

ABSTRACT
Type I interferon (IFN) release by irradiated cancer cells is paramount for radiation therapy to elicit anticancer immunity. Our findings demonstrate that mitochondrial outer membrane permeabilization (MOMP) triggered by RT enables exposure of mitochondrial DNA to the cytosol, hence setting off CGAS-driven type I IFN synthesis. These data point to the existence of a therapeutically actionable mitochondrial checkpoint that restricts innate immune signaling in irradiated cancer cells.
KEYWORDS: Abscopal responses, autophagy, caspases, immunogenic cell death, micronuclei, STING, venetoclax
Accumulating preclinical and clinical evidence suggests that the ability of radiation therapy (RT) to mediate robust anticancer effects with local and system outreach largely depends on the (re-)activation of tumor-targeting immune responses orchestrated by type I interferon (IFN).1 Although dendritic cells are expected to be the main producers of type I interferon (IFN) in the irradiated tumor microenvironment,2 previous results from the Demaria lab elegantly demonstrated that systemic anticancer immune responses elicited by RT require type I IFN secretion by cancer cells.3 In line with this notion, mouse mammary carcinoma TSA cells engineered with an inducible construct for the depletion of cyclic GMP-AMP synthase (CGAS) or stimulator of interferon response cGAMP interactor 1 (STING1), two key components of the molecular machinery for type I IFN secretion downstream of cytosolic double-stranded (ds)DNA sensing,4 lose their capacity to initiate anticancer immune responses with systemic outreach upon focal irradiation.3 In this context, micronuclei were considered as the main source of CGAS-activating dsDNA,5 at least in part reflecting the ability of RT to cause DNA damage associated with mitotic failure and micronucleation.6 However, chromatin (i.e., nuclear dsDNA complexed with histones) appears to be a poor CGAS activator.7 Our recent findings suggest that mitochondrial DNA (mtDNA) exposed to the cytosol of cancer cells undergoing mitochondrial outer membrane permeabilization (MOMP) in response to clinically relevant RT doses is a potent driver of type I IFN secretion,8 pointing to the existence of a mitochondrial immune checkpoint that can be therapeutically actioned to boost the immunogenicity of RT.
Inspired by an abundant literature on the ability of autophagy (an evolutionary conserved mechanism that disposes of potentially dangerous cytosolic entities through lysosomal degradation) to limit type I IFN secretion in a variety of settings,9 we set out to investigate the impact of autophagy on innate immune signaling by irradiated cancer cells. In line with previous results, we found that TSA cells lacking either of the key autophagy genes Atg5 or Atg7, as well as TSA cells exposed to the lysosomal inhibitor hydroxychloroquine (HCQ), secrete increased levels of type I IFN downstream of CGAS-STING1 signaling upon exposure to a single RT dose of 8 Gy, correlating with the accrued accumulation of cytosolic dsDNA. Moreover, autophagy-incompetent TSA lesions established in immunocompetent syngeneic mice not only exhibited intact (if not increased) sensitivity to a single RT dose of 20 Gy, but also were more proficient than their autophagy-proficient counterparts at eliciting a type I IFN-dependent, tumor-targeting immune response with systemic outreach when subjected to focal RT in the presence of an otherwise systemically inactive immune checkpoint blocker (ICB).8
Next, we focused on the identity of dsDNA species accumulating in the cytosol of TSA cells 24–48 hours after a single RT dose of 8 Gy. We found that the dsDNA molecules accruing in the cytosol of irradiated TSA cells do not colocalize with (micro)nuclear markers such as lamin B (LMNB), but exist in the close proximity of mitochondrial markers such as cytochrome c oxidase subunit 4I1 (COX4I1), and co-localize with the mitochondrial transcription factor transcription factor A, mitochondrial (TFAM). Moreover, TSA cells depleted of mtDNA upon long-term exposure to ethidium bromide (so-called rho0 cells) largely failed to secrete type I IFN upon irradiation, as they exhibited little cytosolic dsDNA accumulation. Further corroborating the implication of MOMP in this process, both TSA cells engineered to express BCL2 apoptosis regulator (BCL2, an endogenous MOMP inhibitor) and human colorectal carcinoma Bax−/- HCT 116 cells (which lack a key MOMP executor) were less proficient than their control counterparts at accumulating cytosolic dsDNA and secreting type I IFN upon irradiation.8 These findings, which are in line with previous results from the Kile and Vaux groups based on the experimental, relatively nonspecific BCL2 inhibitor ABT-737,10,11 point to the existence of a BCL2-dependent mitochondrial checkpoint that restrict innate immune signaling in cancer cells responding to RT at clinically relevant doses.
