

ABSTRACT
Chimeric antigen receptor (CAR) T cells show remarkable therapeutic effects in some hematological malignancies. However, CAR T cells can also cause life-threatening side effects. In order to minimize off-target and on-target/off-tumor reactions, improve safety, enable controllability, provide high flexibility, and increase tumor specificity, we established a novel humanized artificial receptor platform termed RevCARs. RevCAR genes encode for small surface receptors lacking any antigen-binding moiety. Steering of RevCAR T cells occurs via bispecific targeting molecules (TMs). The small size of RevCAR-encoding genes allows the construction of polycistronic vectors. Here, we demonstrate that RevCAR T cells efficiently kill tumor cells, can be steered by TMs, flexibly redirected against multiple targets, and used for combinatorial targeting following the “OR” and “AND” gate logic.
KEYWORDS: Chimeric antigen receptor (CAR), T cell therapy, tumor immunotherapy, adaptor CAR platform, combinatorial gated targeting
Introduction
Chimeric antigen receptors (CARs) can be used for redirection of T cells against tumor cells in a MHC-independent manner.1,2 Conventional second-generation CARs consist of an extracellular domain (ECD), which is commonly an antigen-binding single-chain fragment variable (scFv) derived from a monoclonal antibody (mAb), a hinge domain (HiD), a transmembrane domain (TMD), and an intracellular domain (ICD) comprising the activation signaling domain of CD3 zeta (CD3z or 3z), and one costimulatory domain (CSD) commonly derived from CD28 (28), 4–1BB, or Ox-40.3–5 T cells genetically engineered with anti-CD19 CARs have demonstrated remarkable therapeutic effects in the clinic and thus were recently approved by the U.S. Food and Drug Administration (FDA) for treatment of relapsed/refractory B cell lymphoblastic leukemia (r/r B-ALL) and diffuse large B cell lymphoma (DLBCL).6–8 Despite impressive responses in some hematological malignancies, conventional CAR T cell therapy also struggles with some drawbacks. In brief, CAR T cells can cause severe to life-threatening side effects in patients like on-target/off-tumor reactions, cytokine release syndrome (CRS), and neurotoxicity.4 Unfortunately, CAR T cells rarely respond against solid tumors.4,9 Since most of the known tumor-associated antigens (TAAs) on solid tumors are not exclusively expressed on malignant cells but also on normal tissues albeit to a lesser extent, CAR T cells redirected against these targets are risky to cause severe to fatal on-target/off-tumor toxicities. For example, the first patient treated with ERBB2/HER2-directed CAR T cells died because of their cytotoxic activation and massive cytokine release after recognition of ERBB2 on lung epithelial cells.10
In case those side effects occur in patients, conventional CAR T cells cannot be controlled or switched off. In order to improve the safety management of CAR T cell therapy, we and others have developed different adaptor CAR platforms allowing a repeatable on/off-switch and steering of CAR T cell activity.11–19 The concept of these adaptor CAR technologies is basically that T cells modified with a universal receptor do not directly recognize a TAA but have to be redirected to tumor cells by separate adaptor molecules that are directed against a tumor target and simultaneously to the universal CAR. By dosing of the adaptor molecules, CAR T cell activity can be controlled. Another advantage of universal adaptor CARs is that they allow a flexible targeting of multiple antigens without re-engineering of T cells simply by using several adaptor molecules possessing different target specificities or adaptors with more than one specificity.18,20 Such “OR” gate combinatorial targeting strategies are helpful to overcome the development of tumor escape variants down-regulating the targeted antigen which frequently arise under the treatment with monospecific conventional CAR T cells having a fixed specificity.7,21
In order to increase tumor specificity and to reduce off-target and on-target/off-tumor toxicities, furthermore, different CAR-based combinatorial targeting strategies were developed following the “AND” gate logic.22,23 Basically, the idea is that one T cell is modified with two separate CARs. On the one hand, a first-generation CAR transmits the first CD3z activation signal after recognition of antigen 1 and, on the other hand, a costimulatory CAR transmits the costimulatory CD28 signal after binding to antigen 2. Recognition of both antigens triggers full activation of Dual-CAR T cells. Proof of concept for this strategy was shown for different tumor entities.23–25 However, conventional CARs were used which are limited by fixed target specificities and strongly depend on well-balanced antigen affinities.23 Thus, using adaptor CARs has the advantage of high flexibility with respect to target selection and targeting strength.18 One obvious problem of gated targeting strategies is the need for manipulation of the effector cells with at least two CAR encoding genes. For this purpose, T cells are commonly transduced with two separate CAR encoding constructs. Consequently, the transduction leads to an unpredictable mixture of cellular products containing either only one or a different ratio of both genes. A fusion of both CAR genes in a single vector could be a solution for this problem. However, it would increase the size of the final vector and, thereby, dramatically reduce the efficacy of transduction.
The final goal of our recent studies was therefore to develop a novel switchable adaptor CAR allowing us to genetically engineer T cells to express more than one CAR gene from a single construct and suitable target modules (TMs) for retargeting of tumor cells according to the rules of Boolean algebra. To achieve this aim we wanted to reduce the size of the genes encoding the respective artificial receptors. In previous studies, we had already validated two modular adaptor CAR platforms known as UniCARs (Figure 1) and alternative UniCARs (alt-UniCARs).3,26 Both UniCAR systems are second-generation CARs recognizing a peptide epitope via their extracellular antibody (Ab) domain. TMs represent different kinds of bispecific fusion molecules consisting of the peptide epitope and a targeting moiety binding to different tumor targets.20,27–35 The peptide epitope recognized by UniCARs consists of 10 amino acids (aa) which are recognized by the mAb 5B9. The peptide epitope recognized by the alt-UniCAR system consists of 18 aa and is recognized by the mAb 7B6. Both UniCAR epitopes are cryptic epitopes present in the primary sequence of the human nuclear autoantigen La/SS-B.3 Our novel modular CAR platform is based on the same peptide epitope/scFv combinations. In order to reduce the size of the gene encoding the artificial receptor, however, we decided to reverse their orientation. Meaning we replaced the respective extracellular anti-peptide epitope scFv of UniCARs by the respective peptide epitope sequence and vice versa the respective peptide epitope sequence in a UniCAR TM by the respective anti-peptide epitope scFv. The resulting adaptor CAR system was therefore named as Reversed CARs or briefly as RevCARs (Figure 1).
Figure 1.
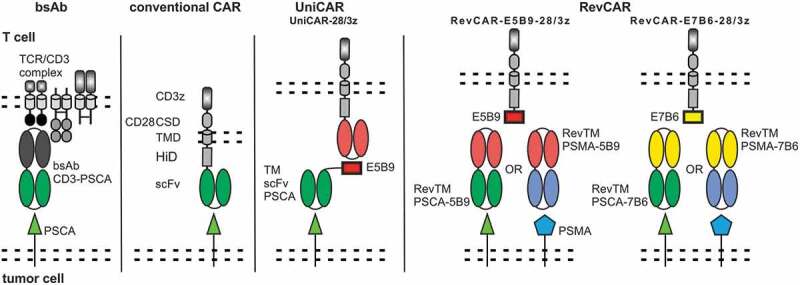
Design of the RevCAR system.
Schematic comparison of the RevCAR design with the bsAb, conventional CAR, and UniCAR system. RevCARs lack the extracellular TAA binding scFv and instead express the peptide epitope E5B9 (10 aa) or E7B6 (18 aa). For the extracellular hinge (HiD), transmembrane (TMD), and intracellular costimulatory domain (CSD) human CD28 was used, followed by three human CD3 zeta chain activating motifs. For redirection of RevCAR T cells to tumor cells an adaptor molecule, termed RevTM, is required. RevTMs consist of two scFvs, one of them targeting a TAA and the other one being directed to the peptide epitope of the RevCAR.
For proof of functionality, in a first attempt, two second-generation RevCARs were established comprising E5B9 or E7B6 as ECD. In addition, we constructed the corresponding Reversed TMs (RevTMs) directed against either E5B9 or E7B6 and simultaneously to the TAA either prostate-specific membrane antigen (PSMA) or prostate stem cell antigen (PSCA). In the second step, we split the activation and costimulatory signaling domains onto two RevCARs. A series of activating and costimulatory RevCARs were established and tested in combination. Finally, we fused the optimal activating and costimulatory RevCAR gene combination in a single vector that allowed us to confirm proof of concept of the RevCAR system including its use for gated targeting.
Materials and methods
Cell lines
HEK 293T, LNCaP, PC3, and 3T3 cells were obtained from the American Type Culture Collection (ATCC) and have not been further authenticated. Culture of cell lines was performed as described elsewhere.28 All cell lines were frequently tested for Mycoplasma infection.
Design of RevCAR constructs
In general, the RevCAR constructs consist of a signal peptide derived from human IL-2 or murine Ig-kappa, the peptide epitope E5B9 or E7B6 derived from the human La/SS-B protein3 fused to the HiD, TMD, and CSD of the human CD28 linked to the human CD3z ICD, connected with the peptide T2A (Thosea asigna virus) or P2A (porcine teschovirus-1) and the marker mCherry or EGFP. The SIG RevCAR-E7B6-3z is composed of the HiD of human IgG4 and the TMD of human CD4. All RevCAR constructs were cloned into the lentiviral vector p6NST70 (Prof. Dr. Dirk Lindemann, Technical University Dresden, Germany) under the control of human elongation factor-1α (EF-1α) promoter for lentiviral transduction of human primary T cells. In addition, we used UniCARs designed as previously described.28
Isolation of human primary T cells
Primary T cells were obtained from buffy coats of anonymous healthy blood donors and provided by the German Red Cross (Dresden, Germany) after written consent of the donors. The research with human T cells was approved by the local ethics committee of the Medical Faculty Carl Gustav Carus, Technical University Dresden (EK27022006). Peripheral blood mononuclear cells (PBMCs) were isolated from buffy coats using Biocoll separating solution (Merck Cat#L6115) for density gradient centrifugation. For further isolation of human primary T cells, negative depletion was used following manual instructions of the Pan T Cell Isolation Kit (Miltenyi Biotec Cat#130-096-535).
