

ABSTRACT
Anti-CD20 treatment represents a therapeutic benefit for patients with B-cell lymphomas, although more efficient therapies are needed for refractory or relapsing patients. Among them, the combination of anti-CD20 and IL-2 that induces T cell response has been hampered by the expansion of FoxP3+ Tregs that strongly express the high affinity IL-2 receptor (IL-2R αβγ). We explore here the anti-tumor effect of an anti-CD20 antibody combined with a mutated IL-2 (no-alpha mutein) which has a disrupted affinity for the IL-2R αβγ. We demonstrate that anti-CD20/no-alpha mutein combination significantly augments the survival rate of mice challenged with huCD20+ cells as compared to animals treated with anti-CD20 ± IL-2. Moreover, the combination with no-alpha mutein but not IL-2 provokes an increase of granzyme B and perforin in splenic NK and CD8+ T cells, a reduction of Tregs and an increase in activated macrophages. The former combination also induces a T helper profile different from that obtained with IL-2, with an earlier polarization to Th1 and no increase in Th17. The therapeutic effect of anti-CD20/no-alpha mutein was accompanied by an expansion of peripheral central (TCM) and effector (TEM) memory CD8+ T cell compartments. Last, as opposed to IL-2, no-alpha mutein administered at the beginning of anti-CD20 treatment did not dampen the long-term protection of surviving mice after tumor rechallenge. Thus, this study shows that the combination of anti-tumor antibodies and no-alpha mutein is a promising approach to improve the therapeutic effect of these antibodies by potentiating NK/macrophage-mediated innate immunity and the adaptive T-cell response.
KEYWORDS: Anti-CD20, IL-2, IL-2 mutein, immunotherapy, lymphoma
Introduction
Rituximab leads the group of anti-CD20 antibodies specific for this molecule in the clinics, with remarkable therapeutic effects in follicular and aggressive B cell non-Hodgkin lymphoma.1,2 Although the efficacy of rituximab is well established, approximately 25–30% of patients do not respond to R-CHOP (rituximab combined with cyclophosphamide, doxorubicin, vincristine and prednisone (CHOP) chemotherapy) as first-line treatment3 and in some patients, the response to rituximab-based regimens ends in a primary disease relapse.4 Regrettably, responses to rituximab are often short-lived and complete responses to rituximab monotherapy are rare.5 Several strategies are currently being investigated to increase the effectiveness of rituximab, some of which include combination treatments.6,7
Interleukin-2 (IL-2) has been considered as a cytokine central for protective immunity because of its potent capacity to induce the proliferation and the cytotoxic capacity of T cells8 and to potentiate antibody-dependent cell cytotoxicity (ADCC) mediated by natural killer (NK) cells.9,10 These properties have encouraged its use at high doses in the treatment of melanoma and renal cell carcinoma, which has led to clinical benefits in a small group of patients but also to a strong toxicity due to a vascular leak syndrome (VLS) related to the dosing. The limited IL-2 therapy efficacy has been related to an IL-2 driven expansion of regulatory T cells (Tregs) which, in turn, inhibit the antitumor immunity11,12 leading to a poor prognosis.13
Several studies have shown that the antitumor effect of anti-CD20 therapies is associated to antibody-dependent cell cytotoxicity (ADCC) involving NK cells and macrophages5 as well as the participation of both CD8+ and CD4+ T cells.14-16 Thus, a combination of rituximab and IL-2 has been tested in preclinical and clinical settings.17,18 Although the first trials showed that some patients with non-Hodgkin lymphoma achieved complete responses,18,19 further trials did not show a significant clinical benefit.20,21 This could be due to the fact that despite IL-2 stimulates T and NK effector cells, it also increases the number of Tregs with a pro-tumor effect.21 Most of the trials have used IL-2 at low doses, which is known to induce Tregs expansion.19,20,22 Although, conflicting results have been reported regarding the prognostic significance of Tregs infiltration in both HL and NHL23,24 other studies have shown a direct correlation between Tregs and bad prognosis in NHL lymphomas.25,26 In addition, the first preclinical experiences in evaluating the combination of IL-2 and anti-CD20 therapy were performed in immunodeficient mice and, hence, made it impossible to assess the role of T cells in the anti-tumor responses observed.17,18 Nonetheless, further experiments have explored the impact of IL-2 in anti-tumor response induced by anti-CD20 treatment in immunocompetent mice through the use of an EL4-huCD20 tumor model.15 This work showed that anti-CD20 antibody allows a long-term protection against tumor cells by inducing a memory T-cell response, a phenomenon termed the “vaccinal” effect of anti-tumor antibodies.15,16,27 This long-lasting protection was found to be dependent on an interferon γ (IFNγ)/interleukin 12 (IL-12) axis and on the presence of CD4+ T cells and NK cells at the initiation of anti-CD20 treatment. In these studies, IL-2 injected at the beginning of the antibody treatment did not improve the survival of the animals induced by anti-CD20 therapy. By contrast, an enhanced survival rate was observed when long-term surviving anti-CD20 treated mice were rechallenged with tumor cells followed by the administration of the cytokine.15 Strikingly, in this preclinical model, Tregs expanded markedly in tumor-bearing mice and their presence was directly associated with tumor-induced animal death. The expansion of these regulatory cells was reverted after anti-CD20 treatment.16 Altogether, these data support the idea that any immunotherapeutic approach to improve the efficiency of anti-CD20-based treatment should tackle Tregs proliferation and expansion.
For that purpose, different strategies have been explored to improve the IL-2-based therapy, some of which relying on the generation of IL-2 variants that do not favor the expansion of Tregs.28–30 The rational design of these variants has been largely based on changes in the IL-2 binding affinity to the different subunits of the high affinity IL-2R αβγ. This trimeric receptor is strongly and constitutively expressed on Tregs although it is also present on activated effector T cells and on a small proportion of NK cells. By contrast, the intermediate affinity IL-2Rββγ is expressed on naïve and memory T cells, as well as on a large percentage of NK cells.31 Engagement of this dimeric receptor by IL-2 also triggers activation and proliferation signals.32-34
A human IL-2 mutant whose interaction with CD25 (IL-2Rα chain) is significantly disrupted,35 that behaves as an IL-2R signaling agonist which expands preferentially CD8+ T lymphocytes and NK cells over Tregs, has been developed at the Center of Molecular Immunology (CIM).28,36 This IL-2 variant, termed no-alpha mutein, has demonstrated a higher anti-metastatic effect than IL-2 in 3LL-D122 and B16 tumor models.28 Of note, this mutated IL-2 displays a reduced toxicity as compared to its wild-type counterpart.28
In the present study, we demonstrate that the injection of no-alpha mutein into naïve mice provokes an increase of splenic central memory CD8+ T cells and a significant decrease of Tregs cells, as compared with IL-2. We also show that no-alpha mutein, unlike wild-type IL-2, improves the survival rate of CD20+ tumor-bearing mice achieved by anti-CD20 treatment and differentially reshapes the adaptive T-cell response with an earlier polarization to Th1, no increase in Th17, a reduction in Tregs, an enhanced cytotoxic potential of NK and CD8+ T cells and an increase in activated macrophages. These findings make no-alpha mutein a promising candidate for combination immunotherapies to improve the therapeutic vaccinal effect of anti-tumor antibodies.
Results
Differential immunomodulatory role of no-alpha mutein
Previous experiments have shown that no-alpha mutein expands preferentially CD8+ and NK cells over Tregs cells28,36 suggesting that it could reinforce the anti-tumor response induced by anti-CD20 antibody treatment that opposes Tregs expansion.16 First, we studied the differential immunomodulatory role of no-alpha mutein as compared with its wild-type counterpart at high doses in naïve C57Bl/6 mice, taking into consideration both cytokines of human origin cross-react with mouse IL-2 receptors. We followed an infusion scheme previously used to evaluate the effect of IL-2 on the antitumor activity of anti-CD20 antibody in an EL4-huCD20 tumor model15 where Tregs are recruited.16 After the intraperitoneal (i.p.) injections of IL-2 or no-alpha mutein, mice were sacrificed at day 6 and spleens were collected. Body weight and temperature were monitored. They remained equivalent as compared to mice receiving PBS and were similar for both groups (data not shown). At the doses assayed, IL-2 and no-alpha mutein did not appear to be immunogenic in mice (data not shown). Spleen T cells and NK cells were then analyzed by flow cytometry.
When compared to the control group, the number of total CD8+ T cells and more specifically of CD8+ central memory T cells (CD44highCD62Lhigh) (TCM), was significantly increased by no-alpha mutein unlike the wild-type IL-2 (Figure 1(a); Supplementary Figure S1A). In contrast, no difference was seen between the number of CD8+ effector memory T cells (CD44highCD62 Llow) (TEM) after administration of either IL-2 or no-alpha mutein, both treatments expanding this cell subset (Figure 1(b)). CD4+ T cells, particularly CD4+ TCM were significantly expanded in mice treated with IL-2 and no-alpha mutein (Figure 1(c) and Supplemental Figure S1B). Similarly, both IL-2 and no-alpha mutein increased the number of CD4+ TEM (Figure 1(d)).
Figure 1.
