

ABSTRACT
Myelofibrosis (MF) is characterized by chronic inflammation and hyper-activation of the JAK-STAT pathway. Infections are one of the main causes of morbidity/mortality. Therapy with Ruxolitinib (RUX), a JAK1/2 inhibitor, may further increase the infectious risk. Monocytes are critical players in inflammation/immunity through cytokine production and release of bioactive extracellular vesicles. However, the functional behavior of MF monocytes, particularly during RUX therapy, is still unclear. In this study, we found that monocytes from JAK2V617F-mutated MF patients show an altered expression of chemokine (CCR2, CXCR3, CCR5) and cytokine (TNF-α-R, IL10-R, IL1β-R, IL6-R) receptors. Furthermore, their ability to produce and secrete free and extracellular vesicles-linked cytokines (IL1β, TNF-α, IL6, IL10) under lipopolysaccharides (LPS) stimulation is severely impaired. Interestingly, monocytes from RUX-treated patients show normal level of chemokine, IL10, IL1β, and IL6 receptors together with a restored ability to produce intracellular and to secrete extracellular vesicles-linked cytokines after LPS stimulation. Conversely, RUX therapy does not normalize TNF-R1/2 receptors expression and the LPS-driven secretion of free pro/anti-inflammatory cytokines. Accordingly, upon LPS stimulation, in vitro RUX treatment of monocytes from MF patients increases their secretion of extracellular vesicles-linked cytokines but inhibits the secretion of free pro/anti-inflammatory cytokines. In conclusion, we demonstrated that in MF the infection-driven response of circulating monocytes is defective. Importantly, RUX promotes their infection-driven cytokine production suggesting that infections following RUX therapy may not be due to monocyte failure. These findings contribute to better interpreting the immune vulnerability of MF and to envisaging strategies to improve the infection-driven immune response.
KEYWORDS: Myelofibrosis, monocyte, ruxolitinib, extracellular vesicles
Introduction
Myelofibrosis (MF) is a clonal disorder of the hematopoietic stem/progenitor cell (HSPC). The molecular pathogenesis relates to driver mutations in 3 genes (namely: Janus Kinase 2 (JAK2); Calreticulin (CALR); myeloproliferative leukemia virus oncogene (MPL)) causing hyper-activation of the JAK-STAT signaling pathway.1,2 More than half of patients with MF carry the JAK2V617F mutation. Beyond “driver” mutations, chronic inflammation is considered to be the MF hallmark.3–7 More recently, it has been demonstrated that cytokine overproduction in MF myeloid cells is driven by multiple signaling pathways (NF-kB and MAPK) beyond JAK-STAT.8 Consistently, NF-kB signaling is hyperactivated in MF and contributes to myeloproliferation and inflammation.8,9
Infections are one of the main causes of morbidity and mortality in MF, representing a cause of death in around 10% of the cases.10–13 Infections are mainly bacterial (78%); however, viral (11%) and fungal infections (2%) were recorded as well.10 The increased risk of infections is thought to arise from deregulation of key mediators of the immune system.10–13 Specifically, numerical and/or functional abnormalities were documented in the monocyte/macrophage compartment, T‐cells, natural killer, and myeloid‐derived suppressor cells.10,14–21 We previously demonstrated that in MF there is a reduced ability of monocytes to differentiate into dendritic cells, decreased plasticity of Th17 lymphocytes and functionally impaired Innate Lymphoid Cells.21
To further aggravate the clinical landscape, prior studies have demonstrated significant inhibitory effects in T cells, natural killer and dendritic cells function after exposure to JAK inhibitors.22–27 Therapy with Ruxolitinib (RUX), a JAK1/2 inhibitor, suppresses clonal myeloproliferation and release of proinflammatory cytokines, reducing splenomegaly and constitutional symptoms in around 50% of MF patients.28,29 Of note, due to the involvement of multiple signaling pathways beyond JAK/STAT, MF cytokine overproduction is reduced but not abrogated by JAK1/2 inhibition with RUX.8,9 Severe infections occur in patients treated with RUX and are mainly bacterial, especially in advanced stage of disease.10
Monocytes are the first line of defense in recognition and clearance of pathogens and play an important role in the primary innate immune response and inflammation.30–32 They are traditionally divided into 3 phenotypically and functionally distinct populations based on differences in expression of CD14 and CD16.31 Recent studies elucidated the sequential differentiation of human monocyte subsets demonstrating that classical monocytes egress from the bone marrow into peripheral blood where they differentiate into intermediate and subsequently non-classical monocytes.32 Therefore, chemokine-driven migration from the bone marrow toward peripheral blood and inflammatory/damaged tissues is a crucial monocyte-related feature.
Following activation, monocytes can release soluble or extracellular vesicles (EVs)-associated, either surface-bound or encapsulated, cytokines. However, the pattern of cytokines secretion was dependent on the infection stimuli.33 EVs, which are composed of exosomes (30–150 nm) and microvesicles (200–1000 nm), are released from a broad variety of cells during homeostasis and cell activation with pleiotropic effects on signaling among cells. EVs affect normal and malignant hemopoiesis34 and are critical players in the regulation of inflammation and immunity.35,36 High serum levels of EVs have been detected in hematological malignancies including myeloproliferative neoplasms (MPN)37,38 and we recently described that circulating monocyte-derived EVs are increased in MF.39
Despite a key role in regulating inflammation and immune response, the role of monocytes in the pathogenesis of MF needs to be fully addressed. Moreover, the impact of RUX inhibition on monocyte behavior has been poorly investigated. Monocytosis may occur in patients with MF (around 15%) and is associated with poor outcome.40 Additionally, there are evidences that monocytes are overactivated,41,42 show inflammatory features,43 and represent the principal cellular source for most inflammatory cytokines after stimulation with PMA/ionomycin.8
To increase knowledge of the pathogenesis of the inflammatory/immune microenvironment of MF and to address the role of monocytes within the infection-related inflammatory network, here we studied the in vitro and ex vivo functional behavior of monocytes from JAK2V617 F mutated MF patients after infectious stimulus and in the presence or absence of RUX inhibition.
Materials and methods
Patients characteristics and cells isolation
EDTA-anticoagulated peripheral blood (PB) was collected from JAK2V617F mutated MF patients (patients) before (n = 30) and after 6 months of RUX therapy (n = 20) and from age/sex-matched healthy donors (n = 30). Patients characteristics are shown in Supplementary Table 1. Patients were at diagnosis or untreated for at least 3 months. The diagnosis of MF was made according to the WHO 2016 criteria. No patients were previously treated with Interferon-α. This study was approved by the local Ethical Committee and was performed according to the declaration of Helsinki. Patients/controls signed informed consent.
PB mononuclear cells (PBMCs) were separated by Lympholyte 1.077 g/cm3 gradient (Cedarlane; CL5020) stratification, cryopreserved in liquid nitrogen and then thawed at 37°C before testing. Of note, to minimize the influence of freezing/thawing, only thawed PBMCs with a survival rate > 80% were used.
CD14+ monocytes isolation
Circulating CD14+ cells were immunomagnetically isolated from thawed PBMCs of MF patients (at baseline and after 6 months of RUX therapy) and healthy donors using a commercially available kit (CD14 Isolation kit, human; Miltenyi Biotec, Bologna, Italy) and purity (95 ± 3%) was routinely checked by flow cytometry.
