

ABSTRACT
The murine MC-38 colorectal cancer model is a commonly used model for cancer with high mutational burden, which is sensitive for immune checkpoint immunotherapy. We set out to analyze endogenous CD8+ T cell responses to MC-38 neo-antigens in tumor-bearing mice and after anti-PD-L1 checkpoint therapy. Through combination of whole-exome sequencing analysis with mass spectrometry of MHC class I eluted peptides we could identify eight candidate epitopes. Of these, a neo-epitope encoded by a point-mutation in the sequence of the ribosomal protein L18 (Rpl18) strongly dominated the CD8+ T cell response to our MC-38 cell-line in comparison to a previously described neo-epitope in the Adpgk protein. Therapeutic vaccination with synthetic peptides induced CD8+ T cell responses against the mutated Rpl18 epitope, which controlled tumor growth in vivo. This immunologically dominant response to mutated Rpl18 is of great importance in the development and optimization of immunotherapeutic strategies with the MC-38 tumor model.
KEYWORDS: Neoantigen, immunotherapy, PD-L1, checkpoint, vaccination, whole exome sequencing
Introduction
In recent years, immunotherapy has received major focus as a novel therapeutic platform for cancer treatment. The field mainly focuses on the (re-)activation of the adaptive immune system that becomes suppressed through cancer-induced immunological changes at systemic- and micro-environmental level.1 Cytotoxic CD8+ T cells are the major immune cells, which have the potential to directly kill cancer cells. Upon alleviation of suppression by anti-PD-1/PD-L1 and/or anti-CTLA4 antibodies, so-called immune checkpoint inhibitors,2,3 these CD8+ T lymphocytes become effector cells that specifically recognize tumor cells and thereby improve tumor control.
Tumor-specific CD8+ T cells have been shown to primarily recognize somatic (bystander) mutations present in the malignant cells. This is supported by the positive-correlation of mutational load of tumors, which increases the chance of expressed and presented epitopes, and thereby therapeutic effectiveness upon immune checkpoint inhibitor treatment.4–8 Moreover, the central role of neo-antigen recognition by the adaptive immune system is reinforced with observations in changing mutational landscapes of tumors through checkpoint inhibition.9–11 Thus, it is now well accepted that epitopes containing mutations are key to the effectivity of immunotherapy.
Improving cytotoxic T cell activity against mutated epitopes is a logical next step to advance immunotherapy. The field is rapidly making steps forward to optimize specific personalized immunotherapy, for instance by therapeutic vaccination.4,12,13 Several issues have to be solved; like the time-consuming determination of relevant epitopes, formulation, and adjuvant inclusions necessary to trigger efficient immune responses in suppressed environments.14 Therefore, good models are required for the future development of personalized treatment regimes.
In this study, we have explored the immunogenic responses of the murine adenocarcinoma MC-38 cancer model targeting neo-antigens. Herewith, we report dominant CD8+ T cell immune responses toward a novel, to our knowledge undescribed, neo-epitope derived from the ribosomal protein L18 (Rpl18). We could show this neo-epitope response in several settings in which mice were exposed to MC-38 tumor cells and treated by immunotherapy. Using synthetic peptides covering the neo-epitope and flanking sequences, we managed to induce tumor control in mice through therapeutic vaccination. Compared with previously described mutation-specific responses against MC-38, our data suggests a major role in tumor control for this neo-epitope in the Rpl18 protein.
Method and materials
Sequencing and mutation analysis
DNA was isolated from the MC-38 cell line and from the tail of inbred C57BL/6JIco mice by use of Nucleospin Tissue kit. Additionally, RNA was isolated from both tissue sources by use of Trizol. SureSelect Mouse All Exon Kit was used for capture of the coding sequences. TruSeq Stranded Total RNA sample preparation kit was used to generate RNAseq libraries. Paired-end, 100bp reads were generated on a HiSeq2000 (Illumina) sequencer, producing approximately 12 Gb of data per sample. Mapping of the sequences was done against the mm10 reference genome with BWA.15 Variant calling was performed using the paired tumor-normal mode of Mutect2,16 where the tail DNA of a C57BL/6J mouse was used as normal. Variants were functionally annotated using the ensembl Variant Effect Predictor.17 Variants were selected as potential neo-antigens if non-synonymous mutations occurred in coding regions and were supported by at least one read at RNA level. To exclude sequencing artifacts, all selected variants were visually inspected using IGV.18,19 MHC binding affinity prediction was performed with NetMHC4.0.20,21 Homology testing was performed with ProteinBlast to eliminate mutant sequences similar to autologous germline sequences in the genome.
