

A highly efficient catalyst for the additive-free dehydrogenation and hydrogenation of amines and alcohols is described.
Abstract
Catalytic dehydrogenation and hydrogenation of amines and alcohols are important in the synthesis of fine chemicals. Despite several efficient homogeneous catalysts having been identified, highly active heterogeneous catalysts remain elusive, although they would meet an unmet need. Here, we show that bimetallic Pd-Au nanoparticles with Pd-to-Au molar ratios of 3:1 immobilized on multiwall carbon nanotubes (Pd3Au1/CNT) display high catalytic activity in the oxidant-free and acceptorless dehydrogenation and hydrogenation of N- and O-containing heterocyclic compounds, amines/imines, and alcohols/ketones. Transmission electron microscopy analysis demonstrates the preferential exposure of Pd3Au1(111) facets on the Pd3Au1/CNT catalyst. Mechanistic insights combining experimental data with density functional theory calculations reveal that the Pd3Au1(111) surface enhances both dehydrogenation and hydrogenation reactions and provides a rationale for the observed enhancements.
INTRODUCTION
Catalytic hydrogenation and, to a lesser extent, dehydrogenation reactions are extensively used in the preparation of a wide and diverse range of chemicals, ranging from simple fuels to complex pharmaceutical products (1, 2). For example, N- and O-containing compounds such as indoles, quinolines, ketones, and their hydrogenated derivatives may be prepared via catalytic hydrogenation and dehydrogenation reactions (3, 4). In addition to affording valuable chemical products, reversible catalytic dehydrogenation and hydrogenation of N- and O-containing compounds could potentially lead to practical hydrogen carriers (2, 5, 6). The introduction of nitrogen or oxygen atoms is considered advantageous for this latter application as they decrease the endothermicity of the dehydrogenation reaction lower than that of hydrocarbons (7).
Dehydrogenation and hydrogenation reactions involving homogeneous catalysts are well developed, although most catalyze either the hydrogenation reaction or the opposite dehydrogenation (7–12). However, catalysts able to both dehydrogenate and hydrogenate N- and O-containing compounds are becoming increasingly attractive, and well-defined molecular catalysts based on Ir (10, 11, 13), Ru (14), Co (15), and Fe (7) have been reported. A recent prominent example comprises homogeneous Ir complexes with substituted triazolylidene ligands that catalyze the reversible, acceptorless dehydrogenation of 1,2,3,4-tetrahydroquinoline to quinoline in water (13). The dehydrogenation and hydrogenation of various heterocycles has also been achieved with visible-light photoredox catalysts and electrocatalysts based on homogeneous Co (15) and Fe (16) complexes.
Despite the excellent performance of these homogeneous catalysts on a laboratory scale, their large-scale implementation is limited by their high cost, stability, and complex recycling protocols. In this respect, heterogeneous catalysts have intrinsic advantages that overcome these limitations, and a number of promising catalysts have been developed (17). However, previous systems required either high catalyst loadings or high temperatures to achieve high efficiency, and the substrate scope was mostly limited to several amines (18). Recently, a Co single-atom catalyst was shown to catalyze the dehydrogenation of N-heterocycles to release H2 and the transfer hydrogenation of N-heterocycles using formic acid as a hydrogen source to afford formamides (19). However, subsequent dehydrogenation of the formamides is problematic and prevented reversible hydrogenation/dehydrogenation cycles. Heterogeneous catalysts that efficiently catalyze both dehydrogenation and hydrogenation of N- and O-containing compounds remain elusive. Notably, limited information is available on the catalytically active sites and the reaction mechanism of heterogeneous catalysis.
Bimetallic catalysts combining Pd and Au are known to be highly active for the oxidative activation of carbon-hydrogen bonds in alcohols, toluene, and methane (20–22) and in hydrogenation reactions (23). However, Pd-Au nanoparticles have not been studied in the acceptorless dehydrogenation of amines and alcohols and the reverse hydrogenation reactions. With the inspiration by these improvements, we report on the discovery of a multiwall carbon nanotube (CNT)–stabilized Pd-Au bimetallic nanoparticle (NP) system that preferentially exposes Pd3Au1(111) facets and displays high catalytic activity in the acceptorless dehydrogenation and reverse hydrogenation of N-heterocycles, amines/imines, and alcohols/ketones. Mechanistic studies were performed, and the high catalytic activity was investigated by DFT calculations, which shows that the Pd3Au1(111) surface lowers energy barriers in each of the elementary steps in both reactions.
RESULTS
Catalyst preparation and characterization
A series of Pd-Au bimetallic NPs using trisodium citrate dihydrate (TCD) as stabilizing reagent to prevent the aggregation of the NPs and immobilized on CNTs were prepared from aqueous solutions of K2PdCl4 (0.02 M) and HAuCl4 (0.015 M) by varying the Pd:Au ratio to generate a series of materials containing a total metal content of 2 weight % (wt %). The CNTs were dispersed in water containing TCD (with a metal:TCD ratio of 1:3), to which the aqueous solutions containing the metal precursors were added dropwise. The resulting suspensions were heated to 30°C for 30 min, excess NaBH4 was subsequently added, and the mixture was maintained at 30°C for 2 hours to afford powders. The powders were filtered, washed with water, and dried under vacuum (see Materials and Methods for full synthetic details). Catalysts with Pd-to-Au molar ratios of 3:1 (Pd3Au1/CNT), 1:1 (Pd1Au1/CNT), and 1:3 (Pd1Au1/CNT) were prepared together with monometallic Pd (Pd/CNT) and Au (Au/CNT) systems as controls. The metal ratios and loadings were measured by an inductively coupled plasma optical emission spectrometer(ICP-OES).
Transmission electron microscopy (TEM) and high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) were used to characterize the catalysts [see Fig. 1 (A to C) for the Pd3Au1 catalyst and figs. S1 and S2 for the other catalysts]. The Pd3Au1 NPs are uniformly dispersed on the CNTs in the Pd3Au1/CNT catalyst with the NPs having a narrow size distribution of 2 to 4 nm with an average diameter of 2.48 ± 0.22 nm (Fig. 1A and fig. S1A). Figure 1B shows the high-resolution TEM (HRTEM) image of a typical Pd-Au bimetallic NP with an interplanar spacing of 2.3 Å, which corresponds to a face-centered cubic (fcc) lattice structure with a (111) surface (21). HAADF-STEM was used to investigate the elemental distribution of the Pd-Au NPs. Figure 1F shows the HAADF-STEM crossline profile of the sample shown in Fig. 1C (corresponding to the red line). The overlap of the Pd and Au profiles demonstrates the coexistence of both metals in the NP and is indicative of local alloying. HAADF-STEM–energy dispersive spectroscopy (EDS) mapping for the Pd3Au1/CNT catalyst (Fig. 1, D and E) corroborates the notion of a Pd-Au alloy. Moreover, HAADF-STEM images of the catalysts with different metal ratios have similar mean particle sizes, ranging from 2.32 ± 0.12 nm to 2.89 ± 0.16 nm, and with similar particle size distributions. Moreover, there is no clear correlation between size and catalytic activity, and since the sizes are similar, the difference in catalytic activities observed cannot be attributed to differences between the size of the NPs (figs. S1 and S2). The size of the Pd3Au1 NPs increases to 2.64 ± 0.25 nm after dehydrogenation (compared to 2.48 nm in the pristine catalyst). Because of the apparent stability of the catalyst in hydrogenation and dehydrogenation reactions, we assumed that the small change in particle size does not strongly affect the catalytic activity (fig. S2).
