

ABSTRACT
Cytokine-Induced Killer (CIK) cells share several functional and phenotypical properties of both T and natural killer (NK) cells. They represent an attractive approach for cell-based immunotherapy, as they do not require antigen-specific priming for tumor cell recognition, and can be rapidly expanded in vitro. Their relevant expression of FcγRIIIa (CD16a) can be exploited in combination with clinical-grade monoclonal antibodies (mAbs) to redirect their lytic activity in an antigen-specific manner. Here, we report the efficacy of this combined approach against triple negative breast cancer (TNBC), an aggressive tumor that still requires therapeutic options. Different primitive and metastatic TNBC cancer mouse models were established in NSG mice, either by implanting patient-derived TNBC samples or injecting MDA-MB-231 cells orthotopically or intravenously. The combined treatment consisted in the repeated intratumoral or intravenous injection of CIK cells and cetuximab. Tumor growth and metastasis were monitored by bioluminescence or immunohistochemistry, and survival was recorded. CIK cells plus cetuximab significantly restrained primitive tumor growth in mice, either in patient-derived tumor xenografts or MDA-MB-231 cell line models. Moreover, this approach almost completely abolished metastasis spreading and dramatically improved survival. The antigen-specific mAb favored tumor and metastasis tissue infiltration by CIK cells, and led to an enrichment of the CD16a+ subset.
Data highlight the potentiality of this novel immunotherapy strategy where a nonspecific cytotoxic cell population can be converted into tumor-specific effectors with clinical-grade antibodies, thus providing not only a therapeutic option for TNBC but also a valid alternative to more complex approaches based on chimeric antigen receptor-engineered cells.
List of abbreviations
ACT, Adoptive Cell Transfer; ADCC, Antibody-Dependent Cell-mediated Cytotoxicity; ADP, Adenosine diphosphate; BLI, Bioluminescence Imaging; CAR, Chimeric Antigen Receptor; CIK, Cytokine Induced Killer cells; CTX, Cetuximab; DMEM, Dulbecco’s Modified Eagle Medium; EGFR, Human Epidermal Growth Factor 1; ER, Estrogen; FBS, Fetal Bovine Serum; FFPE, Formalin-Fixed Paraffin-Embedded; GMP, Good Manufacturing Practices; GVHD, Graft Versus Host Disease; HER2, Human Epidermal Growth Factor 2; HRP, Horseradish Peroxidase; IFN-γ, Interferon-γ; IHC, Immunohistochemistry; IL-2, Interleukin-2; ISO, Irrelevant antibody; i.t., intratumoral; i.v., intravenous, mAbs, Monoclonal Antibodies; mIHC, Multiplex Fluorescence Immunohistochemistry; MHC, Major Histocompatibility Complex; NK, Natural Killer; NKG2D, Natural-Killer group 2 member D; NSG, NOD/SCID common γ chain knockout; PARP, Poly ADP-ribose polymerase; PBMCs, Peripheral Blood Mononuclear Cells; PBS, Phosphate-buffered saline; PDX, Patient-derived xenograft; PR, Progesterone; rhIFN-γ, Recombinant Human Interferon-γ; RPMI, Roswell Park Memorial Institute; STR, Short tandem Repeat; TCR, T Cell Receptor; TNBC, Triple Negative Breast Cancer; TSA, Tyramide Signal Amplification
KEYWORDS: Cytokine-induced killer cells (CIK), adoptive cells therapy (ACT), triple negative breast cancer (TNBC), TNBC mouse models, cetuximab
Introduction
Triple negative breast cancer (TNBC) is an aggressive tumor that occurs in approximately 15% of all breast cancer patients, and is defined by the lack of expression of estrogen (ER) and progesterone (PR) receptors, and human epidermal growth factor receptor 2 (HER2). Thus, patients cannot be treated with hormone blockade or HER2-specific monoclonal antibodies,1 and generally receive neoadjuvant or adjuvant systemic chemotherapy. Nonetheless, TNBC is more likely to spread and recur than other types of breast cancer, and the 5-year survival is remarkably reduced.2 In this context, the development of additional therapeutic approaches is still an unmet clinical need.
Cancer immunotherapy has shown promising results against different tumors. Currently, enthusiasm on adoptive cell transfer (ACT) approaches mainly relies on the excellent results reported for B-cell malignancies with the infusion of CD19 chimeric antigen receptor (CAR)-T cells.3,4 However, the laborious and expensive production of such effector cells, along with all the technical and safety hurdles associated with genetic modification of autologous T lymphocytes, constitutes an important disadvantage for this ACT therapy and an impediment to its wide diffusion.
These obstacles might be overcome by using CIK cells, a population of effectors that can be obtained from peripheral blood mononuclear cells (PBMCs) after ex vivo stimulation, by the timed addition of IFN-γ, interleukin-2 (IL-2) and mAb directed against CD3. CIK cells share phenotypical and functional characteristics with both NK and T cells. Indeed, their main features are the expression of both CD3 and CD56, and the MHC-unrestricted NKG2D-mediated antitumor activity, which is exerted against a broad range of tumor histotypes but not against normal tissues and hematopoietic precursors, without requiring prior antigen exposure or priming.5 Preclinical and clinical studies have assessed the feasibility and the therapeutic efficacy of CIK cell infusions, and demonstrated that they almost completely lack Graft Versus Host Disease (GVHD) activity, even in a fully allogeneic setting.6-9 Moreover, CIK cell activity can be further empowered by the combination with clinical-grade mAbs, due to their relevant expression of FcγRIIIa (CD16a) that fosters potent antibody-dependent cell–mediated cytotoxicity (ADCC).10 Here we report an extensive in vivo analysis showing that the combination of CIK cells and cetuximab (CTX), an epidermal growth factor receptor (EGFR)-specific chimeric IgG1 antibody,11 can provide a highly efficient ACT therapeutic option for metastatic TNBC, where EGFR is largely overexpressed.12
Material and methods
Ethics approval and consent to participate
Anonymized human buffy coats were obtained from the Blood Bank of Padova Hospital, and donors provided their written informed consents to participate in this study, in accordance with The Code of Ethics of the World Medical Association (Declaration of Helsinski). Procedures involving animals and their care were in conformity with national (D.L. 26/2014 and subsequent implementing circulars) and international (EU Directive 2010/63/EU for animal experiments) laws and policies, and the experimental protocol (Authorization n. 1143/2015-PR) was approved by the Italian Ministry of Health.
Generation and characterization of CIK cells
CIK cells were obtained from PBMCs of healthy donors isolated by means of Ficoll-Paque PLUS (GE Healthcare) density gradient centrifugation, according to standard protocols.10 PBMCs were plated in RPMI 1640 (Euroclone) supplemented with 10% heat-inactivated FBS (Gibco), 1% Ultraglutamine, 1% Hepes buffer, 1% penicillin/streptomycin (all from Lonza), at 37°C and 5% CO2, and stimulated with rhIFN-γ (PeproTech) at 1000 U/ml at day 0. Twenty-four hours later, anti-CD3 mAb (OKT-3, Ortho Biotech Inc) at 50 ng/ml and rhIL-2 (Proleukin, Novartis) at 500 IU/ml were added to the culture medium; every 2–3 days medium was replenished and fresh rhIL-2 at 500 IU/ml was added. CIK cells phenotype was analyzed by multi-color flow cytometry, using the following antibodies: CD3-BV510 (clone UCHT1), IL-2-BV421 (clone 5344.111), CD25-APC (IL-2 Rα, clone M-A251), CD122-BV650 (IL-2Rβ, clone Mik-β3), CD132-BV786 (IL-2Rγ, clone AG184), from BD Bioscience; CD56-PE (clone HCD56), CD16a-FITC (clone 3G8), from BioLegend. Flow cytometry analysis was performed on either LSRII or Celesta, using DIVA software (BD Bioscience). Data analyzes were performed using FlowJo software (Treestar).
TNBC cell lines
TNBC MDA-MB-231 and MDA-MB-468 cell line were authenticated by single tandem repeats (STR) sequences analysis. The cells were analyzed for the EGFR expression by flow cytometry using the chimeric anti-EGFR monoclonal IgG1 antibody cetuximab (CTX, MerckSerono) and a PE-conjugated anti IgG1 antibody (Miltenyi Biotec) as secondary antibody. MDA-MB-231 were transduced with a lentiviral vector coding for the Firefly Luciferase reporter gene (MDA-MB-231_Luc) for the in vivo experiments.13 All cell lines were maintained and expanded in DMEM medium (Euroclone) supplemented with 10% heat-inactivated FBS (Gibco), 1% Ultraglutamine, 1% Hepes buffer, 1% penicillin/streptomycin (all from Lonza).
