

ABSTRACT
Recurrence of advanced epithelial ovarian cancer is common despite optimal surgical debulking and initial favorable responses to chemotherapy. Evidences suggest that cancer stem cells (CSCs) have inherent resistance to conventional therapies such as chemotherapy and play a decisive role in cancer recurrence. Cancer stem cells are also believed to be able to evade immunological attack. However, this study showed a different scenario in which cancer stem-like cells are more vulnerable to immunosurveillance. Our study demonstrated that isolated murine cancer stem-like cells, stem cell antigen (SCA)-1+ ID8 and CD133+ HM-1 cells, were susceptible to phagocytosis by macrophages and consequent CD8+ T cell immunity. The increased phagocytosis of these stem cell-like cells is attributed to low CD47 protein expression. SCA-1+ ID8 cells were able to grow in syngeneic mice but were soon rejected. Restoring CD47 expression delayed this immune-mediated rejection. SCA-1+ ID8 cells showed rapid growth by mixing with bulk ID8 cells. These results suggest that stem-like cells could be protected by surrounding non-stem cancer cells from immune attack. Similarly, both isolated human CD24−/low SKOV3 stem-like cells and spheroid OVCAR3 cells expressed lower CD47 levels. Our study provided novel insights into the immune characteristics of CSCs within a tumor microenvironment. The results might lead to the design of more effective treatment strategies for ovarian cancer.
KEYWORDS: Cancer stem cell, CD47, tumor microenvironment, ovarian cancer
Introduction
Epithelial ovarian cancer is the leading cause of death from gynecologic malignancy worldwide because a large proportion of patients with this cancer are diagnosed at advanced stages. Currently, appropriate ovarian cancer treatment includes optimal surgical debulking whenever possible, followed by adjuvant chemotherapy, if indicated. Despite the high chemotherapy response rate, ovarian cancer treatment is still challenging because of frequent disease relapse. For most of these patients, cancer will recur at gradually shortened time intervals, and they eventually develop drug resistance. Drug resistance is thought to occur due to the presence of a subpopulation of dormant cancer cells within the tumor mass that can promote and sustain tumor growth and impart drug resistance, leading to cancer recurrence after chemotherapy.1 These dormant cells are believed to possess stemness properties such as self-renewal, differentiation potential, and resistance to conventional chemotherapies and radiotherapies.
Based on data from clonality, genomic, epigenomic, and proteomic studies, ovarian cancer is a heterogeneous and complex disease. Ovarian cancer stem cells (CSCs) have been extensively studied for a long time. It is generally believed that these tumor-initiating cells survive during chemotherapy and play a decisive role in later tumor recurrence. Theoretically, treatment strategies should focus on targeting these cells to eradicate ovarian cancer. However, no universally definitive biomarker of ovarian CSCs exists so far. Phenotypically diverse ovarian CSC populations have been isolated from both patient specimens and immortalized cell lines; these cells have been characterized, and variable markers have been identified. It is unclear whether different CSC subsets isolated using diverse isolation protocols exhibit varying biological functions. A series of stemness markers, such as CD44/CD24, CD133, octamer-binding transcription factor 4 (Oct4), NANOG, and aldehyde dehydrogenase (ALDH), have been used to isolate CSCs from primary ovarian cancer cells or cell lines.2–5 In addition, different approaches, such as side population (SP) sorting and ALDH and hypoxia-inducing assays, have been used to enrich potential CSCs.6,7 These isolated cells generally demonstrate high tumorigenicity, clonal growth, and chemo- or radio-resistance. Current CSC studies have primarily used immunocompromised mouse models to examine the tumorigenesis of these cells in vivo. Previous studies have neglected the immune reactivity of these cells. Cells that transform in vivo are usually recognized by immune cells and are eliminated through immune reactions before forming tumors; this process is called immunosurveillance. On the other hand, tumor cells might develop a mechanism to escape immunologic attack, and the tumor microenvironment is usually immunosuppressive. Whether CSCs share the same immune escape mechanisms remains unknown.
A recent study showed that chemotherapy usually leaves behind an entity of CSC-like cells, which are more invasive and induce disease relapse.8 Conversely, recurrent ovarian cancers are enriched with CSCs, indicating that CSCs might contribute to cancer recurrence.9 Residual CSCs that survive chemotherapy might provide a favorable microenvironment to facilitate the growth of residual cells. This environment provides not only autocrine and paracrine signaling but also has a complex immune network interacting with surrounding cells. Understanding CSC immunoreactivity is important to improve the treatment and prevention of ovarian cancer recurrence.
In the present study, we isolated murine and human ovarian cancer stem-like cells from murine and human cell lines, respectively. Surviving cells were treated with either cisplatin or taxol in nonattachment culture flasks. Selected cells exhibited stemness properties such as high clonogenic capacity, enriched proportion of SP cells, tumorigenesis, and increased stem cell-related surface protein expression. This approach enabled the evaluation of the immune reaction of these stem-like cells in an immunocompetent mouse model.
Materials and methods
Animals, cells, and antibodies
NOD-SCID, C57BL/6, and C57BL/6 × C3/He F1 female mice were purchased from BioLASCO, Taiwan. Animals were maintained under specific pathogen-free conditions. This study has been approved by the Institutional Review Board (IRB No. 14MMHIS119) and Institutional Animal Care and Use Committee (IACUC No. MMH-A-S-102-57) of MacKay Memorial Hospital, Taipei, Taiwan. All procedures were conducted in accordance with approved protocols and recommendations for the proper care and use of laboratory animals. Murine ovarian cancer cell lines, ID8 (from C57BL/B6 mice) and HM-1 (from C57BL/6 × C3/He F1 mice), were cultured as previously described.10 The mouse ID8-luc cells were derived from mouse ovarian cancer cell line MOSEC-luc (C57BL/6 origin and engineered expression of firefly luciferase) with VEGF overexpression. The murine BALB/c macrophage cell line RAW 264.7 was cultured in Corning® Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with HycloneTM 10% fetal bovine serum and 100 U/mL penicillin–streptomycin solution (Biological Industries, CT). T cells and splenocytes were cultured in CTL media (an RPMI-1640 medium supplemented with 2 mM GibcoTM 2-mercaptoethanol) plus 10% fetal bovine serum, 100 U/mL penicillin, 100 μg/mL streptomycin, and 10 U/mL mIL-2 (PeproTech, NJ). SKOV3 and OVCAR-3 cells were obtained from the American Type Culture Collection and were maintained according to the manufacturer’s recommendations. U937 monocytic cells were maintained in CTL medium. For macrophage differentiation, cells (a density of 5 × 105/mL) were cultured in RPMI-1640 with 10% fetal bovine serum containing 100 nM phorbol 12-myristate 13-acetate (PMA) for 2 days. Antibodies used for labeling the stem-like cells included anti-mouse stem cell antigen (SCA)-1 (1:50, eBioscience, CA), anti-human CD24 (1:20, Biolegend, CA), anti-human CD44 (1:20, Biolegend, CA), anti-mouse CD133 (1:50, eBioscience), and anti-human EpCAM (1:20, Biolegend, CA) antibodies.
Isolation of stem-like cells from ovarian cancer cell lines
Murine ovarian cancer cells, ID8 and HM-1, were cultured with serially increasing concentrations of cisplatin (0.25 – 0.5 – 1 – 2 µg/mL) or taxol (5 – 10 – 15 – 20 µM) and were then maintained at the highest concentration for 1 month before cells were cultured in spheroid culture systems. Spheroid cultures were performed in methylcellulose-based culture medium. ID8 cells (5 × 103 cells/mL) were cultured in 0.6% methylcellulose (Sigma-Aldrich, MD) MEM medium in ultra-low attachment 6-well plates (Corning, MA) for 10 days to form spheroid cells. Cells were then resuspended as a monolayer in normal culture dishes containing MEM medium for 2 days, after which these cells were re-cultured in ultra-low attachment plates. These steps were repeated four times to obtain stem-like cells. Stem-like cells of the SKOV3 and OVCAR3 human ovarian cancer cell lines were obtained using the same procedure. Cells with defined markers were isolated through flow sorting (BD FACSVantageTM).
Sphere and soft agar colony-forming assays
For the sphere formation assay, cells at a density of 5 × 103/mL were suspended in DMEM/F12 media (Corning, VA) supplemented with 0.8% methylcellulose (Sigma-Aldrich), 2% B27 (Invitrogen, NY), 4 µg/mL insulin (Sigma-Aldrich), 20 ng/mL human EGF (ProSpec, Ness-Ziona, Israel), and 10 ng/mL human β-FGF (ProSpec, Ness-Ziona, Israel). Two milliliters of cells were then seeded into each well of a 6-well ultra-low attachment plate. After 3-week culture, the number of spheres formed was quantified directly using an inverted light microscope. For the soft agar colony formation assay, 2 mL of 0.5% agarose in complete media was added to each well of a 6-well plate as the bottom layer. Cells (5 × 103) from each group were then mixed with 1 mL of 0.35% agarose in complete media; the mixture was then added over the bottom agarose layer. Cells were cultured until colonies formed and were then fixed in pure ethanol with 0.05% crystal violet. The colonies were quantified using an inverted microscope.
SP analysis
Cells were trypsinized and resuspended at a density of 1 × 106/mL. For the SP assay, 5 μL of 1 mg/mL Hoechst 33342 (Sigma-Aldrich) was added to 1 mL of resuspended cells and incubated at 37°C for 2 h. To inhibit dye pumping, cells were incubated with 50 μM verapamil (Sigma-Aldrich) or 10 μM of the ABCG2 inhibitor fumitremorgin (FTC) (Sigma-Aldrich) 10 min before incubation with Hoechst 33342. Cells were washed with cold media and stained with PI to exclude dead cells. SPs were further analyzed through flow cytometry.
Immunogenicity and tumor rejection assay
In Figure 3a, C57BL/6 female mice were injected i.p. with different numbers of either bulk ID8-luc or SCA-1+ ID8-Luc cells (1 × 102, 5 × 102, 2.5 × 103, 1.5 × 104 cells/mouse) on day 0. Tumor growth was measured by noninvasive bioluminescence Imaging on D10, D14, D25, and D32. In Figure 3b, analysis of splenocytes from those mice injected with 2.5 × 103 bulk or SCA-1+ ID8 cells (as immunized) and were re-stimulated with irradiated bulk or SCA-1+ ID8 cells (5 × 104 cells) on D32. In Figure 3c, the immunogenicity of bulk or SCA-1+ ID8 cells was evaluated by analysis of splenocytes from mice immunized by these cells. C57BL/6 female mice were injected i.p. with irradiated bulk ID8 or SCA-1+ ID8 cells (5 × 104/mouse) on day 0, 7, and 14. On day 33, splenocytes (106 cells/mL) from these mice were harvested and cocultured with (bulk) ID8 cells or SCA-1+ ID8 cells (5 × 104 cells/mL) in CTL media and GolgiPlug (1 μL/mL). On the following day, cells were harvested and stained with the APC-conjugated monoclonal rat anti-mouse CD8α antibody followed by the FITC-conjugated rat anti-mouse IFN-γ antibody; cells were then subjected to flow cytometric analysis. To evaluate tumor rejection, cells were obtained by peritoneal lavage with HBSS and were stained with PE-conjugated rat anti-mouse CD45.2 (1:50, eBioscience) and APC-conjugated rat anti-mouse SCA-1 antibodies; CD45.2−/SCA-1+ cells were then subjected to flow cytometric analysis.
Figure 3.
