment and histological scoring of hematoxylin and eosin (H&E)–stained wound cross sections (fig. S5). Similar to studies assessing the efficacy of exogenously delivered therapeutics on murine and leporine dermal wound healing (21–23), this scoring system aimed to capture the stage of wound resolution as an endpoint analysis, focusing on degree of wound closure, appearance, and continuity of stratified dermis and epidermis, as well as resolution of granulation tissue (fig. S5).
Fig. 4. MCM-mediated mRNA delivery increases wound closure rate and improves final wound resolution.

(A) Table of treatment groups. (B) Representative gross and histological images show improved wound closure and resolution for CM-mRNA treatment with MCMs relative to no treatment and recombinant bFGF with MCMs. Scale bar, 5 mm. (C) Percent wound perimeter reduction versus time show that MCM-mediated mRNA, but not recombinant bFGF delivery, improves wound closure relative to no treatment. Mean closure and SE of n = 8 wounds. *P < 0.05 by two-way ANOVA with Dunnett’s post hoc analysis relative to the no-treatment control. (D) Scoring of H&E-stained wound cross sections show mRNA, but not bFGF delivery, with MCMs improved wound resolution relative to no treatment. Mean score and 95% confidence interval of n = 8 wounds. ***P < 0.001 by one-way ANOVA with Dunnett’s post hoc analysis relative to no treatment. (E) Analysis of epidermal keratin content by colorimetric analysis of Masson’s trichrome and DAB density from immunohistochemistry for collagen I (Col I). Mean intensity and 95% confidence interval of n = 8 wounds. *P < 0.05 by one-way ANOVA with Dunnett’s post hoc analysis relative to no treatment.
A single treatment of 3-μg bFGF CM-mRNA delivered via MCMs (CM-mRNA + MCM) decreased wound perimeter relative to no treatment by 59.1% after 19 days, while 5-μg rhbFGF protein adsorbed and delivered via MCMs (bFGF + MCM) resulted in no improvement (Fig. 4C). CM-mRNA + MCM additionally improved the wound resolution score (5.44) relative to the no-treatment control (3.19), on a scale of 1 to 6 (Fig. 4D). This score of 5.44 in the CM-mRNA + MCM condition represented a mean healing response of complete reepithelization, resolution of granulation tissue, and near-complete return to wound thickness and matrix deposition. In contrast, the bFGF + MCM treatment resulted in a wound resolution score (2.88) not significantly different from no treatment (Fig. 4D). There were no observed differences in total collagen deposition, epidermal keratin content, or endothelial cell infiltration between treatment groups (figs. S6 and S7) via analysis of red and blue staining intensity, respectively, of Masson’s trichrome and diaminobenzidine (DAB) density from immunohistochemistry for CD31. The CM-mRNA + MCM group resulted in 25.8% reduction in collagen type I relative to the no-treatment control (Fig. 4E). CM-mRNA + MCM and bFGF + MCM trended toward increased epidermal keratin content by 246.3 and 222.0%, respectively (Fig. 4E), although these results were not statistically significant.
To specifically examine the effect of MCMs on CM-mRNA–mediated wound healing, we next compared a single, lower dose of 0.7-μg bFGF CM-mRNA delivered with MCMs (CM-mRNAlow + MCM) or without MCMs (CM-mRNAlow-MCM; fig. S8, A and B). This lower dose was required as the CM-mRNA (high) dose involved first binding mRNA lipoplexes to MCMs and then concentrating them into a volume compatible with the wound geometry (20 μl), making comparisons at 3 μg CM-mRNA without MCMs not possible. At this lower dose, neither delivery with nor without MCMs improved the wound perimeter reduction rate relative to no treatment (fig. S8C). However, the CM-mRNAlow + MCMs resulted in significantly improved wound resolution score (4.5) relative to the no-treatment control (3.2), while delivery of the CM-mRNAlow without MCMs (3.9; figs. S8D and S9) and MCMs alone (3.7) (fig. S10, A to D) provided no significant improvement in wound resolution. There were no observed differences in total collagen deposition or endothelial cell infiltration between treatment groups (figs. S6, S7, S8E, S9, S10E, and S11) via analysis of blue staining intensity of Masson’s trichrome and DAB density from immunohistochemistry for CD31. The CM-mRNAlow treatments with and without MCMs reduced collagen type I deposition by 72.3 and 47.2%, respectively, relative to the no-treatment control (fig. S8E). The CM-mRNAlow + MCM additionally increased the amount of epidermal keratin content by 513.8% (fig. S8E).
The CM-mRNA + MCM treatment also resulted in the fewest mean days to complete wound closure (23.2 ± 2.6 days) when compared to no treatment (28.3 ± 2.3 days), bFGF + MCM delivery (32.2 ± 6.2 days), CM-mRNAlow + MCMs (29.5 ± 2.6 days), or CM-mRNAlow-MCMs (34.3 ± 13.0 days) (fig. S12, A and B). Thus, the expected wound closure time with CM-mRNA + MCM treatment was reduced by 18.1% relative to no treatment and was the only treatment condition that resulted in a significant improvement (fig. S12A).
MCM-mediated codelivery of WT-mRNA with B18R improved biological outcomes
In vitro transfection of hDFs with bFGF-encoding CM-mRNA resulted in a 2.28-fold increase in bFGF production relative to WT-mRNA (fig. S13A). Treatment of cells with the viral interferon-γ (IFNγ) suppressing protein B18R has been previously shown (10, 24) to reduce the inflammatory signaling pathways responsible for lower transgene expression in WT-mRNA relative to CM-mRNA transfection. In our experiments, codelivery of B18R via MCMs increased luciferase expression 1.56-fold in FLuc WT-mRNA–transfected hDFs relative to cells treated with soluble B18R alone (fig.S 13B). In vitro, the bFGF production after WT-mRNA + B18R + MCM delivery was equivalent to bFGF production after CM-mRNA + MCM delivery (fig. S13C). In vivo, a single treatment of 3-μg WT-mRNA codelivered with 400-ng B18R delivered via MCMs (WT-mRNA + B18R + MCM; Fig. 5A) increased dermal wound perimeter reduction relative to no treatment by 87.9% after 19 days, while the same amount of WT-mRNA delivered via MCMs without B18R (WT-mRNA + MCM) resulted in no improvement (Fig. 5, B and C). WT-mRNA + B18R + MCM also improved the wound resolution score (4.75) relative to the no-treatment control (3.19), on a scale of 1 to 6 (Fig. 5D), while WT-mRNA + MCM (2.95) showed no improvement (Fig. 5D). These data demonstrate that in this experimental context, codelivery of mRNA with B18R obviates the need for chemical modification of mRNA. There were no observed differences in total collagen deposition or endothelial cell infiltration between treatment groups (figs. S14 and S15) via analysis of blue staining intensity of Masson’s trichrome and DAB density from immunohistochemistry for CD31. The WT-mRNA + B18R + MCM treatment reduced collagen type I deposition by 33.3% and increased the amount of epidermal keratin content by 545.0% relative to the no-treatment control (Fig. 5E).
Fig. 5. Codelivery of B18R via MCMs increases wound closure rate and improves final wound resolution with WT-mRNA treatment.