Generation and expansion of RevCAR T cells
RevCAR T cells and UniCAR T cells were generated by lentiviral transduction. Therefore, viral particles were produced in HEK 293 T cells as described.36 Activation, transduction, sorting, and maintenance of human primary T cells were performed as described elsewhere.20,36 Transduction efficiency of T cells was evaluated based on the marker (EGFP or mCherry) expression. Transduced T cells were sorted to >99% purity of EGFP+ or mCherry+ cells. Alternatively, a shortened protocol for RevCAR T cell transduction using T Cell TransAct™ (Miltenyi Biotec Cat#130-111-160) and TexMACS™ Medium (Miltenyi Biotec Cat#170-076-307) was used following the manufacturer’s instructions. In brief, 24 h after TransAct™ activation T cells were transduced with a multiplicity of infection of 2 for 2–3 times in 24 h intervals using spin infection for 2 h. T cells were then expanded over the weekend and used without sorting in the second week.
Determination of the receptor density
Receptor density on T cells was determined using the QIFIKIT (Agilent Cat#K007811-8) according to the manufacturer’s instructions. Briefly, 2*105 RevCAR T cells were incubated for 30 min at 4°C with 10 µl (15 µg/ml) of an unconjugated monoclonal antibody either directed against E5B9 or E7B6. As negative control, the unconjugated antibody was replaced by the respective isotype control mouse IgG1 (BD BioSciences Cat#555746, RRID:AB_396088) or mouse IgG2a (Miltenyi Biotec Cat#130-106-546, RRID:AB_2661589). As secondary antibody 100 µl of Pacific Blue-conjugated goat-anti-mouse IgG (1:50) (Thermo Fisher Scientific Cat#P31582, RRID:AB_10374586) was added and incubated for 45 min at 4°C.
Design, expression and purification of RevTMs
In general, the RevTM constructs consist of the murine Ig-kappa signal peptide, scFv PSCA or PSMA, scFv 5B9 or 7B6 and 6x Histidine tag for purification. The scFvs PSCA, PSMA, 5B9, and 7B6 were taken from previously published constructs.20,26,37,38 The RevTMs were cloned into the lentiviral vector p6NST50 to establish TM producing permanent 3T3 cell lines and purified using Ni-NTA affinity chromatography as previously described.37,38 In addition, we used the UniCAR TM PSCA-E5B9 designed and produced as previously described.28
Generation of target cell lines
LNCaP cells were modified to express PSCA using lentiviral transduction as described previously.28 Likewise, PC3 cells were transduced for expression of PSCA and PSMA.28 For in vivo experiments, PSCA-expressing LNCaP cells were additionally transduced with the open reading frame encoding firefly luciferase.28
Flow cytometry analysis
For binding analysis, 2*105 T cells or target cells were seeded in a 96-well plate and incubated with 50 µl RevTM (5 µg/ml) in PBS at 4°C for 60 min. For detection, the mAb mouse anti-His/PE (Miltenyi Biotec Cat#130-092-691, RRID:AB_1103227) was used according to the manufacturer’s manual. For analysis of the killing mechanism, T cells were stained with fluorochrome-labeled mAbs directed against CD178 (FasL) (Miltenyi Biotec Cat#130-096-458, RRID:AB_10827748) and CD253 (TRAIL) (BD BioSciences Cat#561784, RRID:AB_10896485) according to the manufacturer’s manuals. For intracellular staining of RevCAR T cells staining was performed using fluorochrome-labeled Abs directed against Granzyme B (Cat#130-116-486, RRID:AB_2727564) and Perforin (Cat#130-123-867, RRID:AB_2811567) with the Inside Stain Kit (Cat#130-090-477), all purchased from Miltenyi Biotec, as instructed in the manufacturer’s manuals.
Chromium-release-based cytotoxicity assay
The cytotoxic potential of T cells was analyzed using the chromium-release assay as described elsewhere.39,40 In short, triplets of 5*103 target cells were seeded in a 96-well cell culture plate and T cells were added as indicated in the figure legends, either testing (I) different effector:target (e:t) ratios in the presence or absence of RevTMs (30 pmoles/ml) or (II) increasing amounts of RevTMs to estimate the EC50 value. Released chromium was measured after 24 or 48 h.
Cytokine-release assay
Triplets of 5*103 tumor cells were seeded in a 96-well cell culture plate and cultivated with RevCAR T cells in the presence or absence of RevTMs. Supernatants of co-culture assays were harvested after 24 or 48 h, as indicated in the figure legends. Analysis of secreted cytokines was performed using the OptEIA Human IFN-γ (Cat#555142), GM-CSF (Cat#555126), IL-2 (Cat#555190), and TNF (Cat#555212) ELISA Sets purchased from BD BioSciences or the human MACSPlex Cytokine 12 Kit (Miltenyi Biotec #130-099-169) as described in the manufacturer’s manuals.
Gene expression analysis
Analysis of gene expression was performed using quantitative PCR analysis as described, with slight modifications.41,42 Briefly, triplets of 1*104 PC3 cells were seeded and cultivated with RevCAR T cells in the presence or absence of RevTMs. After incubation for 24 h, co-transduced RevCAR T cells were sorted for EGFP+/mCherry+ expression under exclusion of PI+ cells. Sorting and analysis of gene expression profile were performed as described earlier.41 The n-fold change of gene expression between incubation with COS RevTM + SIG RevTM versus w/o RevTM was calculated using the ΔΔCP method.
Tumor xenograft model and optical imaging
In order to evaluate the cytotoxic potential of RevCAR T cells, co-injection experiments were performed in vivo using six weeks old male NMRI-nude mice (Rj:ATHYM-Foxn1nu/nu) obtained by Janvier Labs. All mice were randomly assigned to experimental groups of five individuals and housed in sterile cages in a pathogen-free facility with a 12 h light/dark cycle. 1*106 LNCaPPSCA+/Luc+ cells were injected subcutaneously in the presence or absence of RevCAR T cells (1*106) and RevTM (102 µg) in a total volume of 100 µl PBS (Gibco™ Cat#10010015). Subsequently, anesthetized mice were i.p. injected with 200 µl D-Luciferin (15 µg/ml in PBS, PerkinElmer Cat#122799) and analyzed for bioluminescence in the planar X-ray (Bruker In-vivo Xtreme) at different time points. Measured luminescence was evaluated using the Bruker MI and Multispectral Software as described previously.43,44 All in vivo studies have been approved by the local ethics committee for animal experiments (Landesdirektion Dresden, 24–9165.40-4/2013, 24.9168.21–4/2004-1) and were performed at the Helmholtz-Zentrum Dresden-Rossendorf (HZDR) in appliance with the terms of German regulations of animal welfare.
Statistical analysis
Data analysis was performed using GraphPad Prism 7.03 (GraphPad Software, RRID:SCR_002798). As indicated in the figure legends either One-Way or Two-Way ANOVA with Tukey’s or Sidak’s multiple comparisons test was used for statistical analysis. P values of less than 0.03 were considered significant (not significant (n.s.) p > 0.03, *p ≤ 0.03, **p ≤ 0.002, ***p ≤ 0.0002). As indicated, obtained values are shown as mean with SD or SEM.
Results
Design and tumor killing efficiency of the RevCAR system
In contrast to conventional CARs that directly recognize a TAA, the switchable RevCAR platform consists of two components that are necessary to redirect T cells to tumor cells: RevCARs and RevTMs (Figure 1). For first proof of functionality, we replaced the extracellular scFv of the second generation UniCAR with the respective epitope sequence E5B9 or E7B6. Thus, the ICD of these first RevCARs is identical to our previously used UniCARs containing three activating motifs derived from human CD3z and one costimulatory domain derived from human CD28. In addition, they consist of the same TMD and HiD. The resulting RevCAR constructs were termed RevCAR-E5B9-28/3z or RevCAR-E7B6-28/3z, respectively. Human T cells were genetically modified to express either RevCAR-E5B9-28/3z or RevCAR-E7B6-28/3z by lentiviral transduction evaluated by the expression of the marker EGFP (Figure S1A-S1B). Transduced RevCAR T cells were sorted on the basis of their EGFP expression (Figure S1B) and RevCAR density was determined as shown in Figure S1C. For redirection of RevCAR T cells, we constructed four different RevTMs recognizing either E5B9 or E7B6 and targeting the TAA either PSCA or PSMA: (I) RevTM PSCA-5B9, (II) RevTM PSCA-7B6, (III) RevTM PSMA-5B9, and (IV) RevTM PSMA-7B6 (Figure S1D-S1H). As target cells we used the two cell lines PC3 and LNCaP co-expressing both PSCA and PSMA. Where indicated, we used PC3 cells expressing either PSCA or PSMA.
For proof of cytotoxic functionality, we co-cultured PC3 tumor cells together with RevCAR-E5B9-28/3z T cells at different e:t ratios in the presence of RevTM PSCA-5B9 in a chromium release cytotoxicity assay. As shown in Figure 2a, RevCAR-E5B9-28/3z T cells could be significantly redirected by RevTM PSCA-5B9 to effectively kill PC3 tumor cells even at low e:t ratios. On the contrary, in the negative controls without any RevTM or in the presence of the irrelevant RevTM PSCA-7B6 no tumor cell lysis occurred. In addition, we tested the cytotoxic potential of RevCAR-E7B6-28/3z T cells against PC3 tumor cells in the presence of RevTM PSCA-7B6, showing similar high and specific anti-tumor effects like redirected RevCAR-E5B9-28/3z T cells (Figure 2a). Moreover, these results were confirmed using LNCaP tumor cells as an additional prostate tumor model (Figure 2a). As PC3 or LNCaP tumor cells co-express PSCA and PSMA, they were eliminated upon redirection of RevCAR-E5B9-28/3z or RevCAR-E7B6-28/3z T cells by RevTM PSMA-5B9 or PSMA-7B6, respectively, equally well (Figure 2b). Additionally, we analyzed the correlation between RevTM concentration and cytotoxicity, in order to prove that the RevCAR platform is tunable by dosing of the RevTM. As shown in Figure 2c–d increasing the RevTM concentration resulted in increasing tumor cell lysis till a plateau is reached. By estimating the half-maximal effective concentration (EC50), we further learned that all RevTMs mediated highly efficient tumor cell killing even at pM range (Figure 2c-d). In order to show that cytotoxicity of RevCAR T cells was dependent on the activating intracellular RevCAR signals, adequate control T cells were generated (Figure S2AI-S2AII). As documented in Figure S2B, neither PC3 nor LNCaP tumor cells were killed by T cells only expressing the marker protein EGFP (vector control) or expressing RevCARs lacking any signaling ICD (RevCAR-E5B9-stop or RevCAR-E7B6-stop). Furthermore, we confirmed that the anti-tumor effect of RevCAR T cells is strictly dependent on the presence of the targeted antigen (Figure S2C). In order to analyze the killing mechanism of the RevCAR system, we measured intracellular granzyme B and perforin as well as extracellular TRAIL and FasL expression of redirected RevCAR T cells. As shown in Figures S2D and S2E, granzyme B and perforin levels increased upon cross-linkage of RevCAR T cells with tumor cells via the corresponding RevTMs. A similar effect was observed for TRAIL. FasL levels only marginally increased.