Phenotype of monocytes
Total monocytes and subsets (Classical- CD14++/CD16−, Intermediate- CD14+/CD16+, Non-Classical-monocytes CD14+/CD16++) were phenotypically characterized by flow cytometry (chemokine receptors: C-C chemokine receptor type 2 (CCR2), C-X3-C motif chemokine receptor 1 (CX3CR1), C-C chemokine receptor type 5 (CCR5); cytokine receptors: Tumor Necrosis Factor (TNF)-αR1, TNF-αR2, Interleukin (IL)10R, IL1βR, IL6R); activation markers: CD86, HLA-DR, CD40, CD163) in PBMCs from MF patients, at baseline and after 6 months of RUX therapy, and healthy donors (Gating Strategy is shown in Figure 1a; monoclonal antibodies panels for chemokine receptors, cytokine receptors, and activation markers evaluation are shown in Supplementary Table 2). Results were expressed as percentage of positive cells and/or Mean Fluorescence Intensity (MFI). All flow cytometry analyzes were performed on a FACSs Canto II (BD Biosciences) and analyzed using FlowJo (FlowJo, LLC).
Figure 1.

Circulating monocyte subsets before and after RUX therapy. Gating strategy for identification of human monocyte subsets (Classical-, Intermediate- and Non-Classical-monocytes), including (from left to right) live cells detection, doublet exclusion, size discrimination, selection of HLA-DR+ cells, exclusion of double-negative CD14−CD16− cells (selection of Total monocytes) and monocytes separation according to expression of CD14 and CD16 (a). Frequency of circulating monocyte subsets (Classical (b), Intermediate (c) and Non-Classical (d)-monocytes, according to CD14/CD16 expression) in PBMCs of healthy donors (HD; n = 30) and MF patients at baseline (MF baseline T0; n = 30) and after 6 months of RUX therapy (MF RUX (T6 m); n = 20). Bars represent the mean percentage of monocyte subsets in total CD14+/HLA-DR+ monocytes ± S.E.M. (Kruskal-Wallis test and Friedman test, as appropriate; *p < .05; Mo = monocytes).
Functional assay of monocytes in the presence/absence of RUX therapy
To investigate the effects of RUX therapy, PBMCs from patients, at baseline and after 6 months of RUX therapy, and healthy donors were stimulated with lipopolysaccharides (LPS, 100 ng/ml) in the presence of 5 ng/ml Brefeldin A (Sigma-Aldrich®) for 4 hours.44 After in vitro stimulation, PBMCs were incubated with anti-CD14 and anti-CD16 monoclonal antibodies for 15 minutes at room temperature. Cells were then fixed/permeabilized according to standard procedures (IntraPrep Permeabilizaton kit, Beckman Coulter® Life Sciences) and IL1β, IL6, IL10 and TNF-α producing monocytes were measured by intracellular flow cytometry analysis (Gating Strategy is shown in Figure 1a; monoclonal antibodies panel for the evaluation of intracellular cytokine production is shown in Supplementary Table 2). Results were expressed as percentage of positive cells. In parallel experiments, immunomagnetically isolated monocytes (106 per well) from patients/healthy donors were stimulated with LPS (100 ng/ml) on the same day. After 4/24 hours, cell viability was tested by apoptosis assay (Annexin-V-FLUOS Staining Kit; Roche) and culture supernatants were obtained after centrifugation at 400 g, collected and stored with 1% dimethylsulfoxide (DMSO) at −80°C for further analysis.
Functional assay of monocytes in the presence/absence of in vitro RUX treatment
To investigate the effects of RUX in vitro, circulating monocytes from MF patients and healthy donors have been immunomagnetically isolated. RUX was dissolved in DMSO and added to the culture media in concentrations varying from 0.2 to 10 µM, which corresponds to serum levels achieved in treated patients. In each case, equal amounts of DMSO were added as a control (vehicle). We analyzed by flow cytometry the effect of titrating doses of RUX (0.2–10 µM) (1) on the monocyte-derived pro-anti/inflammatory cytokines production (TNF-α, IL6, IL10, and IL1β) after 24 hours of incubation in the presence or absence of LPS (100 ng/mL); (2) on the surface-bound inflammatory cytokines expression of monocyte-derived-EVs in the supernatants of cultures in the presence or absence of LPS stimulation (24 hours).
Cytokine concentration of the monocyte cultures supernatants
Supernatants from monocytes cultures in the presence/absence of LPS stimulation (4/24 hours) and before/after 6 months of RUX therapy or with/without in vitro RUX treatment were harvested (centrifugation at 400 g) and frozen at −80°C with 1% of DMSO until assays were performed. Cytokine concentration was determined by commercially available MACSPlex Cytokine 12 Kit (Miltenyi Biotec, Bologna, Italy) for human IL6, TNF-α and IL10 according to the manufacturer’s instruction. IL1β was determined by Human IL1 beta/IL1F2 Quantikine ELISA Kit (R&D Systems, Milan, Italy).
Flow cytometric analysis of the inflammatory cytokine’s expression on the surface of EVs from monocyte culture supernatants
Cytofluorimetric analysis and staining method were developed to study the expression of surface-attached pro/anti-inflammatory cytokines in EVs from monocyte culture supernatants in the presence/absence of LPS stimulation and before/after 6 months of RUX therapy or with/without in vitro RUX treatment. Supernatants (50 μl) were incubated with anti-CD14 (5 μl), IL1β (5 μl), IL6 (5 μl), TNF-α (5 μl) and IL10 (5 μl) monoclonal antibody for 30 minutes at room temperature and then EVs were analyzed by flow cytometry (Navios, Beckman Coulter, Milan, Italy) (Monoclonal antibodies panel for Cytokine-bound EVs evaluation is shown in Supplementary Table 2). To detect EVs the instrument was calibrated with MegaMix Beads (Stagò, Marseille, France). Fluorescence gated polystyrene beads of different sizes were used to determine the gates identifying big (500–900 nm), small (200–300 nm) and nano (100–160 nm) EVs. The Violet Side Scatter laser (VSSC) is used as a trigger signal to discriminate the noise. Our analysis was focused on big EVs which were identified by using size and ability to bind specific monoclonal antibodies. Matched isotype controls were used to select the cutoff. Using the defined gate for big EVs, all events positive for markers staining were recorded. The expression of the pro/anti-inflammatory cytokines was expressed as percentage of positive EVs.
Statistical analysis
Statistical analyses were performed with GraphPad (GraphPad Software Inc., La Jolla, USA). Data are expressed as mean ± SEM. P values were considered significant when ≤ 0.05 (2-tailed). The differences between the groups were analyzed with Mann Whitney, Kruskal Wallis, one-way, and two-way ANOVA tests as appropriate.
Results
The profile of circulating MF monocyte subsets is altered and not affected by RUX therapy
We firstly analyzed the proportion of total/subsets monocytes. Based on the expression of CD14/CD16, Classical-CD14++/CD16−, Intermediate-CD14+/CD16+ and Non-Classical-CD14+/CD16++ monocytes were investigated before and after RUX therapy (Figure 1). At baseline, only the mean percentages of Intermediate/Non-Classical MF monocytes (Figure 1c and d) were significantly increased/reduced as compared with the normal counterparts (p < .05). Of note, no significant differences were observed between patients and healthy donors when total monocytes were analyzed (data not shown). RUX therapy did not significantly modify the monocyte subsets profile. These results demonstrate that the profile of circulating monocyte subsets is altered in MF at baseline and after RUX therapy.
Chemokine receptors repertoire of MF monocytes is altered but normalized by RUX therapy
To evaluate the differentiation program and migration ability, we then studied the expression of crucial chemokine receptors on total circulating monocytes and subsets from patients and healthy donors.
At baseline, only the percentage of CCR2+ total MF monocytes was significantly increased as compared with the normal counterpart (Figure 2a; p < .05). This is mainly due to a significant increased percentage of CCR2+ Intermediate and Non-Classical monocytes (Figure 2a; p < .01 and p < .001, respectively). Additionally, we detected a reduced percentage of CX3CR1+ Non-Classical monocytes only (Figure 2b; p < .01). No differences between patients and healthy donors were observed when the percentage of CCR5+ monocytes was analyzed (Figure 2c). Taking into consideration the MFI, CCR2 expression was significantly increased in total monocytes and in both Intermediate and Non-Classical monocytes (Supplementary Figure 1a; p < .05). Conversely, CX3CR1 expression was reduced in Intermediate and Non-Classical monocytes only (Supplementary Figure 1b; p < .05, p < .01, respectively). CCR5 expression was reduced in all three monocyte subpopulations (Supplementary Figure 1c; p < .05).