Tumor cell-line, elution, and mass-spectrometry
MC-38 (kindly provided by Mario P. Colombo) was cultured in IMDM medium (Lonza) supplemented with 8% Fetal Calf Serum (Greiner), 100 IU/mL penicillin/streptomycin (Gibco), 2 mM L-glutamine (Gibco) and 25 µM 2-mercaptoethanol (culture medium). Cell lines were mycoplasma and MAP-tested before the start of experiments. Approximately 8 × 108 MC-38 cells were lysed in 10 mL lysis buffer (50 mM Tris-Cl pH 8.0, 150 mM NaCl, 5 mM EDTA, 0.5% Zwittergent 3–12 (N-dodecyl-N,N-dimethyl-3-ammonio-1-propanesulfonate) and protease inhibitor (Complete, Roche Applied Science)) for 2 h at 0°C (ref A). Lysates were successively centrifuged for 10 min at 2500 × g and for 45 min at 31,000 x g to remove nuclei and other insoluble material, respectively. Next, lysates were passed through an 800 µL CL-4B Sepharose column to preclear the lysate. The cleared lysate was passed through a 800 µL column containing 2 mg anti-Kb (Y3) IgG coupled to protein A Sepharose22 and subsequently through a 800 µl column containing 2 mg anti-Db (28-14-8) IgG coupled to protein A Sepharose. The antibody columns were washed with 1 mL of lysis buffer, 3 mL of low salt buffer (20 mM Tris-Cl pH 8.0, 120 mM NaCl), 1 mL of high salt buffer (20 mM Tris-Cl pH 8.0, 1 M NaCl), and finally with 3 mL of low salt buffer. Peptides were eluted with 5 mL of 10% acetic acid, diluted with 10 mL of 0.1% TFA and purified by SPE (Oasis HLB, Waters) using 20% and 30% acetonitrile in 0.1% TFA to elute the peptides. Peptides were lyophilized, dissolved in 95/3/0.1 v/v/v water/acetonitrile/formic acid and subsequently analyzed by on-line C18 nanoHPLC MS/MS with a system consisting of an Easy nLC 1200 gradient HPLC system (Thermo, Bremen, Germany), and a LUMOS mass spectrometer (Thermo). Fractions were injected onto a homemade precolumn (100 μm × 15 mm; Reprosil-Pur C18-AQ 3 μm, Dr. Maisch, Ammerbuch, Germany) and eluted via a homemade analytical nano-HPLC column (30 cm × 75 μm; Reprosil-Pur C18-AQ 3 um). The gradient was run from 2% to 36% solvent B (20/80/0.1 water/acetonitrile/formic acid (FA) v/v) in 120 min. The nano-HPLC column was drawn to a tip of ∼5 μm and acted as the electrospray needle of the MS source. The LUMOS mass spectrometer was operated in data-dependent MS/MS mode for a cycle time of 3 seconds, with a HCD collision energy at 32 V and recording of the MS2 spectrum in the orbitrap. In the master scan (MS1) the resolution was 60,000, the scan range 300–1400, at an AGC target of 400,000 at maximum fill time of 50 ms. Dynamic exclusion after n = 1 with exclusion duration of 20 s. Charge states 1–4 were included. For MS2 precursors were isolated with the quadrupole with an isolation width of 1.2 Da. First mass was set to 110 Da. The MS2 scan resolution was 30,000 with an AGC target of 50,000 at maximum fill time of 100 ms. In a post-analysis process, raw data were first converted to peak lists using Proteome Discoverer version 2.1 (Thermo Electron), and then submitted to the Uniprot Mus Musculus database (52015 entries), using Mascot v. 2.2.04 (www.matrixscience.com) for protein identification. Mascot searches were with 10 ppm and 0.02 Da deviation for precursor and fragment mass, respectively, and no enzyme was specified. Methionine oxidation was set as a variable modification. Peptides with MASCOT scores <35 were generally discarded. For PRM analyses, the samples were lyophilized and resuspended in buffer A. The ligandome of 100 E6 cell equivalents was injected together with a mix of 20 fmol of each heavy labeled peptide. The Orbitrap Fusion LUMOS mass spectrometer was operated in PRM-mode. Peptides ASMTNMELM and KILTFDRL were monitored. The normalized collision energy was set to 32%. Selected peptides and the transitions can be found in Supplementary table 2. The isolation width of Q1 was 1.2 Da. MS2 resolution was 35,000 at an AGC target value of 1 million at a maximum fill time of 100 ms. LCMS conditions and gradients were the same as for the data-dependent analyses. PRM data analysis and data integration were performed in Skyline 3.6.0.10493. Peptide abundances were calculated by comparing the peak area of the eluted (light) and the peak area of the spiked-in heavy peptides.
Mice and vaccination
C57BL/6J mice were purchased from Envigo, Harlan laboratories (The Netherlands) at an age of 8–10 weeks and housed under specified pathogen-free conditions in animal facilities of the Leiden University Medical Center. All animal experiments were approved by and implemented according to ethical guidelines of the Dutch Central Animal Experiment Committee and the Federation of European Laboratory Animal Science Associations (FELASA). Mice vaccinated with irradiated (15000 rads) MC-38 where twice injected S.C. in the right flank with 5 × 106 cells in 200 µL of PBS with a two-week interval. Animals involved in tumor outgrowth experiments were S.C. injected in the right flank with 3 × 105 live MC-38 cells in 200 µL PBS. Tumor sizes where measured 2–3 times a week and 1500 mm3 was maintained as humane endpoint. The method used for the mice treated with anti-PD-L1 antibodies can be found in the publication of Sow, H.S. et al.23 Vaccinated mice were injected once with 20 nmol Adpgk or Rpl18 synthetic peptides intradermally in 50 µL PBS adjuvanted with 20 µg of CpG (ODN 1826 – TLR9 ligand InvivoGen tlrl-1826-1) or 10 nmol Rpl18-conjugated with equimolar Upam-ligand (peptides and Upam where conjugated as described previously in).24–26
In vitro (re-)stimulation
T cell cultures were started with splenocytes from several experiments. Therefore, spleens were mashed on single cell strainers with the blunt end of a 5 mL syringe and washed with culture medium. Cellular precipitates after centrifugation were treated with 5 mL of lysis buffer for 3 min. at room temperature to remove erythrocytes and subsequently washed with culture medium. For the initial restimulation and expansion of tumor-antigen specific T cells, 5 × 106 splenocytes per well were plated in a 24-wells and co-cultured with 1 × 105 irradiated (15000 rads) MC-38 cells. After 7 days, debris was separated from living cells with ficol and living cells were plated at 1 × 105 cells/well in a 24 wells plate in culture medium reconstituted with 5 Cetus Units (ca. 35 IU) of recombinant human IL-2 per mL. Consecutive re-stimulations and expansions were initiated after two weeks of rest with 2 × 106 fresh, irradiated (3000 rads) splenocytes from naïve mice and 1 × 105 irradiated MC-38 cells. Read-out of T cell specificities was performed through o/n pre-culture of 5 × 104 DC’s of the growth-factor dependent cross-presenting dendritic cell-line D127 with 5 µM synthetic peptide, whereupon T cells and 2 µg/mL brefeldin A (Sigma-Aldrich) were added. Synthetic “long” peptides were designed for optimal synthesis and dissolvability with the mutation located at the central region. Therefore, N- and C-terminal wild-type sequences were added resulting in peptides of approximately 20 amino-acids long. The following peptides were designed and synthesized: SFMAPIDHTTMSDDARTE (Aatf), HLELASMTNMELMSSIVHQ (Adpgk), GSNGDPSSPYSLHYLSPTGVNE (Cpne1), FRRPSTYVIPRLERILLAK (Gtf2i), RVLELFRAAQLANDVVLQIME (Reps1), KAGGKILTFDRLALESPK (Rpl18), ARCAQFPRALDKVHYYIKLKD (Tdg), QYFDAVKNAQHLEVESIPLPD (Wbp11). Direct tumor-recognition assays were done through co-culture of T cells with 1 × 105 live tumor cells in 48-wells flat-bottom in culture medium supplemented with 2 µg/mL brefeldin A.