Cytotoxic assay
CIK cell cytotoxicity was tested against MDA-MB-231 and MDA-MB-468 using a calcein-acetoxymethyl (AM) release assay. Briefly, target cells were labeled with calcein-AM (3.5 µM, Sigma) for 30 minutes at 37°C, and added to CIK cells in U-bottom 96-well plates at an effector:target (E/T) ratio of 50:1 in the presence of 1 µg/ml of Cetuximab (CTX) or a IgG1 isotype control antibody (ISO). After 4 h at 37°C, 100 µL supernatant was transferred on a 96-well ViewPlateTM plates (PerkinElmer) and measured using a VICTOR Multilabel Plate Reader. Each test was performed in triplicate. The results are expressed as the percentage of lysis, which is calculated as follows: % Specific Lysis = (experimental release – spontaneous release)/(maximum release–spontaneous release) x 100. Maximum and spontaneous release were obtained by incubating target cells with RPMI containing 3% Triton X-100 (Sigma) or complete RPMI growth medium, respectively.
Co-culture in vitro assay
MDA-MB-231 were plated overnight in a 24-well plate. The day after, CIK cells were added at an E/T ratio of 10:1 in the presence of 1 µg/ml of Cetuximab (CTX) or IgG1 isotype control (ISO). To determine the intracellular expression of IL-2, CIK cells were incubated for 4 hours in the presence of 2 µM monensin (BD GolgiStop™ protein transport inhibitor), and treated by using the Cytofix/Cytoperm™ Fixation/Permeabilization Kit (BD Bioscience). The IL-2 receptors were analyzed in CIK cells after 24 hours of co-culture by flow cytometry, while the IL-2 released in the supernatants was measured by using ProQuantum High-Sensitivity Immunoassay (Thermo Fisher Scientific).
In vivo studies
Six-to-eight week-old female NOD/SCID common γ chain knockout (NSG, Charles River) mice were used for all the experiments. Animals received either 105 MDA-MB-231_Luc cells by tail vein injection, or 106 cells by fat pad inoculation. In the fat pad model, tumor growth was monitored by caliper measurements, and the volume was calculated using the formula: Tumor volume (mm3) = D× d2/2, where D and d are the longest and the shortest diameters, respectively. To monitor systemically administered cells and metastasis, bioluminescence in vivo imaging was carried out at different time points in anesthetized animals (1–3% isoflurane, Merial Italia S.p.A), after intraperitoneal injection of the substrate D-Luciferin (Biosynth AG) at 150 mg/kg in PBS (Sigma). The light emitted from the bioluminescent primary tumors or metastases was detected using the IVIS Lumina II Imaging System (PerkinElmer). Regions of interest from the displayed images were identified around the tumor sites or metastasis regions, such as the lymph nodes and lungs, and quantified as total photon counts (photon/s) using Living Image® software (PerkinElmer). In the presence of both a primary tumor in the fat pad and metastases in the lungs or lymph nodes, the lower portion of each animal was covered before reimaging, to ensure that the bioluminescence signals from the metastatic regions could be observed. Where indicated, the primary tumor was surgically excised from the fat pad 10 days after the injection,14 and metastasis dissemination and growth in lymph nodes and lungs were monitored by bioluminescence.
Patient-derived tumor xenograft (PDX)
The EGFR-positive TNBC PDX was established at the Istituto Oncologico Veneto (IOV-IRCCS, Padua, Italy) after patient informed consent. The sample was cut into 3 × 3 mm pieces in RPMI 1640 medium supplemented with 100 U/mL penicillin/streptomycin, and grafted subcutaneously in the fat pad area of NSG mice, and subsequently transplanted and expanded from mouse to mouse. Xenografts were considered established after 3 passages. At each passage, a PDX tissue fragment was fixed in formalin and embedded in paraffin (FFPE), and the EGFR expression was evaluated by immunohistochemistry (IHC). In the PDX models, the intratumoral (i.t.) or intravenous (i.v.) treatments started when the tumor became palpable. Tumor growth was monitored by caliper measurement.
CIK cell therapy
Tumor-bearing mice were treated by co-administration for five consecutive days, either i.t. or i.v., of 107 CIK cells resuspended with 50 mg/kg of the anti-EGFR mAb CTX; control animals were left untreated or received CTX only or CIK cells resuspended with 50 mg/kg of the isotype control mAb (ISO). Mice were sacrificed when they showed signs of suffering, such as weight loss, ruffling of hair or difficulties in movements, for systemic tumors, and when subcutaneous masses from PDX in i.t. therapy exceeded a 1500 mm3 volume or subcutaneous masses from PDX and from MDA-MB-231_Luc in i.v. therapy, exceeded a 700 mm3 volume or appeared ulcerated.
Immunohistochemistry analysis (IHC)
Metastatic lung tissues from mice were collected, and 3–4 µm-thick FFPE sections were stained with anti-human cytokeratin mAb (clone MNF116, Dako), or with anti-human CD3 mAb (PS1, Leica Biosystem). Images were acquired with Aperio Scan Scope (Leica Biosystem) and automatically quantified using a custom-made algorithm created with VisiopharmTM software version 4.5.6.5 (Visiopharm, Hoersholm, Denmark).
Multiplex fluorescence immunohistochemistry (mIHC)
mIHC analysis was carried out on 4 µm-thick FFPE tissue sections, using the Opal 7-colors manual IHC kit (PerkinElmer, NEL811001KT). The slides were stained with CD3 (clone F.7.2.38, Dako), CD56 (clone 123 C3, Dako), CD16 (clone 2 H7, Leica Biosystems), pan-cytokeratin (clone AE1/AE3, Dako) antibodies and DAPI. Multiplex stained slides were scanned at 20X using the Mantra Quantitative Pathology Workstation (PerkinElmer), and analyzed with InForm Image Analysis Software (PerkinElmer, v2.4.2).
Statistics
Results were analyzed for statistical significance by using paired or unpaired Student t test, as appropriate, and ANOVA (****P < .0001, ***P < .001, **P < .01, *P < .05). Mice survival was compared using log-rank (Mantel-Cox) survival statistics. Statistical analysis was performed using GraphPad Prism 8.0.1 Software.
Results
The combined therapy with CIK cells and cetuximab restrains the growth of patient-derived TNBC xenografts
Bulk cultures of CIK cells were obtained from healthy donor PBMC, and consisted of 23.38 ± 12.66% CD3+CD56+ cells, of which 20.85 ± 20.63% were CD3+CD56+CD16+; the CD3−CD56+ (NK) cell subset represented only 1.3 ± 0.7% of the bulk population (Figure 1a). Upon incubation with MDA-MB-231 and MDA-MB-468 target cell lines, which express EGFR (Figure 1b), CIK effector cells exerted a strong ADCC following addition of CTX, while cytotoxicity was negligible either when cells were tested alone or in the presence of an isotype control mAb (Figure 1c). CTX alone was not able to induce any target cells lysis in the four hours of the assay, and the values of spontaneous release were comparable to the spontaneous release in absence of the antibody (data not shown). Aiming to assess whether such combination could translate into a potential therapy in the clinical setting, we decided to evaluate its role and impact in vivo against a patient-derived TNBC xenograft. To this end, an established EGFR-positive TNBC PDX (Figure 2a) was implanted in the mammary fat pad area of NSG mice. When the tumor volume reached 500 mm3, mice were randomly divided into four experimental groups, namely CIK cells with CTX (CIK+CTX), CIK cells plus an irrelevant antibody (CIK+ISO), cetuximab alone (CTX), or untreated mice. Treatment protocol consisted in daily i.t. administrations for five days, and tumor growth was monitored by caliper measurement (Figure 2b).
Figure 1.

