

SUMMARY
53BP1 plays a central role in dictating DNA repair choice between non-homologous end joining (NHEJ) and homologous recombination (HR), which is important for the sensitivity to poly(ADP-ribose) polymerase inhibitors (PARPis) of BRCA1-deficient cancers. In this study, we show that FOXK1 associates with 53BP1 and regulates 53BP1-dependent functions. FOXK1–53BP1 interaction is significantly enhanced upon DNA damage during the S phase in an ATM/CHK2-dependent manner, which reduces the association of 53BP1 with its downstream factors RIF1 and PTIP. Depletion of FOXK1 impairs DNA repair and induces compromised cell survival upon DNA damage. Overexpression of FOXK1 diminishes 53BP1 foci formation, which leads to resistance to PARPis and elevation of HR in BRCA1-deficient cells and decreased telomere fusion in TRF2-depleted cells. Collectively, our findings demonstrate that FOXK1 negatively regulates 53BP1 function by inhibiting 53BP1 localization to sites of DNA damage, which alters the DSB-induced protein complexes centering on 53BP1 and thus influences DNA repair choice.
In Brief
53BP1 plays a critical role in DNA double-strand break repair choice. Tang et al. report that FOXK1 acts together with 53BP1 and participates in proper DNA repair pathway choice during various cell cycle phases.
Graphical Abstract
INTRODUCTION
Chromosomes are under constant assault as cells encounter endogenous lesions or are exposed to various DNA-damaging agents. Among all of the DNA lesions, DNA double-strand breaks (DSBs) are considered the most genotoxic because unrepaired DSBs prevent the completion of DNA replication and transcription. Cells respond to DSBs by blocking cell cycle progression and initiating DNA repair. Usually, DSBs can be repaired via two major pathways: non-homologous end joining (NHEJ) (Lieber, 2010) and homologous recombination (HR) (Heyer et al., 2010).
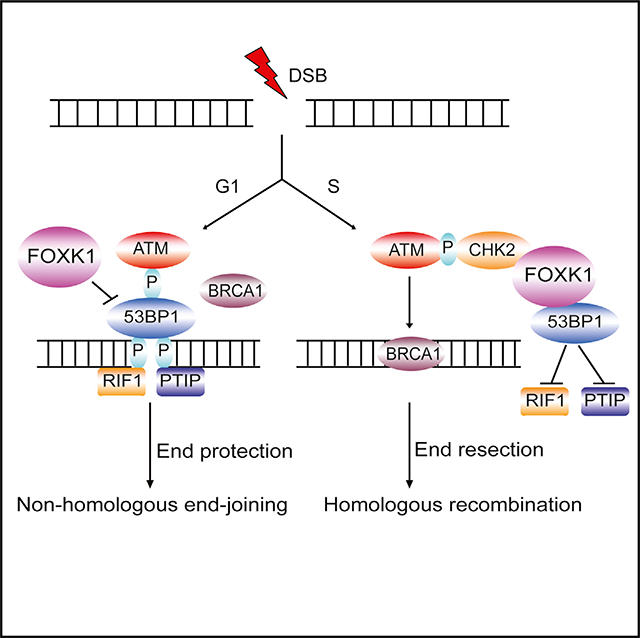
53BP1 is a key regulator of DNA damage response and is required for DNA repair and tumor suppression (Schultz et al., 2000; Ward et al., 2003). 53BP1 plays critical roles in the regulation of class-switch recombination in B lymphocytes (Manis et al., 2004; Ward et al., 2004), end joining of dysfunctional telomeres in TRF2-depleted cells (Dimitrova et al., 2008), and sensitivity to poly(ADP-ribose) polymerase inhibitors (PARPis) in BRCA1-deficient cancers (Bouwman et al., 2010; Bunting et al., 2010). Upon DSB induction, 53BP1 can rapidly form damage-induced foci near DNA lesions. The minimal region in 53BP1 that controls its localization to DSBs contains an oligomerization domain (Zgheib et al., 2009), a tandem Tudor domain that recognizes histone H4 Lys 20 dimethylation (H4K20me2) (Charier et al., 2004), and a ubiquitin-dependent recruitment (UDR) motif that recognizes histone H2A(X) Lys-15 ubiquitination (H2AK15ub) (Fradet-Turcotte et al., 2013). 53BP1 coordinates the two major DSB repair pathways; while it promotes NHEJ repair, it inhibits HR repair (Bunting et al., 2010). In the G1 phase, ATM-mediated 53BP1 phosphorylation recruits the downstream factors RIF1 and PTIP to sites of DNA damage to suppress BRCA1-mediated 5′-to-3′ DNA end resection; in the S/G2 phase, BRCA1 can antagonize 53BP1 signaling to promote HR repair and inhibit NHEJ by inhibiting 53BP1 phosphorylation and preventing the translocation of RIF1 to DSBs (Chapman et al., 2013; Daley and Sung, 2013; Di Virgilio et al., 2013; Escribano-Díaz et al., 2013; Feng et al., 2013; Munoz et al., 2007; Wang et al., 2010).
The role of 53BP1 in dictating DNA repair choice between NHEJ and HR is critically important for the treatment of BRCA1-deficient breast and ovarian cancers. BRCA1- and BRCA2-mutated cancers, which are deficient in HR repair, are hypersensitive to PARPis through the mechanism of synthetic lethality (Farmer et al., 2005; Helleday et al., 2005). Recent studies demonstrated that BRCA1-deficient tumors may acquire resistance to PARPis by partially restoring HR repair, either through reversion mutations in BRCA1 or through “synthetic viability” due to a loss of 53BP1 or 53BP1-related proteins (Bunting et al., 2010; Cao et al., 2009). These findings indicate that 53BP1 may play an important role in determining the outcome of PARPi-based cancer therapy, which is being adopted rapidly in the clinic for the treatment of breast, ovarian, prostate, and other cancers that have defects in the HR pathway.
Thus, identification of novel 53BP1 regulators would help us better understand the regulation of 53BP1 function in DSB repair choice and design more efficient anticancer strategies. To this end, we used CRISPR-Cas9 technology to insert a tag at the C terminus of 53BP1 at its genomic loci and analyzed endogenous 53BP1-associated proteins using a tandem affinity purification (TAP) approach. In this study, we identified that forkhead box K1 (FOXK1) associates with 53BP1 and regulates 53BP1-dependent functions in DNA damage response.
FOXK1 belongs to the family of forkhead box class K (FOXK) transcription factors that contain a forkhead-associated (FHA) domain, which is required for recognition of phosphopeptides, and a winged helix (WH) DNA-binding domain, which is required for their transcriptional regulation (Clark et al., 1993; Durocher and Jackson, 2002). FOXK1 mediates a wide spectrum of biological processes that are mostly dependent on its transcriptional activity; for example, FOXK1 activates myogenic progenitors by interacting with transcription repression complex Sin3/Sds3 (Shi et al., 2010; Shi and Garry, 2012), promotes cell growth by activating the wnt/β-catenin pathway (Ji et al., 2018; Wang et al., 2015), and acts as an important regulator that reprograms cellular metabolism to induce aerobic glycolysis (Sukonina et al., 2019).
Although multiple roles of FOXK1 in cell proliferation, cell growth, and metabolism have been documented, whether FOXK1 also has a role in DNA damage response remains unknown. In this study, we found that FOXK1 specifically associates with 53BP1 and regulates 53BP1 foci formation upon DNA damage. ATM-dependent phosphorylation events significantly enhanced FOXK1–53BP1 interaction in soluble nuclear fraction upon DNA damage during the S phase, which reduced the association of 53BP1 with its downstream factors RIF1 and PTIP. Depletion of FOXK1 increased γH2AX and 53BP1 foci accumulation and compromised cell survival in the G1 and S phases and aberrant cell cycle signaling upon DNA damage. Conversely, overexpression of FOXK1 diminished 53BP1 foci formation, which led to resistance to PARPis and elevation of HR in BRCA1-deficient cells and decreased telomere fusion in TRF2-depleted cells. Collectively, our data identified a cell-cycle-dependent action of FOXK1 that negatively regulates 53BP1 function by inhibiting 53BP1 localization to sites of DNA damage and alters the DSB-induced protein complexes centering on 53BP1.
RESULTS
FOXK1 Is a Novel 53BP1-Associated Protein
To identify endogenous 53BP1-associated proteins, we used an HR-based CRISPR knockin strategy to insert an SFB tag at the C terminus of 53BP1 in 293T cells (Figure S1A). Correctly edited clones were identified by genomic PCR and immunoblotting (Figures S1B and S1C) and confirmed by analysis of their localization to ionizing radiation (IR)-induced foci (Figure S1D). Then, we carried out TAP to analyze 53BP1-associated proteins at an endogenous level (Figure 1A). We carefully analyzed our purification protein list with multiple controls available in our laboratory, and we identified 19 high-confidence interacting proteins (HCIPs) in the soluble fraction and 27 HCIPs in the chromatin fraction (Figures 1B and 1C; Table S1). Among those HCIPs, four proteins (TP53, USP28, TIRR, and CDK1) of 19 HCIPs from the soluble fraction and five proteins (MDC1, RIF1, CDK1, TP53, and MCPH1) of 26 HCIPs from the chromatin fraction were known 53BP1-binding proteins. Interestingly, we uncovered a novel 53BP1-binding protein, FOXK1, in the soluble fraction (Figures 1B and 1C).
Figure 1. FOXK1 Is a Novel 53BP1-Associated Protein.

(A) Strategy for tandem affinity purification (TAP) in 293T 53BP1 SFB knockin cells.