Fig. 1. Characterization of the Pd3Au1/CNT catalyst.
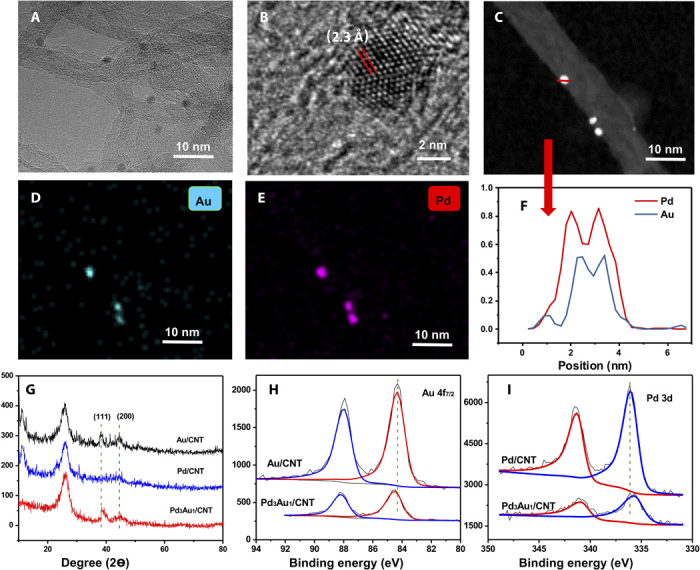
(A) Bright-field TEM image; (B) HRTEM image; (C) HAADF-STEM image. HAADF-TEM-EDS mapping images of the Pd3Au1/CNT catalyst: (D) Au; (E) Pd; (F) crossline profiles of elemental composition for the Pd-Au NP. (G) XRD patterns of the Au/CNT, Pd/CNT, and Pd3Au1/CNT catalysts. XPS of (H) Au 4f spectra of the Au/CNT and Pd3Au1/CNT catalysts and (I) Pd 3d spectra of the Pd/CNT and Pd3Au1/CNT catalysts.
X-ray diffraction (XRD) pattern of the Au/CNT catalyst (Fig. 1G) exhibits crystalline features corresponding to Au(111) and Au(200). The Au(111) diffraction peak is slightly shifted from 38.34 ± 0.03 eV in the Au/CNT material to 38.56 ± 0.03 eV in the Pd3Au1/CNT catalyst, further supporting an alloy structure with the (111) facet exposed preferentially. Note that larger Au NPs are formed at high Au loadings (figs. S1 and S2). The electronic structures of the supported NPs with different Pd:Au ratios were probed with x-ray photoelectron spectroscopy (XPS). In the Pd3Au1/CNT catalyst, the peaks located at 84.48 ± 0.02 eV and 88.15 ± 0.02 eV (Fig. 1H) may be attributed to Au 4f, and the peaks located at 335.69 and 340.96 eV correspond to Pd 3d (Fig. 1I), and confirm the presence of metallic Au0 and Pd0 in an alloy structure. The binding energies (BEs) of Pd 3d in Pd3Au1/CNT shift to lower values compared to the Pd 3d BE in Pd/CNT (Pd 3d5/2, 336.08 ± 0.02 eV; Pd 3d3/2, 341.29 ± 0.02 eV), while the Au 4f BEs in Pd3Au1/CNT move to higher values related to the Au 4f BE in Au/CNT (Au 4f7/2, 84.32 ± 0.02 eV; Au 4f5/2, 87.99 ± 0.02 eV). This change indicated the alloying of Pd-Au NPs and the electronic modification of Pd with Au. XPS analysis of the Pd3Au1/CNT catalyst reveals a Pd:Au ratio of 3.2:1 with the Pd predominantly in the metallic state, consistent with Pd-Au alloys immobilized on carbon and TiO2 surfaces (21). Furthermore, the O 1s and C 1s signals are similar for both the Pd3Au1/CNT and CNT materials, and sodium is not observed on the Pd3Au1/CNT, indicating that TCD is not retained on the surface of the catalyst. This is confirmed by 1H and 13C nuclear magnetic resonance (NMR) spectroscopy of the filtrate of the catalyst after ultrasonication in D2O. After 5 cycles, the BEs of the Pd3Au1/CNT catalyst for Pd 3d5/2 and Au 4f7/2 are 335.76 ± 0.01 eV and 84.39 ± 0.01 eV (fig. S3), respectively, which are similar to the BEs of the fresh catalyst, confirming the stability of the bimetallic catalyst.
Catalytic dehydrogenation and hydrogenation studies
The catalytic dehydrogenation of 1,2,3,4-tetrahydro-quinoline (1a) was chosen as the benchmark reaction to determine the optimum catalyst and conditions under oxidant-free and acceptorless conditions. Xylene was used as a solvent as it should be inert under the reaction conditions used. The monometallic Pd/CNT and Au/CNT catalysts afford the dehydrogenated product in only 30 and 8% yields, respectively (table S1, entries 2 and 3). In contrast, the bimetallic catalysts are considerably more active; the Pd3Au1/CNT catalyst, by heating in xylene at 140°C for 12 hours using 0.3 mole percent (mol %) of metals relative to 1a, displays the highest activity with quinoline (2a) obtained in 96% yield (table S1, entry 4) with 321 of TON (turnover number; moles of product per mole of Pd and Au) and 26.8 of TOF (turnover frequency; TON/reaction time). Under the optimal conditions, the Pd3Au1/CNT catalyst is also superior to commercial catalysts (table S1, entries 7 and 8) and various homogeneous Pd catalysts (table S1, entries 9 to 12). Changing the solvents and reaction time did not improve the activity (table S1, entries 13 to 15). Moreover, the Pd1Au1 NPs supported on charcoal and Al2O3 showed lower yields on this dehydrogenation (table S1, entries 16 and 17).