CIK cell cytotoxicity against TNBC cell lines is enhanced when combined with CTX. (a) CIK cell bulk cultures were characterized by flow cytometry for the content of CD3+CD56+ and CD3+CD56+CD16a+ subsets, and CD3−CD56+ cells. Dot plot of one representative donor is showed (upper panels). Histograms refer to the mean expression ± SD of 15 independent expansions from distinct healthy donors (lower panels). (b) MDA-MB-231 (left panel) and MDA-MB-468 (right panel) were analyzed for EGFR expression (pink) by flow cytometry. A fluorochrome-conjugated secondary antibody alone (gray) served as negative control. (c) CIK cells were assessed for cytotoxicity against MDA-MB-231 (left panel) and MDA-MB-468 (right panel) tumor cell lines. Lytic activity was measured in the absence (white boxes) or in the presence of an isotype control mAb (ISO, light gray boxes) or cetuximab (CTX, dark gray boxes) at an E/T ratio of 50:1; boxes indicate mean value ± SD of 15 independent experiments. ***P < .001, ****P < .0001.
Figure 2.

CIK cells and Cetuximab combined treatment is therapeutically efficient against TNBC patient-derived xenografts. (a) Representative image of a TNBC patient-derived xenograft IHC-stained for EGFR (brown). (b) The experimental protocol consisted in the implantation of PDX in the fat pad area of NSG mice, followed by i.t. or i.v. daily treatments for five days, starting when tumors reached about 500 mm3. (c) Mice received i.t. CIK cells in combination with CTX (n = 16; inverted triangle) or ISO (n = 13; diamond), CTX alone (n = 10; triangle), or were left untreated (n = 7; circle). Tumor volume was measured by caliper measurement. Data are expressed as mean ± SD (Repeated measures ANOVA). (d) Representative images of anti-human CD3 IHC staining (brown) of tumors excised from i.t.-treated mice, and the correspondent CD3+ cell density quantification (right panel). Data are expressed as mean ± SD (Student’s T-test). (e) Quantification of the CD3+CD56+ density (left panel) and CD3+CD56+CD16+ (right panel) of PPFE tumors from i.t. mouse model, analyzed by mIHC. Data are presented as mean ± SD. (f) Mice received i.v. CIK cells in combination with CTX (n = 7; inverted triangle) or ISO (n = 5; diamond), CTX alone (n = 5; triangle), or were left untreated (n = 4; circle). Tumor volume was measured by caliper measurement. Data are expressed as mean ± SD (Repeated measures ANOVA). (g) Representative images of anti-human CD3 IHC staining (brown) of tumors excised from i.v.-treated mice, and the correspondent CD3+ cell density quantification (right panel). (h) Quantification of the CD3+CD56+ density (left panel) and CD3+CD56+CD16+ (right panel) of PPFE tumors from i.v. mouse model, analyzed by mIHC. Data are presented as mean ± SD (Student’s T-test). *P < .05; **P < .01; ***P < .001, ****P < .0001.
Mice treated with CIK+CTX or CIK+ISO showed a delay in tumor growth already 5 days after the beginning of the therapy, but only the combination of CIK cells with the tumor-specific antibody succeeded in completely arresting neoplastic development (Figure 2c). When mice were sacrificed, tumors were extracted and analyzed by IHC with anti-human CD3 to evaluate the immune cell infiltrate. The CD3 density was found significantly higher in PDX tissues from the CIK+CTX group (668 ± 673 cells/mm2) as compared to CIK+ISO counterpart (135 ± 134 cells/mm2; P = .0247) (Figure 2d). For a more accurate analysis of the phenotype of immune cells infiltrating the tumor and to quantify the density of CD3+CD56+ CIK cells, FFPE tissue sections of tumors were analyzed by mIHC. The density of CD3+CD56+ CIK cells in tumor tissues of mice receiving CIK+CTX appeared significantly more pronounced as compared to counterpart tissues from control mice treated with CIK+ISO (24.15 ± 23 vs 5.85 ± 8.4 cells/mm2, respectively; P < .0001) (Figure 2e, left panel). Moreover, most of these infiltrating CIK cells also expressed CD16 and their amount significantly differed from the density of CD3+CD56+CD16+ cells in the CIK+ISO control group (17.59 ± 19.8 vs 4.2 ± 7.9 cells/mm2, respectively; P < .0001) (Figure 2e, right panel).
To study the efficacy of CIK+CTX therapy according to a more clinically-compliant route of administration, the PDX was implanted in the fat pad of mice and the treatment was administered i.v. instead of i.t. In this case, the CIK+CTX combined treatment significantly delayed the tumor growth as compared to all other treatments. Interestingly, the efficacy of CIK+CTX combination was even more evident than the previous model, since the CIK+ISO treatment did not induce any apparent inhibition of tumor growth (figure 2f). As performed in mice treated intratumorally, at the end of the experiment the PDX tumors were analyzed by IHC for cell infiltrate. CIK+CTX therapy led to a higher infiltration of CD3+ cells (999 ± 395 cells/mm2), as compared to mice receiving CIK+ISO (340 ± 238 cells/mm2; P = .039) (Figure 2g). Moreover, PDX tissues from the CIK+CTX group appeared deeply infiltrated, whereas in mice treated with CIK+ISO the CD3+ component was mostly confined at the borders of the tumor mass (Figure 2g). Also in this model, the mIHC density of CD3+CD56+ CIK cells was significantly higher in tumor tissues of mice receiving CIK+CTX, as compared to animals treated with CIK+ISO (11.44 ± 8.2 vs 3.8 ± 6.66 cells/mm2, respectively; P = .0064) (Figure 2h, left panel). Additionally, the amount of the CD16+ component within the infiltrating CIK cells resulted more represented in tumor tissues from mice inoculated with CIK+CTX than CIK+ISO (6.6 ± 4.3 vs 3.1 ± 4.9 cells/mm2, respectively; P = .0356) (Figure 2h, right panel), as already observed in the i.t. mouse model. In FFPE tumor tissue sections from either models, NK cells were virtually absent.
CIK+CTX therapy impairs the development of experimental and spontaneous lung metastases
The PDX mouse model that we established did not disseminate to form distant metastases. Thus, to assess the ability of the CIK+CTX combined therapy to affect the metastatic process, we used a model employing the TNBC cell lines.
To mimic the formation of lung metastasis, MDA-MB-231_Luc cells were injected in the tail vein; 24 hours later, mice were treated daily for five days with i.v. injections of CIK+CTX, CIK+ISO, CTX alone, or were left untreated (Figure 3a). BLI monitoring disclosed that treatments significantly reduced tumor growth by week 3 (Figure 3b, upper panels), while in a single additional week only the CIK+CTX combination maintained a therapeutic effect as compared to CIK+ISO or CTX alone, and demonstrated to be capable of almost completely arresting metastasis outgrowth (Figure 3b, lower panels). When animals were sacrificed, lungs were analyzed by BLI showing that CIK with the irrelevant mAb exerted a therapeutic effect; nonetheless, the CIK+CTX group had a significantly lower signal, thus indicating a highly reduced metastasis growth in these mice (Figure 3c). Human cytokeratin staining fully confirmed the BLI results, as a reduced presence of human breast cancer cells was detected following the CIK+CTX treatment in comparison to the lungs from the other experimental groups (Figure 3d), with an overall tumor eradication of more than 95%. Additionally, even in this setting the analysis of the immune infiltrate revealed an increased density of CD3+ cells in the lungs of mice treated with CIK+CTX in comparison to animals receiving CIK+ISO (3768 ± 1650 vs 1787 ± 654 cells/mm2, respectively; P = .0109) (Figure 3e). Most importantly, the target-specific combined therapy significantly improved overall survival (P < .0001, Figure 3f); in particular, the CIK+CTX combination led to a longer median survival than the CIK+ISO treatment (median survival = 42 days vs 28 days; P = .0427), which in turn was partly effective when compared to untreated mice (median survival = 25 days; P = .0027).
Figure 3.