(B) Selected lists of 53BP1-associated proteins in soluble and chromatin fractions analyzed by mass spectrometry.
(C) Interaction network of the 53BP1 soluble and chromatin-associated complexes illustrated by Cytoscape. Red, known 53BP1-binding protein; blue, putative 53BP1-binding protein.
(D) Selected lists of FOXK1-associated proteins in soluble and chromatin fractions analyzed by mass spectrometry.
(E) 293T cells were transfected with SFB-tagged mock control, FOXK1, or FOXK2 and then immunoprecipitated with S-protein beads and subjected to immunoblot with indicated antibodies.
The FOXK family contains two proteins, FOXK1 and FOXK2, that share very similar protein domain structures (Figure S1E) and may have redundant functions. Thus, we investigated whether these two proteins could interact with 53BP1. Previously, we performed proteomics analyses of transcription factors including FOXK1 and FOXK2 (Li et al., 2015) as well as TAP of SFB-tagged FOXK1 and FOXK2 in 293T cells. Interestingly, we found that 53BP1 peptides were present in both FOXK1 and FOXK2 purification (Figures 1D and S1F), and we recovered more 53BP1 peptides in FOXK1 purification than in FOXK2 purification. These unbiased reciprocal purification results strongly suggest that FOXKs (mainly FOXK1) associate with 53BP1.
We then performed pull-down experiments by overexpressing FOXK1 and FOXK2 and immunoblotting for endogenous 53BP1 in 293T cells. As shown in Figure 1E, both FOXK1 and FOXK2 could pull down 53BP1, and FOXK1 showed stronger binding to 53BP1 than FOXK2 did, which is consistent with the purification results. These data suggest a potential role of FOXKs—mainly FOXK1—in responding to DNA damage through their interaction with 53BP1.
Mapping the Interaction Domain between FOXK1 and 53BP1
We then examined which domain of 53BP1 interacts with FOXK1. We found that deletion of either the 53BP1 N terminus or its Tudor domain did not affect the interaction between FOXK1 and 53BP1, whereas deletion of the 53BP1 C terminus, especially the IR-induced foci domain or the oligomerization domain of 53BP1, abolished its interaction with FOXK1 (Figures 2A and 2B), suggesting that the 53BP1 oligomerization domain is required for 53BP1’s interaction with FOXK1. Next, we generated various truncation mutants of FOXK1 (Figure 2C). As shown in Figure 2D, two conserved domains—the FHA domain and the WH domain—were not required for the binding of FOXK1 to 53BP1, but a region between these two domains (residues 175–305) appeared to be required for the interaction with 53BP1.
Figure 2. Mapping the Binding Regions on FOXK1 and 53BP1.

(A) 53BP1 full-length (FL) and deletion mutants used in this study.
(B) 293T cells co-transfected with SFB-FOXK1 and HA-53BP1 WT or mutants were immunoprecipitated with S-protein beads. Western blot was conducted with indicated antibodies.
(C) FOXK1 FL and deletion mutants used in this study.
(D) 293T cells co-transfected with HA-53BP1 and SFB-FOXK1 WT or deletion mutants were immunoprecipitated with S-protein beads and subjected to immunoblot with indicated antibodies.
(E) 293T cells co-transfected with HA-53BP1 and SFB-FOXK1 WT and point-mutation mutants were immunoprecipitated with S-protein beads and subjected to immunoblot with indicated antibodies.
(F) Left: Coomassie blue staining shows the bacterially purified MBP, MBP FOXK1 175–305 fusion protein, and MBP FOXK1 175–305 F201A fusion protein. Right: cell lysates containing exogenously expressed HA-tagged 53BP1 were pulled down with amylose resin coated with indicated purified protein. Immunoblot was conducted with indicated antibodies.
Considering that the region containing residues 175–305 is not a conserved domain, we decided to further map the minimal region required for FOXK1–53BP1 interaction. Additional deletion mutants revealed that deletion of residues 201–205 of FOXK1 abolished its interaction with 53BP1 (Figures S2A and S2B). We mutated each of these five residues to alanine and found that the phenylalanine 201-to-alanine mutant of FOXK1 abolished the interaction between FOXK1 and 53BP1 (Figure 2E). Furthermore, using a bacterially expressed and purified maltose-binding protein (MBP)-fused FOXK1 protein incubated with cell lysates expressing exogenous HA-53BP1, we showed that the FOXK1 region comprising residues 175–305 was sufficient to bind to 53BP1, while FOXK1 175–305 F201A disrupted this in vitro binding (Figure 2F). Taken together, these data suggest that FOXK1 is a bona fide 53BP1-binding protein, with a critical residue phenylalanine 201 that is essential for the binding.
FOXK1 Participates in DNA Damage Response and Is Important for Efficient DSB Repair
The interaction between FOXK1 and 53BP1 suggests that FOXK1 may be involved in DNA damage response and repair. We first examined whether FOXK1 can localize to sites of DNA damage. As shown in Figure S3A, both endogenous FOXK1 and FOXK2 could not localize to UV-laser-induced DNA damage sites. We then used a proximity ligation assay (PLA) to determine 53BP1-FOXK1 co-localization and interaction. We found that under normal conditions, similar to the weak RIF1–53BP1 interaction, the FOXK1–53BP1 interaction was too weak to see the PLA signal. However, upon DNA damage, both FOXK1–53BP1 and RIF1–53BP1 interaction significantly increased, and the RIF1–53BP1 interaction was stronger than that of FOXK1–53BP1 (Figures 3A and 3B). These findings suggest that FOXK1–53BP1 interaction is regulated by DNA damage. To further determine whether FOXK1 is as involved in DNA damage response as 53BP1 is, we generated FOXK1 knockout (KO), FOXK2 KO, and FOXK1/FOXK2 double KO (DKO) HeLa cells (Figure 3C). As shown in Figure 3D, FOXK1 KO cells showed increased sensitivity to IR compared with wild-type (WT) cells, whereas FOXK2 KO cells showed no increased sensitivity. FOXK1/FOXK2 DKO cells showed slightly more sensitivity to IR than FOXK1 KO cells did, which suggests that FOXK1 and FOXK2 have redundant functions, whereas FOXK1 plays a major role in promoting cell survival after IR.
Figure 3. FOXK1 Depletion Impairs DNA Damage Response.

(A) Proximity ligation assay (PLA) showed the interaction of endogenous 53BP1 and FOXK1 (red spots) in non-treated (NT) and IR (10 Gy, released for 1 h)-treated cells. RIF1–53BP1 interaction was included as a positive control.
(B) Quantification of PLA from (A). More than 80 cells of each group were quantified. Data are represented as mean ± SD (n = 2); ***p < 0.001, Student’s t test.
(C) Generation of FOXK1 KO, FOXK2 KO, and FOXK1/2 DKO in HeLa cells. Immunoblot was performed with indicated antibodies.
(D) Clonogenic survival of indicated cells after exposure to indicated doses of IR. Results are the means (± SD) of three independent experiments.
(E) Cell cycle profile in WT, FOXK1 KO, FOXK2 KO, and FOXK1/2 DKO HeLa cells in NT and IR (2 Gy)-treated cells for different time points after IR.
(F) Statistical quantification of the percentages of cells in different cell cycles from (E). Data are represented as mean ± SD (n = 3); **p < 0.01, ***p < 0.001, Student’s t test.
This difference in IR sensitivity may be related to altered cell cycle distribution and checkpoint signaling upon DNA damage. Upon analysis of cell cycle profiles, we found that FOXK2 KO cells displayed the same profile as WT cells, whereas FOXK1 KO and FOXK1/FOXK2 DKO cells showed significant S and G2/M accumulation in an untreated condition. Upon DNA damage, FOXK1 KO and DKO cells showed more significant G2/M accumulation (Figures 3E and 3F). We then monitored cell cycle progression in WT and FOXK1 KO cells by first synchronizing them in the M phase and then releasing them into the cell cycle. We found that FOXK1 KO cells have normal cell progression from the M phase (M0h) to G2 phase (M18h); however, after M18h, FOXK1 KO cells progressed slower than WT cells, with significant G2/M accumulation (Figure S3B), suggesting that FOXK1 may have a role in progression from G2 to M. We therefore examined the activation of DNA-damage-induced checkpoint pathways, measured by CHK1 and CHK2 phosphorylation before and after DNA damage. We noticed an increase in phospho-CHK1 levels in FOXK1 KO and FOXK1/FOXK2 DKO cells at both 1 h and 4 h after release from IR treatment, whereas FOXK2 KO cells behaved the same as WT cells (Figure S3C). The 53BP1 protein level did not change in any of these KO cells, compared with WT cells either before or after IR treatment (Figure S3C), suggesting that although FOXK1 regulates DNA damage response, it does not affect 53BP1 expression or protein stability.
We also performed cell fractionation in WT and FOXK1 KO cells to examine the distribution of 53BP1 in the soluble and chromatin fractions. We found that 53BP1 protein in the chromatin fraction was increased slightly in FOXK1 KO cells (Figure S3D). We detected FOXK1 protein mainly in the soluble fraction, which is consistent with FOXK1 being found mainly in the soluble fraction of 53BP1-associated proteins (Figures 1B and 1C).
