

Abstract
Bile acids (BAs) comprise heterogenous amphipathic cholesterol-derived molecules that carry out physicochemical and signaling functions. A major site of BA action is the terminal ileum, where enterocytes actively reuptake BAs and express high levels of BA-sensitive nuclear receptors. BA pool size and composition are affected by changes in metabolic health, and vice versa. One of several factors that differentiate BAs is the presence of a hydroxyl group on C12 of the steroid ring. 12α-Hydroxylated BAs (12HBAs) are altered in multiple disease settings, but the consequences of 12HBA abundance are incompletely understood. We employed mouse primary ileum organoids to investigate the transcriptional effects of varying 12HBA abundance in BA pools. We identified Slc30a10 as one of the top genes differentially induced by BA pools with varying 12HBA abundance. SLC30A10 is a manganese efflux transporter critical for whole-body manganese excretion. We found that BA pools, especially those low in 12HBAs, induce cellular manganese efflux and that Slc30a10 induction by BA pools is driven primarily by lithocholic acid signaling via the vitamin D receptor. Administration of lithocholic acid or a vitamin D receptor agonist resulted in increased Slc30a10 expression in mouse ileum epithelia. These data demonstrate a previously unknown role for BAs in intestinal control of manganese homeostasis.
Keywords: intestine, bile acids, manganese, organoid, metal transport, bile acid, gene expression, nuclear receptor
Bile acids (BAs) are cholesterol catabolites that regulate many biological functions, including multiple aspects of macronutrient metabolism. One of the mechanisms by which they do so is by promoting lipid emulsification and absorption (1–3). A second mechanism is by acting as a ligand for BA receptors, which can regulate lipid and glucose metabolism (1–4). It is underappreciated that there is structural diversity among BAs that results in variable capacities to activate BA receptors (5–10). This structural diversity arises from the number and position of hydroxyl groups and the conjugation of the molecule to glycine, taurine, or neither (11, 12). Thus, the composition of the BA pool may affect the activity of BA receptors. However, the biological consequences of altered BA pool composition are not fully known.
One key determinant of BA composition is the hepatic enzyme sterol 12α-hydroxylase (encoded by CYP8B1). By adding a 12α-hydroxylation to an intermediate of the BA synthesis pathway, CYP8B1 determines the hepatic synthesis of cholic acid (CA) instead of chenodeoxycholic acid (CDCA) (Fig. 1A) (12, 13). In settings of insulin resistance, there is an increased proportion of CA, its bacterial metabolite deoxycholic acid (DCA), and their conjugates—collectively termed 12α-hydroxylated BAs (12HBAs) in the BA pool (14, 15). Rates of 12HBA synthesis and CA conversion to DCA are also higher in insulin resistance and type 2 diabetes (16, 17). Moreover, even in healthy subjects, increases in 12HBAs are correlated with the characteristic metabolic abnormalities of insulin resistance (14). In contrast, Cyp8b1–/– mice, which lack 12HBAs, are protected from Western-type diet–induced weight gain and atherosclerosis compared with WT mice (18–20). Cyp8b1–/– mice also show improved glucose tolerance, which has been proposed to be due to increased secretion of glucagon-like peptide-1 (GLP-1) (21). Mice subjected to vertical sleeve gastrectomy, a common weight loss surgery, exhibit reduced Cyp8b1 mRNA expression as well as lower ratios of 12HBAs:non-12HBAs in circulation (22). Furthermore, siRNA against Cyp8b1 improved nonalcoholic steatohepatitis in mice (23). Thus, CYP8B1 inhibition is a potential therapeutic target for metabolic diseases. However, the biological processes that are regulated by 12HBAs are incompletely understood.
Figure 1.
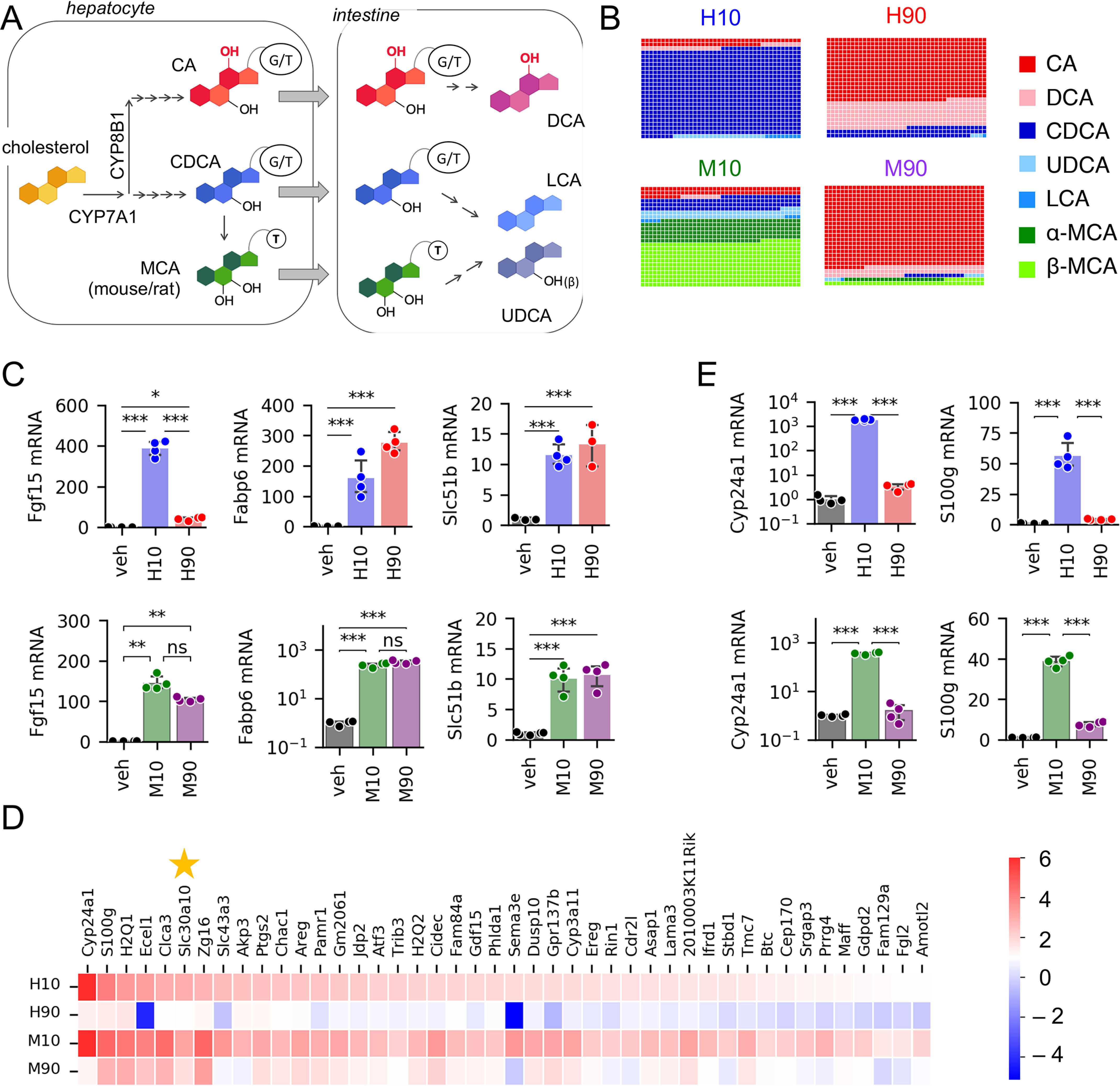
Differential regulation of gene expression by BA pool composition. A, simplified BA synthesis pathway. B, composition of BA pools used in this study. Glycine-conjugated BAs were used in human pools, whereas taurine-conjugated BAs were used in mouse pools, except LCA, which was unconjugated. C, expression of FXR target genes measured by qPCR. n = 3–4 wells of organoids/condition. D, genes preferentially induced by H10 and M10 compared with H90 and M90 identified by RNA-Seq (log2 -fold change > 1 and padj < 0.05). n = 3/condition. E, expression of VDR target genes measured by qPCR (n = 3–4 wells/condition). Full result tables for one-way ANOVA (C and E) and post hoc pairwise comparisons are shown in Table S4. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ns, not significant. Error bars, S.D.
A major site of BA signaling is the intestine, which encounters high BA concentrations, ∼2–12 mm after a meal (24, 25). The intestine epithelium expresses at least three BA receptors. The transcription factor FXR regulates BA transport and feedback suppression of hepatic BA synthesis and also modulates lipid and glucose metabolism (1). The membrane receptor TGR5 regulates glucose homeostasis and colonic motility by promoting the secretion of GLP-1 and serotonin (26–28). Another intestinal receptor responsive to certain BAs is the vitamin D receptor (VDR), whose canonical role is to promote calcium and phosphate absorption (29). For each of these receptors, the best reported endogenous BA agonists are non-12HBAs. For FXR, it is CDCA (5, 6, 9), and for TGR5 and VDR, it is lithocholic acid (LCA) (7, 8, 10). LCA is formed by 7α-dehydroxylation of CDCA by bacterial enzymes in the gut. Thus, BA composition is predicted to impact signaling through multiple receptors in the intestine.
Investigating the effects of BA composition on intestinal BA signaling in vivo is challenging because of the continued presence of endogenous BAs. This is particularly important for experiments in mice, as mice contain a class of BAs—muricholic acids, which are non-12HBAs—that are not found in healthy adult humans (although we note that, conversely, all known BAs in the human BA pool are also present in mice and are transported through the mouse intestinal epithelium to undergo enterohepatic circulation). To fill the gap, we used primary murine intestinal organoids, also called enteroids. These organoids are generated from stem cells of the intestinal crypts and contain all known cell types of the intestinal epithelium (30). We investigated the effects of BA pools of different compositions on ileal organoids, with a particular focus on the effects of lowering 12HBAs (to mimic CYP8B1 inhibition). Furthermore, we addressed the interspecies differences in BAs by using BA pools that we designed to mimic the effects of CYP8B1 inhibition in humans and mice.
We unexpectedly found that varying 12HBA proportions modulates expression of Slc30a10, a manganese efflux transporter critical for whole-body Mn excretion. Cellular Mn levels are tightly regulated, as Mn is essential for numerous cellular processes, yet its excess is toxic (31). Our data demonstrate a previously unknown role of BAs in intestinal control of metal homeostasis.