Importantly, MOMP generally enables the release of the mitochondrial protein cytochrome c, somatic (CYCS) into the cytosol, hence driving the sequential activation of caspase 9 (CASP9) and CASP3 to precipitate apoptotic cell death.12 Both CASP9 and CASP3 have been shown to mediate a plethora of immunosuppressive effects, encompassing the inhibition of type I IFN release13-15 upon CGAS cleavage16 as well as the exposure of immunosuppressive signals including CD274 (best known as PD-L1)17 and phosphatidylserine on the cell surface.18,19 In line with this notion, type I IFN secretion by cancer cells exposed to ABT-737, which experience massive caspase activation, can only be documented upon pharmacological or genetic caspase inhibition. However, TSA cells responding to clinically relevant RT doses of 8–20 Gy do not experience robust caspase activation, and their clonogenic survival after irradiation is indeed insensitive to the presence of the pan-caspase inhibitor Z-VAD-fmk or the deletion of Casp3.15 Moreover, the ability of RT to trigger type I IFN secretion (and anticancer immune responses with systemic outreach) has been documented for various caspase-proficient tumors beyond the TSA model.3,17 That said, we have previously documented that Casp3−/- TSA cells,15 but not wild-type TSA cells exposed to Z-VAD-fmk (unpublished observations), exhibit superior type I IFN secretion and cytosolic dsDNA accumulation not only in response to RT doses that associated with minimal caspase activation, but also at baseline.15 Thus, CASP3 may also limit type I IFN secretion by mechanisms that dot not rely on its proteolytic activity.
Irrespective of these and other open issues, our most recent findings delineate the existence of a mitochondrial immune checkpoint that limit the ability of irradiated cells to secrete type I IFN and initiate therapeutically relevant immune responses. Importantly, the selective BCL2 inhibitor venetoclax is currently approved for the treatment of chronic lymphocytic leukemia,20 and has demonstrated promising clinical activity in patients with luminal breast cancer,21 suggesting that such the mitochondrial immune checkpoint (at odds with autophagy) may be clinically actionable (Figure 1). Clinical trials testing the safety and efficacy of focal RT plus venetoclax in breast cancer patients are urgently awaited.
Figure 1.
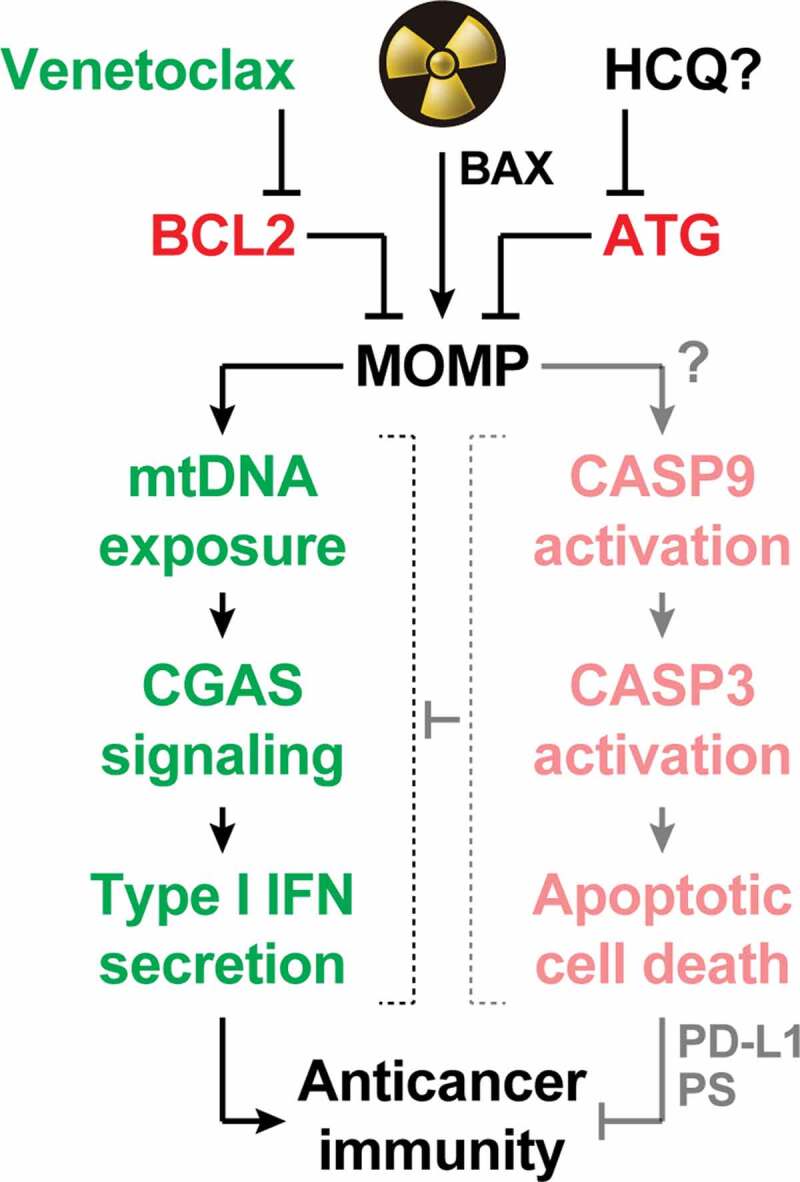
Mitochondrial apoptosis operates as a central rheostat to control the immunogenicity of irradiated cells. At least in some circumstances, radiation therapy (RT) can trigger abundant mitochondrial outer membrane permeabilization (MOMP) upon activation of BCL2 associated X, apoptosis regulator (BAX). In irradiated cells, MOMP is associated with the exposure of mitochondrial DNA (mtDNA) to the cytosol, which drives type I interferon (IFN) secretion via cyclic GMP-AMP synthase (CGAS). Conversely, it seems that RT-driven MOMP does not elicit potent caspase 9 (CASP9) and caspase 3 (CASP3) activation. This explains why type I IFN secretion and the consequent initiation of adaptive immune responses against irradiated cells has been documented in a variety of caspase-proficient models, despite the established capacity of active caspases to mediate immunosuppressive effects including CGAS cleavage, as well as CD274 (best known as PD-L1) and phosphatidylserine (PS) exposure. Both autophagy (ATG) and the anti-apoptotic protein BCL2 apoptosis regulator (BCL2) potently inhibit MOMP and its immunological consequences, delineating potential strategies for enhancing the immunogenicity (and hence the efficacy) of RT. While the safety and efficacy of ATG suppression with systemic hydroxychloroquine (HCQ) remain unclear, BCL2 inhibition with the clinically approved agent venetoclax stands out as a promising approach to clinical translation.