Figure 2.

Chemokine receptors expression of monocytes before and after RUX therapy. Percentages of CCR2+ (a), CX3CR1+ (b) and CCR5+ (c) cells in total monocytes and of the three subsets (Classical-, Intermediate- and Non-Classical-monocytes) from PBMCs of healthy donors (HD; n = 30) and MF patients at baseline (MF baseline T0; n = 30) and after 6 months of RUX therapy (MF RUX (T6 m); n = 20) were analyzed. Bars represent the mean percentage of chemokine receptors-positive monocytes ± S.E.M. (Kruskal-Wallis test and Friedman test, as appropriate; *p < .05; **p < .01; ***p < .001; Mo = monocytes). Below, representative histograms of CCR2, CX3CR1 and CCR5 expression of the monocyte subsets of one MF patient (MF baseline (T0)) and one healthy donors (HD) are shown. Isotype control (black), Classical- (dark blue), intermediate- (blue) and Non-Classical-monocytes (light blue) are shown superimposed on each other.
Interestingly, RUX therapy normalized the percentage of CCR2+ Intermediate and Non-Classical monocytes (Figure 2a; p < .05 and p < .01, respectively) and the percentage of CX3CR1+ Non-Classical monocytes only (Figure 2b; p < .05).
The analysis of the MFI revealed that RUX therapy restored CX3CR1 expression in both Intermediate and Non-Classical monocytes (Supplementary Figure 1b; p < .05) and CCR5 expression in total monocytes as well as in all monocyte subsets (Supplementary Figure 1c; p < .05).
Collectively, these data demonstrated that in MF circulating monocytes show an altered phenotype. Interestingly, RUX therapy normalizes the expression of crucial chemokine receptors.
MF monocytes subsets show an abnormal cytokine receptors expression and RUX therapy partially modifies their repertoire
To evaluate how circulating monocytes sense the inflammatory microenvironment, we tested the expression of crucial pro/anti-inflammatory cytokine receptors on patients and healthy donors.
Figure 3 showed a decreased percentage of TNF-αR1+ cells (A; p < .001) together with an increased percentage of TNF-αR2+ and IL10-R+ total MF monocytes (B and C; p < .01) as compared with the normal counterpart. The percentages of TNF-αR1+ cells were significantly reduced in all the monocyte subsets (Figure 3a; p < .001). Conversely, the percentages of IL10-R+ cells were significantly increased in all subsets (Figure 3b; p < .01 (Classical monocytes), p < .05 (Intermediate and Non-Classical monocytes)). No differences were observed regarding the percentage of TNF-αR2+ cells in monocyte subsets.
Figure 3.

Inflammatory cytokine receptors expression of monocytes before and after RUX therapy. Percentages of TNF-α-R1+ (a), TNF-α-R2+ (b), IL10-R+ (c) cells in total monocytes (identified by CD14 and HLA-DR expression) and of the Classical-, Intermediate- and Non-Classical-monocytes from PBMCs of healthy donors (HD; n = 30) and MF patients (n = 30) at baseline (MF baseline T0) and after 6 months of RUX therapy (MF RUX (T6 m); n = 20) is shown. Bars represent the mean percentage of the cytokine receptors-positive monocytes ± S.E.M. (Kruskal-Wallis test and Friedman test, as appropriate; *p < .05; **p < .01, ***p < .001; Mo = monocytes).
RUX therapy normalized the percentages of IL10-R+ Intermediate and Non-Classical monocytes (Figure 3c; p < .05) while the percentage of TNF-αR1+ and TNF-αR2+ monocytes was not affected.
Of note, the percentages of IL1β-R+ and IL6-R+ cells in total monocytes and monocyte subsets of patients were not significantly different from the normal counterparts (data not shown). However, as shown in Supplementary Figure 2, the analysis of the MFI revealed that total monocytes of MF patients showed a significantly increased expression of IL1β-R and IL6-R (p < .05) as compared with the normal counterparts. When monocytes subsets were analyzed, the IL1β-R MFI (Intermediate and Non-Classical monocytes; p < .05 and p < .001, respectively) and IL6-R MFI (Classical and Non-Classical monocytes; p < .05) were significantly increased as compared with the normal counterparts. RUX therapy reduced the IL1β-R (MFI; total and Intermediate monocytes; p < .05) and IL6-R expression (MFI; total and Classical monocytes (p < .05) (Supplementary Figure 2) but the percentages of IL1β-R+ and IL6-R+ cells were not affected (data not shown).
To further evaluate the phenotype of MF monocytes before and after RUX therapy, we analyzed the expression of HLA-DR, CD86, CD40, and CD163 (both percentages and MFI). No significant differences were observed between healthy donors and MF monocytes and before and after RUX therapy (both percentages and MFI; data not shown).
These results demonstrate that, due to altered expression of crucial inflammatory cytokine receptors, MF monocytes may abnormally sense the inflammatory microenvironment. RUX therapy partially fails in restoring their cytokine receptors profile.
The ability of MF monocytes to produce cytokines under LPS stimulation is reduced and RUX therapy normalizes this pattern
Cytokine production represents the main mechanism used by monocytes to respond to the external stimuli. To investigate the ability of MF monocytes to produce inflammatory cytokines we ex vivo analyzed the percentages of IL1β, TNF-α, IL10, and IL6 producing monocytes with and without LPS stimulation (4 hours) in PBMC from healthy donors and patients before and after 6 months of RUX therapy.
As shown in Figure 4, in the absence of LPS stimulation the mean percentages of IL1β+ (A), TNF-α+ (B), and IL6+ (C) cells were very low and no differences were observed between healthy donors and patients. LPS stimulation failed to increase the percentages of IL1β+, TNF-α+ and IL6+ monocytes in MF, which were therefore significantly reduced as compared with the normal counterparts (Figure 4a–d; p < .001). As IL10 is produced by monocytes only after 7/8 hours of stimulation with LPS,45 no IL10+ cells were detected after 4 hours of incubation in the presence/absence of LPS stimulation in MF and healthy donors monocytes (data not shown). Interestingly, RUX therapy restored the quote of IL1β+, TNF-α+, and IL6+ monocytes after LPS stimulation.
Figure 4.

Intracellular monocyte production of inflammatory cytokines before and after RUX therapy. Intracellular production of pro-inflammatory cytokines in total monocytes from PBMC of healthy donors (HD; n = 30) and MF (n = 30) PBMCs (at baseline (MF baseline T0) and after 6 months of RUX therapy (MF RUX (T6 m); n = 20) with or without LPS stimulation (4 hours) is shown. Above, representative histograms identify the IL1β (a), TNF-α (b) and IL6 (c) positive total monocyte of one healthy donors (HD) and one MF patient (at baseline (T0) and after 6 months of RUX therapy (T6 m)) in the presence or absence of LPS stimulation. Unstimulated (blue) and LPS-stimulated (light blue) monocytes are shown superimposed on each other. Below, bars represent the mean percentage of monocytes producing IL1β (A), TNF-α (B) and IL6 (C) ± S.E.M. (Two-way ANOVA test; ***p < .001).
These data demonstrate an impaired ability of MF monocytes to produce inflammatory cytokines in response to an infectious stimulus and show that RUX therapy restores normal cytokine production ability.