(A) Table of treatment groups. (B) Representative gross and histological images show improved wound closure and resolution for WT-mRNA delivered via MCMs with and without B18R. Scale bar, 5 mm. (C) Percent wound perimeter reduction versus time for wounds treated with WT-mRNA delivered via MCMs with and without B18R compared to no treatment. Mean closure and SE of n = 8 to 10 wounds. **P < 0.01 by two-way ANOVA with Dunnett’s post hoc analysis relative to the no-treatment control. (D) Scoring of H&E-stained wound cross sections shows that MCMs delivering WT-mRNA with, but not without, B18R improved wound resolution relative to no treatment. Mean score and 95% confidence interval of n = 8 to 10 wounds. *P < 0.05 by one-way ANOVA with Dunnett’s post hoc analysis relative to no treatment. (E) Analysis of epidermal keratin content by colorimetric analysis of Masson’s Trichrome and DAB density from immunohistochemistry for Col I. Mean intensity and 95% confidence interval of n = 8 wounds. *P < 0.05 and **P < 0.01 by one-way ANOVA with Dunnett’s post hoc analysis relative to no treatment.
DISCUSSION
Conventional nonviral pDNA delivery is largely dependent on passive nuclear transport during mitosis and is therefore an ineffective gene therapy strategy for cells not active in the cell cycle (Fig. 1), such as most cells found in vivo (25, 26). Delivery of mRNA overcomes this limitation but is critically limited in therapeutic efficacy by its short intracellular half-life that necessitates repeated dosing (7). We demonstrated this paradigm here, where mRNA delivery resulted in higher mRNA transfection efficiency and transgene expression than pDNA in mitotically inactivated primary human fibroblasts (Fig. 1, C and E), but expression was lost after 24 hours (Fig. 1D and fig. S1) To overcome current limitations in nonviral mRNA delivery, we explored the use of MCMs.
Although MCMs increased the number of EGFP+ cells from mRNA transfection, they had no impact on the duration of transgene expression (fig. S1). This finding illustrated the need for a new mRNA delivery approach for therapeutic applications focused on extending the effects of mRNA transfection. We hypothesized that MCMs could potentially extend the therapeutic effects of cell-secreted gene products, as they simultaneously afford both highly efficient localized nonviral gene delivery, and they can efficiently sequester and release therapeutic proteins (13, 14, 27–30). Specifically, we aimed to use MCMs to locally deliver mRNA with high gene transfer efficiency and reduced toxicity (13) while also sequestering the overexpressed protein and thereby extending the therapeutic effects (14). Our results here provide a new “overexpress and sequester” approach for nonviral mRNA delivery that uses a synthetic material (MCMs) to achieve single-dose mRNA delivery to improve wound healing in a murine model of diabetic ulcers and obviate the need for chemical modification of therapeutic mRNA.
We demonstrated that MCMs can improve the biological effects of mRNA encoding for the therapeutic protein bFGF (Fig. 2, A and B). Using proliferation of serum-starved hDF as an assay provided us with a model to measure this improvement, as previous studies have established that bFGF has a short half-life in cell culture media (~7 hours) (31), and a cellular response provides an unambiguous readout of protein bioactivity. In addition, we have previously examined the effects of MCMs on primary cells including hDFs (13, 14) in delivering nucleic acids and recombinant bFGF. We have observed that delivery of nucleic acids via MCMs does not increase the metabolic activity above baseline (13) and does not promote proliferation without active bFGF (14). At the 2-day time point of this experiment, we expected that little or no remaining bFGF from the initial overexpression would be present or remain bioactive, given the short bFGF half-life and the low level of additional transgene expression after the first 24 hours (Fig. 1C and fig. S1). Our observation that MCM delivery of bFGF mRNA resulted in biological activity at the 2-day time point provided initial evidence that the MCMs were likely sequestering cell-secreted bFGF and thereby prolonging bFGF bioactivity. To visualize and validate our proposed mechanism (Fig. 2C), we delivered mRNA encoding for a secreted mCherry fluorescent protein (Fig. 2D) and directly demonstrated that the mCherry had indeed been “overexpressed,” secreted by the cells, and “sequestered” in the mineral coating of the MCMs.
In this study, our plasmid template contained the coding sequence for not just a single isoform, but multiple bFGF isoforms found in the endogenous bFGF locus, including the additional upstream noncanonical CUG nucleotide sequence start sites for the HMW bFGF isoforms (fig. S2A). These HMW bFGF isoforms could be important, as they have been shown to promote cell proliferation in concert with low–molecular weight isoforms in low-serum conditions by translocating to the nucleus (32). Here, we found that mRNA delivery increased nuclear bFGF relative to either no treatment or recombinant bFGF delivery. This finding illustrates a fundamental distinction between gene delivery and protein delivery, where the use of a properly designed transcript can allow for activation of endogenous autocrine and paracrine signaling pathways (33–35) that would not be achieved via a single, exclusively extracellular isoform of a recombinant protein. In a clinical regenerative medicine setting, this may translate to lower effective doses of the therapeutic agent while maintaining or increasing the therapeutic effect. This would be a potential advantage over recombinant protein–based therapies, which can lead to untoward heterotopic effects due to the supraphysiological doses required (2, 3, 36).
To explore the potential therapeutic utility of our approach, we observed an increase in wound closure in a murine diabetic model using mice with homozygous mutations to the leptin receptor (db+/db+) and treatment with bFGF CM-mRNA delivered via MCMs. db+/db+ mice have been previously used (37) as a model for chronic diabetic wounds when the wound perimeter is restricted in place and wound healing is limited to only re-epithelialization and regrowth of the dermal tissue. Previously, researchers have demonstrated that either 5 μg/cm2 daily doses(37) or a sustained release of 7 to 14 μg/cm2 of bFGF from a scaffold (38–40) was required to improve outcomes in this murine model of diabetic wound healing and that increased bFGF dosage showed no improvement or worse outcomes (38, 39). As we have previously demonstrated that these mineral coatings are capable of sustained delivery of a variety of growth factors (27, 28, 41–43), including bFGF (14), we selected 5 μg of sustained delivery from MCMs as an experimental condition to allow for comparison of single-dose mRNA therapy versus recombinant protein therapy.
The CM-mRNA dose was selected on the basis of our previous reports using MCMs for lipoplex delivery (13), which showed peak transfection efficiency of fibroblasts in vitro achieved at a dosage of approximately 3-μg mRNA/cm2. We observed a significant increase in wound perimeter reduction in the CM-mRNA + MCM treatment relative to no treatment after 7 days, which continued to increase for 19 days (Fig. 4C). In addition, we fit exponential curves to each wound perimeter over time to estimate the time until complete wound closure (fig. S12). On the basis of previous studies in human patients with diabetes (44), this type of wound closure modeling has predictive value for the expected overall wound resolution and patient outcome. Decreased expected time to complete closure with CM-mRNA + MCM treatment in this model indicated that MCMs offer significant increase in potential therapeutic utility of mRNA delivery. As we have previously demonstrated, the promiscuous electrostatic binding of mineral coatings allows for sustained release of a range of other growth factors, cytokines, and enzymes, including bone morphogenetic protein–2 (27, 45), vascular endothelial growth factor (14, 27, 43), interleukin-1RA (IL-1RA) (41), IL-10 (46), NT-3 (neurotrophin-3) (42), and chondroitinase ABC (47). Therefore, our MCM-mediated overexpress and sequester approach may have broad potential in therapeutic applications where in situ production and retention of highly bioactive secreted proteins would be beneficial.