Figure 2.
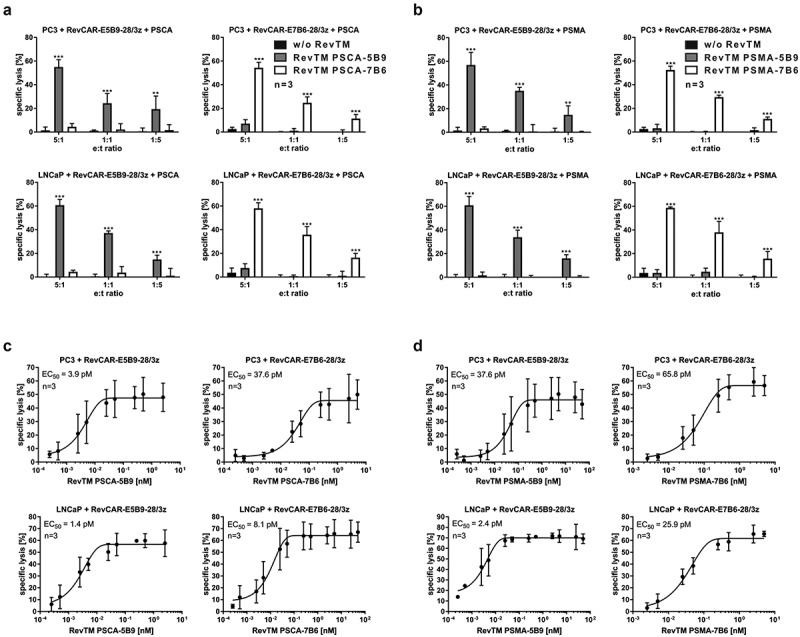
Tumor killing efficiency of the RevCAR system.
(a–b) Specific lysis of PSCA/PSMA-double-positive PC3 or LNCaP tumor cells by RevCAR-E5B9-28/3z or RevCAR-E7B6-28/3z T cells after 24 h co-incubation with or without RevTMs targeting either PSCA (a) or PSMA (b) (n = 3, mean ± SD, Two-Way ANOVA with Tukey’s multiple comparisons test. Significance is shown versus controls w/o RevTM or with irrelevant RevTM.).(c–d) Specific lysis of PC3 or LNCaP target cells was measured after 24 h co-culture with RevCAR-E5B9-28/3z or RevCAR-E7B6-28/3z T cells and increasing amounts of RevTMs targeting PSCA (c) or PSMA (d) to estimate the EC50 (n = 3, mean ± SD). See also Figure S2.
Cytokine release from redirected RevCAR T cells
On the one hand, the CRS is one of the most critical side effects of CAR T cell therapy and on the other hand, pro-inflammatory cytokines may support the anti-tumor effect especially against solid tumors. Thus, it was of special interest, whether redirected RevCAR T cells are able to release cytokines upon their cross-linkage with tumor cells. In order to address this question, we incubated RevCAR-E5B9-28/3z T cells together with PC3 or LNCaP tumor cells with the addition of RevTM PSCA-5B9 or RevTM PSMA-5B9. Moreover, RevCAR-E7B6-28/3z T cells were co-cultured with PC3 or LNCaP tumor cells together with RevTM PSCA-7B6 or RevTM PSMA-7B6. As controls, we incubated (I) tumor cells together with RevCAR T cells without any RevTM, (II) RevCAR T cells with their epitope-matching RevTM without tumor cells or (III) tumor cells plus RevCAR T cells together with an irrelevant RevTM that could not bind to the RevCAR epitope. As shown in Figure 3 and S3, among tested 12 human cytokines, GM-CSF, IFN-gamma, IL-2, and TNF-alpha were significantly released. Most importantly, significant cytokine release occurred only upon specific cross-linkage of RevCAR T cells with PSCA/PSMA-co-expressing tumor cells via the matching RevTMs. In contrast, no significant cytokine release was induced in all of the control settings.
Figure 3.
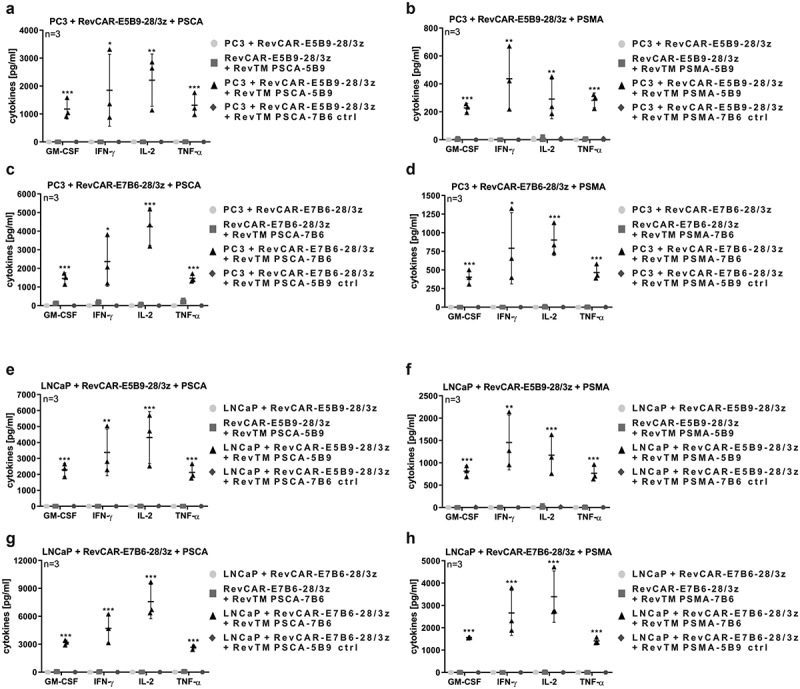
Cytokine release from redirected RevCAR T cells.
(a-h) Release of GM-CSF, IFN-gamma, IL-2, and TNF-alpha from RevCAR-E5B9-28/3z or RevCAR-E7B6-28/3z T cells after their incubation with PSCA/PSMA-double-positive PC3 or LNCaP tumor cells (e:t ratio 5:1) and/or RevTMs (n = 3, mean ± SD, One-Way ANOVA with Tukey’s multiple comparisons test. Significance versus all controls is shown.). See also Figure S3.
Tumor cell killing by RevCAR system in mice
In order to investigate whether the RevCAR platform is able to kill tumor cells in vivo, luciferase-expressing LNCaP tumor cells together with RevCAR-E5B9-28/3z T cells and RevTM PSCA-5B9 or RevTM PSMA-5B9 were injected in immunodeficient NMRI-Foxn1nu/nu mice (Figure 4a,c). Additionally, LNCaP tumor cells were administered together with RevCAR-E7B6-28/3z T cells in the presence of RevTM PSCA-7B6 or RevTM PSMA-7B6 (Figure 4b,d). As controls, tumor cells were injected alone or in addition to RevCAR T cells without any RevTM. All groups indicated in Figure 4 were analyzed in parallel in one experiment whereas the LNCaP group was performed only once and is identical in Figure 4a-d. Optical imaging of the luciferase signal revealed that, in contrast to the negative controls, tumor cells were significantly killed by redirected RevCAR T cells in the presence of the appropriate RevTM (Figure 4a-d). Effects of RevCAR T cells alone without RevTMs can most likely be explained by donor-dependent allogeneic reactions of RevCAR T cells upon recognition of tumor alloantigens via the endogenous T cell receptor (TCR) complex.
Figure 4.
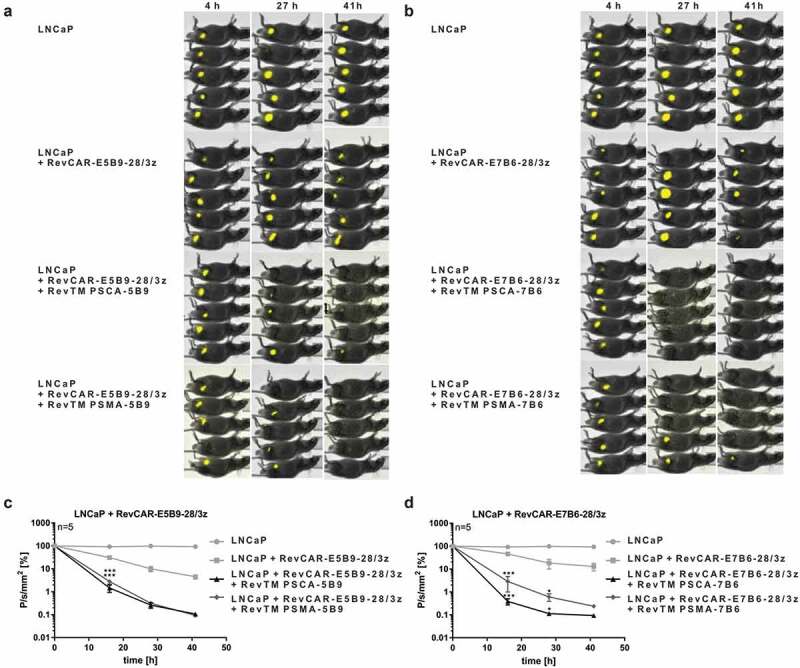
Tumor cell killing by RevCAR system in mice.