Results
Distinct BA pool compositions differentially induce gene expression
To test the effects of a low-12HBA pool on intestinal gene expression, we designed four distinct BA pools with which to stimulate primary murine ileal organoids. The differences between the pools were due to two key features: (i) the proportion of 12HBAs, either 10% (low) or 90% (high), and (ii) the BA pool of the species we modeled, either human or mouse (Table 1 and Fig. 1B). In the human pools, there was a larger proportion of DCA, and the BAs were glycine-conjugated, whereas in the mouse pools, BAs were taurine-conjugated, to mimic the natural abundance in those species (4, 32, 33). Mouse BA pools contained muricholic acids, whereas these were not included in the human pools. Thus, the four BA pools are labeled human low 12HBA (H10), human high 12HBA (H90), mouse low 12HBA (M10), and mouse high 12HBA (M90). Importantly, the total BA pool concentration was the same across all treatment groups. We prepared the four BA pools in mixed micelles containing oleic acid, 2-palmitoyl glycerol, phosphatidylcholine, and free cholesterol to mimic conditions of the intestinal lumen. We used the four lipid-emulsified BA pools to treat primary murine ileal organoids. The vehicle control contained all micelle components except BAs. We performed bulk RNA-Seq after 24 h of treatment.
Table 1.
Composition of BA pools. BAs were in their glycine-conjugated forms for human BA pools and taurine-conjugated forms for mouse BA pools, except LCA, which was unconjugated
| BA group | BA species | In H10 | In H90 | In M10 | In M90 |
|---|---|---|---|---|---|
| mol % | mol % | mol % | mol % | ||
| 12HBA | Cholic acid | 7 | 63 | 9.0 | 81.0 |
| 12HBA | Deoxycholic acid | 3 | 27 | 1.0 | 9.0 |
| Non-12HBA | Chenodeoxycholic acid | 86.85 | 9.65 | 13.5 | 1.5 |
| Non-12HBA | Ursodeoxycholic acid | 2.25 | 0.25 | 8.1 | 0.9 |
| Non-12HBA | Lithocholic acid | 0.90 | 0.10 | 0.9 | 0.1 |
| Non-12HBA | α-Muricholic acid | 22.5 | 2.5 | ||
| Non-12HBA | β-Muricholic acid | 45.0 | 5.0 |
We focused on the effects of the low-12HBA pools, as this would mimic the effects of CYP8B1 inhibition. We examined all genes that were significantly induced compared with vehicle, setting thresholds of log2FC > 1.0 and padj < 0.05 for differential expression. The low-12HBA pools collectively induced 1361 genes. Of these, 516 reached those thresholds for both H10 and M10 pools, 95 reached those thresholds for H10 only, and 750 reached the thresholds for M10 only (Fig. S1A). Pathway analysis indicates enrichment of genes involved in lipid metabolism, consistent with known effects of BAs (Table 2).
Table 2.
Pathway analysis of 516 genes induced by both human low-12HBA and mouse low-12HBA pools
| GO term | Description | Count | Percentage | p | Genes | List total | Population hits | Population total | -Fold enrichment | False discovery rate |
|---|---|---|---|---|---|---|---|---|---|---|
| GO:0006629 | Lipid metabolic process | 39 | 7.88 | 1.60E−11 | HSD17B11, ACOX1, CHKA, PTGS2, ALOXE3, CHKB, EHHADH, ABHD3, ASAH2, APOB, INSIG2, APOBR, ACOT12, ETNK1, MGLL, PLCD1, HSD17B4, PLCB1, ACAA1B, ACSL5, SCD1, SOAT2, MOGAT2, CUBN, NCEH1, LIPA, PLB1, E PHX2, ACACB, LPIN2, LPCAT3, LPCAT4, GDPD2, MTTP, PCX, CLPS, HMGCS2, LIPH, VLDLR | 425 | 459 | 18,082 | 3.61500961 | 2.70E−08 |
| GO:0016491 | Oxidoreductase activity | 40 | 8.08 | 1.57E−08 | HSD17B11, ME1, ACOX1, CYP24A1, ALDH1L1, CYP3A25, PTGS2, ALOXE3, EHHADH, OSGIN1, ALDH1L2, ALDH3A2, MTHFD2, FMO5, ADH1, ALDH1A3, ADH4, GPX3, ALDH1A7, SMOX, HSD17B4, NOS2, QSOX1, CYP3A44, ADH6A, SCD1, SUOX, CYP3A13, CYP3A11, DHRS9, POR, AKR1B8, CYP4A10, DHRS1, AKR1B7, RDH10, HSDL2, TPH1, BCO2, RETSAT | 415 | 604 | 17,446 | 2.78401021 | 2.34E−05 |
| GO:0052689 | Carboxylic ester hydrolase activity | 13 | 2.63 | 4.39E−07 | NCEH1, ACOT2, ABHD3, ACOT1, ACOT5, ACOT4, ACOT3, ACOT12, CES2A, CES1G, MGLL, LIPH, CES2B | 415 | 81 | 17,446 | 6.74692845 | 6.56E−04 |
| GO:0055114 | Oxidation-reduction process | 38 | 7.68 | 1.66E−06 | HSD17B11, ME1, ACOX1, CYP24A1, ALDH1L1, CYP3A25, PTGS2, ALOXE3, EHHADH, ALDH1L2, ALDH3A2, MTHFD2, FMO5, ADH1, ALDH1A3, ADH4, GPX3, ALDH1A7, SMOX, HSD17B4, NOS2, QSOX1, CYP3A44, SCD1, SUOX, CYP3A13, CYP3A11, DHRS9, POR, AKR1B8, DHRS1, CYP4A10, AKR1B7, RDH10, HSDL2, TPH1, BCO2, RETSAT | 425 | 676 | 18,082 | 2.39163244 | 0.00281558 |
| GO:0005102 | Receptor binding | 27 | 5.45 | 6.36E−06 | PVR, ACOX1, FGF15, EHHADH, ACOT2, ACOT4, SCT, ANG, GSTK1, CCDC129, NOS2, HSD17B4, FGF3, MATK, H2-Q2, PLAT, ICOSL, H2-Q1, BTNL2, EPHX2, CD160, PLAUR, LAMA3, GM8909, LAMA5, H2-BL, VEGFA | 415 | 412 | 17,446 | 2.7549538 | 0.00950826 |
| GO:0047617 | Acyl-CoA hydrolase activity | 6 | 1.21 | 8.08E−06 | ACOT12, ACOT2, ACOT1, ACOT5, ACOT4, ACOT3 | 415 | 13 | 17,446 | 19.4024096 | 0.01207519 |
| GO:0006631 | Fatty acid metabolic process | 15 | 3.03 | 1.89E−05 | SCD1, ACOX1, LIPA, PTGS2, ALOXE3, EHHADH, ACACB, LPIN2, CYP4A10, ACOT12, MGLL, FABP2, HSD17B4, ACAA1B, ACSL5 | 425 | 156 | 18,082 | 4.09095023 | 0.03207602 |
| GO:0000038 | Very long–chain fatty acid metabolic process | 6 | 1.21 | 2.44E−05 | ACOX1, ACOT2, HSD17B4, ACOT5, ACOT4, ACOT3 | 425 | 16 | 18,082 | 15.9547059 | 0.04137758 |
Next, we focused on the subset of these genes that are differentially regulated by low- versus high-12HBA pools, with a particular focus on those for which the effects were shared between human and mouse pools. We used a threshold of log2FC > 1.0 and padj < 0.05 for differential expression. The majority of genes did not reach this threshold, indicating that they are similarly regulated by both low- and high-12HBA pools or are differentially regulated by low versus high 12HBAs in human pools only or in mouse pools only (Fig. S1B). These genes included canonical FXR targets such as Fgf15, Fabp6 (encoding the ileal bile acid–binding protein, Ibabp), and Slc51b (encoding the basolateral BA efflux transporter Ostβ), and we validated these by qPCR (Fig. 1C).
There were 44 genes that were preferentially induced by H10 and M10 compared with H90 and M90, respectively (Fig. 1D). Among these, we noted that several are known transcriptional targets of VDR. These included Cyp24a1, S100g, and Cyp3a11, and we validated these by qPCR (Fig. 1E). This is consistent with the concepts that (i) certain BAs, especially the non-12HBA LCA and its conjugates, can activate VDR in the micromolar range (10), and (ii) the low-12HBA pools (i.e. H10 and M10) contain more LCA (9 μm, as opposed to 1 μm in the high-12HBA pools).
Next, we validated the RNA-Seq findings in multiple experimental systems. Using organoids derived from multiple mice, we confirmed that all BA pools induced expression of FXR targets Fgf15 and Fabp6 (Supp. Fig. S1C). We also confirmed that VDR targets Cyp24a1 and S100g were preferentially induced by low-12HBA pools (Fig. S1D). We found that delivery in micelles was not required and that BAs per se were sufficient to induce Fgf15, Fabp6, and S100g (Fig. S1E). Last, to determine whether differential responses to BA pools also occur in human cells, we performed experiments in Caco-2 cells, which are enterocyte-like cells derived from a human epithelial colorectal tumor. We observed that FXR and VDR targets were robustly induced by H10, but not H90 (Fig. S1, F and G).
BA composition regulates Slc30a10 expression and cellular Mn efflux
Among the genes identified to be differentially expressed between BA pools with varying 12HBA abundance, one of the most robust was Slc30a10 (Fig. 1D). This gene encodes a Mn efflux transporter critical for whole-body Mn excretion (34–41). Humans with mutations in SLC30A10 develop hypermanganesemia, accompanied by parkinsonism and cirrhosis (34, 35, 42–44).
In gut organoids, low-12HBA pools were superior to high-12HBA pools in inducing Slc30a10 (Fig. 2, A and B). This preferential induction was consistent across multiple independent batches of organoids (Fig. 2, C and D). This differential expression of Slc30a10 was also observed when BA pools were delivered without micelles (Fig. 2E), indicating a direct effect of BAs. We also validated that the H10 pool induces SLC30A10 in human Caco-2 cells (Fig. 2F).
Figure 2.