Monocytes from MF patients show reduced ability to secrete selected cytokines and in vitro RUX treatment inhibits their secretion
In parallel experiments, we tested the concentrations of selected pro/anti-inflammatory cytokines produced in the supernatants by isolated MF and healthy donor monocytes upon LPS stimulation and before and after RUX therapy or in vitro RUX treatment. As shown in Figure 5, the concentrations of IL1β (Figure 5a), TNF-α (Figure 5b), IL6 (Figure 5c) and IL10 (Figure 5d) were significantly increased after LPS stimulation in the supernatants of healthy donor monocytes. Conversely, upon LPS stimulation, no significant differences were observed between stimulated and unstimulated MF monocytes. As a consequence, at baseline the concentrations of IL1β (Figure 5a; 4 and 24 hours; p < .001), TNF-α (B; 4 hours; p < .01), IL6 (C; 4 and 24 hours; p < .01) and IL10 (D; 24 hours; p < .001) were significantly reduced in the supernatants of MF monocytes as compared with the normal counterparts. Of note, RUX therapy did not significantly modify the concentrations of the four selected cytokines in the supernatants of MF monocytes (Figure 5a–d).
Figure 5.

Monocyte secretion of free inflammatory cytokines before and after RUX therapy or in vitro RUX treatment. Concentrations of crucial cytokines (IL1β, TNF-α, IL6, IL10) in the supernatants of immunomagnetically isolated monocytes from healthy donors (HD; n = 20) and MF patients at baseline ((MF baseline T0; n = 20) and after 6 months of RUX therapy (MF RUX (T6 m); n = 20) in vitro cultured for 4/24 hours in the presence or absence of LPS stimulation. Bars represent the mean concentration of IL1β (a), TNF-α (b), IL6 (c) and IL10 (d) ± S.E.M (Two-way ANOVA test; *p < .05, **p < .01, ***p < .001). “-LPS” represents the concentrations of cytokines after 4 or 24 hours without LPS. Panels (e-h) show the concentrations of crucial cytokines (TNF-α and IL10) in the supernatants of immunomagnetically isolated monocytes from healthy donors (HD; n = 20; panels E, G) and MF patients (n = 20; panels F, H) in vitro cultured for 24 hours in the presence or absence of LPS stimulation and titrating doses of RUX (0.2–10 µM). Bars represent the mean concentration of TNF-α and IL10 ± S.E.M (one-way ANOVA test; *p < .05, **p < .01, ***p < .001).
We also functionally analyzed the in vitro ability of isolated MF (at baseline) and healthy donor monocytes to release cytokines upon LPS stimulation in the presence of titrating doses of RUX (0.2, 0.5, 1, 5, 10 µM). As shown in Figure 5, low dose RUX treatment (0.2, 0.5, 1 µM) significantly increased the concentration of TNF-α in the supernatants from healthy donors (Figure 5e) and MF (figure 5f) monocytes (p < .05, respectively). Conversely, high dose RUX (10 µM) significantly decreased the concentration of TNF-α in the supernatants from both healthy donors and MF monocytes (p < .05). Interestingly, RUX reduced the IL10 concentration in the supernatants from healthy donors (Figure 5g) and MF (Figure 5h) monocytes in a dose-dependent way. By contrast, IL1β and IL6 were not affected by RUX (data not shown).
Of note, when we tested the viability of MF and healthy donor monocytes after 4/24 hours in the presence/absence of LPS stimulation, no significant differences were observed between MF and healthy donors monocytes. Additionally, RUX therapy/in vitro RUX treatment did not significantly influence the viability of MF and healthy donor monocytes (data not shown).
Collectively, these results indicate a reduced ability of MF monocytes to secrete inflammatory-related signals in response to an infectious stimulus. In vitro RUX treatment inhibits the cytokine secretion of both normal and MF monocytes.
The expression of cytokines on the surface of MF monocyte-derived EVs is reduced and RUX normalizes their profile
To investigate whether cytokine-bound EVs are produced by monocytes and whether RUX may affect their profile, we analyzed the presence of selected inflammatory cytokines on the surface of EVs produced by healthy donors and MF monocytes (at baseline and after in vitro RUX treatment/RUX therapy) in the supernatants after in vitro culture with/without LPS (Supplementary Figure 3). As shown in Figure 6(a–d), in the absence of LPS stimulation, no significant differences were observed between patients and healthy donors or before and after RUX therapy. When monocytes were stimulated with LPS, the percentages of IL1β, TNF-α, IL6, and IL10 positive EVs from MF patients were significantly decreased as compared with the healthy donor counterpart (Figure 6a–d; p < .001). After in vitro RUX treatment and LPS stimulation, the percentages of IL1β, TNF-α, IL6, and IL10 positive EVs in the supernatants of isolated monocytes from MF patients at baseline significantly increased (Figure 7a–d). Similarly, albeit non-statistically significant, RUX treated patients showed the same trend toward increased percentages of cytokine-positive EVs (Figure 6a–d).
Figure 6.

Cytokine-positive EVs secretion of monocytes before and after RUX therapy. Gating strategy, based on beads calibration and size, for identification of monocyte-derived EVs (BIG EVs: 500–900 nm) by flow cytometry and representative dot plot of the IL1β, TNF-α, IL6 and IL10 positive EVs in the supernatants of monocytes from one healthy donors (HD) and one MF patient (at baseline (MF T0) and after 6 months of RUX therapy (MF T6 m) after LPS stimulation are shown (a). Histograms show the expression of surface-bound IL1β (b), TNF-α (c), IL6 (d) and IL10 (e) of EVs in the supernatants of immunomagnetically isolated monocytes from healthy donors (HD; n = 20) and MF patients at baseline (MF baseline T0; n = 20) and after 6 months of RUX therapy (MF RUX (T6 m); n = 20), in the presence/absence of LPS stimulation (24 hours). Bars represent the mean percentage of monocyte-derived cytokine-positive EVs ± S.E.M. (Two-way ANOVA test; ***p < .001).
Figure 7.
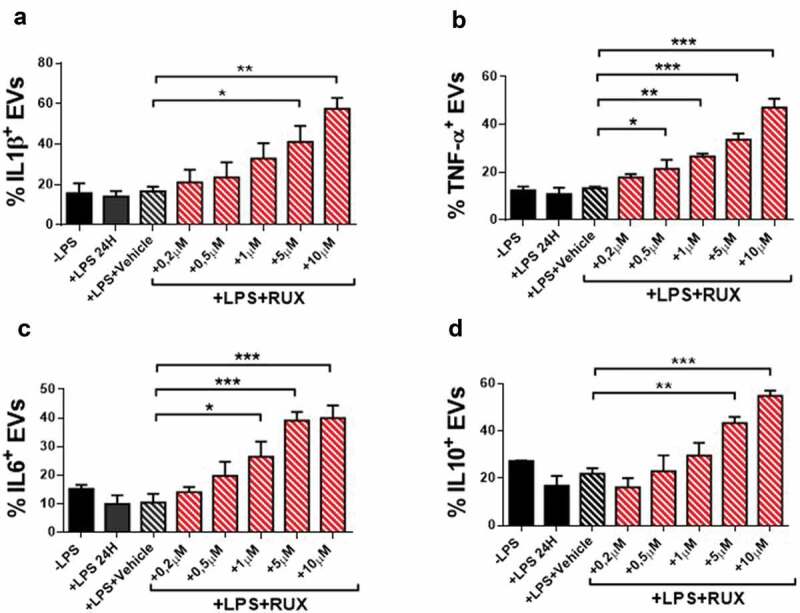
Cytokine-positive EVs secretion of monocytes in the presence or absence of in vitro RUX treatment. Surface-bound IL1β (a), TNF-α (b), IL6 (c) and IL10 (d)-positive EVs in the supernatants of immunomagnetically isolated monocytes from MF patients at baseline (n = 10) in the presence or the absence of LPS stimulation (24 hours) and titrating doses of in vitro RUX treatment or vehicle (DMSO) is shown. Bars represent the mean percentage of monocyte-derived cytokine-positive EVs ± S.E.M. (One-way ANOVA test; *p < .05).