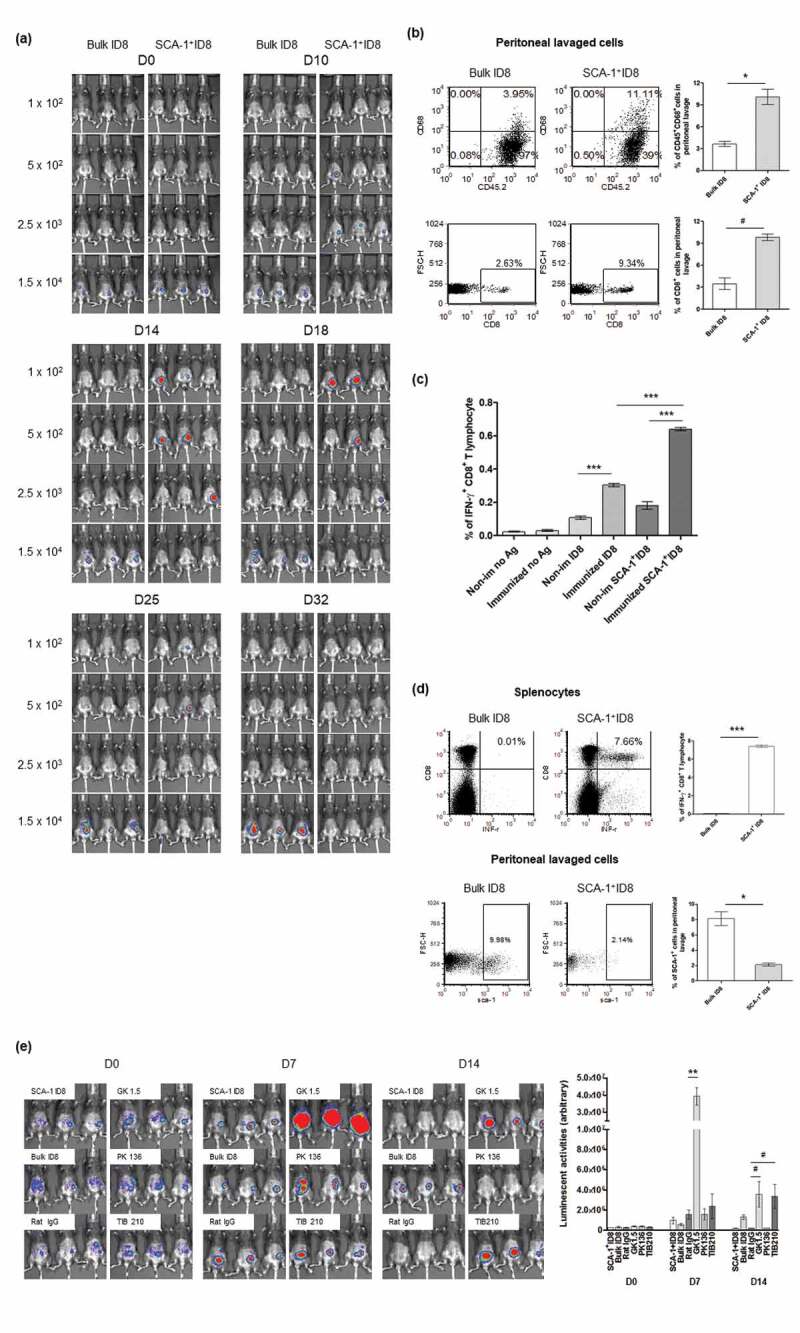
SCA-1+ ID8 cells were immunogenic and being rejected in vivo by anti-tumor immunity involving CD4+ and CD8+ T lymphocytes. (a) SCA-1+ ID8 cells showed stronger tumorigenesis initially by D18 but disappeared later. Bulk ID8 cells grew slower but did form tumor eventually by day 25. (b) Representative flow analysis of CD8+ and CD68+ cells in peritoneal lavage of mice injected with 2.5 × 103 bulk ID8 and SCA-1+ ID8 cells on D22. The results show significantly increased percentages of CD45+CD68+ cells and CD8+ cells in peritoneal lavage from mice implanted with SCA-1+ ID8, comparing to mice with bulk ID8 cells. (c) Analysis of splenocytes demonstrated the highest number of IFN-γ+ CD8+ T lymphocytes was found in mice in which SCA-1+ tumors disappeared (splenocytes from mice injected with 2.5 × 103 SCA-1+ cells on D32, as immunized) and were re-stimulated with irradiated SCA-1+ ID8 cells (p < .0001, splenocytes re-stimulated by irradiated bulk ID8 cells [5 × 104 cell] [labeled as immunized ID8] vs splenocytes re-stimulated by irradiated SCA-1+ ID8 cells [5 × 104 cells] [labeled as immunized SCA-1+ ID8]). Splenocytes from those mice with SCA-1+ tumor disappeared also showed higher percentages of IFN-γ+ CD8+ T lymphocytes than that from mice injected with 2.5 × 103 cells bulk ID8 cells, irrespective of the re-stimulated antigen (p < .0001, immunized ID8 vs non-immunized ID8 and immunized SCA-1+ ID8 vs non-immunized SCA-1+ ID8). (d) Representative data show that mice immunized with 3 doses of irradiated SCA-1+ID8 cells exhibited strong tumor-specific CD8+ T lymphocyte response (p < .0001) and rejected the growth of SCA-1+ cells inside the peritoneum (p < .01). Rejection of SCA-1+ tumor was more obviously seen in mice immunized with irradiated SCA-1+ ID8 cells demonstrated by less SCA-1+ lavaged cells in peritoneal cavity. (e) Rejection of SCA-1+ ID8 tumor was abolished by CD4+ and CD8+ lymphocyte depletion. All C57BL/6 mice were implanted with SCA-1+ ID8 cells, except those labeled with “Bulk ID8” were implanted with bulk ID8 cells. Lymphocyte depletion was performed through i.p. injection of rat monoclonal antibody GK1.5 (anti-CD4), 2.43 (anti-CD8), or PK136 (anti-NK1.1) described in Materials and Methods. Signal of SCA-1+ tumor peaked on D7 if CD4+ lymphocyte was neutralized by GK 1.5 antibody. The signals were reduced on D14, possibly through the immune response elicited by rapidly growing tumor. SCA-1+ tumor became obvious on day 14 if CD8+ lymphocyte was neutralized by TIB 210 antibody. Administration of rat IgG or PK136 antibody did not alter the rejection of SCA-1+ tumors. These data suggest involvement of CD4+ and CD8+ immune reactions in the attack of SCA-1+ ID8 stem-like cells. #p < .05, *p < .01, **p < .001, ***p < .0001.
Lymphocyte depletion assay
Mice were injected i.p. with either ID8 cells or CD133+ HM-1-ova cells (5 × 104/mouse for ID8 and SCA-1+ ID8 cells and 106 for CD133+ HM-1-ova cells). The elimination of lymphocyte subpopulations was achieved through injections with 100 mg of GK1.5 (anti-CD4), 2.43 (anti-CD8), and PK136 (anti-NK1.1) rat monoclonal antibodies. Depletion was initiated 1 week before the tumor challenge and continued every other day for the duration of tumor growth. Depletion was assessed through flow cytometric analysis of spleen cells 1 day after the fourth antibody was administered to confirm the depletion of CD8, CD4, or natural killer cells. Tumor growth was evaluated either using a Xenogen IVIS luminescent imaging system or through subcutaneous tumor size measurement.
SCA-1+ ID8-specific T cell generation
C57BL/6 female mice were injected intraperitoneally (i.p.) with SCA-1+ ID8 cells (106/mouse) on days 0 and 10 as immunization. Ten days later (day 20), splenocytes (106 cells/mL) were harvested and cocultured with irradiated SCA-1+ ID8 cells as feeder cells for immunogen re-stimulation (5 × 104 cells/mL) in CTL media. Surviving T cells emerged after 2 months of culture. Specific T cells were surface stained with an APC-conjugated anti-mouse CD8α antibody (1:100, eBioscience, CA) and were intracellularly stained with an FITC-conjugated anti-mouse IFN-γ antibody (1:50, eBioscience). Subsequently, these cells were assayed through flow cytometric analysis (BD FACS Calibur).
In vitro phagocytosis assay and anti-CD47 antibody treatment
CFSE-labeled target cells (105) were cocultured with RAW 264.7 murine macrophage cells (5 × 105), differentiated human U937 cells (5 × 105), or DC98 dendritic cells (5 × 105) for 1 h at 37°C in a humidified atmosphere containing 5% CO2. Cells were stained with CD11b or CD11 c and were subjected to flow cytometric analysis. To block CD47, CFSE-labeled target cells (106 cells/mL) were treated with an anti-mouse CD47 antibody (LEAFTM purified anti-mouse CD47 antibody, Biolegend, CA) for 20 min at 37ºC. Cells were washed, resuspended in culture medium, and cocultured with macrophages and DCs.
In vitro macrophage cross-presentation assay
Target cells (106) were cocultured with peritoneal macrophages (5 × 106) from C57BL/6 female mice in flow tubes for 4.5 h. Macrophages were isolated using MACS CD11b microbeads and were then mixed with resting SCA-1+ ID8-specific T cells (a ratio of 2 × 105 macrophages to 106 T cells) in 2 mL of CTL media supplemented with mIL-2 (10 U/mL) and a BD GolgiPlugTM protein transport inhibitor (1 µL/mL). Sixteen hours later, cells were stained with the APC-conjugated monoclonal rat anti-mouse CD8α (1:100; eBioscience) antibody for 20 min, fixed with Cytofix/Cytoperm (BD Pharmingen, CA), and then intracellularly stained with the FITC-conjugated rat anti-mouse IFN-γ antibody (1:50; eBioscience) for 20 min. Thereafter, flow cytometric analysis was performed.
Quantitative polymerase chain reaction (PCR)
Two micrograms of total RNA extracted from bulk and spheroid cells was reverse transcribed to generate cDNA by using the RevertAid First Strand cDNA Synthesis Kit (Fermentas). Human (h)CD47-F (5ʹ-GGCAATGACGAAGGAGGT TA-3ʹ) and hCD47-R (5ʹ-ATCCGGTGGTATGGATGAGA-3ʹ) and mouse mCD47-F (5ʹ- GGC GCA AAG CAC CGA AGA AAT GTT-3ʹ) and mCD47-R (5ʹ-CCA TGG CAT CGC GCT TAT CCA TTT −3ʹ) primer sets were designed for quantitative PCR on the CFX96 touch real-time PCR detection system (Bio-Rad).
Statistical analysis
All data are expressed as means ± statistical errors (SEs) and represent at least two experiments. Comparisons between individual data points were made using the Student t test and ANOVA.