The reverse reaction, i.e., the hydrogenation of quinoline, was studied under 1 bar of hydrogen. The reaction did not proceed in the presence of the Au/CNT catalyst (table S2, entry 1), whereas full conversion was achieved using the Pd/CNT catalyst, but the selectivity of reaction was low owing to the over-hydrogenation of quinoline to decahydroquinoline (table S2, entry 2). The bimetallic Pd3Au1/CNT catalyst resulted in the near-quantitative formation of tetrahydroquinoline by heating the mixture in xylene at 80°C and 1 bar H2 for 12 hours using 0.3 mol % of metals relative to 2a (table S2, entry 3) without conversion for xylene, whereas the Pd1Au1/CNT and Pd1Au3/CNT catalysts were both less active and selective (table S2, entries 4 and 5), and yields were reduced under more ambient conditions (table S2, entries 6 and 7). In addition, using Pd3Au1/C and Pd3Au1/Al2O3, 2a was hydrogenated to 1a with 71 and 84% yields, respectively.
Following removal of the Pd3Au1/CNT catalyst from the reaction (hot filtration test), reactivity was suppressed, implying that the active sites in heterogeneous CNT-supported Pd3Au1 NPs might be stable and absent in the filtrate (fig. S4A for the dehydrogenation). In addition, a mercury poisoning experiment (24) was performed, with mercury reducing the yield of 2a to only 10%. The lower yield obtained in the presence of mercury further indicates that the active catalyst corresponds to the Pd3Au1(111) NPs. The stability and recyclability of the Pd3Au1/CNT catalyst were also established, with continuous switching between hydrogenation and dehydrogenation modes. After each step, the Pd3Au1/CNT catalyst was recovered by centrifugation, and the product was quantified. The reversible catalytic transformation between 1a and 2a could be repeated multiple times with high catalytic activity retained (fig. S4B).
Dehydrogenation of N-heterocycles
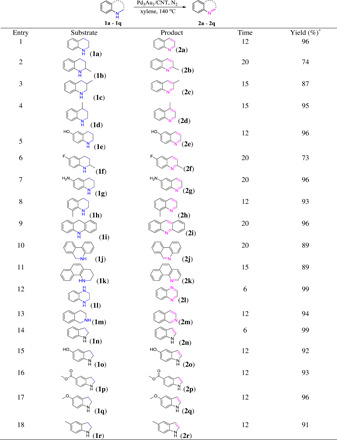
The Pd3Au1/CNT catalyst was evaluated in the dehydrogenation of various N-heterocyclic compounds and displayed outstanding activity. As shown in Table 1, functional groups at the 2-, 3-, and 4-positions influenced the dehydrogenation reaction with the yield increasing (up to 95%) with the distance of the substituent from the N atom (Table 1, entries 2 to 4). Quinolines with CH3, OH, F, or NH2 groups at the 6- and 8-positions on the aromatic ring were transformed in 73 to 96% yields (Table 1, entries 5 to 8). N-heteroarenes such as 9,10-dihydroacridine, 5,6-dihydrophenanthridine, and 1,2,3,4-tetrahydrobenzo[h]quinoline were dehydrogenated selectively, and in all cases, the desired products were obtained in good to excellent yields (Table 1, entries 9 to 11). Hydroxyl-, methoxy-, and methyl carboxylate–substituted indolines were transformed under the optimized conditions in yields of 91 to 99% (Table 1, entries 14 to 18), confirming the high selectivity/tolerance of the catalyst.
Table 1. Dehydrogenation of N-heterocycle derivatives using the Pd3Au1/CNT catalyst.
Reaction conditions: Pd3Au1/CNT (0.3 mol % of metals to substrate), N-heterocyclic substrate (0.2 mmol), xylene (1 ml), N2 atmosphere, and 140°C. *Isolated yield.
Dehydrogenation of N-alkylated amines
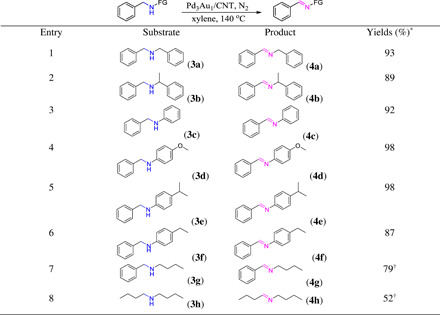
Imines and amines are key intermediates in the synthesis of fine chemicals, pharmaceuticals, and natural products (25), obtained from condensation reactions of amines with active carbonyl compounds and self-coupling of primary amines (26, 27). Imines can also be obtained from the oxidation of amines, and the dehydrogenation of amines to imines is particularly attractive as fewer side products tend to be generated (28). Thus, the Pd3Au1/CNT catalyst was evaluated in the dehydrogenation of N-alkylated amines to imines under the optimized conditions and was found to selectively provide the desired imines in high yields (Table 2, entries 1 and 2). Notably, substituted N-phenylbenzylamines with different substituent groups were converted to their respective imines in excellent yields (Table 2, entries 3 to 6). Moreover, N-alkyl substrates were successfully dehydrogenated to their corresponding imines in moderate yields (Table 2, entries 7 and 8). These imine/amine couples can be considered as liquid organic hydrogen carriers but with lower hydrogen storage capacities than heterocycles (28).
Table 2. Dehydrogenation of N-alkylated amines using the Pd3Au1/CNT catalyst.
Reaction conditions: Pd3Au1/CNT (0.3 mol % of metals to substrate), N-alkylated amine (0.2 mmol), xylene (1 ml), N2 atmosphere, 140°C, and 24 hours. *Isolated yield. †NMR yields with diphenylmethanol as standard.
Dehydrogenation of amines and alcohols
The acceptorless dehydrogenation of primary amines to imines by heterogeneous catalysts provides an opportunity to produce imines and molecular hydrogen (29). The Pd3Au1/CNT serves as an efficient catalyst for the acceptorless dehydrogenation of primary amines under oxidant-free and base-independent conditions. Using the same reaction conditions used in the dehydrogenation of secondary amines, benzylamine was converted to the corresponding imine in 94% yield (table S3, entry 1). Nitrile formation was not detected (fig. S4E) (30), suggesting that the transamination step is much faster than the second dehydrogenation of the aldimine, which differs from the reactivity observed for homogeneous catalysts (31). Other substituted benzylamines with methyl and methoxy groups were efficiently and selectively dehydrogenated to the desired products (table S3, entries 2 to 4). 2-Picolylamine was transformed into the corresponding imine in 93% yield (table S3, entry 5), and an aliphatic amine afforded the dehydrogenated imine in 64% yield without the formation of a nitrile or N-alkylated amine (table S3, entry 6). In the presence of the Pd3Au1/CNT catalyst, imines are selectively obtained, which may be attributed to the combination of the two metals. Pd is very active for hydrogenations, whereas Au is much less active, and when alloyed, the Pd3Au1 facet has the ideal electronic properties to dehydrogenate the amine but not further hydrogenate the imine to alkylating amines.