Collectively, these results demonstrate that the EVs-related inflammatory signals of MF monocytes are impaired and that RUX has the potential to normalize the EVs-bound cytokines repertoire.
Discussion
Monocytes are key components of the innate immune system and their function is principally based on cytokine production. CCR2, CX3CR1, and CCR5 are the main chemokine receptors involved in their recruitment in circulation and inflamed tissues. They are differentially expressed on the 3 monocyte subpopulations. CCR2, which plays a role in the mobilization of monocytes from bone marrow to peripheral blood, is primarily expressed by Classical monocytes and is progressively downregulated in the Intermediate and Non-Classical monocytes. Conversely, CX3CR1, which favors migration of circulating monocytes from peripheral blood toward inflamed/damaged tissues, is upregulated in Intermediate and Non-Classical monocytes.32 CCR5, also involved in the recruitment of monocytes in inflamed tissues, is expressed by all three subsets but mainly by Intermediate monocytes.31,46
Here we demonstrated that in JAK2V617 F mutated MF patients the monocyte differentiation program is dysregulated as referred to chemokine/cytokine receptors subsets profile. MF patients show an altered chemokine receptors profile due to a failed CCR2 downregulation and CX3CR1 upregulation. This suggests an impaired capacity of monocytes to migrate from peripheral blood into damaged tissues, as confirmed by a higher percentage of Intermediate monocytes in peripheral blood. Of note, RUX therapy normalized monocyte phenotype, suggesting that RUX-driven JAK1/2 inhibition, reducing plasma inflammatory cytokines, may indirectly support monocyte differentiation and homing to inflamed/damaged tissues through CX3CR1 and CCR5 upregulation and CCR2 downregulation.
MF patients also showed a higher percentage of circulating IL10-R+ and TNF-αR2+ monocytes while the percentage of TNF-αR1+ monocytes was reduced. It is interesting to note that RUX therapy reduced the percentages of IL10-R+ monocytes but did not modify the percentages of TNF-αR1/2+ monocytes. Binding of IL10 to its receptor leads to JAK1-STAT3 pathway activation.47,48 Therefore, our results suggest that JAK/STAT pathway, along with a role in the IL10-R signaling, may be also involved in the IL10-R expression. Moreover, since RUX reduces their expression, it is likely that also the IL1β-R and IL6-R exposition is Indirectly linked by the JAK/STAT pathway.
We have also found that the cytokine production of unstimulated MF circulating monocytes was superimposable to the normal counterparts, suggesting that steady-state monocytes of MF are not hyperactivated. Upon LPS stimulation, the intracellular production and secretion of free pro/anti-inflammatory cytokines by monocytes of JAK2V617 F mutated MF patients were severely impaired.
Our findings are consistent with a prior study demonstrating that isolated circulating monocytes from MF fail to respond to LPS and the levels of pro-inflammatory cytokines such as IL23, IL12, and TNF-α remain very low or fail to increase in the case of IL8.43 Recently, Lai YH et al.49 showed that monocytes from MPN patients have a defective negative regulation in the Toll-like receptor (TLR) signaling pathway leading to unrestrained production of TNF-α after activation. Specifically, they demonstrated that monocytes do not respond to IL10 which negatively regulates TLR-induced TNF-α production. This discrepancy might be related to the fact that they studied, in addition to MF, also Essential Thrombocythemia and Polycythemia Vera patients. Nevertheless, according to our study, they also demonstrated that, upon LPS stimulation, MPN patients appeared to be less responsive to inflammatory stimulus initially, as the percentage of TNF-α positive monocytes were significantly decreased. Additionally, Fisher DAC et al.8 showed that 14 out of 15 cytokines, measured by mass cytometry, were found to be overproduced in MF patients after PMA/ionomycin stimulation, with the principal cellular source for most cytokines being monocytes. Keeping in mind our results with LPS stimulation, we can draw the conclusion that circulating MF monocytes may have the potential to produce/secrete inflammatory cytokines; however, this varies according to the nature of the stimulus.
The reduced ability of MF monocytes to produce inflammatory cytokines in response to infectious stimuli (LPS) might have several explanations. First, it may be due, at least in part, to the reduced frequency of circulating TNF-αR1+ monocytes; this receptor mediates the autocrine capacity of the LPS-induced TNF to sustain the upregulation of pro-inflammatory cytokines. Indeed, a prior study has shown that the binding of TNF-α leads to a sustained upregulation of pro-inflammatory cytokines via TNF-αR1, and the binding through TNF-αR2 upregulates the anti-inflammatory cytokine (IL10), in the absence of any upregulation of pro-inflammatory cytokines.50 Since circulating inflammatory cytokines are elevated in MF, this may explain why, although MF patients show high levels of TNF-αR2+ monocytes, they do not produce the anti-inflammatory IL10 in the presence of an infectious stimulus. In addition, IL10-R overexpression in MF monocytes might play a crucial role as well. Prior experimental evidence demonstrated that IL10-R-targeted deletion in monocytes leads to enhanced susceptibility to an LPS-induced production of cytokines.51 Indeed, in our study, RUX therapy decreases IL10-R expression in MF monocytes and restores their intracellular cytokine production under LPS stimulation. Of note, this mechanism could be the basis for the development of new therapeutic approaches targeting IL10-R (anti-IL10-R monoclonal antibodies). Finally, we cannot also rule out the hypothesis that, in MF, monocytes might be exhausted by the continuous stimulus of the inflammatory microenvironment.
Moreover, we provided also evidence that, upon LPS stimulus, the frequency of monocyte-derived EVs expressing surface pro-/anti-inflammatory cytokines is strongly reduced in MF patients.
Interestingly, RUX therapy promoted the LPS-driven intracellular cytokines production of monocytes from MF patients but failed to normalize their ability to secrete free cytokines. Importantly, both in vitro RUX treatment and RUX therapy increased the proportion of cytokines-associated EVs released by MF monocytes in response to LPS stimulus. However, since the LPS-driven cytokines production of monocytes is mediated by the MyD88 signaling pathway,52 we can hypothesize that JAK1/2 inhibition might indirectly favor the MyD88 pathway activation and cytokines production.53 It is also likely that the decrease of plasma inflammatory cytokines levels after RUX therapy may promote MF monocyte function. Thus, RUX-linked JAK1/2 inhibition restores intracellular cytokine production and promotes EV-driven inflammatory signaling of MF monocytes rather than free cytokines secretion, suggesting that the JAK-STAT pathway might be involved in the mechanisms of inflammatory cytokines secretion in monocytes. It is also likely that upon JAK1/2 inhibition, monocytes from MF patients preferentially secrete the EV-linked inflammatory cytokines instead of secreting free cytokines, being the former more advantageous. Indeed, as suggested by Fitzgerald et al., EV-entrapment/association can be a mechanism to protect cytokines from environmental degradation. In addition, EVs can facilitate delivery and targeting of cytokines to distant target cells by binding EV-surface cytokines to cells expressing specific cytokine receptors.33 Whether this is due to the fact that JAK1/2 inhibition promotes the expression of genes which are involved in vesiculation of inflammatory cytokines or to the fact that off-target JAK1/2 inhibition includes genes important for the secretion of free inflammatory cytokines remains a matter of speculation.
