

Abstract
Ectodomain shedding is a post-translational modification mechanism by which the entire extracellular domain of membrane proteins is liberated through juxtamembrane processing. Because shedding rapidly and irreversibly alters the characteristics of cells, this process is properly regulated. However, the molecular mechanisms governing the propensity of membrane proteins to shedding are largely unknown. Here, we present evidence that negatively charged amino acids within the stalk region, an unstructured juxtamembrane region at which shedding occurs, contribute to shedding susceptibility. We show that two activated leukocyte cell adhesion molecule (ALCAM) protein variants produced by alternative splicing have different susceptibilities to ADAM metallopeptidase domain 17 (ADAM17)-mediated shedding. Of note, the inclusion of a stalk region encoded by a 39-bp-long alternative exon conferred shedding resistance. We found that this alternative exon encodes a large proportion of negatively charged amino acids, which we demonstrate are indispensable for conferring the shedding resistance. We also show that the introduction of negatively charged amino acids into the stalk region of shedding-susceptible ALCAM variant protein attenuates its shedding. Furthermore, we observed that negatively charged amino acids residing in the stalk region of Erb-B2 receptor tyrosine kinase 4 (ERBB4) are indispensable for its shedding resistance. Collectively, our results indicate that negatively charged amino acids within the stalk region interfere with the shedding of multiple membrane proteins. We conclude that the composition of the stalk region determines the shedding susceptibility of membrane proteins.
Keywords: ectodomain shedding, ADAM, metalloprotease, alternative splicing, membrane protein, immunoglobulin-like domain, stalk region, posttranslational modification, Erb-B2 receptor tyrosine kinase 4 (ERBB4), activated leukocyte cell adhesion molecule (ALCAM)
Ectodomain shedding, also simply called shedding, is a post-translational modification mechanism that releases the extracellular domain from membrane proteins through a juxtamembrane processing event. This process is executed mainly by a family of membrane-anchored metalloproteases, called ADAMs (a disintegrin and metalloproteases) (1–4). Shedding is involved in a wide variety of biological phenomena, as membrane proteins having distinct functions, including growth factors, cytokines, cytokine receptors, and adhesion molecules, are affected by this process. For example, shedding can release growth factors and cytokines that comprise the extracellular domains of membrane proteins, which drastically potentiates their functions. Shedding can also decrease the amount of cell-surface receptors and adhesion molecules in parallel with the release of soluble extracellular domains, which effectively reduces the reactivity of cells to their cognate ligands. The remaining membrane-anchored products of shedding are often subjected to γ-secretase–dependent intramembrane proteolysis (3–5). This generates soluble intracellular domains, some of which translocate to the nucleus and regulate gene expression. Note that γ-secretase cannot cleave membrane proteins having bulky extracellular domains; thus, shedding can be a rate-limiting step for a sequence of processing events leading to various protein functions. Taken together, shedding is a unique mechanism exerting pleiotropic effects at a variety of cellular locations, including the extracellular space, cell surface, cytoplasm, and nucleus, through a simple processing event.
Because shedding is such an effective mechanism, it must be properly regulated. Additionally, because shedding is an adaptive response, physiologically relevant substrate proteins should be selectively shed, depending on the type of extracellular stimuli (i.e. pro-inflammatory cytokine should be shed in response to lipopolysaccharide (LPS)) (3). Whereas only one-tenth of membrane proteins are thought to be subjected to shedding (6, 7), the molecular mechanisms ensuring the specificity of shedding are largely unknown. To date, membrane proteins known to be shedding targets have no consensus cleavage site sequence, and two major sheddases, ADAM10 and ADAM17, have remarkably relaxed substrate specificity (8, 9). Thus, a comprehensive analysis of shedding target proteins is required to understand the molecular mechanisms that determine membrane protein susceptibility to shedding (shedding susceptibility).
To perform such an analysis, we developed a proteomic screening system to identify multiple membrane proteins that are simultaneously shed upon LPS stimulation of macrophages (10, 11). We found that these membrane proteins have no consensus sequence in their stalk regions, the region between the membrane-proximal extracellular domain and the cell surface. However, detailed analysis of one identified shedding target, CADM1 (cell adhesion molecule 1), an Ig superfamily cell adhesion molecule, revealed unexpected features that determined shedding susceptibility: alternative splicing of the murine Cadm1 mRNA gives rise to protein variants that are either shedding-susceptible or shedding-resistant (12). The inclusion of an alternative exon, only 33 bp long, is necessary and sufficient to render CADM1 variants susceptible to shedding. We further showed that O-glycans attached to the amino acids adjacent to the shedding cleavage site interfere with CADM1 shedding. The alternative exon confers shedding susceptibility to CADM1 through the insertion of five amino acids that cannot be glycosylated between the interfering O-glycans and the shedding cleavage site. These results indicate that the shedding susceptibility of membrane proteins can be determined by two steps in their biosynthesis: post-transcriptional alternative splicing and post-translational O-glycosylation.
In the present study, we characterize the shedding susceptibility of ALCAM (activated leukocyte cell adhesion molecule, also known as CD166), an Ig superfamily cell adhesion molecule (13, 14), which we identified as another shedding target in our previous study (12). Similar to CADM1, we demonstrate that two ALCAM splice variants differ in shedding susceptibility. We further show that negatively charged amino acids encoded by an alternative exon confer shedding resistance to an ALCAM variant protein. Taken together, we conclude that a short amino acid sequence located in the stalk region and containing specific biochemical properties determines the susceptibility of membrane proteins to shedding.
Results
Alternative splicing gives rise to both shedding-susceptible and shedding-resistant ALCAM variant proteins
To confirm the results of our previous screening study (12), we first evaluated the shedding of endogenous ALCAM protein in the murine macrophage cell line, RAW 264.7. RAW 264.7 cells were treated with or without LPS, cell extracts were prepared, and extracts as well as culture media were subjected to Western blotting using an antibody recognizing the extracellular domain of ALCAM. Soluble ALCAM was detected in the culture medium of LPS-stimulated cells (Fig. 1A). Note that soluble ALCAM is only slightly lower in apparent molecular weight compared with full-length ALCAM detected in the cell extracts. Because the cytoplasmic domain of ALCAM is short, the result indicates that almost the entire extracellular domain is released. The release of soluble ALCAM was completely suppressed by the metalloprotease inhibitor, BB94 (Fig. 1A). These results indicate that endogenous ALCAM is shed via the action of metalloproteases in an LPS-dependent manner.
Figure 1.