The catalytic dehydrogenation of alcohols represents an attractive route to aldehydes and ketones that can be further transformed into imines, amides, esters, carboxylic acids, and various heterocycles (3, 32). Consequently, we evaluated the Pd3Au1/CNT catalyst in the acceptorless dehydrogenation of alcohols. Secondary benzylic alcohols were successfully dehydrogenated to their corresponding acetophenone derivatives in excellent yields (table S3, entries 7 and 8). Moreover, diphenylmethanol was converted to the corresponding product in 98% yield (table S3, entry 9), and 1-indanol and 1,2,3,4-tetrahydro-1-naphthol react much faster with high yields after 12 hours as the ring systems facilitate catalytic dehydrogenation (table S3, entries 10 and 11).
Catalytic hydrogenation of N-heterocycles, imines, and ketones
Catalytic hydrogenation of a range of N- and O-containing compounds was studied using the Pd3Au1/CNT catalyst under the optimized conditions. As shown in Table 3, quinoline derivatives with electron-donating (−OH and −NH2) and electron-withdrawing (−F) substituents were converted in high yields to the corresponding hydrogenated derivatives (Table 3, 1a to 1h). Methyl groups on different positions of the quinoline framework appeared to have little impact on the catalytic hydrogenation (Table 3, 1b and 1c). N-heteroarenes such as 9,10-dihydroacridine and 1,2,3,4-tetrahydrobenzo[h]quinoline were obtained in 87 and 88% yields, respectively (Table 3, 1i and 1k). In addition, quinoxaline and isoquinoline were hydrogenated in near-quantitative yields to the expected products (Table 3, 1l and 1m), whereas the hydrogenation of indole derivatives required a higher hydrogen pressure of 5 bars to achieve high yields, presumably due to their highly resonance-stabilized aromatic structure (Table 3, 1n and 1r). The activity displayed by the Pd3Au1/CNT catalyst is superior to other heterogeneous catalysts that tend to require harsher reaction conditions, which lowers selectivity (17).
Table 3. Hydrogenation of unsaturated N-heterocycles, imines, and ketones.
Reaction conditions: Pd3Au1/CNT (0.3 mol % of metals to substrate), substrate (0.2 mmol), xylene (0.5 ml), 80°C, H2 (1 bar), and 12 hours. Isolated yield. *H2 (5 bar). †Toluene.
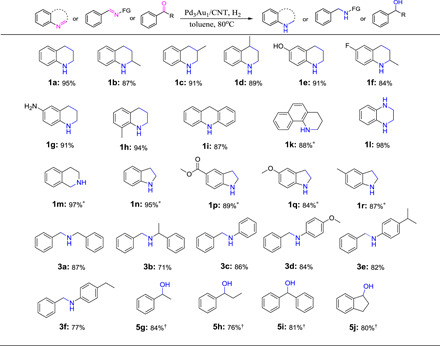
Prompted by the promising results obtained with N-heterocycles, we investigated the hydrogenation of imines and carbonyls. The imine hydrogenation reactions were performed in toluene at 80°C under 1 bar of H2 in the presence of the Pd3Au1/CNT catalyst. Dibenzylamine and N-benzyl-α-methylbenzylamine were obtained following the hydrogenation of the corresponding imines (Table 3, 3a and 3b, respectively). N-benzylideneaniline, 4-methoxy-N-benzylideneaniline, 4-isopropyl-N-benzylideneaniline, and 4-ethyl-N-benzylideneaniline were efficiently reduced within 12 hours to afford their corresponding N-benzylanilines in excellent yields (Table 3, 3c to 3f). However, debenzylation takes place, lowering the yield (71 to 86%) of the desired products. The Pd3Au1/CNT catalyst also catalyzes the hydrogenation of carbonyls to their corresponding alcohols, demonstrated by the synthesis of 1-phenylethanol, 1-phenyl-1-propanol, benzhydrol, and 1-hydroxyindan from their ketone precursors (Table 3, 5g to 5j).
DISCUSSION
Control experiments and mechanistic studies
A tentative mechanism for the dehydrogenation and hydrogenation reaction, i.e., interconversion between 1a and 2a, using the Pd3Au1/CNT catalyst is illustrated in Fig. 2A. For the dehydrogenation reaction, 1a is adsorbed on the Pd3Au1(111) surface (I in Fig. 2A), activating the N─H and C─H bonds and leading to the liberation of the first H2 and the formation of intermediate 1a″ (II in Fig. 2A). Next, the intermediate in 1a″ undergoes tautomerization to afford 2a″. A second dehydrogenation step takes place, involving the same N─H and C─H bonds, to complete the process with the release of the second H2 and the formation of 2a (III in Fig. 2A and fig. S4F).
Fig. 2. Mechanism studies.
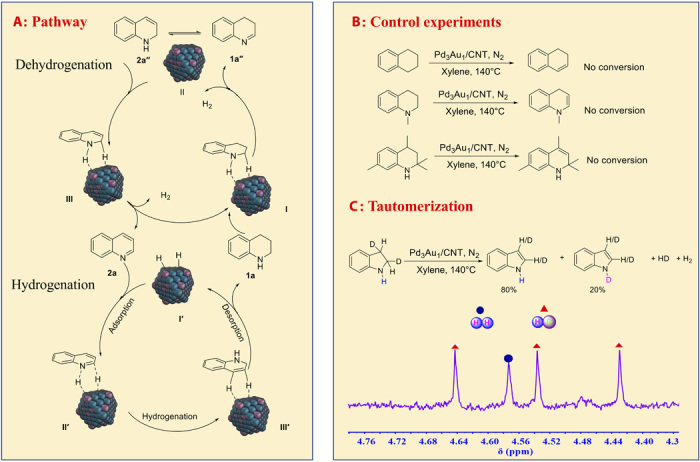
(A) Proposed mechanism of the dehydrogenation and hydrogenation cycles. (B) Control experiments for the dehydrogenation. (C) Tautomerization study under conditions: Pd3Au1/CNT (0.3 mol % of metals to 1a), 1a (0.2 mmol), xylene (1 ml), N2 atmosphere, and 140°C.
To substantiate the proposed mechanism, a number of control experiments were performed. No conversion was observed under dehydrogenation conditions using 1,2,3,4-tetrahydronaphthalene, 1-methyl-1,2,3,4-tetrahydroquinoline, or 2,2,4,7-tetramethyl-1,2,3,4-tetrahydroquinoline as the starting materials (Fig. 2B). These experiments all indicated that a N─H bond is essential for the reaction to proceed and indirectly supports the tautomerization step. Isomerization of C═N to C═C appears to be rapid, as 1a″ was not observed and the dehydrogenation of 1,2-dihydroquinoline (2a″) was very fast (fig. S4C). The dehydrogenation of indoline-d2, used as a model compound, was studied in situ by 1H NMR spectroscopy, confirming that the tautomerization step as indoline-d2 was quantitatively converted to indole with 20% of the N-D product resulting from the tautomerization process (Fig. 2C and scheme S2). Moreover, the formation of HD was also observed (33).