Histological examinations demonstrated that delivery of bFGF CM-mRNA was superior for final wound resolution when compared to recombinant bFGF (Fig. 4D). In addition, we observed that MCMs improved the wound resolution, while delivery without MCMs did not (fig. S8D). We did not observe any differences in total collagen deposition or endothelial cell infiltration between any treatment groups (figs. S6, S7, S8E, S9, S10E, S11, S14, and S15). However, we did observe a decrease in collagen type I deposition with CM-mRNA treatment, which was observed to a greater extent when mRNA was delivered via MCMs (Fig. 4D and fig. S8E) or when WT-mRNA was codelivered with B18R via MCMs (Fig. 5E). A decrease in dermal collagen type I without an overall decrease in total collagen content suggests a less fibrotic healing response (48), supporting the assertion that CM-mRNA delivered via MCMs improves the healing outcomes of the dermis. In addition, we observed that CM-mRNA and WT-mRNA + B18R treatments, when delivered via MCMs, trended toward increased epidermal keratin content, whereas other treatments or no treatment did not (Figs. 4E and 5E, and figs. S6, S7, S8E, S9, S10E, and S11). Although this effect was less pronounced, the observed increase in epidermal keratin deposition is indicative of a more mature epidermis and earlier onset of epidermal healing (49, 50), supporting the observed increase in wound closure rate with CM-mRNA delivered via MCMs relative to bFGF, delivery without MCMs, or the no-treatment and vehicle controls (Figs. 4, C and E, and 5, C and E; and figs. S6E, S7, S8E, S9, S10E, and S11).
Chemical modification of mRNA has been shown to reduce the Toll-like receptor (TLR) and retinoic acid-inducible gene I (RIG-I) viral RNA receptor pathways that decrease the transgene expression efficiency of WT-mRNA (51). However, this chemical modification step adds cost and unpredictability with regard to potential antigenicity of the chemically modified ribonucleobases. In addition, some emerging mRNA design strategies of potential therapeutic value, such as mRNA capable of cytoplasmic replication (24) and the design of 3′ untranslated regions to provide tissue-specific control of transgene translation (52), depend on protein-mRNA interactions that are likely to be removed upon mRNA chemical modification. Here, we demonstrated that we could increase transgene expression levels (fig. S13) and in vivo biological outcomes (Figs. 4 and 5) from WT-mRNA to equivalency with CM-mRNA by codelivering B18R via MCMs. Recognition of delivered WT-mRNA is part of the ubiquitous innate antiviral response pathway mediated via the TLR- and RIG-I–mediated signaling pathways (53–56). The resulting cytotoxicity from WT-mRNA results from downstream induction of the IFNγ–nuclear factor κB (NFκΒ) transcriptional program after TLR and RIG-I activation, and B18R inhibits this antiviral state via competitive binding of IFNγ (57). As the IFNγ-NFκΒ transcriptional program is part of a ubiquitous innate antiviral response to exogenously delivered unmodified mRNA (54, 56), our dual delivery method of mRNA along with B18R eliminated the need for mRNA chemical modification in the context of bFGF mRNA delivery in vivo. The fundamental mechanism by which B18R inhibits the ambitious innate immune response to exogenously delivered therapeutic mRNA suggests that our dual delivery approach could potentially represent an alternative to chemical modification in future studies as well.
CONCLUSIONS
Here, we describe a biomaterials-based approach for a single-dose, nonviral therapeutic mRNA delivery. MCMs enhanced biological outcomes from mRNA delivery by improving mRNA lipofection and by locally sequestering overexpressed proteins in situ. We also found that in situ produced protein had greater bioactivity than a recombinantly produced growth factor due, in part, to activity of multiple growth factor isoforms. This higher bioactivity of proteins produced via mRNA delivery may be beneficial to biologic therapies by lowering the dose required for efficacy and therefore reducing the risk of adverse effects. mRNA delivery via MCMs improved the rate of wound closure and final resolution in a murine model of diabetic ulcers. MCM codelivery of WT-mRNA with B18R obviated the need for mRNA chemical modification, and this codelivery provides an alternative therapeutic strategy in scenarios where chemical modification would reduce or eliminate proper mRNA processing or expression. In summary, we have described a single-dose mRNA treatment regimen enabled by intrinsic biomaterial properties that affords efficient overexpression and sequestration of a highly bioactive protein in vivo, offering a broadly applicable strategy for therapeutic mRNA delivery.
MATERIALS AND METHODS
Fabrication of MCMs
Hydroxyapatite powder (Plasma Biotal Limited) was used as a microparticle core material. The powder was suspended at concentrations of 1 mg/ml in mSBF (modified simulated body fluid) containing concentrations of 141 mM NaCl, 4 mM KCl, 0.5 mM MgSO4●7H2O, 1 mM MgCl2●6H2O, 4.2 mM NaHCO3, 20 mM Hepes, 5 mM CaCl2●2H2O, 2 mM KH2PO4, and 1 mM NaF. The suspension was rotated at 37°C for 24 hours, at which point the microparticles were centrifuged at 2000g for 2 min, and the supernatant was decanted and replaced with freshly made mSBF. We repeated this process daily for 5 days, at which point the MCMs were washed three times with 50-ml deionized (DI) water, filtered through a 40-μm-pore cell strainer, suspended in 15-ml distilled water, frozen in liquid nitrogen, and lyophilized for 48 hours. The lyophilized MCMs were then analyzed for nanotopography and calcium release as previously described (27, 30).
Culture of hDFs
Dermal fibroblasts from human foreskin (hDF; American Type Culture Collection) were grown on tissue culture–treated polystyrene (TCPS) flasks (Corning) in Dulbecco’s modified Eagle’s medium (DMEM; Corning) with 10% (v/v) fetal bovine serum (FBS; Gibco) and penicillin/streptomycin (100 U/ml; Gibco; growth media). Cells were passaged at 80% confluence using 0.05% (v/v) trypsin/EDTA (Corning). Cells were cryopreserved after three passages in DMEM with 20% FBS and 10% dimethylsulfoxide (DMSO; Thermo Fisher Scientific) at 500,000 cells/ml. Cells were frozen at −80°C overnight and then transferred to storage in liquid nitrogen. Cells used for experiments were thawed quickly at 37°C, diluted in growth media, and centrifuged at 200g for 5 min, and the supernatant was replaced with fresh growth media to remove DMSO from the freezing process. Thawed cells were seeded at 500 to 1000 cells/cm2 and used for subsequent experiments or further passaging. Cells were not expanded beyond seven passages.