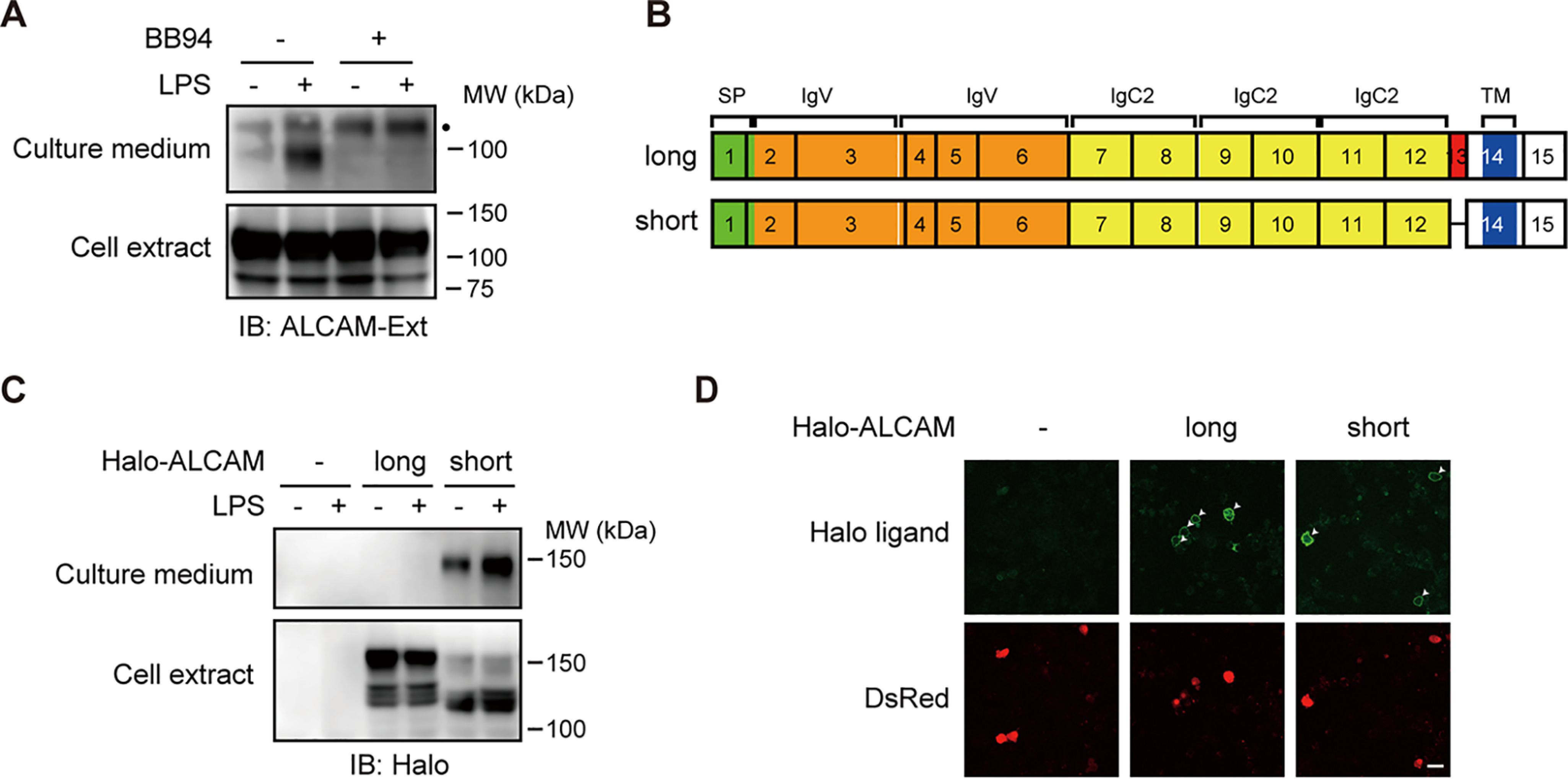
Alternative splicing of Alcam gene gives rise to both shedding-susceptible and shedding-resistant ALCAM variant proteins. A, RAW 264.7 cells were treated with (+) or without (−) 1 μg/ml LPS and/or 10 μm BB94 for 60 min as indicated. Both cell extracts (Cell extract) and culture supernatants (Culture medium) were subjected to Western blotting with an anti-ALCAM antibody. Ext, extracellular domain. A black dot indicates a nonspecific band. B, schematic diagrams of the molecular structure of two splice variants of ALCAM. Exons are indicated by black rectangles with numbers. SP, signal peptide (green); IgV, immunoglobulin (Ig)-like variable-type domain (orange); IgC2, Ig-like constant 2–type domain (yellow); TM, transmembrane domain (blue). The additional exon in the long splice variant is indicated in red. C, RAW 264.7 cells expressing N-terminally Halo-tagged long or short ALCAM variants were treated with or without LPS for 60 min, and cell extracts and culture media were subjected to Western blotting with an anti-HaloTag antibody. D, RAW 264.7 cells expressing Halo-tagged long or short ALCAM variants were stained using cell-impermeable HaloTag Alexa Fluor 488 ligand (Halo ligand). Arrowheads, transfected DsRed-positive cells (DsRed). Bar, 25 μm.
ALCAM protein is encoded by 15 exons in mice. Two splice variants, long and short ALCAM, are generated by the inclusion or skipping of a 39-bp alternative exon, exon 13 (Fig. 1B). The invariable exon 12 encodes the C-terminal half of the membrane-proximal Ig-like C2 domain, whereas the invariable exon 14 encodes the transmembrane domain. Accordingly, the alternative exon 13 encodes the stalk region of ALCAM, where shedding would occur. As we had previously demonstrated that alternative splicing of a 33-bp exon encoding the CADM1 stalk region generates both shedding-susceptible and shedding-resistant CADM1 variant proteins (12), we compared the shedding susceptibility of the ALCAM variant proteins. RAW 264.7 cells expressing N-terminally Halo-tagged ALCAM variant proteins were treated with or without LPS, and the cell extracts and culture media were subjected to Western blotting using an anti-HaloTag antibody. Importantly, Halo-tagged soluble ALCAM was only detected in the culture medium of cells expressing the short ALCAM variant, even though both ALCAM variants were equivalently expressed in the cell extracts (Fig. 1C). These results clearly show that the short ALCAM variant is shedding-susceptible, whereas the long ALCAM variant is shedding-resistant. In other words, the inclusion of exon 13 of murine Alcam confers shedding resistance to the long ALCAM variant protein.
Because in many cases localization on the cell surface is required for shedding of membrane proteins, we next assessed the localization of ALCAM variant proteins. Staining with a cell-impermeable HaloTag fluorescent ligand showed comparable amounts of fluorescent signal on the surface of cells expressing both ALCAM variants (Fig. 1D), indicating that cell-surface localization of ALCAM variants is not the determinant of their shedding susceptibility. These results suggest that the amino acid sequence encoded by exon 13 determines the resistance of the long ALCAM variant to shedding and that this resistance is independent of cellular localization.
Shedding susceptibility of endogenous ALCAM protein is determined by the alternative splicing of exon 13
RT-PCR analysis showed that the long ALCAM mRNA is roughly 10-fold more abundant than the short ALCAM mRNA in RAW 264.7 cells (data not shown). This indicates that a large proportion of endogenous ALCAM protein is the shedding-resistant long ALCAM variant. Despite this, however, we detect shedding of endogenous ALCAM (Fig. 1A). To investigate the shedding susceptibility of endogenous long ALCAM, we generated exon 13–knockout RAW 264.7 cells using lentivirus vectors and CRISPR/Cas9-based genome editing. Western blotting of the cell extracts showed that exon 13–knockout cells express approximately equivalent amounts of ALCAM protein of a slightly lower molecular weight compared with control cells (Fig. 2). These results indicate that, whereas control cells mainly express long ALCAM, exon 13–knockout cells mainly express short ALCAM. In addition, Western blotting of the culture media showed that the shedding of endogenous ALCAM protein is considerably potentiated in exon 13–knockout cells (Fig. 2). On the other hand, the shedding of endogenous SIRPα (signal-regulatory protein α) protein, another shedding target of LPS-stimulated macrophages (12), showed no difference between control and exon 13–knockout cells (Fig. 2), indicating that the potentiation of ALCAM shedding is not due to the activation of sheddases. These results show that the inclusion of exon 13 attenuates shedding of endogenous ALCAM protein and indicate that endogenous long ALCAM is shedding-resistant.