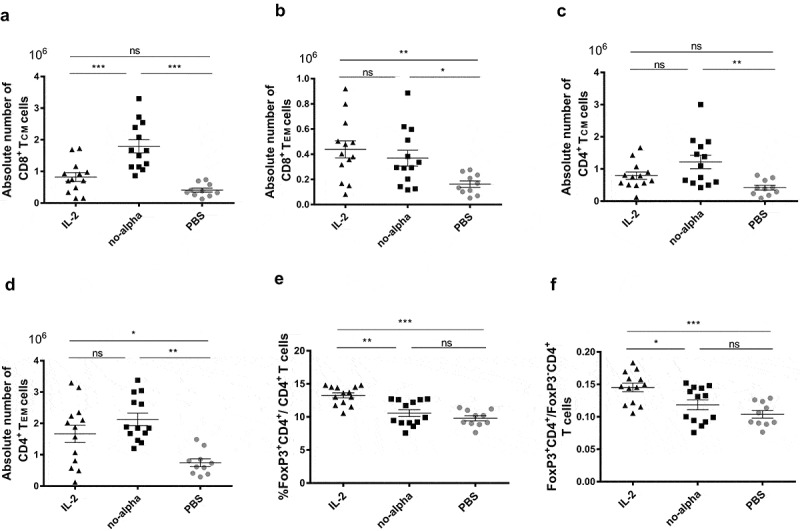
Effects of IL-2 and no-alpha mutein on splenic T cells in naïve C57Bl/6. (a–d), absolute numbers of CD8+ central memory T cells (TCM) (CD8+CD44high CD62L+) (a), CD8+ effector memory T cells (TEM) (CD8+CD44highCD62L−) (b), CD4+ TCM cells (CD4+CD44highCD62L+) (c), and CD4+ TEM cells (CD4+CD44highCD62L−). (d) of spleens from C57Bl/6 mice, injected with PBS, IL-2 or no-alpha mutein at day 0 and day 4, and analyzed by flow cytometry at day 6. (e), No-alpha mutein kept the percentages of FoxP3+CD4+ T cells to the level of control mice. (f), Mice receiving no-alpha mutein showed lower ratio of FoxP3+CD4+ T cells/FoxP3− CD4+ T cells. Data correspond to three independent pooled experiments; IL2 (n = 13), no-alpha (n = 13), and PBS (n = 10). (a–f), Each symbol represents an individual mouse. Horizontal bars represent the mean ± SEM (*, P <.05; **, P <.01; ***, P <.001; ns, not significant).
Regarding the splenic NK cells, IL-2 and no-alpha mutein treatments increased markedly the number of the cells (Supplementary Figure S1C). Furthermore, both IL-2 cytokines enriched equally the subset of activated NK cells (NK1.1+B220+)37 (Supplementary Figure S1D).
We then compared the effect of IL-2 and no-alpha mutein on splenic Tregs (FoxP3+CD4+). IL-2 treatment increased significantly the percentage of Tregs among CD4+ T cells (Figure 1(e)). By contrast, the no-alpha mutein treatment kept this percentage to the level of control mice (Figure 1(e)).
It is noteworthy that a lower ratio of FoxP3+CD4+/CD8+ cells was observed in animals receiving no-alpha mutein as compared to those injected with IL-2 (Supplementary Figure S1E). These results evidence for the first time that no-alpha mutein, in this treatment scheme, shifts the balance toward the effector cells rather than regulatory T cells, in concordance with previous in vitro findings or with another administration schedule in vivo.28,36
No-alpha mutein, and not IL-2, improves the antitumor effect of anti-CD20 antibody
We then evaluated the effect of a combination of no-alpha mutein with the anti-CD20 CAT-13 mAb (a type 1 anti-CD20 mouse IgG2a) in an immunocompetent mouse model where C57Bl/6 mice are intravenously injected with EL4-huCD20 cells.15,16,27 First, we examined whether IL-2 or no-alpha mutein treatment induces per se an anti-tumor effect in C57Bl/6 mice injected with EL4-huCD20 tumor cells. Mice received intraperitoneally two doses of IL-2 or no-alpha mutein at day −1 and day 3, with respect to tumor cells inoculation (Figure 2(a)). Neither IL-2 or no-alpha mutein monotherapies increased the survival of the animals (Figure 2(b)). Based on these results, we then treated EL4-huCD20 tumor-bearing mice with the anti-CD20 CAT-13 mAb.15 In these experiments, immunocompetent C57Bl/6 mice were injected intravenously with 2 × 105 EL4-huCD20 cells on day 0 followed by CAT-13 (200 μg intraperitoneal injections on days 1, 4, 7, 10, and 13) alone or in combination with IL-2 or no-alpha mutein as described above (Figure 2(c)). Noteworthy, the responses to anti-CD20 therapy achieved about a 60% protection, in agreement with previous studies. As previously reported,15 the survival rate was similar between mice treated only with anti-CD20 and those that received antibody therapy combined with IL-2 (Figure 2(d)). In contrast, a significant increase of the survival rate of mice treated with the no-alpha mutein and the anti-CD20 antibody was observed (Figure 2(d)). In all experiments, mice that received anti-CD20 + no-alpha mutein combined treatment exhibited the highest anti-tumor protection. These data show that combining no-alpha mutein with antitumor therapies inducing adaptive cellular responses treatment could be of clinical therapeutic significance.
Figure 2.
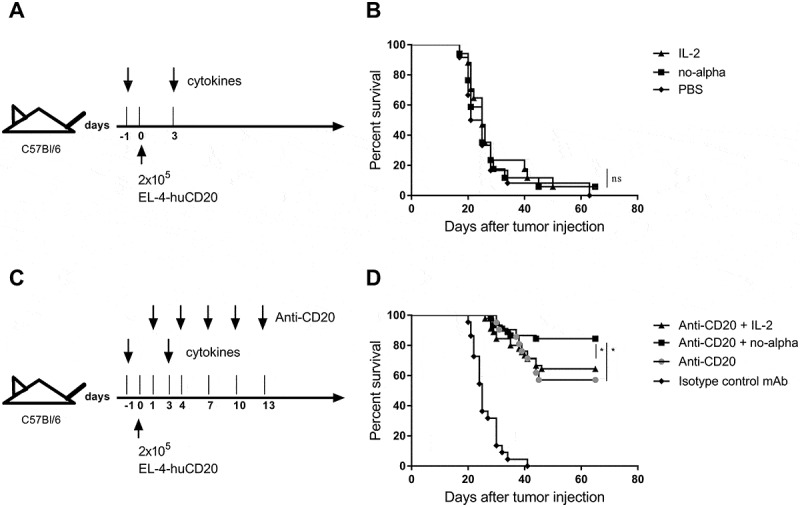
Potentiation of the anti-tumor effect of anti-CD20 antibody upon combination with no-alpha mutein. (a), Schematic representation of cytokines monotherapy treatment schedule. (b), IL-2 and no-alpha mutein have no antitumor effect in C57Bl/6 mice injected with EL4-huCD20 cells; IL-2 (n = 17), no-alpha (n = 17), and PBS (n = 12). (c), Schema of combined treatment schedule. (d), Survival curves of mice with the different treatments as indicated; CD20 + IL-2 (n = 45), CD20 + no alpha (n = 45), CD20 (n = 21) and IgG2a (n = 21). Data correspond to two (b) and three (d) independent pooled experiments (log-rank test; *, P < .05; **, P < .01; ***, P < .001; ns: not significant).
Anti-CD20 + no-alpha mutein combined treatment modulates the balance between Th1 and Tregs induced by the antibody alone
To investigate the underlying immune mechanisms that may be related to the increased survival of mice treated with anti-CD20 and no-alpha mutein, we then studied the evolution of T-cell compartment. Of note, the absolute number of splenic CD8+ and CD4+ T cells at days 14 and 21 were not significantly different between animals receiving the anti-CD20 therapy alone, or combined with any of IL-2 molecules (Supplementary Figure S2A,B). We previously reported that anti-CD20 therapy induces a Th1 response and prevents protumor Tregs expansion detectable at day 21 in the same tumor model.16 Here, a significantly increased percentage of Th1 cells in mice treated with anti-CD20 + no-alpha mutein was observed at day 14 when compared to animals receiving anti-CD20 alone or anti-CD20 + IL-2 (Figure 3(a)). This increase was also observed at day 21, although it was non-significant when compared with animals receiving anti-CD20 + IL-2. No difference in the Th2 percentages was seen between the three groups of animals (Supplementary Figure S3A). Noteworthy, a rise in the percentages of Th17 cells was observed for IL-2-based treatment, at day 21 (Figure 3(b)). On the contrary, no change in this cell subset resulted upon anti-CD20 therapy alone or combined with no-alpha mutein (Figure 3(b)).
Figure 3.