Flow cytometrics
For in vitro peptide stimulation assays, analysis of intracellular cytokine production (ICS) was started through extracellular staining of CD3ε (EF450, clone 17A2, eBioscience), CD4 (BV605, clone RM4-5, BioLegend), and CD8α (APC-R700, clone 53–6.7, BDBiosciences) in FACS buffer (0,5% Bovine Serum Albumin, 0,02% Sodium Azide). Afterward, cells were washed and o/n treated with 0.5% paraformaldehyde to subsequently be stained with TNFα (FITC, clone MP6-XT22, eBioscience) and IFNγ (APC, clone XMG1.2, Invitrogen) through the use of perm/wash (BioLegend). For the analysis of tumor-infiltrated lymphocytes, whole tumors were resected from the flank of mice and mashed in 3 mL of serum-free medium (IMDM medium (Lonza) supplemented with 100 IU/mL penicillin/streptomycin (Gibco), 2 mM L-glutamine (Gibco) and 25 µM 2-mercaptoethanol). 500 µL tumor-mash was incubated with 77 µL (stock conc. 2,5 mg/mL) of Liberase TL (Roche) for 30 min. at 37°C. A single cell suspension was made through the use of a cell strainer and washed with culture medium. Tumor material and respective splenocytes were similarly stimulated and stained as described in previous ICSs, although extracellular and intracellular staining markers now included CD45 (BV605, clone 30-F11, BioLegend), CD3ε (EF450, clone 17A2, eBioscience), CD4 (PE, clone RM4-5, BioLegend), CD8α (APC-R700, clone 53–6.7, BDBiosciences), TNFα (FITC, clone MP6-XT22, eBioscience), and IFNγ (PE-Cy7, clone XMG1.2, BDBiosciences). Tumor-recognition samples were stained at room temperature for 15 min. with PE and APC tetramers for Adpgk and Rpl18, respectively (in-house production with predicted epitope). Extracellular markers included CD3ε (EF450, clone 17A2, eBioscience), CD4 (BV605, clone RM4-5, BioLegend), and CD8α (APC-R700, clone 53–6.7, BDBiosciences) were added to tetramer stained samples for 5 min. and kept on ice, while intracellular markers TNFα (FITC, clone MP6-XT22, eBioscience) and IFNγ (PE-Cy7, clone XMG1.2, BDBiosciences) were stained according to previous staining methods. Conjugate vaccinated, tumor-bearing mice were analyzed on blood tetramer staining. Blood from the tail was kept on ice in heparin tubes to prevent clotting, prior to the lysis of erythrocytes. Tetramer staining was done according to the previous tetramer staining protocol, except for a smaller extracellular staining panel whereby only CD8α (FITC, clone 53–6.7, BioLegend) was used, and without intracellular staining. All samples were analyzed on a BD Bioscience LSR-II with FACSDiva for data-capture software. FlowJo vX (FlowJo, LLC) was used for sample analysis and processing.
Statistical analysis
Graphpad prism v8 was used for statistical analysis. Respective statistical tests are mentioned in the legends of the figures. Significance was determined when p-values reached 0.05 or less. In general, group comparisons were done with (multiple) student’s t tests, and survival with Gehan-Breslow-Wilcoxon or log-rank tests.
Results
Identification of potential epitopes
Recent studies have identified several neo-peptide sequences of the MC-38 adenocarcinoma colorectal cell-line. However, in explorative experiments with this model (not shown) we could only detect very low frequencies of endogenous T cell responses to these neo-epitopes. To identify other potential neo-antigens underlying the CD8+ T cell response, we independently analyzed MHC class I presented neo-epitopes in our MC-38 cell line by combination of whole-exome/RNA sequencing and MHC class I peptide elution/mass-spectrometry (Figure 1).
Figure 1.

Strategy to identify MC-38 colorectal tumor specific neo-peptides. MC-38 DNA was isolated and its sequenced exome was compared to DNA from C57BL/6 wild-type mice for genetic variants. Corresponding amino acid sequences were used to predict potential binders to MHC class I Kb and Db by NetMHC4.0 prediction algorithm. A combination of immunoprecipitation of MHC class I from MC-38 lysate and subsequent mass-spectrometry of eluted peptides was performed to establish MHC class I presentation.
DNA exome sequencing data derived from MC-38 cells was compared to the wild-type DNA sequence obtained from the tail of C57BL/6J mice. We have identified a total of 16,143 genomic mutations, of which 3,808 resulted in amino acid coding changes. By subsequent RNA sequencing we could define a total of 1,661 expressed variants. MHC class I binding prediction algorithms resulted in a ranking of 1,501 mutated peptide-epitopes with potential binding affinity for either Kb or Db molecules (suppl. table 1). This set of potential neo-peptides was analyzed for MHC class I presentation through specific pulldown of both H-2b alleles, followed by mass spectrometry of eluted peptides. Sequence length distribution analysis of all peptides derived from separate H-2Kb and −2Db isolates showed the expected binding preferences of 8-, 9- or 10-mer peptides and the well-established anchor-binding residues of these MHC class I molecules (suppl. Fig. 1). Identity of the eluted peptides were confirmed by spectral similarity of synthetically produced analogues of these sequences (suppl. Fig 2).
Intersection of sequencing and elution databases resulted in the identification of eight possible neo-epitopes (Table 1). These identified neo-antigens ranged from very low to very high predicted affinities toward MHC class I and, based on sequencing data, were mostly occurring heterozygously. Relative expression frequencies of variant RNA followed the frequencies seen in DNA. Presentation of the wild-type sequences by MHC class I could not be determined through elution in most cases. In conclusion, we confirmed the presence of most of the mutations reported previously28 but we also detected four novel candidate neo-epitopes.
Table 1.