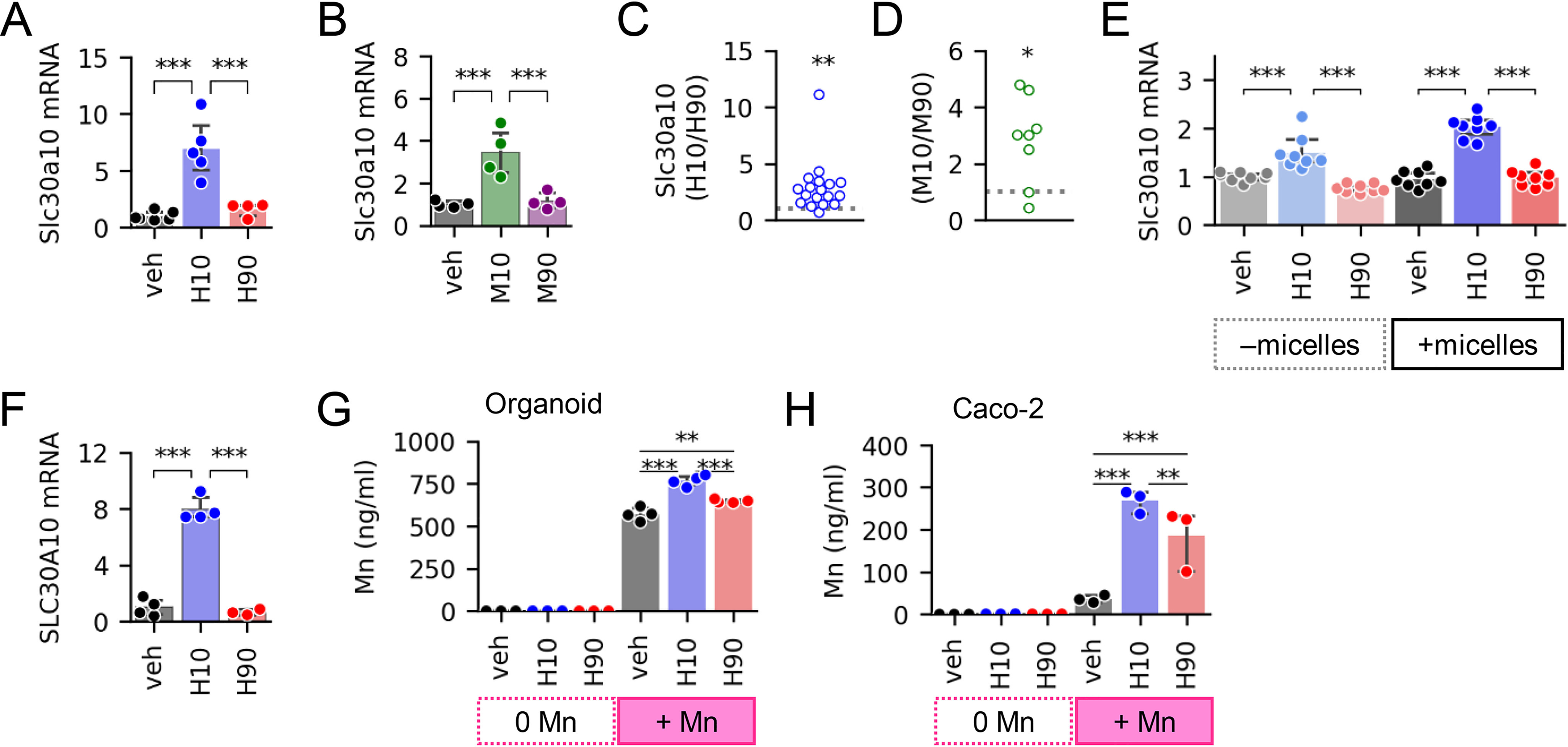
Low-12HBA pools regulate Slc30a10 expression and Mn efflux. Shown are Slc30a10 mRNA levels in gut organoids treated with human BA pools (A) and mouse BA pools (B); n = 4–6 wells of organoids/condition. C and D, aggregate -fold change differences in Slc30a10 induction across different batches of organoids. Each point represents the data derived from a different mouse (n = 8–18). The data point plotted is the ratio of (average mRNA levels in low-12HBA–treated organoids (n = 3–8 well))/(average mRNA levels in high-12HBA–treated organoids (n = 3–8 wells)). Ratios >1 signify higher expression in the low-12HBA–treated group, ratios <1 signify higher expression in the high-12HBA–treated group, and ratios = 1 signify no difference between low- and high-12HBA–treated groups. The gray dotted line marks ratio = 1. *, p < 0.05; **, p < 0.01, one-sample t test with µ = 1 (expected ratio for no preferential induction). E, Slc30a10 mRNA levels in ileal organoids treated with human BA pools delivered without or with micelles. n = 8 wells/condition. F, SLC30A10 mRNA levels in Caco-2 treated with human BA pools; n = 3–4 wells of cells/condition. G and H, Mn concentrations in efflux media of organoids (G) and Caco-2 cells (H); n = 3–4 wells/condition. Full result tables for one-way ANOVA (A, B, and F), two-way ANOVA (E and G), one-sample t tests (C and D), and pairwise comparisons are provided in Table S5. *, p < 0.05; **, p < 0.01; ***, p < 0.001. Error bars, S.D.
Next, we sought to determine whether BA-induced changes in Slc30a10 transcript levels yield functional cellular consequences. We predicted that the induction of Slc30a10 expression by low-12HBA pools would increase Mn efflux from organoids. To test this, we carried out Mn efflux assays, where we preloaded cells with Mn, washed away unabsorbed Mn, then treated the cells with vehicle or BA pools, and measured Mn levels in efflux media. Organoids treated with H10 had higher Mn in their efflux media compared with organoids treated with vehicle or H90 (Fig. 2G). Consistently, Caco-2 cells receiving H10 also showed higher Mn efflux compared with vehicle- and H90-treated cells (Fig. 2H). Altogether, these data show that BAs, particularly pools low in 12HBAs, induce cellular Mn efflux, consistent with their induction of Slc30a10.
Slc30a10 expression is driven by LCA-to-VDR signaling
Recently, SLC30A10 mRNA expression was found to be induced by the VDR agonist 1α,25-dihydroxyvitamin D3 in Caco-2 cells (45). LCA is a potent VDR ligand, with an EC50 comparable with CDCA's EC50 for human FXR (10). Although LCA made up <1% of the BA pools in our studies, the LCA concentration in the low-12HBA pools was 9 μm. As a point of reference, in the duodenum of healthy adult humans, the total bile acid concentration is 20 mm, and LCA makes up 1–3% of the pool, ∼200-600 μm (25, 46, 47). Although LCA is partly sulfated and excreted, a portion is taken up into enterocytes (48, 49). Thus, we investigated whether the differential Slc30a10 induction by low-12HBA pools was due to LCA-to-VDR signaling.
To test whether LCA is the differentiating factor between H10 and H90 in their induction of Slc30a10, we treated ileal organoids with H10 and H90 pools lacking LCA (Table S1). In pools lacking LCA, we compensated for its removal by increasing other non-12HBAs such that the total BA pool concentrations matched. Without LCA, H10 induction of Slc30a10 was significantly blunted (Fig. 3A). This pattern was similar to that of VDR targets Cyp24a1 and S100g (Fig. 3B). On the other hand, the presence or absence of LCA had no effect on FXR targets Fgf15 and Fabp6, consistent with the observation that LCA is not a potent FXR agonist (Fig. 3C).
Figure 3.
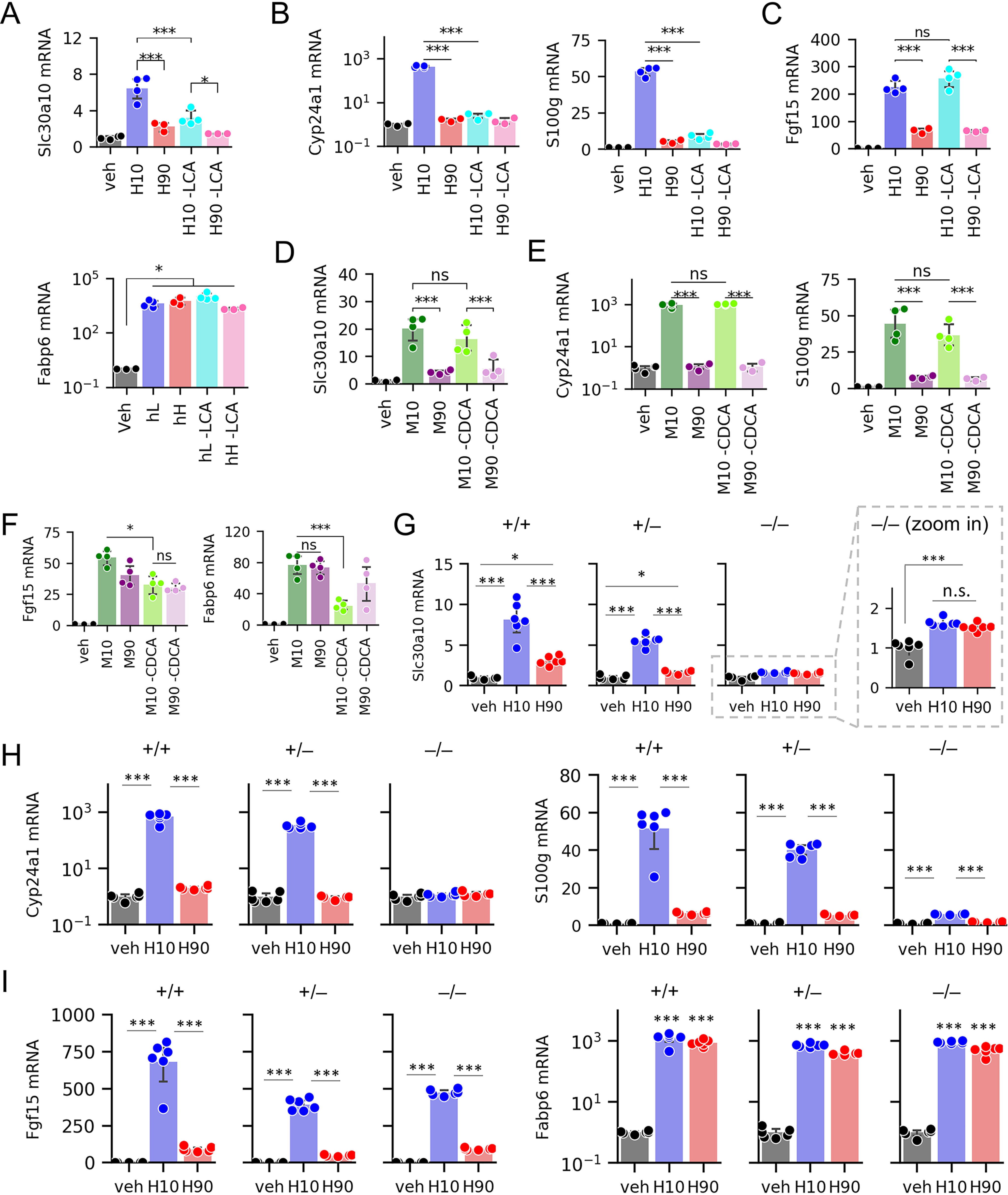
Slc30a10 expression is regulated by LCA–VDR signaling. A–F, mRNA levels of Slc30a10, VDR targets, and FXR targets in WT ileal organoids after treatment with human BA pools containing or lacking LCA (A–C) or mouse BA pools containing or lacking CDCA (D–F); n = 3–4 wells of organoids/condition. G–I, expression of Slc30a10, VDR targets, and FXR targets in organoids derived from Vdr+/+, Vdr+/–, and Vdr–/– littermates after exposure to vehicle or human BA pools; n = 6 wells of organoids/condition. Full result tables for two-way ANOVA (A–F), one-way ANOVA (G–I), and pairwise comparisons are provided in Table S6. *, p < 0.05; ***, p < 0.001; ns, not significant. Error bars, S.D.