Results
Identification of cancer stem-like cells from two murine ovarian cancer cell lines (ID8 and HM-1)
To investigate the immunogenicity of ovarian CSCs, we isolated stem-like cells from two murine ovarian cancer cell lines (ID8, which originated from B6 mice and HM-1, which originated from C57BL/6 × C3/He F1 mice) through drug survival selection and spheroid culture. After analyzing various putative stemness markers, we found that SCA-1+ cells were gradually enriched through continuous culture in cisplatin-containing media for ID8. The proportion of SCA-1+ ID8 cells was also higher in a spheroid formation-inducing anchorage-independent culture system and was much higher after taxol and cisplatin selection (Figure 1a). These data implicate the possible stemness characteristic of the SCA-1+ protein in ID8 cells. Similarly, we documented gradual SCA-1+ cell enrichment after the sequential passage of spheroid cultures (Figure 1b, P2 to P3 to P4). SCA-1+ cell percentages decreased abruptly once suspended spheroid ID8 cells adhered to type IV collagen-coated dishes (Figure 1b, P4 vs attached). To further assess whether SCA-1+ ID8 cells are resistant to chemotherapeutic drugs, NOD-SCID mice were implanted with ID8 cells i.p. (1 × 106 cells/mouse) and treated with cisplatin plus taxol (3 mg/kg and 5 mg/kg, i.p., every 3 days). During the treatment course, the proportion of surviving peritoneal SCA-1+ cells was progressively enriched (Figure 1c). We further tested whether SCA-1+ ID8 cells exhibit CSC characteristics, including the ability to grow in anchorage-independent soft agar and form multicellular tumor spheroids. As shown in Figure 1d, a greater number of colonies and spheres were formed by SCA-1+ ID8 cells than by (bulk) ID8 cells. Reflecting previous data in identifying stem cell-like cells, cisplatin- and taxol-surviving spheroid cells were also able to form more spheres (Figure 1d). Furthermore, cells were assayed for the expression of one universal stem cell marker – ATP binding cassette transporter G2 (ABCG2), which is responsible for the pumping of a range of compounds out of cells. It is recognized as a mechanism of drug resistance in stem cells by reducing the intracellular chemotherapeutic agents. We documented that ABCG2 mRNA expression was higher in SCA-1+ ID8 cells than in bulk ID8 cells. The expression level was also higher when ID8 cells were under spheroid culture (Figure 1e). By side population (SP) phenotypic analysis, a significant SP fraction (20.83%) was found in the SCA-1+ ID8 cells, whereas only 1.2% of the bulk ID8 cells was found as the fraction (Figure 1e). This SP proportion within SCA-1+ ID8 cells was diminished after adding the ABCG2 inhibitor FTC or the ABC transporter inhibitor verapamil. Most importantly, compared with the bulk ID8 cells, SCA-1+ ID8 cells exhibited stronger in vivo tumorigenesis in NOD-SCID mice (Figure 1f). This result is consistent with the prior data, indicating that SCA-1+ ID8 cells have stem cell-like characteristics.
Figure 1.
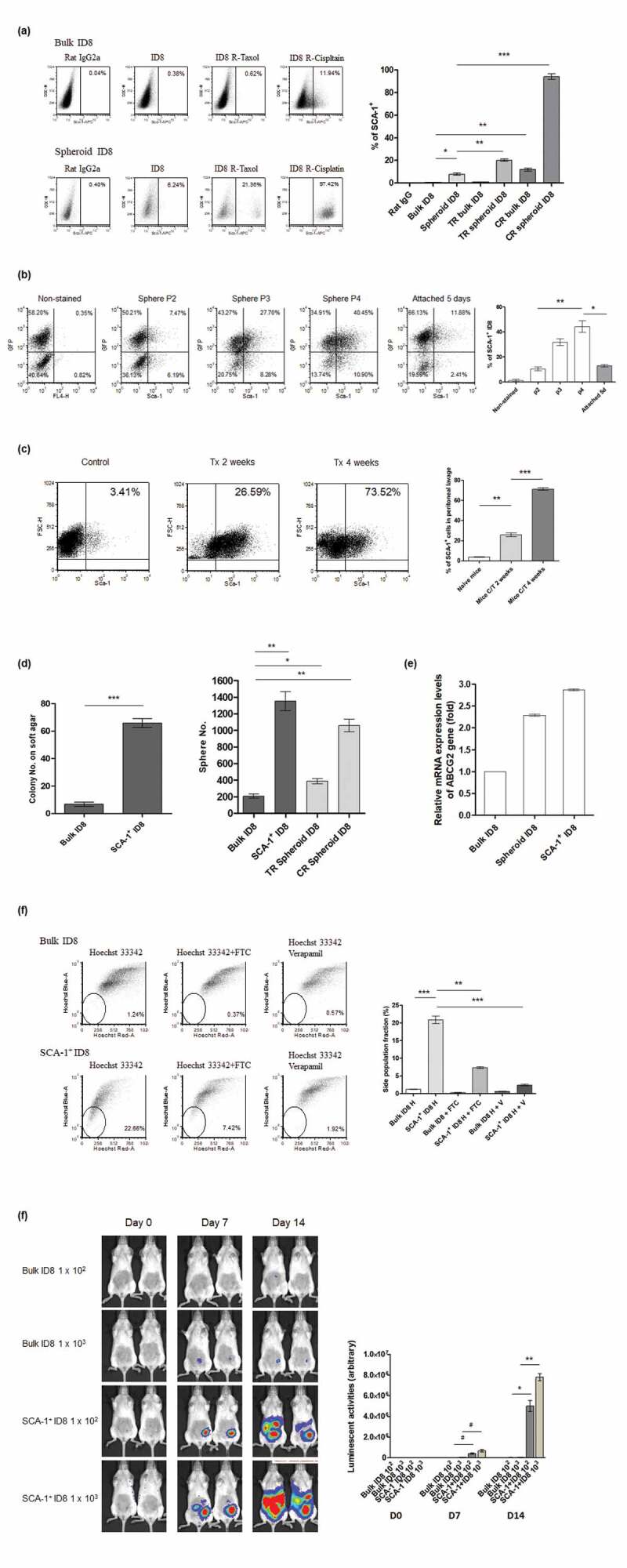
Identification and characterization of cancer stem-like cells from a murine ovarian cancer cell line- ID8. (a) Representative flow data and depicted statistical bars shows that SCA-1+ ID8 cells was enriched following long-term culture with chemotherapeutic agent (p < .001, bulk cells in normal [Bulk ID8] vs cisplatin-containing medium [CR bulk ID8]) or spheroid culture (p < .01, bulk vs spheroid culture [Spheroid ID8]), and both (p < .001 and p < .0001, spheroid cells in normal [Spheroid ID8] vs taxol [TR Spheroid ID8] vs cisplatin [CR Spheroid ID8] containing medium). (b) One representative flow data and depicted statistical bars indicated the proportion of SCA-1+ cells was increased by sequential spheroid culture of ID8 cells, and cell attachment to type IV collagen-coated dishes reversed the enrichment of SCA-1+ cells (bar graphs: average values of triplicate experiments; 10.46% to 31.82% to 44.25%, P2 to P3 to P4, respectively, p < .001, one-way ANOVA). SCA-1+ cell percentages decreased abruptly once suspended spheroid ID8 cells adhered to type IV collagen-coated dishes (44.25% vs. 12.99%, P4 vs. attached culture, p < .01, Figure 1b). (c) Enrichment of SCA-1+ cells by short- and long-term chemotherapies in vivo is shown in the representative flow data. Proportions of SCA-1+ cells were increased after cisplatin and taxol treatment (p < .0001, 2 weeks versus 4 weeks after chemotherapy). (d) SCA-1+ ID8 cells exhibited substantially higher tumorigenic ability than bulk cells did, in both clonogenic and sphere-forming assays (p < .0001 and p < .001, bulk vs SCA-1+ ID8). Note that cells originated from taxol and cisplatin-containing spheroid culture also exhibited higher sphere-forming abilities (p < .01, bulk vs taxol spheroid culture; p < .001, bulk vs cisplatin spheroid culture). (e) Relative mRNA expression levels of ABCG2 gene was highest on SCA-1+ ID8 cells. It was nearly 3 folds of mRNA level on bulk ID8 cells. mRNA level on ID8 under spheroid culture was also higher than that on bulk ID8 cells. (f) Representative data in Hoechst 33342 (h) exclusion assay reveals significantly higher SP fraction in SCA-1+ ID8 cells than in bulk cells (20.83% vs 1.2%, bulk vs SCA-1+ ID8, p < .0001). This distinct dim population in SCA-1+ ID8 cells was diminished in the presence of 10 mM FTC (p < .001), or verapamil (v) (p < .0001). (g) SCA-1+ ID8 cells exhibited much stronger in vivo tumorigenesis in NOD-SCID mice by day 14, both in low or high number of cell implantation (p < .01 and p < .001, on day 7 and day 14). #p < .05, *p < .01, **p < .001, ***p < .0001.
We tested another murine ovarian cancer cell line, HM-1, and found that CD133+ HM-1 cells were significantly enriched through concomitant spheroid and chemotherapeutic drug selection (Figure 2a). CD133+ HM-1 cells formed more cell colonies and tumor spheroids than (bulk) HM-1 cells did (Figure 2b). We also detected a greater proportion of CD133+ HM-1 cells in the dim SP, and these cells could form tumors from as little as 100 cells (in 2 of 5 mice) compared with the bulk HM-1 cells, which demonstrated the stronger tumorigenesis of the CD133+ HM-1 cells (Figure 2c and d). All data consistently indicated that CD133 is the stemness marker of HM-1 cells.
Figure 2.
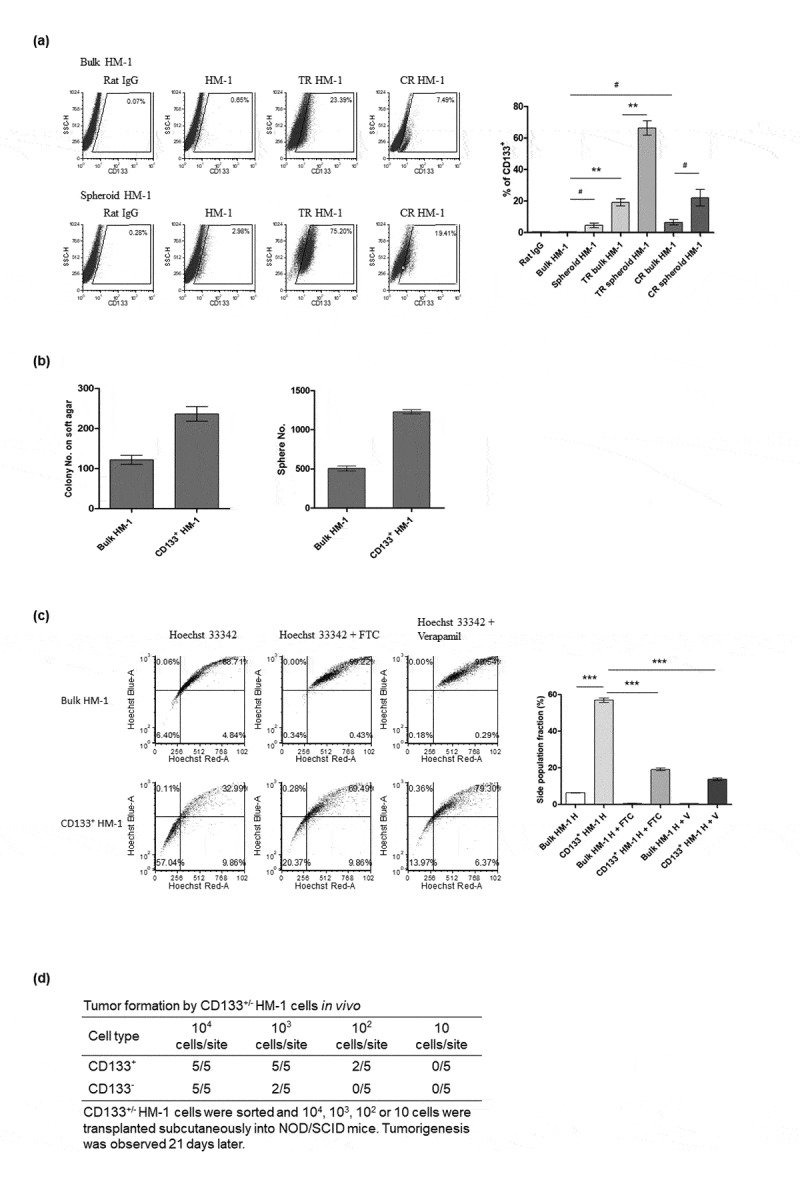
Identification and characterization of cancer stem-like cells from a murine ovarian cancer cell line, HM-1. (a) Representative flow data and statistical bars show that CD133+ cells were enriched by either cisplatin (CR bulk HM-1) or taxol (TR bulk HM-1) (p < .05 and p < .001, in taxol- and cisplatin-containing culture) or further spheroid culture (p < .001, p < .05, Taxol-containing culture, TR bulk HM-1 vs TR spheroid HM-1 or cisplatin-containing culture, CR bulk HM-1 vs CR spheroid HM-1). (b) Comparing to bulk HM-1 cells, CD133+ HM-1 cells exhibited substantially higher tumorigenic ability, indicated by both clonogenic assay and sphere-forming assay (p < .01 and p < .0001). (c) Representative data in Hoechst 33342 staining disclose significantly higher SP fraction in CD133+ HM-1 cells than in bulk cells (p < .0001, bulk HM-1 H vs CD133+HM-1 H). This SP fraction was diminished in the presence of 10 mM FTC or verapamil (p < .0001, CD133+HM-1 H vs CD133+HM-1 H + FTC and CD133+HM-1 H + V). (d) CD133+ HM-1 cells exhibited stronger in vivo tumorigenesis by day 21, starting from both 102 and 103 cells. #p < .05, *p < .01, **p < .01, ***p < .001.
