

Abstract
The transcription factor AHR (aryl hydrocarbon receptor) drives the expression of genes involved in detoxification pathways in cells exposed to pollutants and other small molecules. Moreover, AHR supports transcriptional programs that promote ribosome biogenesis and protein synthesis in cells stimulated to proliferate by the oncoprotein MYC. Thus, AHR is necessary for the proliferation of MYC-overexpressing cells. To define metabolic pathways in which AHR cooperates with MYC in supporting cell growth, here we used LC–MS–based metabolomics to examine the metabolome of MYC-expressing cells upon AHR knockdown. We found that AHR knockdown reduced lactate, S-lactoylglutathione, N-acetyl-l-alanine, 2-hydroxyglutarate, and UMP levels. Using our previously obtained RNA sequencing data, we found that AHR mediates the expression of the UMP-generating enzymes dihydroorotate dehydrogenase (quinone) (DHODH) and uridine monophosphate synthetase (UMPS), as well as lactate dehydrogenase A (LDHA), establishing a mechanism by which AHR regulates lactate and UMP production in MYC-overexpressing cells. AHR knockdown in glioblastoma cells also reduced the expression of LDHA (and lactate), DHODH, and UMPS but did not affect UMP levels, likely because of compensatory mechanisms in these cells. Our results indicate that AHR contributes to the regulation of metabolic pathways necessary for the proliferation of transformed cells.
Keywords: MYC (c-Myc), oncogene, aryl hydrocarbon receptor (AHR), metabolism, pyrimidine, glycolysis, glioblastoma, metabolomics, cancer, gene regulation
To obtain the energy and macromolecules necessary for cell growth and division, cancer cells adapt their metabolism to maximize the utilization of the nutrients available in their environment (1–3). The oncoprotein MYC is a master regulator of cellular metabolism (3, 4), coordinating the anabolic and catabolic needs of proliferating cells. For example, MYC directly activates the expression of genes involved in metabolic pathways such as the glycolytic enzyme lactate dehydrogenase A (LDHA) (5) or the pyrimidine synthesis enzymes 1-carbamoyl-phosphate synthetase 2, 2-aspartate transcarbamylase, and 3-dihydroorotase (CAD) (6) and dihydroorotate dehydrogenase (quinone) (DHODH) (7), among others (3, 8). Furthermore, MYC promotes the expression of transporters such as the solute carrier 2A1 (SLC2A1) (9), which imports glucose, or the solute carrier 1A5 (SLC1A5) (10, 11), which transports glutamine, thereby increasing the intracellular availability of nutrients (12). In addition to directly regulating the expression of metabolic enzymes and transporters, MYC promotes the expression of transcription factors that in turn modulate metabolic pathways such as the α subunit of hypoxia-inducible factor-1 or the lymphoid enhancer–binding factor 1 (13, 14).
Recently, our laboratory discovered that MYC promotes the expression of the aryl hydrocarbon receptor (AHR) (15). AHR is a cytoplasmic receptor that responds to exogenous ligands, such as the pollutant 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) (16, 17), or to endogenous ligands including the tryptophan catabolite kynurenine, which is elevated in cancer cells (10, 18). In the presence of its ligands, AHR shuttles to the nucleus where it dimerizes with aryl hydrocarbon receptor nuclear translocator (ARNT) (19) to bind to DNA and activate the expression of genes that contain xenobiotic responsive elements (XREs) in their promoters including genes involved in detoxification such as CYP1A1 (20). We found that AHR also regulates the expression of a subset of genes that are necessary for the growth of MYC-expressing cells such as NOLC1 and OGFOD1, which are involved in rRNA production and protein synthesis (15, 21, 22).
To determine the contribution of AHR to metabolic pathways regulated by MYC, we performed metabolomics of rat1 fibroblasts myc−/− with or without MYC upon AHR knockdown. By combining metabolomics and transcriptomics, we found that AHR contributes to the regulation of glycolysis and the de novo pyrimidine biosynthesis pathway in MYC-expressing cells. In addition, to identify metabolic pathways supported by AHR in glioblastoma (GBM), an aggressive brain tumor dependent on AHR (18), we examined the metabolome of GBM cells upon AHR knockdown and found that AHR knockdown affected several metabolic pathways in these cells.
Results and discussion
AHR regulates metabolic pathways in MYC-expressing cells
To define AHR-regulated metabolic pathways downstream of MYC, we performed an LC–MS metabolomics screening in the myc −/− rat fibroblasts HO15.19 that were stably reconstituted by retroviral infection of MYC or empty vector, in which AHR was silenced for 48 h, using two different siRNA (each in triplicate) (Fig. 1, A and B). Although AHR knockdown was efficient 48 h after AHR siRNA transfection, cell proliferation was modestly affected (15), making this an appropriate time point to identify metabolic events that cause reduced cell growth.
Figure 1.
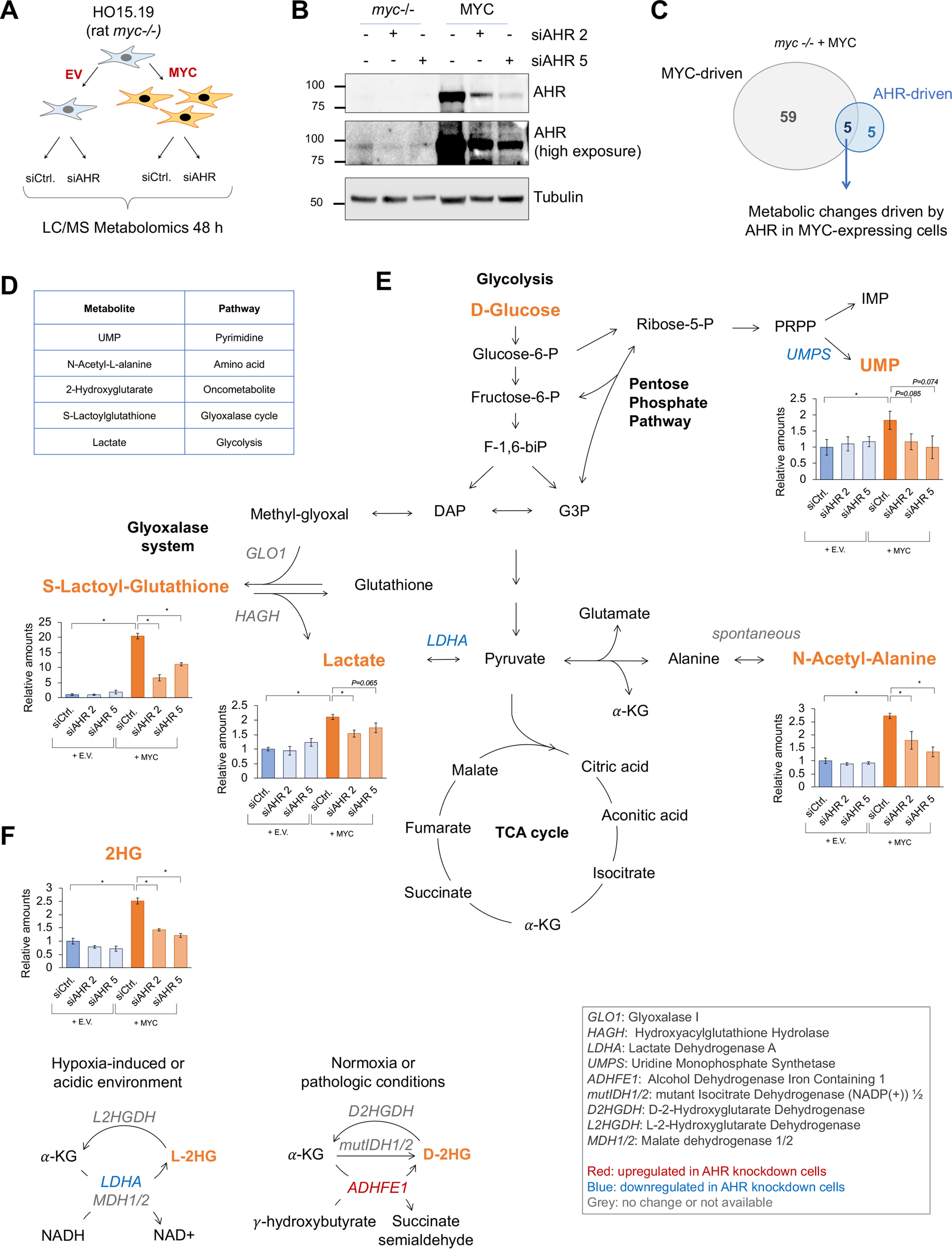