To test whether CDCA contributes to differential Slc30a10 induction between low- and high-12HBA pools, we treated gut organoids with M10 and M90 pools containing or lacking T-CDCA (Table S2). In pools lacking T-CDCA, we compensated for its removal by increasing other non-12HBAs such that the total BA pool concentrations matched. Removing T-CDCA had no significant effect on Slc30a10 mRNA levels (Fig. 3D), suggesting that CDCA is dispensable for Slc30a10 induction. Similarly, there was no effect of removing T-CDCA on Cyp24a1 or S100g expression (Fig. 3E). CDCA and its conjugates are potent agonists of FXR (5, 6, 9). Consistent with this, removing T-CDCA from M10 and M90 pools resulted in blunted Fgf15 and Fabp6 induction (Fig. 3F). Together, these findings indicate that LCA, but not CDCA, is the BA primarily responsible for the differential expression of Slc30a10 between low- versus high-12HBA pools.
To directly test the requirement of VDR for BA-induced Slc30a10 mRNA expression, we carried out experiments on gut organoids derived from VDR+/+, VDR+/−, and VDR–/– littermate mice. Strikingly, VDR–/– gut organoids showed substantially blunted Slc30a10 mRNA expression in response to H10 pool (Fig. 3G). Furthermore, VDR+/– gut organoids increased Slc30a10 mRNA expression to intermediate levels between that in VDR+/+ and VDR–/– organoids, suggesting a gene dosage effect. As expected, ablating VDR diminished the induction of Cyp24a1 and S100g (Fig. 3H) but had little effect on Fgf15 and Fabp6 (Fig. 3I). We noted that VDR–/– gut organoids still showed a small but significant increase in Slc30a10 upon H10 and H90 treatment, suggesting possible residual activation by another transcription factor. Thus, we conclude that the induction of Slc30a10 by low-12HBA pools is largely mediated by VDR.
Effects of BA composition on other Mn transporters
In addition to SLC30A10, enterocytes express at least two other Mn transporters. The divalent metal transporter 1 (DMT1, encoded by SLC11A2) transports Mn from the intestinal lumen into enterocytes (31). SLC39A14 takes up Mn from the basolateral side of enterocytes (50), and it has been reported that SLC39A14 acts synergistically with SLC30A10 to mediate whole-body Mn excretion (50–52). However, we found that BA treatments had no effects on Slc11a2 or Slc39a14 expression in murine organoids (Fig. 4A) and no effects on SLC11A2 or SLC39A14 in Caco-2 cells (Fig. 4B).
Figure 4.

BA effects on additional Mn transporters. A–D, expression of Slc11a2 and Slc39a14 (A), Slc39a8 in organoids (C), and their corresponding human genes in Caco-2 cells (B and D) (n = 4–8 wells of cells or organoids per group). E and F, Slc39a8 expression in organoids exposed to human BA pools with or without LCA (E) and mouse BA pools with or without CDCA (n = 4 wells of organoids/group) (F). G, Slc39a8 expression in organoids generated from Vdr+/+, Vdr+/–, and Vdr–/– littermates after exposure to human BA pools (n = 6 wells of organoids/group). Full result tables for one-way ANOVA (A–D and G) and two-way ANOVA (E and F) and post hoc pairwise comparisons are shown in Table S7. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ns, not significant. Error bars, S.D.
Another Mn transporter is SLC39A8. This protein is highly expressed in the liver, where it is critical for Mn uptake from bile into hepatocytes (53). The localization and physiological relevance of SLC39A8 in the intestine has not been reported. We found that gut organoids treated with H10 and M10 pools up-regulated Slc39a8 mRNA expression more strongly than H90 and M90 pools (Fig. 4C). Caco-2 cells also showed increased SLC39A8 expression upon H10, but not H90, treatment (Fig. 4D). Further analyses showed that, similar to Slc30a10, Slc39a8 mRNA expression was blunted upon removal of LCA from BA pools and in gut organoids from VDR–/– mice, whereas removal of CDCA from BA pools had no effect (Fig. 4, E–G). These data support the possibility that both Slc30a10 and Slc39a8 are regulated by LCA-to-VDR signaling.
LCA and vitamin D induce Slc30a10 expression in mouse ileum
To determine the distribution of Slc30a10 and Slc39a8 expression in vivo, we quantified mRNA expression in murine enterohepatic tissues. We found that all segments of the small intestine expressed Slc30a10 mRNA at levels higher than the liver (Fig. 5A). We also detected Slc39a8 expression in the small intestine, at levels ∼10–30% of those in the liver (Fig. 5B).
Figure 5.

VDR activation increases Slc30a10 mRNA levels in vivo. A and B, expression of Slc30a10 (A) and Slc39a8 (B) in the liver and gastrointestinal tissues in C57BL/6 mice (n = 7). The liver was used as a reference tissue. C–E, ileal expression of Slc30a10 (C), VDR targets Cyp24a1 and S100g (D), and Slc39a8 (E) after oral gavage with corn oil supplemented with vehicle, LCA, calcitriol, or CA. n = 5–7 mice/group. F, Slc30a10 expression in the liver, duodenum, and jejunum after gavage experiment. Full result tables of one-way ANOVA and post-hoc pairwise comparisons are shown in Table S8. *, p < 0.05; ***, p < 0.001. Error bars, S.D.
Next, we tested whether LCA and VDR signaling induces expression of Mn transporters in vivo. We administered (i) vehicle, (ii) LCA, (iii) the VDR agonist calcitriol, or (iv) the 12HBA CA to C57BL/6 mice by oral gavage and analyzed mRNA expression in ileal epithelia. Mice receiving LCA and calcitriol showed on average 4–6-fold increases in Slc30a10 expression in the ileum (Fig. 5C). The increase in Slc30a10 expression was accompanied by increased Cyp24a1 and S100g (Fig. 5D), consistent with activation of VDR. Notably, CA did not induce Slc30a10, Cyp24a1, or S100g (Fig. 5, C and D). In contrast to the results of ex vivo and in vitro experiments, Slc39a8 was not induced by any of the treatments (Fig. 5E). Interestingly, Slc30a10 was also induced by LCA in the jejunum, but not in the duodenum or liver (Fig. 5F). These data show that in vivo, LCA promotes Slc30a10 expression in the ileum and jejunum.
Discussion
Our analyses reveal that BA composition regulates the expression of Slc30a10 and cellular Mn efflux. SLC30A10 is one of three transporters identified thus far to be critical for whole-body Mn homeostasis in humans and mice (34, 35, 39, 40, 52, 54–56). We found that BA pools low in 12HBAs, which have increased abundance of the non-12HBA LCA, act primarily via VDR to promote Slc30a10 expression. We also found that ileal Slc30a10 was inducible by LCA and the VDR agonist calcitriol, indicating that LCA and VDR signaling modulate intestinal Mn transport. A model is shown in Fig. 6
Figure 6.

Low-12HBA pools promote expression of Slc30a10 mRNA and Mn efflux. Induction of Slc30a10 expression is driven by LCA-to-VDR signaling.
Similar to BAs, a significant portion of Mn undergoes enterohepatic cycling (57, 58). Thus, enterocytes are exposed to luminal Mn from two sources: bile and diet. In adult humans, only about 3–5% of ingested Mn is absorbed (31), indicating robust regulation of intestinal Mn absorption. Obstructing the bile duct results in reduced excretion rates of intravenously administered 54Mn in rats (59), highlighting the importance of Mn clearance by the liver and final excretion in the feces. But the intestine appears to play an important role in Mn homeostasis as well, especially when liver excretion has been saturated (60). Whereas deletion of Slc30a10 only in hepatocytes is insufficient to cause hypermanganesemia, combined deletion in the liver and gastrointestinal tract causes severe hypermanganesemia and neurotoxicity (39, 41), mimicking the whole-body Slc30a10 knockout mice, although less severe (39, 40). These findings support the notion that intestine and hepatobiliary excretion systems both participate in Mn homeostasis.
Mn is a cofactor for numerous metalloproteins involved in many cellular processes (31, 61). However, Mn in excess amounts is toxic. Hypermanganesemia results in dystonia, parkinsonism, polycythemia, and cirrhosis (31, 34, 35, 42, 52, 62, 63). Most cases of Mn toxicity are due to occupational and environmental exposures (31, 62). Mn toxicity has also been reported in cases of severe liver diseases, in patients receiving total parenteral nutrition, and in rare genetic loss-of-function mutations (34, 35, 44, 64–67). Current treatment for Mn toxicity is with chelation therapy, which increases urinary Mn excretion (44) but is nonspecific. Since the identification of disease-causing mutations of SLC30A10, there has been considerable progress in understanding how the transporter works and where it is localized in cells (36, 68). However, the regulation of the transporter is incompletely understood.
We identified BA composition, especially LCA abundance, as a modulator of Slc30a10 expression in vitro, ex vivo, and in the mouse ileum. We also showed that VDR is the primary mediator of this signaling pathway. This is consistent with a previous report showing VDR activation inducing SLC30A10 in Caco-2 cells and that this requires the VDR element in SLC30A10's promoter region (45). Claro da Silva et al. (45) previously reported increased SLC30A10 mRNA levels in half of duodenal biopsies from volunteers administered oral calcitriol (0.5 µg for 10 days). However, ileal biopsies were not reported, and the ileum is expected to be the primary site of uptake of bile acids into enterocytes (49). Our data show that Slc30a10 expression is induced by LCA and calcitriol in vivo in the mouse ileum. Published data from in vitro studies using Caco-2 cells and the neuroblastoma cell line SH-SY5Y have also proposed that the ER stress response factor ATF4 and the zinc-sensitive transcription factor ZNF658 contribute to SLC30A10 regulation (69–71). The identification of this new BA-sensitive pathway may shed light on a known phenomenon: Mn poisoning has been reported in patients on total parenteral nutrition (65–67), suggesting that the lack of intestinal BA signaling leads to reduced SLC30A10 and impaired efflux of Mn from enterocytes into the intestinal lumen, consequently reducing Mn excretion.