Vulnerability of isolated SCA-1+ ID8 and CD133+ HM-1 ovarian cancer stem-like cells to immune attack
Cancer stem-like cell tumorigenesis was investigated in syngeneic mice. Small number of SCA-1+ ID8 cells (1 × 102 and 5 × 102) initially grew faster than bulk ID8 tumor cells (Figure 3a, D14 and D18). However, they were soon rejected once the tumor developed (Figure 3a, D25 and D32). Conversely, bulk ID8 tumor cells grew slower but could stably form tumors compared with SCA-1+ ID8 cells (Figure 3a, D25 and D32). We speculated that tumor rejection was immune-related. SCA-1+ ID8 tumor cells grew to a number high enough to elicit an antitumor immune response, indicating immunogenicity against SCA-1+ ID8 cells. This was supported by the findings of higher percentage of CD45+CD68+ and CD8+ cells in peritoneal lavage of mice implanted with SCA-1+ ID8 cells, indicating increased macrophage and CD8+ lymphocytes within the tumor microenvironment (Figure 3b). Furthermore, highest number of IFN-γ+ CD8+ T lymphocytes was found in the splenocytes of mice, in which SCA-1+ tumors disappeared (2.5 × 103 on D32, regarded as immunized) and were re-stimulated with irradiated SCA-1+ ID8 cells, comparing with those splenocytes stimulated with bulk ID8 tumor cells or splenocytes from mice injected with bulk ID8 cells (2.5 × 103 on D32, regarded as nonimmunized) (Figure 3c). We further confirmed that mice immunized with three doses of irradiated SCA-1+ ID8 cells exhibited stronger tumor-specific CD8+ T cell responses; thus, the growth of SCA-1+ ID8 cells in the peritoneal cavity was inhibited (i.e., tumor rejection; shown by lavaged cells in Figure 3d). To elucidate the role of lymphocyte subpopulation involving the SCA-1+ ID8 tumor in immunocompetent mice, we performed lymphocyte depletion assay through i.p. injection with 100 μg of rat monoclonal antibody GK1.5 (anti-CD4), TIB210 clone 2.43 (anti-CD8), or PK136 (anti-NK1.1). In Figure 3d, the signal of SCA-1+ tumor peaked on D7 if CD4+ lymphocyte was neutralized by GK 1.5 antibody. The signals were reduced on D14, possibly through the immune response elicited by rapidly growing tumor. On the other hand, SCA-1+ tumor became obvious on day 14 if CD8+ lymphocyte was neutralized by TIB 210 antibody. Administration of PK136 antibody did not alter the rejection of SCA-1+ tumors (Figure 3e). These data suggest the involvement of cell-mediated immune reactions in the attack of SCA-1+ ID8 stem-like cells. Similarly, enhanced immunogenicity of cancer stem-like cells was noted in another murine ovarian cancer cell line, HM-1 (Figure 4). As previously described, CD133+ HM-1 cells possess stemness characteristics. Based on the findings of ID8 and SCA-1+ ID8 cells, we speculated CD133+ HM-1 cell is more immunogenic than bulk HM-1 cell. However, the difference of immunogenicity might not strong enough to lead to two distinct tumor growth curves. To further disclose the immune vulnerability of CD133+ HM-1 cells, we tried to use ovalbumin to enhance the immunogenicity of HM-1 and CD133+ HM-1 cell and see if there is a discrepancy on tumor growth. Cells were engineered to carry a foreign antigen-chick ovalbumin (bulk HM-1-ova or CD133+ HM-1-ova). The expression levels of ovalbumin were similar on both bulk and CD133+ HM-1 cells on flow analysis. After 3 weeks of implantation, CD133+ HM-1-ova cells failed to form tumors in syngeneic mice, whereas bulk HM-1 cells expressing this protein did (HM-1-ova) (Figure 4a). When CD8+ and CD4+ T lymphocytes were depleted, CD133+ HM-1-ova tumor rejection was abolished, suggesting the involvement of immune reactions (Figure 4b). This was further proven by the analysis of splenocytes from mice immunized with bulk HM-1 or CD133+ HM-1 cells. Mice immunized with CD133+ HM-1 cells showed more CD8+IFN-γ+ T lymphocytes induced by the same stimulating cells, whereas mice immunized with bulk HM-1 cells showed fewer activated CD8+ T lymphocytes (Figure 4c). In addition, we found the increased phagocytosis of live and irradiated CD133+ HM-1 cells by syngeneic CD11c+ cells compared with that of bulk HM-1 cells (Figure 4d).
Figure 4.
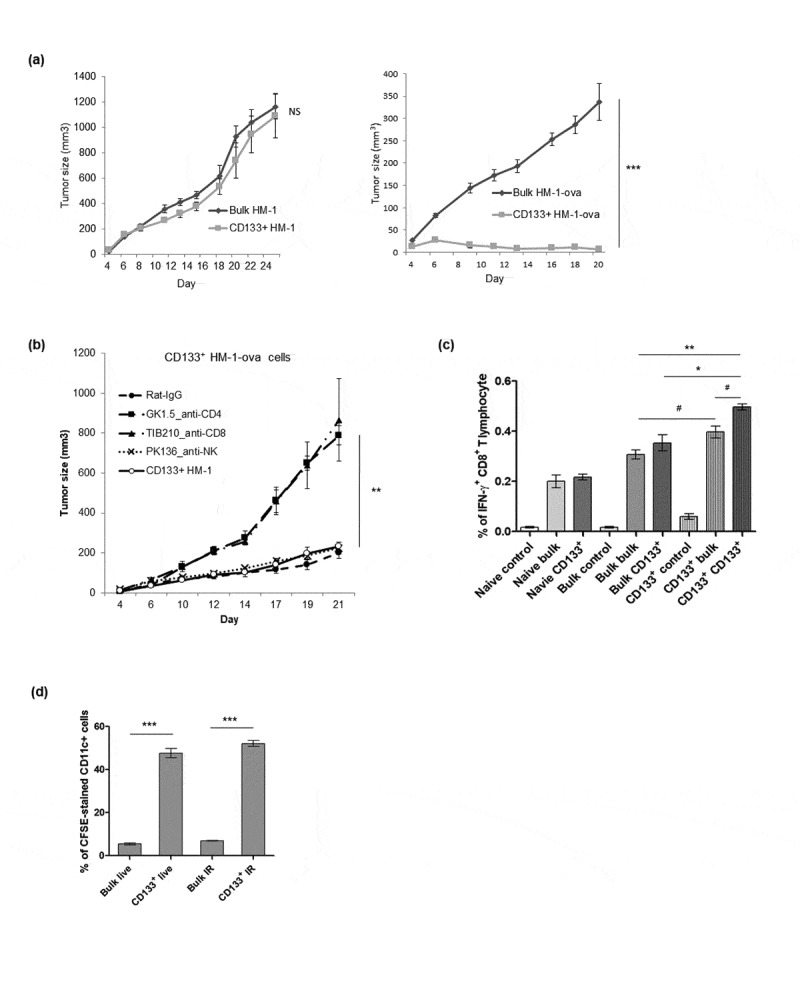
Enhanced immunogenicity of cancer stem-like cells CD133+ HM-1 cells. (a) Tumor growth curve of bulk and CD133+ HM-1 cells showed similar growth pattern (left). However, CD133+ HM-1 cells expressing chicken ovalbumin (CD133+ HM-1-ova) were rejected. Bulk HM-1 cells expressing ovalbumin grew slowly but could still form the tumors (p < .0001, right). (b) Suppression of CD133+ HM-1-ova tumor growth was abolished through depleting CD8+ and CD4+ T lymphocytes by TIB210 (anti-CD8) and GK1.5 (anti-CD4) neutralizing antibodies (p < .001), suggesting rejection of CD133+ HM-1-ova tumor involved CD8+ and CD4+ T cell immune response. (c) Mice were either not immunized (naïve), immunized with bulk HM-1 or CD133+ HM-1 cells. The re-stimulated antigens were either nothing (control), bulk or CD133+ HM-1 cells. Splenocytes from mice immunized with CD133+ HM-1 cells are associated with more CD8+ IFN-γ+ T lymphocyte by the same stimulating cells (p < .001, Bulk bulk vs CD133+ CD133+; p < .05, Bulk bulk vs CD133+ bulk, the former is cells for immunization, the latter is cells for re-stimulation in assay). Vaccination with bulk cells induced less activated CD8+ T lymphocytes. (d) CFSE-pulsed bulk and CD133+ HM-1 cells were incubated with syngeneic CD11c+ cells for 6 hours. Phagocytosis was analyzed by flow cytometry for CFSE-stained CD11c+ cells. Increased phagocytosis of CD11c+ cells was observed on both live and irradiated CD133+ HM-1 cells (p < .0001 on both). #p < .05, *p < .01, **p < .001, ***p < .0001, NS: not significant.
Increased phagocytosis of stem cell-like cells is attributed to low CD47 protein expression
To explore the possible mechanisms responsible for the increased phagocytosis of stem-like cells by CD11c+ cells, we profiled differential gene expression in both sets of bulk/SCA-1+ ID8 and bulk HM-1/CD133+ HM-1 cells through the murine phagocytosis PCR array. Among the 84 genes involved in phagocytosis, an integrin-associated protein, CD47, was found to be significantly downregulated in both SCA-1+ ID8 and CD133+ HM-1 stem-like cells (data not shown). Real-time PCR revealed 3-fold lower CD47 mRNA expression on SCA-1+ ID8 cells than in bulk ID8 cells, which was supported by the lower level of CD47 protein surface expression on SCA-1+ ID8 cells through flow cytometric analysis. Similarly, CD47 expression was lower in CD133+ HM-1 cells than in bulk HM-1 cells (Figure 5a). The association between CD47 levels on tumor cells and phagocytosis was supported by the observation of the increased phagocytosis of both live and irradiated SCA-1+ ID8 cells by CD11c+ cells (Figure 5b) compared with that of bulk ID8 cells. We further elucidated this in a murine macrophage cell line (RAW 264.7) through the manipulation of CD47 gene expression in ID8 cells. RAW 264.7 macrophages exhibited better phagocytosis of SCA-1+ ID8 cells than that of bulk ID8 tumor cells. Suppression of CD47 expression on bulk SCA-1− ID8 tumor cells by using the anti-CD47 antibody markedly enhanced RAW 264.7 phagocytic activity, whereas CD47 overexpression on SCA-1+ ID8 cells (SCA-1+ ID8 CD47) inhibited phagocytosis (Figure 5c). To determine whether T lymphocytes are involved in this phenomenon, we developed a SCA-1+ ID8-specific T cell line from SCA-1+ ID8 cell-immunized mice. Similar results were obtained in cross-presentation of phagocytic antigens to SCA-1+ -specific T cells with/without anti-CD47 antibody treatment: increased CD47 expression decreases the number of activated CD8+ T cells (Figure 5d). Our in vivo study also showed that SCA-1+ ID8 cell tumor rejection was hampered when CD47 was overexpressed. SCA-1+ ID8 and SCA-1+ ID8 cells overexpressing CD47 could grow on the sixth day of culture, but SCA-1+ ID8 cells were mostly cleared by the 10th day. CD47 expression delayed the immune-mediated rejection of SCA-1+ ID8 tumors (Figure 5e), which was further proven by the observation shown in Figure 5f and g. On day 20, a significantly higher percentage of CD8+ lymphocyte and F4/80+ cells was noted in peritoneal lavage from mice with SCA-1+ID8 cells (SCA-1+ID8 vehicle) than mice with SCA-1+ID8 cell overexpressing CD47 (SCA-1+ID8 CD47). However, the percentage of CD45+CD68+ cells was significantly lower in mice with SCA-1+ ID8 vehicle cells (Figure 5f). Moreover, a significantly decreased number of activated CD8+IFN-γ+ cells were found in splenocytes obtained from mice immunized with SCA-1+ ID8 CD47 cells compared with mice immunized with SCA-1+ ID8 cells (Figure 5g).