To further understand the differences in catalytic performance between the Pd and Pd3Au1 NP catalysts, DFT calculations were carried out. The (111) surface of both catalysts was chosen for all simulations as the structural data strongly suggest that it is the main facet orientation [Fig. 1 (B and G) for Pd3Au1 and fig. S2A for Pd]. The mechanism for the hydrogenation of quinoline involves sequential addition of H to the nitrogen and carbon atoms of quinoline. However, there are several possibilities for the first H addition, i.e., to the N atoms or to the C atoms of quinoline. We therefore calculated the energy barriers of the first H addition to N and the different C atoms and found that the formation of the N─H bond is the most favorable pathway with the lowest energy barrier. Next, for the addition of the second H to the partially hydrogenated quinoline, we calculated the energy barriers for H addition on the C atoms, allowing the most favorable pathway for the second H addition to be identified. Following this procedure, we identified the most favorable pathway for the hydrogenation. Other potential pathways include ring opening via C─C bond scission, which was not considered in this work because no such products were detected experimentally.
Figure 3A depicts the potential-free energy diagrams for the dehydrogenation of 1a at 140°C. The Pd3Au1(111) surface leads to lower-energy barriers for each dehydrogenation step compared to the Pd(111) facet. The energy barriers of the four dehydrogenation steps are 0.91, 0.57, 0.85, and 0.25 eV on Pd3Au1(111) compared to values of 1.15, 0.92, 1.07, and 0.36 eV on the Pd(111) surface. Here, the energetic span model (34) was applied to compare the differences in activity of the two catalysts, which provides an elegant way to evaluate the TOF for a reaction. In this model, only one transition state, i.e., the TOF-determining transition state (TDTS), and one intermediate, i.e., the TOF-determining intermediate (TDI), determine the TOF in catalytic cycles. The TDTS-TDI energy difference and the reaction driving force (∆Gr) define the energetic span (δE) of the reaction, and a lower δE indicates higher activity. On the basis of the Gibbs free energy diagram in Fig. 3A, the δE values for the dehydrogenation of 1a on the Pd3Au1(111) and Pd(111) surfaces are 2.04 and 2.69 eV, respectively. This indicates the superior activity of the Pd3Au1 catalyst in dehydrogenation reactions, which is in line with the experimental data.
Fig. 3. Potential-free energy diagrams.
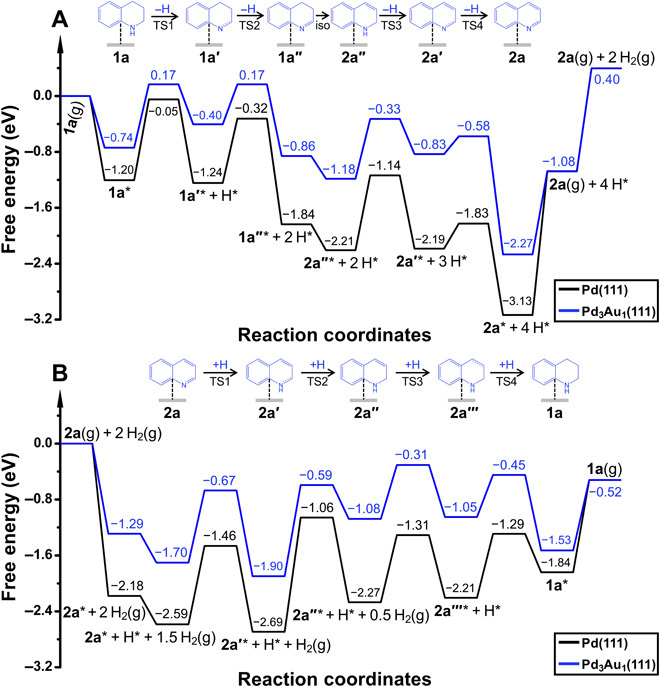
(A) Dehydrogenation of 1a at 140°C and (B) hydrogenation of 2a at 80°C on the Pd(111) and Pd3Au1(111) surfaces.
In addition, the reaction mechanisms for the selective hydrogenation of 2a to 1a on the Pd3Au1 (111) and Pd(111) surfaces were also calculated, and the associated free energy profile is shown in Fig. 3B. As expected, initial hydrogenation takes place at the C═N bond to afford intermediate 2a′, which is subsequently converted to 2a″. The energy barrier for the hydrogenation of the C═C in 2a″ is much lower than that of the C═N bond in 2a, and in a control experiment, it was shown that 2a″ is quantitatively hydrogenated to 1a in 3 hours (fig. S4D). Notably, the Pd3Au1(111) surface leads to lower-energy barriers than those observed on the Pd(111) surface for each of the elementary step, due to modulation of the electronic properties of the active Pd atoms by the Au atoms and the CNT surface. Furthermore, the δE values for the hydrogenation of 2a to 1a on Pd3Au1(111) and Pd(111) surfaces are 1.59 and 1.63 eV, respectively. This indicates the better activity of Pd3Au1 compared to the Pd catalyst in this hydrogenation reaction. To better understand the differences in selectivity of these two catalysts, we further calculated the reaction mechanism for the further hydrogenation of aromatic ring in 1a. As shown in the potential-free diagram (fig. S5), the δE values for hydrogenation of aromatic ring of 1a to 1a4H on the Pd3Au1(111) and Pd(111) surfaces are 1.58 and 1.45 eV, respectively, which shows that Pd is more active in the hydrogenation of aromatic ring than the Pd3Au1 catalyst. In this respect, the calculations indicate that the Pd3Au1 catalyst has higher activity and selectivity for the selective hydrogenation of 2a to 1a, whereas the Pd(111) catalyst tends to over-hydrogenate the product, lowering the selectivity to 1a. All these results are in good agreement with our experimental findings.