In our experiments, Slc30a10 induction was not completely abrogated upon removal of LCA from BA pools or deletion of VDR. Thus, other BAs and nuclear receptors might also contribute to BA-dependent induction of Slc30a10. A published ChIP-Seq data set identified two potential FXR-binding sites within 1 kb of the first exon of mouse Slc3010 and another potential site within 5 kb (72). Although these binding peaks are approximately an order of magnitude smaller than those seen for classical intestinal FXR targets Fgf15 and Slc51a (72), it is possible that FXR participates in Slc30a10 regulation at low levels or under certain conditions. Consistently, analysis of the human SLC30A10 promoter region revealed potential FXR and FXR::RXRA binding sites at four sites within 2 kb upstream of the transcription start site (Fig. S2). Another BA receptor that could modulate Slc30a10 induction is TGR5, although its effects might be indirect (e.g. through increasing cAMP and facilitating recruitment of transcription factors to cAMP response elements), as the receptor is largely membrane-bound (7, 8).
Does BA-dependent regulation of Mn homeostasis have any impact on cardiometabolic disease? Some researchers have reported that patients with type 2 diabetes have plasma or serum Mn at levels higher than controls, whereas others have reported the opposite (73–76). Because Mn levels must be maintained within a small range (31, 44), it makes sense that both deficiency and excess of Mn would lead to negative consequences. Accordingly, a recent report showed a U-shaped relationship between plasma Mn and the odds ratio for type 2 diabetes (77). When interpreting the previous findings, it should be noted that the most accurate circulating Mn levels are obtained from whole blood, not serum or plasma, because over 60% of blood Mn is found in erythrocytes (78). Interestingly, intravenous glucose tolerance tests in individuals with chronic manganism revealed reactionary hypoglycemia, which was proposed to be due to dysregulation of the hypothalamus-pituitary-adrenal axis (79). Human genetic variants in SLC39A8 are linked to blood Mn levels as well as body mass index, total cholesterol, and high-density lipoprotein cholesterol (80–83). In an unusual case of diabetes, a patient showed hypoglycemia in response to oral MnCl2, which was abolished upon a partial pancreatectomy (84). Thus, although severe Mn deficiency and toxicity lead to overt neurological and liver disorders, the role of Mn in common cardiometabolic diseases might be more nuanced.
It has been well-established that BAs and BA pool composition regulate macronutrient metabolism. This work demonstrates that BAs and BA pool composition also regulate metal homeostasis, which may be relevant for health and disease.
Experimental procedures
Reagents
Taurine-conjugated α- and β-muricholic acids were purchased from Cayman Chemical. All other BAs, micelle components, corn oil, and MnCl2 were obtained from Sigma. Calcitriol was obtained from Selleck Chemicals. Unless noted otherwise, all cell culture media and supplements were purchased from Gibco.
Crypt isolation and gut organoid culture
Mouse ileal crypts were isolated following methods developed by the Clevers Laboratory(30), with minor modifications. We euthanized adult (6–12-week-old) mice using CO2 inhalation followed by cervical dislocation. With the exception of the growth medium, all solutions and Matrigel were kept ice-cold during handling of crypts and organoids. We divided the small intestine into three parts of equal length and collected the last segment, adjacent to the cecum. We cut open the ileum longitudinally and rinsed it in PBS. We minced the tissue and washed the pieces by vigorously pipetting in PBS, allowing the pieces to settle, and removing the cloudy supernatant. We repeated this wash step until the supernatant was clear, 4–6 times. We then transferred the pieces into PBS containing 2 mm EDTA and incubated the mixture at 4 °C for 30–60 min with gentle shaking. Next, we removed the EDTA-PBS and replaced it with PBS containing 10% fetal bovine serum (FBS). We harvested crypts by vigorously pipetting, allowing the segments to settle, and collected the supernatant in a new tube. We repeated this harvesting procedure with additional FBS-PBS mixture, 3–4 times, pooling the supernatant from the same mouse. We then pelleted the crypts by centrifuging the crypts-FBS-PBS mixture at 900 rpm, 4 °C for 5 min. We discarded the supernatant, resuspended the crypts in base medium (Advanced DMEM/F-12 supplemented with 2 mm GlutaMax, 100 units/ml penicillin-streptomycin, and 10 mm HEPES), and pelleted the crypts by centrifuging at 720 rpm at 4 °C for 5 min. We discarded the supernatant, resuspended the crypts in base medium, passed the crypts-medium mixture through a 70-µm cell strainer, and centrifuged the crypts-medium mixture at 900 rpm at 4 °C for 5 min. Finally, we resuspended the crypts in Matrigel and seeded them on prewarmed 24-well Nunclon Delta surface-treated plates. After a 15–20-min incubation at 37 °C, we added IntestiCult Mouse Organoid Growth Medium (STEMCELL Technologies) to the wells. Medium was replaced twice weekly. Mature organoids were passaged every 7–10 days by dissolving the Matrigel dome with vigorous pipetting in PBS, passing the organoids-Matrigel-PBS mixture through a 27½-gauge needle, centrifuging to pellet the organoid pieces, resuspending them in new Matrigel, and distributing the organoid-Matrigel mixture into new prewarmed multiwell plates.
Caco-2 cell culture
Cells were cultured in DMEM containing GlutaMAX and high glucose, supplemented with 10% FBS and 100 units/ml penicillin-streptomycin, and incubated in a 37 °C, 5% CO2 chamber. Medium was replenished 3 times/week, and cells were passaged by trypsinization every 5–7 days. After confluence was achieved, cells were matured for an additional 5–7 days prior to treatment, a time point at which we validated that FXR, VDR, and their targets were expressed and that established FXR targets were induced by BAs (see Fig. S1F).
Preparation of micelles
We prepared micelles as described previously (85) with minor modifications. Individual stock solutions of oleic acid, 2-palmitoyl glycerol, phosphatidylcholine, cholesterol, glycoursodeoxycholic acid, and LCA were prepared in chloroform. Individual stock solutions of all other BAs were prepared in PBS. We mixed lipophilic components (those in chloroform stocks) in a glass vial and allowed them to dry under an N2 stream. We mixed BAs to prepare four distinct BA pools: human low (10%) 12HBA (H10), human high (90%) 12HBA (H90), mouse low (10%) 12HBA (M10), and mouse high (90%) 12HBA (M90). After the lipophilic components dried, we added BA pools to the vials and vortexed the mixture. We further diluted the mixture with DMEM containing 0.2% BSA (base medium), yielding final concentrations of 0.6 mm oleic acid, 0.2 mm 2-palmitoyl glycerol, 0.2 mm phosphatidylcholine, 0.05 mm cholesterol, and 1 mm total BAs. Compositions of BA pools are detailed in Table 1 and illustrated in Fig. 1B.
BA treatments of organoids and cells
We removed gut organoids from Matrigel and exposed the lumens by pipetting in ice-cold PBS followed by centrifugation at 150 × g at 4 °C for 10 min. Following a second PBS wash, we placed exposed gut organoids to micelle components without BAs (vehicle), or mixed micelles containing BAs (H10, H90, M10, or M90) in base medium (DMEM, 0.2% BSA) for 24 h. For BA treatments in the absence of micelles, BA pools were prepared in PBS and diluted in base medium for a final concentration of 1 mm, to match the concentration in the mixed micelle treatments. For Caco-2 cells, we seeded 200,000 cells/well into 12-well plates and allowed cells to reach confluence. We replaced the growth medium, washed cells with PBS, and added test media to the cells as indicated in the experiments.
RNA-Seq
We extracted RNA using the RNeasy Mini Kit (Qiagen). RNA concentration and quality were determined by Qubit Bioanalyzer. RNA from 3 samples/treatment group were submitted to the JP Sulzberger Columbia Genome Center for library preparation (standard poly(A) pulldown for mRNA enrichment), RNA-Seq (30M depth, single read), and processing of raw data. Differential expression analysis on counts data were done using the DESeq2 package in R.
Mn efflux assays
Mn efflux assays were done as described previously (36). We collected organoids by dissolving the Matrigel dome with ice-cold PBS, pipetting, and centrifuging at 150 × g for 10 min. Organoids were first incubated in “exposure medium” (DMEM supplemented with 0.2% BSA containing 0 or 500 μm MnCl2) for 16 h at 37 °C, 5% CO2. Following exposure, organoids were washed with PBS, given medium containing micelle components (vehicle), 1 mm H10, or 1 mm H90, and incubated for another 20 h. Organoids were pelleted by centrifugation, and efflux media were collected in metal-free tubes. For the Mn efflux assay in Caco-2 cells, we followed the same procedure, with the exception of 100 μm MnCl2 for exposure. Mn was measured by inductively coupled plasma MS.
Mouse experiments
12-Week-old male C57BL/6 mice (Taconic) were given AIN-93G with vitamin D adjusted at 50 IU/kg (Envigo TD.97184) for 3 days before the experiment. The vitamin D level was adjusted to minimize the contribution of dietary vitamin D to VDR signaling, which might interfere with the effects of LCA. This dose of vitamin D has been previously shown to cause no significant changes in body weight, serum calcium, serum parathyroid hormone, cortical or trabecular bone density, or parathyroid gland morphology, even after 4 months on the diet (86, 87). We observed no overt defects in the mice after 3 days on this diet. Mice were housed in a facility with a 12-h light/12-h dark cycle and had free access to food at all times. Reagent preparation, doses, and time points for gavage experiments were based on previous studies by Ishizawa et al. (88). LCA, calcitriol, and CA stocks were prepared in ethanol (vehicle), diluted 10-fold in corn oil, and vigorously vortexed. We gavaged 10 µl/g of body weight such that the final doses were 10% ethanol, 0.8 mmol/kg LCA, 50 nmol/kg calcitriol, and 0.8 mmol/kg CA. Oral gavages were done at 14 and 2 h prior to euthanasia. Experiments were approved by the Institutional Animal Care and Use Committee of Columbia University Medical Center. We collected ileum epithelia by cutting open the segment longitudinally and scraping the mucosa layer away from the muscle wall. Tissues were stored at –80 °C until they were processed for RNA extraction.