We then monitored the kinetics of γH2AX and 53BP1 foci formation in WT and FOXK1 KO cells at various time points after IR treatment (Figures 4A–4C). In untreated WT and untreated FOXK1 KO cells, both γH2AX and 53BP1 foci were barely detectable, suggesting no significant difference in endogenous DNA damage. When cells were treated with 2 Gy of IR and released after 1 h, γH2AX and 53BP1 rapidly formed DNA-damage-induced foci in more than 90% of both WT and FOXK1 KO cells. After that, the numbers of DNA-damage-induced foci began to taper off; in WT cells, the percentage of γH2AX foci of >10 cells decreased to about 45% at 6 h and to 10% at 24 h after release from IR, indicating successful repair. However, 75% of FOXK1 KO cells showed strong staining for both γH2AX and 53BP1 foci at 6 h after release from IR, and about 50% of cells still showed γH2AX and 53BP1 accumulation at DSBs at 24 h after release from IR.
Figure 4. FOXK1-Depleted Cells Accumulate More DSBs and Show Defects in DNA Repair.

(A) Immunofluorescence for γH2AX and 53BP1 in WT and FOXK1 KO cells treated with 2 Gy of IR and incubated for the indicated time points.
(B) Statistical quantification of γH2AX foci formation from (A). Data are represented as mean ± SD (n = 3); ***p < 0.001, Student’s t test.
(C) Statistical quantification of 53BP1 foci formation from (A). Data are represented as mean ± SD (n = 3); ***p < 0.001, Student’s t test.
(D) Representative images of neutral comet assay performed in WT and FOXK1 KO cells treated with 10 Gy of IR.
(E) Statistical quantification of the olive tail moment from (D). Data are represented as mean ± SD (n = 3); ***p < 0.001, Student’s t test.
(F) Statistical quantification of 53BP1 foci formation in synchronized WT and FOXK1 KO cells in NT and IR (2 Gy)-treated conditions for different time points after IR. Data are represented as mean ± SD (n = 3); **p < 0.01, ***p < 0.001, Student’s t test.
(G) Clonogenic survival of WT and FOXK1 KO cells after exposure to indicated doses of IR at different cell cycle phases. Data are represented as mean ± SD (n = 3).
(H and I) U2OS NHEJ and HR reporter cell lines transfected with indicated small interfering RNA (siRNA) and I-SceI plasmids were collected for flow cytometry analysis to determine NHEJ (H) and HR (I) repair efficiency. Quantification data are represented as mean ± SD (n = 3); **p < 0.01, ***p < 0.001, Student’s t test.
We also performed a neutral comet assay to evaluate DSB repair in WT and FOXK1 KO cells. As shown in Figures 4D and 4E, both WT and FOXK1 KO cells showed similar background comet tails in the untreated condition. Similar comet tail lengths between the two groups were also observed at 1 h after release from 10 Gy of IR. However, at 24 h of recovery after IR, comet tail lengths of WT cells recovered to the basal level of the untreated condition, whereas FOXK1 KO cells still had longer tails than WT cells did. These observations indicate that depletion of FOXK1 prevents efficient DNA repair.
Since FOXK1 KO cells showed significant cell cycle accumulation in S and G2, we sought to determine whether the observed DNA repair defect is cell cycle regulated. As shown in Figure 4F, we synchronized WT and FOXK1 KO cells at the G1, S, and G2 phases and treated the cells with IR. We then compared the percentage of 53BP1-positive cells at various times after recovery following IR. We observed no differences in non-treated (NT) cells or in cells at 1 h after release from IR in any phase of the cell cycle. However, we did notice that ~50% of FOXK1 KO cells still had strong 53BP1 foci at 24 h after recovery following IR at the G1 and S phases, whereas only <20% of WT cells did; in comparison, ~20% of FOXK1 KO cells following G2 irradiation remained 53BP1 positive, whereas 11% of WT cells did (Figure 4F).
We also analyzed cell survival upon IR in WT and FOXK1 KO cells when radiation was delivered at different phases of the cell cycle (Figure 4G). We found that FOXK1 KO cells were sensitive to IR when they were irradiated at the G1 and S phases, with cells irradiated in G1 showing more sensitivity than cells irradiated in S. These data suggest that FOXK1 KO cells are defective in the repair of DSBs generated in both G1 and S and that FOXK1 is probably more important for DNA repair in cells irradiated in G1.
We also used the I-SceI-based U2OS NHEJ and U2OS DR-GFP HR reporter systems to determine the effect of FOXK1 knockdown (KD) on NHEJ and HR repair. As shown in Figure 4H, KD of FOXK1 increased NHEJ efficiency, which is contrary to 53BP1 depletion. However, unlike KD of BRCA1, which was associated with dramatically decreased HR efficiency, FOXK1 depletion was not associated with a significant HR defect (Figure 4I). The increased NHEJ repair efficiency assessed by these reporter assays seem to be inconsistent with the significantly compromised repair in the G1 phase observed in FOXK1 KO cells. We speculate that the increased NHEJ repair is due to the finetuned 53BP1 level in the chromatin fraction in FOXK1 KO cells; however, the accumulation of 53BP1 at DSBs and inefficient removal of 53BP1 during repair led to the aberrant repair and comprised survival in G1. Taken together, these data suggest that FOXK1 is required for the proper control of NHEJ repair at different cell cycle phases, but it does not appear to directly affect HR.
Because FOXK1 is known as a transcription factor, we investigated whether DNA repair defects observed in cells with FOXK1 depletion could be due to dysregulated transcription of some DNA damage regulators. As shown in Figure S4A, KD of FOXK1 or depletion of FOXK1 in HeLa cells was not associated with any difference in mRNA levels of 53BP1, BRCA1, MDC1, RAD51, or RPA1. We performed RNA sequencing (RNA-seq) analysis to determine whether any DNA-damage-related pathways were affected in FOXK1 KO, FOXK2 KO, or FOXK1 FOXK2 DKO cells. K-means clustering analysis was conducted to separate differentially expressed genes (DEGs) into four subgroups based on expression across all experimental groups (Figure S4B), and we found that FOXK1 KO and DKO groups have significantly overlapping DEGs, while the FOXK2 KO group has similar gene expression to that of the WT group (Figure S4B). Gene Ontology analysis did not uncover any dysregulated DNA-damage-related pathways upon FOXK KO (Figure S4C;Table S2). We also listed the top 30 significantly changed genes from each cluster (Figure S4D). For example, the top changed genes—ENO1, PGM1, and HIF1A from cluster 3, which function mainly in gluconeogenesis, canonical glycolysis, and the glycolytic process (Choi et al., 2005; Ji et al., 2016; Jin et al., 2018) — were significantly downregulated in the FOXK1 KO and DKO groups. Conversely, several top changed genes—ERRFI1, ARHGAP29 and DUSP1 from cluster 4, which are involved mainly in intracellular signaling transduction and positive regulation of GTPase activity (Calvisi et al., 2008; Post et al., 2013; Zhang and Vande Woude, 2007)—were significantly upregulated in the FOXK1 KO and DKO groups (Figure S4D; Table S2). These results suggest that the role of FOXK1 in DNA damage response is likely independent of its role in transcriptional regulation of DNA-damage-related pathways and genes.
FOXK1 Overexpression Impairs 53BP1 Functions
As shown in Figure S5A, higher FOXK1 expression was found in various types of tumors, which suggests that its overexpression has a role in facilitating tumor progression; such a role would concord with the known functions of FOXK1 in promoting cell proliferation and growth. Here, we sought to determine whether FOXK1 overexpression would affect DNA damage response. Indeed, overexpression of FOXK1 significantly decreased 53BP1 foci formation upon DNA damage but did not appear to alter γH2AX foci (Figure S5B). We also assessed the effects of overexpression of FOXK1 F201A, FOXK1 H355A, and FOXK1 F201A&H355A mutants on 53BP1 foci formation. While the FOXK1 F201A mutant abolished the interaction of FOXK1 with 53BP1, the FOXK1 H355A mutant was defective in DNA binding (Freddie et al., 2007; Grant et al., 2012), and the FOXK1 F201A&H355A mutant abolished both 53BP1 association and DNA binding of FOXK1. Overexpression of FOXK1 F201A and FOXK1 F201A&H355A did not affect 53BP1 foci formation upon DNA damage (Figures 5A and 5B), whereas FOXK1 WT and FOXK1 H355A overexpression significantly decreased 53BP1 foci formation. As shown in Figure 5C, FOXK1 WT and FOXK1 H355A associated with 53BP1 and CHK2, whereas FOXK1 F201A and FOXK1 F201A&H355A failed to do so, suggesting that FOXK1–53BP1 interaction is required for the suppression of 53BP1 foci formation.
Figure 5. DNA-Damage-Dependent ATM Phosphorylation Enhances FOXK1–53BP1 Interaction and Inhibits 53BP1 Foci Formation.

(A) Immunofluorescence for 53BP1 and FLAG in HeLa cells transfected with HA-FLAG-tagged WT or three mutants of FOXK1 treated with IR (10 Gy, 2 h). Representativa images of cells with strong, weak, or no 53BP1 foci are shown. Strong, ≥10 53BP1 foci; weak, <10 53BP1 foci.
(B) Statistical quantification of 53BP1 foci formation from (A).
(C) 293T cells co-transfected with HA-53BP1 and SFB-FOXK1 WT or mutants were immunoprecipitated with S-protein beads and subjected to immunoblot with indicated antibodies.
(D) 293T cells co-transfected with HA-53BP1 and GFP-FOXK1 were irradiated (10 Gy, 2 h) and collected for immunoprecipitation with GFP-trap beads. Western blot was conducted with indicated antibodies. Quantification analysis was done with ImageJ, and the ratio of FOXK1–53BP1 interaction is shown.