AHR regulates metabolic pathways in MYC-expressing cells. A, schematic representation of the LC–MS metabolomics analyses in the myc−/− rat fibroblasts HO15.19 infected with either a MYC-expressing or an empty vector (EV). B, Western blotting showing the silencing of AHR in the myc−/− cells expressing MYC or empty vector 48 h after transfection with two independent AHR siRNAs (siAHR) or control siRNA (siCtrl). C, Venn diagram of metabolites regulated by MYC, AHR, or both. D, description of the metabolites shown in C. E and F, representation of the relative amounts of the five metabolites found to be driven by AHR in the MYC-expressing cells (C and D) normalized to myc−/− conditions and the interconnections between the pathways in which they are directly involved. Asterisks represent p values of <0.05.
We first defined the metabolic signature regulated by MYC by comparing the abundance of metabolites in myc −/− fibroblasts expressing MYC and myc −/− expressing empty vector using a cutoff p value of ≤0.05. MYC regulated the abundance of 64 metabolites (Fig. 1C and Fig. S1), including metabolites in pathways previously attributed to MYC function such as glycolysis, TCA cycle, and nucleotide biosynthesis (3) (Fig. 1C and Fig. S1). To identify AHR-mediated metabolic changes in the MYC-expressing cells, we considered the metabolites for which abundance was affected with both AHR siRNAs using a cutoff p value of ≤0.01. AHR knockdown altered the levels of 10 metabolites in the myc−/− fibroblasts reconstituted with MYC (Fig. 1C and Figs. S2, A and B). Five of the ten metabolites were induced by MYC and reduced upon AHR knockdown, suggesting that MYC depended on AHR to increase the abundance of those metabolites (Fig. 1, D and E). AHR knockdown also altered the abundance of seven metabolites in the myc−/− fibroblasts (Fig. S2, C and D). With the goal of identifying novel AHR-mediated, growth-promoting pathways, we focused this study on the metabolites increased by MYC and decreased by AHR knockdown.
The five metabolites affected by AHR knockdown in MYC-expressing cells were lactate, S-lactoylglutathione, N-acetyl-l-alanine, 2-hydroxyglutarate, and UMP (Fig. 1, E and F). Lactate is the end product of glycolysis and therefore is used as a readout of glycolytic activity (3). S-lactoylglutathione is metabolized by glyoxalase II through the glyoxalase system, producing the antioxidant GSH and lactate in the mitochondria (23). N-Acetyl-l-alanine is a derivative of alanine. UMP can be produced by metabolizing ribose-5-phosphate through the de novo pyrimidine biosynthesis pathway or by phosphorylating uridine through the salvage pathway (24). UMP gives rise to other pyrimidines such as UTP, CTP, and TTP, which are needed for RNA and DNA synthesis (25). Our experiment did not distinguish between the two 2-hydroxyglutarate (2HG) enantiomers, d-2HG or l-2HG (26). d-2HG is a reduced form of α-ketoglutarate produced by the aberrant activity of the mutant enzyme isocitrate dehydrogenase 1/2 (mutIDH1/2) present in cancer cells (27) (Fig. 1F). However, IDH1/2 were not mutated in the rat fibroblast used in this experiment. d-2HG can be produced/accumulated by additional mechanisms such as decreased expression of d-2-hydroxyglutarate dehydrogenase (28) or increased expression of ADHFE1 (alcohol dehydrogenase iron-containing 1) (29) (Fig. 1F). l-2HG in contrast, is mainly produced in hypoxic or acidic environments by LDHA or MDH1/2 reactions (26, 30, 31). l-2-Hydroxyglutarate dehydrogenase is responsible for decreasing the abundance of l-2HG by converting it back to α-ketoglutarate (32) (Fig. 1F). Because of their ability to inhibit hydroxylases such as the Jumonji histone demethylases, the TET cytosine demethylases, and other prolyl hydroxylases, d- and l-2-HG are known as oncometabolites (33).
AHR regulates expression of metabolic genes
Using our previously published RNA-seq data, which was performed under the same conditions as the metabolomic screening (15), we examined the expression of the enzymes directly involved in the production of the five metabolites that were induced by MYC and reduced upon AHR knockdown (Fig. 1, E and F, and Fig. S2, E–I). The mRNA levels of three genes encoding for enzymes related to the metabolites discovered in our metabolomics (Fig. 1E) were altered upon AHR knockdown. ADHFE1, which produces d-2-hydroxyglutarate (D2HG), was up-regulated upon AHR knockdown and thus did not align with the observed decreased in abundance of the metabolite (Fig. 1F and Fig. S2F). UMPS (Fig. S2H), which converts orotate into UMP, and LDHA (Fig. S2I), which catalyzes the conversion of pyruvate into lactate, were down-regulated upon AHR knockdown, which is consistent with the reduced levels of UMP and lactate, respectively (Fig. 1E). The decrease in LDHA could also account for the observed decrease in 2-HG (Fig. 1F) (26). We validated LDHA reduction upon AHR knockdown by qPCR (Fig. S2J) and reported that the rat LDHA promoter contained MYC-binding (E-box: CACGTG) and AHR-binding (XRE: GCGTG) sites (Fig. S2K).
Although there were no AHR-regulated genes that could account for the reduction in S-lactoylglutathione, 2-hydroxyglutarate, and N-acetyl-l-alanine (Fig. 1E and Fig. S2, E–H), we observed that glucose and pyruvate were also lower upon AHR silencing in MYC-expressing cells (Fig. S3B). Because S-lactoylglutathione, 2-hydroxyglutarate, and N-acetyl-l-alanine are derived from glycolytic intermediates (Fig. 1, E and F), a decrease in glucose and pyruvate could contribute, at least in part, to their lower abundance upon AHR knockdown (Fig. 1E).