Selected candidate neo-epitopes. All eight neo-peptides shown were selected by exome and RNA sequencing and confirmed with synthetic peptide mass-spectrometry for MHC class I presentation. IC50 of presented peptide affinity was predicted with NetMHC (ANN) for respective MHC alleles. Candidate neo-epitopes indicated with (*) were previously described in Yadav M. et al. (2014). Context of these candidate neo-epitopes was included in the form of variant frequencies in DNA and RNA, whole gene expression levels in MC-38 (in transcripts per million), and the observed presence of wild-type (WT) peptides in the elution.
| # | Name | Selected peptide | WT AA | IC50 (nM) | MHC allele | Variant occurance | Variant DNA freq. | Variant RNA freq. | Expression in MC-38 | Eluted WT |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Aatf* | MAPIDHTTM | A | 90,2 | Db | Heterozygous | 0.49 | 0.43 | 5,321 | Y |
| 2 | Adpgk* | ASMTNMELM | R | 4,3 | Db | Heterozygous | 0.33 | 0.40 | 1,391 | Y |
| 3 | Cpne* | SSPYSLHYL | D | 182,3 | Db | Heterozygous | 0.39 | 0.40 | 6,399 | N |
| 4 | Gtf2i | STYVIPRL | G | 16,5 | Kb | Heterozygous | 0.39 | 0.53 | 1,530 | N |
| 5 | Reps1 | AQLANDVVL | P | 16,2 | Db | Heterozygous | 0.24 | 0.18 | 2,486 | Y |
| 6 | Rpl18 | KILTFDRL | Q | 33,2 | Kb | Heterozygous | 0.35 | 0.31 | 15,546 | Y |
| 7 | Tdg | RALDKVHYYI | Q | 4656,2 | Db | Heterozygous | 0.45 | 0.48 | 1,869 | N |
| 8 | Wbp11 | DAVKNAQHL | V | 892,3 | Db | Heterozygous | 0.36 | 0.36 | 4,875 | Y |
Endogenous induction of specific t cells upon tumor-vaccination
To test whether our neo-epitope candidates were relevant and immunogenic, we vaccinated “wild-type” C57BL/6J mice with irradiated MC-38 tumor cells and analyzed tumor-specific CD8+ T cell responses (Figure 2(a)). For readout, we synthesized long peptides of ca. 20 amino-acid residues of all candidate neo-epitopes with the mutation located in the central region of the amino acid sequence. Splenocytes from MC-38-vaccinated mice were cultured with peptide-pre-incubated DC’s and analyzed by intracellular cytokine staining of CD8+ T cells. Three out of eight candidate neo-peptides induced TNFα and IFNγ poly-functional activation of CD8+ T cells (Figure 2(b)).
Figure 2.

Irradiated MC-38 immunization induces CD8+ T cells specific for candidate neo-epitopes. (a) Mice were prime-boost vaccinated with subcutaneous injection of 5 × 106 irradiated MC-38 tumor cells. (b) Cytokine production of CD8α+ T lymphocytes through stimulation with synthetic “long” peptide-loaded dendritic cells ex vivo and (c) after restimulation with irradiated MC-38 cells. (b) and (c) are from two independent experiments, n = 3 for each. Shown are representative cytokine (IFNγ and TNFα) staining plots from CD8α+ populations (left) and plotted percentages of double-positive IFNγ and TNFα cytokine producing populations of individual mice (right).
Candidate epitope six, derived from a point-mutated sequence of ribosomal protein L18 (Rpl18), elicited robust activation of up to 5 percent of CD8+ T cells ex vivo. The second largest responses were observed by stimulation with the previously described neo-epitope from ADP-dependent Glucokinase (Adpgk). A third mutation in Copine-1 (Cpne-1) also elicited an observable activation of CD8+ T cells in one mouse. Upon restimulation of splenocytes with irradiated MC-38 cells in vitro strongly boosted the expansion of Rpl18-specific CD8+ T cells up to 70 percent (Figure 2(c)). In addition, activation of CD8+ T cells was observed to a lower extent for Adpgk-specific T cells, but not for the third candidate Cpne1.
Analysis of poly-functional T cell populations in tumor-bearing mice
To follow up on endogenously induced CD8+ T cells by irradiated MC-38, we analyzed endogenous CD8+ T cell specificities in tumor bearing mice. Therefore we inoculated untreated mice with MC-38 and eight days after inoculation, palpable and growing tumors were established subcutaneously. These mice were used for analysis of both splenic and intratumoral T cell populations (Figure 3(a)).
Figure 3.
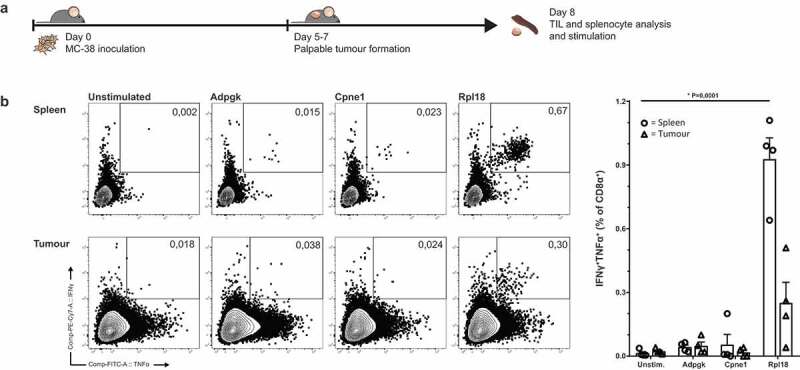
Tumor-bearing mice have circulating and tumor-infiltrating CD8+ lymphocytes specific for Rpl18 neo-peptide. (a) Mice were inoculated with 3 × 105 live MC-38 tumor cells. Lymphocytes isolated from spleen and tumor-infiltrating lymphocytes from the established tumors were stimulated with neo-peptides eight days post inoculation (n = 4). (b) IFNγ and TNFα cytokine production of CD8α+ T lymphocytes through stimulation with synthetic “long” peptides-loaded dendritic cells; from a single mouse (left) and all double-cytokine producing summarized (right). Where indicated, statistical significance was determined with student’s t test.