Figure 2.
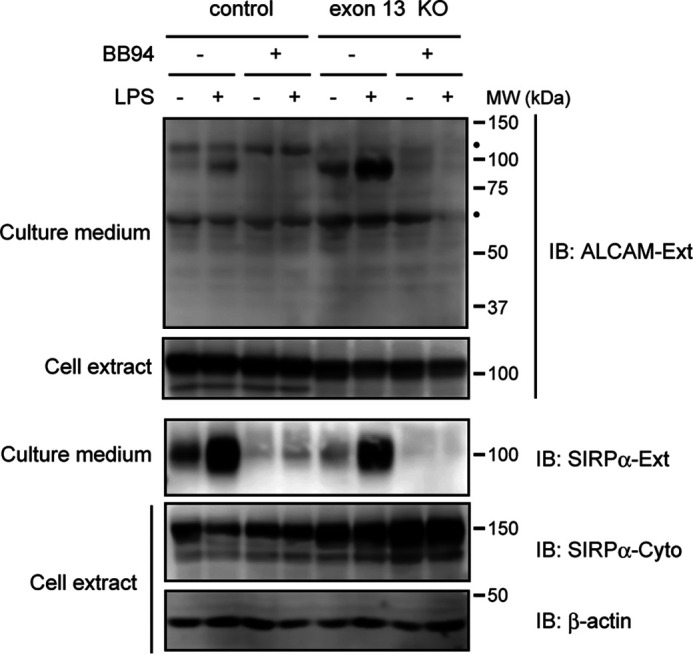
Disruption of Alcam gene exon 13 potentiates the shedding of endogenous ALCAM protein. RAW 264.7 cells expressing exon 13-sgRNA/Cas9 (exon 13 del) or Cas9 only (control) were treated with or without LPS and/or BB94 for 60 min, and cell extracts and culture supernatants were subjected to Western blotting (IB) using the indicated antibodies. β-Actin was used as a loading control. Black dots, nonspecific bands. Ext, extracellular domain. Cyto, cytoplasmic domain.
Negatively charged amino acids encoded by exon 13 interfere with shedding of long ALCAM
To elucidate the molecular mechanisms by which the amino acid sequence encoded by exon 13 interferes with shedding of long ALCAM, we first investigated the shedding cleavage site of short ALCAM. When cytoplasmic PA-tagged long and short ALCAM were expressed in human embryonic kidney (HEK) 293T cells, a protein of ∼15 kDa was detected by an anti-PA antibody in cells expressing short PA-ALCAM (Fig. 3A). Because both a universal shedding activator, 12-O-tetradecanoylphorbol 13-acetate (TPA), and a γ-secretase inhibitor, DAPT, caused accumulation of the ∼15-kDa protein (Fig. 3A, arrowhead), this protein must be the membrane-anchored shedding product. The N-terminal sequence of the ∼15-kDa protein is Val-Ser-Ala-Asn-Glu, indicating that short ALCAM is cleaved between asparagine 499 and valine 500 (Fig. 4A, arrowhead). Because the transmembrane domain of short ALCAM begins with leucine 515 in UniProt database, the membrane-anchored shedding product has 15-amino acid-long extracellular domain.
Figure 3.
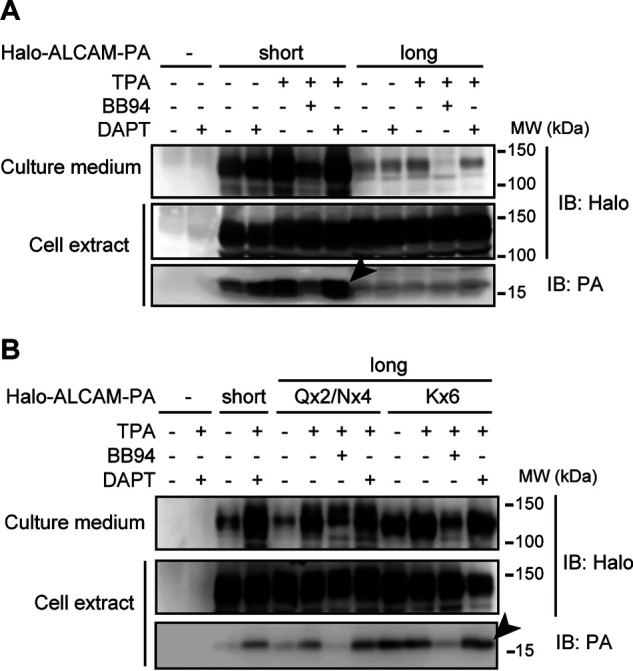
Accumulation of the membrane-anchored shedding product of ALCAM proteins. HEK 293T cells expressing N-terminally Halo-tagged and cytoplasmic PA-tagged long or short ALCAM (A) or substitution mutants (see “Results” and Fig. 4 for details of mutants) of long ALCAM (B) were treated with or without 200 ng/ml TPA, 10 μm BB94, and/or 10 μm DAPT for 90 min as indicated, and cell extracts and culture supernatants were subjected to Western blotting (IB) using the indicated antibodies. Arrowheads, ∼15-kDa membrane-anchored shedding products.
Figure 4.