Generation of plasmids and mRNA transcripts
bFGF-encoding plasmid (pbFGF; OriGene), EGFP-encoding plasmid (Addgene), FLuc plasmid encoding (pFLuc) (Addgene), and an empty vector with a phosphoglycerate kinase (PGK) promoter and neomycin selection cassette (pPGK; Addgene) were purchased from their respective suppliers. Upon receiving the plasmids, they were transformed in DH5α-chemically competent Escherichia coli (Invitrogen) and spread on the appropriate antibiotic selection agar plate. Colonies were isolated after ~24 hours and expanded in the appropriate antibiotic-containing lysogeny broth. Plasmids were purified from these cultures using GeneJET plasmid mini- or maxiprep plasmid purification kits (Thermo Fisher Scientific) according to the manufacturer’s instructions.
Templates for generation of messenger RNA transcripts were either linearized plasmids, linear double-stranded DNA (dsDNA) sequences that were commercially synthesized (Integrated DNA Technologies), or polymerase chain reaction (PCR) products from plasmids or dsDNA sequences. For bFGF mRNA transcripts, the pbFGF plasmid was linearized with MsuI at 37°C overnight and then purified via column purification (Zymo Research DNA Cleaning & Concentrator-5). dsDNA sequences were used directly as templates, while PCR products were also purified via column purification. In vitro transcription was done using the T7 HiScribe ARCA mRNA Transcription Kit (with tailing; New England Biosciences). CM-mRNA was generated according to the T7 HiScribe Kit’s manufacturer’s protocol using 5-meC and ψU (TriLink BioTechnologies). Denatured mRNA was run in 1.5 weight % agarose gels in Mops running buffer (Thermo Fisher Scientific) at 5 V/cm and 4°C with a circulating pump constantly circulating buffer between chambers. These steps were critical to resolve mRNA gels (fig. S2B).
Transfection of mitotically inactivated hDFs
hDFs were seeded in 96-well TCPS plates at 15,000 cells/cm2 36 to 48 hours before transfection in growth media. 2.5 hours before transfection, hDFs were inactivated with mitomycin C [Alfa Aesar (5 mg/ml) in ethanol stock solution]. The mitomycin C solution was diluted 500× to 10 μg/ml in growth media. The growth medium on the cells was replaced with the 1× mitomycin C growth media and incubated at 37°C for 2.5 hours. After the incubation, the mitotically inactivated cells were returned to growth media. WT- and CM-mRNA encoding for EGFP and FLuc (TriLink BioTechnologies) were complexed with Lipofectamine Messenger Max (Invitrogen) at 30 μg/ml in Opti-MEM (Gibco) with a ratio of 3 μl of Lipofectamine Messenger Max per microgram of mRNA. pDNA encoding for EGFP (Addgene) and FLuc (Addgene) were complexed with Lipofectamine 2000 at 30 μg/ml with a ratio of 2 μl/μg of pDNA. The mRNA and DNA complex solutions were incubated for 20 min at room temperature to allow for complexes to form.
For MCM-mediated delivery, nucleic acid lipoplexes were adsorbed onto the MCM surface out of solution via electrostatic incubations as previously described (13). Briefly, after lipoplex formation, MCMs and lipoplexes were incubated together for 30 additional minutes at room temperature while rotating. After rotating, the solution was centrifuged and lipoplex-laden MCMs were resuspended for downstream use. Lipofectamine 2000 was used for all pDNA transfections, and Lipofectamine Messenger Max was used for all mRNA transfections. Cells were transfected with 100 ng of mRNA complexes (by mass of mRNA) unless otherwise specified. WT-mRNA denotes mRNA synthesized with naturally occurring ribonucleobases. CM-mRNA denotes mRNA with uracil substituted with ψU and cytosine substituted with 5-meC.
Cells transfected with EGFP mRNA and pDNA were examined for green fluorescence using epifluorescence microscopy (Nikon Ti Eclipse with fluorescein isothiocyanate filter cube) 12 and 24 hours after transfection. FLuc-transfected cells were lysed using Passive Lysis Buffer (Promega) at 6- and 24-hour time points, and their supernatant immediately collected for measuring of bioluminescence. To measure bioluminescence, 100-μl supernatant was added to a microwell plate and placed in a Fluoroskan Microplate Reader (Thermo Fisher Scientific) with single-well injection capabilities. The injection port was loaded with 5 mM MgCl2, 150 μM adenosine triphosphate (New England Biosciences), and d-Luciferin (150 μg/ml; BD Biosciences) in 100 mM tris-HCl (pH 7.8; Luciferin buffer) buffer. The plate was set to read with injection of 100 μl of Luciferin buffer, followed by 10 s of shaking to mix, 30 s of incubation, and then photodetection with 10 s of integration. Expression of FLuc transgene was reported as relative luminescence units.
In vitro bFGF mRNA delivery efficacy in serum-starved hDFs
hDFs were cultured in 10% DMEM with penicillin/streptomycin (P/S) and seeded into cyclic-olefin high-content half-area 96-wells (Corning) at 1000 cells per well 48 hours before transfection. Twenty-four hours before transfection, the cells were transferred into starvation media composed of DMEM plus P/S and 0.5% FBS. We starved fibroblasts to remove the effects of mitogens contained within serum, including bFGF (58), and to synchronize the cells in the G0 state to isolate the specific proliferative response to overexpressed bFGF (59). Cells were transfected with 150 ng of CM-mRNA as described above. In addition, cells were treated with recombinant bFGF (either 1 or 10 ng/ml), with or without MCMs. For the recombinant bFGF conditions with MCMs, these cells were also transfected with the secreted mCherry-encoding mRNA transcript. Six hours after transfection, the transfected cells were quenched with 10% FBS DMEM for 1 hour to promote survival. After the 1-hour quenching, the cells were returned to starvation medium.
Forty-eight hours after transfection, proliferation was measured using a Click-iT EdU assay (Thermo Fisher Scientific) according to the manufacturer’s protocol. Briefly, the starvation medium was replaced with fresh starvation media containing 10 mM EdU and incubated for 6 hours. After the 6-hour incubation, the cells were fixed with 10% neutral-buffered formalin and rinsed 3× with 1× PBS. The incorporated Edu in the fixed cells was then labeled with Alexa Fluor 594 according to the manufacturer’s instructions, while the nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI). Total and EdU-containing nuclei were quantified via epifluorescence microscopy on a Nikon Ti-Eclipse using DAPI and tetramethyl rhodamine isothiocyanate (TRITC) filter cubes, respectively. The object count selection feature in Nikon Elements was used to automatically select and count DAPI and EdU+ nuclei, with the percent of EdU of total nuclei reported as the % S phase+ cells.
bFGF production and sequestration was quantified 24 hours posttransfection using an enzyme-linked immunosorbent assay (R&D Technologies) according to the manufacturer’s instructions. Briefly, the media was collected from each well 24 hours after transfection to determine the “free” produced bFGF. MCMs were then dissolved via incubation in 100 mM EDTA in PBS at pH 7.4 for 30 min at 37°C to determine the sequestered produced bFGF.