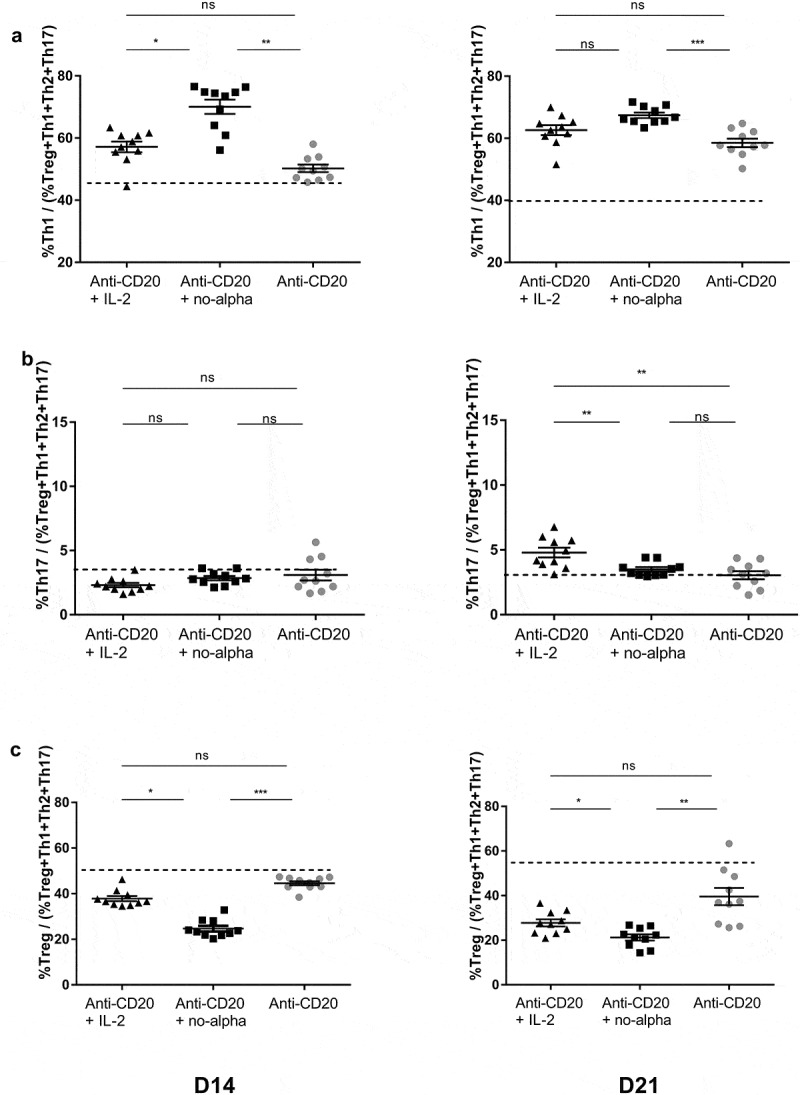
Effect of anti-CD20 + no-alpha mutein combination therapy on CD4+ T cell subsets. The anti-CD20 + no-alpha mutein combination therapy increased the percentages of Th1/(IFNγ+CD4+CD3+) (a), kept the percentages of Th17/(IL-17A+CD4+CD3+) to the level of anti-CD20 mAb-treated mice (b) and induced a decrease of the percentage of Treg/(FoxP3+CD4+CD3+), (c) of stimulated splenocytes from C57Bl/6 mice injected with EL4-huCD20 cells, evaluated by flow cytometry, at days 14 and 21. Dotted lines represent the mean values obtained with isotype control-treated C57Bl/6 mice for the indicated population. Data correspond to two independent pooled experiments; n = 10. Each symbol represents the relative percentage of the indicated cell subset among the total (Treg+Th1+Th2+Th17) CD4+ T-cell compartment for one individual mouse. Horizontal bars represent the mean ± SEM (*, P < .05; **, P < .01; ***, P < .001; ns: not significant).
Importantly, the evaluation of splenocytes showed that the combined therapy with no-alpha mutein induced a significant decrease of the percentage of Tregs in comparison with treatment with anti-CD20 alone or in combination with IL-2, both at days 14 and 21 (Figure 3(c)). Furthermore, the highest %Th1/%Tregs ratio in mice receiving no-alpha mutein points out a preferential expansion of effector CD4+ T cells over regulatory lymphocytes (Supplementary Fig. S3B). Overall, it should be noted that the combination of no-alpha mutein with anti-CD20 therapy positively modulates the Th1/Tregs balance induced by the antibody while the combination with IL-2 increases the percentage of Th17 cells.
The beneficial effects of IL-2 based treatments have been linked to the enhancement of CD8+ T cell function. Thus, we examined the impact of the combination treatments on these cells (Figure 4). Similar increases in the percentages of splenic IFNγ+ CD8+ cells were found in the combination groups with IL-2 or no-alpha mutein treatments at day 14 (Figure 4(a)). Nonetheless, a noticeable higher proportion of this cell subset was observed a week later in mice receiving anti-CD20 + no-alpha mutein (Supplementary Figure S4A). Additionally, taken into consideration the Th1 biased response as early as day 14, the production of perforin and granzyme B by CD8+ T cells was examined at that moment. Both the percentage (Figure 4(b,c)) and the absolute number (Supplementary Figure S4B,C) were significantly higher in the anti-CD20 + no-alpha mutein combination, indicating a potentiation of the killing capacity of these cells. This increase in the killing potential of T cells is likely associated with the polarization of Th1 induced by the combination of the anti-CD20 treatment with no-alpha mutein.
Figure 4.
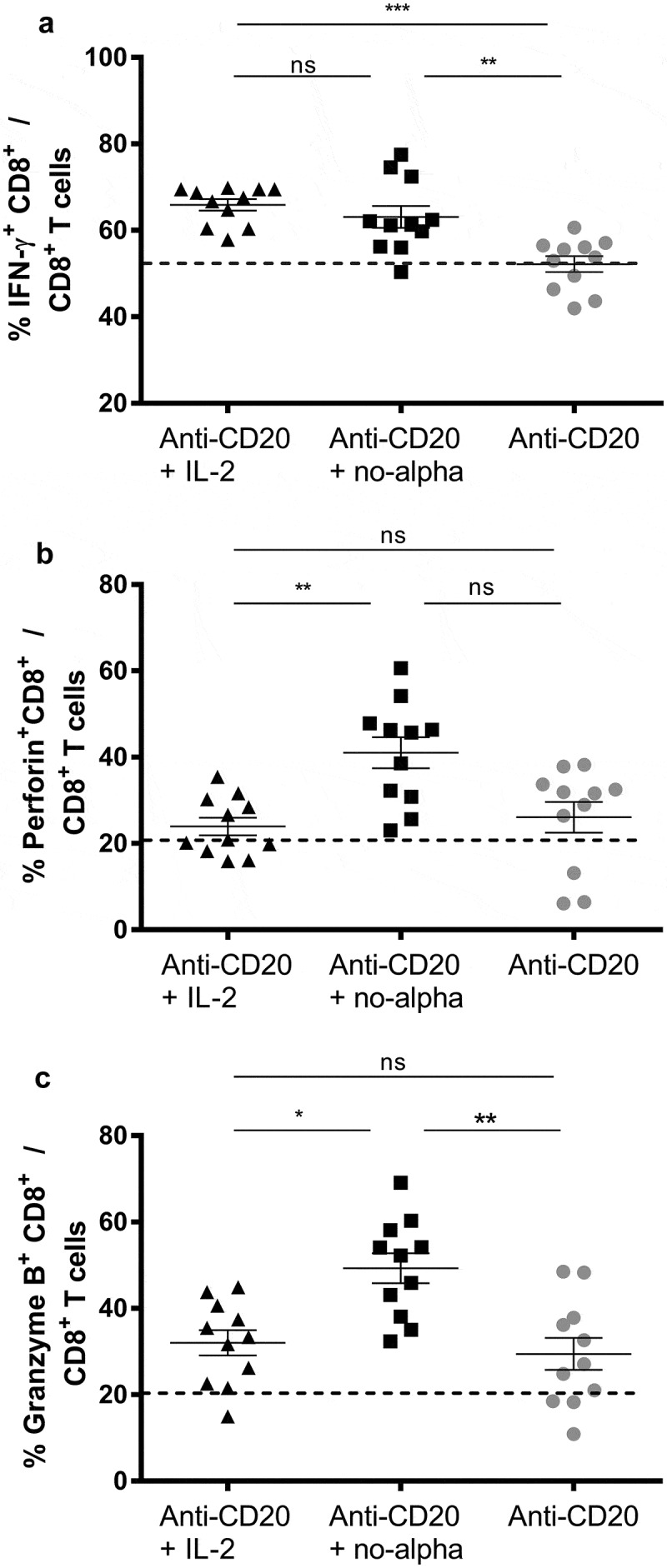
Anti-CD20 + no-alpha mutein combination therapy activates CD8+ T cell subset. Percentage of CD8+ T cells expressing IFNγ+ (a), perforin+ (b), and granzyme B+ (c) of spleens from C57Bl/6 mice analyzed by flow cytometry 14 days after challenge with 2 × 105 EL-4-huCD20 cells and treated with the described different treatments. Dotted lines represent the mean values obtained with isotype control-treated C57Bl/6 mice for the indicated population. Data correspond to two independent experiments (n = 4–7 per group). Horizontal bars represent the mean ± SD (Kruskal-Wallis, Dunn’s post hoc test, *, P < .05; **, P < .01; ***, P < .001; ns, not significant).