(a–d) As indicated, seven different groups each consisting of five NMRI-Foxn1nu/nu mice were analyzed in parallel in one experiment. One control group was transplanted with luciferase-expressing LNCaP tumor cells alone (identical in a–d). Additional control mice were injected with LNCaP tumor cells together with either RevCAR-E5B9-28/3z (a, c) or RevCAR-E7B6-28/3z (b, d) T cells without RevTM. In the treatment groups, RevCAR-E5B9-28/3z (a, c) or RevCAR-E7B6-28/3z (b, d) T cells were co-injected with luciferase-expressing LNCaP tumor cells with indicated RevTMs. (a–b) After administration of D-Luciferin, optical imaging was performed at different time points. (c–d) Measured luminescence was evaluated using the Bruker MI and Multispectral Software (n = 5, mean ± SEM, Two-Way ANOVA with Tukey’s multiple comparisons test. Significance versus tumor cells with RevCAR T cells w/o RevTM is shown.).
In summary, these data confirm proof of functionality for the RevCAR platform both in vitro and in vivo by using two types of RevCARs, either containing E5B9 or E7B6, and by targeting of two different TAAs, either PSCA or PSMA.
Design and functionality of RevCARs for “AND” gate targeting
After showing proof of functionality of the RevCAR system, we tested whether the RevCAR system can be used for “AND” gate targeting. In a first proof of concept study, we wanted to follow our previously described combinatorial CAR T cell-based targeting strategy.23 For this purpose, two independent RevCAR-encoding vectors were required: One RevCAR has to mediate the activation signal, the other one the costimulatory signal. As a prerequisite, we firstly designed two RevCARs that do not dimerize in order to avoid cross-talk signaling and to guarantee a separate transmission of both signals. The RevCAR constructs above described, RevCAR-E5B9-28/3z and RevCAR-E7B6-28/3z, represent second-generation CARs comprising identical CD28 HiD, TMD, and CSD (Figure 5a). As CD28 is known to dimerize, we expected that RevCAR-E5B9 forms homodimers or even heterodimers with RevCAR-E7B6, when they are simultaneously transduced in the same T cell (Figure 5b). In order to confirm this experimentally, we integrated either the signaling RevCAR-E7B6-28/3z or the non-signaling RevCAR-E5B9-stop lacking any intracellular signaling domains or both RevCARs into one T cell (Figure 5c). As expected, the STOP RevTM PSMA-5B9 could not trigger cytotoxic activity of single-transduced Mono-RevCAR-T cells expressing either the signaling RevCAR-E7B6-28/3z or the non-signaling RevCAR-E5B9-stop (Figure 5d). Interestingly, the same STOP RevTM PSMA-5B9 was able to induce cytotoxic potential of Bi-RevCAR-T cells expressing both RevCARs (Figure 5d). This cytotoxic activation of Bi-RevCAR-T cells in the presence of STOP RevTM (capable to bind to the RevCAR-E5B9-stop) would only occur if there was a cross-talk between the non-signaling RevCAR-E5B9-stop and the signaling RevCAR-E7B6-28/3z. Thus, we concluded that both RevCARs dimerized most likely via their identical CD28 portions (HiD and TMD). Consequently, to strictly separate the activating and costimulatory signal for reliable “AND” gate targeting, we had to identify different, non-dimerizing RevCAR structures. A series of second-generation RevCARs containing different TMD and HiD were constructed and tested in analogy to Figure 5 (data not shown). In addition, we tested the receptor density on the surface of RevCAR T cells after transduction with the respective vector (see also Figure 6c and data not shown). Finally, we generated on the one hand, the signaling SIG RevCAR-E7B6-3z (SIG 3z) comprising a CD4 TMD and an IgG4 HiD and on the other hand, the costimulatory COS RevCAR-E5B9-28 (COS 28) containing a CD28 TMD and a CD28 HiD (Figure 5e). Obviously, Bi-RevCAR-T cells, expressing both SIG 3z and COS 28 RevCARs, were not cytotoxic in the presence of the COS RevTM PSCA-5B9 but, as expected, only by adding the SIG RevTM PSMA-7B6 (Figure 5f). Thus, no dimerization and cross-talk signaling occurred between the SIG 3z and COS 28 RevCARs qualifying them as a suitable combination for “AND” gate targeting.
Figure 5.
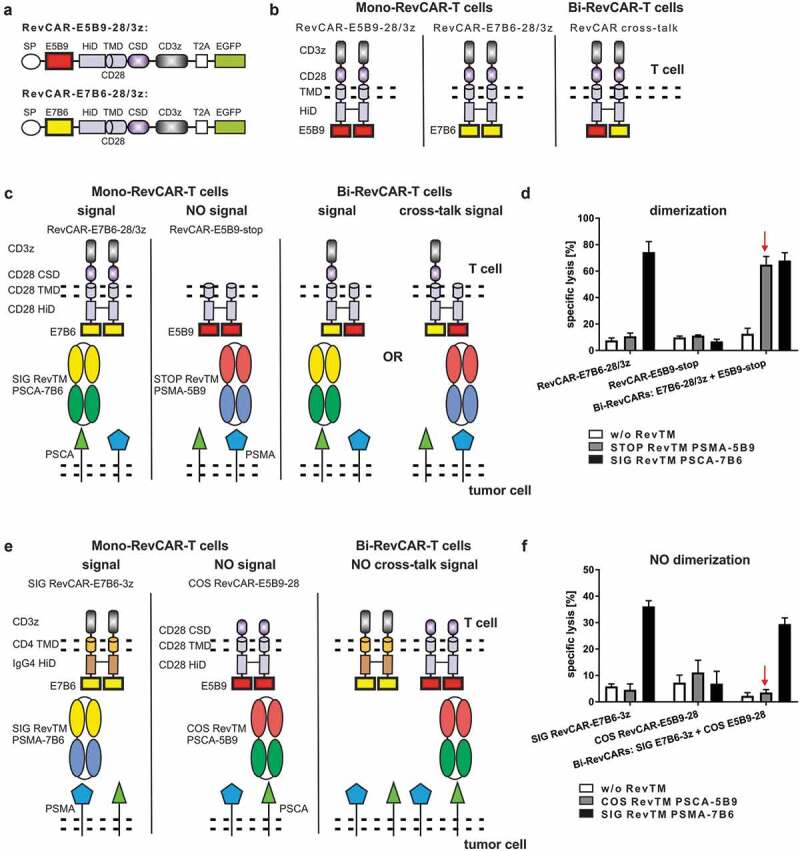
Design of RevCARs for “AND” gate targeting.
(a) Schematic structure of RevCAR-E5B9-28/3z and RevCAR-E7B6-28/3z. They consist of signal peptide (SP), peptide epitope E5B9 or E7B6, hinge (HiD), transmembrane (TMD), and costimulatory (CSD) domain. T2A (Thosea asigna virus) peptide enables EGFP expression. (b) Schematic display of RevCAR dimerization. RevCARs form homodimers, when separately transduced in one T cell. Transduction of both RevCAR-E5B9-28/3z and RevCAR-E7B6-28/3z in one T cell results in heterodimers. (c–f) Test for RevCAR dimerization via killing assay. (c, e) Experimental setup is shown. (c) T cells were transduced with either RevCAR-E7B6-28/3z, comprising CD28 and CD3 signaling domains, or RevCAR-E5B9-stop, lacking any signaling domains, or both together. Both RevCAR-E7B6-28/3z and RevCAR-E5B9-stop contain identical HiD and TMD. When co-transduced, the RevCAR-E5B9-stop can trigger T cell mediated tumor cell killing upon co-cultivation with the STOP RevTM by mediating a cross-talk signal via RevCAR-E7B6-28/3z. (e) In addition, T cells were transduced with either signaling SIG RevCAR-E7B6-3z or costimulatory COS RevCAR-E5B9-28 or both together. Both RevCARs differ with respect to HiD, TMD, and intracellular domain. In contrast to (c), cross-talk signaling by COS RevCAR-E5B9-28 via SIG RevCAR-E7B6-3z is prevented. (d, f) Mono- or Bi-RevCAR-T cells were incubated with LNCaP (d) or PC3 (f) tumor cells with or without indicated RevTMs in a chromium-release assay. Specific lysis is shown for one representative donor.
Figure 6.
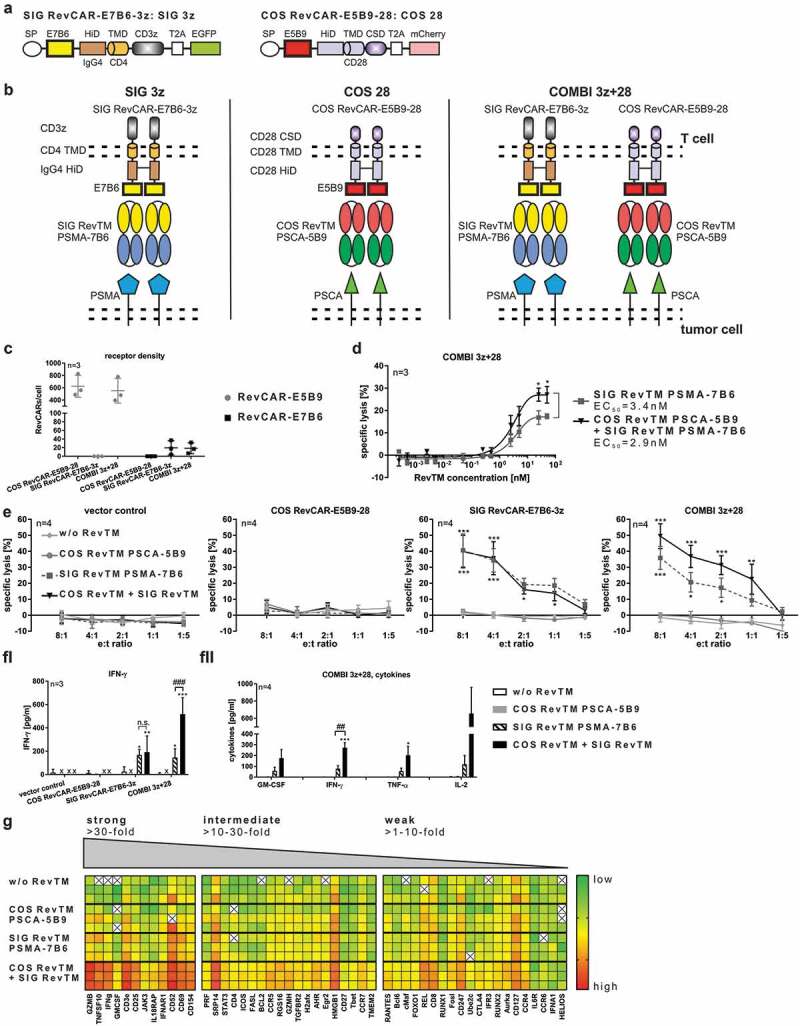
Functional “AND” gate logic of RevCAR system.