Our RNA-seq data indicated that AHR knockdown caused a modest reduction in the mRNA levels of additional glycolytic (ALDOA, GAPDH, PGK1, and ENO1) (Fig. S3C) and TCA cycle (ACO2, IDH1, and MDH2) enzymes (Fig. S4C). The glycolytic products of those enzymes were not detected in our metabolomics screening, likely because of their low abundance in these samples. Moreover, the levels of the products of those TCA cycle enzymes did not change upon AHR knockdown. It is possible that compensatory mechanisms maintained the levels of most TCA intermediates upon AHR knockdown.
AHR regulates the expression of enzymes in the de novo pyrimidine biosynthesis pathway in fibroblasts
UMP can be synthesized through the salvage pathway in which nucleotides are recycled by either phosphorylating/dephosphorylating reactions or the de novo pathway in which ribose-5-phosphate and amino acids are processed by the enzymes CAD, DHODH, and UMPS (Fig. 2A). CAD and DHODH were previously found to be up-regulated by MYC (6). Our RNA-seq data indicated that although the expression of the genes involved in the salvage pathway were not affected by AHR knockdown, mRNA levels of CAD, DHODH, and UMPS were reduced upon AHR knockdown (Fig. 2B and Fig. S4E). These data and our metabolomic data showing a decrease in UMP (Fig. 1E) suggest that the de novo pyrimidine biosynthesis pathway is regulated by AHR. Because AHR silencing decreased the expression of the enzymes involved in the de novo pyrimidine biosynthesis, we further examined the role of AHR in the regulation of this pathway.
Figure 2.
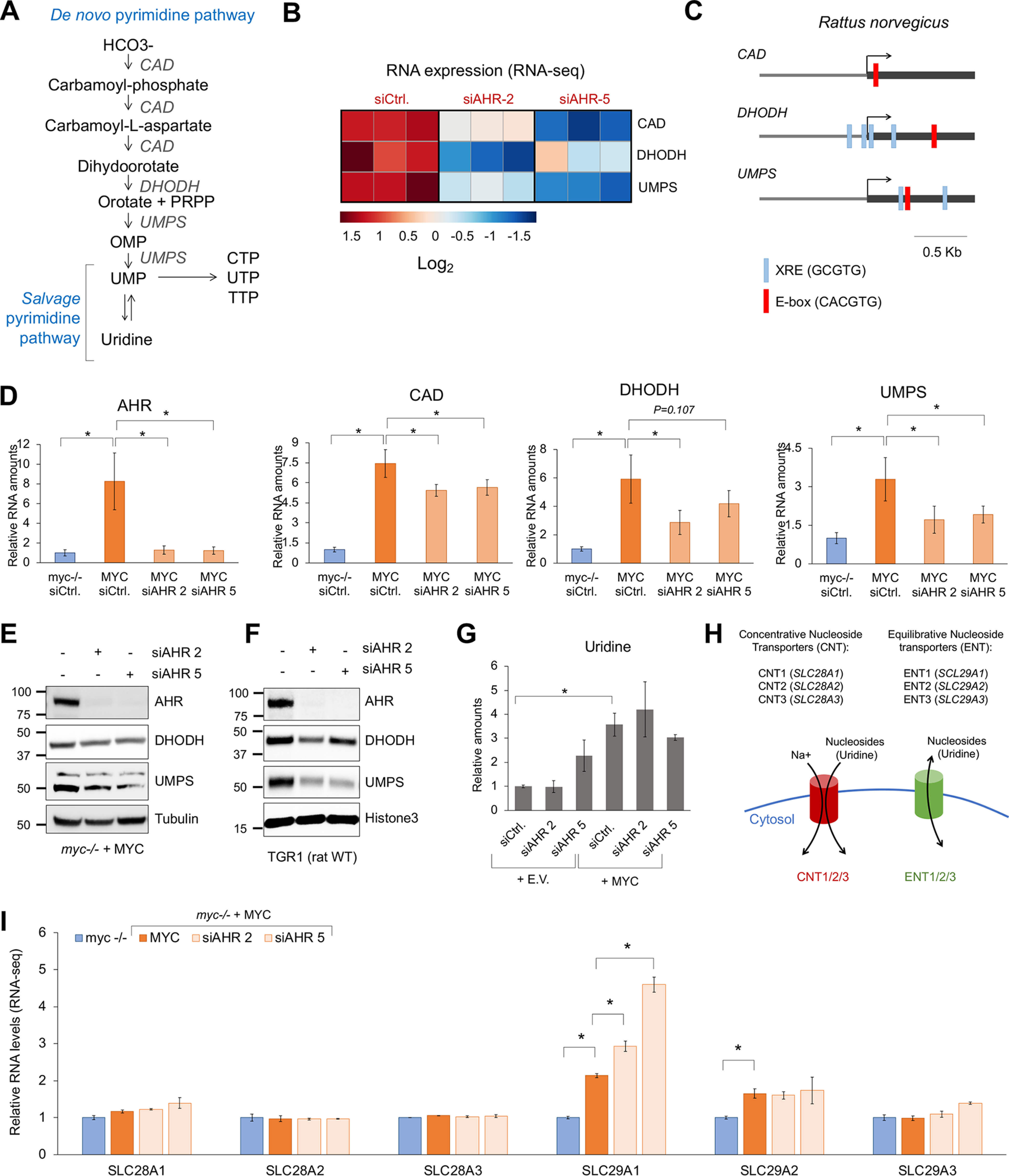
AHR regulates the expression of enzymes in the de novo pyrimidine biosynthesis pathway in fibroblasts. A, schematic representation of the de novo and salvage pyrimidine biosynthesis pathways. B, heat map of the expression levels of CAD, DHODH, and UMPS in myc−/− and MYC cells 48 h after transfection with control or AHR siRNA. C, schematic representation of the rat CAD, DHODH, and UMPS gene promoter regions displaying the presence of XRE (AHR-binding sites) or E-box (MYC-binding sites). D, qPCR for AHR, CAD, DHODH, and UMPS in myc−/− cells expressing MYC or empty vector (E.V.) 72 h after transfection with control siRNA (siCtrl) or AHR siRNA (siAHR). E, Western blotting for the indicated proteins in myc−/− rat fibroblasts reconstituted with MYC 72 h after transfection with control or AHR siRNAs. F, Western blotting of the WT rat fibroblasts TGR1 72 h after transfection with control or AHR siRNAs. G, relative amounts of uridine levels found in the metabolomic screening, normalized to the myc−/− siCtrl conditions. H, schematic representation of the nucleoside transporters. I, relative mRNA levels of the nucleoside transporters found in our RNA-seq in myc−/− and MYC cells 48 h after transfection with control or AHR siRNA. Asterisks represent p values of <0.05.
Bioinformatic analysis of the rat CAD, DHODH, and UMPS promoter regions within ± 1 kb of the transcription start site showed that all three genes have one MYC-binding site and that DHODH and UMPS, but not CAD, have several AHR-binding sites (Fig. 3E). Supporting these data, DHODH and UMPS mRNA levels were significantly lower than those of CAD in MYC-expressing cells upon AHR silencing (Fig. 2D). DHODH and UMPS protein levels were also reduced 72 h after AHR silencing (Fig. 2, E and F). Altogether, these data suggest that AHR regulates the transcription of DHODH and UMPS and to a lesser extent CAD.