(E) 293T cells stably expressing FOXK1-SFB were first treated with DMSO or ATMi (AZD0156, 1 μM), ATRi (AZD6738, 1 μM), DNA-PKi (NU7441, 5 μM), or ATMi+DNA-PKi (AZD0156, 1 μM + KU7441, 5 μM) for 1 h; cells were then irradiated at 10 Gy and harvested after 2 h. Immunoprecipitation was conducted with streptavidin beads and subjected to immunoblot with indicated antibodies.The ratio of FOXK1–53BP1 interaction was normalized to the NT group and calculated as an average of three independent experiments.
(F) Determination of FOXK1–53BP1 interaction in 293T WT and CHK2 KO cells in NT or IR (10 Gy, 2 h)-treated condition. The ratio was normalized to the WT NT group and calculated as an average of two independent experiments.
(G) Statistical quantification of 53BP1 foci formation at different cell cycle phases in HeLa mock and overexpressing HA-FLAG FOXK1 cells.
(H) 293A cells stably expressing FOXK1-SFB were synchronized and collected at different phases of the cell cycle for immunoprecipitation with streptavidin beads and subjected to immunoblot with indicated antibodies. Quantification analysis of FOXK1–53BP1 and FOXK1-pCHK2 interaction was done with ImageJ. Both ratios were calculated as an average of two independent experiments
(I) Cell cycle profile of 293A cells stably expressing FOXK1-SFB at different cell cycle phases.
Previously, we observed that depletion of FOXK1 affected cell cycle progression (Figures 3E and 3F) and sensitized cells to irradiation (Figure 3D). Here, we performed reconstitution assays with FOXK1 WT and the three mutants of FOXK1. FOXK1 WT and FOXK1 H355A were able to rescue cell cycle progression as well as IR sensitivity in FOXK1 KO cells, whereas FOXK1 F201A and FOXK1 F201A&H355A could not (Figures S6A–S6D).
We also performed RNA-seq analysis in FOXK1 KO cells reconstituted with FOXK1 WT and mutants. We found that cells reconstituted with any of the three FOXK1 mutants could largely rescue dysregulated gene expression in FOXK1 KO cells, and our observations were similar in the cells reconstituted with FOXK1 WT (Figure S6E). This finding suggested that the previously reported DNA-binding mutant H355A is probably not fully defective in transcription regulation. Nevertheless, the 53BP1-binding-deficient mutant F201A could rescue transcription, but it could not rescue cell cycle progression or cell survival upon IR in FOXK1 KO cells (Figure S6), suggesting that the DNA repair function of FOXK1 depends on its interaction with 53BP1. Together, these findings suggest that FOXK1–53BP1 interaction is required for regulating 53BP1 localization to sites of DNA damage and for the choice of pathways for efficient DNA repair.
DNA-Damage-Dependent ATM Phosphorylation Enhances FOXK1–53BP1 Interaction in the Soluble Fraction
We further investigated how FOXK1–53BP1 interaction may be regulated after DNA damage. As we showed previously, FOXK1–53BP1 interaction occurs mainly in the soluble fraction (Figures 1B–1D). We performed co-immunoprecipitation of 53BP1 and FOXK1 with NETN lysis buffer containing a low concentration of NaCl to collect the soluble fraction of cell lysate with or without IR treatment. The soluble fraction showed no histone H3 and abundant tubulin, as well as significantly enhanced FOXK1–53BP1 interaction upon DNA damage (Figure 5D). We also showed that the FOXK1 H355A mutant, which is defective in DNA binding, still binds to 53BP1 at levels similar to those of FOXK1 WT (Figure 5C). These data suggest that FOXK1–53BP1 association occurs mainly in the soluble fraction and is enhanced in the soluble fraction upon DNA damage. The enhanced association of FOXK1 with 53BP1 was dependent on ATM kinase but not on ATR or DNA-dependent protein kinase (DNA-PK) (Figure 5E), suggesting that DNA-damage-dependent ATM phosphorylation events may promote the association of FOXK1 and 53BP1 in the soluble fraction and may help inhibit 53BP1 foci formation. Since FOXK1 mutants that were disrupted in 53BP1 binding also abolished their binding with CHK2 (Figure 5C), and the CHK2-FOXK axis was recently found to play an important role in DNA-damage-mediated transcriptional control of autophagy (Chen et al., 2020), we sought to determine whether FOXK1–53BP1 interaction is dependent on CHK2. As shown in Figure 5F, depletion of CHK2 decreased the FOXK1–53BP1 interaction in both the NT and IR-treated conditions, with a larger decrease upon IR, suggesting that DNA-damage-enhanced FOXK1–53BP1 interaction partially depends on CHK2. Moreover, we synchronized cells at different cell cycle phases and assessed whether the inhibition of 53BP1 foci formation by FOXK1 is cell cycle regulated. As shown in Figure 5G, overexpression of FOXK1 significantly blocked 53BP1 foci in both G1- and S-phase cells, with a stronger inhibitory effect on 53BP1 in G1 than in S phase. We also found cell-cycle-regulated enhancement of FOXK1–53BP1 interaction upon DNA damage. As shown in Figures 5H and 5I, the increase in FOXK1–53BP1 interaction in S-phase cells was similar to that of FOXK1-pCHK2 interaction. However, in G1-phase cells, the FOXK1-pCHK2 interaction decreased, and the FOXK1–53BP1 interaction did not change significantly. These data suggest that FOXK1–53BP1 interaction is CHK2 dependent in S-phase cells and that CHK2 probably also coordinates with FOXK1 in regulating 53BP1 foci formation in G1-phase cells. Taken together, our results support that FOXK1 acts with 53BP1 and participates in proper DNA repair pathway choice during various cell cycle phases, at least partly because of the cell-cycle-regulated interaction between FOXK1 and 53BP1.
FOXK1 Participates in the Regulation of Telomere Fusion in TRF2-Deficient Cells and PARPi Sensitivity in BRCA1-Deficient Cells
53BP1 plays critical roles in the regulation of end joining of dysfunctional telomeres in TRF2-depleted cells (Dimitrova et al., 2008) and PARPi sensitivity in BRCA1-deficient cancers (Bouwman et al., 2010; Bunting et al., 2010). We sought to determine whether FOXK1 overexpression affects these 53BP1-mediated functions. As shown in Figures 6A–6C, upon TRF2 depletion, telomere fusion was significantly decreased by overexpression of FOXK1 WT or FOXK1 H355A in TRF2-depleted HeLa cells, compared with empty vector control, but overexpression of FOXK1 F201A or FOXK1 F201A&H355A did not show any effect. Furthermore, we examined RAD51 foci formation and PARPi sensitivity in BRCA1 KD cells with or without overexpression of WT or mutant FOXK1 (Figures 6D–6F). In BRCA1 KD HeLa cells, overexpression of FOXK1 WT or FOXK1 H355A significantly increased survival of cells treated with the PARPi olaparib (Figure 6E) and significantly increased RAD51 foci formation (Figure 6F), whereas these effects were not observed in cells with overexpression of FOXK1 F201A or FOXK1 F201A&H355A. Similar PARPi sensitivity was found in BRCA1 KD MCF10A cells with overexpression of FOXK1 WT or FOXK1 H355A but not with FOXK1 F201A or FOXK1 F201A&H355A (Figures 6G and 6H). Moreover, using the U2OS DR-GFP reporter system, we found that HR repair was partially recovered in BRCA1 KD cells with overexpression of FOXK1 WT or FOXK1 H355A, but overexpression of FOXK1 F201A or FOXK1 F201A&H355A did not lead to such rescue (Figures 6I and 6J).
Figure 6. FOXK1–53BP1 Interaction Is Required for Regulation of 53BP1-Mediated Functions.

(A) Western blot for expression of HA-FLAG FOXK1 WT and mutants as well as TRF2 KO efficiency in inducible TRF2 KO HeLa cells.
(B) Representative images of normal telomeres and fused telomeres by metaphase DNA fluorescence in situ hybridization (FISH) with telomere probe.
(C) Statistical quantification of percentage of chromosome fusions from (A). Data are represented as mean ± SD (n = 3); ***p < 0.001, Student’s t test.
(D) Western blot to determine BRCA1 siRNA KD efficiency as well as the expression of FOXK1 WT and mutants in HeLa cells.
(E) Statistical quantification of percentage of survival in colonies from (D) treated with olaparib at different concentrations. Data are represented as mean ± SD (n = 3); ***p < 0.001, Student’s t test.
(F) Statistical quantification of percentage of cells with >10 RAD51 foci from (D). Cells were treated with 10 Gy IR and allowed to recover for 4 h. At least 200 cells were counted in each group. Data are represented as mean ± SD (n = 3); ***p < 0.001, Student’s t test.
(G) Statistical quantification of percentage of survival in colonies treated with olaparib in MCF10A cells. Data are represented as mean ± SD (n = 3); **p < 0.01, Student’s t test.
(H) Western blot was conducted to determine BRCA1 siRNA KD efficiency as well as expression of FOXK1 WT and mutants in MCF10A cells stably expressing GFP-tagged FOXK1 WT and mutants.
(I) Determined HR repair efficiency in FOXK1 WT and mutant overexpression condition with or without BRCA1 depletion in U2OS DR-GFP reporter cells.
(J) Western blot was conducted to determine BRCA1 siRNA KD efficiency as well as expression of FOXK1 WT and mutants from (I).