Anti-CD20 + no-alpha mutein combined treatment increases the number of activated NK cells
An increase in the percentage of splenic NK1.1+CD3− cells was observed in mice treated with anti-CD20 alone or in combination with IL-2 or no-alpha mutein at days 14 and 21 (data no shown). In addition, both IL-2-based therapies incremented the number of splenic NK1.1+B220+ cells, that are considered as activated NK cells in mouse,37 at the same time point (Supplementary Figure S5). Of note, only the co-administration of no-alpha mutein enhanced significantly the production of granzyme B by these cells (Figure 5(a)). Also, an increase in the number of NK1.1+ cells producing perforin was observed in the mice that received anti-CD20 + no-alpha mutein therapy as compared with those receiving anti-CD20 + IL-2 (Figure 5(b)). A higher number of IFNγ+NK1.1+ producing cells as compared with the antibody monotherapy was also triggered by the no-alpha mutein combination treatment (Figure 5(c)). We previously reported that the induction of anti-tumor response by anti-CD20 treatment requires NK cells.15 Thus, the increase in the number of activated NK cells observed here could be involved in the improvement of the survival rate by the combination of anti-CD20 with no-alpha mutein.
Figure 5.
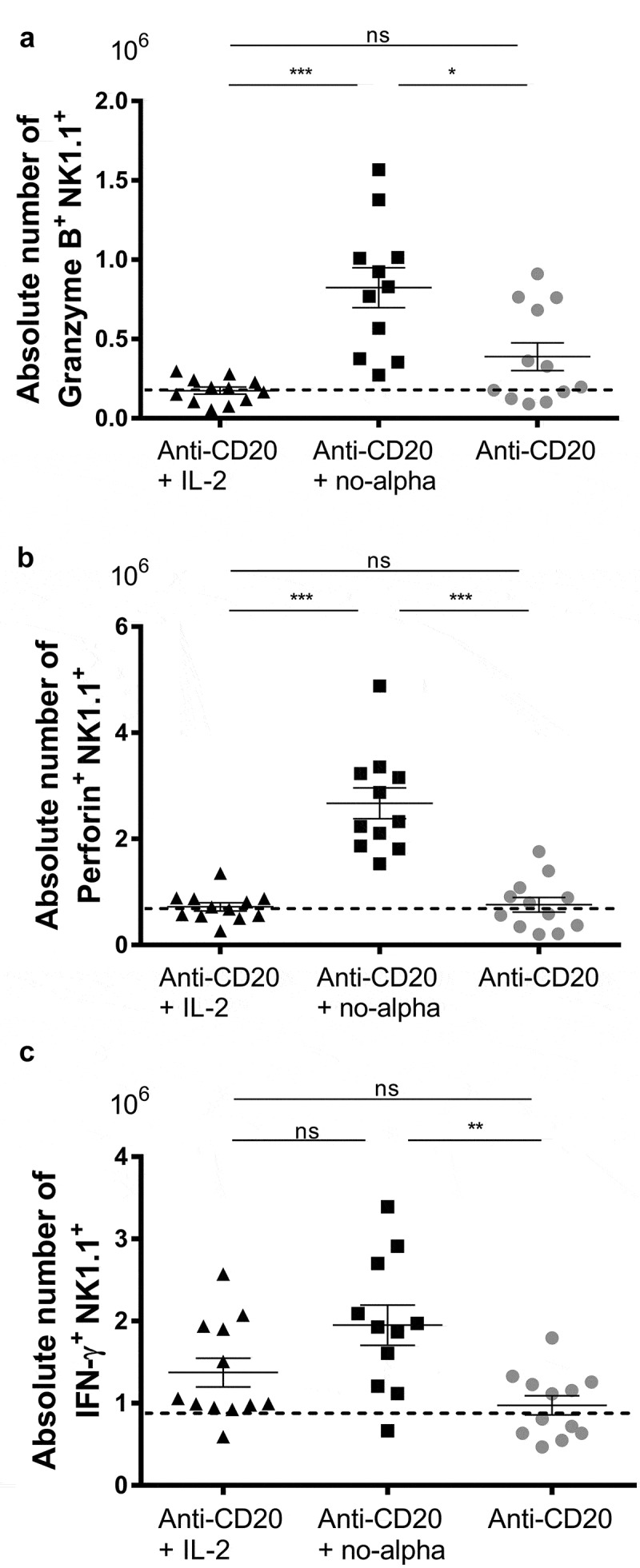
Increase of activated NK cells by anti-CD20 + no-alpha mutein combination therapy. Absolute number of granzyme B+ NK1.1+ (a), perforin+NK1.1+ (b), and IFNγ+NK1.1+ (c) of splenocytes from C57Bl/6 mice analyzed by flow cytometry 14 days after challenge with 2 × 105 EL-4-huCD20 cells and treated with different therapies. Dotted lines represent the mean values obtained with isotype control-treated C57Bl/6 mice for the indicated population. Data correspond to two independent experiments (n = 5–7 per group). Horizontal bars represent the mean ± SD (Kruskal-Wallis, Dunn’s post hoc test, *, P < .05; **, P < .01; ns, not significant).
Anti-CD20 + no-alpha mutein combined treatment increases the number of activated macrophages
We next explored the effect of combination treatment with anti-CD20 antibody and IL-2 or no-alpha mutein on macrophages (MΦ, defined as CD11b+CD11c−Gr1lowF4/80+), at day 14. These cells have been shown to be important effectors following anti-CD20 therapy38 and are also involved in the generation of anti-tumor T-cell memory response.27 A significant increase in the number of MΦ was observed in the group treated with anti-CD20 and no-alpha mutein, but not with the IL-2 treatment (Figure 6(a)). Furthermore, in these animals, an increased expression of the major histocompatibility complex class II (MHCII) molecules and of CD16/CD32 (mouse FcγRII, FcγRIII) and FcγRIV on MΦ (Figure 6(b,c)) was detected. The rise in the expression of these molecules are a hallmark of macrophage activation.
Figure 6.
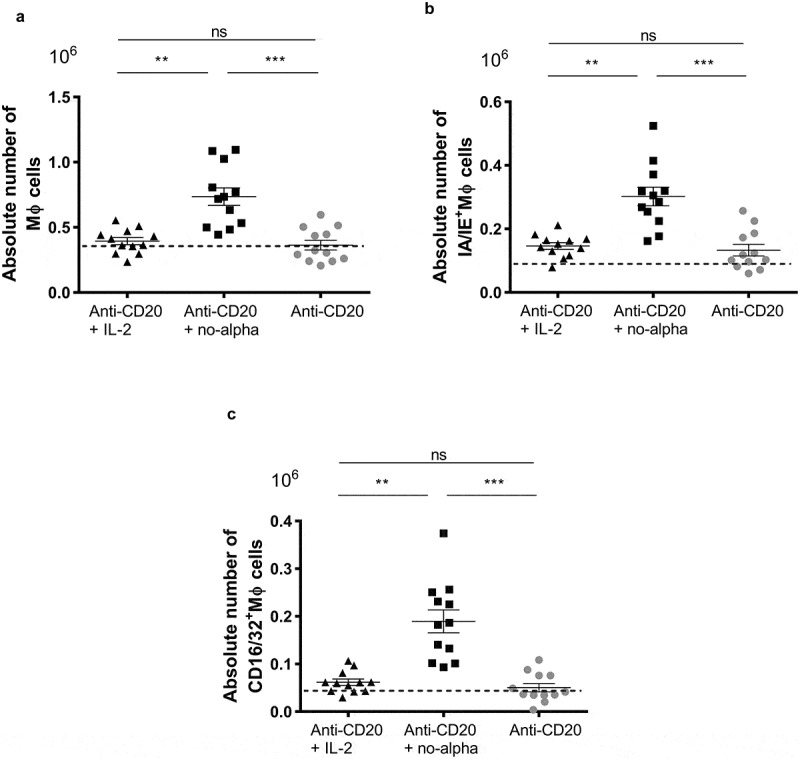
Increase in the macrophages number in animals receiving the anti-CD20 + no-alpha mutein combination therapy. (a), Mice treated with anti-CD20 + no-alpha mutein increased the absolute number of MΦ (CD11b+CD11c−Gr1lo F4/80+) cells. Absolute numbers of spleen MΦ expressing IA/IE class II molecules (b), and FcγRII, FcγRIII (CD16/32) and FcγRIV (c) from C57Bl/6 mice analyzed by flow cytometry 14 days after challenge with 2 × 105 EL-4-huCD20 cells and receiving different treatments. Dotted lines represent the mean values obtained with isotype control-treated C57Bl/6 mice for the indicated population. Data correspond to two independent experiments (n = 5–7 per group). Horizontal bars represent the mean ± SD (Kruskal-Wallis, Dunn’s post hoc test, *, P < .05; ***, P < .001; ns: not significant).