Figure 5.
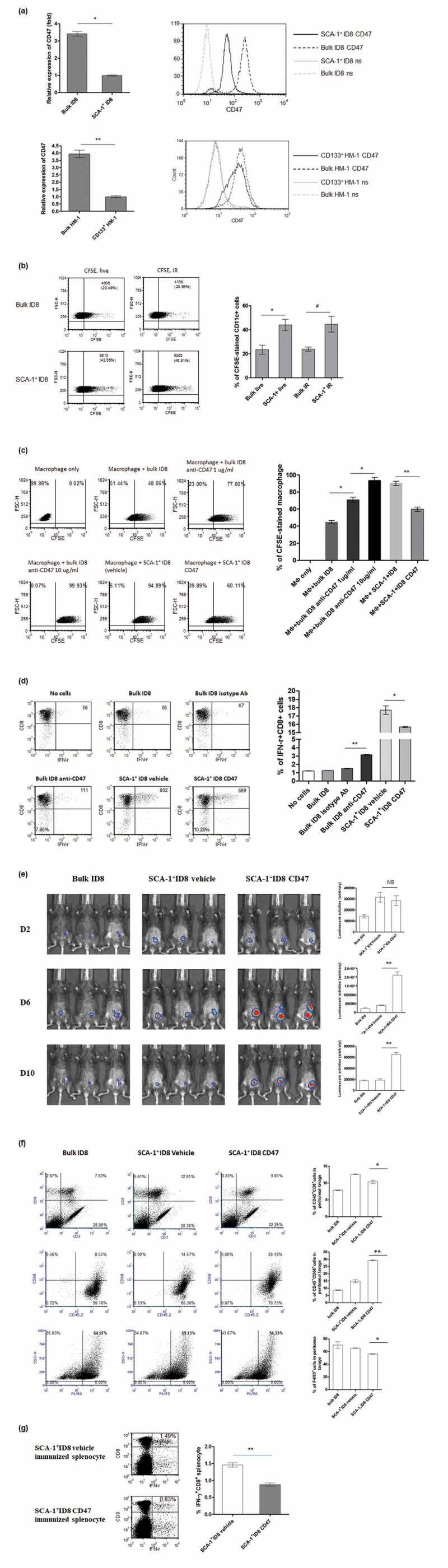
Immunogenicity of cancer stem-like cells results from lower CD47 expression and increased macrophage phagocytosis. (a) CD47 expression levels quantified by quantitative PCR demonstrated 3-fold lower expression of CD47 on SCA-1+ ID8 cells (p < .01, bulk vs SCA-1+ ID8, left). Histogram in flow cytometry showed lower CD47 protein level on SCA-1+ ID8 cells (right, ns: no staining). Similarly, expression of CD47 is lower on CD133+ HM-1 cells comparing to the expression on bulk population (p < .001, value in folds). Histogram in flow cytometry also showed lower CD47 protein level on CD133+ HM-1 cells (right, ns: no staining). (b) CFSE-pulsed bulk and SCA-1+ ID8 cells were incubated with syngeneic primary CD11c+ cells for 1 hour. Increased phagocytosis (indicated by CD11c+ gated CFSE-stained cells) was observed on both live (p < .01) and irradiated (IR) (p < .05) SCA-1+ ID8 cells (bar data). (c) Analysis on RAW 264.7 cell revealed better phagocytosis on SCA-1+ ID8 cells than on bulk ID8 cells. SCA-1+ ID8 cells were stably transfected with either pcDNA3.1 empty vector (SCA-1+ ID8 vehicle) or pcDNA3.1-CD47 (SCA-1+ ID8 CD47). Suppression of CD47 on bulk ID8 cells by anti-CD47 antibody markedly enhances the phagocytic activities, while forced expression of CD47 on SCA-1+ ID8 cells (SCA-1+ ID8 CD47) hampered the phagocytosis. (d) Cross-presentation of phagocytic antigens to a SCA-1+ ID8-specific T lymphocyte line developed from SCA-1+ ID8 immunized mice showed the same pattern. To a lesser degree, anti-CD47 in bulk ID8 cells increased the number of activated CD8+ T cells, while forced expression of CD47 decreased the number activated T cells. (e) Image study showed that tumor rejection of SCA-1+ ID8 cells was abrogated by forced expression of CD47. Both SCA-1+ ID8 (vehicle) and SCA-1+ ID8 CD47 (started from 10000 cells) grew on 6th day image but the former was mostly cleared on 10th day while the latter retained their growth longer. (f) Representative analysis of peritoneal lavaged cells showed highest percentage of CD3+CD8+ cells in mice with SCA-1+ ID8 vehicle cells (day 20). Mice with SCA-1+ID8 CD47 cells had less CD3+CD8+ and F4/80 cells (p < .01). However, these mice exhibited significantly higher percentage of CD45+CD68+ cells in peritoneal lavage than mice with SCA-1+ID8 vehicle did (p < .001). (g) Significantly less activated CD8+IFN-γ+ splenocytes were seen from mice immunized with SCA-1+ ID8 CD47 cells compared to those from mice immunized with SCA-1+ ID8 vehicle (p < .001). *p < .01, **p < .001, NS: not significant.
Stem-like cells are protected by neighboring differentiated cancer cells from attack of immune effector cells
Our data indicated that the enhanced immunogenicity of SCA-1+ ID8-Luc cells is attributed to the susceptibility of these cells to phagocytic attack. Next, we established an in vivo coculture model to investigate how a small proportion of CSCs can survive in a tumor microenvironment if they are vulnerable to immune attack; we focused on the interaction of CSCs with other non-stem cancer cells. We used the luciferase gene to label our target cell population (SCA-1+ ID8-Luc or bulk ID8-Luc, 1000 cells) and then cocultured these cells with or without bulk ID8 cells (no luciferase gene, 5000 cells). SCA-1+ ID8-Luc cells could grow in C57BL/6 mice but soon disappeared. However, when mixed with bulk ID8 cells, SCA-1+ ID8-Luc cells grew rapidly by day 8. By contrast, the growth of bulk ID8-Luc tumor cells in these mice was very low (Figure 6a). The surrounding bulk (mostly SCA-1− & non-stem) ID8 tumor cells seemed to protect cancer stem-like (SCA-1+ ID8) cells from elimination by immune effector cells, which might be due to the immunosuppressive microenvironment driven by non-stem cancer cells. This finding was supported by the observation that the percentage of myeloid-derived suppressor cells (MDSC, Gr-1+CD11b+cells) in SCA-1+ ID8 cell-generated tumor–bearing mice was significantly less than that in mice bearing tumors generated by bulk ID8 cells. Similar trends were noted in mice injected with SCA-1+ ID8 mixed with bulk ID8 cells and mice injected with bulk ID8 mixed the same bulk ID8 cells, although the tumor loading in the former was much higher indicated by imaging (Figure 6b). Likewise, enhanced macrophage phagocytosis of SCA-1+ ID8 cells was compromised by neighboring bulk (mostly SCA-1− & non-stem) ID8 tumor cells in an amount-dependent pattern (Figure 6c). Another assay to determine apoptotic tumor cells revealed a similar phenomenon; that is, surrounding bulk SCA-1− ID8 tumor cells dose-dependently exerted protective effects on SCA-1+ ID8 cells, preventing their phagocytosis (CFSE+ Annexin V stained apoptotic cells in Figure 6d).
Figure 6.
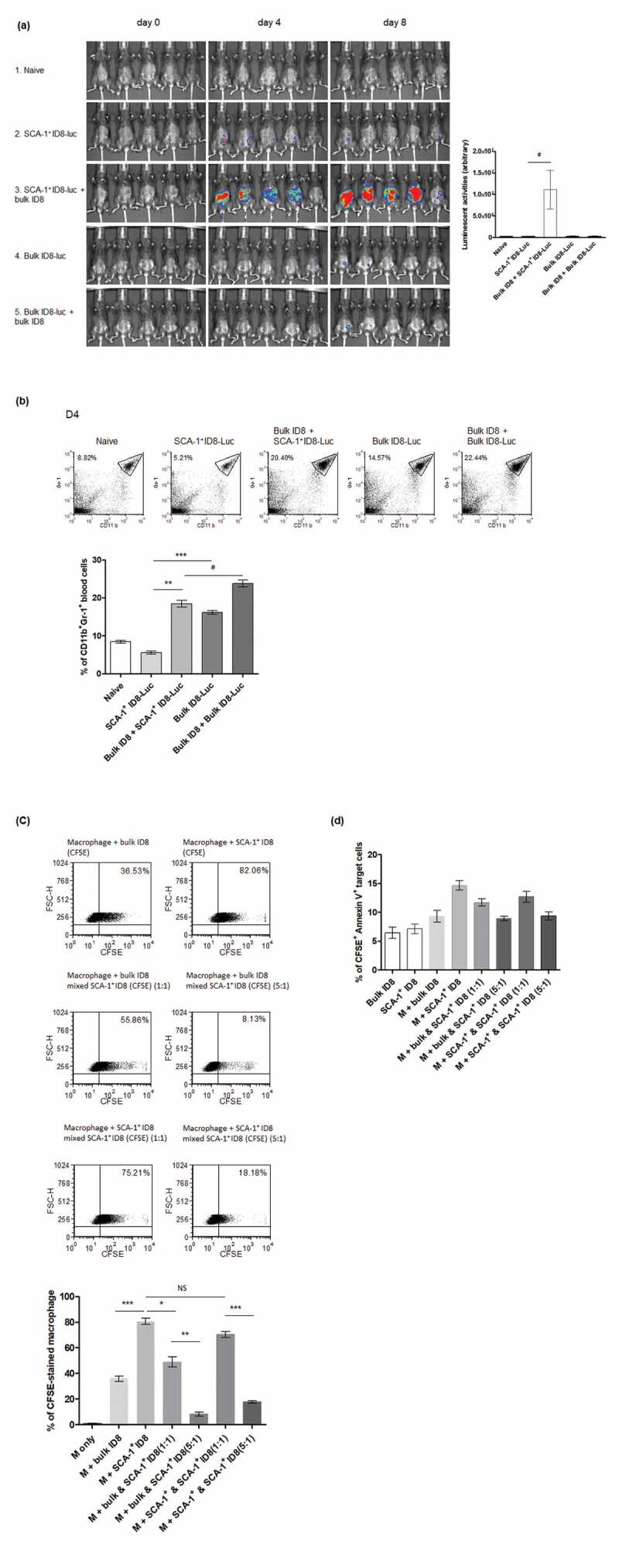
SCA-1+ ID8 cells were susceptible to the attack of phagocytes, and surrounding bulk ID8 (mostly SCA-1− & non-stem) cells could protect SCA-1+ ID8 cells from elimination of immune effectors cells. (a) SCA-1+ ID8-Luc cells (started from 1000 cells) were able to grow inside the C57BL/6 mice on day 4 but soon disappeared on day 8 (by luminescent signal activities, second row). When mixed with bulk ID8 cells (5X, 5000 cells), SCA-1+ ID8-Luc cells rapidly propagated and grew to large amount of tumor cells on day 8 (third row). The growth of bulk ID8-luc cells in these mice is hardly seen (row 4, 5). Bar graph indicates significant growth difference in SCA-1+ ID8-Luc cells with or without bulk ID8 cells around. p < .05 (b) Representative flow analysis of CD11b+Gr-1+ MDSC shows significantly less systemic MDSC in SCA-1+ ID8-Luc bearing mice (5.6% vs 16.19%, SCA-1+ ID8-luc vs bulk ID8-Luc, P < .0001), or in mice bearing SCA-1+ ID8-luc mixed with bulk ID8 cells (18.48% vs 23.83%, SCA-1+ ID8-luc mixed with bulk ID8 vs bulk ID8-luc with the same bulk ID8 cells, p < .05). Note that there is an abrupt increase in the percentage of MDSC when SCA-1+ ID8-luc was mixed by bulk ID8 cells (5.6% vs 18.48%, p < .001). (c) In vitro culture of SCA-1+ ID8 cells with bulk ID8 cells showed the engulfment of macrophage was interfered by surrounding bulk cancer cells. Representative flow and statistical bar graph showed enhanced phagocytosis of SCA-1+ ID8 by macrophages (indicated by CFSE-stained macrophage) is abolished by co-culture with bulk ID8 cells (averaged 80.65% vs 49.02% in SCA-1+ ID8 [stained] vs SCA-1+ ID8 cells [stained] mixed with bulk ID8 cells [unstained], p < .01). The number of CFSE-stained macrophage disproportionally decreased when more bulk ID8 cells was added (49.02% to 8.29%, 1:1 vs 5:1, p < .001). The percentage of CFSE-stained macrophage indicating phagocytic activities was displayed as bar graph. (d) Assay of tumor cell apoptosis labeled by CFSE & Annexin V-stained cells revealed a similar phenomenon that co-culture with bulk ID8 cells relatively protected the SCA-1+ ID8 cells from being engulfed (averaged 14.69% vs 11.72%, SCA-1+ ID8 [CFSE-stained] vs SCA-1+ ID8 cells [CFSE-stained] mixed with bulk ID8 cells [CFSE-unstained], p < .05). The number of CFSE & Annexin V-stained cells was further lessened when more bulk ID8 cells was added (11.72% to 8.93%, 1:1 vs 5:1, p < .05). #p < .05, *p < .01, **p < .001, ***p < .0001, NS: not significant.