To establish the origin of the superior performance of the Pd3Au1 catalyst compared to the Pd1Au1, Pd1Au3, Pd, and Au systems, we considered the Sabatier principle (35), in which the interactions between the adsorbate and catalyst should be neither too strong nor too weak but ideally poised to activate the substrate and release the product. As shown in Fig. 3A, the adsorption of quinoline (2a*) corresponds to the TDI of this reaction, which also indicates its important role in determining the performance of the catalysts. We therefore chose the adsorption energy of 2a as the descriptor to compare the intrinsic differences among the catalysts. As shown in table S4, Pd(111) and Au(111) have the strongest (−3.03 eV) and weakest (−0.91 eV) adsorption energies for 2a, indicating their less promising performance in dehydrogenation reactions based on the Sabatier principle. However, the adsorption energy of 2a in the Pd3Au1(111) surface is −2.14 eV, stronger than that of Pd1Au1(111), Pd1Au3(111), and Au(111) but weaker than that of Pd(111). Combined with the catalytic performance, the Pd3Au1(111) displays optimal adsorption energies of 2a, leading to the superior performance of this catalyst. In this respect, the reason for the superior performance of Pd3Au1(111) to other Pd-Au–based catalysts could be attributed to its optimal adsorption strengths of adsorbates as intrinsically driven by the Sabatier principle.
In summary, we described the development of a highly efficient bimetallic Pd3Au1/CNT catalyst for the dehydrogenation and hydrogenation of N- and O-containing compounds. The mechanism of the interconversion between 1,2,3,4-tetrahydroquinoline and quinoline was probed as a model reaction using experimental methods and consolidated by DFT calculations, which show that the Pd3Au1(111) facet leads to lower-energy barriers than those observed on the Pd(111) surface for all of the elementary steps in both dehydrogenation and hydrogenation reactions. The high selectivity of the Pd3Au1/CNT catalyst allows it to be used in the synthesis of a wide range of N- and O-containing compounds offering a practical strategy for the synthesis of N-heterocycles, amines/imines, and alcohols/ketones substrates, tolerating a wide range of functional groups via dehydrogenation and hydrogenation reactions, in the absence of additives.
MATERIALS AND METHODS
Preparation of the Pd3Au1/CNT catalyst
First, multiwalled CNTs were pretreated under 10 M HNO3 for 12 hours at 80°C. After purification treatment, the Fe and Co content in the CNT was 0.04 and 0.01 wt %, respectively, and other metals such as Ni, Cr, and Pt are lower than the limit of detection of ICP-OES. CNT (200 mg) was added to an aqueous solution (10 ml) containing HAuCl4·3H2O (3.14 mg/0.008 mmol) and K2PdCl4 (7.77 mg/0.024 mmol, Pd:Au molar ratio of 3:1). Under vigorous stirring (~700 rpm), an aqueous solution (10 ml) containing TCD (28.2 mg/0.096 mmol, 3 eq relative to the metals) was added. The solution was stirred for 30 min at 30°C and NaBH4 (6.08 mg/0.16 mmol) was added, and the mixture was maintained at 30°C for 2 hours. The resulting black powder was filtered and washed with distilled water to reach a pH value of 7. After drying under vacuum for 12 hours, the catalyst was stored under N2 and used for the dehydrogenation and hydrogenation without further treatment.
Preparation of the Pd1Au1/CNT, Pd1Au3/CNT, Pd/CNT, and Au/CNT catalyst
These catalysts were prepared using a similar procedure to the one mentioned above, but the amount of the Pd and Au was changed, affording 2.0 wt % of metals.
General procedure for dehydrogenation reactions
Pd3Au1/CNT (5 mg), starting material (0.2 mmol), and xylene (1.5 ml) were placed in an oven-dried Schlenk tube and charged with nitrogen three times. The sealed Schlenk tube was heated to 140°C for 24 hours. After reaction, the flask was cooled to room temperature, and the reaction mixture was diluted using ethyl acetate and analyzed by gas chromatography–mass spectrometry (GC-MS) or NMR spectroscopy. To isolate the products, the volatiles were removed under vacuum, and the residues were further purified by column chromatography. All the dehydrogenated products were characterized by NMR spectroscopy in CDCl3 (see fig. S6 for 1H and 13C NMR of 1a and 2a).
General procedure of hydrogenation reactions
Pd3Au1/CNT (5 mg), starting material (0.2 mmol), and xylene or toluene (1.5 ml) were added to a 25-ml stainless steel pressure reactor. The reactor was sealed, flushed three times with H2, and placed under an appropriate H2 pressure (1 to 5 bar). The mixture was heated to 80°C and stirred for 12 hours. After reaction, the reactor was cooled to room temperature, and the solution was diluted with ethyl acetate. After analysis by GC and GC-MS, the mixture was filtered through a silica gel column, and the volatiles were removed under vacuum. The residues were further purified by column chromatography. All the dehydrogenated products were characterized by NMR spectroscopy in CDCl3.
Computational details
DFT computations were carried out using the plane wave–based method as implemented in the Vienna ab initio simulation package (36). Periodic slab models were used to simulate the Pd and Pd-Au alloy catalysts. The projector augmented wave (37) method was applied to describe the electron-ion interaction, and the electron exchange and correlation energy were treated within the generalized gradient approximation in the Perdew-Burke-Ernzerhof formalism. To obtain accurate energies with errors less than 1 meV per atom, a cutoff energy of 400 eV was chosen for all computations. A second-order Methfessel-Paxton electron smearing with σ = 0.2 eV was used. Structure optimization was performed when the force tolerance and energy difference became <0.02 eV/Å and 10−5 eV, respectively. A vacuum layer of 15 Å was set between the periodically repeating slabs to avoid strong interactions. The density-dependent dDsC method was used for the dispersion correction (38). To elucidate the intrinsic differences of the Pd and Pd-Au systems in hydro-treating reactions, we chose fcc Pd and Pd3Au1 for further reaction mechanism investigations and comparisons. Computations of the fcc-bulk Pd and Pd3Au1 crystal structures with a k-point mesh of 19 × 19 × 19 give lattice constants of 3.91 and 3.96 Å, respectively. The most stable close-packed (111) surfaces of these two catalysts as confirmed by the surface energy (J/m2) calculations [Pd(100), 1.53; Pd(110), 1.57; Pd(111), 1.34; Pd(210), 1.63; Pd(211), 1.61; Pd(310), 1.63; Pd(321), 1.59; Pd3Au1(100), 1.04; Pd3Au1(110), 1.33; Pd3Au1(111), 1.02; Pd3Au1(210), 1.48; Pd3Au1(211), 1.28; Pd3Au1(310), 1.47; Pd3Au1(321), 1.42] were applied for reaction mechanism simulations, where extensive model tests were performed to validate our choices of models. To have a balance between the computational accuracy and cost, a p(4 × 4)-3L supercell was eventually used to simulate the Pd(111) and Pd3Au1(111) surfaces (fig. S5), and the first layer was fully relaxed, while the bottom two layers were constrained. In addition, k-point mesh for sampling the Brillouin zone was also tested, and a (3 × 3 × 1) k-point mesh is accurate enough to acquire converged adsorption energies. Regarding the free energies in this work, the hydrogenation of quinoline in gas phase was used to benchmark the method for thermal corrections to gas phase species. We noticed that the results within ideal gas approximation were in very good agreement with experimental results (i.e., −1.29 eV[theory] versus −1.24 eV[experiment] for changes in reaction enthalpy at 298K). Therefore, the ideal gas approximation was applied to evaluate the free energies of gas phase species. The harmonic approximation as implemented in the Atomic Simulation Environment python library (39) was used to describe the thermodynamic corrections for adsorbates.