(a) Schematic structure of SIG RevCAR-E7B6-3z and COS RevCAR-E5B9-28. They contain a signal peptide (SP), differ with respect to peptide epitope (E5B9 or E7B6), hinge (HiD), transmembrane (TMD), and intracellular domains. T2A peptide enables expression of marker protein EGFP or mCherry. (b) Experimental setup for “AND” gate targeting. The signaling SIG RevCAR-E7B6-3z and the costimulatory COS RevCAR-E5B9-28 were transduced either separately or both together (COMBI 3z+28) in T cells. SIG RevCAR-E7B6-3z is redirected via SIG RevTM and RevCAR-E5B9-28 via COS RevTM. (c) Receptor density of E5B9- and E7B6-RevCARs on transduced T cells (n = 3, mean ± SD). (d) EC50 determination of COMBI 3z+28 RevCAR T cells incubated with PC3 tumor cells and either SIG RevTM alone or both SIG RevTM and COS RevTM for 48 h (e:t ratio 5:1, n = 3, mean ± SD, Two-Way ANOVA with Sidak’s multiple comparisons test. Significance is shown versus SIG RevTM PSMA-7B6.). (e) Specific lysis of PC3 target cells after 48 h incubation with indicated RevCAR T cells in the absence or presence of COS RevTM and/or SIG RevTM (n = 4, mean ± SD, Two-Way ANOVA with Tukey’s multiple comparisons test. Significance versus the controls w/o RevTM and COS RevTM PSCA-5B9 is indicated.). (f) Specific release of IFN-gamma, GM-CSF, IL-2, and TNF-alpha of RevCAR T cells after 48 h incubation with PC3 in the absence or presence of indicated RevTMs. (e:t ratio 5:1, “*” indicates significance versus controls w/o RevTM and COS RevTM PSCA-5B9, “#” indicates significance versus SIG RevTM PSMA-7B6). (fi) ELISA, n = 3, mean ± SD, Two-Way ANOVA with Tukey’s multiple comparisons test. (fii) MACSPlex Cytokine 12 Kit, n = 4, mean ± SEM, One-Way ANOVA with Tukey’s multiple comparisons test. (g) According to their n-fold change of gene expression, analyzed genes were classified into three different groups, showing either strong (≥30-fold), intermediate (≥10 – 30-fold), or weak (≥1 – 10-fold) increase of gene expression. Below, calculated Ct-values of COMBI 3z+28 RevCAR T cells after 24 h incubation with PC3 target cells in the absence or presence of indicated RevTMs are plotted in a heat map (Mean, e:t ratio 5:1, n = 3). Gene expression level ranged from low (green) to high (red) expression. White (X) indicates no expression detectable.
Taken together, from these experiments we learned that (i) RevCARs consisting of a CD4 TMD and an IgG4 HiD do not form heterodimers with RevCARs based on the TMD and HiD of CD28 and thus avoid any kind of cross-talk signaling, (ii) the receptor density depends on the used TMD and HiD, and (iii) the signaling receptor density should be low for “AND” gate targeting approaches.
Based on our findings, for “AND” gate targeting we decided to use the (i) signaling RevCAR SIG 3z which is based on the CD4 TMD and IgG4 HiD and the (ii) costimulatory RevCAR COS 28 which is based on the CD28 TMD and HiD (Figure 6a-b). As ECD, the peptide epitope E7B6 was used in SIG 3z, whereas E5B9 was chosen for the COS 28 in order to strictly separate RevCAR specificities. As already mentioned, both RevCAR constructs were separately encoded on two different lentiviral vectors (Figure 6a), we therefore double-transduced T cells to gain RevCAR T cells expressing both SIG 3z and COS 28 (COMBI 3z+28) for “AND” gate targeting (Figure 6b). As controls, T cells were transduced either with SIG 3z or COS 28 alone. For redirection of SIG 3z, we used the SIG RevTM PSMA-7B6. On contrary, COS 28 was redirected via the COS RevTM PSCA-5B9. Firstly, RevCAR expression was detected (data not shown) and receptor densities were determined on transduced T cells (Figure 6c). As intended, the selected RevCAR SIG 3z showed a lower expression than the selected COS 28. Interestingly, we observed that even marginal numbers of RevCARs (approximately 20 for SIG 3z and around 600 for COS 28) were sufficient for T cell activation. In order to show proof of concept for “AND” gate logic of the RevCAR system, we co-cultured PC3 tumor cells together with COMBI 3z+28 T cells co-expressing both RevCARs SIG 3z and COS 28 in the presence or absence of COS RevTM PSCA-5B9 and SIG RevTM PSMA-7B6. Most importantly, we have successfully proven that the combination of both the activation and costimulatory signal significantly increased maximal tumor cell lysis in comparison to the activation signal alone (Figure 6d). Bearing in mind that the RevCAR SIG 3z represents per definition a first-generation CAR, it is not unexpected that its activation signal in the presence of the SIG RevTM also induced a detectable tumor cell lysis. However, there is a clearly superior anti-tumor effect upon “AND” gate targeting at all tested RevTM concentrations and e:t ratios (Figure 6d-e). Thus, providing costimulatory signals can further enhance the cytotoxic potential of COMBI 3z+28 T cells. No tumor cell lysis occurred without any RevTM, in the presence of COS RevTM PSCA-5B9 alone or when using T cells expressing only COS 28 or lacking any RevCARs (Figure 6e). Cytotoxicity of redirected COMBI 3z+28 T cells was tunable by RevTM dosing as tumor cell lysis increased with increasing RevTM concentrations (Figure 6d). Furthermore, “AND” gate logic of the RevCAR system was proven with respect to cytokine release. In detail, redirected COMBI 3z+28 T cells released more pro-inflammatory cytokines after transmitting both signals than in the presence of the SIG RevTM alone (Figure 6fI-fII). No cytokines were detectable without any RevTMs or in the presence of COS RevTM alone. Moreover, we learned that COMBI 3z+28 T cells considerably upregulated the expression of several genes upon combinatorial targeting of both PSCA and PSMA on the same tumor cells (Figure 6g). Mainly affected were genes involved in T cell cytotoxicity, activation, and cytokine release such as granzyme B, CD25, CD69, IFN-gamma, and GM-CSF. Beside in vitro anti-tumor effects, we could observe the functionality of COMBI 3z+28 T cells in experimental mice (data not shown).
Dual-RevCAR expression for “AND” gate targeting
As RevCARs lack the extracellular scFv domain commonly used in conventional CARs, they are characterized by minimal size, thus fulfilling the original aim of the study. Their small size now allowed us to fuse the two RevCAR genes encoding the RevCARs SIG 3z and COS 28 in one lentiviral vector, in order to facilitate genetic engineering of T cells expressing both RevCARs needed for “AND” gate targeting. The resulting Dual-RevCAR vector, as schematically shown in Figure 7a, could now be tested for lentiviral transduction and bicistronic expression of SIG 3z and COS 28 in Dual-RevCAR T cells. As an advantage, only one single vector was used to generate Dual-RevCAR T cells, whereas COMBI 3z+28 T cells had to be transduced with two separate vectors encoding either SIG 3z or COS 28. Similar to COMBI 3z+28 T cells, Dual-RevCAR T cells presented clearly more COS 28 than SIG 3z receptors on their cell surface (Figure 7b). Cytotoxic analysis considerably demonstrated that this Dual-RevCAR T cells enabled tumor cell lysis exclusively in the presence of both the SIG RevTM PSMA-7B6 and COS RevTM PSCA-5B9, as shown for one representative donor in Figure 7c. True “AND” gate targeting for Dual-RevCAR T cells was further confirmed using Dual-RevCAR T cells derived from three individual donors (Figure 7d-e). In this case, we used Dual-RevCAR T cells generated by a shortened transduction protocol which was established in order to accelerate and optimize the T cell engineering process. Confirming first proof of concept results (Figure 7c), these Dual-RevCAR T cells can significantly kill PC3 tumor cells exclusively in the presence of both the SIG RevTM PSMA-7B6 and COS RevTM PSCA-5B9 (Figure 7dI-dII). In contrast, no significant increase in tumor cell lysis was observed in the presence of either SIG RevTM or COS RevTM alone compared to the control setting without any RevTMs. Obviously, compared to COMBI 3z+28 T cells in Dual-RevCAR T cells the activation signal transmitted by the SIG 3z was diminished. Furthermore, true “AND” gate targeting by Dual-RevCAR T cells was proven with respect to cytokine release (Figure 7e). Indicated cytokines were significantly released exclusively after triggering both the activating and costimulatory signals. Upon the activating or costimulatory signal alone, tested cytokines were not significantly secreted.
Figure 7.