Ex vivo CD8+ T cell frequencies in both spleen and tumor of these mice displayed a similar dominant response toward the mutated Rpl18 epitope. Presence of Adpgk- and Cpne1-specific T cells was observed as well, although to a lower degree as observed in the MC-38-vaccinated mice (Figure 3(b)).
Rpl18 dominance in checkpoint-inhibitor protected mice
MC-38 is a well-known tumor model for its reliance on PD-L1 expression, both on the tumor and its microenvironment, to suppress immunological pressure.29,30 Treatment of MC-38 bearing mice through anti-PD-L1 checkpoint-inhibition has thus a strong therapeutic effect on tumor sizes, and often leads to complete regression and cure. We analyzed several mice that were cured from established MC-38 tumors by PD-L1 blockade therapy and had acquired immunological protection for rechallenge with live MC-38 cells (Figure 4(a) top).
Figure 4.

Dominant Rpl18-specific CD8+ populations in immunologically protected mice through PD-L1 checkpoint blockade therapy. (a) Mice bearing palpable tumors 6 days after inoculation with MC-38 tumor cells were injected thrice with anti-PD-L1 in a three-day interval to induce complete tumor control. At eighty days post inoculation, mice were re-challenged with MC-38 to confirm immunological protection. Tumor-specific T cells were expanded through co-culture with irradiated MC-38 tumor cells for one week and analyzed by peptide-loaded dendritic cell stimulation o/n (n = 6). (b) A shortened treatment schedule was used to analyze Rpl18-specific T cells in mice during tumor regression (left panel). At twelve days post challenge, lymphocytes specific for Rpl18 neo-epitope were observed in spleens and LN’s through tetramer staining (middle panel) and ICS (right panel). Anti-PD-L1 treatment increased Rpl18-specific T cell frequencies in both lymphoid organs (n = 7, statistical significance was determined with student’s t test, representative results of two independent experiments).
We restimulated splenocytes of these mice with irradiated MC-38 cells and analyzed CD8+ T cell specificities. Again, we observed a dominant polyfunctional response to mutated Rpl18 peptide in all samples (Figure 4(a) bottom). In addition, we could detect the presence of polyfunctional responses against mutated Adpgk as well, although to a lesser magnitude than Rpl18. In conclusion, we have identified in multiple settings major CD8+ T cell anti-tumor responses against two predicted and eluted neo-antigens.
Thereupon, we have analyzed the binding affinity of Rpl18 minimal epitope KILTFDRL to H-2Kb and observed a similar affinity compared to the chicken ovalbumin epitope SIINFEKL (suppl. Fig. 3). This allowed us to produce MHC Kb-KILTFDRL-tetramers in-house to identify Rpl18-specific T cell populations prior to PD-L1 blockade-induced regression of tumors (Figure 5(b) left panel, gating strategies in suppl. Fig 4A). In treated mice, Rpl18-specific T cells were increased more than two-fold on average in the spleen (Figure 5(b) middle panel). Resection of the lymph nodes from the tumor-matched flank revealed a similar trend. In addition, reactivity of Rpl18-specific T cells matched tetramer+ population sizes (Figure 5(b) right panel). We could not observe a similar trend for Adpgk- or Cpne1-specific T cells (Suppl. Fig 4B).
Figure 5.

Specificity to mutated peptide and direct recognition of tumor cells. (a) Minimal synthetic neo-peptide was compared to N- and C-terminally extended “long” peptides in intracellular cytokine staining of activated T cells upon coculture with peptide-loaded dendritic cells. (n = 6, error bars depict SEM) (b) Rpl18-specific T cell bulks were stimulated with either mutated or wild-type extended peptides to determine specificity to the single amino-acid mutation. (c) In vitro recognition of live MC-38 vs B16F10 melanoma control tumor cells by tetramer-stained T cells in Rpl18+ (5 out of 6 original bulks) and Adpgk+ (3 out of 6) cultures. A representative gating-plot of a single culture is shown (top). Tumor-specific single and double cytokine production for both Adpgk and Rpl18-specific T cells is shown below.
Specific T cell recognition of novel neo-epitopes on MC-38
We next analyzed the specificities of cultured bulk T cells from protected mice (Figure 4a) and their reactivity toward live MC-38 tumor cells. Coculture of bulk CD8+ T cells from protected mice were able to specifically recognize Rpl18 and Adpgk synthetic peptide pre-loaded dendritic cells. Recognition to both the minimally presented peptide epitope or elongated long synthetic peptides was shown by cytokine production of specific CD8+ T cells (Figure 5(a)). In addition, pre-incubation of dendritic cells with a wild-type sequence of the Rpl18 peptide, fully negated the responses induced by the mutated variant (Figure 5(b)). Thereby authenticating the specificity of these T cells for a single amino acid substitution of the Rpl18 protein.
Furthermore, both Rpl18- and Adpgk-specific populations produced significant levels of cytokines upon co-culture with live cells MC-38 and not to control B16F10 melanoma tumor cells (Figure 5(c)). However, it should be noted that these presented neo-epitopes were not observed in a commercially available MC-38 cell-line (suppl. fig 5A), nor was there any recognition by our cultured bulk T cell (suppl. fig 5B). In summary, T cells derived from PD-L1 cured mice recognize two sequenced and eluted candidate neo-epitopes derived from the Rpl18 and Adpgk genes in our MC-38, and are able to specifically recognize live tumor cells.
Tumor growth control in peptide-vaccinated mice
Since the CD8+ T cell response to this novel neo-epitope was capable of direct tumor cell recognition, we aimed to therapeutically vaccinate mice against the neo-epitope of Rpl18. Therefore, we synthesized peptides covering the neo-epitope flanked by natural amino acid sequences.
We tested the effectivity of subcutaneous injection of synthetic long peptide adjuvanted with CpG, eight days post-inoculation of MC-38 tumor cells when all mice had developed palpable tumors (Figure 6(a) top). In mice receiving the Rpl18 peptide vaccination, we observed a significantly lower tumor burden twelve and fourteen days after vaccination compared to CpG control-injected mice. Tumor burden in mice receiving Adpgk peptide vaccination was lower than control but did not result in improved survival (Figure 6(b) bottom). To determine if the difference between neo-epitope vaccinations was the result of altering presentation levels, we performed parallel reaction monitoring (PRM) of peptides ASMTNMELM and KILTFDRL which revealed that both were present at the same level in our MHC class I elution (suppl. table 2 and suppl. fig. 6).