We also examined the impact of FOXK1 downregulation on telomere fusion in TRF2-depleted cells and HR recovery in BRCA1-depleted cells. As shown in Figures S7A and S7B, KD of FOXK1 did not affect telomere fusion in TRF2-depleted cells. We also investigated RAD51 foci formation and PARPi sensitivity in WT, FOXK1 KO, 53BP1 KO, and FOXK1&53BP1 DKO cells with or without BRCA1 KD (Figures S7C–S7E). As shown in Figures S7D and S7E, among BRCA1 KD cells, FOXK1 KO cells and WT cells showed similar decreases in RAD51 foci formation and PARPi sensitivity, whereas 53BP1 KO rescued levels of RAD51 foci formation and PARPi sensitivity, approaching levels in cells without BRCA1 KD. We also noticed among BRCA1 KD cells that FOXK1&53BP1 DKO cells showed the same RAD51 foci formation and PARPi sensitivity as 53BP1 KO cells (Figures S7D and S7E). Similar PARPi sensitivity was found in BRCA1 KD MCF10A cells with KD of FOXK1 (Figures S7F and S7H) and FOXK1 KD MCF10A cells sensitized to IR (Figure S7G), similar to FOXK1 KO HeLa cells.
Taken together, these data indicate that the FOXK1–53BP1 association can suppress 53BP1-dependent repair functions and restore HR in BRCA1-deficient cells; however, depletion of FOXK1 has no further effect when enough 53BP1 is present or 53BP1 is absent.
FOXK1 Is Involved in the Regulation of 53BP1’s Association with Its Downstream Factors upon DNA Damage
We then assessed how FOXK1 binding could regulate 53BP1 function by preventing 53BP1 localization to sites of DNA damage. We conducted a co-immunoprecipitation assay with overexpression of HA-53BP1 in 293T 53BP1 SFB knockin control and FOXK1 KD cells. We found that KD of FOXK1 did not alter the oligomerization of 53BP1 or the recognition of histone H4K20me2 and H2AK15ub by 53BP1 with or without DNA damage; however, FOXK1 depletion modestly increased the association of 53BP1 with RIF1 and PTIP upon DNA damage (Figure 7A).
Figure 7. FOXK1 Is Involved in the Regulation of 53BP1’s Association with Its Downstream Factors upon DNA Damage.

(A) Immunoblot of HA-53BP1 binding partners pulled down from control (ctrl) and FOXK1 KD cells in 293T 53BP1 SFB knockin cells without or with IR (10 Gy, 2 h) treatment. Quantification of the interaction ratio was normalized to the sictrl without IR treatment group and calculated as an average of two independent experiments.
(B) Immunoblot of HA-53BP1 binding partners pulled down from 293T mock and FOXK1-SFB stably expressing cells without or with IR (10 Gy, 2 h) treatment. Quantification of the interaction ratio was normalized to the mock without IR treatment group and was calculated as an average of two independent experiments.
(C) Model of FOXK1 in DSB repair.
We also noticed that overexpression of FOXK1 reduced the interaction of 53BP1 with RIF1 and PTIP upon DNA damage, with a slightly more deduction in 53BP1-PTIP interaction, whereas 53BP1 oligomerization and recognition of histone H4K20me2 and H2AK15ub by 53BP1 with or without DNA damage were not altered (Figure 7B). Thus, our findings indicate that through cell-cycle-regulated binding of FOXK1 with 53BP1 upon DNA damage, FOXK1 is involved in the regulation of 53BP1 association with its downstream factors upon DNA damage, especially PTIP and RIF1, which may alter DNA repair.
DISCUSSION
53BP1 must be recruited to sites of DNA damage to participate in DNA repair and to antagonize BRCA1-mediated end resection. Several identified proteins can regulate 53BP1 accumulation at DSB sites. For example, the recently discovered 53BP1-associated Tudor-interacting repair regulator (TIRR) binds directly to the 53BP1 Tudor domain, masks its H4K20me2 binding motif, and thereby negatively regulates 53BP1 foci formation (Drané et al., 2017; Zhang et al., 2017). Also, RNF169, which is significantly similar to RNF168, can antagonize the ubiquitin-dependent signaling cascade at DSBs and thus represses 53BP1 accumulation at DNA damage sites (An et al., 2017; Chen et al., 2012; Poulsen et al., 2012).
In this study, with the use of CRISPR-Cas9 technology and TAP of endogenously tagged 53BP1, we identified a new 53BP1-binding protein, FOXK1. FOXK1 depletion led to an aberrant cell cycle profile and compromised cell survival in both G1 and S phases upon DNA damage, potentially because of the accumulation of 53BP1 at DNA damage sites. FOXK1–53BP1 interaction is required for cell cycle progression and cell survival in WT cells and is involved in PARPi resistance in BRCA1-deficient cells and telomere protection in TRF2-depleted cells. FOXK1–53BP1 interaction is enhanced in the soluble fraction upon DNA damage in ATM/CHK2-dependent manner during the S phase. Moreover, FOXK1 is involved in the regulation of 53BP1 association with RIF1 and PTIP. On the basis of these observations, we propose that in the G1 phase upon DNA damage, ATM-mediated 53BP1 phosphorylation recruits RIF1 and PTIP to sites of DNA damage to suppress DNA end resection and favors NHEJ. When FOXK1 is absent, the increased accumulation of 53BP1 foci at the DNA damage site significantly enhances the recruitment of RIF1 and PTIP, which leads to aberrant NHEJ repair and compromised cell survival; thus, FOXK1 probably is required for the inhibition of NHEJ repair during the G1 phase. In the S phase, the increased FOXK1–53BP1 interaction mediated by ATM (probably also by CHK2) phosphorylation in the soluble faction helps reduce 53BP1 association with RIF1 and PTIP and thus promotes DNA end resection and HR repair (Figure 7C).
Because FOXK1 contains an FHA domain, which is required for recognition of phosphopeptides, it is possible that the FOXK1 FHA domain recognizes ATM phosphorylation substrates and thereby regulates 53BP1 functions in DNA damage response. We noticed that FOXK1 could associate with CHK2 in a manner similar to that of its association with 53BP1 (Figure 5C). We also found that CHK2 depletion reduced FOXK1–53BP1 association with or without DNA damage (Figure 5F). Thus, CHK2 and other ATM substrates may be involved in the regulation of FOXK1–53BP1 interaction, which warrants further investigation.
53BP1 has received attention for the past decade because of its role in dictating DNA repair pathway choice and its relevance to the treatment of BRCA1-deficient breast and ovarian cancers. The status of 53BP1 and its related proteins is important in determining the outcome of PARPi-based cancer therapy, which is being used to treat various cancers with defects in the HR pathway. In this study, we uncovered a novel 53BP1-FOXK1 complex involved in DNA repair. Considering that FOXK1 expression has been found to be significantly upregulated in a variety of human cancers and that FOXK1 overexpression suppresses 53BP1 foci formation and 53BP1-dependent DNA repair functions, FOXK1 could serve as a novel target for cancer therapy, especially in combination with PARPis in BRCA1-deficient or HR-deficient cancers. Future studies will be conducted to test this possibility.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Junjie Chen (jchen8@mdanderson.org).
Materials Availability
Plasmids and cell lines generated in this study are available upon request from the lead contact.
Data and Code Availability
RNA-seq data generated in this study are available at NCBI GEO database with the accession number GSE151029. The 53BP1 mass spectrometry data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD020090. The accession number for the FOXK1 and FOXK2 MS data reported in this paper is PRIDE: PXD001383
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
HEK293A (293A) cell line was purchased from Thermo Fisher Scientific. HEK293T (293T), HeLa S3, and MCF10A cell lines were purchased from the American Type Culture Collection. HEK293A (293A), HEK293T (293T), and HeLa S3 were cultured in Dulbecco modified Eagle medium supplemented with 10% fetal calf serum. MCF10A cells were cultured in DMEM/F-12 medium with 5% horse serum, 20 ng/ml human EGF, 0.5 μg/ml hydrocortisone, 100 ng/ml cholera toxin, and 10 μg/ml recombinant human insulin.
METHOD DETAILS
Generation of stable cell lines
The FOXK1 KO, FOXK2 KO, FOXK1/2 DKO, 53BP1 KO, and 53BP1&FOXK1 DKO HeLa cell lines and the CHK2 KO 293T cell line were established by transfecting the plenti-V2 gRNA vector into cells. Single clones were sorted in 96-well plates. Individual KO clones were isolated and confirmed by both western blot and DNA sequencing.
Inducible TRF2 KO HeLa cells were generated by infecting two sgRNAs into inducible Cas9-SFB G418 HeLa cells. Two sgRNAs were ligated into pLenti-blasticidin or pLenti-hygromycin gRNA vectors. The two sgRNA sequences were used as previously reported (Kim et al., 2017).
Plasmids
The human FOXK1, FOXK2 ORF was synthesized by Integrated DNA Technologies. Full-length FOXK1 and the indicated domain-deletion or point-mutation mutants were subcloned into the pBabe-SFB vector, Pcl-T7-GFP and pLenti HA-Flag vectors by Gateway recombination cloning technology. 53BP1 full-length and truncated constructs were used as previously reported (Feng et al., 2013).
SFB-tagging of endogenous 53BP1
The CRISPR/Cas9 technology was used to knock in the SFB tag, which consists of S-tag, Flag tag, and streptavidin-binding tag, to the C terminus of endogenous 53BP1. 293T cells were co-transfected with PX330 plasmid and a donor vector (PUC19 backbone) containing SFB, P2A self-cleavage site, and puromycin resistance selection gene flanked by approximately 1 kb of homology arms. Positive clones were first screened by genomic PCR and further validated by western blotand immunofluorescence staining.