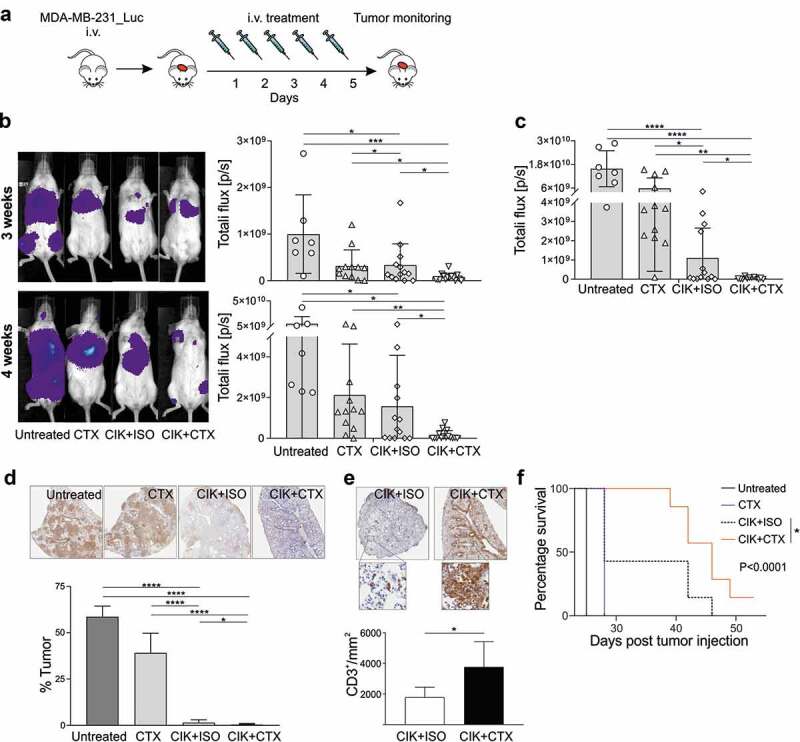
Assessment of CIK+CTX therapeutic activity against TNBC lung metastases. (a) The experimental protocol consisted in the i.v. inoculation of MDA-MB-231_Luc cells, followed the day after by the i.v. administration of CIK cells in combination with CTX (n = 15) or ISO (n = 13), or CTX alone (n = 12); control mice were left untreated (n = 7). (b) Metastasis growth was assessed weekly by BLI on all animals, and one representative mouse for each group is shown at week 3 (upper panel) and week 4 (lower panel) after tumor injection. Overall quantifications of BLI signals are reported as mean ± SD in the right histogram graphs, where symbols refer to individual mice. (c) At the sacrifice time, lungs were excised and BLI signals were quantified and reported as mean ± SD. (d) Representative images of anti-human cytokeratin IHC staining (brown) of lungs recovered from treated mice (upper panels), and the correspondent quantification of the percentage of tumor area (lower panel). Data are presented as mean ± SD. (e) Representative images of anti-human CD3 IHC staining (brown) of lungs as in D, and the correspondent CD3+ cell density quantification. Data are presented as mean ± SD. (f) Kaplan–Meier survival curves of one experiment involving MDA-MB-231_Luc tumor-bearing mice that received CIK+CTX (n = 9), CIK+ISO (n = 6), CTX alone (n = 6), or were left untreated (n = 3). Statistical analysis was performed using the Log-rank (Mantel-Cox) test. *P < .05; **P < .01; ***P < .001; ****P < .0001.
Although the described model effectively confirmed that the CTX antigen-specific mAb empowers the therapeutic activity of CIK cells, it did not recapitulate the naturally occurring steps of tumor cell dissemination from the primary tumor, and the spontaneous metastasis outgrowth at different distant sites. Hence, an orthotopic model was adopted that consisted in the injection into the mammary fat pad of MDA-MB-231_Luc cells, which are known to spontaneously develop distant metastases.15,16 One week after tumor injection, tumors became palpable and mice started i.v. daily treatments for five days (Figure 4a). Primary tumor growth was evaluated weekly by caliper measurement (Figure 4b), while metastasis formation was monitored by BLI (Figure 4c). As it occurred for the PDX model (figure 2f), the administration of CIK+CTX significantly delayed the growth of the tumor at the primary site, in contrast to the treatments with CTX alone or CIK+ISO that did not affect tumor growth in the fat pad (Figure 4b). On the other hand, at 4 weeks after tumor injection both the treatments with CIK+CTX and CIK+ISO were apparently able to restrain the development of distant metastases (Figure 4c, upper panels), but one week later only the treatment with CIK cells and CTX proved to be capable of almost completely controlling metastatic growth (Figure 4c, lower panels). At sacrifice, the lungs were analyzed by BLI confirming that the CIK+CTX group had a barely detectable BLI signal, which was significantly lower than all other groups (Figure 4d). This data was confirmed by the human cytokeratin staining of lungs where mice treated with CIK+CTX had a tumor elimination of more than 95% (Figure 4e). Additionally, mice left untreated or that received CIK+ISO or CTX alone had significantly larger homolateral axillary lymph nodes as compared to mice treated with CIK+CTX (Figure 4f). This superior therapeutic activity resulted in a significantly prolonged overall survival (P < .0001); indeed, mice treated with CIK+CTX performed better than the control group receiving CIK+ISO (median survival = 52 days vs 39 days; P = .012) (Figure 4g), which nonetheless was partly effective versus the untreated animals (median survival = 32 days; P = .0085).
Figure 4.
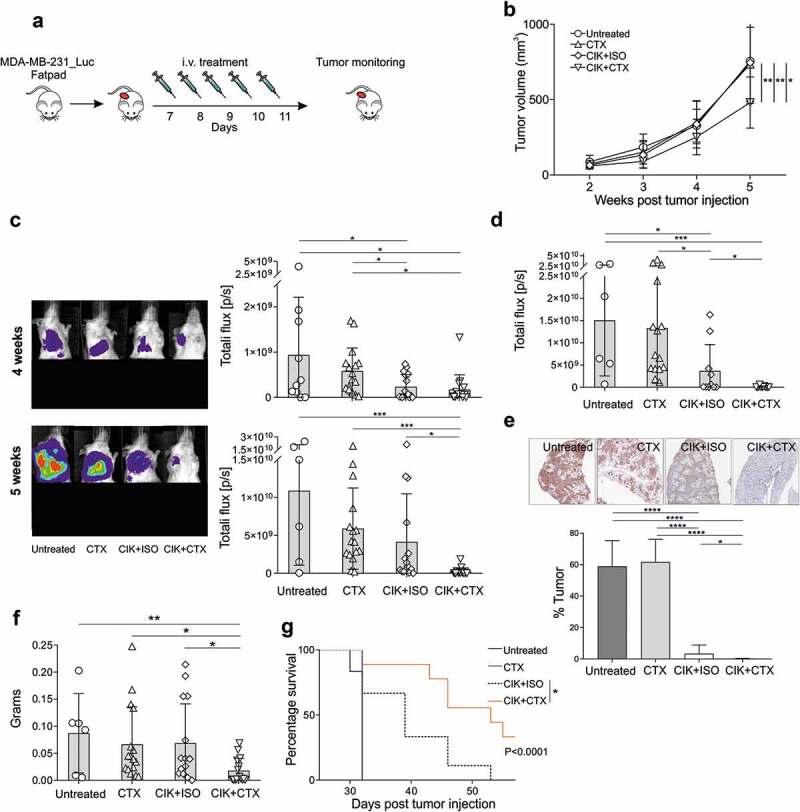
Assessment of CIK+CTX therapeutic activity against TNBC primary tumor and metastases. (a) The experimental protocol consisted in the fat pad injection of MDA-MB-231_Luc cells, followed one week after by the i.v. administration of CIK cells in combination with CTX (n = 16) or ISO (n = 16), or CTX alone (n = 16); control mice were left untreated (n = 10). (b) The tumor growth was monitored weekly by caliper measurement and data are presented as mean ± SD (Repeated measures ANOVA). (c) Metastasis growth was assessed weekly by BLI on all animals, and one representative mouse for each group is shown at week 4 (upper panel) and week 5 (lower panel) after tumor injection. Overall quantifications of BLI signals are reported as mean ± SD in the right histogram graphs, where symbols refer to individual mice. Here, the lower portion of each animal was covered before reimaging to minimize the bioluminescent signal from the primary tumor. (d) At the sacrifice time, lungs were excised and BLI signals were quantified and reported as mean ± SD, and stained for anti-human cytokeratin IHC (E, upper panels, brown) where the percentage of tumor area was quantified (lower panel). Data are presented as mean ± SD. (f) The lymph nodes were weighted, as considered as a sign of distant metastatic colonization, and data are presented as mean ± SD. (g) Kaplan–Meier survival curves of one experiment involving MDA-MB-231_Luc tumor-bearing mice that received CIK+CTX (n = 9), CIK+ISO (n = 9), CTX alone (n = 9), or were left untreated (n = 6). Statistical analysis was performed using the Log-rank (Mantel-Cox) test. *P < .05; **P < .01; ***P < .001; ****P < .0001.