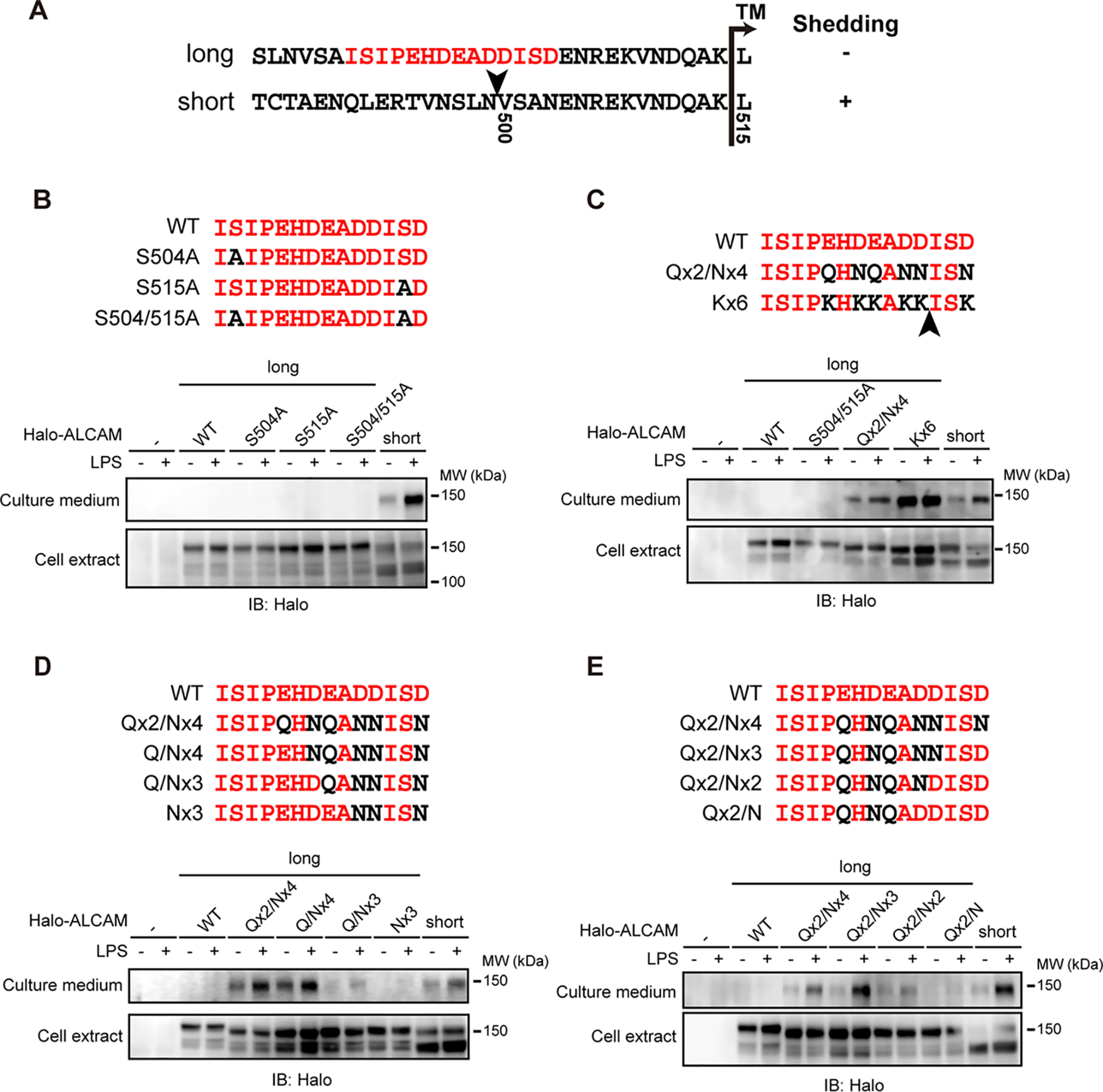
Negatively charged amino acids encoded by exon 13 interfere with the shedding of long ALCAM. A, the amino acid sequences of the stalk regions (area between the membrane-proximal IgC2 domain and transmembrane domain) of ALCAM variant proteins. The amino acids encoded by exon 13 are colored in red. An arrowhead indicates the shedding cleavage site of short ALCAM. TM, transmembrane domain. B–E, RAW 264.7 cells expressing N-terminally Halo-tagged substitution mutants of long ALCAM were treated with or without LPS for 60 min, and cell extracts and culture supernatants were subjected to Western blotting (IB) with an anti-HaloTag antibody. The substituted amino acid residues are colored in black. Arrowhead, shedding cleavage site of Kx6 long ALCAM (C).
The inclusion of exon 13 replaces asparagine 503 of short ALCAM with the 14-amino acid sequence, ISIPEHDEADDISD (Fig. 4A, red letters). Thus, the exon 13 sequence in long ALCAM replaces the position corresponding to the shedding cleavage site in short ALCAM. We therefore hypothesized that the amino acid sequence encoded by exon 13 creates a structural barrier(s) for shedding.
Because we previously reported that O-glycans adjacent to the shedding cleavage site interfere with the shedding of CADM1 (12), we assessed whether O-glycosylation of the amino acids encoded by exon 13 could also have this effect. Alanine substitutions of the two serine residues in this portion of the protein, either one at a time or both, did not alter shedding of long ALCAM (Fig. 4B), indicating that O-glycans are not the structural barrier in this sequence. We then noticed that the sequence is particularly abundant in aspartic and glutamic acids (42.9%, 6 of 14), which are negatively charged amino acids. To determine whether the negatively charged amino acids affected shedding, we constructed additional substitution mutants. Intriguingly, substitutions of the negatively charged amino acids with uncharged polar amino acids potentiated shedding of long ALCAM (Fig. 4C, Qx2/Nx4). Strikingly, substitutions with positively charged amino acids potentiated the shedding even more strongly (Fig. 4C, Kx6). These results indicate that the negatively charged amino acids encoded by exon 13 interfere with shedding of long ALCAM.
We constructed additional substitution mutants to delineate which negatively charged amino acid(s) is critical in interfering with the shedding of long ALCAM. The reverse stepwise substitution of uncharged polar amino acids with negatively charged amino acids gradually attenuated the shedding of Qx2/Nx4 long ALCAM regardless of the direction (Fig. 4, D and E). Of note, both Nx3 and Qx2/N long ALCAM were almost completely shedding-resistant, although they each contain three negatively charged amino acids in completely different positions. These results suggest that it is the number, not the position, of negatively charged amino acids that is critical for interfering with shedding of long ALCAM.
Distance between the shedding cleavage site and the cell surface is comparable between Kx6 long ALCAM and short ALCAM
We next investigated the shedding cleavage site of the substitution mutants of long ALCAM. When cytoplasmic PA-tagged Qx2/Nx4 and Kx6 long ALCAM were expressed in HEK 293T cells, an ∼15-kDa membrane-anchored shedding product was detected (Fig. 3B). Because the shedding product of Kx6 long ALCAM was more abundant than that of Qx2/Nx4 long ALCAM (Fig. 3B, arrowhead), the shedding product of Kx6 long ALCAM was affinity-purified and sequenced. N-terminal sequencing revealed the sequence Ile-Ser-Lys-Glu-Asn, indicating that Kx6 long ALCAM is cleaved between lysine 513 and isoleucine 514 (Fig. 4C, arrowhead). Because the transmembrane domain of long ALCAM starts at leucine 528, the membrane-anchored shedding product of Kx6 long ALCAM has a 14-amino acid-long extracellular domain. As described above, the shedding product of short ALCAM has a 15-amino acid-long extracellular domain. These results indicate that the distance between the shedding cleavage site and the cell surface is comparable for Kx6 long ALCAM and short ALCAM.
Negatively charged amino acids can interfere with the shedding of short ALCAM
We next investigated whether negatively charged amino acids can interfere with the shedding of short ALCAM. Substitutions of two amino acids bracketing the shedding cleavage site of short ALCAM, asparagine 499 and valine 500 (Fig. 5, WT, highlighted), with negatively charged aspartic acids, attenuated the shedding of short ALCAM (Fig. 5, DD). On the other hand, substitution at these positions with positively charged lysines had no effect on the shedding of short ALCAM (Fig. 5, KK). Thus, substitution at the shedding cleavage site with negatively charged amino acids interferes with the shedding of short ALCAM, confirming the inhibitory effect of negatively charged amino acids on shedding.
Figure 5.

Negatively charged amino acids bracketing the shedding cleavage site interfere with the shedding of short ALCAM. RAW 264.7 cells expressing N-terminally Halo-tagged substitution mutants of short ALCAM were treated with or without LPS for 60 min, and cell extracts and culture supernatants were subjected to Western blotting (IB) with an anti-HaloTag antibody. The amino acid sequences of the stalk regions of WT and substitution mutants (DD and KK) of short ALCAM are shown. Arrowhead, shedding cleavage site of short ALCAM. The substituted amino acid residues are highlighted.