Tandem affinity purification and co-immunoprecipitation
53BP1 SFB knockin 293T cells were used for tandem affinity purification. Cells were lysed in NETN buffer (20 mM Tris-HCl [pH 8.0], 1 mM EDTA, 100 mM NaCl, 0.5% NP-40, and 1 mM DTT, with phosphatase and proteinase inhibitors) for 30 min and centrifuged at 13,000 rpm for 15 min at 4°C. We collected the soluble fraction (i.e., the supernatant) and the chromatin fraction (i.e., pellets suspended in NETN without EDTA and then sonicated and treated with turbonuclease) for tandem affinity purification. The soluble fraction and the chromatin fraction were incubated with streptavidin-conjugated beads (Amersham) for 2 h at 4°C. The beads were then washed with NETN buffer twice and eluted with 2 mg/mL biotin (Sigma-Aldrich) for 1 h. The eluates were then incubated with S-protein beads (Novagen) for 2 h at 4°C. After being washed with NETN five times, the bound proteins were resolved by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and analyzed by mass spectrometry.
For pull-down of 53BP1 binding partners (Figures 7A and 7B), cells were lysed in NETN buffer (20 mM Tris-HCl [pH 8.0], 1 mM EDTA, 250 mM NaCl, 0.5% NP-40, and 1 mM DTT, with phosphatase, proteinase inhibitors, and 50 U turbonuclease) for 30 min and centrifuged at 13,000 rpm for 15 min at 4°C. The supernatant was incubated with HA beads (A2095, Sigma-Aldrich) for 2 h at 4°C. After being washed with NETN three times, the bound proteins were resolved by SDS-PAGE and blotted with indicated antibodies. For other FOXK1-related pull-downs, co-immunoprecipitation was done in NETN buffer (20 mM Tris-HCl [pH 8.0], 1 mM EDTA, 100 mM NaCl, 0.5% NP-40, and 1 mM DTT, with phosphatase and proteinase inhibitors) with indicated beads (streptavidin-conjugated beads [Amersham], S-protein beads [Novagen], GFP-Trap beads [Chromotek]).
Immunofluorescence staining
The immunostaining procedures were similar to those described previously (Feng et al., 2013). Briefly, cells grown on glass coverslips were fixed in 4% formaldehyde for 10 min at room temperature, then permeabilized for 15 min in 0.5% Triton X-100, followed by blocking with 3% bovine serum albumin for 1 h. Cells were then incubated with primary antibodies diluted in 3% bovine serum albumin for 2 h at 37°C and secondary antibody for 1 h at room temperature. Coverslips were mounted with use of DAPI solution (Thermo Fisher Scientific).
Analysis of protein localization to UV laser–induced sites of DNA damage
HeLa cells were seeded onto 35-mm glass-bottomed dishes, which were placed under a Nikon TE200 inverted microscope coupled with a 365-nm UV laser MicroPoint system to induce UV laser damage. After irradiation, cells were immediately fixed with 4% paraformaldehyde and processed for immunostaining with indicated antibodies.
Colony formation assay
Equal amounts of the cells used in the study were seeded onto six-well plates in triplicate, treated with various doses of irradiation or PARPi (olaparib), and then incubated for 2 weeks. Colonies were fixed and stained with 0.5% crystal violet. Colonies were counted manually or using ImageJ. Results were obtained from three independent experiments.
Real-time quantitative reverse transcription PCR (qRT-PCR)
Real-time qRT-PCR was carried out as previously described (He et al., 2015). Briefly, total RNA was isolated with TRIzol (Thermo Fisher Scientific), and the same amount of RNA was used for reverse transcription with the iScript cDNA Synthesis Kit (Bio-Rad). Real-time qRT-PCR amplification reactions were performed with use of the SYBR Green Mix (Thermo Fisher Scientific) and the ABI 7500 Real-Time PCR system (Applied Biosystems).
Neutral comet assay
A neutral comet assay, similar to that described previously (Olive and Banáth, 2006), was performed to detect DSBs. Briefly, cells subjected to IR (10 Gy, 1-h or 24-h recovery) were digested with trypsin and diluted to a density of 5 × 104 cells/mL in ice-cold phosphate-buffered saline. Next, 400 μL of untreated cells or IR-treated cells were mixed with 1.2 mL of 1% (wt/vol) low-gelling-temperature agarose (Sigma-Aldrich, A4018) and spread over slides precoated with 1% (wt/vol) agarose LE (Thomas Scientific After the agarose solidified, slides were placed into ice-cold N1 neutral lysis buffer (2% sarkosyl, 0.5 M Na2-EDTA, 0.5 mg/mL proteinase K [pH 8.0]) and were incubated at 37°C overnight in the dark. After overnight lysis, slides were submerged in N2 rinse and electrophoresis solution buffer (90 mM Tris buffer, 90 mM boric acid, 2 mM Na2-EDTA [pH 8.5]) for 30 min at room temperature. Slides were then subjected to electrophoresis at 20 V for 25 min. After electrophoresis, slides were carefully removed and neutralized with distilled water for 20 min and stained with 2.5 μg/mL propidium iodide solution. Comet assay images were captured using a Nikon 90i microscope at 20 × magnification. Comet tail moments were measured with ImageJ software with the plugin OpenComet (Gyori et al., 2014). At least 50 cells were analyzed for each sample. Results were obtained from three independent experiments.
Telomere fusion analysis
TRF2 inducible KO HeLa cells with either FOXK1 KD by siRNA or FOXK1 (HA-tagged WT or mutant) stable expression were first incubated in medium with or without 0.5 μg/mL doxycycline for 6 days to induce TRF2 KO. Before they were harvested, the cells were treated with nocodazole (0.5 μg/mL) for 3 h and then collected and suspended in 0.075 M KCl for 30 min at 37°C. After being fixed in methanol and acetic acid (3:1) three times, the cells were spread on glass slides to obtain the metaphase spreads, which were hybridized with TelC-FITC telomere probes (Panagene, F1009). Images were captured using a Nikon Eclipse E800 fluorescence microscope. For each group, a minimum of 1,500 chromosomes were counted for analysis.
Cell fractionation
Indicated cells were collected and lysed in NETN buffer (20 mM Tris-HCl [pH 8.0], 1 mM EDTA, 100 mM NaCl, 0.5% NP-40, and 1 mM DTT, with phosphatase and proteinase inhibitors) for 15 min on ice. Cell lysates were centrifuged at 13,000 rpm for 10 min at 4°C. We collected the supernatant as a soluble fraction, which contained both cytoplasmic and nuclear fractions. The remaining pellets (chromatin fraction) were washed with NTN buffer (NETN buffer without EDTA) twice and then suspended in NTN buffer followed by sonication and TurboNuclease digestion. The soluble fraction and chromatin fraction were boiled at 95°C for 10 min in 2 × Laemmli buffer, and proteins were resolved by SDS-PAGE.
Cell-cycle synchronization and analysis
For cell cycle synchronization of HeLa cells, cells were first treated with nocodazole (100 μg/mL) for 16 h and then released at different times to monitor cell cycle progression. For cell cycle synchronization of 293A cells, cells were synchronized by double thymidine block. Cells were first treated with 2 mM thymidine for 16 h and then released for 9 h. After 2 mM thymidine treatment for another 16 h, cells were again released from thymine treatment and collected at different time points (G1: 0 h release, S: 4 h release, G2/M: 8 h release). For collection of cells with IR treatment at different phases of the cell cycle, cells were subjected to IR (2 Gy or 10 Gy) with a 1-h or a 24-h recovery time.
Indicated cells were collected and fixed in 70% ethanol overnight. After two washes with phosphate-buffered saline, cells were incubated in propidium iodide solution (50 μg/mL) with RNase A (100 μg/mL) for 30 min. Cell cycle profiles were analyzed by flow cytometry.
Proximity Ligation Assay
Proximity ligation assays were performed in HeLa cells by using Duolink In Situ Red Starter Kit Mouse/Rabbit (DUO92101–1KT, Sigma-Aldrich) according to the manufacturer’s protocol.
HR-GFP and NHEJ-GFP reporter assays
U2OS cells stably expressing HR reporter DR-GFP or NHEJ-GFP (EJ5-GFP) reporter were gifts from the Albert C. Koong laboratory at MD Anderson Cancer Center. For detecting NHEJ and HR repair efficiency in a FOXK1 KD background, cells were first transfected with 50 pmol of the indicated siRNA with Lipofectamine RNAiMAX (Thermo Fisher Scientific). Twenty-four hours later, 2 μg of I-SceI expression vector was transfected into the cells with Lipofectamine 3000 (Thermo Fisher Scientific). After culture for an additional 48 h, cells were collected and subjected to flow cytometry analysis to determine percentages of GFP-positive cells.
For detecting HR repair efficiency in FOXK1 overexpression background, 1 × 106 cells were first electrotransfected with 5 μg of pLenti-HA-Flag–tagged FOXK1-related plasmids by the Neon Transfection System (Thermo Fisher Scientific). Twenty-four hours later, cells were transfected with 50 pmol sictrl or siBRCA1 with Lipofectamine RNAiMAX (Thermo Fisher Scientific). After another 24 h, 2 μg of I-SceI expression vector were transfected into the cells with Lipofectamine 3000 (Thermo Fisher Scientific). After culture for an additional 48 h, cells were collected and subjected to flow cytometry analysis to determine percentages of GFP-positive cells. Results were obtained from three independent experiments.
RNA-seq and data analysis
HeLa WT and FOXK1 KO, FOXK2 KO, and DKO cells and FOXK1 KO HeLa cells reconstituted with GFP-tagged FOXK1 WT, F201A, H355A, and F201AH355A cells (each with two biological replicates) were collected; total RNA was then extracted by using the RNeasy Mini Kit (QIAGEN, 74104) according to the manufacturer’s instructions. The library was prepared with the Illumina TruSeq Stranded Total RNA Library Prep Kit including rRNA depletion and sequencing at NextSeq 550 (Illumina) to generate 75-bp pairedends.