Overall, data confirm the ability of CIK+CTX therapy to both control tumor growth at the primary site and arrest metastases formation at different distant organs, such as lymph nodes and lungs.
CIK cells and CTX combination acts as an excellent adjuvant therapy to limit metastatic spread after the removal of the primary tumor
To study the impact of CIK+CTX therapy on the metastatic growth after surgical removal of the primitive cancer, as it occurs in the clinical practice, MDA-MB-231_Luc cells were injected in the mammary fat pad and ten days later the primary tumor mass was surgically excised. The next day, mice were treated i.v. daily for five days with CIK+CTX, CIK+ISO, CTX alone, or were left untreated (Figure 5a). At the end of treatment, BLI imaging showed a comparable signal from metastases in the lymph nodes and in the lungs among all experimental groups. Nonetheless, in one additional week the signal from mice left untreated or receiving CIK+ISO or CTX alone markedly increased, while it appeared significantly more controlled in animals treated with CIK+CTX (Figure 5b). The density of CD3+CD56+ CIK cells in FFPE lungs sections analyzed by mIHC (Figure 5c, left panel) of mice receiving CIK+CTX was significantly higher as compared to counterpart tissues from control mice treated with CIK+ISO (76 ± 90 vs 34 ± 65 cells/mm2, respectively) (Figure 5c, middle panel). Moreover, most of these infiltrating CIK cells also expressed CD16 and their amount significantly differed from the density of CD3+CD56+CD16+ cells in the CIK+ISO control group (42.31 ± 53 vs 16.10 ± 17 cells/mm2, respectively; P < .0001) (Figure 5c, right panel). As observed in the other models, residual NK cells could not be detected. The efficacy of CIK+CTX to arrest the metastatic process was confirmed by the prolonged overall survival (P < .0001). Mice receiving this treatment had indeed a benefit in comparison to control animals administered with CIK+ISO (median survival = 51 days vs 42 days; P = .0016) (Figure 5d), which in turn disclosed a therapeutic activity over the untreated mice (median survival = 35 days; P = .0244).
Figure 5.
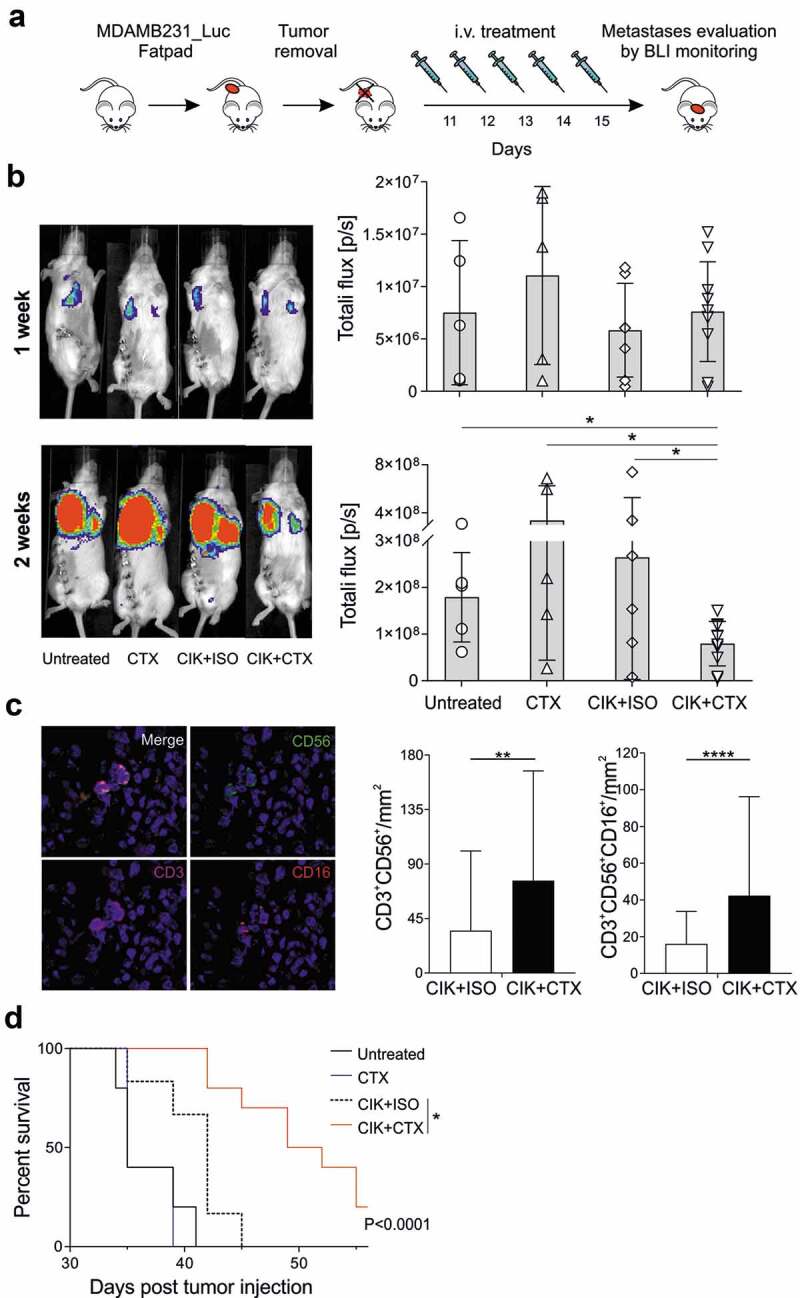
Inhibition of lung metastases development by CIK+CTX therapy. (a) The experimental protocol consisted in the fat pad injection of MDA-MB-231_Luc cells, followed ten days after by tumor removal. On the next day (day 11) mice were i.v. treated with CIK cells in combination with CTX (n = 10) or ISO (n = 6), or CTX alone (n = 5); control mice were left untreated (n = 5). (b) Metastasis growth was assessed weekly by BLI on all animals, and one representative mouse for each group is shown at week 1 (upper panel) and week 2 (lower panel) after surgery. Overall quantifications of BLI signals are reported as mean ± SD in the right histogram graphs, where symbols refer to individual mice. (c) Representative images (x20, left panels) of paraffine embedder lungs, stained with CD3 (magenta), CD56 (green), CD16 (red), pan-cytokeratin (orange) and nuclei (blue), analyzed by mIHC. The correspondent quantification of the CD3+CD56+ and CD3+CD56+CD16+ densities (right panels) are presented as mean ± SD. (d) Kaplan–Meier survival curves of experimental mice. Statistical analysis was performed using the Log-rank (Mantel-Cox) test. *P < .05; **P < .01; ***P < .001; ****P < .0001.
CD16 engagement by tumor-specific mAb leads to CIK cell activation and increased CD25 expression
Results of mIHC indicate that the engagement of CD16 by a tumor-targeting mAb is followed by an enrichment of the CD3+CD56+CD16+ component in the infiltrate; this is not observed with an irrelevant mAb. Based on the hypothesis that CD16 engagement may be involved in CIK cell activation and it may provide a proliferative advantage, in vitro experiments were set up in which MDA-MB-231 cells were co-cultured with CIK cells in the presence of CTX or ISO. After 4 hours of incubation, CIK cell intracellular staining showed a comparable expression of IL-2 between all groups (Figure 6a, left panel), and also within the CD3+CD56+CD16+ fraction (data not shown). This data was confirmed by measuring the IL-2 released after 24 hours in the cell supernatants, where the IL-2 amount detected was only slightly but not significantly higher in the CIK+CTX group, as compared to all other groups (Figure 6a, right panel). Overall, these results are suggestive of a constitutive synthesis of the cytokine by CIK cells that is not further fostered by CD16a engagement. On the other hand, previous reports indicate that CD16a stimulation in NK cells is followed by a positive modulation of the high affinity IL-2 receptor (CD25), leading us to assess this matter in our CIK cell co-cultures. Overall, CD25 expression and intensity tended to increase in CD3+CD56+ CIK cells upon CTX addition, although not significantly (Figure 6b). However, the expression and intensity of the high affinity IL-2 receptor turned out clearly and significantly upregulated in the CD3+CD56+CD16+ fraction (Figure 6c), conversely to what occurred in the CD16 negative CIK cell subset (Figure 6d), where CD25 remained comparable between the groups. The expression of the other two IL-2 receptor chains (CD122 and CD132) did not modify irrespective of the presence of either CTX or ISO (Figure 6e,f).