Both human ovarian CD24−/low SKOV3 cancer stem-like cells and spheroid OVCAR3 cells have low CD47 expression
We also verified whether the same phenomenon is applicable to human ovarian stem-like cells. We isolated stem-like cells from a human ovarian cancer cell line – SKOV3 and found CD24−/low SKOV3 possesses stemness characteristics (Figure S2). A significant decrease in CD47 expression was detected on CD24−/low SKOV3 cells (on both mRNA and surface protein expression levels, Figure 7a). Figure S3 also depicts that another human ovarian cancer cell line, OVCAR3, derived from spheroid cultures exhibited stemness characteristics. Both quantitative PCR and flow cytometric analysis also demonstrated that CD47 expression is down regulated on spheroid OVCAR3 cells compared with their bulk counterpart (Figure 7b). Moreover, the increased phagocytosis of CD24−/low SKOV3 cells by human macrophages was also abrogated by CD24+ SKOV3 cells in a dose-dependent manner (Figure 7c), and surrounding CD24+ SKOV3 tumor cells dose-dependently protected CD24−/low SKOV3 cells from being killed (CFSE+ Annexin V-stained apoptotic cells in Figure 7d).
Figure 7.
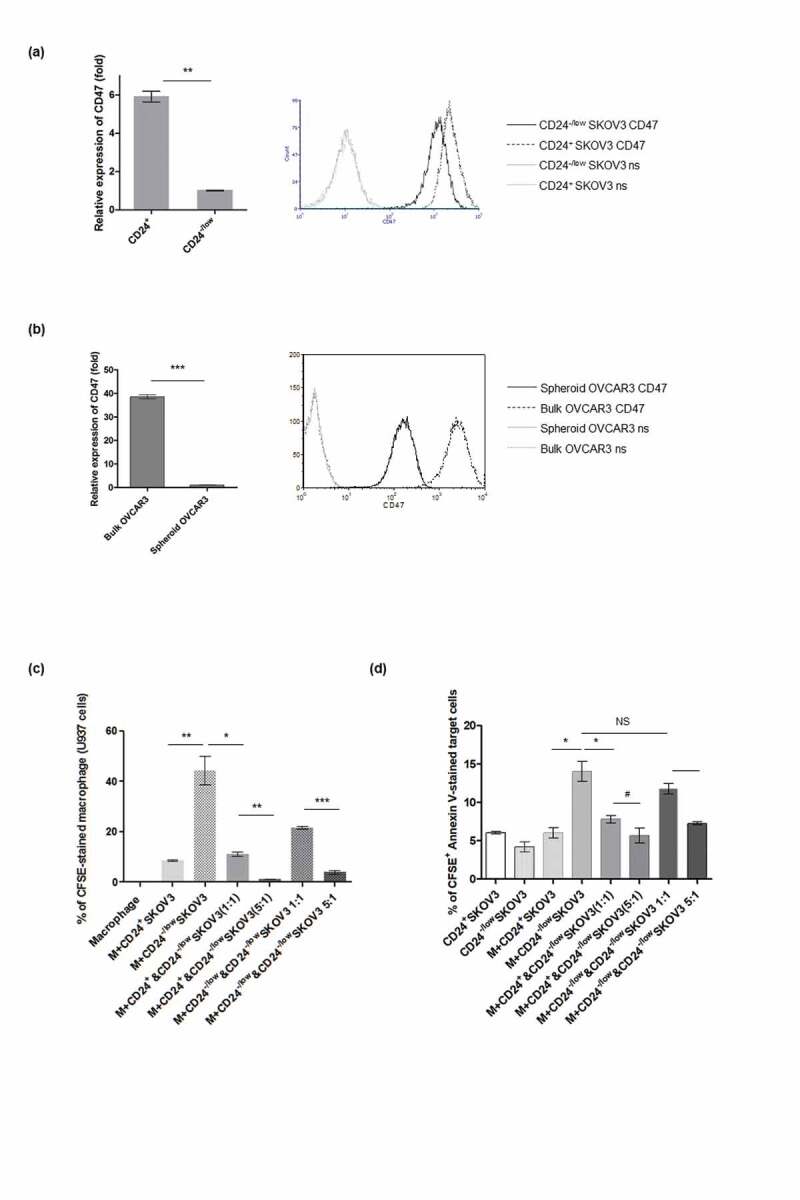
Down-regulation of CD47 expression in human ovarian cancer cells showing stemness rendered the vulnerability to macrophage engulfment. (a) Significant decrease on CD47 expression was noted in isolated CD24−/low human ovarian cancer cell SKOV3 (for mRNA, p < .001 and surface protein), which exhibited stem cell-like characteristics (see Fig S2). (b) Quantitative PCR and flow analysis demonstrated CD47 expression is down regulated in spheroid OVCAR3 cells with stemness characteristics (shown in figure S3). p < .0001, CD47: stained with CD47 antibody. (c) Data shows that increased phagocytosis of macrophage (m) on CD24−/low SKOV3 cells (indicated by CFSE-stained human macrophage cell U937; 9.67% vs 43.35%, CD24+ vs CD24−/low SKOV3 cells, p < .01) was abrogated by CD24+ SKOV3 cells (43.35% vs 11.94% in CD24−/low [CFSE-stained] versus CD24−/low SKOV3 cells [CFSE-stained] mixed with CD24+ SKOV3 [CFSE-unstained], p < .01). The stained macrophage was disproportionally decreased when more CD24+ SKOV3 cells were added (11.94% to 1.45%, 1:1 versus 5:1, p < .001). (d) Statistical bar graph shows percentage of apoptotic CD24−/low SKOV3 cells (CFSE+ Annexin V+ cells) was much more than that of CD24+ SKOV3 cells when they were co-cultured with macrophage (M) (14.04% vs 6.05%, p < .01), and apoptotic CD24−/low SKOV3 cells significantly decreased when mixed with CD24+ SKOV3 cells (14.04% vs 7.85%, p < .01). The number of CFSE & Annexin V-stained cells were further lessened when more CD24+ SKOV3 cells was added (7.85% to 5.01%, 1:1 versus 5:1, p < .05). #p < .05, *p < .01, **p < .001, ***p < .0001, NS: not significant.
Collectively, our data indicated that cancer stem-like cells are susceptible to macrophage phagocytosis through downregulation of CD47 expression, and this immune vulnerability was protected by surrounding non-stem cancer cells. This observation implicates a potential scenario in which cancer cells and CSCs interact in the tumor microenvironment.
Discussion
In this study, we successfully enriched mouse murine and human ovarian cancer stem-like cells through escalating concentrations of chemotherapeutic drug selection and attachment-free spheroid cultures. Many reports supported that a self-renewing subpopulation of cancer-initiating cells could be enriched among established human tumor-derived cell lines through similar approaches.8,11-13 The proportion of ovarian stem cells was is increased in response to platinum- and taxane-based chemotherapies and to a combination of both drugs.8,14 There are also different approaches to identify and purify CSCs. Although those CSCs successfully met the criteria of stemness both in vitro and in vivo in most studies, it remains to be determined whether these isolated cells possess identical characteristics in other biological or molecular aspects. On the other hand, gene expression profiles are considerably heterogenous in the published data on cancer stem or stem-like cells, even though the same isolation protocols have been used.
The finding of higher rates of tumor development in immunocompromised patients and animal models supports the concept of immunosurveillance.15,16 Transformed cells might be eliminated by the host immune system to prevent tumorigenesis. It is generally believed that CSCs are more capable of escaping host immune-mediated rejection in an immunocompetent host, which might due to the low expression of tumor-associated antigens (TAAs) associated with undifferentiated cancer-initiating cells,17,18 or cancer-initiating cells that lack or have downregulated MHC class I molecules.19,20 Other possible mechanisms for the immune resistance of CSCs include the activation of TGF-β signaling or the induction of immunosuppressive microenvironments by regulatory T lymphocytes,21–23 which inhibit the maturation or function of antigen-presenting cells or causes T cell anergy through the expression of negative costimulatory molecules.24–26 In parallel with immune escape during tumor development, it is speculated that cancer-initiating cells within tumors may be inherently resistant to antitumor immune responses. However, most of the conclusions were inferred based on studies of physiologic stem cells.
In our study, SCA-1 and CD133 surface protein expression was used to isolate the stem-like populations in two murine ovarian cancer lines, ID8 (C57BL/6 origin) and HM-1 (originated from B6 × C3/He origin), with CSC characteristics. Strong in vivo tumorigenesis of these two cell lines was observed in NOD-SCID mice; however, these cells were unexpectedly rejected in immune-competent mice. This was attributed to the increased number of activated (IFN-γ+) CD8+ T lymphocytes against SCA-1+ ID8 cells. Furthermore, C57BL/6 mice immunized with irradiated SCA-1+ ID8 cells exhibited stronger tumor-specific CD8+ T cell responses and rejection of SCA-1+ ID8 tumors. In parallel, B6 × C3/He mice immunized with CD133+ HM-1 cells had stronger tumor-specific CD8+ T cell immunity and subsequent tumor rejection. Both CD4+ and CD8+ T cells induced tumor rejection. Our data clearly demonstrate the immune vulnerability of cancer stem-like cells; the results are inconsistent with the current dogma in published reports.
