

Abstract
BACKGROUND:
Germline DICER1 mutations increase the risk of developing a wide variety of generally uncommon tumors. We describe a case of DICER1-related embryonal rhabdomyosarcoma (ERMS) of the uterine corpus in a prepubertal girl.
CASE:
A ten-year-old- girl with history of cystic nephroma presented with a three week history of vaginal bleeding. A 3-cm mass filling the uterine cavity was detected and histopathologic examination of hysteroscopy-guided biopsies revealed ERMS. Molecular genetic sequencing of the tumor sample revealed a DICER1 mutation.
SUMMARY AND CONCLUSION:
This report highlights the importance of screening for DICER1 mutations in the presence of the early-onset features of this syndrome and extends the spectrum of DICER1-related tumors by showing the mutation in a case of ERMS of the uterine corpus.
Keywords: DICER1, Embryonal rhabdomyosarcoma, Uterus
INTRODUCTION
Germline pathogenic variation in DICER1 is associated with increased risk of developing a wide variety of generally uncommon tumors that occur at relatively younger ages. This condition is most commonly linked with pleuropulmonary blastoma (PPB), ovarian Sertoli-Leydig cell tumors (SLCT), cystic nephroma and neoplasms of thyroid gland1,2. In the female genital tract, DICER1 variation may be associated with embryonal rhabdomyosarcoma of the cervix and ovary2,3.
Rhabdomyosarcoma (RMS) is a malignant mesenchymal tumor, and is the most common soft tissue tumor in childhood. The genitourinary (GU) tract (bladder, prostate, vagina, uterus, and paratesticular regions) is the second most common site in the pediatric population, after the head and neck region. Most of RMS seen in the female genital tract is histologically of embryonal type. Vaginal ERMS are commonly of the botryoid variant histologically and are mostly found in very young children4. Cervical and uterine sarcomas are diagnosed more commonly in older girls and present with a mass, with or without vaginal discharge. Cervical ERMS (cERMS), has been reported to be associated with pathogenic DICER1 variation5,6. Primary ERMS of the uterine corpus is very rare in prepubertal girls and there is limited information about its clinicopathological features in the literature. We report a case of ERMS of uterine corpus in a ten-year-old prepubertal girl who was diagnosed with cystic nephroma as an infant. Genetic testing of the tumor revealed a DICER1 mutation.
CASE
A ten-year-old-prepubertal girl presented with three weeks history of vaginal bleeding. There was no history of trauma or sexual abuse. Her previous history was significant for cystic nephroma of the right kidney treated with total nephrectomy at age 1. Family history revealed that her paternal uncle had a nephrectomy for Wilms’ tumor. On physical examination, she had Tanner stage 1 breast and pubic hair development. The inspection of vulva, hymen and the distal part of the vagina revealed normal anatomy of the external genitalia and no mass or foreign body. There was also no sign of vulvovaginitis or trauma.
Abdominal ultrasonography showed a markedly distended uterus with an echogenic mass which was suspicious for hematometra or a tumoral lesion. Magnetic resonance imaging (MRI) revealed a mass with necrosis and peripheral contrast enhancement in the uterine cavity (Figure 1). No myometrial invasion or adjacent organ involvement was observed. Hysteroscopy under general anesthesia was performed, which showed a three cm irregular friable mass attached to the anterior uterine wall by an elongated pedicle (Figure 2). Multiple biopsy samples were taken with hysteroscopy scissors and grasper. Histopathologic and immunohistochemical examination of samples revealed ERMS. Due to the patient’s history of cystic nephroma of right kidney, molecular genetic sequencing was performed by polymerase chain reaction amplification of genomic DNA (gDNA) extracted from the formalin-fixed, paraffin embedded tumor, followed by Sanger sequencing and revealed a missense mutation, c.5113G>A (p.E1705K), in the RNase IIIb domain of DICER1. A subcentimeter suspicious right external iliac lymph node was also detected by positron emission tomography/computed tomography (PET/CT). CT of chest and MRI of abdomen were otherwise unremarkable.
Figure 1.