Funding Statement
The LG lab is supported by a Breakthrough Level 2 grant from the US Department of Defense (DoD), Breast Cancer Research Program (BRCP) (#BC180476P1), by the 2019 Laura Ziskin Prize in Translational Research (#ZP-6177, PI: Formenti) from the Stand Up to Cancer (SU2C), by a Mantle Cell Lymphoma Research Initiative (MCL-RI, PI: Chen-Kiang) grant from the Leukemia and Lymphoma Society (LLS), by a startup grant from the Department of Radiation Oncology at Weill Cornell Medicine (New York, US), by a Rapid Response Grant from the Functional Genomics Initiative (New York, US), by industrial collaborations with Lytix (Oslo, Norway) and Phosplatin (New York, US), and by donations from Phosplatin (New York, US), the Luke Heller TECPR2 Foundation (Boston, US) and Sotio a.s. (Prague, Czech Republic).
Disclosure of potential conflicts of interest
TY has no conflicts of interest to disclose. LG received consulting fees from OmniSEQ, Astra Zeneca, Inzen and the Luke Heller TECPR2 Foundation, and he is member of the Scientific Advisory Committee of Boehringer Ingelheim, The Longevity Labs and OmniSEQ.
References
- 1.Rodriguez-Ruiz ME, Vitale I, Harrington KJ, Melero I, Galluzzi L.. Immunological impact of cell death signaling driven by radiation on the tumor microenvironment. Nat Immunol. 2020;21:120–3. doi: 10.1038/s41590-019-0561-4. [DOI] [PubMed] [Google Scholar]
- 2.Sprooten J, Agostinis P, Garg AD. Type I interferons and dendritic cells in cancer immunotherapy. Int Rev Cell Mol Biol. 2019;348:217–262. [DOI] [PubMed] [Google Scholar]
- 3.Vanpouille-Box C, Alard A, Aryankalayil MJ, Sarfraz Y, Diamond JM, Schneider RJ, Inghirami G, Coleman CN, Formenti SC, Demaria S, et al. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat Commun. 2017;8(1):15618. doi: 10.1038/ncomms15618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vanpouille-Box C, Demaria S, Formenti SC, Galluzzi L. Cytosolic DNA sensing in organismal tumor control. Cancer Cell. 2018;34:361–378. doi: 10.1016/j.ccell.2018.05.013. [DOI] [PubMed] [Google Scholar]
- 5.Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, Greenberg RA. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature. 2017;548:466–470. doi: 10.1038/nature23470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McLaughlin M, Patin EC, Pedersen M, Wilkins A, Dillon MT, Melcher AA, Harrington KJ. Inflammatory microenvironment remodelling by tumour cells after radiotherapy. Nat Rev Cancer. 2020;20:203–217. doi: 10.1038/s41568-020-0246-1. [DOI] [PubMed] [Google Scholar]
- 7.Zierhut C, Yamaguchi N, Paredes M, Luo JD, Carroll T, Funabiki H. The cytoplasmic DNA sensor cGAS promotes mitotic cell death. Cell. 2019;178:302–15 e23. doi: 10.1016/j.cell.2019.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yamazaki T, Kirchmair A, Sato A, Buqué A, Rybstein M, Petroni G, Bloy N, Finotello F, Stafford L, Navarro Manzano E, et al. Mitochondrial DNA drives abscopal responses to radiation that are inhibited by autophagy. Nat Immunol. 2020. In press. [DOI] [PubMed] [Google Scholar]