Figure 6.

Therapeutic vaccination with Rpl18 neo-peptide-based vaccinations improves tumor control. (a) Schematic representation of therapeutic vaccination protocol. 3 × 105 tumor cells were subcutaneously injected in the right flank. Eight days post inoculation, tumors established and a single synthetic neo-peptide vaccination was given intradermally at the tail-base. (b) Therapeutic vaccination with 10 nmol of extended synthetic peptide harboring neo-epitopes of Adpgk or Rpl18 delayed outgrowth of MC-38 tumors. At day 20, average tumor size in Adpgk-vaccinated mice was two-fold reduced (279 vs. 563 mm3, P = 0,1894), while Rpl18 vaccination reduced tumor size nearly 10-fold (60 mm3, P = 0,0163). This trend was maintained 22 days post inoculation (Adpgk: 574 vs. 817 mm3, P = 0,4098; Rpl18: 102 vs. 817 mm3, P = 0,0083). Subsequent median survival was extended in Rpl18 vaccinated mice (37 vs 23,5 days, P = 0,0445), while no significant extension was observed for Adpgk-vaccinated mice (26,5 days, P = 0,4244). (Eight mice per group; error bars indicate SEM; multiple t tests and Gehan-Breslow-Wilcoxon test were used for statistical analysis of tumor size and survival, respectively) (representative results of two independent experiments). (c) To improve vaccination efficacy we conjugated the Rpl18 neo-peptide to Upam TLR2-ligand (also known as Amplivant®). Therapeutic vaccination with 10 nmol of Rpl18-conjugate against established tumor significantly improved tumor control and survival compared to Upam-control (P = 0,0174; n = 8 vs n = 7 in control group; log rank tested) (depicted results are from a single experiment).
Since overall survival was modest in Rpl18-vaccinated mice, we decided to improve the formulation of our vaccination through a conjugation with Toll-like receptor (TLR) 2-ligand Upam, a Pam3CSK4 derivative with improved affinity for TLR-2.25,31 Similarly as the previous setting, we gave tumor-bearing mice a single injection of this peptide-based vaccine 8 days post inoculation. Six weeks after vaccination with Upam-Rpl18 synthetic peptide conjugate, we observed a significantly improved survival in the vaccinated group compared to a Upam control-injected group (Figure 6(c)). Complete regression of tumors in mice 44 days after tumor challenge was maintained as no palpable tumors could be observed until 127 days post inoculation. Concludingly, these results show that a dominant CD8+ T cell population targeting the neo-epitope of Rpl18 is relevant for anti-tumor immunity to MC38.
Discussion
Here we report a detailed analysis of the CD8+ immune response to the highly mutated MC-38 colorectal tumor cell line. This cell line is frequently used as an appropriate model for optimizing and development of new immunotherapeutic strategies as it is sensitive for most immune checkpoint therapies. We have studied spontaneous CD8+ T cell responses in tumor-bearing mice, response induction after tumor cell vaccination, and after effective PD1 immune checkpoint therapy. In all cases we observed that the CD8+ response was dominated by the point-mutated Rpl18 neo-epitope.
In these three different ex vivo settings of MC-38 exposure in vivo, we could distinguish a large fraction of CD8+ T cells to respond to a mutation derived from 60S ribosomal protein L18 (Rpl18), and to a lesser extent to previously described epitopes derived from ADP-dependent Glucokinase (Adpgk) and Copine-1 (Cpne1).28 Upon restimulation, the majority of samples obtained from all ex vivo settings demonstrated a striking dominance of Rpl18-specific CD8+ T cells, suggesting a central role in anti-MC-38 immunity. This is supported by analysis of tumor-infiltrating lymphocytes, in which primarily Rpl18-specific T cells appeared to be present and can be activated to polyfunctional cytokine production.
The MC-38 model is well-described for its expression of PD-L1 on both the tumor cells and the tumor microenvironment.29,30 Thereby providing an explanation for the lack of tumor control despite the presence of specific-CD8+ T cells in the tumors. Splenocytes obtained from several of the cured and immunologically protected mice were challenged with our peptides, whereby all samples were dominated by Rpl18-specific T cells, although Adpgk-specific T cells were substantially present as well. Our analysis showed that the reactivity against our Rpl18-peptide was specific for the single amino-acid substitution caused by the somatic mutation in this gene. Cultured Rpl18-specific T cells were able to directly recognize MC-38 tumor cells. Moreover, we could ex vivo stain CD8+ T cells with Rpl18-H-2Kb tetramers in tumor-bearing mice. This suggested a significant contribution of Rpl18-specific cytotoxic T cells to immunological control of MC-38.
The MC-38 model is commonly used to optimize immunotherapeutic approaches to prime or re-activate anti-tumor cytotoxic T cells which can overcome tumor-suppressive environments and “exhausted” conditions. We here report a detailed characterization of the CD8+ neo-epitopes of this tumor model, in which Rpl18-specific T cells appears to determine a dominant immune-response accompanied by lower frequencies against mutated Adpgk and Cpne1. Albeit speculative, the dominant endogenous immune responses against the Rpl18-derived neo-epitope may originate from the constitutive expression of this ribosome associated protein being part of the protein synthesis machinery. Indeed we did observe a high gene expression level in our MC-38, although this did not translate to an equally higher presentation in MHC class I compared to Adpgk. Nevertheless, the previously described Adpgk epitope is beyond doubt relevant for vaccination-induced MC-38 tumor control as is observed by several groups,28,32,33 including our own experimental data with specific DNA vaccination.34 In addition, care should be taken with the origin of the MC-38 cell line, as different lines may contain other neo-epitopes.
Therapeutic vaccination against Rpl18 using synthetic peptides induced delay of tumor outgrowth and extended survival although overall survival was not affected. We hypothesized that a conjugation to an optimized TLR-2 ligand Upam or Amplivant®25,26,31,35,36 would improve immunogenicity and survival. As expected, vaccination of MC-38 tumor-bearing mice with this construct resulted in a significant improvement of survival when compared to adjuvant alone, thus underscoring the relevance of the Rpl18 CD8+ neo-epitope in the MC-38 colorectal cancer model.