Negatively charged amino acids of JM-b ErbB4 are indispensable for shedding resistance
ErbB4 receptor tyrosine kinase has both shedding-susceptible (JM-a) and shedding-resistant (JM-b) forms. These derive from splice variants generated by the switching between two mutually exclusive exons (75 and 45 bp, respectively), both of which encode a stalk region (15, 16). The shedding cleavage site of human JM-a ErbB4 is between histidine 641 and serine 642 (Fig. 6, arrowhead) (17). Cleavage at this site leaves 10 amino acids at the cell surface. Our analysis revealed that shedding-resistant JM-b ErbB4 has two negatively charged amino acids, glutamic acid 631 and aspartic acid 632 (Fig. 6, JM-b WT, highlighted), at the 11th and 10th amino acid counting from the cell surface. Given our results above with ALCAM, we hypothesized that these negatively charged amino acids interfere with the shedding of JM-b ErbB4. Indeed, substitutions of the negatively charged amino acids with uncharged polar amino acids substantially potentiated the shedding of JM-b ErbB4 (Fig. 6, QN), indicating that these negatively charged amino acids interfere with shedding of the protein. Substitutions with positively charged amino acids only modestly potentiated the shedding of JM-b ErbB4 (Fig. 6, KK), whereas the substitutions with positively charged amino acids markedly potentiated the shedding of long ALCAM (Fig. 4C, Kx6). Thus, negatively charged amino acids have an inhibitory effect on the shedding of multiple membrane proteins.
Figure 6.

Two negatively charged amino acids of JM-b ErbB4 are indispensable for shedding resistance. HEK 293T cells expressing N-terminally Halo-tagged JM-a ErbB4 or substitution mutants of JM-b ErbB4 were treated with or without TPA for 60 min, and cell extracts and culture supernatants were subjected to Western blotting (IB) with an anti-HaloTag antibody. The amino acid sequences of the stalk regions of JM-a and WT and substitution mutants of JM-b are shown. Arrowhead, shedding cleavage site of JM-a ErbB4. The two negatively charged amino acids and substituted amino acids are highlighted.
Dependence on ADAM17 is different between short ALCAM and ErbB4 proteins
The differential effects of the substitution with positively charged amino acids on the shedding of ALCAM and ErbB4 raised the possibility that different a sheddase(s) is responsible for cleaving these two proteins. The two major sheddases are ADAM17 and ADAM10; ADAM17 is a stimulus-dependent enzyme, whereas ADAM10 is relatively constitutive (2, 18). As we reported previously, ADAM17 catalyzes the LPS-dependent shedding of both CADM1 and SIRPα in RAW 264.7 cells (12). We therefore evaluated the contribution of ADAM17 to the shedding of ALCAM and ErbB4 using ADAM17-knockout mouse embryonic fibroblasts (MEFs) (19). Similar to RAW 264.7 cells, short ALCAM is shedding-susceptible in control MEFs, whereas long ALCAM is shedding-resistant (Fig. 7A, WT). Thus, the responsible sheddase for short ALCAM is most likely identical in these two cell lines. In ADAM17-knockout MEFs, the shedding of short ALCAM was completely abolished (Fig. 7A, ADAM17−/−), indicating that ADAM17 is the responsible sheddase for short ALCAM. In contrast, the shedding of both JM-a and JM-b QN ErbB4 in ADAM17-knockout MEFs was approximately half of that in control MEFs (Fig. 7B), indicating that ADAM17 only partially participates in the shedding of ErbB4 proteins. Taken together, these results indicate that ADAM17 plays a different role in the shedding of short ALCAM versus ErbB4 proteins.
Figure 7.
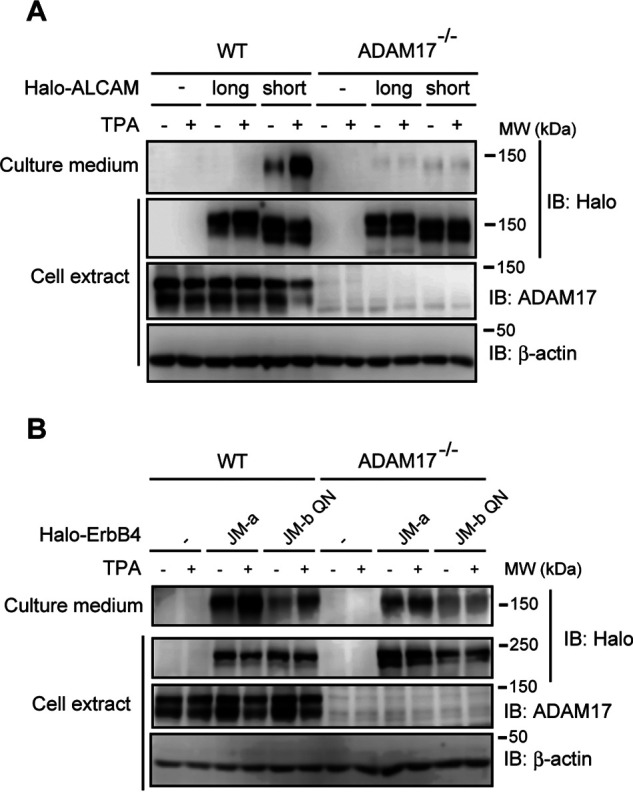
The shedding of ALCAM and ErbB4 are differentially affected by deletion of ADAM17. WT and ADAM17−/− MEFs expressing N-terminally Halo-tagged ALCAM (A) or ErbB4 (B) were treated with or without TPA for 60 min, and cell extracts and culture supernatants were subjected to Western blotting (IB) using the indicated antibodies.
Discussion
In this study, we show that alternative splicing of the murine Alcam gene generates both shedding-susceptible and shedding-resistant ALCAM variant proteins. The inclusion of a 39-bp alternative exon, exon 13, encoding the stalk region, makes the long ALCAM protein shedding-resistant. Because the exon encodes <3% of the full-length protein, the inclusion/skipping of the exon is likely to generate variant proteins having different shedding susceptibilities without affecting the overall structure and function. Our detailed analyses of the amino acid sequence encoded by the exon have substantially expanded our understanding of the molecular mechanisms determining shedding susceptibility.
Herein we reveal that the inclusion of exon 13 of murine Alcam confers shedding resistance to ALCAM variant protein through the insertion of negatively charged amino acids. Substitutions of the negatively charged amino acids encoded by exon 13 with uncharged polar amino acids substantially potentiated the shedding of long ALCAM. We speculate that the negatively charged amino acids interfere with the shedding of long ALCAM simply through the accumulation of negative electric charges on the basis of the following observations: 1) the number, not the positions, of the negatively charged amino acids determines shedding susceptibility; 2) substitutions with positively charged amino acids potentiate the shedding further; 3) secondary structure prediction analyses of WT and Kx6 long ALCAM using two algorithms, CRNPRED and Jpred, show no obvious differences (data not shown). We further show that substitutions at the shedding cleavage site with two negatively charged amino acids interfere with the shedding of short ALCAM. In addition, substitutions of two negatively charged amino acids with uncharged polar amino acids potentiate the shedding of JM-b ErbB4. In support of these findings, an SNP in the human IL-6 receptor gene, substituting alanine for aspartic acid adjacent to the shedding cleavage site, potentiates ADAM17-mediated shedding of the IL-6 receptor protein (20). It was also reported that substitutions of the shedding cleavage site with negatively charged amino acids interfered with the shedding of the IL-6 receptor protein (21). Collectively, these observations indicate that negatively charged amino acids around the shedding cleavage site generally interfere with the shedding of membrane proteins. In agreement with these observations, neither ADAM17 nor ADAM10 prefers negatively charged amino acids around their cleavage sites; the only exception is that glutamic acid is somewhat preferred at the P2 position (2 amino acids upstream of the cleavage site) (8, 9).