Although monocytes are the main cellular component in inflamed tissues, they are not the only cells responsible for the clearance of the infections. In this scenario, different cells of the innate and adaptive immunity are involved. Here we have shown that MF monocytes exhibit phenotypic and functional alterations that may contribute to the increased susceptibility to infections in MF. Furthermore, in our previous study,21 we demonstrated that MF monocytes failed to fully differentiate in DCs showing a macrophage-like phenotype. This finding was supported by a reduced amount of circulating myeloid and plasmacytoid DCs. Our data on the reduced ability of MF monocytes to differentiate were consistent with those of Wang et al. showing an increased level of myeloid-derived suppressor cells in MPN and confirming that the increased release of immature myeloid cells leads to the reduction of differentiated myeloid subsets like DCs.19
This study highlights that in MF, RUX-driven JAK1/2 inhibition leads to a partial restoration of the immune function of monocytes, enhancing their ability to produce/release cytokines in response to an infectious stimulus. However, other key players in the setting of infection clearance are negatively affected by JAK1/2 inhibition. DCs differentiation can be further exacerbated during RUX therapy as this drug affects normal monocytes differentiation and DCs migration both in vitro and in vivo.25,54 The inhibition of IL-4 signaling (key factor for DC differentiation) by RUX is likely to drive the reduced differentiation capacity of monocytes into DCs. In line with this, mice with induced deletion of JAK2 have impaired DC but not macrophage compartment.54 Also, Stalder R et al., evaluating the effects of tofacitinib (JAK1 inhibitor) on monocyte-derived DCs, observed a reduced differentiation of normal monocytes into immature DCs.55 Macrophages have the potential to activate T cells but not at the same extent as DCs. Altogether, these findings suggest that the failure of T cell priming by DCs following RUX therapy might be one of the main risk factors responsible for an increased infection rate. T-helper cells are essential in the clearance of the infections and their differentiation and mechanism of action are strictly linked to the JAK/STAT pathway. In line with this, RUX therapy in MPN has been linked with inhibition of T-cell function, cytokines secretion and with reduced number of circulating regulatory T cells.22,24,26 Additionally, JAK inhibition impairs NK (Natural killer) cell function in MPN. Specifically, RUX-treated patients show a reduction in NK cell numbers due to their reduced maturation capacity. Further, in vitro and in vivo results, show a reduction in cytokine-induced NK cell activation and a reduced killing activity associated with an impaired capacity to form lytic synapses with NK target cells.23
On these bases, the RUX-mediated effect on DC, T, and NK cells may be the cause of the increased infection rates and possible long-term side effects associated with the treatment with RUX.
In conclusion, here we demonstrated that circulating monocytes from JAK2V617F mutated MF patients are dysregulated and show a reduced in vitro ability to produce/secrete inflammatory cytokines in response to an infectious stimulus. These findings suggest that this monocyte dysfunction may favor altered immune surveillance against infectious complications. Importantly, RUX ameliorates their cytokines production and promotes the EVs-based inflammatory cytokine signaling. Therefore, infectious events in MF occurring following exposure to RUX therapy may not be due to monocyte compartment dysregulation. Our findings may have therapeutic implications because they contribute to better interpreting the immune vulnerability of MF and off-target efficacy of JAK1/2 inhibition to envisaging strategies aimed at facilitating the immunosurveillance in MF. Specifically, the IL10-R pathway might be a novel therapeutic target to improve monocyte function and infection-driven immune response.
Supplementary Material
Acknowledgments
We greatly thank Novartis Pharma AG for providing the active drug substance for the in vitro experiments. This work was funded by AIL Bologna and RFO 2017/2018 (L.C.). D.F. was supported by AIRC (fellowship 20930-2018) and by SIE-Società Italiana di Ematologia- e Associazione “Amici di Beat Leukemia Dr. Alessandro Cevenini ONLUS”.
Funding Statement
This work was supported by AIL Bologna; SIE-Società Italiana di Ematologia e Associazione “Amici di Beat Leukemia Dr. Alessandro Cevenini ONLUS”; AIRC [fellowship 20930-2018]; University of Bologna [RFO 2017/2018].
Author contributions
M.B., F.R., M.R., D.F., and L.C. contributed to study design, statistical analysis, and data interpretation. F.P., G.A., and N.V. managed patients and collected blood samples. M.B. and F.R. performed monocytes, extracellular vesicles, cytokine analysis, and data interpretation. E.O. performed molecular analysis. M.B. and D.B. were involved in statistical analysis. M.B., M.R., D.F., F.P. and L.C. wrote or contributed to the writing of the manuscript. P.L.T. and M.C. reviewed and corrected the manuscripts. All Authors read and contributed to the final version of the manuscript.
Disclosure of potential conflicts of interest
The authors declare that they have no competing interests.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Tefferi A, Vainchenker W.. Myeloproliferative neoplasms: molecular pathophysiology, essential clinical understanding, and treatment strategies. J Clin Oncol. 2011. February 10;29(5):573–14. doi: 10.1200/JCO.2010.29.8711. [DOI] [PubMed] [Google Scholar]
- 2.Rampal R, Al-Shahrour F, Abdel-Wahab O, Patel JP, Brunel JP, Mermel CH, Bass AJ, Pretz J, Ahn J, Hricik T, et al. Integrated genomic analysis illustrates the central role of JAK-STAT pathway activation in myeloproliferative neoplasm pathogenesis. Blood. 2014. May 29;123(22):e123–33. doi: 10.1182/blood-2014-02-554634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tefferi A, Vaidya R, Caramazza D, Finke C, Lasho T, Pardanani A. Circulating interleukin (IL)-8, IL-2R, IL-12, and IL-15 levels are independently prognostic in primary myelofibrosis: a comprehensive cytokine profiling study. J Clin Oncol. 2011. April 1;29(10):1356–1363. doi: 10.1200/JCO.2010.32.9490. [DOI] [PubMed] [Google Scholar]
- 4.Hasselbalch HC. Perspectives on chronic inflammation in essential thrombocythemia, polycythemia vera, and myelofibrosis: is chronic inflammation a trigger and driver of clonal evolution and development of accelerated atherosclerosis and second cancer? Blood. 2012. April 5;119(14):3219–3225. doi: 10.1182/blood-2011-11-394775. [DOI] [PubMed] [Google Scholar]