Sequestration of model secreted fluorescent protein
A dsDNA template was designed in the online molecular biology suite Benchling that contained a noncoding lead sequence of 60 base pairs followed by a T7 promoter, a 5′ untranslated region derived from β-globin, a Kozak sequence, a signal peptide derived from mouse matrix metalloproteinase 9, and then mCherry coding sequence. The mRNA was synthesized from this template as described above containing the ψU and 5-meC nucleobases to generate CM-mRNA encoding for a secreted mCherry protein. hDFs were cultured as described above and seeded into cyclin-olefin high-content half-area 96-well plates (Corning) at 1000 cells per well. The cells were transfected with 75 ng of the CM-mCherry mRNA in Opti-MEM as described above and then quenched with 10% FBS DMEM after 6 hours. After 1 hour of quenching, the media was exchanged with 5% FBS in FluoroBrite DMEM (Gibco). Twelve hours after transfection, the medium was collected and exchanged with fresh 5% FBS in FluoroBrite. The cells were then imaged via phase and epifluorescence microscopy using a Nikon Ti-Eclipse and a TRITC filter. Fluorescence in the collected medium was measured for red fluorescence using a Fluoroskan Multiplate reader (590ex/630em; Thermo Fisher Scientific). Media fluorescence was reported as mean relative fluorescence between three technical replicates (S4.6).
In vivo localization of secreted mCherry
Male C57BLKS/J mice with a homozygous mutation at the leptin receptor (db+/db+) were given bilateral circular dermal wounds, approximately 1 cm in diameter and approximately 0.5 cm apart across the upper back. Briefly, the animals were treated with isoflurane at 5% until reaching anesthetic depth and then maintained under anesthesia with 2.5% isoflurane during the procedure. The hair from the back was removed from the skin using Nair hair removal cream, followed by skin sanitization with 70% ethyl alcohol immediately before injury. The skin was pulled away from the underlying tissue using forceps, and surgical scissors were used to remove the full-thickness skin, including the epidermis, subcutaneous fat, dermis, and underlying fascia.
Before treatment, the animals were imaged for red fluorescence using an IVIS (PerkinElmer) to provide a baseline level of autofluorescence of the wounds. Briefly, the animals were anesthetized with 5% isoflurane and then maintained with 2.5% while imaging. The preset imaging protocol for mCherry was selected with a focal height of 1.5 cm and the exposure settings automatically controlled. Within 1 hour of injury, the treatment group (left wound) was transfected with 3-μg CM-mCherry mRNA with MCMs as described above, while the right wound received a saline control. Six hours after transfection, the surgical dressing was removed, and the animal was imaged again for red fluorescence via IVIS. The animals were subsequently imaged at 1, 2, 3, 4, and 5 days after transfection. Regions of interest were set on each wound, and red fluorescence over time was reported as mean radiant efficiency between wounds with the SE.
Dermal wound model in diabetic mice
Animals were injured as described above (in vivo localization of secreted mCherry). Within 1 hour of injury, animals were treated with 3 μg of CM-mRNA with 4-μg MCM, 5-μg recombinant bFGF with 4-μg MCMs, 3 μg of WT-mRNA with 4-μg MCMs with and without 0.4-μg B18R recombinant protein, 0.7 μg of CM-mRNA (low) with and without 0.9-μg MCMs, 4-μg MCMs alone, 4-μg MCMs with 0.4-μg B18R recombinant protein, and Opti-MEM (saline control) alone. The mRNA conditions were condensed with Lipofectamine Messenger Max as described above in the in vitro experiments. For MCM-containing conditions, 4 μg of MCMs were used and incubated with the mRNA lipoplexes for 30 min at room temperature while rotating. For the 3-μg mRNA with MCM conditions, the mRNA/MCM solution was pelleted after 30 min of incubation and resuspended at 150 ng/μl mRNA. The 3-μg mRNA (with and without B18R), bFGF, MCMs only (with and without B18R), and Opti-MEM–only conditions were delivered in 20 μl of total solution per wound, while the 0.7 mRNA conditions were delivered in 30 μl per wound, the maximum volume the wounds were able to hold without the fluid spilling over. There was no 3-μg mRNA condition without MCMs as the volume required to deliver 3-μg mRNA without the pelleting and concentrating step afforded by the MCMs was greater than the volume capacity of the wounds. The treatments were left on the wounds for 1 min and then dressed with the Transpore surgical tape (3M) to provide a gas-permeable wound barrier that aided in reducing wound infection and to physically restrict the wound perimeter to promote reepithelization over wound constriction as the method of wound healing. Each treatment consisted of four to five animals with two wounds per animal (n = 8 to 10 wounds) with treatments divided evenly among separate surgical cohorts. All animal care and procedures were carried out under Institutional Animal Care and Use Committee–approved protocols overseen by the Research Animal Resources and Compliance Department at the University of Wisconsin-Madison.
Dermal wound closure measurements
The wound perimeters were measured on days 1, 3, 5, 7, 13, 16, and 19. Briefly, the animals were anesthetized via isoflurane as described above and the wounds photographed. A glass petri dish with a 1-cm scale cross was gently pressed on to the Transpore-dressed wound to produce a flat-image plane that reduced distortions and artifacts. The Transpore tape was replaced as necessary to provide clear images of the wound perimeter and ensure that proper wound edge physical restriction was maintained. Special care was taken during this wound dressing change to minimize disturbing the wound. After 19 days of wound measurements, the wound dressing was removed for a final image and gross assessment. The animals were euthanized via asphyxiation with CO2 the dermal tissue collected for later histological analysis. The wounds were analyzed in ImageJ using the 1-cm scale cross from the petri dish and perimeter-tracing tool. The wound perimeters were reported as mean wound perimeter as a percentage of the starting wound perimeter (n = 8 to 10) with the SEM.
Histological assessment of wound resolution
Dermal tissues collected after 19 days of wound perimeter measurements were placed in between tissue foam pads in a tissue cassette holder (one wound per holder) and fixed in 10% neutral-buffered formalin for ~36 hours. After fixation, the samples were stored in 70% ethanol while remaining in the tissue cassette holders. The fixed tissues were then dehydrated in 100% ethanol, cut laterally through the center (full diameter of the wound area), and embedded in paraffin with the full dermal thickness transverse to the sectioning plane. The embedded tissues were sectioned in 5-μm sections and mounted on microscope slides for staining.
After sectioning and slide mounting, the samples were cleared with 5× 30-s washes in Xylenes and then rehydrated in subsequent washes in 100, 95, 90, and 85% ethanol for 10 min each. The samples were then washed in DI water 3× for 1 min each. Samples were treated with Harris hematoxylin with glacial acetic acid for 3 min and then rinsed 3× in DI water and then a 5-min incubation in DI water. The hematoxylin-stained samples were then treated in Eosin Phloxine stain for 30 s and then rinsed 3× for 1 min in 95% ethanol and then again in 100% ethanol. The samples were then covered with CitriSolv mounting media and coverslipped. Color-image micrographs of H&E-stained sections at 10× resolution were taken using a Nikon Ti-Eclipse and Nikon DU-S5 color camera. Large format images were stitched together automatically using the Nikon Elements ND-acquisition feature. The samples were cropped to show the entire wound cross section and uninjured wound edges. The samples were deidentified and scored by two referees for scoring the level of wound resolution. The score criteria defined six stages of wound resolution beginning with (i) incomplete wound closure as evidenced by clear gaps in the dermal tissue micrograph; (ii) complete wound closure with no presence of dermal/epidermal stratification; (iii) apparent epidermal/dermal stratification, but not contiguous across the wound diameter; (iv) complete wound closure and stratification with presence of granulation tissue; (v) complete closure and stratification with apparent keratinization and minimal granulation tissue; and (vi) complete wound closure, dermal stratification, keratinization, no granulation tissue, deposition of adipose tissue within wound perimeter, and presence of underlying connective fascia (S4.4). The scores were reported as scatter plots with 95% confidence intervals (n = 8 to 10).