The adsorption energies of surface species were defined as Eads = E(adsorbate/surface) − E(surface) − E(adsorbate), where E(adsorbate/surface), E(surface), and E(adsorbate) are the total electronic energies of species adsorbed on the Pd(111) and Pd3Au1(111) surfaces, clean surfaces, and species in the gas phase, respectively. The climbing image nudged elastic band method was used to locate transition-state structures of each primary step (40). The energy barrier (Ea) is defined as Ea = ETS − EIS, and the reaction energy is defined as Er = EFS − EIS, where EIS, EFS, and ETS are the total energies of the initial state (IS), final state (FS), and transition state (TS) of each elementary step, respectively. All transition states are validated by vibration analyses with only one imaginary frequency.
Supplementary Material
Acknowledgments
We thank P. Mettraux and L. Thomas for analytical support. Funding: We thank the EPFL, Swiss National Science Foundation, and the Swiss Competence Center for Energy Research (SCCER) on Heat and Electricity Storage for financial support. Author contributions: X.C. and P.J.D. conceived and designed the experiments. X.C. performed the experiments. Z.H. contributed to TEM analysis. T.W. finished the DFT calculation. X.C., Z.F., A.P.M., T.W., and P.J.D. prepared the manuscript. All authors contributed to discussions. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/27/eabb3831/DC1
REFERENCES AND NOTES
- 1.K. Weissermel, H.-J. Arpel, in Industrial Organic Chemistry (Wiley-VCH, 2003), pp. 59–89. [Google Scholar]
- 2.Eberle U., Felderhoff M., Schüth F., Chemical and physical solutions for hydrogen storage. Angew. Chem. Int. Ed. 48, 6608–6630 (2009). [DOI] [PubMed] [Google Scholar]
- 3.Crabtree R. H., Homogeneous transition metal catalysis of acceptorless dehydrogenative alcohol oxidation: Applications in hydrogen storage and to heterocycle synthesis. Chem. Rev. 117, 9228–9246 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Zhang Y., Cui X., Shi F., Deng Y., Nano-gold catalysis in fine chemical synthesis. Chem. Rev. 112, 2467–2505 (2012). [DOI] [PubMed] [Google Scholar]
- 5.Sartbaeva A., Kuznetsov V. L., Wells S. A., Edwards P. P., Hydrogen nexus in a sustainable energy future. Energ. Environ. Sci. 1, 79–85 (2008). [Google Scholar]
- 6.Jessop P., Reactions with a reverse gear. Nat. Chem. 1, 350–351 (2009). [DOI] [PubMed] [Google Scholar]
- 7.Chakraborty S., Brennessel W. W., Jones W. D., A molecular iron catalyst for the acceptorless dehydrogenation and hydrogenation of N-heterocycles. J. Am. Chem. Soc. 136, 8564–8567 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Talwar D., Gonzalez-de-Castro A., Li H. Y., Xiao J. L., Regioselective acceptorless dehydrogenative coupling of N-heterocycles toward functionalized quinolines, phenanthrolines, and indoles. Angew. Chem. Int. Ed. 54, 5223–5227 (2015). [DOI] [PubMed] [Google Scholar]
- 9.Yao W. B., Zhang Y., Jia X., Huang Z., Selective catalytic transfer dehydrogenation of alkanes and heterocycles by an iridium pincer complex. Angew. Chem. Int. Ed. 53, 1390–1394 (2014). [DOI] [PubMed] [Google Scholar]
- 10.Fujita K., Tanaka Y., Kobayashi M., Yamaguchi R., Homogeneous perdehydrogenation and perhydrogenation of fused bicyclic N-heterocycles catalyzed by iridium complexes bearing a functional bipyridonate ligand. J. Am. Chem. Soc. 136, 4829–4832 (2014). [DOI] [PubMed] [Google Scholar]
- 11.Yamaguchi R., Ikeda C., Takahashi Y., Fujita K.-i., Homogeneous catalytic system for reversible dehydrogenation-hydrogenation reactions of nitrogen heterocycles with reversible interconversion of catalytic species. J. Am. Chem. Soc. 131, 8410–8412 (2009). [DOI] [PubMed] [Google Scholar]
- 12.Moores A., Poyatos M., Luo Y., Crabtree R. H., Catalysed low temperature H2 release from nitrogen heterocycles. New J. Chem. 30, 1675–1678 (2006). [Google Scholar]
- 13.Vivancos A., Beller M., Albrecht M., NHC-based iridium catalysts for hydrogenation and dehydrogenation of N-heteroarenes in water under mild conditions. ACS Catal. 8, 17–21 (2018). [Google Scholar]
- 14.Muthaiah S., Hong S. H., Acceptorless and base-free dehydrogenation of alcohols and amines using ruthenium-hydride complexes. Adv. Synth. Catal. 354, 3045–3053 (2012). [Google Scholar]
- 15.He K.-H., Tan F.-F., Zhou C.-Z., Zhou G.-J., Yang X.-L., Li Y., Acceptorless dehydrogenation of N-heterocycles by merging visible-light photoredox catalysis and cobalt catalysis. Angew. Chem. Int. Ed. 56, 3080–3084 (2017). [DOI] [PubMed] [Google Scholar]
- 16.Bonitatibus P. J. Jr., Chakraborty S., Doherty M. D., Siclovan O., Jones W. D., Soloveichik G. L., Reversible catalytic dehydrogenation of alcohols for energy storage. Proc. Natl. Acad. Sci. U.S.A. 112, 1687–1692 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Deraedt C., Ye R., Ralston W. T., Toste F. D., Somorjai G. K., Dendrimer-stabilized metal nanoparticles as efficient catalysts for reversible dehydrogenation/hydrogenation of N-heterocycles. J. Am. Chem. Soc. 139, 18084–18092 (2017). [DOI] [PubMed] [Google Scholar]
- 18.Forberg D., Schwob T., Zaheer M., Friedrich M., Miyajima N., Kempe R., Single-catalyst high-weight% hydrogen storage in an N-heterocycle synthesized from lignin hydrogenolysis products and ammonia. Nat. Commun. 7, 13201 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Han Y. H., Wang Z. Y., Xu R. R., Zhang W., Chen W. X., Zheng L. R., Zhang J., Luo J., Wu K. L., Zhu Y. Q., Chen C., Peng Q., Liu Q., Hu P., Wang D. S., Li Y. D., Ordered porous nitrogen-doped carbon matrix with atomically dispersed cobalt sites as an efficient catalyst for dehydrogenation and transfer hydrogenation of N-heterocycles. Angew. Chem. Int. Ed. 57, 11262–11266 (2018). [DOI] [PubMed] [Google Scholar]