- 9.Galluzzi L, Yamazaki T, Kroemer G. Linking cellular stress responses to systemic homeostasis. Nat Rev Mol Cell Biol. 2018;19:731–745. doi: 10.1038/s41580-018-0068-0. [DOI] [PubMed] [Google Scholar]
- 10.McArthur K, Whitehead LW, Heddleston JM, Li L, Padman BS, Oorschot V, Geoghegan ND, Chappaz S, Davidson S, San CH, et al. BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science. 2018:359:eaao6047. [DOI] [PubMed] [Google Scholar]
- 11.Lindqvist LM, Frank D, McArthur K, Dite TA, Lazarou M, Oakhill JS, Kile BT, Vaux DL. Autophagy induced during apoptosis degrades mitochondria and inhibits type I interferon secretion. Cell Death Differ. 2018;25:784–796. doi: 10.1038/s41418-017-0017-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Singh R, Letai A, Sarosiek K. Regulation of apoptosis in health and disease: the balancing act of BCL-2 family proteins. Nat Rev Mol Cell Biol. 2019;20:175–193. doi: 10.1038/s41580-018-0089-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rongvaux A, Jackson R, Harman CC, Li T, West AP, de Zoete MR, Wu Y, Yordy B, Lakhani S, Kuan C-Y, et al. Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell. 2014;159:1563–1577. doi: 10.1016/j.cell.2014.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.White MJ, McArthur K, Metcalf D, Lane RM, Cambier JC, Herold MJ, van Delft M, Bedoui S, Lessene G, Ritchie M, et al. Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell. 2014;159(7):1549–1562. doi: 10.1016/j.cell.2014.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rodriguez-Ruiz ME, Buque A, Hensler M, Chen J, Bloy N, Petroni G, Sato A, Yamazaki T, Fucikova J, Galluzzi L, et al. Apoptotic caspases inhibit abscopal responses to radiation and identify a new prognostic biomarker for breast cancer patients. Oncoimmunology. 2019;8(11):e1655964. doi: 10.1080/2162402X.2019.1655964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ning X, Wang Y, Jing M, Sha M, Lv M, Gao P, Zhang R, Huang X, Feng J-M, Jiang Z, et al. Apoptotic caspases suppress type I interferon production via the cleavage of cGAS, MAVS, and IRF3. Mol Cell. 2019;74(1):19–31 e7. doi: 10.1016/j.molcel.2019.02.013. [DOI] [PubMed] [Google Scholar]
- 17.Han C, Liu Z, Zhang Y, Shen A, Dong C, Zhang A, Moore C, Ren Z, Lu C, Cao X, et al. Tumor cells suppress radiation-induced immunity by hijacking caspase 9 signaling. Nat Immunol. 2020;21:546–554. doi: 10.1038/s41590-020-0641-5. [DOI] [PubMed] [Google Scholar]
- 18.Segawa K, Kurata S, Yanagihashi Y, Brummelkamp TR, Matsuda F, Nagata S. Caspase-mediated cleavage of phospholipid flippase for apoptotic phosphatidylserine exposure. Science. 2014;344:1164–1168. doi: 10.1126/science.1252809. [DOI] [PubMed] [Google Scholar]
- 19.Suzuki J, Denning DP, Imanishi E, Horvitz HR, Nagata S. Xk-related protein 8 and CED-8 promote phosphatidylserine exposure in apoptotic cells. Science. 2013;341:403–406. doi: 10.1126/science.1236758. [DOI] [PubMed] [Google Scholar]
- 20.Jain N, Keating M, Thompson P, Ferrajoli A, Burger J, Borthakur G, Takahashi K, Estrov Z, Fowler N, Kadia T, et al. Ibrutinib and Venetoclax for First-Line Treatment of CLL. N Engl J Med. 2019;380:2095–2103. doi: 10.1056/NEJMoa1900574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lok SW, Whittle JR, Vaillant F, Teh CE, Lo LL, Policheni AN, Bergin ART, Desai J, Ftouni S, Gandolfo LC, et al. A phase Ib dose-escalation and expansion study of the BCL2 inhibitor venetoclax combined with tamoxifen in ER and BCL2-positive metastatic breast cancer. Cancer Discov. 2019;9:354–369. doi: 10.1158/2159-8290.CD-18-1151. [DOI] [PubMed] [Google Scholar]