For immunohistochemistry of collagen type I, collagen type III, and CD31, paraffin-embedded samples were cut at a 5-μm thickness and mounted on ColorFrost Plus microscope slides. Samples were deparaffinized, rehydrated, exposed to 3% hydrogen peroxide to eliminate endogenous peroxidase activity, and fixed with 10% neutral-buffered formalin to prevent tissue removal from slides. Heat-induced epitope retrieval was performed using 10 mM sodium citrate at 80°C for 1.5 hours. Samples were then blocked with Rodent Block M (Biocare Medical, Pacheco, CA) and incubated overnight at 4°C with rabbit primary polyclonal antibodies to detect mouse CD31+ endothelial cells (1:100; Abcam, Raleigh, NC), type I collagen (1:200; Abcam, Raleigh, NC), and type III collagen (1:150; Abcam, Raleigh, NC). After primary antibody incubation, samples were exposed to Rabbit on Rodent HRP Polymer (Biocare Medical, Pacheco, CA). The bound antibody complex was visualized using DAB (Innovex Biosciences, Richmond, CA). Stained sections were dehydrated, cleared, coverslipped, and viewed using light microscopy. After immunohistochemistry (IHC), micrographs were collected using a camera-assisted microscope (Nikon Eclipse microscope, model E6000 with an Olympus camera, model DP79). Approximately four dermal sections were captured and counted per wound. If sections exhibited any processing flaws (not faced, wrinkled, torn, etc.), then samples were omitted from further analyses. All images were captured at 40× resolution and quantified via ImageJ (National Institutes of Health, Bethesda, MD). Density measurements of the wound were collected and normalized to the area measured. Dermal samples were also subjected to Masson’s trichrome stain and used to quantify wound closure. Samples were imaged at 40× resolution and quantified via FIJI (National Institutes of Health, Bethesda, MD) using the color deconvolution function to measure the red and blue channels. All IHC and histology results were expressed as density/mm2.
Statistical analysis
For each protein analyzed via IHC, four sections per wound were quantified and averaged. A total of 8 to 10 wounds were examined per treatment. A one-way analysis of variance (ANOVA) with Dunnett’s post hoc analysis (relative to no-treatment control) was used to examine treatment differences. Results were considered significant if the overall P value for the F test in ANOVA was P ≤ 0.5. Experimental data were presented as the means ± SEM. Computations and figures were performed using KaleidaGraph, version 4.03 (Synergy Software Inc., Reading, PA).
Figure generation and statistics
All schematic graphics were created by the author using Adobe Illustrator and Photoshop except for the microparticle, which was commissioned from a professional graphic designer. Figures were generated in Adobe Illustrator and graphs were generated in GraphPad Prism. Statistics were done in GraphPad Prism with statistical tests, replicates, and significance for each comparison as denoted in the methods and in each figure.
Supplementary Material
Acknowledgments
Funding: The authors would like to acknowledge the NIH (grants R01AR059916 and R21EB019558), Environmental Protection Agency (S3.TAR grant 83573701), the National Science Foundation (grant DMR 1105591), and Michael and Mary Sue Shannon for funding provided for this research, and National Institute of General Medical Sciences’ Biotechnology Training Program (grant NIH 5 T32 GM008349) and National Science Foundation (grant DGE-1256259) for funding provided to A.S.K. In addition, the authors would like to acknowledge use of facilities and instrumentation supported by the National Science Foundation through the University of Wisconsin-Madison Materials Research Science and Engineering Center (grants DMR-1121288, 0079983, and 0520057), as well as the University of Wisconsin-Madison Nanoscale Science and Engineering Center (grants DMR-0832760 and 0425880), and the Department of Surgery Histology Core at the University of Wisconsin. X.Y. acknowledges current financial support from the National Natural Science Foundation of China (31870959). Author contributions: A.S.K., X.Y., and W.L.M. designed the study. A.S.K., X.Y., and J.M.U. synthesized materials, performed in vitro experiments, and analyzed data. C.S.C. and L.A.W. performed histological characterizations and analysis. G.M.D. and T.A.H. developed the dermal wound healing model and performed animal surgeries and care. A.S.K. performed animal treatments, in vivo studies, and data analysis. A.S.K. and W.L.M. wrote the manuscript. All authors reviewed and approved the manuscript. Competing interests: W.L.M. is a cofounder and Chief Science Officer at Stem Pharm Inc. and Dianomi Therapeutics. A.S.K. is a consultant to Stem Pharm and Dianomi Inc. The authors declare that they have no remaining competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/27/eaba2422/DC1
REFERENCES AND NOTES
- 1.Jones A. L., Bucholz R. W., Bosse M. J., Mirza S. K., Lyon T. R., Webb L. X., Pollak A. N., Golden J. D., Valentin-Opran A.; BMP- Evaluation in Surgery for Tibial Trauma-Allgraft (BESTT-ALL) Study Group , Recombinant human BMP-2 and allograft compared with autogenous bone graft for reconstruction of diaphyseal tibial fractures with cortical defects. A randomized, controlled trial. J. Bone Joint Surg. Am. 88, 1431–1441 (2006). [DOI] [PubMed] [Google Scholar]
- 2.Burkus J. K., Heim S. E., Gornet M. F., Zdeblick T. A., Is INFUSE bone graft superior to autograft bone? An integrated analysis of clinical trials using the LT-CAGE lumbar tapered fusion device. J. Spinal Disord. Tech. 16, 113–122 (2003). [DOI] [PubMed] [Google Scholar]
- 3.Shields L. B. E., Raque G. H., Glassman S. D., Campbell M., Vitaz T., Harpring J., Shields C. B., Adverse effects associated with high-dose recombinant human bone morphogenetic protein-2 use in anterior cervical spine fusion. Spine 31, 542–547 (2006). [DOI] [PubMed] [Google Scholar]
- 4.Ruponen M., Honkakoski P., Rönkkö S., Pelkonen J., Tammi M., Urtti A., Extracellular and intracellular barriers in non-viral gene delivery. J. Control. Release 93, 213–217 (2003). [DOI] [PubMed] [Google Scholar]
- 5.Yamamoto A., Kormann M., Rosenecker J., Rudolph C., Current prospects for mRNA gene delivery. Eur. J. Pharm. Biopharm. 71, 484–489 (2009). [DOI] [PubMed] [Google Scholar]