For RNA-seq data analysis, reads were adaptor-trimmed and preprocessed with the Cutadapt (v1.15) software for quality control and data filtering. Genome mapping was conducted with use of STAR (v2.5.3a) (Dobin et al., 2013) and the human reference genome (UCSC hg38). Uniquely mapped reads overlapping genes were counted by HTseq-count with default parameters by using annotation from ENSEMBL v83. Only genes with > 5 reads in at least one sample were retained. The raw read counts of retained genes were submitted for differential expression analysis of cases compared with controls with DESeq2 software (Anders and Huber, 2010). Resulting P values were adjusted by using the Benjamini and Hochberg approach (Benjamini and Hochberg, 1995) to control for false discovery rate (FDR). Genes with fold change (FC) >1.2 (or FC < 0.83) and FDR < 0.05 were assigned as differentially expressed genes (DEGs). Standard gene set enrichment analysis was performed with a hypergeometric test using RDAVID WebService (v1.19.0) (Fresno and Fernández, 2013). The resulting P values were also adjusted by using the Benjamini and Hochberg approach. Expression of the genes or transcripts were normalized as FPKM (fragments per kilobase of transcript per million mapped reads) for accurate quantification using RSEM (v1.3.0) (Li and Dewey, 2011). K-means clustering analysis was conducted to separate differentially expressed genes into four subgroups based on expression across all experimental groups.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed with use of Student t tests or one-way ANOVA. All results are presented as means (±SD) of experiments repeated at least two times or more unless indicated otherwise. P value less than 0.05 were considered statistically significant.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| FOXK1 | Cell Signaling Technology | Cat#12025; RRID: https://antibodyregistry.org/AB_2797801 |
| FOXK2 | Bethyl Laboratories | Cat# A301-729A; RRID: https://antibodyregistry.org/AB_1211448 |
| Flag | Sigma-Aldrich | Cat#F3165; RRID:AB_262044 |
| HA | Sigma-Aldrich | Cat#H3663; RRID: https://antibodyregistry.org/AB_262051 |
| GFP | Santa Cruz Biotechnology | Cat#sc-9996; RRID: https://antibodyregistry.org/AB_627695 |
| 53BP1 | Novus Biologicals | Cat#NB100-304; RRID: https://antibodyregistry.org/AB_10003037 |
| 53BP1 | Santa Cruz Biotechnology | Cat#sc-515841 |
| Vinculin | Sigma-Aldrich | Cat#V9131; RRID:AB_477629 |
| BRCA1 | Santa Cruz Biotechnology | Cat#sc-6954; RRID: https://antibodyregistry.org/AB_626761 |
| CHK1 | Cell Signaling Technology | Cat#2360; RRID: https://antibodyregistry.org/AB_2080320 |
| pCHK1 (S317) | Cell Signaling Technology | Cat#2344; RRID: https://antibodyregistry.org/AB_331488 |
| CHK2 | Cell Signaling Technology | Cat#6334; RRID: https://antibodyregistry.org/AB_11178526 |
| pCHK2 (T68) | Cell Signaling Technology | Cat#2197; RRID: https://antibodyregistry.org/AB_2080501 |
| γH2A.X | Millipore | Cat#05-636; RRID: https://antibodyregistry.org/AB_309864 |
| RAD51 | Abcam | Cat#ab63801; RRID: https://antibodyregistry.org/AB_1142428 |
| H3 | Cell Signaling Technology | Cat#9715; RRID:AB_331563 |
| RIF1 | Cell Signaling Technology | Cat#95558S; RRID: https://antibodyregistry.org/AB_2800249 |
| PTIP | Abcam | Cat#ab70434; RRID: https://antibodyregistry.org/AB_1270106 |
| pDNA PKcs (S2056) | Abcam | Cat#ab18192; RRID: https://antibodyregistry.org/AB_869495 |
| Histone H4 (dimethyl K20) | Abcam | Cat#ab9052; RRID: https://antibodyregistry.org/AB_1951942 |
| H2AK15ub | Millipore | Cat#MABE1119; |
| β-tubulin | Sigma-Aldrich | Cat#T5168; RRID: https://antibodyregistry.org/AB_477579 |
| β-tubulin | Santa Cruz Biotechnology | Cat#sc-9104; RRID: https://antibodyregistry.org/AB_2241191 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Olaparib | BioVision | Cat#1952-5 |
| AZD0156 | Selleck Chemicals | Cat# S8375 |
| AZD6738 | Selleck Chemicals | Cat# S7693 |
| NU7441 | Selleck Chemicals | Cat#S2638 |
| Deposited Data | ||
| 53BP1 raw mass spectrometry data | This paper | ProteomeXchange Consortium PXD020090 |
| RNA-seq data | This paper | GSE151029 |
| FOXK1 and FOXK2 raw mass spectrometry data | (Li et al., 2015) | ProteomeXchange Consortium PXD001383 |
| Experimental Models: Cell Lines | ||
| HEK293T | ATCC | Cat#CRL-3216 |
| HEK293A | Thermo Fisher Scientific | Cat# R70507 |
| HeLa S3 | ATCC | Cat#CCL-2.2 |
| MCF10A | ATCC | Cat#CRL-10317 |
| Oligonucleotides | ||
| sgRNA sequences listed in Table S3 | This study | N/A |
| sequences of primers listed in Table S4 | This study | N/A |
| siFOXK1 | QIAGEN | SI04770801 |
| siBRCA1 | QIAGEN | SI02664361 |
| si53BP1 | QIAGEN | SI02653168 |
| sictrl | QIAGEN | SI04380467 |
| Software and Algorithms | ||
| Gepia | Gepia | http://gepia.cancer-pku.cn/index.html |
| Graphpad Prism | Graphpad Software | https://www.graphpad.com/scientific-software/prism/ |
| OpenComet | Gyori et al., 2014 | http://www.cometbio.org/ |
| ImageJ | ImageJ | https://imagej.nih.gov/ij/ |
Highlights.
FOXK1 interacts with 53BP1 and regulates 53BP1 localization to DSBs
FOXK1–53BP1 interaction is enhanced in S phase in an ATM/CHK2-dependent manner
FOXK1 regulates the association of 53BP1 with RIF1 and PTIP upon DNA damage
ACKNOWLEDGMENTS
We thank all members of the Chen laboratory for their help and constructive discussion. We also thank Sarah Bronson and Tamara K. Locke in Scientific Publications, Research Medical Library, MD Anderson, for help with the scientific editing of the manuscript. This work was supported in part by NIH R01CA216911 to J.C. In addition, many reagents used in this study were generated in part with the support of CPRIT award RP160667and NIH grants P01CA193124, R01CA210929, and R01CA216437 to J.C. J.C. also received support from the Pamela and Wayne Garrison Distinguished Chair in Cancer Research. We thank the staff of MD Anderson’s Flow Cytometry and Cellular Imaging Facility (supported by MD Anderson’s NIH Cancer Center Support grant P30CA016672) for their help with the flow cytometry experiments and the UTHealth Cancer Genomics Core for RNA-seq data generation and analysis (supported by CPRIT grant RP180734).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108018.