No-alpha mutein conserves the long-lasting protection conferred by anti-CD20 antibody
We previously reported that anti–CD20 therapy provides a long-lasting protection against huCD20+ tumor cells, allowing mice to survive to a subsequent tumor challenge.15 Also, Th1 polarization and reduction of Treg compartment have been shown to be involved in the generation of functional CD8+ memory T cells,39 an important end-point to consider in adaptive responses induced by antibodies. In addition, we have previously shown that the anti-CD20 treatment induces CD4+ effector memory T cells in the EL4-huCD20 model.16
Thus, we evaluated the impact of the combined treatments on blood memory T cells. The analysis made at day 21 showed no significant difference in the percentages of peripheral CD8+ and CD4+ T cells (data not shown). However, a marked increase in the percentages of blood central memory CD8+ T cells (CD8+ TCM) among CD8+ T cells was observed in the animals receiving anti-CD20 + no-alpha mutein when compared to the antibody alone (Figure 7(a)). Anti-CD20 in combination with IL-2 induced a weaker increase of these cells as compared to no-alpha mutein (Figure 7(a)). Also, the proportion of effector memory CD8+ T cells (CD8+ TEM) increased upon co-administration with no alpha mutein as compared to the IL-2 combination therapy (Figure 7(b)). On the contrary, there was no change at day 21 in the peripheral CD4+ TCM and TEM cells compartment after the different therapies (Supplementary Figure S6A,B).
Figure 7.
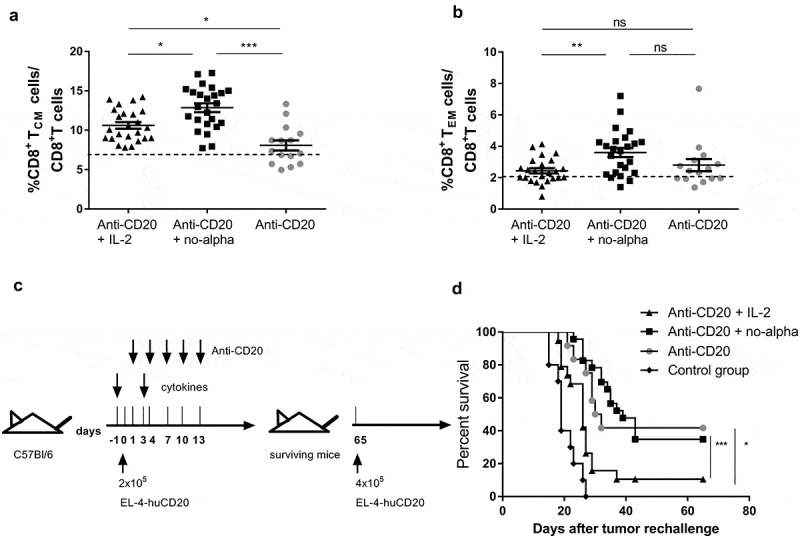
No-alpha mutein maintains the long-lasting protection conferred by the anti-CD20 mAb. Induction of CD8+ TCM cells (a) and CD8+ TEM cells (b) in the peripheral blood of mice injected with 2 × 105 EL4-huCD20 tumor cells and treated with anti-CD20 + no-alpha mutein measured by flow cytometry, at day 21 after tumor injection. Horizontal bars represent the mean ± SD of percentages and the dotted lines represent the mean value obtained with isotype control-treated C57Bl/6 mice for the indicated population. Data correspond to two independent experiments (n = 5–10 per group) (Kruskal-Wallis, Dunn’s post hoc test, *, P < .05; **, P < .01; ***, P < .001; ns: not significant). (c), Schematic representation of treatment schedule. (d), No-alpha mutein maintained the long-term protection conferred by anti-CD20 therapy when surviving animals were rechallenged with tumor cells. Age-matched naïve mice were injected with equal number of cells and used as controls (n = 5 per control group). Data correspond to two independent experiments (log-rank test; *, P < .05; ***, P < .001; ns: not significant).
Finally, we evaluated the long-term effect on mouse survival induced by the use of IL-2 or no-alpha mutein in combination with anti-CD20 therapy in the early phase of the experiment. The surviving animals were rechallenged at day 65 with a higher number of EL4-huCD20 tumor cells (4 x 105/mouse) than at the time of treatment initiation, with no further antibody treatment (Figure 7(c)). Interestingly, while the no-alpha mutein did not impact the ability of mice to resist tumor rechallenge, the administration of IL-2 with anti-CD20 impaired the protection conferred by the anti-CD20 therapy (Figure 7(d)). Thus, the remodeling in the subsets of adaptive and innate cells induced by IL-2 during the induction phase of the treatment negatively impact the long-lasting effect of the anti-CD20 antibody upon tumor rechallenge as opposed to the no-alpha mutein.
Discussion
Although efficacy and success of rituximab are undeniable, a substantial number of NHL patients fail to respond and show signs of resistance to the therapy. The probability of responding to the first treatment with rituximab is about 65–70%.3 Strategies to improve the clinical effectiveness of rituximab are therefore being explored, including combination with cytokines to increase the capacity of the antibody to exert one of its key effector functions, ADCC,40 as well as the induction of adaptive immunity.15,41,42
Addition of IL-2 to rituximab therapy led to NK cell expansion that correlated with a better clinical outcome.17 However, no clinical study has shown a clear signal of greater efficacy of combination than mAb monotherapy.19 Most of clinical trials have combined the rituximab with low IL-2 dose regimens, which have been described to favor the expansion of Tregs.43 The manipulation of these suppressor cells has become the focus of attention as Tregs are significantly increased in peripheral blood of NHL patients receiving or not chemotherapy44 and have been associated with a poor prognosis.26
Different strategies have been developed to reduce the toxic side effects of IL-2 and its capacity to activate negative immune regulators. The generation of IL-2 variants able to expand preferentially immune effector cells has been explored.29,30,45 In particular, the no-alpha mutein, a rationally engineered IL-2 that can activate NK and memory CD44+CD8+ T cells, minimizes the Tregs expansion.28 Here, we demonstrate that the administration of this mutant protein in naïve mice expands the central memory CD8+ T-cell compartment, whose relevance in antitumor immunity has been pinpointed.46 In addition, we show that the no-alpha mutein controls Tregs proliferation, which in turn favors the immune effector players. In agreement with previous results,36 our work confirms the differential immunoregulatory role of the no-alpha mutein as compared to IL-2, which makes it an attractive candidate to be combined with other immunotherapies, including those based on anti-CD20 antibodies.
In the present study, we also show that an induction regimen with anti-huCD20 mAb combined with no-alpha mutein, and not IL-2, increases the overall survival obtained with the antibody monotherapy in mice injected with EL4-huCD20 tumor cells (Figure 2(d)). This is the first demonstration that the combined use of an anti-CD20 mAb with an optimized variant IL-2, administered both at the beginning of treatment, makes it possible to potentiate the in vivo antitumor activity of the antibody. This is an important result as, in a similar setting, Abes et al.15 evidenced no effect of IL-2 on mice protection. It suggests therefore that the combination of an anti-CD20 mAb with an optimized variant IL-2 could be used in the clinic to increase the number of patients that respond to R-CHOP treatment in the first line. The absence of any antitumor effect when the no-alpha mutein was infused alone, at the doses assayed, indicates an increased anti-tumor potency of the combination. It may be due to the no-alpha mutein-driven reinforcement of the immune effectors mobilized by the anti-CD20 antibody and to additional mechanisms directly induced by the cytokine.
In an experimental scenario similar to the one used here, it has been demonstrated that anti-CD20 treatment leads to Th1 polarization that largely overcome the potent protumor effect of Tregs.16 The present study shows that the combination of anti-CD20 + no-alpha mutein accelerates the induction of Th1 response and modulates the Th1/Tregs balance induced by the antibody in favor of the Th1 cell subset, whereas the combination with IL-2 also increases the percentage of Th17 cells (Figure 3(c)). The combination with no alpha mutein prevents the Tregs expansion earlier than the anti-CD20 monotherapy or than the anti-CD20 + IL-2 combined treatment. The importance of the Th1 cell subset in orchestrating the activity of multiple cell types to eradicate tumor cells through the secretion of various cytokines, including IFNγ, is well documented.47 Moreover, in patients and in preclinical murine models, the frequency of tumor-associated CD4+ Th1 cells correlates with therapy success and tumor rejection.48,49 It has been previously evidenced that a Th17 phenotype is associated with a poor survival.16 Contradictory results in the clinics have correlated the increase of Th17 with either a good50 or a bad prognosis.51 Interestingly, in B-NHL, we found that circulating Th17 frequencies in relapsed patients are significantly higher than those in untreated patients or normal individuals.52
Our results also show that the use of no-alpha mutein in the combined treatment stimulates CD8+ T-cell activation as indicated by their enhanced lytic ability, represented by the exacerbated production of perforin and granzyme B (Figure 4).