- 5.Mendez Luque LF, Blackmon AL, Ramanathan G, Fleischman AG. Mo key role of inflammation in myeloproliferative neoplasms: instigator of disease initiation, progression. and symptoms. Curr Hematol Malig Rep. 2019. June;14(3):145–153. doi: 10.1007/s11899-019-00508-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sollazzo D, Forte D, Polverelli N, Romano M, Perricone M, Rossi L, Ottaviani E, Luatti S, Martinelli G, Vianelli N et al. Crucial factors of the inflammatory microenvironment (IL-1β/TNF-α/TIMP-1) promote the maintenance of the malignant hemopoietic clone of myelofibrosis: an in vitro study. Oncotarget. 2016. July 12;7(28):43974–43988. doi: 10.18632/oncotarget.9949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mondet J, Hussein K, Mossuz P. Circulating cytokine levels as markers of inflammation in philadelphia negative myeloproliferative neoplasms: diagnostic and prognostic interest. Mediators Inflamm. 2015;2015:670580. doi: 10.1155/2015/670580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fisher DAC, Miner CA, Engle EK, Hu H, Collins TB, Zhou A, Allen MJ, Malkova ON, Oh ST. Cytokine production in myelofibrosis exhibits differential responsiveness to JAK-STAT, MAP kinase, and NFκB signaling. Leukemia. 2019. February 4;33(8):1978–1995. doi: 10.1038/s41375-019-0379-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heaton WL, Senina AV, Pomicter AD, Salama ME, Clair PM, Yan D, Bell RN, Gililland JM, Prchal JT, O’Hare T et al. Autocrine Tnf signaling favors malignant cells in myelofibrosis in a Tnfr2-dependent fashion. Leukemia. 2018. November;32(11):2399–2411. doi: 10.1038/s41375-018-0131-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Polverelli N, Breccia M, Benevolo G, Martino B, Tieghi A, Latagliata R, Sabattini E, Riminucci M, Godio L, Catani L et al. Risk factors for infections in myelofibrosis: role of disease status and treatment. A multicenter study of 507 patients. Am J Hematol. 2017. January;92(1):37–41. doi: 10.1002/ajh.24572. [DOI] [PubMed] [Google Scholar]
- 11.Cervantes F, Dupriez B, Pereira A, Passamonti F, Reilly JT, Morra E, Vannucchi AM, Mesa RA, Demory J-L, Barosi G et al. New prognostic scoring system for primary myelofibrosis based on a study of the International working group for myelofibrosis research and treatment. Blood. 2009. March 26;113(13):2895–2901. doi: 10.1182/blood-2008-07-170449. [DOI] [PubMed] [Google Scholar]
- 12.Passamonti F, Cervantes F, Vannucchi AM, Morra E, Rumi E, Pereira A, Guglielmelli P, Pungolino E, Caramella M, Maffioli M et al. A dynamic prognostic model to predict survival in primary myelofibrosis: a study by the IWG-MRT (International Working Group for Myeloproliferative Neoplasms Research and Treatment). Blood. 2010. March 4;115(9):1703–1708. doi: 10.1182/blood-2009-09-245837. [DOI] [PubMed] [Google Scholar]
- 13.Hultcrantz M, Wilkes SR, Kristinsson SY, Andersson TM, Derolf ÅR, Eloranta S, Samuelsson J, Landgren O, Dickman PW, Lambert PC et al. Risk and cause of death in patients diagnosed with myeloproliferative neoplasms in Sweden between 1973 and 2005: a population-based study. J Clin Oncol. 2015. July 10;33(20):2288–2295. doi: 10.1200/JCO.2014.57.6652. [DOI] [PubMed] [Google Scholar]
- 14.Froom P, Aghai E, Kinarty A, Lahat N. Decreased natural killer (NK) activity in patients with myeloproliferative disorders. Cancer. 1989. September 1;64(5):1038–1040. doi:. [DOI] [PubMed] [Google Scholar]
- 15.Cervantes F, Hernández-Boluda JC, Villamor N, Serra A, Montserrat E. Assessment of peripheral blood lymphocyte subsets in idiopathic myelofibrosis. Eur J Haematol. 2000. August;65(2):104–108. doi: 10.1034/j.1600-0609.2000.90262.x. [DOI] [PubMed] [Google Scholar]
- 16.Briard D, Brouty-Boyé D, Giron-Michel J, Azzarone B, Jasmin C, Le Bousse-Kerdilès C. Impaired NK cell differentiation of blood-derived CD34+ progenitors from patients with myeloid metaplasia with myelofibrosis. Clin Immunol. 2003. March;106(3):201–212. doi: 10.1016/S1521-6616(02)00046-3. [DOI] [PubMed] [Google Scholar]
- 17.Barosi G. An immune dysregulation in MPN. Curr Hematol Malig Rep. 2014. December;9(4):331–339. doi: 10.1007/s11899-014-0227-0. [DOI] [PubMed] [Google Scholar]
- 18.Wang JC, Sindhu H, Chen C, Kundra A, Kafeel MI, Wong C, Lichter S. Immune derangements in patients with myelofibrosis: the role of Treg, Th17, and sIL2Rα. PLoS One. 2015. March 20;10(3):e0116723. doi: 10.1371/journal.pone.0116723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang JC, Kundra A, Andrei M, Baptiste S, Chen C, Wong C, Sindhu H. Myeloid-derived suppressor cells in patients with myeloproliferative neoplasm. Leuk Res. 2016. April;43:39–43. Epub 2016 Feb 16. doi: 10.1016/j.leukres.2016.02.004. [DOI] [PubMed] [Google Scholar]
- 20.Massa M, Campanelli R, Fois G, Villani L, Bonetti E, Catarsi P, Poletto V, Viarengo G, De Amici M, Rosti V et al. Reduced frequency of circulating CD4+CD25brightCD127lowFOXP3+ regulatory T cells in primary myelofibrosis. Blood. 2016. September 22;128(12):1660–1662. doi: 10.1182/blood-2016-03-704577. [DOI] [PubMed] [Google Scholar]
- 21.Romano M, Sollazzo D, Trabanelli S, Barone M, Polverelli N, Perricone M, Forte D, Luatti S, Cavo M, Vianelli N, et al. Mutations in JAK2 and Calreticulin genes are associated with specific alterations of the immune system in myelofibrosis. Oncoimmunology. 2017. July 5;6(10):e1345402. eCollection 2017. doi: 10.1080/2162402X.2017.1345402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Parampalli YS, Stübig T, Cornez I, Alchalby H, Schönberg K, Rudolph J, Triviai I, Wolschke C, Heine A, Brossart P et al. JAK1/2 inhibition impairs T cell function in vitro and in patients with myeloproliferative neoplasms. Br J Haematol. 2015. June;169(6):824–833. doi: 10.1111/bjh.13373. [DOI] [PubMed] [Google Scholar]
- 23.Schönberg K, Rudolph J, Vonnahme M, Parampalli YS, Cornez I, Hejazi M, Manser AR, Uhrberg M, Verbeek W, Koschmieder S et al. JAK Inhibition Impairs NK Cell Function in Myeloproliferative Neoplasms. Cancer Res. 2015. June 1;75(11):2187–2199. doi: 10.1158/0008-5472.CAN-14-3198. [DOI] [PubMed] [Google Scholar]
- 24.Keohane C, Kordasti S, Seidl T, Perez Abellan P, Thomas NSB, Harrison CN, McLornan DP, Mufti GJ. JAK inhibition induces silencing of T Helper cytokine secretion and a profound reduction in T regulatory cells. Br J Haematol. 2015. October;171(1):60–73. doi: 10.1111/bjh.13519. [DOI] [PubMed] [Google Scholar]
- 25.Rudolph J, Heine A, Quast T, Kolanus W, Trebicka J, Brossart P, Wolf D. The JAK inhibitor ruxolitinib impairs dendritic cell migration via off-target inhibition of ROCK. Leukemia. 2016. October;30(10):2119–2123. doi: 10.1038/leu.2016.155. [DOI] [PubMed] [Google Scholar]
- 26.Massa M, Rosti V, Campanelli R, Fois G, Barosi G. Rapid and long-lasting decrease of T-regulatory cells in patients with myelofibrosis treated with ruxolitinib. Leukemia. 2014. February;28(2):449–451. doi: 10.1038/leu.2013.296. [DOI] [PubMed] [Google Scholar]
- 27.Schönberg K, Rudolph J, Yajnanarayana SP, Wolf D. Get a grip on immune cells by inhibiting JAKs. Oncoimmunology. 2015. August 12;5(2):e1071009. eCollection 2016 Feb. doi: 10.1080/2162402X.2015.1071009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tefferi A, Pardanani A. Myeloproliferative neoplasms: a contemporary review. JAMA Oncology. 2015;1:97–105. doi: 10.1001/jamaoncol.2015.89. [DOI] [PubMed] [Google Scholar]
- 29.Vainchenker W, Leroy E, Gilles L, Marty C, Plo I, Constantinescu SN. JAK inhibitors for the treatment of myeloproliferative neoplasms and other disorders. F1000Res. 2018. January 17;7:82. eCollection 2018. doi: 10.12688/f1000research.13167.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Narasimhan PB, Marcovecchio P, Hamers AAJ, Hedrick CC. Nonclassical monocytes in health and disease. Annu Rev Immunol. 2019. April 26;37:439–456. doi: 10.1146/annurev-immunol-042617-053119. [DOI] [PubMed] [Google Scholar]