RNA extraction, cDNA synthesis, and qPCR
We extracted RNA from gut organoids using the RNeasy Mini Kit (Qiagen) and used 200 ng for cDNA synthesis. We extracted RNA from Caco-2 cells and ileum epithelium scrapings using TRIzol (Thermo Fisher Scientific) and used 1 µg for cDNA synthesis. cDNA synthesis was done using the High Capacity cDNA reverse transcription kit (Thermo Fisher). Quantitative real-time PCRs were carried out using iTaq Universal SYBR Green Supermix (Bio-Rad) and the CFX96 real-time PCR detection system (Bio-Rad). Mouse 36b4 or B2m was used as a reference gene for samples from mouse gut organoids and tissues. Human RPLP0 (primers from Qiagen) was used as a reference gene for cDNA from Caco-2 samples. Primer sequences are listed in Table S3.
Statistical analysis
Unless noted otherwise, we used one-way ANOVA followed by Benjamini–Hochberg correction. Adjusted p values of <0.05 were considered significant, with p < 0.05 (*), p < 0.01 (**), and p < 0.001 (***) between groups as marked.
Data availability
Raw RNA-Seq data have been deposited at the Gene Expression Omnibus (GEO), accession number GSE144398. All other data are contained within the article.
Acknowledgments
We thank Ana Flete and Thomas Kolar for technical assistance. This research utilized the Genomics and High Throughput Screening Shared Resource.
This article contains supporting information.
Author contributions—T. R. A., S. H., E. B., and R. A. H. conceptualization; T. R. A., S. H., E. B., A. H., N. S., N. C. G., and J. R. G. data curation; T. R. A., S. H., E. B., A. H., N. S., N. C. G., J. R. G., and R. A. H. formal analysis; T. R. A., A. H., N. S., N. C. G., and J. R. G. validation; T. R. A. and R. A. H. investigation; T. R. A. and R. A. H. visualization; T. R. A. and R. A. H. methodology; T. R. A. and R. A. H. writing-original draft; T. R. A., S. H., E. B., A. H., N. S., N. C. G., J. R. G., and R. A. H. writing-review and editing; R. A. H. resources; R. A .H. supervision; R. A .H. funding acquisition.
Funding and additional information—This work was supported by National Institutes of Health Grants DK115825 (to R. A. H.) and T32DK007328 (to T. R. A.) and the Russell Berrie Foundation. This work was also funded in part through NCI, National Institutes of Health, Cancer Center Support Grant P30CA013696. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- BA
- bile acid
- 12HBA
- 12α-hydroxylated bile acid
- CA
- cholic acid
- CDCA
- chenodeoxycholic acid
- DCA
- deoxycholic acid
- FXR
- farnesoid X receptor
- LCA
- lithocholic acid
- VDR
- vitamin D receptor
- qPCR
- quantitative PCR
- FBS
- fetal bovine serum
- DMEM
- Dulbecco's modified Eagle's medium
- ANOVA
- analysis of variance.
References
- 1. de Aguiar Vallim T. Q., Tarling E. J., and Edwards P. A. (2013) Pleiotropic roles of bile acids in metabolism. Cell Metab. 17, 657–669 10.1016/j.cmet.2013.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chávez-Talavera O., Tailleux A., Lefebvre P., and Staels B. (2017) Bile acid control of metabolism and inflammation in obesity, type 2 diabetes, dyslipidemia, and nonalcoholic fatty liver disease. Gastroenterology 152, 1679–1694.e3 10.1053/j.gastro.2017.01.055 [DOI] [PubMed] [Google Scholar]
- 3. de Boer J. F., Bloks V. W., Verkade E., Heiner-Fokkema M. R., and Kuipers F. (2018) New insights in the multiple roles of bile acids and their signaling pathways in metabolic control. Curr. Opin. Lipidol. 29, 194–202 10.1097/MOL.0000000000000508 [DOI] [PubMed] [Google Scholar]
- 4. Ahmad T. R., and Haeusler R. A. (2019) Bile acids in glucose metabolism and insulin signalling—mechanisms and research needs. Nat. Rev. Endocrinol. 15, 701–712 10.1038/s41574-019-0266-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Parks D. J., Blanchard S. G., Bledsoe R. K., Chandra G., Consler T. G., Kliewer S. A., Stimmel J. B., Willson T. M., Zavacki A. M., Moore D. D., and Lehmann J. M. (1999) Bile acids: natural ligands for an orphan nuclear receptor. Science 284, 1365–1368 10.1126/science.284.5418.1365 [DOI] [PubMed] [Google Scholar]
- 6. Makishima M., Okamoto A. Y., Repa J. J., Tu H., Learned R. M., Luk A., Hull M. V., Lustig K. D., Mangelsdorf D. J., and Shan B. (1999) Identification of a nuclear receptor for bile acids. Science 284, 1362–1365 10.1126/science.284.5418.1362 [DOI] [PubMed] [Google Scholar]
- 7. Maruyama T., Miyamoto Y., Nakamura T., Tamai Y., Okada H., Sugiyama E., Nakamura T., Itadani H., and Tanaka K. (2002) Identification of membrane-type receptor for bile acids (M-BAR). Biochem. Biophys. Res. Commun. 298, 714–719 10.1016/S0006-291X(02)02550-0 [DOI] [PubMed] [Google Scholar]
- 8. Kawamata Y., Fujii R., Hosoya M., Harada M., Yoshida H., Miwa M., Fukusumi S., Habata Y., Itoh T., Shintani Y., Hinuma S., Fujisawa Y., and Fujino M. (2003) A G protein-coupled receptor responsive to bile acids. J. Biol. Chem. 278, 9435–9440 10.1074/jbc.M209706200 [DOI] [PubMed] [Google Scholar]
- 9. Wang H., Chen J., Hollister K., Sowers L. C., and Forman B. M. (1999) Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol. Cell 3, 543–553 10.1016/S1097-2765(00)80348-2 [DOI] [PubMed] [Google Scholar]
- 10. Makishima M., Lu T. T., Xie W., Whitfield G. K., Domoto H., Evans R. M., Haussler M. R., and Mangelsdorf D. J. (2002) Vitamin D receptor as an intestinal bile acid sensor. Science 296, 1313–1316 10.1126/science.1070477 [DOI] [PubMed] [Google Scholar]
- 11. Hofmann A. F., and Roda A. (1984) Physicochemical properties of bile acids and their relationship to biological properties: an overview of the problem. J. Lipid Res. 25, 1477–1489 [PubMed] [Google Scholar]
- 12. Russell D. W. (2009) Fifty years of advances in bile acid synthesis and metabolism. J. Lipid Res. 50, S120–S125 10.1194/jlr.R800026-JLR200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li-Hawkins J., Gåfvels M., Olin M., Lund E. G., Andersson U., Schuster G., Björkhem I., Russell D. W., and Eggertsen G. (2002) Cholic acid mediates negative feedback regulation of bile acid synthesis in mice. J. Clin. Invest. 110, 1191–1200 10.1172/JCI0216309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Haeusler R. A., Astiarraga B., Camastra S., Accili D., and Ferrannini E. (2013) Human insulin resistance is associated with increased plasma levels of 12α-hydroxylated bile acids. Diabetes 62, 4184–4191 10.2337/db13-0639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Choucair I., Nemet I., Li L., Cole M. A., Skye S. M., Kirsop J. D., Fischbach M., Gogonea V., Brown J. M., Tang W. H. W., and Hazen S. L. (2019) Quantification of bile acids: a mass spectrometry platform for studying gut microbe connection to metabolic diseases. J. Lipid Res. 61, 159–177 10.1194/jlr.ra119000311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Brufau G., Stellaard F., Prado K., Bloks V. W., Jonkers E., Boverhof R., Kuipers F., and Murphy E. J. (2010) Improved glycemic control with colesevelam treatment in patients with type 2 diabetes is not directly associated with changes in bile acid metabolism. Hepatology 52, 1455–1464 10.1002/hep.23831 [DOI] [PubMed] [Google Scholar]
- 17. Haeusler R. A., Camastra S., Nannipieri M., Astiarraga B., Castro-Perez J., Xie D., Wang L., Chakravarthy M., and Ferrannini E. (2016) Increased bile acid synthesis and impaired bile acid transport in human obesity. J. Clin. Endocrinol. Metabol. 101, 1935–1944 10.1210/jc.2015-2583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Slätis K., Gåfvels M., Kannisto K., Ovchinnikova O., Paulsson-Berne G., Parini P., Jiang Z.-Y., and Eggertsen G. (2010) Abolished synthesis of cholic acid reduces atherosclerotic development in apolipoprotein E knockout mice. J. Lipid Res. 51, 3289–3298 10.1194/jlr.M009308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bonde Y., Eggertsen G., and Rudling M. (2016) Mice abundant in muricholic bile acids show resistance to dietary induced steatosis, weight gain, and to impaired glucose metabolism. PLoS ONE 11, e0147772 10.1371/journal.pone.0147772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bertaggia E., Jensen K. K., Castro-Perez J., Xu Y., Di Paolo G., Chan R. B., Wang L., and Haeusler R. A. (2017) Cyp8b1 ablation prevents Western diet-induced weight gain and hepatic steatosis because of impaired fat absorption. Am. J. Physiol. Endocrinol. Metab. 313, E121–E133 10.1152/ajpendo.00409.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kaur A., Patankar J. V., de Haan W., Ruddle P., Wijesekara N., Groen A. K., Verchere C. B., Singaraja R. R., and Hayden M. R. (2015) Loss of Cyp8b1 improves glucose homeostasis by increasing GLP-1. Diabetes 64, 1168–1179 10.2337/db14-0716 [DOI] [PubMed] [Google Scholar]
- 22. McGavigan A. K., Garibay D., Henseler Z. M., Chen J., Bettaieb A., Haj F. G., Ley R. E., Chouinard M. L., and Cummings B. P. (2017) TGR5 contributes to glucoregulatory improvements after vertical sleeve gastrectomy in mice. Gut 66, 226–234 10.1136/gutjnl-2015-309871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chevre R., Trigueros-Motos L., Castaño D., Chua T., Corlianò M., Patankar J. V., Sng L., Sim L., Juin T. L., Carissimo G., Ng L. F. P., Yi C. N. J., Eliathamby C. C., Groen A. K., Hayden M. R., et al. (2018) Therapeutic modulation of the bile acid pool by Cyp8b1 knockdown protects against nonalcoholic fatty liver disease in mice. FASEB J. 32, 3792–3802 10.1096/fj.201701084RR [DOI] [PubMed] [Google Scholar]