Since the rapid establishment of immune-checkpoint therapy for cancer patients, an increasing amount of studies determine the prerequisites and mechanisms underlying application of these antibodies. It has now been recognized that patients bearing tumors with high mutational loads benefit the most, which appears to be driven by neo-antigen specific T cells.5–8,11,37,38 Despite the positive outcome for many of these patients, a large fraction of patients remains unresponsive to these therapies while risking autoimmune-related disorders. Our characterization of endogenous, mutation-specific, responses against MC-38 tumor cells will contribute to investigations and optimizations of immunotherapeutic strategies against cancer.
Supplementary Material
Acknowledgments
The authors would like to thank Jan-Wouter Drijfhout and Willemien Benckhuijsen for the synthesis of peptides and Nico Meeuwenoord for his expert assistance with the synthesis of UPam-Rpl18 conjugate. Likewise, the Animal Facility of the LUMC for their good care of the animals, as well as the Flow Cytometry Facility of the LUMC for their efforts to maintain the facility and protocols at professional standards.
Funding Statement
B.J.H, F.O. and D.V.F. have received funding by The Netherlands Organization for Scientific Research (NWO) through the gravitational program 2013 granted to the Institute of Chemical Immunology (ICI-024.002.009). E.T. is funded by the University Leiden Profiling Area Bioscience: the Science Base of Health. J.v.d.B, D.R. and N.F.C.C.d.M. are funded through the. G.M.C.J. and P.A.v.V received funding by the research programme Investment Grant NWO Medium with project number 91116004, which is (partly) financed by ZonMw. T.C.v.d.E has been funded through Molecule-To-Man program (jointly by LUMC and Leiden University).
Author contribution
B.J.H was the primary designer, performer, interpreter of experiments, and writer of this manuscript. M.G.M.C. and E.T. provided help for the design and execution of experiments. J.v.d.B, D.R. and N.F.C.C.d.M. contributed to this manuscript through the analysis and processing of tumor- and mouse sequencing data and prediction analysis. G.M.C.J., A.d.R and P.A.v.V were vital in the elution, mass-spectrometry, and identification of MHC binding neo-peptides. T.C.v.d.E and D.M.F for their design and synthesis of peptide-conjugates. K.F. was involved for the production of tetramers. R.A. provided feedback and input for experimental design and manuscript. F.O. was the principle investigator responsible for supervision during each step of this project.
Declaration of interest statement
The authors declare there was no potential conflict of interest.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Chen DS, Mellman I.. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39(1):1–11. doi: 10.1016/j.immuni.2013.07.012. [DOI] [PubMed] [Google Scholar]
- 2.Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. 2017;541(7637):321–330. doi: 10.1038/nature21349. [DOI] [PubMed] [Google Scholar]
- 3.Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer. 2016;16(5):275–287. doi: 10.1038/nrc.2016.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yarchoan M, Johnson BA 3rd, Lutz ER, Laheru DA, Jaffee EM. Targeting neoantigens to augment antitumour immunity. Nat Rev Cancer. 2017;17(4):209–222. doi: 10.1038/nrc.2016.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348(6230):69–74. doi: 10.1126/science.aaa4971. [DOI] [PubMed] [Google Scholar]
- 6.Łuksza M, Riaz N, Makarov V, Balachandran VP, Hellmann MD, Solovyov A, Rizvi NA, Merghoub T, Levine AJ, Chan TA, et al. A neoantigen fitness model predicts tumour response to checkpoint blockade immunotherapy. Nature. 2017. doi: 10.1038/nature24473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372(26):2509–2520. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348(6230):124–128. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS, Hodi FS, Martin-Algarra S, Mandal R, Sharfman WH, et al. Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell. 2017;171(4):934–49 e15. doi: 10.1016/j.cell.2017.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Efremova M, Rieder D, Klepsch V, Charoentong P, Finotello F, Hackl H, Hermann-Kleiter N, Lower M, Baier G, Krogsdam A, et al. Targeting immune checkpoints potentiates immunoediting and changes the dynamics of tumor evolution. Nat Commun. 2018;9(1):32. doi: 10.1038/s41467-017-02424-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Verdegaal EM, de Miranda NF, Visser M, Harryvan T, van Buuren MM, Andersen RS, Hadrup SR, van der Minne CE, Schotte R, Spits H, et al. Neoantigen landscape dynamics during human melanoma-T cell interactions. Nature. 2016;536(7614):91–95. doi: 10.1038/nature18945. [DOI] [PubMed] [Google Scholar]
- 12.Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, Zhang W, Luoma A, Giobbie-Hurder A, Peter L, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. 2017;547(7662):217–221. doi: 10.1038/nature22991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sahin U, Derhovanessian E, Miller M, Kloke BP, Simon P, Lower M, Bukur V, Tadmor AD, Luxemburger U, Schrors B, et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature. 2017. doi: 10.1038/nature23003. [DOI] [PubMed] [Google Scholar]
- 14.van der Burg SH, Arens R, Ossendorp F, van Hall T, Melief CJ. Vaccines for established cancer: overcoming the challenges posed by immune evasion. Nat Rev Cancer. 2016;16(4):219–233. doi: 10.1038/nrc.2016.16. [DOI] [PubMed] [Google Scholar]