Figure 3.
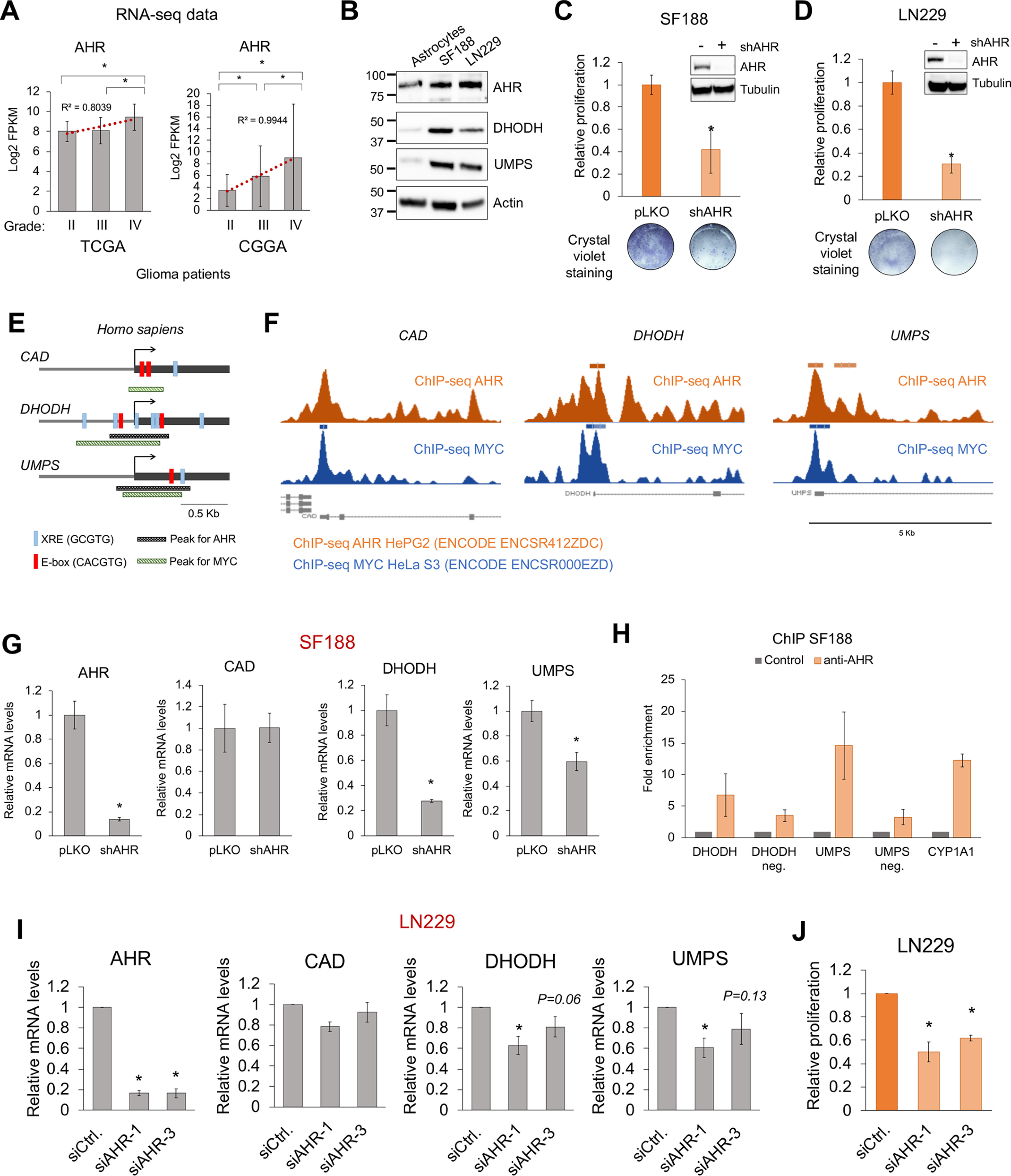
AHR regulates the expression of the de novo pyrimidine biosynthesis pathway enzymes in glioblastoma. A, mRNA levels for AHR in grade II, III, or IV/GBM glioma patients derived from the TCGA and CGGA databases. B, Western blotting displaying the expression of AHR, DHODH, and UMPS in the glioblastoma SF188 and LN229 cells and in immortalized human p14/ARF−/− astrocytes (cell of origin of glioblastoma). C and D, relative proliferation of SF188 (C) and LN229 (D) cells 72 h after infection with a lentiviral vector containing an AHR shRNA. Upper panels show AHR silencing by Western blotting. Lower panels show crystal violet staining of infected cultures. Glioblastoma cells were sensitive to AHR silencing. E, schematic representation of the human CAD, DHODH, and UMPS gene promoter regions showing the presence of XRE (AHR-binding sites) or E-box (MYC-binding sites) as well as the presence of MYC and AHR ChIP-seq peaks found in the ENCODE database. F, representation of the AHR and MYC ChIP-seq obtained from the ENCODE database for the human CAD, DHODH, and UMPS genes. G, qPCR for AHR, CAD, DHODH, and UMPS in SF188 72 h after infection with a lentiviral vector containing an AHR shRNA. H, ChIP of AHR on DHODH, UMPS, and CYP1A1 promoter regions. DHODH-negative and UMPS-negative (neg.) sites with no XRE were used as negative controls. n = 1 for DHODH, DHODH-negative, and UMPS-negative sites; n = 2 for UMPS and CYP1A1. I, qPCR for AHR, CAD, DHODH, and UMPS in LN229 72 h after AHR siRNA transfection. The expression of DHODH and UMPS but not CAD was decreased when AHR was silenced. J, relative proliferation of LN229 72 h after AHR siRNA transfection. Asterisks represent p values of <0.05. siCtrl, control siRNA; siAHR, AHR siRNA.
The nucleoside uridine can be converted into UMP and vice versa (Fig. 2A) (25). Our metabolomics screening indicated that uridine abundance was not affected by AHR knockdown (Fig. 2G), thus suggesting that the uridine pool could be maintained by the import of extracellular uridine (Fig. 2H). Interestingly, of the six nucleoside transporters (Fig. 2H): concentrative nucleoside transporters (CNT1–3 encoded by SLC28A1–3) and equilibrative nucleoside transporters (ENT1–3 encoded by SLC29A1–3) (34), the expression of SLC29A1(ENT1) was elevated by MYC and further induced upon AHR knockdown (Fig. 2I). SLC29A2 (ENT2) was also elevated in the MYC-expressing cells but was not affected by AHR knockdown (Fig. 2I). These data suggest that when the de novo pyrimidine biosynthesis is impaired by AHR silencing, cells attempt to maintain the intracellular pyrimidine pool by importing uridine from the extracellular environment to compensate for the reduction in the de novo biosynthesis of UMP.