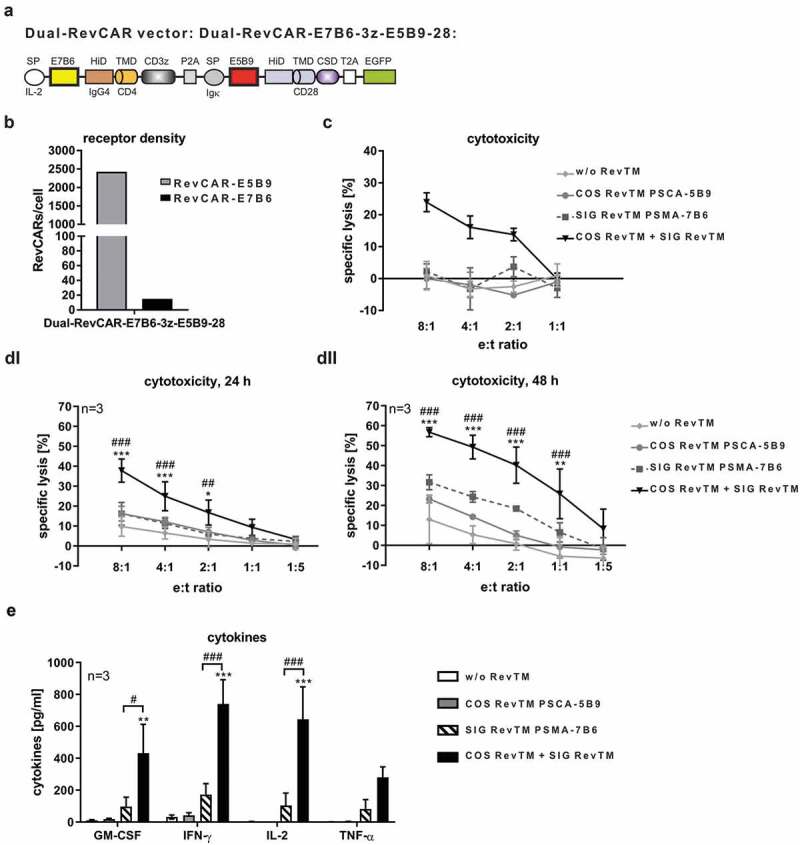
Dual-RevCAR expression for “AND” gate targeting.
(a) Schematic structure of the Dual-RevCAR-E7B6-3z-E5B9-28. It contains both the SIG RevCAR-E7B6-3z flanked by the IL-2 derived signal peptide (SP) and P2A peptide and the COS RevCAR-E5B9-28 flanked by the Ig-kappa signal peptide, T2A peptide, and EGFP. (b) Receptor density of E5B9- and E7B6-RevCARs on Dual-RevCAR-E7B6-3z-E5B9-28 T cells. (c) Cytotoxicity of Dual-RevCAR-E7B6-3z-E5B9-28 T cells after 48 h incubation with PSCA/PSMA-co-expressing PC3 cells in the absence or presence of indicated RevTMs, shown for one representative donor (Mean ± SD). (b-c) T cells were transduced with the traditional protocol. (d) Specific lysis of PSCA+/PSMA+ PC3 target cells in the absence or presence of indicated RevTMs after incubation with Dual-RevCAR-E7B6-3z-E5B9-28 T cells for 24 h (di) and 48 h (dii) (n = 3, Mean ± SD, Two-Way ANOVA with Tukey’s multiple comparisons test). (e) Release of indicated cytokines from redirected Dual-RevCAR-E7B6-3z-E5B9-28 T cells after their incubation with PSCA+/PSMA+ PC3 target cells in the absence or presence of indicated RevTMs (n = 3, mean ± SEM, One-Way ANOVA with Tukey’s multiple comparisons test). (d-e) T cells were transduced with the shortened protocol. “*” indicates significance versus controls w/o RevTM and COS RevTM PSCA-5B9, “#” indicates significance versus SIG RevTM PSMA-7B6.
Discussion
In spite of the great success of clinical approved anti-CD19 CAR T cells,45,46 several hurdles of conventional CAR T cell therapy still exist. Major problems are antigen loss of tumor escape variants, on-target/off-tumor toxicities, and uncontrollability of conventional CAR T cells that unpredictably expand in patients. In order to improve the safety management and tumor specificity of CAR T cells, adaptor CAR platforms and “AND” gate targeting strategies were developed. Here, we introduce the novel switchable and controllable RevCAR adaptor system facilitating combinatorial tumor targeting characterized by minimal RevCAR size allowing a bicistronic expressing of two RevCARs with different specificity.
In this system, genetically engineered T cells armed with RevCARs are redirected against tumor targets via RevTMs which are bsAbs based on scFvs similar to the bispecific T cell engager (BiTE) format. These small recombinant adaptor components are easy to produce, able to penetrate tumor tissues, and have a short half-life in vivo which favors a fast on/off-switch and steering of RevCAR T cell activity.3,47,48 As shown in Figure 2, RevTMs redirect RevCAR T cells to efficiently kill tumor cells at low RevTM concentrations (pM range) and e:t ratios. Side-by-side comparison of the cytotoxic potential in vitro revealed that redirected UniCAR and RevCAR T cells mediate comparably high maximal tumor cell lysis (Figure S4). As we have already published that compared to conventional second-generation CARs the UniCAR system performs equally well,28 conventional CAR T cells were not included. Remarkably, in the tested setting the RevCAR approach was superior with respect to killing efficiency as the calculated EC50 value for the RevCAR system was lower than for the UniCAR system (Figure S4). However, both adaptor CAR platforms are highly efficient, specific, and flexible and have their inherent advantages and limitations. Whether one approach holds an essential benefit has to be proven in more detail in vitro and finally in humans in future studies. Analyzing the killing mechanism of the completely humanized RevCAR system in a human in vitro model, we learned that mainly the granzyme/perforin pathway but also TRAIL were involved in tumor cell killing by redirected RevCAR T cells (Figure S2D-S2E). In agreement, these data were confirmed by gene expression analysis revealing that redirected RevCAR T cells strongly upregulate the genes encoding for granzyme B and perforin, while the expression of the FasL-encoding gene was upregulated on intermediate levels (Figure 6g). Our data are in line with previously published findings showing that the killing mechanism of conventional CARs and the UniCAR system is mainly mediated via the granzyme/perforin pathway, whereas the Fas/FasL axis is also involved.49,50 Surprisingly, Karches et al.19 published that tumor cell killing caused by their adaptor CAR platform termed SAR was independent from granzyme and perforin release but mainly mediated by FasL. Different killing mechanisms of RevCAR versus SAR that both trigger CD28 and CD3z signals, might maybe also be explained by differences between mice and humans. Besides cytotoxicity, redirected RevCAR T cells significantly release pro-inflammatory cytokines (Figure 3) which might be helpful to overcome the immunosuppressive barrier and to enhance their anti-tumor effect in solid tumors. For safety reasons it is worthwhile to mention that RevCAR T cells are functionally activated only after their cross-linkage with tumor cells via appropriate RevTMs simultaneously binding to RevCARs and target antigens (Figures 2-4, S2C). So, we did not observe an antigen-unspecific tumor cell killing induced by RevCAR T cells after binding of RevTMs in the presence of target-negative tumor cells (Figure S2C). As no specific tumor cell lysis and cytokine release were observed in the absence of any RevTMs or in the presence of irrelevant RevTMs targeting a non-matching peptide epitope or TAA, we conclude that the RevCAR system works strictly antigen- and epitope-specific as well as RevTM-dependent. Furthermore, we showed that RevCAR T cells can be flexibly redirected against different TAAs (here either PSCA or PSMA) simply by replacing RevTMs possessing diverse specificities. In addition to PSCA and PSMA, we have proven that RevCAR T cells can be efficiently redirected to several other TAAs presented on hematological as well as solid malignancies (data not shown). Importantly, replacement of the anti-TAA domain did not dramatically impair RevTM functionality. Thus, we confirmed that the RevCAR system can be used for “OR” gate targeting to overcome antigen lacking tumor escape variants.
In order to reduce off-tumor side reactions and to increase tumor specificity, “AND” gate targeting strategies have been developed. Despite the first proof of concept was demonstrated, this technology still struggles with many challenges.22–25 At the beginning “AND” gate targeting was only shown with respect to cytokine release and proliferation, while the problem was to strictly limit bi-specific Dual-CAR T cells to “AND” gate tumor cell killing.24,25 One solution for this major hurdle could be to reduce the strength of the activation CD3z signal triggered by the signaling CAR in a way that it is not sufficient to fully activate T cells without receiving CD28 costimulatory signals. Indeed, we successfully demonstrated true “AND” gate targeting by reducing the affinity of the CD3z signaling CAR, revealing that the balance between the first activation and second costimulatory signal is crucial for “AND” gate targeting.23 However, “AND” gate reactivity of Dual-CAR T cells depends not only on scFv affinity but also on many other parameters such as receptor design, signal strength, transduction efficiency, receptor expression, receptor dimerization, and antigen density level. From this point of view, our modular adaptor RevCAR system has the advantage of high flexibility. In contrast to conventional CAR approaches, where specificities and affinities are strictly fixed, universal RevCAR T cells can be redirected to multiple antigens simply by replacing or adding several RevTMs that can be easily and rapidly re-engineered and adapted to multiple antigens with different affinities. Another advantage in light of “AND” gate targeting by split CARs is the small size of RevCARs facilitating the transduction of several CARs with different specificities in one T cell. Here, we have successfully proven that the RevCAR system is programmable and can be used for “AND” gate targeting shown with respect to tumor cell killing, cytokine release, and upregulation of gene expression (Figures 6d-6g and 7c-e). True “AND” gate targeting is realized by redirected Dual-RevCAR T cells, where SIG RevCAR and COS RevCAR were both encoded by one vector (Figure 7a). True “AND” gate targeting means that only the combination of the activating and costimulatory signal results in a significantly increased tumor cell lysis as well as cytokine release, whereas in the absence of either one or both signals no significant effects occur (Figure 7c-e). In contrast, specific tumor cell lysis was also induced by COMBI-RevCAR T cells triggering only the CD3z signal alone, where both RevCARs were encoded on separate vectors (Figure 6a). However, tumor cell lysis was clearly increased by COMBI-RevCAR T cells after transmitting both CD3z and CD28 signals (Figure 6d-e). Obviously, the balance between the CD3z activation and CD28 costimulatory signal was more favorable for true “AND” gate targeting in single-transduced Dual-RevCAR T cells in comparison to double-transduced COMBI-RevCAR T cells. Thus, we conclude that the total numbers of SIG RevCARs and COS RevCARs expressed on transduced T cells and furthermore the ratio between both receptors could be critical for “AND” gate targeting. We hypothesized and confirmed that marginal numbers of RevCAR SIG 3z, that are lower in comparison to COS 28, are favorable for “AND” gate targeting because most probably this results in a reduction of the CD3z signal strength. Based on the receptor numbers and ratio as well as on non-dimerization properties, we selected the herein presented receptor combination from a series of different tested RevCAR constructs as most suitable for gated targeting.