- 6.McIvor R. S., Therapeutic delivery of mRNA: The medium is the message. Mol. Ther. 19, 822–823 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sahin U., Karikó K., Türeci Ö., mRNA-based therapeutics — Developing a new class of drugs. Nat. Rev. Drug Discov. 13, 759–780 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Kirschman J. L., Bhosle S., Vanover D., Blanchard E. L., Loomis K. H., Zurla C., Murray K., Lam B. C., Santangelo P. J., Characterizing exogenous mRNA delivery, trafficking, cytoplasmic release and RNA – Protein correlations at the level of single cells. Nucleic Acids Res. 45, e113 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lv H., Zhang S., Wang B., Cui S., Yan J., Toxicity of cationic lipids and cationic polymers in gene delivery. J. Control. Release 114, 100–109 (2006). [DOI] [PubMed] [Google Scholar]
- 10.Kormann M. S. D., Hasenpusch G., Aneja M. K., Nica G., Flemmer A. W., Herber-Jonat S., Huppmann M., Mays L. E., Illenyi M., Schams A., Griese M., Bittmann I., Handgretinger R., Hartl D., Rosenecker J., Rudolph C., Expression of therapeutic proteins after delivery of chemically modified mRNA in mice. Nat. Biotechnol. 29, 154–157 (2011). [DOI] [PubMed] [Google Scholar]
- 11.Thess A., Grund S., Mui B. L., Hope M. J., Baumhof P., Fotin-Mleczek M., Schlake T., Sequence-engineered mRNA without chemical nucleoside modifications enables an effective protein therapy in large animals. Mol. Ther. 23, 1456–1464 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roundtree I. A., Evans M. E., Pan T., He C., Dynamic RNA modifications in gene expression regulation. Cell 169, 1187–1200 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Khalil A. S., Yu X., Xie A. W., Fontana G., Umhoefer J. M., Johnson H. J., Hookway T. A., McDevitt T. C., Murphy W. L., Functionalization of microparticles with mineral coatings enhances non-viral transfection of primary human cells. Sci. Rep. 7, 14211 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yu, A. H. Biedrzycki, A. S. Khalil, D. Hess, J. M. Umhoefer, M. D. Markel, W. L. Murphy X., Nanostructured mineral coatings stabilize proteins for therapeutic delivery. Adv. Mater. 1404, 11 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khalil A. S., Xie A. W., Johnson H. J., Murphy W. L., Sustained release and protein stabilization reduce the growth factor dosage required for human pluripotent stem cell expansion. Biomaterials 248, 120007 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Warren L., Manos P. D., Ahfeldt T., Loh Y.-H., Li H., Lau F., Ebina W., Mandal P. K., Smith Z. D., Meissner A., Daley G. Q., Brack A. S., Collins J. J., Cowan C., Schlaeger T. M., Rossi D. J., Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell 7, 618–630 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Karikó K., Muramatsu H., Welsh F. A., Ludwig J., Kato H., Akira S., Weissman D., Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Mol. Ther. 16, 1833–1840 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Karikó K., Buckstein M., Ni H., Weissman D., Suppression of RNA recognition by Toll-like receptors: The impact of nucleoside modification and the evolutionary origin of RNA. Immunity 23, 165–175 (2005). [DOI] [PubMed] [Google Scholar]
- 19.Makino T., Jinnin M., Muchemwa F. C., Fukushima S., Kogushi-Nishi H., Moriya C., Igata T., Fujisawa A., Johno T., Ihn H., Basic fibroblast growth factor stimulates the proliferation of human dermal fibroblasts via the ERK1/2 and JNK pathways. Br. J. Dermatol. 162, 717–723 (2010). [DOI] [PubMed] [Google Scholar]
- 20.Yu P.-J., Ferrari G., Galloway A. C., Mignatti P., Pintucci G., Basic fibroblast growth factor (FGF-2): The high molecular weight forms come of age. J. Cell. Biochem. 100, 1100–1108 (2007). [DOI] [PubMed] [Google Scholar]
- 21.Zhang Z., Zheng Y., Li Y., Bai H., Ma T., Song X., Zhao J., Gao L., The effects of sodium usnic acid by topical application on skin wound healing in rats. Biomed. Pharmacother. 97, 587–593 (2018). [DOI] [PubMed] [Google Scholar]
- 22.Pelizzo G., Avanzini M. A., Icaro Cornaglia A., Osti M., Romano P., Avolio L., Maccario R., Dominici M., de Silvestri A., Andreatta E., Costanzo F., Mantelli M., Ingo D., Piccinno S., Calcaterra V., Mesenchymal stromal cells for cutaneous wound healing in a rabbit model: Pre-clinical study applicable in the pediatric surgical setting. J. Transl. Med. 13, 219 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tsuboi R., Rifkin D. B., Recombinant basic fibroblast growth factor stimulates wound healing in healing-impaired Db/Db mice. J. Exp. Med. 172, 245–251 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yoshioka N., Gros E., Li H.-R., Kumar S., Deacon D. C., Maron C., Muotri A. R., Chi N. C., Fu X.-D., Yu B. D., Dowdy S. F., Efficient generation of human iPSCs by a synthetic self-replicative RNA. Cell Stem Cell 13, 246–254 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rubin H., The disparity between human cell senescence in vitro and lifelong replication in vivo. Nat. Biotechnol. 20, 675–681 (2002). [DOI] [PubMed] [Google Scholar]
- 26.Lechardeur D., Lukacs G. L., Intracellular barriers to non-viral gene transfer. Curr. Gene Ther. 2, 183–194 (2002). [DOI] [PubMed] [Google Scholar]
- 27.Yu X., Khalil A., Dang P. N., Alsberg E., Murphy W. L., Multilayered inorganic microparticles for tunable dual growth factor delivery. Adv. Funct. Mater. 24, 3082–3093 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Suárez-González D., Lee J. S., Diggs A., Lu Y., Nemke B., Markel M., Hollister S. J., Murphy W. L., Controlled multiple growth factor delivery from bone tissue engineering scaffolds via designed affinity. Tissue Eng. Part A 20, 2077–2087 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jongpaiboonkit L., Franklin-Ford T., Murphy W. L., Growth of hydroxyapatite coatings on biodegradable polymer microspheres. ACS Appl. Mater. Interfaces 1, 1504–1511 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Choi S., Yu X., Jongpaiboonkit L., Hollister S. J., Murphy W. L., Inorganic coatings for optimized non-viral transfection of stem cells. Sci. Rep. 3, 1567–1593 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen G., Gulbranson D. R., Yu P., Hou Z., Thomson J. A., Thermal stability of fibroblast growth factor protein is a determinant factor in regulating self-renewal, differentiation, and reprogramming in human pluripotent stem cells. Stem Cells 30, 623–630 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sørensen V., Nilsen T., Wiȩdłocha A., Functional diversity of FGF-2 isoforms by intracellular sorting. Bioessays 28, 504–514 (2006). [DOI] [PubMed] [Google Scholar]
- 33.M. T. Ramsey, A Review of the Main Viral and Non-Viral Vectors for Gene Therapy (2016).