AHR regulates the expression of enzymes in the de novo pyrimidine biosynthesis pathway in glioblastoma cells
The RNA-seq data derived from grade II, III, and IV/GBM patient data deposited in The Cancer Genome Atlas (TCGA) and the Chinese Glioma Genome Atlas (CGGA) databases indicated that the expression of AHR positively correlated with higher grade gliomas (Fig. 3A). Furthermore, AHR, DHODH, and UMPS protein levels were higher in the SF188 and LN229 GBM cells than in the nontransformed human immortalized p14/ARF−/− astrocytes (cell of origin of GBM) (Fig. 3B). The expression of ARNT (AHR heterodimeric partner (19)), MYC, and MAX (MYC heterodimeric partner (35)) in gliomas did not consistently correlate with disease grade (Fig. S5, A and B). In agreement with data from previous studies (18), our data showed that the proliferation of the GBM cells SF188 and LN229 was reduced upon infection with AHR shRNA-containing lentiviral particles (Fig. 3, C and D). This was due to cell cycle arrest, as indicated by higher levels of the cell cycle inhibitor p27 and lower levels of the cell cycle protein cyclin A1, as well as to the activation of apoptosis as shown by an increase in cleaved caspase 3 and cleaved poly(ADP-ribose) polymerase 1 (Fig. S5C).
Bioinformatic analysis of the human CAD, DHODH, and UMPS promoter regions within ± 1 kb of the transcription start site found that these genes exhibited MYC- and AHR-binding sites (Fig. 3E). Furthermore, ChIP-seq data deposited from the ENCODE database (processed with the hg19/reference genome) demonstrated that MYC significantly binds to the promoter regions of CAD, DHODH, and UMPS. AHR binds significantly to DHODH and UMPS (Fig. 3, E and F, and Fig. S6A). Even though a peak for AHR on CAD was not detected, the AHR ChIP-seq signal visualization shows an enrichment for AHR on the CAD promoter as compared with surrounding areas, suggesting that AHR binds to the promoter of CAD with lower affinity (Fig. 3, E and F, and Fig. S6A). ARNT and MAX ChIP-seq data (processed with the hg19/reference genome) indicated that MAX but not ARNT also binds to the promoter regions of CAD, DHODH, and UMPS (Fig. S6B). It is likely that MYC and MAX form a complex to bind these gene promoters. In contrast, it is possible that AHR binds to the promoter of these genes with another, unknown, partner or that the ChIP-seq for ARNT was not sensitive enough to identify ARNT-positive peaks in the CAD, DHODH, and UMPS promoters.
As positive controls for AHR-ARNT and MYC-MAX, the regions of the CYP1A1 (bona fide AHR-ARNT target gene (20)) and LDHA (bona fide MYC-MAX target gene (5)) were analyzed. As expected, AHR and ARNT bind to the CYP1A1 promoter, and MYC and MAX bind to the LDHA promoter (Fig. S6B), thus validating the reliability of the ChIP-seq results. Interestingly, AHR displayed a positive peak in the LDHA promoter (Fig. S6B), which supports our metabolomic and transcriptional data on lactate (Fig. 1E) and LDHA expression (Fig. S2, I and J).
Consistent with the ENCODE ChIP-seq data, our data show that DHODH and UMPS mRNA levels but not CAD mRNA levels were reduced in the SF188 and LN229 cells upon AHR knockdown with AHR shRNA-containing lentiviral particles (Fig. 3G and Fig. S5E). Moreover, ChIP experiments revealed that AHR bound to the promoter regions of DHODH and UMPS in SF188 cells (Fig. 3H). Distant regions on the DHODH and UMPS genes that did not contain XRE elements were used as negative controls. The CYP1A1 promoter, a canonical AHR target gene, was used as a positive control for AHR binding (Fig. 3H). We also confirmed that silencing AHR with siRNA reduced the levels of DHODH and UMPS, as well as the proliferation of LN229 (Fig. 3, I and J) and GBM9 cells (Fig. S5D). These data indicate that AHR regulates the expression of the enzymes involved in the de novo pyrimidine biosynthesis in GBM cells.
AHR regulates metabolic pathways in GBM cells
To define AHR-regulated metabolic pathways specific to GBM cells, we performed LC–MS metabolomics in SF188 cells infected with lentiviral particles containing two different AHR shRNAs (named 84 and 86) or a control vector (pLKO) (Fig. 4A). AHR was efficiently silenced 72 h after infection (Fig. 3B). Applying a cutoff p value of ≤0.01 in both AHR shRNA conditions, we found 26 metabolites affected by AHR knockdown in the SF188 cells (Fig. 4C). Of those, 2 of them changed inconsistently with the 2 AHR shRNAs, probably because of off-target effects, 13 of them were increased, and 11 were decreased. These metabolites were part of key pathways, such as fatty acid and nucleotide metabolism and redox equilibrium (Fig. 4D), suggesting that AHR may be involved in maintaining the metabolic homeostasis of GBM cells. We then examined the abundance of the metabolites found to be regulated by AHR in fibroblasts (Fig. 1) and found that although there was a trend toward decreased lactate, S-lactoylglutathione, d-2-hydroxyglutarate, and N-acetyl-alanine, the results were not significant with both shRNAs (Fig. 4E).
Figure 4.
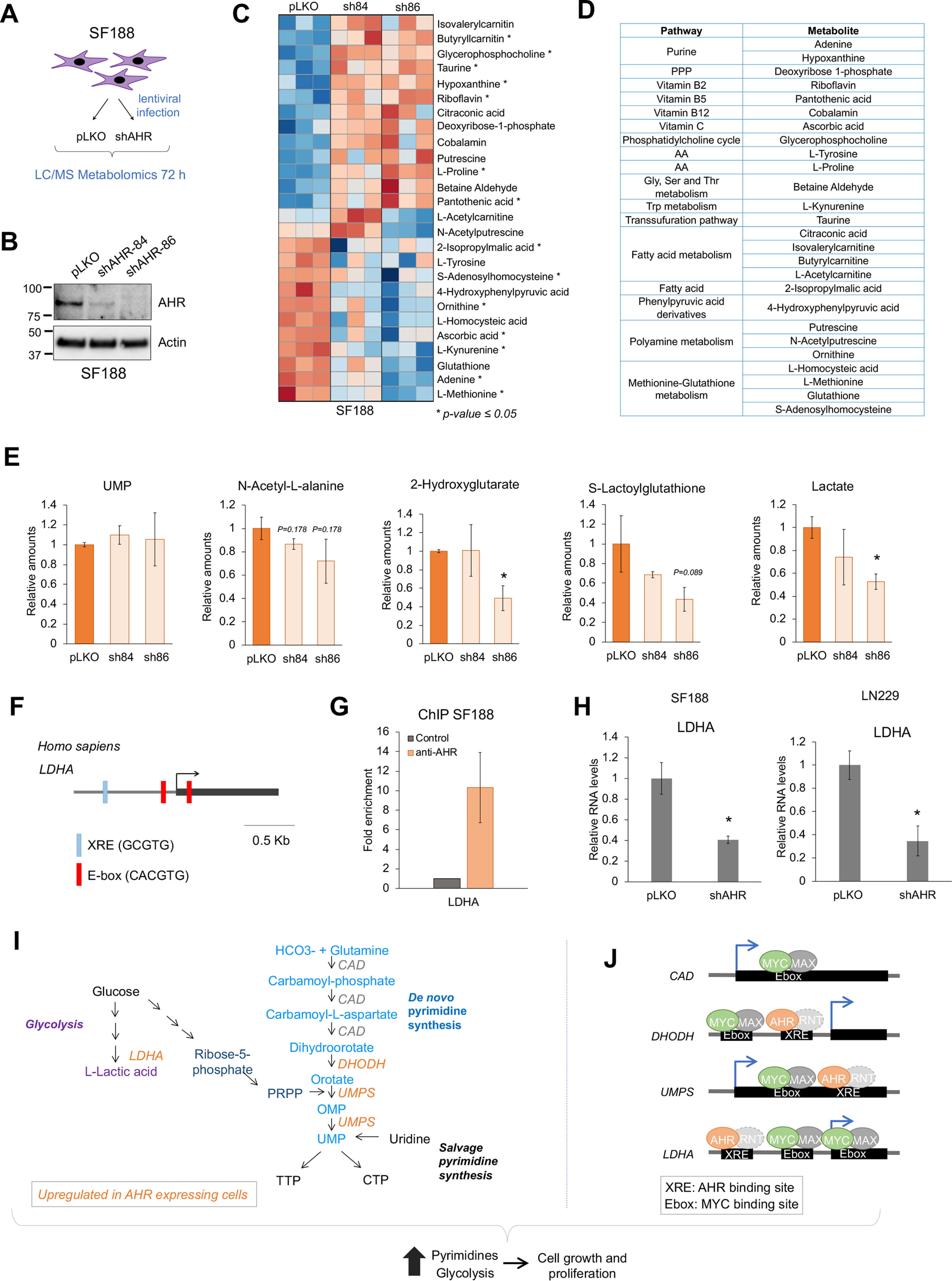