Hebron et al. (22) also previously reported the processing of ALCAM variant proteins. They showed that short ALCAM is more susceptible to processing than long ALCAM, as we show herein. However, they report that short ALCAM is processed by another metalloprotease, MMP14, releasing a much smaller extracellular fragment into the culture medium. Because we could not detect this smaller fragment in the culture medium of our exon 13–knockout cells, which express mainly short ALCAM, we consider this discrepancy to be a result of differences in experimental conditions. We collected the culture media incubated with LPS-stimulated macrophage cells for 60 min, whereas Hebron et al. (22) collected the culture media incubated with nonstimulated cancer cells for 24 h. Because ADAM17 is a stimulus-dependent enzyme (2, 18), ADAM17-mediated shedding would be more prominent in our study. On the other hand, because the cancer cell line expressed high levels of MMP14 (23, 24), MMP14-mediated processing might be emphasized in their study. Further studies are needed to evaluate this possibility.
To explain the selectivity of shedding, previous studies have focused on looking for favorable amino acid sequences as substrates for ADAM family metalloproteases. Our own previous studies as well as this report, however, raise the possibility that “unfavorable factors” may contribute to the selectivity of shedding. As we show here, negatively charged amino acids within the stalk region comprise an unfavorable factor for the shedding of ALCAM and ErbB4 (Fig. 8, green circles). As we showed previously, the presence of O-glycans adjacent to the shedding cleavage site is also an unfavorable factor (Fig. 8, broken lines). Because a majority of membrane proteins are glycosylated on their extracellular domain as protection from the environment, quite a few of them should have shedding-interfering glycans. In addition, glycosaminoglycans, highly negatively charged components of the extracellular matrix, might interact with the stalk region of membrane proteins and also protect them from the shedding. In sum, it is conceivable that the majority of membrane proteins are protected from shedding by the presence of unfavorable factors in or around their stalk region. On the other hand, we have elucidated that the insertion or deletion of short amino acid sequences encoded by an alternative exon can switch shedding susceptibility, indicating that the position of unfavorable factors is a critical factor in their ability to interfere with the shedding. We now hypothesize that the shedding susceptibility of membrane proteins is determined by the number of unfavorable factors within the narrow window of their stalk region, and only membrane proteins having little or no unfavorable factor in the window can be shedding-susceptible.
Figure 8.

Shedding susceptibility of ALCAM and CADM1 is determined by the unfavorable factors in their stalk region. A schematic diagram shows the structure of the stalk region of ALCAM and CADM1 variant proteins and their shedding susceptibility. Orange, IgV domain; yellow, IgC2 domain; red, amino acids encoded by alternative exon; green, negative charge in ALCAM; broken lines, O-glycans; blue scissors, ADAM17. Red circle, cleavage by ADAM17; black X, no cleavage.
What factors determine the position of the window? We speculate that the responsible sheddase controls this. We show here that ADAM17 is the active sheddase for short ALCAM, and the membrane-anchored shedding product of short ALCAM has a 15-amino acid-long extracellular domain. We previously showed that ADAM17 is the sheddase that acts on CADM1, and the membrane-anchored shedding product of CADM1 has a 14-amino acid-long extracellular domain. In addition, the membrane-anchored shedding product of Kx6 long ALCAM has a 14-amino acid-long extracellular domain. These observations indicate that ADAM17 prefers to cleave at the 15th amino acid counting from the cell surface, at least in RAW 264.7 cells. In support of this notion, long ALCAM is shedding-resistant even though it contains an amino acid sequence identical to the shedding cleavage site of short ALCAM at around the 30th amino acid counting from the cell surface. Given these findings, we speculate that the shedding susceptibility of ALCAM and CADM1 variant proteins is determined by what we term “unfavorable factors” (i.e. negatively charged amino acids and O-glycans, respectively) located around the 15th amino acid counting from the cell surface, the cleavage location preferred by ADAM17 (Fig. 8, blue scissors). ADAM17 is not a major sheddase for JM-a ErbB4, and JM-a ErbB4 is cleaved between the 11th and 10th amino acid counting from the cell surface. The substitutions of the 11th and 10th negatively charged amino acids with uncharged polar amino acids potentiates the shedding of JM-b ErbB4, supporting the hypothesis that the active sheddase determines the position of the window. In addition, we speculate that the difference in sheddases should explain the difference in the effects of the substitution with positively charged amino acids on the shedding of long ALCAM and JM-b ErbB4.
The existence of ALCAM variant proteins having different susceptibilities to shedding means that individual cells can simultaneously express these variants at different ratios through cell type–specific alternative splicing. As described above, in RAW 264.7 cells, shedding-susceptible short ALCAM is presumably a minor population; however, considering the mechanism of action of ALCAM proteins, a small amount of short ALCAM has the potential to act as a key regulator of ALCAM functions. ALCAM is an Ig superfamily adhesion molecule mediating both homophilic (ALCAM-ALCAM) and heterophilic (ALCAM-CD6) cell-cell interactions (14). Stable interactions are dependent on oligomerization through the extracellular Ig domain (25). Therefore, shedding of ALCAM can effectively attenuate cell-cell interactions through three mechanisms: 1) a decrease in the level of functional full-length protein, 2) the release of interfering soluble extracellular domain, and 3) the disruption of adhesive ALCAM oligomers. Because shedding is an immediate mechanism, scattered short ALCAM can serve as a molecular switch to break ALCAM-mediated interactions, and the amount of short ALCAM present can regulate the adhesive properties of the cell. The alternative splicing of the Alcam gene might regulate cell-cell interactions through the modulation of the ratio of long/short ALCAM in a variety of biological contexts. Because ALCAM plays pivotal roles in axon guidance and fasciculation, immune responses, B cell migration in multiple sclerosis, and cancer progression (26–29), the biological significance of the effects of alternative splicing on these phenomena will be a rich source of future studies.
In summary, we propose that it is the absence of “unfavorable factors” rather than the presence of favorable factors that determines the shedding susceptibility of membrane proteins. These findings provide important new insights into the molecular mechanisms controlling the high specificity of ectodomain shedding. Indeed, we have already identified additional membrane proteins whose shedding susceptibility is regulated through the inclusion/skipping of an alternative exon encoding their stalk regions. Further investigations may reveal additional unfavorable factors that affect shedding and thus lead to the development of the methods by which particular shedding processes may be modified. Such developments may lead to therapies for diseases associated with shedding, such as autoimmune diseases, neurodegenerative diseases, and cancer (1–5).
Experimental procedures
Cell line, transfection, and sample preparation for Western blotting
A murine macrophage cell line, RAW 264.7, was cultured in high-glucose Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 50 μm 2-mercaptoethanol, and antibiotics. MEFs established from WT and ADAM17−/− mouse embryos (19) and HEK 293T cells were cultured in low-glucose Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum and antibiotics. Transfections were performed using FuGENE HD (Promega) for RAW 264.7 cells and MEFs. Viofectin (Viogene) was used for transfection to HEK 293T cells. For Western blotting, cell extracts and proteins of conditioned culture media were prepared as described previously (11).