- 24. Clarysse S., Tack J., Lammert F., Duchateau G., Reppas C., and Augustijns P. (2009) Postprandial evolution in composition and characteristics of human duodenal fluids in different nutritional states. J. Pharm. Sci. 98, 1177–1192 10.1002/jps.21502 [DOI] [PubMed] [Google Scholar]
- 25. Northfield T. C., and McColl I. (1973) Postprandial concentrations of free and conjugated bile acids down the length of the normal human small intestine. Gut 14, 513–518 10.1136/gut.14.7.513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Thomas C., Gioiello A., Noriega L., Strehle A., Oury J., Rizzo G., Macchiarulo A., Yamamoto H., Mataki C., Pruzanski M., Pellicciari R., Auwerx J., and Schoonjans K. (2009) TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab. 10, 167–177 10.1016/j.cmet.2009.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Alemi F., Poole D. P., Chiu J., Schoonjans K., Cattaruzza F., Grider J. R., Bunnett N. W., and Corvera C. U. (2013) The receptor TGR5 mediates the prokinetic actions of intestinal bile acids and is required for normal defecation in mice. Gastroenterology 144, 145–154 10.1053/j.gastro.2012.09.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bunnett N. W. (2014) Neuro-humoral signalling by bile acids and the TGR5 receptor in the gastrointestinal tract. J. Physiol. 592, 2943–2950 10.1113/jphysiol.2014.271155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bouillon R., Carmeliet G., Verlinden L., van Etten E., Verstuyf A., Luderer H. F., Lieben L., Mathieu C., and Demay M. (2008) Vitamin D and human health: lessons from vitamin D receptor null mice. Endocr. Rev. 29, 726–776 10.1210/er.2008-0004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sato T., Vries R. G., Snippert H. J., van de Wetering M., Barker N., Stange D. E., van Es J. H., Abo A., Kujala P., Peters P. J., and Clevers H. (2009) Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 459, 262–265 10.1038/nature07935 [DOI] [PubMed] [Google Scholar]
- 31. Chen P., Bornhorst J., and Aschner M. (2018) Manganese metabolism in humans. Front. Biosci. 23, 1655–1679 10.2741/4665 [DOI] [PubMed] [Google Scholar]
- 32. Wahlström A., Kovatcheva-Datchary P., Ståhlman M., Khan M.-T., Bäckhed F., and Marschall H.-U. (2017) Induction of farnesoid X receptor signaling in germ-free mice colonized with a human microbiota. J. Lipid Res. 58, 412–419 10.1194/jlr.M072819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li J., and Dawson P. A. (2019) Animal models to study bile acid metabolism. Biochim. Biophys. Acta 1865, 895–911 10.1016/j.bbadis.2018.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tuschl K., Clayton P. T., Gospe S. M., Gulab S., Ibrahim S., Singhi P., Aulakh R., Ribeiro R. T., Barsottini O. G., Zaki M. S., Del Rosario M. L., Dyack S., Price V., Rideout A., Gordon K., et al. (2012) Syndrome of hepatic cirrhosis, dystonia, polycythemia, and hypermanganesemia caused by mutations in SLC30A10, a manganese transporter in man. Am. J. Hum. Genet. 90, 457–466 10.1016/j.ajhg.2012.01.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Quadri M., Federico A., Zhao T., Breedveld G. J., Battisti C., Delnooz C., Severijnen L.-A., Di Toro Mammarella L., Mignarri A., Monti L., Sanna A., Lu P., Punzo F., Cossu G., Willemsen R., et al. (2012) Mutations in SLC30A10 cause parkinsonism and dystonia with hypermanganesemia, polycythemia, and chronic liver disease. Am. J. Hum. Genet. 90, 467–477 10.1016/j.ajhg.2012.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Leyva-Illades D., Chen P., Zogzas C. E., Hutchens S., Mercado J. M., Swaim C. D., Morrisett R. A., Bowman A. B., Aschner M., and Mukhopadhyay S. (2014) SLC30A10 is a cell surface-localized manganese efflux transporter, and parkinsonism-causing mutations block its intracellular trafficking and efflux activity. J. Neurosci. 34, 14079–14095 10.1523/JNEUROSCI.2329-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nishito Y., Tsuji N., Fujishiro H., Takeda T., Yamazaki T., Teranishi F., Okazaki F., Matsunaga A., Tuschl K., Rao R., Kono S., Miyajima H., Narita H., Himeno S., and Kambe T. (2016) Direct comparison of manganese detoxification/efflux proteins and molecular characterization of ZnT10 protein as a manganese transporter. J. Biol. Chem. 291, 14773–14787 10.1074/jbc.M116.728014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zogzas C. E., Aschner M., and Mukhopadhyay S. (2016) Structural elements in the transmembrane and cytoplasmic domains of the metal transporter SLC30A10 are required for its manganese efflux activity. J. Biol. Chem. 291, 15940–15957 10.1074/jbc.M116.726935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mercadante C. J., Prajapati M., Conboy H. L., Dash M. E., Herrera C., Pettiglio M. A., Cintron-Rivera L., Salesky M. A., Rao D. B., and Bartnikas T. B. (2019) Manganese transporter Slc30a10 controls physiological manganese excretion and toxicity. J. Clin. Invest. 129, 5442–5461 10.1172/JCI129710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hutchens S., Liu C., Jursa T., Shawlot W., Chaffee B. K., Yin W., Gore A. C., Aschner M., Smith D. R., and Mukhopadhyay S. (2017) Deficiency in the manganese efflux transporter SLC30A10 induces severe hypothyroidism in mice. J. Biol. Chem. 292, 9760–9773 10.1074/jbc.M117.783605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Taylor C. A., Hutchens S., Liu C., Jursa T., Shawlot W., Aschner M., Smith D. R., and Mukhopadhyay S. (2019) SLC30A10 transporter in the digestive system regulates brain manganese under basal conditions while brain SLC30A10 protects against neurotoxicity. J. Biol. Chem. 294, 1860–1876 10.1074/jbc.RA118.005628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Stamelou M., Tuschl K., Chong W. K., Burroughs A. K., Mills P. B., Bhatia K. P., and Clayton P. T. (2012) Dystonia with brain manganese accumulation resulting from SLC30A10 mutations: a new treatable disorder. Mov. Disord. 27, 1317–1322 10.1002/mds.25138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mukhtiar K., Ibrahim S., Tuschl K., and Mills P. (2016) Hypermanganesemia with dystonia, polycythemia and cirrhosis (HMDPC) due to mutation in the SLC30A10 gene. Brain Dev. 38, 862–865 10.1016/j.braindev.2016.04.005 [DOI] [PubMed] [Google Scholar]
- 44. Anagianni S., and Tuschl K. (2019) Genetic disorders of manganese metabolism. Curr. Neurol. Neurosci. Rep. 19, 33 10.1007/s11910-019-0942-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Claro da Silva T., Hiller C., Gai Z., and Kullak-Ublick G. A. (2016) Vitamin D3 transactivates the zinc and manganese transporter SLC30A10 via the vitamin D receptor. J. Steroid Biochem. Mol. Biol. 163, 77–87 10.1016/j.jsbmb.2016.04.006 [DOI] [PubMed] [Google Scholar]
- 46. Mallory A., Kern F., Smith J., and Savage D. (1973) Patterns of bile acids and microflora in the human small intestine. Gastroenterology 64, 26–33 10.1016/S0016-5085(73)80088-5 [DOI] [PubMed] [Google Scholar]
- 47. Ridlon J. M., Kang D.-J., and Hylemon P. B. (2006) Bile salt biotransformations by human intestinal bacteria. J. Lipid Res. 47, 241–259 10.1194/jlr.R500013-JLR200 [DOI] [PubMed] [Google Scholar]
- 48. Kramer W., Stengelin S., Baringhaus K.-H., Enhsen A., Heuer H., Becker W., Corsiero D., Girbig F., Noll R., and Weyland C. (1999) Substrate specificity of the ileal and the hepatic Na+/bile acid cotransporters of the rabbit. I. Transport studies with membrane vesicles and cell lines expressing the cloned transporters. J. Lipid Res. 40, 1604–1617 [PubMed] [Google Scholar]
- 49. Dawson P. A., and Karpen S. J. (2015) Intestinal transport and metabolism of bile acids. J. Lipid Res. 56, 1085–1099 10.1194/jlr.R054114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Scheiber I. F., Wu Y., Morgan S. E., and Zhao N. (2019) The intestinal metal transporter ZIP14 maintains systemic manganese homeostasis. J. Biol. Chem. 294, 9147–9160 10.1074/jbc.RA119.008762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Liu C., Hutchens S., Jursa T., Shawlot W., Polishchuk E. V., Polishchuk R. S., Dray B. K., Gore A. C., Aschner M., Smith D. R., and Mukhopadhyay S. (2017) Hypothyroidism induced by loss of the manganese efflux transporter SLC30A10 may be explained by reduced thyroxine production. J. Biol. Chem. 292, 16605–16615 10.1074/jbc.M117.804989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mukhopadhyay S. (2018) Familial manganese-induced neurotoxicity due to mutations in SLC30A10 or SLC39A14. Neurotoxicology 64, 278–283 10.1016/j.neuro.2017.07.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lin W., Vann D. R., Doulias P.-T., Wang T., Landesberg G., Li X., Ricciotti E., Scalia R., He M., Hand N. J., and Rader D. J. (2017) Hepatic metal ion transporter ZIP8 regulates manganese homeostasis and manganese-dependent enzyme activity. J. Clin. Invest. 127, 2407–2417 10.1172/JCI90896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Park J. H., Hogrebe M., Grüneberg M., DuChesne I., von der Heiden A. L., Reunert J., Schlingmann K. P., Boycott K. M., Beaulieu C. L., Mhanni A. A., Innes A. M., Hörtnagel K., Biskup S., Gleixner E. M., Kurlemann G., et al. (2015) SLC39A8 deficiency: a disorder of manganese transport and glycosylation. Am. J. Hum. Genet. 97, 894–903 10.1016/j.ajhg.2015.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Xin Y., Gao H., Wang J., Qiang Y., Imam M. U., Li Y., Wang J., Zhang R., Zhang H., Yu Y., Wang H., Luo H., Shi C., Xu Y., Hojyo S., et al. (2017) Manganese transporter Slc39a14 deficiency revealed its key role in maintaining manganese homeostasis in mice. Cell Discov. 3, 17025 10.1038/celldisc.2017.25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Aydemir T. B., Kim M.-H., Kim J., Colon-Perez L. M., Banan G., Mareci T. H., Febo M., and Cousins R. J. (2017) Metal transporter Zip14 (Slc39a14) deletion in mice increases manganese deposition and produces neurotoxic signatures and diminished motor activity. J. Neurosci. 37, 5996–6006 10.1523/JNEUROSCI.0285-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Cikrt I. (1973) Enterohepatic circulation of 64Cu, 52Mn, and 203Hg in rats. Arch. Toxikol. 31, 51–59 10.1007/BF00330296 [DOI] [PubMed] [Google Scholar]