- 20.Agarwal N., Freakley S. J., McVicker R. U., Althahban S. M., Dimitratos N., He Q., Morgan D. J., Jenkins R. L., Willock D. J., Taylor S. H., Kiely C. J., Hutchings G. J., Aqueous Au-Pd colloids catalyze selective CH4 oxidation to CH3OH with O2 under mild conditions. Science 358, 223–226 (2017). [DOI] [PubMed] [Google Scholar]
- 21.Kesavan L., Tiruvalam R., Ab Rahim M. H., bin Saiman M. I., Enache D. I., Jenkins R. L., Dimitratos N., Lopez-Sanchez J. A., Taylor S. H., Knight D. W., Kiely C. J., Hutchings G. J., Solvent-free oxidation of primary carbon-hydrogen bonds in toluene using Au-Pd alloy nanoparticles. Science 331, 195–199 (2011). [DOI] [PubMed] [Google Scholar]
- 22.Xie S. H., Tsunoyama H., Kurashige W., Negishi Y., Tsukuda T., Enhancement in aerobic alcohol oxidation catalysis of Au25 clusters by single Pd atom doping. ACS Catal. 2, 1519–1523 (2012). [Google Scholar]
- 23.El Kolli N., Delannoy L., Louis C., Bimetallic Au-Pd catalysts for selective hydrogenation of butadiene: Influence of the preparation method on catalytic properties. J. Catal. 297, 79–92 (2013). [Google Scholar]
- 24.Lamblin M., Nassar-Hardy L., Hierso J. C., Fouquet E., Felpin F. X., Recyclable heterogeneous palladium catalysts in pure water: Sustainable developments in Suzuki, Heck, Sonogashira and Tsuji-Trost reactions. Adv. Synth. Catal. 352, 33–79 (2010). [Google Scholar]
- 25.Sonobe T., Oisaki K., Kanai M., Catalytic aerobic production of imines en route to mild, green, and concise derivatizations of amines. Chem. Sci. 3, 3249–3255 (2012). [Google Scholar]
- 26.Cui X. J., Li Y. H., Bachmann S., Scalone M., Surkus A. E., Junge K., Topf C., Beller M., Synthesis and characterization of iron–nitrogen-doped graphene/core–shell catalysts: Efficient oxidative dehydrogenation of N-heterocycles. J. Am. Chem. Soc. 137, 10652–10658 (2015). [DOI] [PubMed] [Google Scholar]
- 27.Chen S. M., Wan Q. Q., Badu-Tawiah A. K., Picomole-scale real-time photoreaction screening: Discovery of the visible-light-promoted dehydrogenation of tetrahydroquinolines under ambient conditions. Angew. Chem. Int. Ed. 55, 9345–9349 (2016). [DOI] [PubMed] [Google Scholar]
- 28.Ainembabazi D., An N., Manayil J. C., Wilson K., Lee A. F., Voutchkova-Kostal A. M., Acceptorless amine dehydrogenation and transamination using Pd-doped hydrotalcites. ACS Catal. 9, 1055–1065 (2019). [Google Scholar]
- 29.Zhai Z.-Y., Guo X.-N., Jin G.-Q., Guo X.-Y., Visible light-induced selective photocatalytic aerobic oxidation of amines into imines on Cu/graphene. Cat. Sci. Technol. 5, 4202–4207 (2015). [Google Scholar]
- 30.Tseng K.-N. T., Rizzi A. M., Szymczak N. K., Oxidant-free conversion of primary amines to nitriles. J. Am. Chem. Soc. 135, 16352–16355 (2013). [DOI] [PubMed] [Google Scholar]
- 31.Hale L. V. A., Malakar T., Tseng K. N. T., Zimmerman P. M., Paul A., Szymczak N. K., The mechanism of acceptorless amine double dehydrogenation by N,N,N-amide ruthenium(II) hydrides: A combined experimental and computational study. ACS Catal. 6, 4799–4813 (2016). [Google Scholar]
- 32.Mitsudome T., Mikami Y., Funai H., Mizugaki T., Jitsukawa K., Kaneda K., Oxidant-free alcohol dehydrogenation using a reusable hydrotalcite-supported silver nanoparticle catalyst. Angew. Chem. Int. Ed. 47, 138–141 (2008). [DOI] [PubMed] [Google Scholar]
- 33.Quinn W. E., Baker J. M., Latourrette J. T., Ramsey N. F., Radio-frequency spectra of hydrogen deuteride in strong magnetic fields. Phys. Rev. 112, 1929–1940 (1958). [Google Scholar]
- 34.Kozuch S., Shaik S., How to conceptualize catalytic cycles? The energetic span model. Acc. Chem. Res. 44, 101–110 (2011). [DOI] [PubMed] [Google Scholar]
- 35.Medford A. J., Vojvodic A., Hummelshøj J. S., Voss J., Abild-Pedersen F., Studt F., Bligaard T., Nilsson A., Nørskov J. K., From the Sabatier principle to a predictive theory of transition-metal heterogeneous catalysis. J. Catal. 328, 36–42 (2015). [Google Scholar]
- 36.Kresse G., Furthmüller J., Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996). [DOI] [PubMed] [Google Scholar]
- 37.Kresse G., Joubert D., From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B. 59, 1758–1775 (1999). [Google Scholar]
- 38.Steinmann S. N., Corminboeuf C., Comprehensive bench marking of a density-dependent dispersion correction. J. Chem. Theory Comput. 7, 3567–3577 (2011). [DOI] [PubMed] [Google Scholar]
- 39.Larsen A. H., Mortensen J. J., Blomqvist J., Castelli I. E., Christensen R., Dułak M., Friis J., Groves M. N., Hammer B., Hargus C., Hermes E. D., Jennings P. C., Jensen P. B., Kermode J., Kitchin J. R., Kolsbjerg E. L., Kubal J., Kaasbjerg K., Lysgaard S., Maronsson J. B., Maxson T., Olsen T., Pastewka L., Peterson A., Rostgaard C., Schiøtz J., Schütt O., Strange M., Thygesen K. S., Vegge T., Vilhelmsen L., Walter M., Zeng Z. H., Jacobsen K. W., The atomic simulation environment—A Python library for working with atoms. J. Phys. Condens. Matter 29, 273002 (2017). [DOI] [PubMed] [Google Scholar]
- 40.Henkelman G., Uberuaga B. P., Jonsson H., A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 113, 9901–9904 (2000). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/27/eabb3831/DC1