The hypothesis that cancer stem-like cells might be susceptible to antitumor immunity is supported by a report that SP cells of several colon cancer cell lines were susceptible to tumor-associated antigen CEP55-specific T lymphocytes both in vitro or in vivo.27 These SP cells showed high expression of serial stem cell markers and thus fulfilled the stemness criteria, and they showed resistance to chemotherapeutic agents. Another report in head and neck squamous cell carcinoma showed stronger immunogenicity in ALDHhigh tumor cells isolated from patient surgical specimens.28 Studies have also used cancer stem-like cells as therapeutic vaccines in mouse models to prove their immune susceptibility. CSCs-pulsed DCs were able to elicit both humoral and T cell antitumor immune responses to directly destroy CSCs.29 CD133+ brain tumor CSCs were also recognized and eliminated by CD8+ cytotoxic T cells in an antigen-specific manner.30 The difference in CSC immune resistance/vulnerability in the literature might be attributed to the diverse methods used to delineate CSCs. It is also likely that CSCs express different biological markers in various microenvironments, and their overall immune scenarios are also different.
CD47 (also known as integrin-associated protein) is a cell surface protein of the immunoglobulin (Ig) superfamily known to interact with integrins and thrombospondin-1.31,32 It has been suggested to be involved in diverse biological processes such as axonal growth,33 neutrophil migration,34 T cell co-stimulation, and macrophage phagocytosis.35,36 In our immunocompetent mouse ovarian cancer model, we noted significantly decreased CD47 protein expression on both SCA-1+ ID8 and CD133+ HM-1 stem-like cells compared with SCA-1− ID8 and CD133− HM-1 tumor cells, and this was further supported by the increased phagocytosis of SCA-1+ ID8 cells by macrophages. CD47 overexpression on SCA-1+ ID8 cells inhibited phagocytosis and delayed immune rejection of SCA-1+ ID8 tumor growth, which was associated with the decreased number of activated CD8+ T cells. Our data indicated that enhanced CSC immunogenicity derived from susceptibility to phagocytic attack was caused by low CD47 expression. This finding is consistent with those of previous studies, which have described the physiologic role of CD47, which signals “don’t eat me.” It also correlated with the loss of CD47 expression, leading to the phagocytosis of aged or damaged cells.36,37
As evidences regarding CD47 continues to accumulate, the overall image emerges that this molecule acts as a signaling receptor and provides biological information to cells regarding neighboring cells and the extracellular microenvironment. An important role of CD47 is to interact with the N-terminus of the extracellular immunoglobulin-like domains of SIRPα on myeloid linage cells (such as macrophage and DCs) to inhibit phagocytosis and other activities of macrophages and DCs. High CD47 expression on cancer cells renders them resistant to macrophage phagocytosis; thus, CD47 is defined as the “don’t eat me” signal. As mentioned before, low CD47 expression on ovarian cancer stem-like cells (SCA-1+ ID8 and CD133+ HM-1 cells) renders them susceptible to attack by macrophages.
Most normal tissues express low levels of CD47, and it is generally highly expressed on tumor cells. The difference in expression between cancer cells and corresponding normal cells can be up to several folds. CD47 expression levels are also correlated with patient prognoses for various cancers.38 High CD47 expression is regarded as an immune escape mechanism of cancer cells. However, CD47 expression on cancer stem-like or stem cells has varied. For example, bladder tumor-initiating cells (CD44+ cells) were found to have higher CD47 expression levels compared with CD44− cells.39 This upregulation is justifiably thought to be related to the immune resistance of tumor-initiating cells. Notably, CD47 is upregulated on circulating hematopoietic stem cells to evade macrophage phagocytosis.40 CD47 expression increased during the mobilization of bone marrow progenitor cells to peripheral hematopoietic stem cells, which was induced by the cyclophosphamide/G-CSF protocol; this indicated the dynamic alteration of CD47 expression in cells with different stemness statuses. However, CD47 expression is low in the most primitive resident marrow cells. Conversely, CD47 expression was decreased in hematopoietic progenitor cells in patients with myelofibrosis.41 Sharrow et al. identified that in ovarian cancer, ALDHhigh cells with stemness properties had approximately 2-fold higher CD47 expression than ALDHlow cells.42 Nevertheless, two public microarray datasets of ovarian cancer cells from the Gene Expression Omnibus (GEO) showed significantly lower CD47 expression on cancer stem-like cells than their controls (GSE28799 and GSE80373).43,44 The difference in CD47 expression levels in ovarian cancer stem-like cells might be due to several factors, such as diverse definition markers, isolation methods of these primitive cells, or even different phases of stemness. Hypoxia-inducible factor 1 (HIF-1) has been reported to directly activate the transcription of the CD47 gene in hypoxic breast cancer cells.45 Hypoxia can induce the CSC phenotype through the HIF-dependent pluripotent stem cell transcriptional program.46 A more direct relationship between CD47 and stemness is addressed in one study that investigated primary murine endothelial cells isolated from CD47-null mice. It was found that the loss of CD47 allowed self-renewal and increased the expression of the stem cell transcription factors c-Myc, Sox2, KLF4, and Oct4. These cells demonstrated increased asymmetric cell division and the formation of spontaneous clusters, which contained pluripotent cells.47 Conversely, re-expression of CD47 in CD47-null endothelial cells inhibited c-Myc, Klf4, Sox2, and nestin expression, indicating a link between the absence of CD47 and stem cell maintenance; this is consistent with our finding that murine ovarian SCA-1+ ID8 and CD133+ HM-1 stem-like cells exhibited very low CD47 expression relative to their SCA-1− ID8 and CD133− HM-1 bulk counterparts.
If ovarian cancer stem-like cells are vulnerable to immune attack, how do these cells survive to maintain tumor growth? Primitive cancer cells within tumors are assumed to adjoin to differentiated bulk cells. In our study, cancer stem-like cells could only grow inside mice when mixed with non-stem bulk cells, which diminished the rejection of cancer stem-like cells. This was probably due to the immunosuppressive microenvironment driven by bulk cancer cells, either by abundant CD47 expression on bulk cells or other immunosuppressive cytokines. Increased MDSC percentages were observed in the presence of differentiated bulk cells, even in smaller tumors. The phagocytosis of stem-like cells by macrophages was also diminished by neighboring bulk cells. Stem cell-like cells undergo asymmetric cell divisions giving rise to new stem cells, but also to more differentiated daughter cells. It is generally thought that CSCs exist under a complex biochemical and physical signal within a tumor microenvironment, called the cancer stem cell niche. Maintaining pure cancer stem cells is unlikely due to spontaneous differentiation once “niche” factors provided by the differentiated cells are lacking. The injection of a sufficiently large number of purified stem-like cells might lead to spontaneous differentiation. Thus, one might argue that the outcome observed in CSC injection could be like that in CSCs co-injected with bulk tumor cells. However, in our experiment the number of differentiated daughter cells produced by 1000 CSCs would be far less than 5000 cells (the number of co-injected bulk cells), and these two environments (pure CSCs or CSCs with bulk tumor cells) would not be the same. Collectively, our study results provide evidence that surrounding differentiated bulk ovarian cancer cells may protect cancer stem-like cells from immune attack and allow immune vulnerable stem-like cells to survive, grow, and differentiate into cancer cells.
In conclusion, our study suggests another scenario for CSCs within tumors, in which primitive tumor cells are susceptible to phagocytosis by macrophages due to low CD47 expression. However, surrounding differentiated bulk tumor cells protect the primitive cells from immune clearance, maintaining their existence. The results may enable more effective clinical treatment strategies for cancer to be designed.
Supplementary Material
Funding Statement
This work was supported by the Ministry of Science and Technology (MOST), Taiwan [103-2314-B-195-011-MY3].
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Allan AL, Vantyghem SA, Tuck AB, Chambers AF.. Tumor dormancy and cancer stem cells: implications for the biology and treatment of breast cancer metastasis. Breast Dis. 2006;26:87–13. doi: 10.3233/BD-2007-26108. [DOI] [PubMed] [Google Scholar]
- 2.Meng E, Long B, Sullivan P, McClellan S, Finan MA, Reed E, Shevde L, Rocconi RP. CD44+/CD24- ovarian cancer cells demonstrate cancer stem cell properties and correlate to survival. Clin Exp Metastasis. 2012;29:939–948. doi: 10.1007/s10585-012-9482-4. [DOI] [PubMed] [Google Scholar]
- 3.Curley MD, Therrien VA, Cummings CL, Sergent PA, Koulouris CR, Friel AM, Roberts DJ, Seiden MV, Scadden DT, Rueda BR, et al. CD133 expression defines a tumor initiating cell population in primary human ovarian cancer. Stem Cells. 2009;27:2875–2883. doi: 10.1002/stem.236. [DOI] [PubMed] [Google Scholar]
- 4.Peng S, Maihle NJ, Huang Y. Pluripotency factors Lin28 and Oct4 identify a sub-population of stem cell-like cells in ovarian cancer. Oncogene. 2010;29:2153–2159. doi: 10.1038/onc.2009.500. [DOI] [PubMed] [Google Scholar]
- 5.Ricci F, Bernasconi S, Porcu L, Erba E, Panini N, Fruscio R, Sina F, Torri V, Broggini M, Damia G. ALDH enzymatic activity and CD133 positivity and response to chemotherapy in ovarian cancer patients. Am J Cancer Res. 2013;3:221–229. [PMC free article] [PubMed] [Google Scholar]