- 15.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, Getz G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31(3):213–219. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A, Flicek P, Cunningham F. The ensembl variant effect predictor. Genome Biol. 2016;17(1):122. doi: 10.1186/s13059-016-0974-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nat Biotechnol. 2011;29(1):24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013;14(2):178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Andreatta M, Nielsen M. Gapped sequence alignment using artificial neural networks: application to the MHC class I system. Bioinformatics. 2016;32(4):511–517. doi: 10.1093/bioinformatics/btv639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nielsen M, Lundegaard C, Worning P, Lauemoller SL, Lamberth K, Buus S, Brunak S, Lund O. Reliable prediction of T-cell epitopes using neural networks with novel sequence representations. Protein Sci. 2003;12(5):1007–1017. doi: 10.1110/ps.0239403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hassan C, Kester MG, de Ru AH, Hombrink P, Drijfhout JW, Nijveen H, Leunissen JA, Heemskerk MH, Falkenburg JH, van Veelen PA. The human leukocyte antigen-presented ligandome of B lymphocytes. Mol Cell Proteomics. 2013;12(7):1829–1843. doi: 10.1074/mcp.M112.024810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sow HS, Benonisson H, Breukel C, Visser R, Verhagen O, Bentlage AEH, Brouwers C, Claassens JWC, Linssen MM, Camps M, et al. FcgammaR interaction is not required for effective anti-PD-L1 immunotherapy but can add additional benefit depending on the tumor model. Int J Cancer. 2019;144(2):345–354. doi: 10.1002/ijc.31899. [DOI] [PubMed] [Google Scholar]
- 24.Khan S, Bijker MS, Weterings JJ, Tanke HJ, Adema GJ, van Hall T, Drijfhout JW, Melief CJ, Overkleeft HS, van der Marel GA, et al. Distinct uptake mechanisms but similar intracellular processing of two different toll-like receptor ligand-peptide conjugates in dendritic cells. J Biol Chem. 2007;282(29):21145–21159. doi: 10.1074/jbc.M701705200. [DOI] [PubMed] [Google Scholar]
- 25.Willems MM, Zom GG, Khan S, Meeuwenoord N, Melief CJ, van der Stelt M, Overkleeft HS, Codee JD, van der Marel GA, Ossendorp F, et al. N-tetradecylcarbamyl lipopeptides as novel agonists for Toll-like receptor 2. J Med Chem. 2014;57(15):6873–6878. doi: 10.1021/jm500722p. [DOI] [PubMed] [Google Scholar]
- 26.Zom GG, Welters MJ, Loof NM, Goedemans R, Lougheed S, Valentijn RR, Zandvliet ML, Meeuwenoord NJ, Melief CJ, de Gruijl TD, et al. TLR2 ligand-synthetic long peptide conjugates effectively stimulate tumor-draining lymph node T cells of cervical cancer patients. Oncotarget. 2016;7(41):67087–67100. doi: 10.18632/oncotarget.11512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Winzler C, Rovere P, Rescigno M, Granucci F, Penna G, Adorini L, Zimmermann VS, Davoust J, Ricciardi-Castagnoli P. Maturation stages of mouse dendritic cells in growth factor-dependent long-term cultures. J Exp Med. 1997;185(2):317–328. doi: 10.1084/jem.185.2.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yadav M, Jhunjhunwala S, Phung QT, Lupardus P, Tanguay J, Bumbaca S, Franci C, Cheung TK, Fritsche J, Weinschenk T, et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature. 2014;515(7528):572–576. doi: 10.1038/nature14001. [DOI] [PubMed] [Google Scholar]
- 29.Kleinovink JW, Marijt KA, Schoonderwoerd MJA, van Hall T, Ossendorp F, Fransen MF. PD-L1 expression on malignant cells is no prerequisite for checkpoint therapy. Oncoimmunology. 2017;6(4):e1294299. doi: 10.1080/2162402X.2017.1294299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Juneja VR, McGuire KA, Manguso RT, LaFleur MW, Collins N, Haining WN, Freeman GJ, Sharpe AH. PD-L1 on tumor cells is sufficient for immune evasion in immunogenic tumors and inhibits CD8 T cell cytotoxicity. J Exp Med. 2017. doi: 10.1084/jem.20160801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zom GG, Willems M, Khan S, van der Sluis TC, Kleinovink JW, Camps MGM, van der Marel GA, Filippov DV, Melief CJM, Ossendorp F. Novel TLR2-binding adjuvant induces enhanced T cell responses and tumor eradication. J Immunother Cancer. 2018;6(1):146. doi: 10.1186/s40425-018-0455-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kuai R, Ochyl LJ, Bahjat KS, Schwendeman A, Moon JJ. Designer vaccine nanodiscs for personalized cancer immunotherapy. Nat Mater. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhu G, Lynn GM, Jacobson O, Chen K, Liu Y, Zhang H, Ma Y, Zhang F, Tian R, Ni Q, et al. Albumin/vaccine nanocomplexes that assemble in vivo for combination cancer immunotherapy. Nat Commun. 2017;8(1):1954. doi: 10.1038/s41467-017-02191-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tondini E, Arakelian T, Oosterhuis K, Camps M, van Duikeren S, Han W, Arens R, Zondag G, van Bergen J, Ossendorp F. A poly-neoantigen DNA vaccine synergizes with PD-1 blockade to induce T cell-mediated tumor control. Oncoimmunology. 1652539. 2019. doi: 10.1080/2162402X.2019.1652539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Khan S, Weterings JJ, Britten CM, de Jong AR, Graafland D, Melief CJ, van der Burg SH, van der Marel G, Overkleeft HS, Filippov DV, et al. Chirality of TLR-2 ligand Pam3CysSK4 in fully synthetic peptide conjugates critically influences the induction of specific CD8+ T-cells. Mol Immunol. 2009;46(6):1084–1091. doi: 10.1016/j.molimm.2008.10.006. [DOI] [PubMed] [Google Scholar]
- 36.Zom GG, Khan S, Britten CM, Sommandas V, Camps MG, Loof NM, Budden CF, Meeuwenoord NJ, Filippov DV, van der Marel GA, et al. Efficient induction of antitumor immunity by synthetic toll-like receptor ligand-peptide conjugates. Cancer Immunol Res. 2014;2(8):756–764. doi: 10.1158/2326-6066.CIR-13-0223. [DOI] [PubMed] [Google Scholar]
- 37.McGranahan N, Furness AJS, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, Jamal-Hanjani M, Wilson GA, Birkbak NJ, Hiley CT, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016. doi: 10.1126/science.aaf1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fehlings M, Simoni Y, Penny HL, Becht E, Loh CY, Gubin MM, Ward JP, Wong SC, Schreiber RD, Newell EW. Checkpoint blockade immunotherapy reshapes the high-dimensional phenotypic heterogeneity of murine intratumoural neoantigen-specific CD8+ T cells. Nat Commun. 2017;8(1):562. doi: 10.1038/s41467-017-00627-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.