Figure 6.

Assessment of IL-2 production and IL-2 receptors expression in CIK cells upon tumor-mAb interaction. (a) CIK cells were co-cultured with MDA-MB-231 cells at an E/T of 10:1 in the presence of CTX or ISO, and IL-2 was measured either after 4 hours by flow cytometry (left panel), or after 24 hours in the culture supernatant (right panel). Figure legends apply also to all other panels. (b-f) At 24 hours of co-culture, CIK cells were analyzed by flow cytometry for the expression of IL-2 receptors (CD25, CD122 and CD132). The percentage (left panel) and the Mean Fluorescence intensity (MFI, right panel) of CD25 expression in (b) CD3+CD56+, (c) CD3+CD56+CD16+, (d) CD3+CD56+CD16− cells are shown as mean ± SD of six independent expansion from distinct healthy donors; *P < .05. In (C), one representative dot plot graph and histogram are shown. Expression of (e) CD122 and (f) CD132 on CD3+CD56+CD16+ cells in the presence of either CTX or ISO.
Discussion
Triple negative breast cancer is an aggressive tumor that tends to affect younger women, undergoes early metastatization, and carries a poor prognosis. Despite the promise of recent pharmacological advancements with new drugs, such as poly (ADP-ribose) polymerase (PARP) inhibitors and immune checkpoint inhibitors, most TNBC metastatic patients remain incurable and hence new therapeutic options are urgently required.
Adoptive cell therapy exploits cells from the immune system to kill cancer. Promising results with ACT have been achieved in metastatic breast cancer patients, where tumor-reactive memory T cells from the bone marrow were activated with dendritic cells pulsed with MCF-7 lysates, expanded and infused in patients who failed other treatments.17 Other ACT therapies, including the novel genetic redirection of T cells following the engineering with TCR or CAR receptors, are being explored to assess the potential activity and safety in TNBC patients.18,19 Recently, indeed, a number of clinical trials are recruiting TNBC patients to assess the potential activity and safety of anti-mesothelin or anti-MUC118 CAR-T cell infusions. Despite the striking outcomes achieved with the infusion of CD19 CAR-T cells in hematological malignances,3 the experience in solid tumors has not yet produced the same encouraging results, due to the difficulties of how to select the appropriate target, to address T cells to the tumor and/or metastases, and to reduce the risk of toxicities. Moreover, the laborious and expensive production of these effector cells is an important obstacle for ACT therapies.
CIK cells can represent an alternative effector population for immunotherapy interventions, because they are easily expandable, safe and relatively inexpensive, as they only require GMP-grade cytokines to obtain a very high number of cytotoxic cells, without any genetic manipulation.20 In the last years, several clinical trials have investigated the capacity of CIK cells to treat different hematological malignancies, and their therapeutic role has been also confirmed against numerous histotypes of solid tumors, such as colorectal, renal, pancreatic, ovarian and lung cancer.21,22 However, few reports, demonstrated an enhancement of the immune function and an improvement of survival time of TNBC cancer patients after treatment with CIK cells in combination with chemotherapy/radiotherapy.6,9 CIK cells present a donor-dependent expression of CD16a, and can be retargeted with clinical-grade mAb to exert potent ADCC in vitro against several tumor histotypes, including TNBC. Data highlighted that there is no correlation between the expression level of CD16a and the cytotoxicity outcome, and that the ADCC is accountable to the CD3+CD56+CD16+ subpopulation and not to the residual NK cells or TCR γ/δ subset.10 This therapeutic concept is exploited here as a novel option to treat primary and metastatic TNBC. The advantages of this combination therapy employs a clinical-grade mAb already widely used in clinics, such as cetuximab, whose usage could now find a much wider range of application than the registered therapeutic indications, because it is mainly exploited for its targeting and opsonizing properties and the capacity to trigger ADCC phenomena upon engagement of CD16a expressing cells.23 In this regard, preclinical in vivo studies in TNBC-bearing mice demonstrated the ability of NK cells combined with a bispecific antibody engaging a tumor antigen and CD16a to increase the overall survival by an ADCC approach.24 Another advantage of this strategy is based on a cytotoxic cell subset that per se is devoid of antigen specificity but can be easily converted into a tumor-specific effector population by the simple combination with a mAb, without requiring complex genetic manipulations. Since both CIK cells and cetuximab have been already widely tested in patients, their combination presents a potential immediate transferability to the clinical setting. The same concept could apply to other clinical-grade mAbs.
To prove the value of the CIK plus mAb therapeutic concept, we decided to mainly focus on an increasingly complex series of in vivo approaches, in order to provide a growing body of evidence of functionality and efficacy against both primary and metastatic TNBC tumors, to support and promote its clinical translation. Therefore, our initial approach tested the combination therapy in a subcutaneous PDX model upon intratumoral or intravenous treatment. Currently, PDXs represent an emerging model for a more reliable assessment of tumor heterogeneity and mechanisms of therapy response and/or resistance,25 because they are available as phenotypically stable and renewable tissues.26 They recapitulate many aspects of the tumor biology and are excellent models for translational research.26 In this setting, antigen-specific retargeting by CTX enhanced CIK cell therapeutic activity not only after local but also systemic administration. In addition, results clearly evidenced the tumor-homing properties of CIK cells and their capacity to infiltrate solid tumors. More interestingly, the combination with the antigen-specific antibody, but not the irrelevant one, led to a more robust infiltration with a CD3+ T cell component that also involved an enrichment in both CD3+CD56+ and CD3+CD56+CD16+ CIK cells, as assessed by co-staining in mIHC.
However, PDXs rarely metastasize and hence this precludes the evaluation of a therapeutic approach against spontaneously arising metastases. Therefore, we moved to models based on the MDA-MB-231 cell line that is endowed with the capacity to establish experimental lung metastases upon i.v. injection, but more importantly to behave physiologically after orthotopic fat pad implantation, giving rise to spontaneous metastases in the lymph nodes and lungs irrespective of the presence or the excision of the primary tumor.16
Also, in all these different contexts, the CIK cells plus antigen-specific mAb combination outperformed results obtained when an irrelevant antibody was used, and proved therapeutic against not only the primary tumor but also distant metastases. In particular, the target-specific combined therapy delayed the experimental or natural metastasis dissemination in the lymph nodes and lungs, and significantly improved the survival. Moreover, and as already observed for the subcutaneous PDX model, the combination therapy promoted CIK cell infiltration also in lungs; in particular, the CD3+CD56+CD16+ component significantly increased when CTX was used, thus suggesting that CD16 engagement by a mAb specifically recognizing the surrounding tumor cells favors the enrichment of the CIK cell subset that specifically bears the Fc binding receptor.
In this regard, both previous27,28 and recent24,29 studies demonstrated that CD16 is a signal-transducing molecule that, upon engagement, induces, in NK cells, the transcription of genes encoding cytokines (IFN-γ and TNF) and surface activation molecules, such as CD25. CD25 is the IL-2 receptor α-chain that interacts with the IL-2Rβ and IL-2Rγ subunits to form the high-affinity IL-2 receptor;30 CD25 overexpression may facilitate the efficacy of cell therapies through the creation of an extracellular reservoir and recycling of IL-2,31 and its upregulation correlates with the cytotoxic activity in NK and CIK cells.32,33 Here we demonstrated that a CD25 upregulation occurred exclusively in the CD3+CD56+CD16+ subset and only in the presence of the antigen-specific mAb that could explain the enrichment of the CD3+CD56+CD16+ fraction within both primary and metastatic tumors when CTX was used.