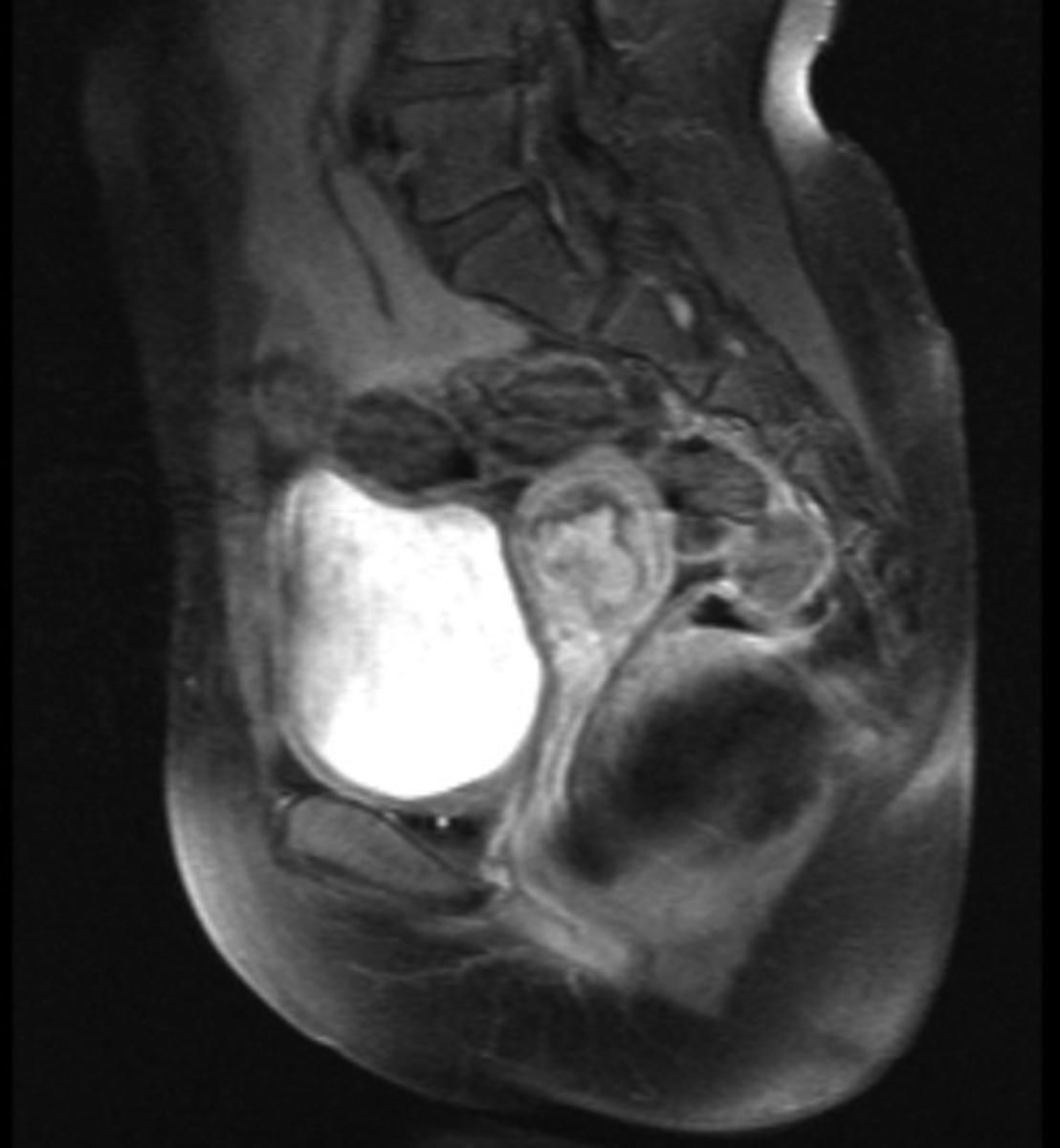
Pelvic MRI showing the mass filling the endometrial cavity
Figure 2.