Antibodies, chemicals, and expression plasmids
Antibodies were purchased from R&D Systems (anti-ALCAM-Ext, AF1172), Promega (anti-HaloTag, G9211), BioLegend (anti-SIRPα-Ext, P84), ProSci (anti-SIRPα-Cyto, 1125), Santa Cruz Biotechnology, Inc. (anti-β-actin, sc-81178), WAKO (anti-PA tag, NZ-1), and Abcam (anti-ADAM17, ab39162). LPS and DAPT were purchased from Sigma–Aldrich. TPA was purchased from Merck Millipore. BB94 was provided by Vernalis. The coding sequence of murine long ALCAM was obtained by PCR from a cDNA library constructed from untreated RAW 264.7 cells. The coding sequence of human JM-a ErbB4 was provided by Dr. Shigeki Higashiyama (Ehime University, Ehime, Japan). Splice variants and substitution mutants of ALCAM and ErbB4 were constructed using a PCR-based method. Cytoplasmic PA tag (WAKO) was attached to C terminus of ALCAM proteins using a PCR-based method. The coding sequences of murine ALCAM and human ErbB4 were subcloned into N-terminally Halo-tagged expression plasmid (11) after removal of their signal sequences (Met1–Gly27 and Met1–Ser25, respectively) by PCR.
Cell-surface staining of halo-tagged proteins
RAW 264.7 cells expressing both N-terminally Halo-tagged ALCAM variant proteins and DsRed were stained with cell-impermeable HaloTag Alexa Fluor 488 Ligand (Promega) according to the manufacturers' instructions. Fluorescence images were taken under a confocal fluorescence microscope (Leica TCS SP5).
CRISPR/Cas9
Lentivirus vectors for CRISPR/Cas9-based genome editing were prepared using pLentiCRISPR v2 (Addgene). sgRNA design and cloning were performed according to a previous report using an online CRISPR design tool (CRISPRdirect) (30). The sequences of oligonucleotides used in this study were 5′-CACCGGATGAGGCAGACGATATAAG-3′ and 5′-AAACCTTATATCGTCTGCCTCATCC-3′. These oligonucleotides were annealed and inserted into BmsBI-digested pLentiCRISPR v2.
N-terminal sequencing of a membrane-anchored shedding product of ALCAM
HEK 293T cells expressing N-terminally Halo-tagged and cytoplasmic PA-tagged ALCAM were treated with 200 ng/ml TPA and 10 μm DAPT for 90 min and extracted. The extract was incubated with anti-PA tag antibody beads (WAKO) at 4 °C overnight, washed by TBS, and eluted by boiling in the saline containing 2% SDS and 40 mm DTT. Eluted proteins were separated by SDS-PAGE, blotted onto polyvinylidene difluoride membrane, and stained with Coomassie Blue. An ∼15-kDa membrane-anchored shedding product of ALCAM was excised and subjected to automated Edman degradation on a protein sequencer, Procise 491 cLC (Thermo Fisher Scientific).
Data availability
All data are included in the article.
Acknowledgments
We thank Shohei Motohashi and Sayana Yoneda for technical assistance and Dr. Takuya Takahashi and Takuya Azami for secondary structure prediction analyses.
Author contributions—R. I., Y. O., and K. S. conceptualization; R. I., R. T., M. S., T. T., N. N., T. U., T. K., J. T., and K. S. investigation; T. K., J. T., Y. O., and K. S. supervision; T. K. and J. T. methodology; Y. O. and K. S. funding acquisition; K. S. validation; K. S. writing-original draft.
Funding and additional information—This work was supported by Japan Society for the Promotion of Science (JSPS) KAKENHI Grant JP18K06911 and a research grant from the Astellas Foundation for Research on Metabolic Disorders. This work was supported by AMED-CREST, AMED under Grant Number JP19gm0610011 (to Y. O.)
Conflict of interest—K. S. was assigned to the Joint Research Department of Tokyo Medical and Dental University and Shionogi & Co. Ltd.
- ADAM
- a disintegrin and metalloproteases
- ALCAM
- activated leukocyte cell adhesion molecule
- LPS
- lipopolysaccharide
- TPA
- 12-O-tetradecanoylphorbol 13-acetate
- MEF
- mouse embryonic fibroblast
- HEK
- human embryonic kidney
- sgRNA
- single-guide RNA.
References
- 1. Seals D. F., and Courtneidge S. A. (2003) The ADAMs family of metalloproteases: multidomain proteins with multiple functions. Genes Dev. 17, 7–30 10.1101/gad.1039703 [DOI] [PubMed] [Google Scholar]
- 2. Blobel C. P. (2005) ADAMs: key components in EGFR signalling and development. Nat. Rev. Mol. Cell Biol. 6, 32–43 10.1038/nrm1548 [DOI] [PubMed] [Google Scholar]
- 3. Huovila A. P., Turner A. J., Pelto-Huikko M., Kärkkäinen I., and Ortiz R. M. (2005) Shedding light on ADAM metalloproteinases. Trends Biochem. Sci. 30, 413–422 10.1016/j.tibs.2005.05.006 [DOI] [PubMed] [Google Scholar]
- 4. Reiss K., and Saftig P. (2009) The “a disintegrin and metalloprotease” (ADAM) family of sheddases: physiological and cellular functions. Semin. Cell Dev. Biol. 20, 126–137 10.1016/j.semcdb.2008.11.002 [DOI] [PubMed] [Google Scholar]
- 5. Lichtenthaler S. F., Lemberg M. K., and Fluhrer R. (2018) Proteolytic ectodomain shedding of membrane proteins in mammals-hardware, concepts, and recent developments. EMBO J. 37, e99456 10.15252/embj.201899456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fong K. P., Barry C., Tran A. N., Traxler E. A., Wannemacher K. M., Tang H. Y., Speicher K. D., Blair I. A., Speicher D. W., Grosser T., and Brass L. F. (2011) Deciphering the human platelet sheddome. Blood 117, e15–e26 10.1182/blood-2010-05-283838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kawahara R., Lima R. N., Domingues R. R., Pauletti B. A., Meirelles G. V., Assis M., Figueira A. C., and Paes Leme A. F. (2014) Deciphering the role of the ADAM17-dependent secretome in cell signaling. J. Proteome Res. 13, 2080–2093 10.1021/pr401224u [DOI] [PubMed] [Google Scholar]
- 8. Caescu C. I., Jeschke G. R., and Turk B. E. (2009) Active-site determinants of substrate recognition by the metalloproteinases TACE and ADAM10. Biochem. J. 424, 79–88 10.1042/BJ20090549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tucher J., Linke D., Koudelka T., Cassidy L., Tredup C., Wichert R., Pietrzik C., Becker-Pauly C., and Tholey A. (2014) LC-MS based cleavage site profiling of the proteases ADAM10 and ADAM17 using proteome-derived peptide libraries. J. Proteome Res. 13, 2205–2214 10.1021/pr401135u [DOI] [PubMed] [Google Scholar]