REFERENCES
- An L, Jiang Y, Ng HH, Man EP, Chen J, Khoo US, Gong Q, and Huen MS (2017). Dual-utility NLS drives RNF169-dependent DNA damage responses. Proc. Natl. Acad. Sci. USA 114, E2872–E2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, and Huber W (2010). Differential expression analysis for sequence count data. Genome Biol. 11, R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, and Hochberg Y (1995). Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. B 57, 289–300. [Google Scholar]
- Bouwman P, Aly A, Escandell JM, Pieterse M, Bartkova J, van der Gulden H, Hiddingh S, Thanasoula M, Kulkarni A, Yang Q, et al. (2010). 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat. Struct. Mol. Biol 17, 688–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunting SF, Callén E, Wong N, Chen HT, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao L, et al. (2010). 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 141, 243–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvisi DF, Pinna F, Meloni F, Ladu S, Pellegrino R, Sini M, Daino L, Simile MM, De Miglio MR, Virdis P, et al. (2008). Dual-specificity phosphatase 1 ubiquitination in extracellular signal-regulated kinase-mediated control of growth in human hepatocellular carcinoma. Cancer Res. 68, 4192–4200. [DOI] [PubMed] [Google Scholar]
- Cao L, Xu X, Bunting SF, Liu J, Wang RH, Cao LL, Wu JJ, Peng TN, Chen J, Nussenzweig A, et al. (2009). A selective requirement for 53BP1 in the biological response to genomic instability induced by Brca1 deficiency. Mol. Cell 35, 534–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman JR, Barral P, Vannier JB, Borel V, Steger M, Tomas-Loba A, Sartori AA, Adams IR, Batista FD, and Boulton SJ (2013). RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double-strand break resection. Mol. Cell 49, 858–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charier G, Couprie J, Alpha-Bazin B, Meyer V, Quéméneur E, Guérois R, Callebaut I, Gilquin B, and Zinn-Justin S (2004). The Tudor tandem of 53BP1: a new structural motif involved in DNA and RG-rich peptide binding. Structure 12, 1551–1562. [DOI] [PubMed] [Google Scholar]
- Chen J, Feng W, Jiang J, Deng Y, and Huen MS (2012). Ring finger protein RNF169 antagonizes the ubiquitin-dependent signaling cascade at sites of DNA damage. J. Biol. Chem 287, 27715–27722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Wu J, Liang G, Geng G, Zhao F, Yin P, Nowsheen S, Wu C, Li Y, Li L, et al. (2020). CHK2-FOXK axis promotes transcriptional control of autophagy programs. Sci. Adv 6, eaax5819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JH, Park MJ, Kim KW, Choi YH, Park SH, An WG, Yang US, and Cheong J (2005). Molecular mechanism of hypoxia-mediated hepatic gluconeogenesis by transcriptional regulation. FEBS Lett. 579, 2795–2801. [DOI] [PubMed] [Google Scholar]
- Clark KL, Halay ED, Lai E, and Burley SK (1993). Co-crystal structure of the HNF-3/fork head DNA-recognition motif resembles histone H5. Nature 364, 412–20. [DOI] [PubMed] [Google Scholar]
- Daley JM, and Sung P (2013). RIF1 in DNA break repair pathway choice. Mol. Cell 49, 840–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Virgilio M, Callen E, Yamane A, Zhang W, Jankovic M, Gitlin AD, Feldhahn N, Resch W, Oliveira TY, Chait BT, et al. (2013). Rif1 prevents resection of DNA breaks and promotes immunoglobulin class switching. Science 339, 711–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrova N, Chen YC, Spector DL, and de Lange T (2008). 53BP1 promotes non-homologous end joining of telomeres by increasing chromatin mobility. Nature 456, 524–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drané P, Brault ME, Cui G, Meghani K, Chaubey S, Detappe A, Parnandi N, He Y, Zheng XF, Botuyan MV, et al. (2017). TIRR regulates 53BP1 by masking its histone methyl-lysine binding function. Nature 543, 211–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durocher D, and Jackson SP (2002). The FHA domain. FEBS Lett. 513, 58–66. [DOI] [PubMed] [Google Scholar]
- Escribano-Díaz C, Orthwein A, Fradet-Turcotte A, Xing M, Young JT, Tkáč J, Cook MA, Rosebrock AP, Munro M, Canny MD, et al. (2013). A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol. Cell 49, 872–883. [DOI] [PubMed] [Google Scholar]
- Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, et al. (2005). Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434, 917–921. [DOI] [PubMed] [Google Scholar]
- Feng L, Fong KW, Wang J, Wang W, and Chen J (2013). RIF1 counteracts BRCA1-mediated end resection during DNA repair. J. Biol. Chem 288, 11135–11143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fradet-Turcotte A, Canny MD, Escribano-Díaz C, Orthwein A, Leung CC, Huang H, Landry MC, Kitevski-LeBlanc J, Noordermeer SM, Sicheri F, and Durocher D (2013). 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature 499, 50–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freddie CT, Ji Z, Marais A, and Sharrocks AD (2007). Functional interactions between the Forkhead transcription factor FOXK1 and the MADS-box protein SRF. Nucleic Acids Res. 35, 5203–5212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fresno C, and Fernández EA (2013). RDAVIDWebService: a versatile R interface to DAVID. Bioinformatics 29, 2810–2811. [DOI] [PubMed] [Google Scholar]
- Grant GD, Gamsby J, Martyanov V, Brooks L 3rd, George LK, Mahoney JM, Loros JJ, Dunlap JC, and Whitfield ML (2012). Live-cell monitoring of periodic gene expression in synchronous human cells identifies Forkhead genes involved in cell cycle control. Mol. Biol. Cell 23, 3079–3093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyori BM, Venkatachalam G, Thiagarajan PS, Hsu D, and Clement MV (2014). OpenComet: an automated tool for comet assay image analysis. Redox Biol. 2, 457–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Q, Kim H, Huang R, Lu W, Tang M, Shi F, Yang D, Zhang X, Huang J, Liu D, and Songyang Z (2015). The Daxx/Atrx Complex Protects Tandem Repetitive Elements during DNA Hypomethylation by Promoting H3K9 Trimethylation. Cell Stem Cell 17, 273–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helleday T, Bryant HE, and Schultz N (2005). Poly(ADP-ribose) polymerase (PARP-1) in homologous recombination and as a target for cancer therapy. Cell Cycle 4, 1176–1178. [DOI] [PubMed] [Google Scholar]
- Heyer WD, Ehmsen KT, and Liu J (2010). Regulation of homologous recombination in eukaryotes. Annu. Rev. Genet 44, 113–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji H, Wang J, Guo J, Li Y, Lian S, Guo W, Yang H, Kong F, Zhen L, Guo L, and Liu Y (2016). Progress in the biological function of alpha-enolase. Anim. Nutr 2, 12–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji ZG, Jiang HT, and Zhang PS (2018). FOXK1 promotes cell growth through activating wnt/β-catenin pathway and emerges as a novel target of miR-137 in glioma. Am. J. Transl. Res 10, 1784–1792. [PMC free article] [PubMed] [Google Scholar]
- Jin GZ, Zhang Y, Cong WM, Wu X, Wang X, Wu S, Wang S, Zhou W, Yuan S, Gao H, et al. (2018). Phosphoglucomutase 1 inhibits hepatocellular carcinoma progression by regulating glucose trafficking. PLoS Biol. 16, e2006483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Li F, He Q, Deng T, Xu J, Jin F, Coarfa C, Putluri N, Liu D, and Songyang Z (2017). Systematic analysis of human telomeric dysfunction using inducible telosome/shelterin CRISPR/Cas9 knockout cells. Cell Discov. 3, 17034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, and Dewey CN (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Wang W, Wang J, Malovannaya A, Xi Y, Li W, Guerra R, Hawke DH, Qin J, and Chen J (2015). Proteomic analyses reveal distinct chromatin-associated and soluble transcription factor complexes. Mol. Syst. Biol 11, 775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieber MR (2010). The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem 79, 181–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manis JP, Morales JC, Xia Z, Kutok JL, Alt FW, and Carpenter PB (2004). 53BP1 links DNA damage-response pathways to immunoglobulin heavy chain class-switch recombination. Nat. Immunol 5, 481–487. [DOI] [PubMed] [Google Scholar]
- Munoz IM, Jowsey PA, Toth R, and Rouse J (2007). Phospho-epitope binding by the BRCT domains of hPTIP controls multiple aspects of the cellular response to DNA damage. Nucleic Acids Res. 35, 5312–5322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olive PL, and Banáth JP (2006). The comet assay: a method to measure DNA damage in individual cells. Nat. Protoc 1, 23–29. [DOI] [PubMed] [Google Scholar]
- Post A, Pannekoek WJ, Ross SH, Verlaan I, Brouwer PM, and Bos JL (2013). Rasip1 mediates Rap1 regulation of Rho in endothelial barrier function through ArhGAP29. Proc. Natl. Acad. Sci. USA 110, 11427–11432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulsen M, Lukas C, Lukas J, Bekker-Jensen S, and Mailand N (2012). Human RNF169 is a negative regulator of the ubiquitin-dependent response to DNA double-strand breaks. J. Cell Biol 197, 189–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz LB, Chehab NH, Malikzay A, and Halazonetis TD (2000). p53 binding protein 1 (53BP1) is an early participant in the cellular response to DNA double-strand breaks. J. Cell Biol 151, 1381–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, and Garry DJ (2012). Sin3 interacts with Foxk1 and regulates myogenic progenitors. Mol. Cell. Biochem 366, 251–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, Bowlin KM, and Garry DJ (2010). Fhl2 interacts with Foxk1 and corepresses Foxo4 activity in myogenic progenitors. Stem Cells 28, 462–469. [DOI] [PubMed] [Google Scholar]
- Sukonina V, Ma H, Zhang W, Bartesaghi S, Subhash S, Heglind M, Foyn H, Betz MJ, Nilsson D, Lidell ME, et al. (2019). FOXK1 and FOXK2 regulate aerobic glycolysis. Nature 566, 279–283. [DOI] [PubMed] [Google Scholar]
- Wang X, Takenaka K, and Takeda S (2010). PTIP promotes DNA double-strand break repair through homologous recombination. Genes Cells 15, 243–254. [DOI] [PubMed] [Google Scholar]
- Wang W, Li X, Lee M, Jun S, Aziz KE, Feng L, Tran MK, Li N, McCrea PD, Park JI, and Chen J (2015). FOXKs promote Wnt/β-catenin signaling by translocating DVL into the nucleus. Dev. Cell 32, 707–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward IM, Minn K, van Deursen J, and Chen J (2003). p53 Binding protein 53BP1 is required for DNA damage responses and tumor suppression in mice. Mol. Cell. Biol 23, 2556–2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward IM, Reina-San-Martin B, Olaru A, Minn K, Tamada K, Lau JS, Cascalho M, Chen L, Nussenzweig A, Livak F, et al. (2004). 53BP1 is required for class switch recombination. J. Cell Biol 165, 459–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zgheib O, Pataky K, Brugger J, and Halazonetis TD (2009). An oligomerized 53BP1 tudor domain suffices for recognition of DNA double-strand breaks. Mol. Cell. Biol 29, 1050–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YW, and Vande Woude GF (2007). Mig-6, signal transduction, stress response and cancer. Cell Cycle 6, 507–513. [DOI] [PubMed] [Google Scholar]
- Zhang A, Peng B, Huang P, Chen J, and Gong Z (2017). The p53-binding protein 1-Tudor-interacting repair regulator complex participates in the DNA damage response. J. Biol. Chem 292, 6461–6467. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq data generated in this study are available at NCBI GEO database with the accession number GSE151029. The 53BP1 mass spectrometry data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD020090. The accession number for the FOXK1 and FOXK2 MS data reported in this paper is PRIDE: PXD001383