NK cells and macrophages can also be targeted by Th1 cytokines.53 Our data evidence that no-alpha mutein administered at the beginning of anti-CD20 treatment is able to successfully expand NK cells and to activate their lytic power. The potentiation of the NK cytotoxic machinery and IFNγ secretion two weeks after starting the antibody therapy is a distinctive feature of the no-alpha mutein and an obvious advantage for the commitment of these cells, whose participation in the antitumor activity of anti-CD20 antibodies has been documented.15 Taking into consideration the administration schedule of both cytokine and antibody (Figure 7(c)), we could presume that different mechanisms could operate and evolve all along and after the treatment. A first wave of direct activation of NK cells by the no-alpha mutein in absence of antibody could be the initial source of IFNγ, that represents a potent inducer of DC maturation and antigen presentation for CD4+ T cell priming.16,53 This early stage could be followed by a second one in which coexist both the anti-CD20 antibody and the IL-2 variant. At this stage, the no-alpha mutein still maintains the proliferation and activation of NK cells,28 while the antibody-mediated engagement of FcγR+ NK cells sustains the IFNγ secretion and killing activity. It leads to the formation of tumor antigen-containing immune complexes that triggers the maturation of DC and the IL-12/IFNγ mediated NK-DC crosstalk.16 Moreover, it has been described that IL-12 and IFNγ favor polarization to Th1 cells, which become the major IFNγ producers and stimulators of T cell cytotoxic responses.16 This might in turn reinforce NK cell activation along the treatment and their contribution to the elimination of tumor cells.
We also demonstrate here that macrophages are stimulated in the combined therapy with no-alpha mutein, as shown by their high expression of MHCII molecules and of FcγRII/III (CD16/32) and FcγRIV, indicative of a possible polarization to an antitumor M1 phenotype (Figure 6). It could lead to increased antigen presentation and antibody-dependent cellular phagocytosis capabilities. Also, M1-type macrophages activate Th1 responses that can further amplify M1/killer-type activity through the production of IFNγ.14 In addition, macrophages are important sources of type I IFNs that favor T cell cross-priming as previously demonstrated during anti-CD20 therapies.14
The relevance of memory T cells in the control of tumor progression has been demonstrated by studies illustrating that their presence within tumors is related to a good prognosis in cancer patients.54 We and others have previously shown that the anti-CD20 treatment triggers an adaptive memory-based anti-tumor immunity,16,27 responsible for the long-term protection in a mouse tumor model.15 Furthermore, CD4+ TEM cells were detected in the spleen two months after antibody treatment and the adoptive transfer of CD4+ T cells from anti-CD20 treated surviving mice to naïve animals enabled resistance to tumor challenge.16 We did not find any impact of the combination with no-alpha mutein or the wild type IL-2 on the CD4+ memory compartment, at least 21 days after initiating the treatment. Nonetheless, the mutated cytokine in the combined treatment did increase CD8+ TCM and TEM cells in the peripheral blood at the same time point (Figure 7). These latter cells are critical mediators of therapeutic antitumor immunity in an administered cancer vaccine.39 Intriguingly, the injection of the no-alpha mutein at the beginning of the treatment maintained, but did not improve, the long-term protection conferred by anti-CD20 therapy when surviving animals were rechallenged with tumor cells while the IL-2-based combined treatment resulted in a reduced survival of mice as compared to animals receiving exclusively the antibody. Thus, although no change in the Tregs number was detected 10 days after the last IL-2 injection, it is tempting to postulate that this cytokine, when not mutated to lower its binding to the high affinity IL-2Rαβγ, triggers a pro-tumor cell network upon tumor cell rechallenge. Future studies should evaluate the memory T cell compartment at the time of the re-challenge, to verify if an exhaustion of the previously expanded memory CD8+ T-cell subset by the no-alpha mutein explains the absence of increased overall survival after a second infusion of tumor cells.
All these results argue strongly in favor of the use of no-alpha mutein rather than IL-2 in combination therapy with rituximab and anti-tumor antibodies.
Materials and methods
Antibodies and cytokines
The mouse CAT-13.6E12 hybridoma was obtained from the Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ, Germany). It produces a type I mouse anti-human CD20 monoclonal antibody (mAb), CAT-13 (IgG2a, κ). CAT-13 was produced and purified as previously described.15 The no-alpha mutein was produced and purified at CIM as previously described.28
Cells
The mouse thymoma EL4 cells expressing human CD20 (EL4-huCD20) were kindly provided by J. Golay (Ospedali Riunti di Bergamo, Bergamo, Italy). These cells are CD25− CD4− CD3+.
Mice
Seven-week-old female C57Bl/6 mice were purchased from Charles River Laboratories France (Saint Germain Nuelles, France). All animal studies were performed in compliance with guidelines from the European Union (EU guideline on animal experiments, European Directive #2010/63/EU) and the French national charter on ethics in animal experiments with the approval of the Charles Darwin Ethics Committee for animal experimentation (Paris, France) (under the reference number 01530.02). In some experiments, body weight and temperature were monitored. The C57Bl/6-EL4-huCD20 tumor model has been previously described.15 C57Bl/6 mice were inoculated intravenously (i.v.) in the tail vein with 2 × 105 EL4-huCD20 cells per mouse in 200 µL PBS (phosphate-buffered saline) on day 0. Human IL-2 (Peprotech, Cat# 200-02) and no-alpha mutein (CIM, Havana) were injected intraperitoneally (i.p.) on days −1 and 3 after tumor inoculation (100,000 IU or 1,000 IU, respectively). The doses were set to maintain a relation of 102 between wild type and no-alpha mutein IU, that has been demonstrated to expand the same number of CD44+ memory T cells (βγ IL2-R expressing cells).28 In other experiments, IL-2 or no-alpha mutein were also injected into naïve animals at day 0 and day 4 to evaluate their impact on spleen cells at day 6. Blood samples were collected at the indicated times for evaluating T-cell subsets by immunofluorescence staining.
Also, surviving treated mice were i.v. challenged in the tail vein with 4 × 105 tumor cells at day 65. Age-matched naïve mice were injected with the same number of cells and used as controls. CAT-13 was administered by i.p. injections (200 µg/injection/mouse) at days 1, 4, 7, 10 and 13. Mice were sacrificed when signs of disease appeared (eg. prostration, paralysis, body weight drop).
Flow cytometry
Surface and intracellular staining was performed according to standard protocols and analyzed with LSRFortessa (BD Biosciences). Surface antibodies were anti-CD3ε-APCeF780 (allophycocyanin-eFluor 780, eBioscience, Cat#47-003282), anti-CD8α-eF660 (eFluor 660, eBioscience, Cat#50-0081-82), anti-CD8α-eF450 (eFluor450, eBioscience, Cat# 480081-82), anti-CD62 L-PECy5.5 (phycoerythrin cyanine-5.5, eBioscience, Cat#35-0621-81), anti-CD11b-eF450 (eFluor450, eBioscience, Cat#48-0112-82), anti-CD44-APC-Alexa750 (allophycocyanin-Alexa Fluor®750, eBioscience, Cat#27-0441-81), anti-CD45-PE-Cy7 (phycoerythrin cyanine-7, BD Biosciences, Cat#552848), anti-CD45 R (B220)-PerCP-Cy5.5 (peridinine-chlorophyll-protein complex cyanine-5.5, BD Biosciences, Cat#552771), anti-CD11 c-BV785 (Brilliant Violet™785, BioLegend, Cat#117335), anti-F4/80-AlexaFluor700 (AlexaFluor®700, BioLegend, Cat#123129), anti-Gr1-PeCy5 (phycoerythrin cyanine™-5, BD Pharmingen, Cat#552093), anti-IA/IE-BV650 (Brilliant Violet™650, BD Pharmingen, Cat# 563415), anti-CD16/CD32-APC (allophycocyanin, BD Pharmingen, Cat#553141), anti-NK1.1-APC (allophycocyanin, BD Pharmingen, Cat#550627), anti-CD3-AF700 (BD Pharmingen, Cat#557984), anti-CD3-PE (phycoerythrin, BD Pharmingen, Cat#553064) and anti-CD4-PE-Texas Red (phycoerythrin -Texas Red, Caltag Medsystems, Cat#MCD0417). Intracellular antibodies were anti-IL-4-PE-Cy7 (phycoerythrin cyanine-7, e-Biosciences, Cat#25-7042-82), anti-Perforin-PE (phycoerythrin, e-Biosciences, Cat#12-93-92), anti-Granzyme B-FITC (fluorescein isothiocyanate, e-Biosciences Cat#11-88-98), anti-IFNγ-Alexa Fluor 647 (Alexa Fluor®647, BD Biosciences, Cat#557735), anti-FoxP3-PE (phycoerythrin, BioLegend, Cat#320007) and anti-IL-17A-Alexa Fluor 700 (Alexa Fluor®700, BioLegend, Cat#506914). Relevant isotype controls were purchased from BioLegend, BD Pharmingen and e-Biosciences. Diva (BD Biosciences) and Kaluza 1.5 (Beckman Coulter) software was used for data analysis.
Intracellular staining
Spleens from anti-CD20, anti CD20 + IL-2, anti-CD20 + no-alpha mutein and isotype control mAb treated mice were collected at days 14 and 21 after tumor injection. CD4+ T-cell subsets were evaluated by flow cytometry after stimulation. Spleen cells (2 x 106) were stimulated for 4 h at 37°C under 5% CO2 in the presence of phorbol 12-myristate 13-acetate (PMA) (25 ng/ml) (Sigma-Aldrich; catalog number P 8139) and ionomycin (5 μg/ml) (Sigma-Aldrich; catalog number I3909) before immunofluorescence analysis. For intracellular staining, cells were incubated with monensin (6.7 μg/ml) (Sigma-Aldrich; Cat#22373-78-0) for the last 2 h of culture.