IL-2 is essential for CIK cell in vitro culture, but clinical trials demonstrated that CIK effector activity is independent of exogenous IL-2 administration,34-40 which is instead required to support the growth and survival of antitumor NK or T cells administered in vivo.41 This is a remarkable feature because it allows to completely eliminate the IL-2-related substantial toxicities, including nausea, hypertension, and pulmonary edema.42 Overall, such observation allows to advance a model where CIK cells can autocrinally sustain their growth in vivo through IL-2 release; on the other hand, when the CD3+CD56+CD16+ subset is engaged by a mAb binding an antigen on the tumor cell surface, specific signals can be delivered through the CD16 leading to upregulation of CD25 and hence the formation of the IL-2 high-affinity receptor. This in turn allows the CD3+CD56+CD16+ subset to increase responsiveness to IL-2 and consequent proliferation and survival, ultimately explaining the enrichment.
This study advances a new therapeutic strategy for incurable metastatic TNBC, which is based on the adoptive transfer of CIK cells in combination with cetuximab. Overall, we propose a new concept of ACT that is less challenging as compared to T cell genetic engineering, and in which the effector cells are obtained in high amounts using a simple and inexpensive expansion protocol, while antigen-specificity and triggering is conferred by the simple combination with a mAb already approved in clinics. Apart the immediate clinical transferability of this approach, the retargeting of CIK cells with antigen-specific mAbs can be regarded as a broad concept, which is valid and applicable to virtually any antibody and tumor histotype and can lead to therapeutic improvements in many different cancer settings.
Acknowledgments
We are grateful to Simone Zorzi and Mattia Bastianello for the technical help in the histology and mIHC analyses.
Funding Statement
The research leading to these results has received funding from Fondazione AIRC under IG 2018 –ID. 21354 project – P.I. Rosato Antonio; 5 per Mille 2019 - ID. 22759 program – P.I. Piccolo Stefano, G.L. Rosato Antonio; BIRD180331/18 from University of Padova to AR; BIGID219SOMM from 5 per Mille 2018, Veneto Institute of Oncology IOV-IRCCS to RS, and the Ministry of Health-Alliance Against Cancer (MoH-ACC) project “Research project on CAR T cells for hematological malignancies and solid tumors” to AR.
Declaration of interest statement
The authors declare that they have no competing interests.
References
- 1.Waks AG, Winer EP.. Breast cancer treatment: a review. JAMA. 2019. January;321(3):288–13. doi: 10.1001/jama.2018.19323. [DOI] [PubMed] [Google Scholar]
- 2.Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. N Engl J Med. 2010. November;363(20):1938–1948. doi: 10.1056/NEJMra1001389. [DOI] [PubMed] [Google Scholar]
- 3.Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, Bader P, Verneris MR, Stefanski HE, Myers GD, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N. Engl. J. Med. 2018. February;378(5):439–448. doi: 10.1056/NEJMoa1709866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lemal R, Tournilhac O. State-of-the-art for CAR T-cell therapy for chronic lymphocytic leukemia in 2019. J Immunother Cancer. 2019;7(1). doi: 10.1186/s40425-019-0686-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pievani A, Borleri G, Pende D, Moretta L, Rambaldi A, Golay J, Introna M. Dual-functional capability of CD3+CD56+ CIK cells, a T-cell subset that acquires NK function and retains TCR-mediated specific cytotoxicity. Blood. 2011;118(12):3301–3310. doi: 10.1182/blood-2011-02-336321. [DOI] [PubMed] [Google Scholar]
- 6.Pan K, Guan -X-X, Li Y-Q, Zhao -J-J, Li -J-J, Qiu H-J, Weng D-S, Wang Q-J, Liu Q, Huang L-X, et al. Clinical activity of adjuvant cytokine-induced killer cell immunotherapy in patients with post-mastectomy triple-negative breast cancer. Clin Cancer Res. 2014;20(11):3003–3011. doi: 10.1158/1078-0432.CCR-14-0082. [DOI] [PubMed] [Google Scholar]
- 7.Sangiolo D, Martinuzzi E, Todorovic M, Vitaggio K, Vallario A, Jordaney N, Carnevale-Schianca F, Capaldi A, Geuna M, Casorzo L, et al. Alloreactivity and anti-tumor activity segregate within two distinct subsets of cytokine-induced killer (CIK) cells: implications for their infusion across major HLA barriers. Int. Immunol. 2008. July;20(7):841–848. doi: 10.1093/intimm/dxn042. [DOI] [PubMed] [Google Scholar]
- 8.Schmeel LC, Schmeel FC, Coch C, Schmidt-Wolf IGH. Cytokine-induced killer (CIK) cells in cancer immunotherapy: report of the international registry on CIK cells (IRCC). J Cancer Res Clin Oncol. 2015. May;141(5):839–849. doi: 10.1007/s00432-014-1864-3. [DOI] [PubMed] [Google Scholar]
- 9.Zhou Z-Q, Zhao -J-J, Pan Q-Z, Chen C-L, Liu Y, Tang Y, Zhu Q, Weng D-S, Xia J-C. PD-L1 expression is a predictive biomarker for CIK cell-based immunotherapy in postoperative patients with breast cancer. J Immunother Cancer. 2019. December;7(1):228. doi: 10.1186/s40425-019-0696-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cappuzzello E, Tosi A, Zanovello P, Sommaggio R, Rosato A. Retargeting cytokine-induced killer cell activity by CD16 engagement with clinical-grade antibodies. Oncoimmunology. 2016. August;5(8):e1199311. doi: 10.1080/2162402X.2016.1199311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ciardiello F, Tortora G. EGFR antagonists in cancer treatment. N Engl J Med. 2008;358(11):1160–1174. doi: 10.1056/NEJMra0707704. [DOI] [PubMed] [Google Scholar]
- 12.da Silva JL, Cardoso Nunes NC, Izetti P, de Mesquita GG, de Melo AC. Triple negative breast cancer: a thorough review of biomarkers. Crit Rev Oncol Hematol. Elsevier Ireland Ltd. 2020;145:102855. doi: 10.1016/j.critrevonc.2019.102855. [DOI] [PubMed] [Google Scholar]
- 13.Keyaerts M, Verschueren J, Bos TJ, Tchouate-Gainkam LO, Peleman C, Breckpot K, Vanhove C, Caveliers V, Bossuyt A, Lahoutte T, et al. Dynamic bioluminescence imaging for quantitative tumour burden assessment using IV or IP administration of d-luciferin: effect on intensity, time kinetics and repeatability of photon emission. Eur J Nucl Med Mol Imaging. 2008. May;35(5):999–1007. doi: 10.1007/s00259-007-0664-2. [DOI] [PubMed] [Google Scholar]
- 14.Schoos A, Gabriel C, Knab VM, Fux DA.. Activation of HIF-1α by δ-Opioid Receptors Induces COX-2 Expression in Breast Cancer Cells and Leads to Paracrine Activation of Vascular Endothelial Cells. J Pharmacol Exp Ther. 2019;370(3):480-489. doi: 10.1124/jpet.119.257501. [DOI] [PubMed] [Google Scholar]
- 15.Tosi A, Dalla Santa S, Cappuzzello E, Marotta C, Walerich D, Del Sal G, Zanovello P, Sommaggio R, Rosato A, . Identification of a HLA-A*0201-restricted immunogenic epitope from the universal tumor antigen DEPDC1. Oncoimmunology. 2017. August;6(8):e1313371. doi: 10.1080/2162402X.2017.1313371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pegoraro S, Ros G, Piazza S, Sommaggio R, Ciani Y, Rosato A, Sgarra R, Del Sal G, Manfioletti G.. HMGA1 promotes metastatic processes in basal-like breast cancer regulating EMT and stemness.. Oncotarget. 2013. August;4(8):1293–1308. doi: 10.18632/oncotarget.1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Domschke C, Ge Y, Bernhardt I, Schott S, Keim S, Juenger S, Bucur M, Mayer L, Blumenstein M, Rom J, et al. Long-term survival after adoptive bone marrow T cell therapy of advanced metastasized breast cancer: follow-up analysis of a clinical pilot trial. Cancer Immunol. Immunother. 2013. June;62(6):1053–1060. doi: 10.1007/s00262-013-1414-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gallo S, Sangiolo D, Carnevale Schianca F, Aglietta M, Montemurro F. Treating breast cancer with cell-based approaches: an overview. Expert Opin Biol Ther. 2017;17(10):1255–1264. doi: 10.1080/14712598.2017.1356816. [DOI] [PubMed] [Google Scholar]