- 58. Tichý M., and Cikrt M. (1972) Manganese transfer into the bile in rats. Arch. Toxicol. 29, 51–58 10.1007/BF00316514 [DOI] [PubMed] [Google Scholar]
- 59. Cotzias G. C., and Papavasiliou P. S. (1964) Primordial homeostasis in a mammal as shown by the control of manganese. Nature 201, 828–829 10.1038/201828a0 [DOI] [PubMed] [Google Scholar]
- 60. Bertinchamps A. J., Miller S. T., and Cotzias G. C. (1966) Interdependence of routes excreting manganese. Am. J. Physiol. 211, 217–224 10.1152/ajplegacy.1966.211.1.217 [DOI] [PubMed] [Google Scholar]
- 61. Hurley L. S. (1981) The roles of trace elements in foetal and neonatal development. Philos. Trans. R. Soc. Lond. B Biol. Sci. 294, 145–152 10.1098/rstb.1981.0095 [DOI] [PubMed] [Google Scholar]
- 62. Andruska K. M., and Racette B. A. (2015) Neuromythology of manganism. Curr. Epidemiol. Rep. 2, 143–148 10.1007/s40471-015-0040-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Tuschl K., Meyer E., Valdivia L. E., Zhao N., Dadswell C., Abdul-Sada A., Hung C. Y., Simpson M. A., Chong W. K., Jacques T. S., Woltjer R. L., Eaton S., Gregory A., Sanford L., Kara E., et al. (2016) Mutations in SLC39A14 disrupt manganese homeostasis and cause childhood-onset parkinsonism–dystonia. Nat. Commun. 7, 11601 10.1038/ncomms11601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Krieger D., Krieger S., Theilmann L., Jansen O., Gass P., and Lichtnecker H. (1995) Manganese and chronic hepatic encephalopathy. Lancet 346, 270–274 10.1016/S0140-6736(95)92164-8 [DOI] [PubMed] [Google Scholar]
- 65. Fell J. M. E., Meadows N., Khan K., Long S. G., Milla P. J., Reynolds A. P., Quaghebeur G., and Taylor W. J. (1996) Manganese toxicity in children receiving long-term parenteral nutrition. Lancet 347, 1218–1221 10.1016/S0140-6736(96)90735-7 [DOI] [PubMed] [Google Scholar]
- 66. Aschner J. L., Anderson A., Slaughter J. C., Aschner M., Steele S., Beller A., Mouvery A., Furlong H. M., and Maitre N. L. (2015) Neuroimaging identifies increased manganese deposition in infants receiving parenteral nutrition. Am. J. Clin. Nutr. 102, 1482–1489 10.3945/ajcn.115.116285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hsieh C.-T., Liang J.-S., Peng S. S.-F., and Lee W.-T. (2007) Seizure associated with total parenteral nutrition–related hypermanganesemia. Pediatr. Neurol. 36, 181–183 10.1016/j.pediatrneurol.2006.10.005 [DOI] [PubMed] [Google Scholar]
- 68. Levy M., Elkoshi N., Barber-Zucker S., Hoch E., Zarivach R., Hershfinkel M., and Sekler I. (2019) Zinc transporter 10 (ZnT10)-dependent extrusion of cellular Mn2+ is driven by an active Ca2+-coupled exchange. J. Biol. Chem. 294, 5879–5889 10.1074/jbc.RA118.006816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Coneyworth L. J., Jackson K. A., Tyson J., Bosomworth H. J., Hagen E., van der, Hann G. M., Ogo O. A., Swann D. C., Mathers J. C., Valentine R. A., and Ford D. (2012) Identification of the human zinc transcriptional regulatory element (ZTRE): a palindromic protein-binding DNA sequence responsible for zinc-induced transcriptional repression. J. Biol. Chem. 287, 36567–36581 10.1074/jbc.M112.397000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ogo O. A., Tyson J., Cockell S. J., Howard A., Valentine R. A., and Ford D. (2015) The zinc finger protein ZNF658 regulates the transcription of genes involved in zinc homeostasis and affects ribosome biogenesis through the zinc transcriptional regulatory element. Mol. Cell Biol. 35, 977–987 10.1128/MCB.01298-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Go S., Kurita H., Yokoo K., Inden M., Kambe T., and Hozumi I. (2017) Protective function of SLC30A10 induced via PERK-ATF4 pathway against 1-methyl-4-phenylpyridinium. Biochem. Biophys. Res. Commun. 490, 1307–1313 10.1016/j.bbrc.2017.07.018 [DOI] [PubMed] [Google Scholar]
- 72. Thomas A. M., Hart S. N., Kong B., Fang J., Zhong X., and Guo G. L. (2010) Genome-wide tissue-specific farnesoid X receptor binding in mouse liver and intestine. Hepatology 51, 1410–1419 10.1002/hep.23450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kaur B., and Henry J. (2014) Micronutrient status in type 2 diabetes: a review. Adv. Food Nutr. Res. 71, 55–100 10.1016/b978-0-12-800270-4.00002-x [DOI] [PubMed] [Google Scholar]
- 74. Mancini F. R., Dow C., Affret A., Rajaobelina K., Dartois L., Balkau B., Bonnet F., Boutron-Ruault M.-C., and Fagherazzi G. (2018) Micronutrient dietary patterns associated with type 2 diabetes mellitus among women of the E3N-EPIC (Etude Epidémiologique auprès de femmes de l'Education Nationale) cohort study. J. Diabetes 10, 665–674 10.1111/1753-0407.12654 [DOI] [PubMed] [Google Scholar]
- 75. Wang X., Zhang M., Lui G., Chang H., Zhang M., Liu W., Li Z., Liu Y., and Huang G. (2016) Associations of serum manganese levels with prediabetes and diabetes among ≥60-year-old Chinese adults: a population-based cross-sectional analysis. Nutrients 8, 497 10.3390/nu8080497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Du S., Wu X., Han T., Duan W., Liu L., Qi J., Niu Y., Na L., and Sun C. (2018) Dietary manganese and type 2 diabetes mellitus: two prospective cohort studies in China. Diabetologia 61, 1985–1995 10.1007/s00125-018-4674-3 [DOI] [PubMed] [Google Scholar]
- 77. Shan Z., Chen S., Sun T., Luo C., Guo Y., Yu X., Yang W., Hu F. B., and Liu L. (2016) U-shaped association between plasma manganese levels and type 2 diabetes. Environ. Health Perspect. 124, 1876–1881 10.1289/EHP176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Milne D. B., Sims R. L., and Ralston N. V. C. (1990) Manganese content of the cellular components of blood. Clin. Chem. 36, 450–452 10.1093/clinchem/36.3.450 [DOI] [PubMed] [Google Scholar]
- 79. Hassanein M., Ghaleb H. A., Haroun E. A., Hegazy M. R., and Khayyal M. A. H. (1966) Chronic manganism: preliminary observations on glucose tolerance and serum proteins. Br. J. Ind. Med. 23, 67–70 10.1136/oem.23.1.67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Teslovich T. M., Musunuru K., Smith A. V., Edmondson A. C., Stylianou I. M., Koseki M., Pirruccello J. P., Ripatti S., Chasman D. I., Willer C. J., Johansen C. T., Fouchier S. W., Isaacs A., Peloso G. M., Barbalic M., et al. (2010) Biological, clinical and population relevance of 95 loci for blood lipids. Nature 466, 707–713 10.1038/nature09270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Speliotes E. K., Willer C. J., Berndt S. I., Monda K. L., Thorleifsson G., Jackson A. U., Allen H. L., Lindgren C. M., Luan J., Mägi R., Randall J. C., Vedantam S., Winkler T. W., Qi L., Workalemahu T., et al. (2010) Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat. Genet. 42, 937–948 10.1038/ng.686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Ng E., Lind P. M., Lindgren C., Ingelsson E., Mahajan A., Morris A., and Lind L. (2015) Genome-wide association study of toxic metals and trace elements reveals novel associations. Hum. Mol. Genet. 24, 4739–4745 10.1093/hmg/ddv190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Waterworth D. M., Ricketts S. L., Song K., Chen L., Zhao J. H., Ripatti S., Aulchenko Y. S., Zhang W., Yuan X., Lim N., Luan J., Ashford S., Wheeler E., Young E. H., Hadley D., et al. (2010) Genetic variants influencing circulating lipid levels and risk of coronary artery disease. Arterioscler. Thromb. Vasc. Biol. 30, 2264–2276 10.1161/ATVBAHA.109.201020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Rubenstein A. H., Levin N. W., and Elliott G. A. (1962) Manganese-induced hypoglycemia. Lancet 280, 1348–1351 10.1016/S0140-6736(62)91022-X [DOI] [PubMed] [Google Scholar]
- 85. Jattan J., Rodia C., Li D., Diakhate A., Dong H., Bataille A., Shroyer N. F., and Kohan A. B. (2017) Using primary murine intestinal enteroids to study dietary TAG absorption, lipoprotein synthesis, and the role of apoC-III in the intestine. J. Lipid Res. 58, 853–865 10.1194/jlr.M071340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Mallya S. M., Corrado K. R., Saria E. A., Yuan F. F., Tran H. Q., Saucier K., Atti E., Tetradis S., and Arnold A. (2016) Modeling vitamin D insufficiency and moderate deficiency in adult mice via dietary cholecalciferol restriction. Endocr. Res. 41, 290–299 10.3109/07435800.2016.1141937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Nehring J. A., Zierold C., and DeLuca H. F. (2007) Lithocholic acid can carry out in vivo functions of vitamin D. Proc. Natl. Acad. Sci. U. S. A. 104, 10006–10009 10.1073/pnas.0703512104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Ishizawa M., Akagi D., and Makishima M. (2018) Lithocholic acid is a vitamin D receptor ligand that acts preferentially in the ileum. Int. J. Mol. Sci. 19, 1975 10.3390/ijms19071975 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Raw RNA-Seq data have been deposited at the Gene Expression Omnibus (GEO), accession number GSE144398. All other data are contained within the article.