AHR regulates metabolism in GBM cells. A, schematic representation of the LC–MS metabolomics experiment in SF188 cells infected with AHR shRNA-containing lentiviral particles. B, Western blotting showing the silencing of AHR in SF188 72 h after infection with two independent AHR shRNA-containing lentiviral particles. C, heat map of the metabolites for which abundance changed upon AHR knockdown with the two shRNAs. D, grouping of the metabolites in C. E, relative amounts of lactate, S-lactoylglutathione, N-acetyl-l-alanine, 2-hydroxyglutarate, and UMP in the SF188 upon AHR knockdown. F, schematic representation of the human LDHA gene promoter region. G, ChIP of AHR on LDHA, n = 2. H, qPCR for LDHA in SF188 and LN229 72 h after infection with a lentiviral vector containing an AHR shRNA. I and J, proposed model for the regulation of metabolism by AHR and its binding of MYC and AHR to the promoters of CAD, DHODH, UMPS, and LDHA.
Despite the decrease in DHODH and UMPS mRNA levels upon AHR knockdown (Fig. 3G), the abundance of UMP or other pyrimidine forms was not affected in SF188 cells upon AHR silencing (Fig. 4E and Fig. S7A), thus indicating that metabolic outputs should not be inferred simply based on the expression of single metabolic enzymes. Interestingly, the levels of uridine in SF188 cells were more than 10 times higher in AHR knockdown cells (Fig. S7A), which could explain why UMP and other pyrimidines levels were not affected by AHR knockdown. To investigate whether uridine was being imported from the culture media, we measured the expression of the nucleoside transporters SLC28A1–3 and SLC29A1–3 upon AHR knockdown (Fig. S7B). Unlike the results observed in the rat fibroblasts (Fig. 2I), the expression of SLC29A1 was decreased, whereas SLC29A3 was modestly increased upon AHR knockdown in SF188 cells, thus suggesting that other mechanisms, such as increased enzymatic activity of the de novo or salvage pathway, may contribute to the increase in uridine levels. The addition of uridine to the culture media did not rescue the effects of AHR knockdown in proliferation (Fig. S7, C and D), likely because of the importance of other growth-promoting pathways regulated by AHR such as protein synthesis (15).
Because LDHA was decreased in the rat fibroblasts upon AHR knockdown (Fig. S2J) and lactate levels were decreased upon AHR knockdown in SF188 cells (Fig. 4E), we analyzed the promoter of the human LDHA. Human LDHA promoter had one XRE, which is consistent with the peak of AHR observed in the ENCODE ChIP-seq data (Fig. S6B) and with our results showing that AHR bound to the promoter of LDHA in SF188 cells (Fig. 4G). Moreover, AHR knockdown decreased LDHA mRNA levels in SF188 and LN229 cells (Fig. 4H).
Based on our results, we proposed a model in which AHR cooperates with MYC to regulate metabolic pathways necessary for the rapid proliferation of MYC-transformed cells including the de novo pyrimidine biosynthesis pathway and glycolysis through the transcriptional regulation of LDHA, DHODH, and UMPS genes (Fig. 4, I and J).
Experimental procedures
Cell cultures
The cells were maintained in Dulbecco's modified Eagle's medium with 10% fetal bovine serum and 100 units/ml penicillin/streptomycin. For transient transfections, the wells were seeded with 150,000 cells, and the cells were transfected with 3 μl of siRNA (20 μm) and 3 μl of Lipofectamine RNAiMAX. The day after transfection, the medium was replaced with fresh medium. After 3 days, the cells were harvested for qPCR or stained with crystal violet to assess proliferation. For viral transduction, 50,000 cells were mixed with 10 μg/ml Polybrene and pLKO or shRNA-containing virus. After the indicated times, the cells were harvested for qPCR, metabolomic analysis, or staining with crystal violet to assess proliferation.
RT-qPCR and Western blotting
Total RNA was extracted with TRI Reagent® solution (Sigma), and the cDNA produced with iScript RT Supermix for RT-qPCR (Bio-Rad). qPCR was performed with the iTaqTM Universal SYBR® Green Supermix (Bio-Rad). Expression levels for each gene were normalized to β-actin expression. For Western blots, the cells were harvested and lysed in RIPA buffer with protease and phosphatase inhibitors. The antibodies, primers, siRNAs, and shRNAs used in this study are indicated in Table S1.
ChIP-qPCR
ChIP was performed essentially as described (36, 37). The cells at 70% confluency were fixed with 1% formaldehyde. The nuclei were purified and then lysed with RIPA buffer. DNA was sonicated to obtain ∼500-bp fragments. Nuclei lysates were diluted 1:10 for immunoprecipitation with AHR antibody. Negative controls lacked antibodies. After immunoprecipitation, DNA was purified and analyzed by qPCR. The primers used in this study are indicated in Table S1.
TCGA and CGGA data analysis
RNA expression data for grade II–IV (GBM) gliomas were obtained from TCGA and the CGGA (RRID:SCR_018802).
LC–MS metabolomic screening
Samples for LC–MS measurements were washed three times with saline, harvested in 80% cold methanol, and snap-frozen three times. Next, the samples were centrifuged at 15,000 rpm for 15 min. The supernatants were collected and dried by SpeedVac. The metabolites were measured as described earlier (38). The metabolomic data are presented in Tables S2 and S3.