Meanwhile, several adaptor CAR platforms have been developed.11–19 However, some of them bear the risk for immunogenicity and cross-reactivity if the interacting components are not carefully selected with respect to species compatibility and natural expression pattern in the recipient.3 Moreover, some of them are not flexible with respect to antigen targeting and guarantee no steering of T cell activity, when the adaptor molecules have a long-term pharmacokinetic half-life.3 To ensure a rapid and reversible on/off-switch as well as fine-tuning of CAR T cell activity, small short-living adaptor molecules showing appropriate pharmacokinetic behavior are required. Especially in this context, it is also important that the interaction/binding properties of the adaptor molecules toward the tumor target and CAR T cells have to be well balanced in a way, that they allow an association as well as dissociation of the complex.3,27,51,52
In summary, here we emphasize the novel RevCAR platform combining all the mentioned features and advantages in one single system. The RevCAR system is completely humanized and based on carefully selected human peptide epitopes minimizing its risk for immunogenicity and cross-reactivity. RevCARs lack the extracellular scFv of conventional CARs. Thus, unspecific binding and tonic signaling effects caused by scFv dimerization are avoided. The RevCAR system can be used for highly efficient killing of tumor cells in an antigen-/epitope-specific and RevTM-dependent manner. As an improvement for the safety management, RevCAR T cell activity can be switched on and off and steered by dosing of the RevTMs. RevTMs can be easily and rapidly re-engineered and adapted to diverse antigens with different affinities. Thus, universal RevCAR T cells can be flexibly redirected to multiple antigens simply by exchanging the RevTMs without T cell re-engineering (“OR” gate logic) to overcome antigen loss of tumor escape variants. As RevCARs have minimal size, they are favorable for genetic modification of T cells and bicistronic expression of Dual-RevCARs. Remarkably, the RevCAR system can be used for true combinatorial targeting following the “AND” gate logic in order to reduce on-target/off-tumor side effects and increase tumor specificity of CAR T cell therapy. The small size of the RevCAR genes might allow to integrate additional receptor genes thus allowing the inclusion of further combinatorial strategies such as “NOT-AND” (NAND) gated targeting. E.g. including an inhibitory RevCAR gene could be used to protect tissues that do express a certain antigen.
Supplementary Material
Author contributions
A.F. and A.H. cloned RevCARs and RevTMs, planned and performed all experiments, analyzed, interpreted the data, generated figures, and wrote the manuscript. A.F. planed conceptual implementation of the project, designed RevCARs and RevTMs. M.B. is the inventor of the RevCAR system, designed the research, secured financing, interpreted the data, and wrote the manuscript. R.B. performed and analyzed the animal imaging experiments. S.K., C.A., L.R.L., N.B., R.B., E.K.B., N.M., C.L., A.K., K.E.G.S., and M.B. supported experiments and provided valuable input and critical materials. All authors reviewed the manuscript.
Acknowledgments
We thank Julia Lagler for her excellent technical assistance.
Disclosure of potential conflicts of interest
M.B. is the inventor of the UniCAR and RevCAR system and a shareholder of the company GEMoaB Monoclonals GmbH, which holds licenses, patents, and has filed patent applications related to the UniCAR and RevCAR system. M.B. holds patents related to the UniCAR and RevCAR system, anti-La mAbs, and the La epitopes used in this paper. M.B. declares no non-financial competing interests. The remaining authors declare no financial and no non-financial conflict of interests.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Kuwana Y, Asakura Y, Utsunomiya N, Nakanishi M, Arata Y, Itoh S, Nagase F, Kurosawa Y.. Expression of chimeric receptor composed of immunoglobulin-derived V resions and T-cell receptor-derived C regions. Biochem Biophys Res Commun. 1987;149(3):960–15. doi: 10.1016/0006-291X(87)90502-X. [DOI] [PubMed] [Google Scholar]
- 2.Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci U S A. 1989;86(24):10024–10028. doi: 10.1073/pnas.86.24.10024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bachmann M. The UniCAR system: A modular CAR T cell approach to improve the safety of CAR T cells. Immunol Lett. 2019;211:13–22. doi: 10.1016/j.imlet.2019.05.003. [DOI] [PubMed] [Google Scholar]
- 4.June CH, O’Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science. 2018;359(6382):1361–1365. doi: 10.1126/science.aar6711. [DOI] [PubMed] [Google Scholar]
- 5.Cartellieri M, Bachmann M, Feldmann A, Bippes C, Stamova S, Wehner R, Temme A, Schmitz M.. Chimeric antigen receptor-engineered T cells for immunotherapy of cancer. J Biomed Biotechnol. 2010;2010:956304. doi: 10.1155/2010/956304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, Braunschweig I, Oluwole OO, Siddiqi T, Lin Y, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. New England J Med. 2017;377(26):2531–2544. doi: 10.1056/NEJMoa1707447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, Bader P, Verneris MR, Stefanski HE, Myers GD, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. New England J Med. 2018;378(5):439–448. doi: 10.1056/NEJMoa1709866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, Jäger U, Jaglowski S, Andreadis C, Westin JR, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. New England J Med. 2019;380(1):45–56. doi: 10.1056/NEJMoa1804980. [DOI] [PubMed] [Google Scholar]
- 9.Majzner RG, Mackall CL. Clinical lessons learned from the first leg of the CAR T cell journey. Nat Med. 2019;25(9):1341–1355. doi: 10.1038/s41591-019-0564-6. [DOI] [PubMed] [Google Scholar]
- 10.Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Molecular Therapy. 2010;18(4):843–851. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Urbanska K, Lanitis E, Poussin M, Lynn RC, Gavin BP, Kelderman S, Yu J, Scholler N, Powell DJ. A universal strategy for adoptive immunotherapy of cancer through use of a novel T-cell antigen receptor. Cancer Res. 2012;72(7):1844–1852. doi: 10.1158/0008-5472.CAN-11-3890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tamada K, Geng D, Sakoda Y, Bansal N, Srivastava R, Li Z, Davila E. Redirecting gene-modified T cells toward various cancer types using tagged antibodies. Clin Cancer Res. 2012;18(23):6436–6445. doi: 10.1158/1078-0432.CCR-12-1449. [DOI] [PubMed] [Google Scholar]
- 13.Ma JS, Kim JY, Kazane SA, Choi S-H, Yun HY, Kim MS, Rodgers DT, Pugh HM, Singer O, Sun SB, et al. Versatile strategy for controlling the specificity and activity of engineered T cells. Proc Natl Acad Sci U S A. 2016;113(4):E450–E458. doi: 10.1073/pnas.1524193113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kudo K, Imai C, Lorenzini P, Kamiya T, Kono K, Davidoff AM, Chng WJ, Campana D. T lymphocytes expressing a CD16 signaling receptor exert antibody-dependent cancer cell killing. Cancer Res. 2014;74(1):93–103. doi: 10.1158/0008-5472.CAN-13-1365. [DOI] [PubMed] [Google Scholar]
- 15.Urbanska K, Lynn RC, Stashwick C, Thakur A, Lum LG, Powell DJ. Targeted cancer immunotherapy via combination of designer bispecific antibody and novel gene-engineered T cells. J Transl Med. 2014;12(1):347. doi: 10.1186/s12967-014-0347-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rodgers DT, Mazagova M, Hampton EN, Cao Y, Ramadoss NS, Hardy IR, Schulman A, Du J, Wang F, Singer O, et al. Switch-mediated activation and retargeting of CAR-T cells for B-cell malignancies. Proc Natl Acad Sci U S A 2016;113(4):E459–E468. doi: 10.1073/pnas.1524155113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koristka S, Cartellieri M, Feldmann A, Arndt C, Loff S, Michalk I, Aliperta R, von Bonin M, Bornhäuser M, Ehninger A, et al. Flexible antigen-specific redirection of human regulatory T cells via a novel universal chimeric antigen receptor system. Blood. 2014;124(21):3494. doi: 10.1182/blood.V124.21.3494.3494 [DOI] [Google Scholar]
- 18.Cho JH, Collins JJ, Wong WW. Universal chimeric antigen receptors for multiplexed and logical control of T cell responses. Cell. 2018;173(6):1426–1438. e11. doi: 10.1016/j.cell.2018.03.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karches CH, Benmebarek M-R, Schmidbauer ML, Kurzay M, Klaus R, Geiger M, Rataj F, Cadilha BL, Lesch S, Heise C, et al. Bispecific antibodies enable synthetic agonistic receptor-transduced T cells for tumor immunotherapy. Clin Cancer Res. 2019;25(19):5890–5900. doi: 10.1158/1078-0432.CCR-18-3927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cartellieri M, Feldmann A, Koristka S, Arndt C, Loff S, Ehninger A, von Bonin M, Bejestani EP, Ehninger G, Bachmann MP, et al. Switching CAR T cells on and off: a novel modular platform for retargeting of T cells to AML blasts. Blood Cancer J. 2016;6(8):e458. doi: 10.1038/bcj.2016.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.O’Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, Martinez-Lage M, Brem S, Maloney E, Shen A, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. 2017;9(399):eaaa0984. doi: 10.1126/scitranslmed.aaa0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ebert LM, Yu W, Gargett T, Brown MP. Logic-gated approaches to extend the utility of chimeric antigen receptor T-cell technology. Biochem Soc Trans. 2018;46(2):391–401. doi: 10.1042/BST20170178. [DOI] [PubMed] [Google Scholar]