- 10. Shirakabe K., Hattori S., Seiki M., Koyasu S., and Okada Y. (2011) VIP36 protein is a target of ectodomain shedding and regulates phagocytosis in macrophage Raw 264.7 cells. J. Biol. Chem. 286, 43154–43163 10.1074/jbc.M111.275586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shirakabe K., Shibagaki Y., Yoshimura A., Koyasu S., and Hattori S. (2014) A proteomic approach for the elucidation of the specificity of ectodomain shedding. J. Proteomics 98, 233–243 10.1016/j.jprot.2014.01.012 [DOI] [PubMed] [Google Scholar]
- 12. Shirakabe K., Omura T., Shibagaki Y., Mihara E., Homma K., Kato Y., Yoshimura A., Murakami Y., Takagi J., Hattori S., and Ogawa Y. (2017) Mechanistic insights into ectodomain shedding: susceptibility of CADM1 adhesion molecule is determined by alternative splicing and O-glycosylation. Sci. Rep. 7, 46174 10.1038/srep46174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bowen M. A., Patel D. D., Li X., Modrell B., Malacko A. R., Wang W. C., Marquardt H., Neubauer M., Pesando J. M., and Francke U. and (1995) Cloning, mapping, and characterization of activated leukocyte-cell adhesion molecule (ALCAM), a CD6 ligand. J. Exp. Med. 181, 2213–2220 10.1084/jem.181.6.2213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Swart G. W. (2002) Activated leukocyte cell adhesion molecule (CD166/ALCAM): developmental and mechanistic aspects of cell clustering and cell migration. Eur. J. Cell Biol. 81, 313–321 10.1078/0171-9335-00256 [DOI] [PubMed] [Google Scholar]
- 15. Elenius K., Corfas G., Paul S., Choi C. J., Rio C., Plowman G. D., and Klagsbrun M. (1997) A novel juxtamembrane domain isoform of HER4/ErbB4: isoform-specific tissue distribution and differential processing in response to phorbol ester. J. Biol. Chem. 272, 26761–26768 10.1074/jbc.272.42.26761 [DOI] [PubMed] [Google Scholar]
- 16. Rio C., Buxbaum J. D., Peschon J. J., and Corfas G. (2000) Tumor necrosis factor-α-converting enzyme is required for cleavage of erbB4/HER4. J. Biol. Chem. 275, 10379–10387 10.1074/jbc.275.14.10379 [DOI] [PubMed] [Google Scholar]
- 17. Cheng Q. C., Tikhomirov O., Zhou W., and Carpenter G. (2003) Ectodomain cleavage of ErbB-4: characterization of the cleavage site and m80 fragment. J. Biol. Chem. 278, 38421–38427 10.1074/jbc.M302111200 [DOI] [PubMed] [Google Scholar]
- 18. Sahin U., Weskamp G., Kelly K., Zhou H. M., Higashiyama S., Peschon J., Hartmann D., Saftig P., and Blobel C. P. (2004) Distinct roles for ADAM10 and ADAM17 in ectodomain shedding of six EGFR ligands. J. Cell Biol. 164, 769–779 10.1083/jcb.200307137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Horiuchi K., Kimura T., Miyamoto T., Takaishi H., Okada Y., Toyama Y., and Blobel C. P. (2007) Cutting edge: TNF-α-converting enzyme (TACE/ADAM17) inactivation in mouse myeloid cells prevents lethality from endotoxin shock. J. Immunol. 179, 2686–2689 10.4049/jimmunol.179.5.2686 [DOI] [PubMed] [Google Scholar]
- 20. Garbers C., Monhasery N., Aparicio-Siegmund S., Lokau J., Baran P., Nowell M. A., Jones S. A., Rose-John S., and Scheller J. (2014) The interleukin-6 receptor Asp358Ala single nucleotide polymorphism rs2228145 confers increased proteolytic conversion rates by ADAM proteases. Biochim. Biophys. Acta 1842, 1485–1494 10.1016/j.bbadis.2014.05.018 [DOI] [PubMed] [Google Scholar]
- 21. Riethmueller S., Somasundaram P., Ehlers J. C., Hung C. W., Flynn C. M., Lokau J., Agthe M., Düsterhöft S., Zhu Y., Grötzinger J., Lorenzen I., Koudelka T., Yamamoto K., Pickhinke U., Wichert R., et al. (2017) Proteolytic origin of the soluble human IL-6R in vivo and a decisive role of N-glycosylation. PLoS Biol. 15, e2000080 10.1371/journal.pbio.2000080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hebron K. E., Li E. Y., Arnold Egloff S. A., von Lersner A. K., Taylor C., Houkes J., Flaherty D. K., Eskaros A., Stricker T. P., and Zijlstra A. (2018) Alternative splicing of ALCAM enables tunable regulation of cell-cell adhesion through differential proteolysis. Sci. Rep. 8, 3208 10.1038/s41598-018-21467-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sato H., Takino T., Okada Y., Cao J., Shinagawa A., Yamamoto E., and Seiki M. (1994) A matrix metalloproteinase expressed on the surface of invasive tumour cells. Nature 370, 61–65 10.1038/370061a0 [DOI] [PubMed] [Google Scholar]
- 24. Sakamoto T., and Seiki M. (2017) Integrated functions of membrane-type 1 matrix metalloproteinase in regulating cancer malignancy: beyond a proteinase. Cancer Sci. 108, 1095–1100 10.1111/cas.13231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. van Kempen L. C., Nelissen J. M., Degen W. G., Torensma R., Weidle U. H., Bloemers H. P., Figdor C. G., and Swart G. W. (2001) Molecular basis for the homophilic activated leukocyte cell adhesion molecule (ALCAM)-ALCAM interaction. J. Biol. Chem. 276, 25783–25790 10.1074/jbc.M011272200 [DOI] [PubMed] [Google Scholar]
- 26. Weiner J. A., Koo S. J., Nicolas S., Fraboulet S., Pfaff S. L., Pourquié O., and Sanes J. R. (2004) Axon fasciculation defects and retinal dysplasias in mice lacking the immunoglobulin superfamily adhesion molecule BEN/ALCAM/SC1. Mol. Cell Neurosci. 27, 59–69 10.1016/j.mcn.2004.06.005 [DOI] [PubMed] [Google Scholar]
- 27. Zimmerman A. W., Joosten B., Torensma R., Parnes J. R., van Leeuwen F. N., and Figdor C. G. (2006) Long-term engagement of CD6 and ALCAM is essential for T-cell proliferation induced by dendritic cells. Blood 107, 3212–3220 10.1182/blood-2005-09-3881 [DOI] [PubMed] [Google Scholar]
- 28. Weidle U. H., Eggle D., Klostermann S., and Swart G. W. (2010) ALCAM/CD166: cancer-related issues. Cancer Genomics Proteomics 7, 231–243 [PubMed] [Google Scholar]
- 29. Michel L., Grasmuck C., Charabati M., Lécuyer M. A., Zandee S., Dhaeze T., Alvarez J. I., Li R., Larouche S., Bourbonnière L., Moumdjian R., Bouthillier A., Lahav B., Duquette P., Bar-Or A., et al. (2019) Activated leukocyte cell adhesion molecule regulates B lymphocyte migration across central nervous system barriers. Sci. Transl. Med. 11, eaaw0475 10.1126/scitranslmed.aaw0475 [DOI] [PubMed] [Google Scholar]
- 30. Ran F. A., Hsu P. D., Wright J., Agarwala V., Scott D. A., and Zhang F. (2013) Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308 10.1038/nprot.2013.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data are included in the article.