Quantification and statistical analysis
All statistical analyses were performed using two-tailed unpaired Student's t test analysis; p values of ≤0.05 were considered statistically significant. To identify MYC-driven metabolic changes, a p value of ≤0.05 was used when comparing myc−/− fibroblasts expressing MYC or empty vector. To identify AHR-driven metabolic changes, a p value ≤ 0.01 was used when comparing AHR siRNA or shRNA with siRNA control conditions. All values are reported as means ± S.D. in each figure.
Data availability
All the data described are contained within the article.
Supplementary Material
Acknowledgments
We are grateful to the Sorrell lab members, Dr. Joachim Seemann, and Dr. Jin Suk Park for valuable input.
This article contains supporting information.
Author contributions—M. C. L.-N. and M. C.-S. conceptualization; M. C. L.-N. data curation; M. C. L.-N. formal analysis; M. C. L.-N. and L. P.-C. validation; M. C. L.-N. and L. P.-C. investigation; M. C. L.-N. visualization; M. C. L.-N. methodology; M. C. L.-N. and M. C.-S. writing-original draft; L. G. Z., S. B., and R. J. D. resources; S. B. software; M. C.-S. supervision; M. C.-S. funding acquisition.
Funding and additional information—This work was supported by Cancer Prevention and Research Institute of Texas Grants RR150059 and RP150596, American Cancer Society Grant IRG-17-174-13, Welch Foundation Grant I-1914, NCI R01CA245548, and a University of Texas Southwestern Circle of Friends Early Investigator Award. M. C.-S. is a Virginia Murchison Linthicum Scholar in Medical Research.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- RNA-seq
- RNA sequencing
- XRE
- xenobiotic responsive element
- GBM
- glioblastoma
- 2HG
- 2-hydroxyglutarate
- qPCR
- quantitative PCR
- TCGA
- The Cancer Genome Atlas
- CGGA
- Chinese Glioma Genome Atlas
- ChIP-seq
- ChIP sequencing
- shRNA
- small hairpin RNA
- AHR
- aryl hydrocarbon receptor
- D2HG
- d-2-hydroxyglutarate.
References
- 1. Faubert B., Solmonson A., and DeBerardinis R. J. (2020) Metabolic reprogramming and cancer progression. Science 368, eaaw5473 10.1126/science.aaw5473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. DeBerardinis R. J., and Chandel N. S. (2016) Fundamentals of cancer metabolism. Sci. Adv. 2, e1600200 10.1126/sciadv.1600200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stine Z. E., Walton Z. E., Altman B. J., Hsieh A. L., and Dang C. V. (2015) MYC, metabolism, and cancer. Cancer Discov. 5, 1024–1039 10.1158/2159-8290.CD-15-0507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Camarda R., Williams J., and Goga A. (2017) In vivo reprogramming of cancer metabolism by MYC. Front. Cell Dev. Biol. 5, 35 10.3389/fcell.2017.00035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shim H., Dolde C., Lewis B. C., Wu C. S., Dang G., Jungmann R. A., Dalla-Favera R., and Dang C. V. (1997) c-Myc transactivation of LDH-A: implications for tumor metabolism and growth. Proc. Natl. Acad. Sci. U.S.A. 94, 6658–6663 10.1073/pnas.94.13.6658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Eberhardy S. R., and Farnham P. J. (2001) c-Myc mediates activation of the cad promoter via a post-RNA polymerase II recruitment mechanism. J. Biol. Chem. 276, 48562–48571 10.1074/jbc.M109014200 [DOI] [PubMed] [Google Scholar]
- 7. Liu Y. C., Li F., Handler J., Huang C. R., Xiang Y., Neretti N., Sedivy J. M., Zeller K. I., and Dang C. V. (2008) Global regulation of nucleotide biosynthetic genes by c-Myc. PLoS One 3, e2722 10.1371/journal.pone.0002722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chalishazar M. D., Wait S. J., Huang F., Ireland A. S., Mukhopadhyay A., Lee Y., Schuman S. S., Guthrie M. R., Berrett K. C., Vahrenkamp J. M., Hu Z., Kudla M., Modzelewska K., Wang G., Ingolia N. T., et al. (2019) MYC-driven small-cell lung cancer is metabolically distinct and vulnerable to arginine depletion. Clin. Cancer Res. 25, 5107–5121 10.1158/1078-0432.CCR-18-4140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Osthus R. C., Shim H., Kim S., Li Q., Reddy R., Mukherjee M., Xu Y., Wonsey D., Lee L. A., and Dang C. V. (2000) Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J. Biol. Chem. 275, 21797–21800 10.1074/jbc.C000023200 [DOI] [PubMed] [Google Scholar]
- 10. Venkateswaran N., Lafita-Navarro M. C., Hao Y. H., Kilgore J. A., Perez-Castro L., Braverman J., Borenstein-Auerbach N., Kim M., Lesner N. P., Mishra P., Brabletz T., Shay J. W., DeBerardinis R. J., Williams N. S., Yilmaz O. H., et al. (2019) MYC promotes tryptophan uptake and metabolism by the kynurenine pathway in colon cancer. Genes Dev. 33, 1236–1251 10.1101/gad.327056.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Marchingo J. M., Sinclair L. V., Howden A. J., and Cantrell D. A. (2020) Quantitative analysis of how Myc controls T cell proteomes and metabolic pathways during T cell activation. Elife 9, e53725 10.7554/eLife.53725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wise D. R., DeBerardinis R. J., Mancuso A., Sayed N., Zhang X. Y., Pfeiffer H. K., Nissim I., Daikhin E., Yudkoff M., McMahon S. B., and Thompson C. B. (2008) Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. U.S.A. 105, 18782–18787 10.1073/pnas.0810199105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Doe M. R., Ascano J. M., Kaur M., and Cole M. D. (2012) Myc posttranscriptionally induces HIF1 protein and target gene expression in normal and cancer cells. Cancer Res. 72, 949–957 10.1158/0008-5472.CAN-11-2371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hao Y. H., Lafita-Navarro M. C., Zacharias L., Borenstein-Auerbach N., Kim M., Barnes S., Kim J., Shay J., DeBerardinis R. J., and Conacci-Sorrell M. (2019) Induction of LEF1 by MYC activates the WNT pathway and maintains cell proliferation. Cell Commun. Signal. 17, 129 10.1186/s12964-019-0444-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lafita-Navarro M. C., Kim M., Borenstein-Auerbach N., Venkateswaran N., Hao Y. H., Ray R., Brabletz T., Scaglioni P. P., Shay J. W., and Conacci-Sorrell M. (2018) The aryl hydrocarbon receptor regulates nucleolar activity and protein synthesis in MYC-expressing cells. Genes Dev. 32, 1303–1308 10.1101/gad.313007.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Denison M. S., and Nagy S. R. (2003) Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Annu. Rev. Pharmacol. Toxicol. 43, 309–334 10.1146/annurev.pharmtox.43.100901.135828 [DOI] [PubMed] [Google Scholar]