- 6.Szotek PP, Pieretti-Vanmarcke R, Masiakos PT, Dinulescu DM, Connolly D, Foster R, Dombkowski D, Preffer F, Maclaughlin DT, Donahoe PK. Ovarian cancer side population defines cells with stem cell-like characteristics and Mullerian Inhibiting Substance responsiveness. Proc Natl Acad Sci U S A. 2006;103:11154–11159. doi: 10.1073/pnas.0603672103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liao J, Qian F, Tchabo N, Mhawech-Fauceglia P, Beck A, Qian Z, Wang X, Huss WJ, Lele SB, Morrison CD, et al. Ovarian cancer spheroid cells with stem cell-like properties contribute to tumor generation, metastasis and chemotherapy resistance through hypoxia-resistant metabolism. PLoS One. 2014;9:e84941. doi: 10.1371/journal.pone.0084941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rizzo S, Hersey JM, Mellor P, Dai W, Santos-Silva A, Liber D, Luk L, Titley I, Carden CP, Box G, et al. Ovarian cancer stem cell-like side populations are enriched following chemotherapy and overexpress EZH2. Mol Cancer Ther. 2011;10:325–335. doi: 10.1158/1535-7163.MCT-10-0788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Steg AD, Bevis KS, Katre AA, Ziebarth A, Dobbin ZC, Alvarez RD, Zhang K, Conner M, Landen CN. Stem cell pathways contribute to clinical chemoresistance in ovarian cancer. Clin Cancer Res. 2012;18:869–881. doi: 10.1158/1078-0432.CCR-11-2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chang CL, Hsu YT, Wu CC, Lai YZ, Wang C, Yang YC, Wu TC, Hung CF. Dose-dense chemotherapy improves mechanisms of antitumor immune response. Cancer Res. 2013;73:119–127. doi: 10.1158/0008-5472.CAN-12-2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Calcagno AM, Salcido CD, Gillet JP, Wu CP, Fostel JM, Mumau MD, Gottesman MM, Varticovski L, Ambudkar SV. Prolonged drug selection of breast cancer cells and enrichment of cancer stem cell characteristics. J Natl Cancer Inst. 2010;102:1637–1652. doi: 10.1093/jnci/djq361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morrison BJ, Steel JC, Morris JC. Sphere culture of murine lung cancer cell lines are enriched with cancer initiating cells. PLoS One. 2012;7:e49752. doi: 10.1371/journal.pone.0049752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lopez J, Poitevin A, Mendoza-Martinez V, Perez-Plasencia C, Garcia-Carranca A. Cancer-initiating cells derived from established cervical cell lines exhibit stem-cell markers and increased radioresistance. BMC Cancer. 2012;12:48. doi: 10.1186/1471-2407-12-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abubaker K, Latifi A, Luwor R, Nazaretian S, Zhu H, Quinn MA, Thompson EW, Findlay JK, Ahmed N. Short-term single treatment of chemotherapy results in the enrichment of ovarian cancer stem cell-like cells leading to an increased tumor burden. Mol Cancer. 2013;12:24. doi: 10.1186/1476-4598-12-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grulich AE, van Leeuwen MT, Falster MO, Vajdic CM. Incidence of cancers in people with HIV/AIDS compared with immunosuppressed transplant recipients: a meta-analysis. Lancet. 2007;370:59–67. doi: 10.1016/S0140-6736(07)61050-2. [DOI] [PubMed] [Google Scholar]
- 16.Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ, Schreiber RD. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature. 2001;410:1107–1111. doi: 10.1038/35074122. [DOI] [PubMed] [Google Scholar]
- 17.Frank NY, Margaryan A, Huang Y, Schatton T, Waaga-Gasser AM, Gasser M, Sayegh MH, Sadee W, Frank MH. ABCB5-mediated doxorubicin transport and chemoresistance in human malignant melanoma. Cancer Res. 2005;65:4320–4333. doi: 10.1158/0008-5472.CAN-04-3327. [DOI] [PubMed] [Google Scholar]
- 18.Schatton T, Frank MH. Antitumor immunity and cancer stem cells. Ann N Y Acad Sci. 2009;1176:154–169. doi: 10.1111/j.1749-6632.2009.04568.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Le Blanc K, Tammik C, Rosendahl K, Zetterberg E, Ringden O. HLA expression and immunologic properties of differentiated and undifferentiated mesenchymal stem cells. Exp Hematol. 2003;31:890–896. doi: 10.1016/S0301-472X(03)00110-3. [DOI] [PubMed] [Google Scholar]
- 20.Carretero R, Romero JM, Ruiz-Cabello F, Maleno I, Rodriguez F, Camacho FM, Real LM, Garrido F, Cabrera T. Analysis of HLA class I expression in progressing and regressing metastatic melanoma lesions after immunotherapy. Immunogenetics. 2008;60:439–447. doi: 10.1007/s00251-008-0303-5. [DOI] [PubMed] [Google Scholar]
- 21.Piccirillo SG, Reynolds BA, Zanetti N, Lamorte G, Binda E, Broggi G, Brem H, Olivi A, Dimeco F, Vescovi AL. Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature. 2006;444:761–765. doi: 10.1038/nature05349. [DOI] [PubMed] [Google Scholar]
- 22.Shipitsin M, Campbell LL, Argani P, Weremowicz S, Bloushtain-Qimron N, Yao J, Nikolskaya T, Serebryiskaya T, Beroukhim R, Hu M, et al. Molecular definition of breast tumor heterogeneity. Cancer Cell. 2007;11:259–273. doi: 10.1016/j.ccr.2007.01.013. [DOI] [PubMed] [Google Scholar]
- 23.Ghannam S, Pene J, Moquet-Torcy G, Jorgensen C, Yssel H. Mesenchymal stem cells inhibit human Th17 cell differentiation and function and induce a T regulatory cell phenotype. J Immunol. 2010;185:302–312. doi: 10.4049/jimmunol.0902007. [DOI] [PubMed] [Google Scholar]
- 24.Beyth S, Borovsky Z, Mevorach D, Liebergall M, Gazit Z, Aslan H, Galun E, Rachmilewitz J. Human mesenchymal stem cells alter antigen-presenting cell maturation and induce T-cell unresponsiveness. Blood. 2005;105:2214–2219. doi: 10.1182/blood-2004-07-2921. [DOI] [PubMed] [Google Scholar]
- 25.Jiang XX, Zhang Y, Liu B, Zhang SX, Wu Y, Yu XD, Mao N. Human mesenchymal stem cells inhibit differentiation and function of monocyte-derived dendritic cells. Blood. 2005;105:4120–4126. doi: 10.1182/blood-2004-02-0586. [DOI] [PubMed] [Google Scholar]
- 26.Augello A, Tasso R, Negrini SM, Amateis A, Indiveri F, Cancedda R, Pennesi G. Bone marrow mesenchymal progenitor cells inhibit lymphocyte proliferation by activation of the programmed death 1 pathway. Eur J Immunol. 2005;35:1482–1490. doi: 10.1002/eji.200425405. [DOI] [PubMed] [Google Scholar]
- 27.Inoda S, Hirohashi Y, Torigoe T, Morita R, Takahashi A, Asanuma H, Nakatsugawa M, Nishizawa S, Tamura Y, Tsuruma T, et al. Cytotoxic T lymphocytes efficiently recognize human colon cancer stem-like cells. Am J Pathol. 2011;178:1805–1813. doi: 10.1016/j.ajpath.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Prince MEP, Zhou L, Moyer JS, Tao H, Lu L, Owen J, Etigen M, Zheng F, Chang AE, Xia J, et al. Evaluation of the immunogenicity of ALDH(high) human head and neck squamous cell carcinoma cancer stem cells in vitro. Oral Oncol. 2016;59:30–42. doi: 10.1016/j.oraloncology.2016.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ning N, Pan Q, Zheng F, Teitz-Tennenbaum S, Egenti M, Yet J, Li M, Ginestier C, Wicha MS, Moyer JS, et al. Cancer stem cell vaccination confers significant antitumor immunity. Cancer Res. 2012;72:1853–1864. doi: 10.1158/0008-5472.CAN-11-1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brown CE, Starr R, Martinez C, Aguilar B, D’Apuzzo M, Todorov I, Shih CC, Badie B, Hudecek M, Riddell SR, et al. Recognition and killing of brain tumor stem-like initiating cells by CD8+ cytolytic T cells. Cancer Res. 2009;69:8886–8893. doi: 10.1158/0008-5472.CAN-09-2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brown EJ, Frazier WA. Integrin-associated protein (CD47) and its ligands. Trends Cell Biol. 2001;11:130–135. doi: 10.1016/S0962-8924(00)01906-1. [DOI] [PubMed] [Google Scholar]
- 32.Gao AG, Lindberg FP, Dimitry JM, Brown EJ, Frazier WA. Thrombospondin modulates alpha v beta 3 function through integrin-associated protein. J Cell Biol. 1996;135:533–544. doi: 10.1083/jcb.135.2.533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dunkle ET, Zaucke F, Clegg DO. Thrombospondin-4 and matrix three-dimensionality in axon outgrowth and adhesion in the developing retina. Exp Eye Res. 2007;84:707–717. doi: 10.1016/j.exer.2006.12.014. [DOI] [PubMed] [Google Scholar]
- 34.Liu Y, Buhring HJ, Zen K, Burst SL, Schnell FJ, Williams IR, Parkos CA. Signal regulatory protein (SIRPalpha), a cellular ligand for CD47, regulates neutrophil transmigration. J Biol Chem. 2002;277:10028–10036. doi: 10.1074/jbc.M109720200. [DOI] [PubMed] [Google Scholar]
- 35.Ticchioni M, Deckert M, Mary F, Bernard G, Brown EJ, Bernard A. Integrin-associated protein (CD47) is a comitogenic molecule on CD3-activated human T cells. J Immunol. 1997;158:677–684. [PubMed] [Google Scholar]
- 36.Chao MP, Weissman IL, Majeti R. The CD47-SIRPalpha pathway in cancer immune evasion and potential therapeutic implications. Curr Opin Immunol. 2012;24:225–232. doi: 10.1016/j.coi.2012.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blazar BR, Lindberg FP, Ingulli E, Panoskaltsis-Mortari A, Oldenborg PA, Iizuka K, Yokoyama WM, Taylor PA. CD47 (integrin-associated protein) engagement of dendritic cell and macrophage counterreceptors is required to prevent the clearance of donor lymphohematopoietic cells. J Exp Med. 2001;194:541–549. doi: 10.1084/jem.194.4.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Willingham SB, Volkmer JP, Gentles AJ, Sahoo D, Dalerba P, Mitra SS, Wang J, Contreras-Trujillo H, Martin R, Cohen JD, et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc Natl Acad Sci U S A. 2012;109:6662–6667. doi: 10.1073/pnas.1121623109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chan KS, Espinosa I, Chao M, Wong D, Ailles L, Diehn M, Gill H, Presti J Jr., Chang HY, van de Rijn M, et al. Identification, molecular characterization, clinical prognosis, and therapeutic targeting of human bladder tumor-initiating cells. Proc Natl Acad Sci U S A. 2009;106:14016–14021. doi: 10.1073/pnas.0906549106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jaiswal S, Jamieson CH, Pang WW, Park CY, Chao MP, Majeti R, Traver D, van Rooijen N, Weissman IL. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell. 2009;138:271–285. doi: 10.1016/j.cell.2009.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nonino A, Nascimento JM, Mascarenhas CC, Mazzeu JF, Pereira RW, Jacomo RH. CD47 expression is decreased in hematopoietic progenitor cells in patients with myelofibrosis. Braz J Med Biol Res. 2018;52:e7784. doi: 10.1590/1414-431x20187784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sharrow AC, Perkins B, Collector MI, Yu W, Simons BW, Jones RJ. Characterization of aldehyde dehydrogenase 1 high ovarian cancer cells: towards targeted stem cell therapy. Gynecol Oncol. 2016;142:341–348. doi: 10.1016/j.ygyno.2016.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang L, Mezencev R, Bowen NJ, Matyunina LV, McDonald JF. Isolation and characterization of stem-like cells from a human ovarian cancer cell line. Mol Cell Biochem. 2012;363:257–268. doi: 10.1007/s11010-011-1178-6. [DOI] [PubMed] [Google Scholar]
- 44.Paullin T, Powell C, Menzie C, Hill R, Cheng F, Martyniuk CJ, Westerheide SD. Spheroid growth in ovarian cancer alters transcriptome responses for stress pathways and epigenetic responses. PLoS One. 2017;12:e0182930. doi: 10.1371/journal.pone.0182930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang H, Lu H, Xiang L, Bullen JW, Zhang C, Samanta D, Gilkes DM, He J, Semenza GL. HIF-1 regulates CD47 expression in breast cancer cells to promote evasion of phagocytosis and maintenance of cancer stem cells. Proc Natl Acad Sci USA. 2015;112:E6215–6223. doi: 10.1073/pnas.1520032112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mathieu J, Zhang Z, Zhou W, Wang AJ, Heddleston JM, Pinna CM, Hubaud A, Stadler B, Choi M, Bar M, et al. HIF induces human embryonic stem cell markers in cancer cells. Cancer Res. 2011;71:4640–4652. doi: 10.1158/0008-5472.CAN-10-3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kaur S, Soto-Pantoja DR, Stein EV, Liu C, Elkahloun AG, Pendrak ML, Nicolae A, Singh SP, Nie Z, Levens D, et al. Thrombospondin-1 signaling through CD47 inhibits self-renewal by regulating c-Myc and other stem cell transcription factors. Sci Rep. 2013;3:1673. doi: 10.1038/srep01673. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.