- 19.Bajgain P, Tawinwung S, D’Elia L, Sukumaran S, Watanabe N, Hoyos V, Lulla P, Brenner MK, Leen AM, Vera JF, et al. CAR T cell therapy for breast cancer: harnessing the tumor milieu to drive T cell activation. J Immunother Cancer. 2018;6(1). doi: 10.1186/s40425-018-0347-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Introna M, Franceschetti M, Ciocca A, Borleri G, Conti E, Golay J, Rambaldi A. Rapid and massive expansion of cord blood-derived cytokine-induced killer cells: an innovative proposal for the treatment of leukemia relapse after cord blood transplantation. Bone Marrow Transplant. 2006. November;38(9):621–627. doi: 10.1038/sj.bmt.1705503. [DOI] [PubMed] [Google Scholar]
- 21.Giraudo L, Gammaitoni L, Cangemi M, Rotolo R, Aglietta M, Sangiolo D. Cytokine-induced killer cells as immunotherapy for solid tumors: current evidence and perspectives. Immunotherapy. 2015. September;7(9):999–1010. doi: 10.2217/imt.15.61. [DOI] [PubMed] [Google Scholar]
- 22.Wang M, Cao J-X, Pan J-H, Liu Y-S, Xu B-L, Li D, Zhang X-Y, Li J-L, Liu J-L, Wang H-B, et al. Adoptive immunotherapy of cytokine-induced killer cell therapy in the treatment of non-small cell lung cancer. PLoS One. 2014;9(11):e112662. doi: 10.1371/journal.pone.0112662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Monteverde M, Milano G, Strola G, Maffi M, Lattanzio L, Vivenza D, Tonissi F, Merlano M, Lo Nigro C. The relevance of ADCC for EGFR targeting: a review of the literature and a clinically-applicable method of assessment in patients.. Crit. Rev. Oncol. Hematol. 2015. August;95(2):179–190. doi: 10.1016/j.critrevonc.2015.02.014. [DOI] [PubMed] [Google Scholar]
- 24.Del Bano J, Florès-Florès R, Josselin E, Goubard A, Ganier L, Castellano R, Chames P, Baty D, Kerfelec B.. A bispecific antibody-based approach for targeting mesothelin in triple negative breast cancer. Front. Immunol. 2019;10:1593. doi: 10.3389/fimmu.2019.01593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dobrolecki LE, Airhart SD, Alferez DG, Aparicio S, Behbod F, Bentires-Alj M, Brisken C, Bult CJ, Cai S, Clarke RB, et al. Patient-derived xenograft (PDX) models in basic and translational breast cancer research. Cancer Metastasis Rev. 2016;35(4):547–573. doi: 10.1007/s10555-016-9653-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meehan TF, Conte N, Goldstein T, Inghirami G, Murakami MA, Brabetz S, Gu Z, Wiser JA, Dunn P, Begley DA, et al. PDX-MI: minimal information for patient-derived tumor xenograft models. Cancer Res. 2017;77(21):e62–e66. doi: 10.1158/0008-5472.CAN-17-0582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Anegón I, Cuturi MC, Trinchieri G, Perussia B. Interaction of Fc receptor (CD16) ligands induces transcription of interleukin 2 receptor (CD25) and lymphokine genes and expression of their products in human natural killer cells. J Exp Med. 1988. February;167(2):452–472. doi: 10.1084/jem.167.2.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Harris DT, Travis WW, Koren HS. Induction of activation antigens on human natural killer cells mediated through the Fc-gamma receptor.. J Immunol. 1989 Oct 1;143(7):2401–2406 [PubMed] [Google Scholar]
- 29.Duggan MC, Campbell AR, McMichael EL, Opheim KS, Levine KM, Bhave N, Culbertson MC, Noel T, Yu L, Carson WE, et al. Co-stimulation of the fc receptor and interleukin-12 receptor on human natural killer cells leads to increased expression of cd25. Oncoimmunology. 2018;7(2):e1381813. doi: 10.1080/2162402X.2017.1381813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Spolski R, Li P, Leonard WJ.. Biology and Regulation of IL-2: From Molecular Mechanisms to Human Therapy. Nat Rev Immunol. 2018. October;18(10):648-659. doi: 10.1038/s41577-018-0046-y. [DOI] [PubMed] [Google Scholar]
- 31.Su EW, Moore CJ, Suriano S, Johnson CB, Songalia N, Patterson A, Neitzke DJ, Andrijauskaite K, Garrett-Mayer E, Mehrotra S, et al. IL-2Rα mediates temporal regulation of IL-2 signaling and enhances immunotherapy. Sci Transl Med. 2015;7(311):311ra170–311ra170. doi: 10.1126/scitranslmed.aac8155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rudnicka K, Matusiak A, Chmiela M. CD25 (IL-2R) expression correlates with the target cell induced cytotoxic activity and cytokine secretion in human natural killer cells. Acta Biochim. Pol. 2015;62(4):885–894. doi: 10.18388/abp.2015_1152. [DOI] [PubMed] [Google Scholar]
- 33.Bremm M, Pfeffermann L-M, Cappel C, Katzki V, Erben S, Betz S, Quaiser A, Merker M, Bonig H, Schmidt M, et al. Improving clinical manufacturing of IL-15 activated Cytokine-Induced Killer (CIK) cells. Front Immunol. 2019;10:1218. doi: 10.3389/fimmu.2019.01218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Linn Y-C, Yong H-X, Niam M, Lim T-J, Chu S, Choong A, Chuah C, Goh Y-T, Hwang W, Loh Y, et al. A phase I/II clinical trial of autologous cytokine-induced killer cells as adjuvant immunotherapy for acute and chronic myeloid leukemia in clinical remission. Cytotherapy. 2012;14(7):851–859. doi: 10.3109/14653249.2012.694419. [DOI] [PubMed] [Google Scholar]
- 35.Introna M, Borleri G, Conti E, Franceschetti M, Barbui AM, Broady R, Dander E, Gaipa G, D’Amico G, Biagi E, et al. Repeated infusions of donor-derived cytokine-induced killer cells in patients relapsing after allogeneic stem cell transplantation: a phase I study1. Haematologica. 2007;92(7):952–959. doi: 10.3324/haematol.11132. [DOI] [PubMed] [Google Scholar]
- 36.Linn Y-C, Niam M, Chu S, Choong A, Yong H-X, Heng -K-K, Hwang W, Loh Y, Goh Y-T, Suck G, et al. The anti-tumour activity of allogeneic cytokine-induced killer cells in patients who relapse after allogeneic transplant for haematological malignancies. Bone Marrow Transplant. 2012;47(7):957–966. doi: 10.1038/bmt.2011.202. [DOI] [PubMed] [Google Scholar]
- 37.Introna M, Gotti E, Bonzi M, Golay J, Algarotti A, Micò C, Grassi A, Boschini C, Ferrari ML, Cavattoni I, et al. A multicenter Phase II study of sequential administration of unmanipulated DLI and donor derived Cytokine Induced Killer (CIK) cells in HSCT patients, relapsed of disease. Blood. 2015;126(23):3160. doi: 10.1182/blood.V126.23.3160.3160. [DOI] [Google Scholar]
- 38.Schmidt-Wolf IG, Negrin RS, Kiem HP, Blume KG, Weissman IL. Use of a SCID mouse/human lymphoma model to evaluate cytokine-induced killer cells with potent antitumor cell activity. J Exp Med. 1991;174(1):139–149. doi: 10.1084/jem.174.1.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lu PH, Negrin RS. A novel population of expanded human CD3+CD56+ cells derived from T cells with potent in vivo antitumor activity in mice with severe combined immunodeficiency. J Immunol. 1994;153:1687–1696. [PubMed] [Google Scholar]
- 40.Nishimura R, Baker J, Beilhack A, Zeiser R, Olson JA, Sega EI, Karimi M, Negrin RS. In vivo trafficking and survival of cytokine-induced killer cells resulting in minimal GVHD with retention of antitumor activity. Blood. 2008;112(6):2563–2574. doi: 10.1182/blood-2007-06-092817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rosenberg SA. IL-2: the first effective immunotherapy for human cancer. J. Immunol. 2014;192(12):5451–5458. doi: 10.4049/jimmunol.1490019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Arenas-Ramirez N, Woytschak J, Boyman O. Interleukin-2: biology, design and application. Trends Immunol. 2015. December;36(12):763–777. doi: 10.1016/j.it.2015.10.003. [DOI] [PubMed] [Google Scholar]