- 31.Sampath P, Moideen K, Ranganathan UD, Bethunaickan R. Monocyte subsets: phenotypes and function in tuberculosis infection. Front Immunol. 2018;9:1726. doi: 10.3389/fimmu.2018.01726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wacleche VS, Tremblay CL, Routy JP, Ancuta P. The biology of monocytes and dendritic cells: contribution to HIV pathogenesis. Viruses. 2018. February;10(2):65. doi: 10.3390/v10020065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fitzgerald W, Freeman ML, Lederman MM, Vasilieva E, Romero R, Margolis L. A system of cytokines encapsulated in extracellular vesicles. Sci Rep. 2018. June 12;8(1):8973. doi: 10.1038/s41598-018-27190-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Butler JT, Abdelhamed S, Kurre P. Extracellular vesicles in the hematopoietic microenvironment. Haematologica. 2018;103:382–394. doi: 10.3324/haematol.2017.183335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Van Niel G, D’Angelo G, Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol. 2018. April;19(4):213–228. doi: 10.1038/nrm.2017.125. [DOI] [PubMed] [Google Scholar]
- 36.Robbins PD, Dorronsoro A, Booker CN. Regulation of chronic inflammatory and immune processes by extracellular vesicles. J Clin Invest. 2016. April 1;126(4):1173–1180. doi: 10.1172/JCI81131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Caivano A, Laurenzana I, De Luca L, La Rocca F, Simeon V, Trino S, D'Auria F, Traficante A, Maietti M, Izzoet T, et al. High serum levels of extracellular vesicles expressing malignancy-related markers are released in patients with various types of hematological neoplastic disorders. Tumor Biology. 2015;36:9739–9752. doi: 10.1007/s13277-015-3741-3. [DOI] [PubMed] [Google Scholar]
- 38.Zhang W, Qi J, Zhao S, Shen W, Dai L, Han W, Huang M, Wang Z, Ruan C, Wu D, et al. Clinical significance of circulating microparticles in Ph- Myeloproliferative Neoplasms. Oncol Lett. 2017;14(2):2531–2536. doi: 10.3892/ol.2017.6459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barone M, Ricci F, Sollazzo D, Ottaviani E, Romano M, Auteri G, Bartoletti D, Reggiani MLB, Vianelli N, Tazzari PL et al. Circulating megakaryocyte and platelet microvesicles correlate with response to ruxolitinib and distinct disease severity in patients with myelofibrosis. Br J Haematol. 2019. June;185(5):987–991. doi: 10.1111/bjh.15682. [DOI] [PubMed] [Google Scholar]
- 40.Elliott MA, Verstovsek S, Dingli D, Schwager SM, Mesa RA, Li CY, Tefferi A. Monocytosis is an adverse prognostic factor for survival in younger patients with primary myelofibrosis. Leuk Res. 2007. November;31(11):1503–1509. doi: 10.1016/j.leukres.2006.12.025. [DOI] [PubMed] [Google Scholar]
- 41.Rameshwar P, Narayanan R, Qian J, Denny TN, Colon C, Gascon P. NF-kappa B as a central mediator in the induction of TGF-beta in monocytes from patients with idiopathic myelofibrosis: an inflammatory response beyond the realm of homeostasis. J Immunol. 2000. August 15;165(4):2271–2277. doi: 10.4049/jimmunol.165.4.2271. [DOI] [PubMed] [Google Scholar]
- 42.Campanelli R, Rosti V, Fois G, Bonetti E, Barosi G, Massa M. CD14(bright)CD16(low) intermediate monocytes expressing Tie2 are increased in the peripheral blood of patients with primary myelofibrosis. Exp Hematol. 2014. April;42(4):244–246. doi: 10.1016/j.exphem.2013.12.002. [DOI] [PubMed] [Google Scholar]
- 43.De la Guardia RD, Correa JG, López-Millán B, Juan M, Bueno C, Cervantes F, Menéndez P. Detection of inflammatory monocytes but not mesenchymal stem/stromal cells in peripheral blood of patients with myelofibrosis. Br J Haematol. 2018. April;181(1):133–137. doi: 10.1111/bjh.14507. [DOI] [PubMed] [Google Scholar]
- 44.Schuerwegh AJ, Stevens WJ, Bridts CH, De Clerck LS. Evaluation of monensin and brefeldin A for flow cytometric determination of interleukin-1 beta, interleukin-6, and tumor necrosis factor-alpha in monocytes. Cytometry. 2001. June 15;46(3):172–176. doi: 10.1002/cyto.1102. [DOI] [PubMed] [Google Scholar]
- 45.de Waal Malefyt R, Abrams J, Bennett B, Figdor CG, de Vries JE. Interleukin 10(IL-10) inhibits cytokine synthesis by human monocytes: an autoregulatory role of IL-10 produced by monocytes. J Exp Med. 1991. November 1;174(5):1209–1220. doi: 10.1084/jem.174.5.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nat Rev Immunol. 2011. October 10;11(11):762–774. doi: 10.1038/nri3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Carey AJ, Tan CK, Ulett GC. Infection-induced IL-10 and JAK-STAT: a review of the molecular circuitry controlling immune hyperactivity in response to pathogenic microbes. JAKSTAT. 2012. July 1;1(3):159–167. doi: 10.4161/jkst.19918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hutchins AP, Diez D, Miranda-Saavedra D. The IL-10/STAT3-mediated anti-inflammatory response: recent developments and future challenges. Brief Funct Genomics. 2013. November;12(6):489–498. Epub 2013 Aug 12. doi: 10.1093/bfgp/elt028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lai HY, Brooks SA, Craver BM, Morse SJ, Nguyen TK, Haghighi N, Garbati MR, Fleischman AG. Defective negative regulation of Toll-like receptor signaling leads to excessive TNF-α in myeloproliferative neoplasm. Blood Adv. 2019. January 22;3(2):122–131. doi: 10.1182/bloodadvances.2018026450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gane JM, Stockley RA, Sapey E. TNF-α autocrine feedback loops in human monocytes: the pro- and anti-inflammatory roles of the TNF-α receptors support the concept of selective TNFR1 blockade in vivo. J Immunol Res. 2016. September 22;2016:1079851. doi: 10.1155/2016/1079851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.houval DS, Ouahed J, Biswas A, Goettel JA, Horwitz BH, Klein C, Muise AM, Snapper SB. Interleukin 10 receptor signaling: master regulator of intestinal mucosal homeostasis in mice and humans. Adv Immunol. 2014;122:177–210. doi: 10.1016/B978-0-12-800267-4.00005-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhengfan J, Philippe G, Xin D, Louis S, Sovath S, Mudd S, Huber M, Kalis C, Keck S, Galanos C, et al. CD14 is required for MyD88-independent LPS signalling. Nat Immunol. 2005;6:565–570. doi: 10.1038/ni1207. [DOI] [PubMed] [Google Scholar]
- 53.Paracha RZ, Ahmad J, Ali A, Hussain R, Niazi U, Tareen SHK, Aslam B. Formal modelling of toll like receptor 4 and JAK/STAT signalling pathways: insight into the roles of SOCS-1, Interferon-β and proinflammatory cytokines in sepsis. PLoS One. 2014;9(9):e108466. doi: 10.1371/journal.pone.0108466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Heine A, Held SA, Daecke SN, Wallner S, Yajnanarayana SP, Kurts C, Wolf D, Brossart P. The JAK-inhibitor ruxolitinib impairs dendritic cell function in vitro and in vivo. Blood. 2013. August 15;122(7):1192–1202. Epub 2013 Jun 14. doi: 10.1182/blood-2013-03-484642. [DOI] [PubMed] [Google Scholar]
- 55.Stalder R, Zhang B, Wrobel LJ, Boehncke WH, Brembilla NC. The Janus Kinase inhibitor tofacitinib impacts human dendritic cell differentiation and favours M1 macrophage development. Exp Dermatol. 2020. January;29(1):71–78. Epub 2019 Nov 26. doi: 10.1111/exd.14059. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.