Statistical analysis
Statistical significance (P values <.05) was determined by one-way ANOVA or non-parametric Kruskal-Wallis tests with the Bonferroni or Dunn’s posttest, respectively, for multiple comparisons. To assess survival differences, Kaplan–Meier curves were produced and analyzed by log-rank tests. Statistical analyses were performed with the Prism software (version 4.0, GraphPad Software Inc.). Data correspond to at least two independent experiments, expressed as mean ± standard error of the mean (SEM).
Supplementary Material
Acknowledgments
Ana Victoria Casadesús was supported by an EMBO short-term fellowship and by grants from the French Embassy in La Havana (Cuba) [ASTF 263-2013 and FE 28/2016]; Béré Kadjdiatou Diallo was supported by a CIFRE fellowship (N°2015/0834).
Funding Statement
This work was supported by Inserm, Sorbonne university and Paris Descartes universiy [UMRS 1135/1138].
Disclosure of potential conflicts of interest
The authors declare no potential conflicts of interest.
Supplementary material
Supplemental data for this article can be accessed through publisher’s website.
References
- 1.Coiffier B, Lepage E, Briere J, Herbrecht R, Tilly H, Bouabdallah R, Morel P, Van Den Neste E, Salles G, Gaulard P, et al. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N Engl J Med. 2002;346(4):235–12. doi: 10.1056/NEJMoa011795. [DOI] [PubMed] [Google Scholar]
- 2.Molina A. A decade of rituximab: improving survival outcomes in non-Hodgkin’s lymphoma. Annu Rev Med. 2008;59(1):237–250. doi: 10.1146/annurev.med.59.060906.220345. [DOI] [PubMed] [Google Scholar]
- 3.Nowakowski GS, Blum KA, Kahl BS, Friedberg JW, Baizer L, Little RF, Maloney DG, Sehn LH, Williams ME, Wilson WH, et al. Beyond RCHOP: a blueprint for diffuse large B cell lymphoma research. J Natl Cancer Inst. 2016;108. doi: 10.1093/jnci/djw257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smith MR. Rituximab (monoclonal anti-CD20 antibody): mechanisms of action and resistance. Oncogene. 2003;22(47):7359–7368. doi: 10.1038/sj.onc.1206939. [DOI] [PubMed] [Google Scholar]
- 5.Salles G, Barrett M, Foa R, Maurer J, O’Brien S, Valente N, Wenger M, Maloney DG. Rituximab in B-cell hematologic malignancies: a review of 20 years of clinical experience. Adv Ther. 2017;34(10):2232–2273. doi: 10.1007/s12325-017-0612-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rummel M, Kaiser U, Balser C, Stauch M, Brugger W, Welslau M, Niederle N, Losem C, Boeck H-P, Weidmann E, et al. Bendamustine plus rituximab versus fludarabine plus rituximab for patients with relapsed indolent and mantle-cell lymphomas: a multicentre, randomised, open-label, non-inferiority phase 3 trial. Lancet Oncol. 2016;17(1):57–66. doi: 10.1016/S1470-2045(15)00447-7. [DOI] [PubMed] [Google Scholar]
- 7.Tedeschi A, Picardi P, Ferrero S, Benevolo G, Margiotta Casaluci G, Varettoni M, Barate C, Motta M, Gini G, Goldaniga MC, et al. Bendamustine and rituximab combination is safe and effective as salvage regimen in Waldenström macroglobulinemia. Leuk Lymphoma. 2015;56(9):2637–2642. doi: 10.3109/10428194.2015.1012714. [DOI] [PubMed] [Google Scholar]
- 8.Smith KA, Lachman LB, Oppenheim JJ, Favata MF. The functional relationship of the interleukins. J Exp Med. 1980;151(6):1551–1556. doi: 10.1084/jem.151.6.1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morgan DA, Ruscetti FW, Gallo R. Selective in vitro growth of T lymphocytes from normal human bone marrows. Science. 1976;193(4257):1007–1008. doi: 10.1126/science.181845. [DOI] [PubMed] [Google Scholar]
- 10.Dunne J, Lynch S, O’Farrelly C, Todryk S, Hegarty JE, Feighery C, Doherty DG. Selective expansion and partial activation of human NK cells and NK receptor-positive T cells by IL-2 and IL-15. J Immunol. 2001;167(6):3129–3138. doi: 10.4049/jimmunol.167.6.3129. [DOI] [PubMed] [Google Scholar]
- 11.Cesana GC, DeRaffele G, Cohen S, Moroziewicz D, Mitcham J, Stoutenburg J, Cheung K, Hesdorffer C, Kim-Schulze S, Kaufman HL. Characterization of CD4+CD25+ regulatory T cells in patients treated with high-dose interleukin-2 for metastatic melanoma or renal cell carcinoma. J Clin Oncol. 2006;24(7):1169–1177. doi: 10.1200/JCO.2005.03.6830. [DOI] [PubMed] [Google Scholar]
- 12.Ahmadzadeh M, Rosenberg SA. IL-2 administration increases CD4+CD25hi Foxp3+ regulatory T cells in cancer patients. Blood. 2006;107(6):2409–2414. doi: 10.1182/blood-2005-06-2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10(9):942–949. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 14.Ren Z, Guo J, Liao J, Luan Y, Liu Z, Sun Z, Liu X, Liang Y, Peng H, Fu Y-X. CTLA-4 limits anti-CD20–mediated tumor regression. Clin Cancer Res. 2017;23(1):193–203. doi: 10.1158/1078-0432.CCR-16-0040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abes R, Gélizé E, Fridman WH, Teillaud J-L. Long-lasting antitumor protection by anti-CD20 antibody through cellular immune response. Blood. 2010;116(6):926–934. doi: 10.1182/blood-2009-10-248609. [DOI] [PubMed] [Google Scholar]
- 16.Deligne C, Metidji A, Fridman W-H, Teillaud J-L. Anti-CD20 therapy induces a memory Th1 response through the IFN-γ/IL-12 axis and prevents protumor regulatory T-cell expansion in mice. Leukemia. 2015;29(4):947–957. doi: 10.1038/leu.2014.275. [DOI] [PubMed] [Google Scholar]
- 17.Hooijberg E, Sein JJ, van den Berk PC, Hart AA, van der Valk MA, Kast WM, Melief CJ, Hekman A. Eradication of large human B cell tumors in nude mice with unconjugated CD20 monoclonal antibodies and interleukin 2. Cancer Res. 1995;55:2627–2634. [PubMed] [Google Scholar]
- 18.Eisenbeis CF, Grainger A, Fischer B, Baiocchi RA, Carrodeguas L, Roychowdhury S, Chen L, Banks AL, Davis T, Young D, et al. Combination immunotherapy of B-cell non-Hodgkin’s lymphoma with rituximab and interleukin-2: a preclinical and Phase I study. Clin Cancer Res. 2004;10(18):6101–6110. doi: 10.1158/1078-0432.CCR-04-0525. [DOI] [PubMed] [Google Scholar]
- 19.Gluck WL, Hurst D, Yuen A, Levine AM, Dayton MA, Gockerman JP, Lucas J, Denis-Mize K, Tong B, Navis D, et al. Phase I studies of interleukin (IL)-2 and rituximab in B-cell non-hodgkin’s lymphoma: IL-2 mediated natural killer cell expansion correlations with clinical response. Clin Cancer Res. 2004;10(7):2253–2264. doi: 10.1158/1078-0432.CCR-1087-3. [DOI] [PubMed] [Google Scholar]
- 20.Khan KD, Emmanouilides C, Benson DM Jr., Hurst D, Garcia P, Michelson G, Milan S, Ferketich AK, Piro L, Leonard JP, et al. A Phase 2 study of rituximab in combination with recombinant interleukin-2 for rituximab-refractory indolent non-Hodgkin’s lymphoma. Clin Cancer Res. 2006;12(23):7046–7053. doi: 10.1158/1078-0432.CCR-06-1571. [DOI] [PubMed] [Google Scholar]
- 21.Bachanova V, Burns LJ, McKenna DH, Curtsinger J, Panoskaltsis-Mortari A, Lindgren BR, Cooley S, Weisdorf D, Miller JS. Allogeneic natural killer cells for refractory lymphoma. Cancer Immunol Immunother. 2010;59(11):1739–1744. doi: 10.1007/s00262-010-0896-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Friedberg JW, Neuberg D, Gribben JG, Fisher DC, Koval M, Poor M, Green LM, Daley J, Soiffer R, Ritz J, et al. Combination immunotherapy with rituximab and interleukin 2 in patients with relapsed or refractory follicular non-Hodgkin’s lymphoma. Br J Haematol. 2002;117(4):828–834. doi: 10.1046/j.1365-2141.2002.03535.x. [DOI] [PubMed] [Google Scholar]
- 23.Pere H, Tanchot C, Bayry J, Terme M, Taieb J, Badoual C, Adotevi O, Merillon N, Marcheteau E, Quillien VR, et al. Comprehensive analysis of current approaches to inhibit regulatory T cells in cancer. Oncoimmunology. 2012;1(3):326–333. doi: 10.4161/onci.18852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.D’Arena G, Vitale C, Coscia M, Festa A, Di Minno NMD, De Feo V, Caraglia M, Calapai G, Laurenti L, Musto P, et al. Regulatory T cells and their prognostic relevance in hematologic malignancies. J Immunol Res. 2017;2017:1832968. doi: 10.1155/2017/1832968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang -Z-Z, Novak AJ, Ziesmer SC, Witzig TE, Ansell SM. Attenuation of CD8+ T-cell function by CD4+CD25+ regulatory T cells in B-cell non-Hodgkin’s lymphoma. Cancer Res. 2006;66(20):10145–10152. doi: 10.1158/0008-5472.CAN-06-1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gunduz E, Sermet S, Musmul A. Peripheral blood regulatory T cell levels are correlated with some poor prognostic markers in newly diagnosed lymphoma patients. Cytometry B Clin Cytom. 2016;90(5):449–454. doi: 10.1002/cyto.b.21330. [DOI] [PubMed] [Google Scholar]