- 23.Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M.. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol. 2013;31(1):71–75. doi: 10.1038/nbt.2459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wilkie S, van Schalkwyk MC, Hobbs S, Davies DM, van der Stegen SJC, Pereira ACP, Burbridge SE, Box C, Eccles SA, Maher J, et al. Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling. J Clin Immunol. 2012;32(5):1059–1070. doi: 10.1007/s10875-012-9689-9. [DOI] [PubMed] [Google Scholar]
- 25.Lanitis E, Poussin M, Klattenhoff AW, Song D, Sandaltzopoulos R, June CH, Powell DJ. Chimeric antigen receptor T Cells with dissociated signaling domains exhibit focused antitumor activity with reduced potential for toxicity in vivo. Cancer Immunol Res. 2013;1(1):43–53. doi: 10.1158/2326-6066.CIR-13-0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koristka S, Ziller-Walter P, Bergmann R, Arndt C, Feldmann A, Kegler A, Cartellieri M, Ehninger A, Ehninger G, Bornhäuser M, et al. Anti-CAR-engineered T cells for epitope-based elimination of autologous CAR T cells. Cancer Immunol Immunother. 2019;68(9):1401–1415. doi: 10.1007/s00262-019-02376-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Albert S, Arndt C, Feldmann A, Bergmann R, Bachmann D, Koristka S, Ludwig F, Ziller-Walter P, Kegler A, Gärtner S, et al. A novel nanobody-based target module for retargeting of T lymphocytes to EGFR-expressing cancer cells via the modular UniCAR platform. Oncoimmunology. 2017;6(4):e1287246. doi: 10.1080/2162402X.2017.1287246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Feldmann A, Arndt C, Bergmann R, Loff S, Cartellieri M, Bachmann D, Aliperta R, Hetzenecker M, Ludwig F, Albert S, et al. Retargeting of T lymphocytes to PSCA- or PSMA positive prostate cancer cells using the novel modular chimeric antigen receptor platform technology “UniCAR”. Oncotarget. 2017;8(19):31368. doi: 10.18632/oncotarget.15572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Loureiro L, Feldmann A, Bergmann R, Koristka S, Berndt N, Arndt C, Pietzsch J, Novo C, Videira P, Bachmann M, et al. Development of a novel target module redirecting UniCAR T cells to Sialyl Tn-expressing tumor cells. Blood Cancer J. 2018;8(9):81. doi: 10.1038/s41408-018-0113-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mitwasi N, Feldmann A, Bergmann R, Berndt N, Arndt C, Koristka S, Kegler A, Jureczek J, Hoffmann A, Ehninger A, et al. Development of novel target modules for retargeting of UniCAR T cells to GD2 positive tumor cells. Oncotarget. 2017;8(65):108584. doi: 10.18632/oncotarget.21017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arndt C, Feldmann A, Koristka S, Schäfer M, Bergmann R, Mitwasi N, Berndt N, Bachmann D, Kegler A, Schmitz M, et al. A theranostic PSMA ligand for PET imaging and retargeting of T cells expressing the universal chimeric antigen receptor UniCAR. OncoImmunology. 2019;8(11):1659095. doi: 10.1080/2162402X.2019.1659095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Albert S, Arndt C, Koristka S, Berndt N, Bergmann R, Feldmann A, Schmitz M, Pietzsch J, Steinbach J, Bachmann M et al. From mono- to bivalent: improving theranostic properties of target modules for redirection of UniCAR T cells against EGFR-expressing tumor cells in vitro and in vivo. Oncotarget. 2018. May 22;9(39):25597–25616. doi: 10.18632/oncotarget.25390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fasslrinner F, Arndt C, Koristka S, Feldmann A, Altmann H, von Bonin M, Schmitz M, Bornhäuser M, Bachmann M. Midostaurin abrogates CD33-directed UniCAR and CD33-CD3 bispecific antibody therapy in acute myeloid leukaemia. Br J Haematol. 2019. September;186(5):735–740. doi: 10.1111/bjh.15975. [DOI] [PubMed] [Google Scholar]
- 34.Mitwasi N, Feldmann A, Arndt C, Koristka S, Berndt N, Jureczek J, Loureiro LR, Bergmann R, Máthé D, Hegedüs N, et al. “UniCAR”-modified off-the-shelf NK-92 cells for targeting of GD2-expressing tumour cells. Sci Rep. 2020. February 7;10(1):2141. doi: 10.1038/s41598-020-59082-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bachmann D, Aliperta R, Bergmann R, Feldmann A, Koristka S, Arndt C, Loff S, Welzel P, Albert S, Kegler A et al. Retargeting of UniCAR T cells with an in vivo synthesized target module directed against CD19 positive tumor cells. Oncotarget. 2018. January 26;9(7):7487–7500. doi: 10.18632/oncotarget.23556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cartellieri M, Koristka S, Arndt C, Feldmann A, Stamova S, von Bonin M, Töpfer K, Krüger T, Geib M, Michalk I, et al. A novel ex vivo isolation and expansion procedure for chimeric antigen receptor engrafted human T cells. PLoS One. 2014;9(4):e93745. doi: 10.1371/journal.pone.0093745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Feldmann A, Arndt C, Topfer K Stamova S, Krone F, Cartellieri M, Koristka S, Michalk I, Lindemann D, Schmitz M, et al. Novel humanized and highly efficient bispecific antibodies mediate killing of prostate stem cell antigen-expressing tumor cells by CD8+ and CD4+ T cells. J Immunol. 2012. September 15;189(6):3249–3259. doi: 10.4049/jimmunol.1200341. [DOI] [PubMed] [Google Scholar]
- 38.Arndt C, Feldmann A, Koristka S, Cartellieri M, Dimmel M, Ehninger A, Ehninger G, Bachmann M. Simultaneous targeting of prostate stem cell antigen and prostate-specific membrane antigen improves the killing of prostate cancer cells using a novel modular T cell-retargeting system. Prostate. 2014. September;74(13):1335–1346. doi: 10.1002/pros.22850. [DOI] [PubMed] [Google Scholar]
- 39.Feldmann A, Stamova S, Bippes CC, Bartsch H, Wehner R, Schmitz M, Temme A, Cartellieri M, Bachmann M. Retargeting of T cells to prostate stem cell antigen expressing tumor cells: comparison of different antibody formats. Prostate. 2011. June 15;71(9):998–1011. doi: 10.1002/pros.21315. [DOI] [PubMed] [Google Scholar]
- 40.Kegler A, Koristka S, Bergmann R, Berndt N, Arndt C, Feldmann A, Hoffmann A, Bornhäuser M, Schmitz M, Bachmann MP, et al. T cells engrafted with a UniCAR 28/z outperform UniCAR BB/z-transduced T cells in the face of regulatory T cell-mediated immunosuppression. Oncoimmunology. 2019;8(9):e1621676. doi: 10.1080/2162402X.2019.1621676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Albert S, Koristka S, Gerbaulet A, Cartellieri M, Arndt C, Feldmann A, Berndt N, Loureiro LR, von Bonin M, Ehninger G, et al. Tonic signaling and its effects on lymphopoiesis of CAR-armed hematopoietic stem and progenitor cells. J Immunol. 2019;202(6):1735–1746. doi: 10.4049/jimmunol.1801004. [DOI] [PubMed] [Google Scholar]
- 42.Heninger A-K, Eugster A, Kuehn D, Buettner F, Kuhn M, Lindner A, Dietz S, Jergens S, Wilhelm C, Beyerlein A et al. A divergent population of autoantigen-responsive CD4 + T cells in infants prior to β cell autoimmunity. Sci Transl Med. 2017. February 22;9(378):378. doi: 10.1126/scitranslmed.aaf8848. [DOI] [PubMed] [Google Scholar]
- 43.Koristka S, Kegler A, Bergmann R, Arndt C, Feldmann A, Albert S, Cartellieri M, Ehninger A, Ehninger G, Middeke JM, et al. Engrafting human regulatory T cells with a flexible modular chimeric antigen receptor technology. J Autoimmun. 2018;90:116–131. doi: 10.1016/j.jaut.2018.02.006. [DOI] [PubMed] [Google Scholar]
- 44.Aliperta R, Welzel PB, Bergmann R, Freudenberg U, Berndt N, Feldmann A, Arndt C, Koristka S, Stanzione M, Cartellieri M, et al. Cryogel-supported stem cell factory for customized sustained release of bispecific antibodies for cancer immunotherapy. Sci Rep. 2017. February;7(1):42855. doi: 10.1038/srep42855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Geyer MB. First CAR to pass the road test: tisagenlecleucel’s drive to FDA approval. Clin Cancer Res. 2019;25(4):1133–1135. doi: 10.1158/1078-0432.CCR-18-3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bouchkouj N, Kasamon YL, de Claro RA, George B, Lin X, Lee S, Blumenthal GM, Bryan W, McKee AE, Pazdur R et al. FDA approval summary: axicabtagene ciloleucel for relapsed or refractory large B-cell Lymphoma. Clin Cancer Res. 2019. March 15;25(6):1702–1708. doi: 10.1158/1078-0432.CCR-18-2743. [DOI] [PubMed] [Google Scholar]
- 47.Zhu M, Wu B, Brandl C, Johnson J, Wolf A, Chow A, Doshi S. Blinatumomab, a bispecific T-cell engager (BiTE®) for CD-19 targeted cancer immunotherapy: clinical pharmacology and its implications. Clin Pharmacokinet. 2016;55(10):1271–1288. doi: 10.1007/s40262-016-0405-4. [DOI] [PubMed] [Google Scholar]
- 48.Slaney CY, Wang P, Darcy PK, Kershaw MH. CARs versus BiTEs: a comparison between T cell–redirection strategies for cancer treatment. Cancer Discov. 2018;8(8):924–934. doi: 10.1158/2159-8290.CD-18-0297. [DOI] [PubMed] [Google Scholar]
- 49.Jureczek J, Bergmann R, Berndt N, Koristka S, Kegler A, Puentes-Cala E, Soto JA, Arndt C, Bachmann M, Feldmann A, et al. An oligo-His-tag of a targeting module does not influence its biodistribution and the retargeting capabilities of UniCAR T cells. Sci Rep. 2019;9(1):1–15. doi: 10.1038/s41598-019-47044-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Benmebarek M-R, Karches CH, Cadilha BL, Lesch S, Endres S, Kobold S. Killing mechanisms of chimeric antigen receptor (CAR) T cells. Int J Mol Sci. 2019. March 14;20(6):6. doi: 10.3390/ijms20061283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Feldmann A, Arndt C, Koristka S, Berndt N, Bergmann R, Bachmann MP. Conventional CARs versus modular CARs. Cancer Immunol Immunother. 2019;68(10):1713–1719. doi: 10.1007/s00262-019-02399-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Arndt C, Bachmann M, Bergmann R Berndt N, Feldmann A, Koristka S. Theranostic CAR T cell targeting: A brief review. J Labelled Comp Radiopharm. 2019. June 30;62(8):533–540. doi: 10.1002/jlcr.3727. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.