- 34.Konishi M., Kawamoto K., Izumikawa M., Kuriyama H., Yamashita T., Gene therapy clinical trials worldwide to 2012– An update. J. Gene Med. 10, 610–618 (2008). [DOI] [PubMed] [Google Scholar]
- 35.Wirth T., Parker N., Ylä-Herttuala S., History of gene therapy. Gene 525, 162–169 (2013). [DOI] [PubMed] [Google Scholar]
- 36.McKay W. F., Peckham S. M., Badura J. M., A comprehensive clinical review of recombinant human bone morphogenetic protein-2 (INFUSE® bone graft). Int. Orthop. 31, 729–734 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Okumura M., Okuda T., Nakamura T., Yajima M., Acceleration of wound healing in diabetic mice by basic fibroblast growth factor. Biol. Pharm. Bull. 19, 530–535 (1996). [DOI] [PubMed] [Google Scholar]
- 38.Kanda N., Morimoto N., Ayvazyan A. A., Takemoto S., Kawai K., Nakamura Y., Sakamoto Y., Taira T., Suzuki S., Evaluation of a novel collagen-gelatin scaffold for achieving the sustained release of basic fibroblast growth factor in a diabetic mouse model. J. Tissue Eng. Regen. Med. 8, 29–40 (2014). [DOI] [PubMed] [Google Scholar]
- 39.Morimoto N., Yoshimura K., Niimi M., Ito T., Aya R., Fujitaka J., Tada H., Teramukai S., Murayama T., Toyooka C., Miura K., Takemoto S., Kanda N., Kawai K., Yokode M., Shimizu A., Suzuki S., Novel collagen/gelatin scaffold with sustained release of basic fibroblast growth factor: Clinical trial for chronic skin ulcers. Tissue Eng. Part A 19, 1931–1940 (2013). [DOI] [PubMed] [Google Scholar]
- 40.Matsumoto S., Tanaka R., Okada K., Arita K., Hyakusoku H., Miyamoto M., Tabata Y., Mizuno H., The effect of control-released basic fibroblast growth factor in wound healing: Histological analyses and clinical application. Plast. Reconstr. Surg. Glob. Open 1, e44 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Clements A. E. B., Groves E. R., Chamberlain C. S., Vanderby R., Murphy W. L., Microparticles locally deliver active interleukin-1 receptor antagonist in vivo. Adv. Healthc. Mater. 7, e1800263 (2018). [DOI] [PubMed] [Google Scholar]
- 42.Hanna A., Thompson D. L., Hellenbrand D. J., Lee J.-S., Madura C. J., Wesley M. G., Dillon N. J., Sharma T., Enright C. J., Murphy W. L., Sustained release of neurotrophin-3 via calcium phosphate-coated sutures promotes axonal regeneration after spinal cord injury. J. Neurosci. Res. 94, 645–652 (2016). [DOI] [PubMed] [Google Scholar]
- 43.Lee J. S., Suarez-Gonzalez D., Murphy W. L., Mineral coatings for temporally controlled delivery of multiple proteins. Adv. Mater. 23, 4279–4284 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Horn S. D., Barrett R. S., Fife C. E., Thomson B., A predictive model for pressure ulcer outcome: The wound healing index. Adv. Skin Wound Care 28, 560–572 (2015). [DOI] [PubMed] [Google Scholar]
- 45.Lee J. S., Lu Y., Baer G. S., Markel M. D., Murphy W. L., Controllable protein delivery from coated surgical sutures. J. Mater. Chem. 20, 8894–8903 (2010). [Google Scholar]
- 46.Hellenbrand D. J., Reichl K. A., Travis B. J., Filipp M. E., Khalil A. S., Pulito D. J., Gavigan A. V., Maginot E. R., Arnold M. T., Adler A. G., Murphy W. L., Hanna A. S., Sustained interleukin-10 delivery reduces inflammation and improves motor function after spinal cord injury. J. Neuroinflammation 16, 93 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hanna A., Kaeppler K. E., Ehlers M. E., Dadsetan M., Yaszemski M. J., Toigo R. D., Kim J., Hwang E., Bogarin-Miranda E., Buchholz M. M., Springer A. R., Hellenbrand D. J., Peripheral nerve grafts and chondroitinase ABC application improves functional recovery after complete spinal cord transection. J. Neurol. Res. 3, 85–95 (2013). [Google Scholar]
- 48.Sidgwick G. P., Bayat A., Extracellular matrix molecules implicated in hypertrophic and keloid scarring. J. Eur. Acad. Dermatol. Venereol. 26, 141–152 (2012). [DOI] [PubMed] [Google Scholar]
- 49.Braiman-Wiksman L., Solomonik I., Spira R., Tennenbaum T., Novel insights into wound healing sequence of events. Toxicol. Pathol. 35, 767–779 (2007). [DOI] [PubMed] [Google Scholar]
- 50.Pastar I., Stojadinovic O., Yin N. C., Ramirez H., Nusbaum A. G., Sawaya A., Patel S. B., Khalid L., Isseroff R. R., Tomic-Canic M., Epithelialization in wound healing: A comprehensive review. Adv. Wound Care 3, 445–464 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ramaswamy S., Tonnu N., Tachikawa K., Limphong P., Vega J. B., Karmali P. P., Chivukula P., Verma I. M., Systemic delivery of factor IX messenger RNA for protein replacement therapy. Proc. Natl. Acad. Sci. U.S.A. 114, E1941–E1950 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Floor S. N., Doudna J. A., Tunable protein synthesis by transcript isoforms in human cells. eLife 5, e10921 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hornung V., Ellegast J., Kim S., Brzózka K., Jung A., Kato H., Poeck H., Akira S., Conzelmann K.-K., Schlee M., Endres S., Hartmann G., 5'-triphosphate RNA is the ligand for RIG-I. Science 314, 994–997 (2006). [DOI] [PubMed] [Google Scholar]
- 54.Diebold S. S., Kaisho T., Hemmi H., Akira S., e Sousa C. R., Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science 303, 1529–1531 (2004). [DOI] [PubMed] [Google Scholar]
- 55.Kawai T., Akira S., The role of pattern-recognition receptors in innate immunity: Update on toll-like receptors. Nat. Immunol. 11, 373–384 (2010). [DOI] [PubMed] [Google Scholar]
- 56.Pfeffer L. M., The role of nuclear factor κB in the interferon response. J. Interferon Cytokine Res. 31, 553–559 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim Y. G., Baltabekova A. Z., Zhiyenbay E. E., Aksambayeva A. S., Shagyrova Z. S., Khannanov R., Ramanculov E. M., Shustov A. V., Recombinant Vaccinia virus-coded interferon inhibitor B18R: Expression, refolding and a use in a mammalian expression system with a RNA vector. PLOS ONE, 1–18 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zheng X., Baker H., Hancock W. S., Fawaz F., McCaman M., Pungor E. Jr., Proteomic analysis for the assessment of different lots of fetal bovine serum as a raw material for cell culture. Part IV. Application of proteomics to the manufacture of biological drugs. Biotechnol. Prog. 22, 1294–1300 (2006). [DOI] [PubMed] [Google Scholar]
- 59.Chen M., Huang J., Yang X., Liu B., Zhang W., Huang L., Deng F., Ma J., Bai Y., Lu R., Huang B., Gao Q., Zhuo Y., Ge J., Serum starvation induced cell cycle synchronization facilitates human somatic cells reprogramming. PLOS Med. 7, e28203 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/27/eaba2422/DC1