- 17. Fernandez-Salguero P. M., Hilbert D. M., Rudikoff S., Ward J. M., and Gonzalez F. J. (1996) Aryl-hydrocarbon receptor-deficient mice are resistant to 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced toxicity. Toxicol. Appl. Pharmacol. 140, 173–179 10.1006/taap.1996.0210 [DOI] [PubMed] [Google Scholar]
- 18. Opitz C. A., Litzenburger U. M., Sahm F., Ott M., Tritschler I., Trump S., Schumacher T., Jestaedt L., Schrenk D., Weller M., Jugold M., Guillemin G. J., Miller C. L., Lutz C., Radlwimmer B., et al. (2011) An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 478, 197–203 10.1038/nature10491 [DOI] [PubMed] [Google Scholar]
- 19. Reyes H., Reisz-Porszasz S., and Hankinson O. (1992) Identification of the Ah receptor nuclear translocator protein (Arnt) as a component of the DNA binding form of the Ah receptor. Science 256, 1193–1195 10.1126/science.256.5060.1193 [DOI] [PubMed] [Google Scholar]
- 20. Rothhammer V., and Quintana F. J. (2019) The aryl hydrocarbon receptor: an environmental sensor integrating immune responses in health and disease. Nat. Rev. Immunol. 19, 184–197 10.1038/s41577-019-0125-8 [DOI] [PubMed] [Google Scholar]
- 21. Yuan F., Xu C., Li G., and Tong T. (2018) Nucleolar TRF2 attenuated nucleolus stress-induced HCC cell-cycle arrest by altering rRNA synthesis. Cell Death Dis. 9, 518 10.1038/s41419-018-0572-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Singleton R. S., Liu-Yi P., Formenti F., Ge W., Sekirnik R., Fischer R., Adam J., Pollard P. J., Wolf A., Thalhammer A., Loenarz C., Flashman E., Yamamoto A., Coleman M. L., Kessler B. M., et al. (2014) OGFOD1 catalyzes prolyl hydroxylation of RPS23 and is involved in translation control and stress granule formation. Proc. Natl. Acad. Sci. U.S.A. 111, 4031–4036 10.1073/pnas.1314482111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Armeni T., Cianfruglia L., Piva F., Urbanelli L., Luisa Caniglia M., Pugnaloni A., and Principato G. (2014) S-d-Lactoylglutathione can be an alternative supply of mitochondrial glutathione. Free Radic. Biol. Med. 67, 451–459 10.1016/j.freeradbiomed.2013.12.005 [DOI] [PubMed] [Google Scholar]
- 24. Van Rompay A. R., Norda A., Lindén K., Johansson M., and Karlsson A. (2001) Phosphorylation of uridine and cytidine nucleoside analogs by two human uridine-cytidine kinases. Mol. Pharmacol. 59, 1181–1186 10.1124/mol.59.5.1181 [DOI] [PubMed] [Google Scholar]
- 25. Lane A. N., and Fan T. W. (2015) Regulation of mammalian nucleotide metabolism and biosynthesis. Nucleic Acids Res. 43, 2466–2485 10.1093/nar/gkv047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Intlekofer A. M., Wang B., Liu H., Shah H., Carmona-Fontaine C., Rustenburg A. S., Salah S., Gunner M. R., Chodera J. D., Cross J. R., and Thompson C. B. (2017) l-2-Hydroxyglutarate production arises from noncanonical enzyme function at acidic pH. Nat. Chem. Biol. 13, 494–500 10.1038/nchembio.2307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Molenaar R. J., Maciejewski J. P., Wilmink J. W., and van Noorden C. J. F. (2018) Correction: wild-type and mutated IDH1/2 enzymes and therapy responses. Oncogene 37, 5810 10.1038/s41388-018-0455-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ye D., Guan K. L., and Xiong Y. (2018) Metabolism, activity, and targeting of d- and l-2-hydroxyglutarates. Trends Cancer 4, 151–165 10.1016/j.trecan.2017.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mishra P., Tang W., Putluri V., Dorsey T. H., Jin F., Wang F., Zhu D., Amable L., Deng T., Zhang S., Killian J. K., Wang Y., Minas T. Z., Yfantis H. G., Lee D. H., et al. (2018) ADHFE1 is a breast cancer oncogene and induces metabolic reprogramming. J. Clin. Invest. 128, 323–340 10.1172/JCI93815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shim E. H., Livi C. B., Rakheja D., Tan J., Benson D., Parekh V., Kho E. Y., Ghosh A. P., Kirkman R., Velu S., Dutta S., Chenna B., Rea S. L., Mishur R. J., Li Q., et al. (2014) l-2-Hydroxyglutarate: an epigenetic modifier and putative oncometabolite in renal cancer. Cancer Discov. 4, 1290–1298 10.1158/2159-8290.CD-13-0696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Intlekofer A. M., Dematteo R. G., Venneti S., Finley L. W., Lu C., Judkins A. R., Rustenburg A. S., Grinaway P. B., Chodera J. D., Cross J. R., and Thompson C. B. (2015) Hypoxia induces production of l-2-hydroxyglutarate. Cell Metab. 22, 304–311 10.1016/j.cmet.2015.06.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rzem R., Veiga-da-Cunha M., Noël G., Goffette S., Nassogne M. C., Tabarki B., Schöller C., Marquardt T., Vikkula M., and Van Schaftingen E. (2004) A gene encoding a putative FAD-dependent l-2-hydroxyglutarate dehydrogenase is mutated in L-2-hydroxyglutaric aciduria. Proc. Natl. Acad. Sci. U.S.A. 101, 16849–16854 10.1073/pnas.0404840101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Reiter-Brennan C., Semmler L., and Klein A. (2018) The effects of 2-hydroxyglutarate on the tumorigenesis of gliomas. Contemp. Oncol. (Pozn.) 22, 215–222 10.5114/wo.2018.82642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pastor-Anglada M., and Perez-Torras S. (2018) Emerging roles of nucleoside transporters. Front. Pharmacol. 9, 606 10.3389/fphar.2018.00606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Blackwood E. M., and Eisenman R. N. (1991) Max: a helix-loop-helix zipper protein that forms a sequence-specific DNA-binding complex with Myc. Science 251, 1211–1217 10.1126/science.2006410 [DOI] [PubMed] [Google Scholar]
- 36. Lafita-Navarro M. C., Liano-Pons J., Quintanilla A., Varela I., Blanco R., Ourique F., Bretones G., Aresti J., Molina E., Carroll P., Hurlin P., Romero O. A., Sanchez-Céspedes M., Eisenman R. N., Delgado M. D., et al. (2020) The MNT transcription factor autoregulates its expression and supports proliferation in MYC-associated factor X (MAX)–deficient cells. J. Biol. Chem. 295, 2001–2017 10.1074/jbc.RA119.010389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Garcia-Sanz P., Quintanilla A., Lafita M. C., Moreno-Bueno G., Garcia-Gutierrez L., Tabor V., Varela I., Shiio Y., Larsson L. G., Portillo F., and Leon J. (2014) Sin3b interacts with Myc and decreases Myc levels. J. Biol. Chem. 289, 22221–22236 10.1074/jbc.M113.538744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mullen A. R., Hu Z., Shi X., Jiang L., Boroughs L. K., Kovacs Z., Boriack R., Rakheja D., Sullivan L. B., Linehan W. M., Chandel N. S., and DeBerardinis R. J. (2014) Oxidation of α-ketoglutarate is required for reductive carboxylation in cancer cells with mitochondrial defects. Cell Rep. 7, 1679–1690 10.1016/j.celrep.2014.04.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the data described are contained within the article.