- 27.DiLillo DJ, Ravetch JV. Differential Fc-receptor engagement drives an anti-tumor vaccinal effect. Cell. 2015;161(5):1035–1045. doi: 10.1016/j.cell.2015.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carmenate T, Pacios A, Enamorado M, Moreno E, Garcia-Martinez K, Fuente D, Leon K. Human IL-2 mutein with higher antitumor efficacy than wild type IL-2. J Immunol. 2013;190(12):6230–6238. doi: 10.4049/jimmunol.1201895. [DOI] [PubMed] [Google Scholar]
- 29.Klein C, Waldhauer I, Nicolini VG, Freimoser-Grundschober A, Nayak T, Vugts DJ, Dunn C, Bolijn M, Benz J, Stihle M, et al. Cergutuzumab amunaleukin (CEA-IL2v), a CEA-targeted IL-2 variant-based immunocytokine for combination cancer immunotherapy: overcoming limitations of aldesleukin and conventional IL-2-based immunocytokines. Oncoimmunology. 2017;6(3):e1277306. doi: 10.1080/2162402X.2016.1277306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Silva D-A, Yu S, Ulge UY, Spangler JB, Jude KM, Labao-Almeida C, Ali LR, Quijano-Rubio A, Ruterbusch M, Leung I, et al. De novo design of potent and selective mimics of IL-2 and IL-15. Nature. 2019;565(7738):186–191. doi: 10.1038/s41586-018-0830-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Malek TR, Castro I. Interleukin-2 receptor signaling: at the interface between tolerance and immunity. Immunity. 2010;33(2):153–165. doi: 10.1016/j.immuni.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Malek TR. The biology of interleukin-2. Annu Rev Immunol. 2008;26(1):453–479. doi: 10.1146/annurev.immunol.26.021607.090357. [DOI] [PubMed] [Google Scholar]
- 33.Boyman O, Krieg C, Homann D, Sprent J. Homeostatic maintenance of T cells and natural killer cells. Cell Mol Life Sci. 2012;69(10):1597–1608. doi: 10.1007/s00018-012-0968-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boyman O, Sprent J. The role of interleukin-2 during homeostasis and activation of the immune system. Nat Rev Immunol. 2012;12(3):180–190. doi: 10.1038/nri3156. [DOI] [PubMed] [Google Scholar]
- 35.Rojas G, Carmenate T, Leon K. Molecular dissection of the interactions of an antitumor interleukin-2-derived mutein on a phage display-based platform. J Mol Recognit. 2015;28(4):261–268. doi: 10.1002/jmr.2440. [DOI] [PubMed] [Google Scholar]
- 36.Leon K, Garcia-Martinez K, Carmenate T, Rojas G. Combining computational and experimental biology to develop therapeutically valuable IL2 muteins. Semin Oncol. 2018;45(1–2):95–104. doi: 10.1053/j.seminoncol.2018.04.001. [DOI] [PubMed] [Google Scholar]
- 37.Blasius AL, Barchet W, Cella M, Colonna M. Development and function of murine B220+CD11c+NK1.1+ cells identify them as a subset of NK cells. J Exp Med. 2007;204(11):2561–2568. doi: 10.1084/jem.20070991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boross P, Jansen JH, de Haij S, Beurskens FJ, van der Poel CE, Bevaart L, Nederend M, Golay J, van de Winkel JG, Parren PW, et al. The in vivo mechanism of action of CD20 monoclonal antibodies depends on local tumor burden. Haematologica. 2011;96(12):1822–1830. doi: 10.3324/haematol.2011.047159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Klebanoff CA, Yu Z, Hwang LN, Palmer DC, Gattinoni L, Restifo NP. Programming tumor-reactive effector memory CD8+ T cells in vitro obviates the requirement for in vivo vaccination. Blood. 2009;114(9):1776–1783. doi: 10.1182/blood-2008-12-192419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kohrt HE, Houot R, Marabelle A, Cho HJ, Osman K, Goldstein M, Levy R, Brody J. Combination strategies to enhance antitumor ADCC. Immunotherapy. 2012;4(5):511–527. doi: 10.2217/imt.12.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hilchey SP, Hyrien O, Mosmann TR, Livingstone AM, Friedberg JW, Young F, Fisher RI, Kelleher RJ Jr., Bankert RB, Bernstein SH. Rituximab immunotherapy results in the induction of a lymphoma idiotype-specific T-cell response in patients with follicular lymphoma: support for a “vaccinal effect” of rituximab. Blood. 2009;113(16):3809–3812. doi: 10.1182/blood-2008-10-185280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Michaud H-A, Eliaou J-F, Lafont V, Bonnefoy N, Gros L. Tumor antigen-targeting monoclonal antibody-based immunotherapy: orchestrating combined strategies for the development of long-term antitumor immunity. Oncoimmunology. 2014;3(9):e955684. doi: 10.4161/21624011.2014.955684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Skrombolas D, Frelinger JG. Challenges and developing solutions for increasing the benefits of IL-2 treatment in tumor therapy. Expert Rev Clin Immunol. 2014;10(2):207–217. doi: 10.1586/1744666X.2014.875856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shi Y-X, Zhang X-S, Liu D-G, Li Y-Q, Guan -Z-Z, Jiang W-Q. CD4+CD25+T regulatory cells in peripheral blood of B-NHL patients with or without chemotherapy. Ai Zheng. 2004;23:597–601. [PubMed] [Google Scholar]
- 45.Levin AM, Bates DL, Ring AM, Krieg C, Lin JT, Su L, Moraga I, Raeber ME, Bowman GR, Novick P, et al. Exploiting a natural conformational switch to engineer an interleukin-2 ‘superkine’. Nature. 2012;484(7395):529–533. doi: 10.1038/nature10975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gattinoni L, Klebanoff CA, Restifo NP. Pharmacologic induction of CD8+ T cell memory: better living through chemistry. Sci Transl Med. 2009;1(11):11ps12. doi: 10.1126/scitranslmed.3000302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Taylor JG, Gribben JG. Microenvironment abnormalities and lymphomagenesis: immunological aspects. Semin Cancer Biol. 2015;34:36–45. doi: 10.1016/j.semcancer.2015.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Haabeth OA, Lorvik KB, Hammarstrom C, Donaldson IM, Haraldsen G, Bogen B, Corthay A. Inflammation driven by tumour-specific Th1 cells protects against B-cell cancer. Nat Commun. 2011;2(1):240. doi: 10.1038/ncomms1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ding Z-C, Huang L, Blazar BR, Yagita H, Mellor AL, Munn DH, Zhou G. Polyfunctional CD4+ T cells are essential for eradicating advanced B-cell lymphoma after chemotherapy. Blood. 2012;120(11):2229–2239. doi: 10.1182/blood-2011-12-398321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hus I, Bojarska-Junak A, Kaminska M, Dobrzynska-Rutkowska A, Szatan K, Szymczyk A, Kukielka-Budny B, Szczepanek D, Rolinski J. Imbalance in circulatory iNKT, Th17 and T regulatory cell frequencies in patients with B-cell non-Hodgkin’s lymphoma. Oncol Lett. 2017;14(6):7957–7964. doi: 10.3892/ol.2017.7232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhong W, Xu X, Zhu Z, Yang L, Du H, Xia Z, Yuan Z, Xiong H, Du Q, Wei Y, et al. Increased interleukin-17A levels promote rituximab resistance by suppressing p53 expression and predict an unfavorable prognosis in patients with diffuse large B cell lymphoma. Int J Oncol. 2018;52(5):1528–1538. doi: 10.3892/ijo.2018.4299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lu T, Yu S, Liu Y, Yin C, Ye J, Liu Z, Ma D, Ji C, Bertolini F. Aberrant circulating Th17 cells in patients with B-cell non-Hodgkin’s lymphoma. PLoS One. 2016;11(1):e0148044. doi: 10.1371/journal.pone.0148044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kaiko GE, Horvat JC, Beagley KW, Hansbro PM. Immunological decision-making: how does the immune system decide to mount a helper T-cell response? Immunology. 2008;123(3):326–338. doi: 10.1111/j.1365-2567.2007.02719.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, Tosolini M, Camus M, Berger A, Wind P, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313(5795):1960–1964. doi: 10.1126/science.1129139. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.