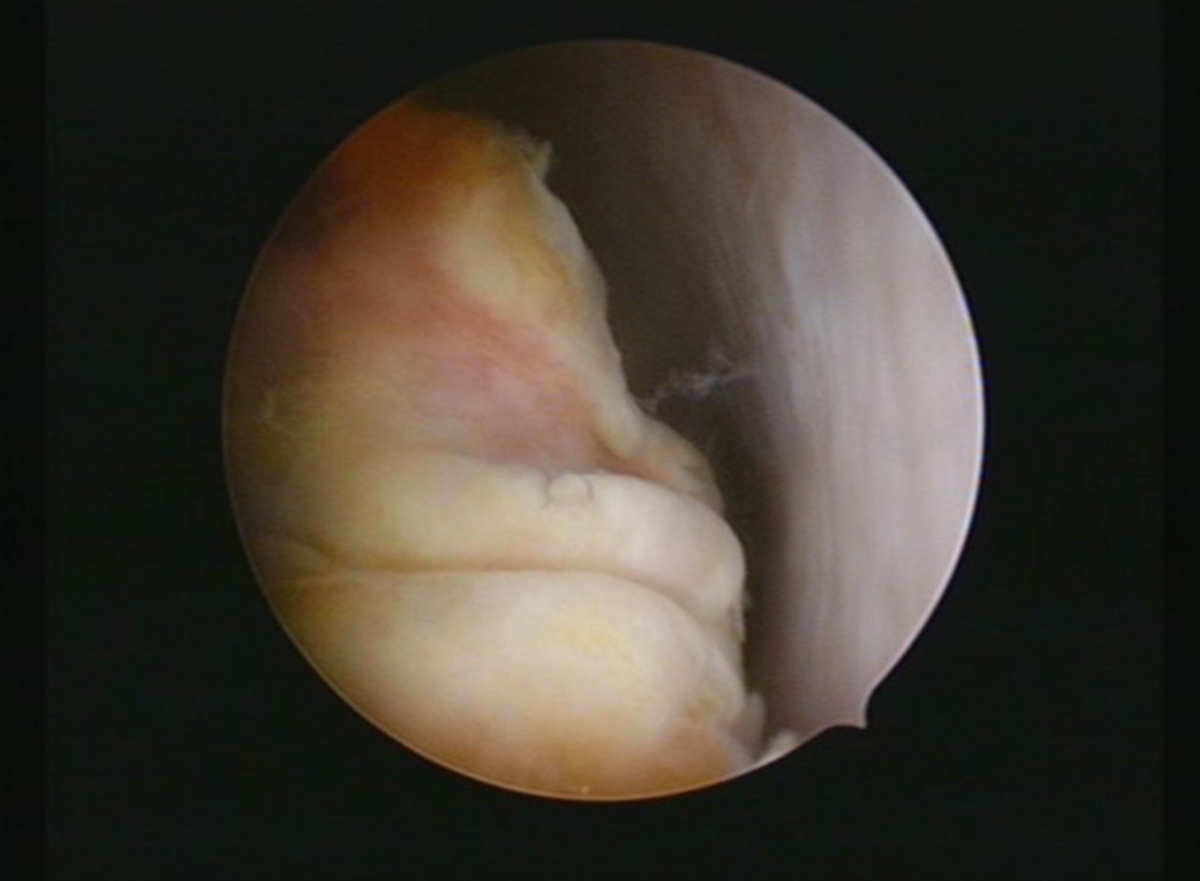
Hysteroscopic appearance of the tumor
She was staged as Group 3A according to the clinical group system (CG) developed by the Intergroup Rhabdomyosarcoma Study Group (IRSG) based on having localized or locally extensive tumor, gross residual disease after biopsy only. She was also staged as having Stage 1 disease according to the tumor, node, metastasis (TNM) system due to favorable disease site (nonbladder/nonprostate genitourinary tumor) without distant metastases. When information from the CG and TNM staging systems combine, it was considered as a low risk subset B tumor based on rhabdomyosarcoma prognostic stratification scheme adopted by the IRSG. She was discussed in the Istanbul University multidisciplinary tumor board and consulted with international experts. She was started on neoadjuvant chemotherapy consisting of vincristine (1.5 mg/m2), actinomycin D (0.045 mg/kg), cyclophosphamide (1.2 g/m2) (VAC) every three weeks (modified COG-ARST0531protocol) due to the presence of a large mass and a small prepubertal uterus in view of the possibility of conservative treatment options following chemotherapy. After seven courses of chemotherapy, pelvic MRI and ultrasound showed no residual tumor and also no pathologic lymph node was observed. The second-look hysteroscopy detected no macroscopic tumor in the uterine cavity, multiple biopsy samples were taken from the area where the tumor had initially originated. Histopathologic examination of these samples revealed microscopic residual tumor cells. Further local treatment options were then discussed in the Istanbul University Tumor Board and with international experts and also with the patient and her parents including external beam radiation therapy (RT) for suspicious pelvic lymph node involvement with intracavitary brachytherapy (BT) for local control and/or surgical treatment options. Hysterectomy with bilateral pelvic lymphadenectomy was performed to reduce the risk of recurrence and prevent the possible long-term complications associated with RT. Histopathologic examination of the uterus showed only microscopic residual tumor cells and no nodal involvement. Postoperatively, she also received seven additional courses of VAC. She is now under regular follow-up.
Genetic testing of the patient and family members was subsequently performed. A heterozygous pathogenic variant in DICER1 (NM_030621.4:c.1525C>T;p.Arg509*) was identified in the patient, her father, her sister, her paternal aunt and her paternal uncle who had a history of Wilms’ tumor as an infant. They were referred for family education and imaging-based surveillance for DICER1 related conditions.
DISCUSSION
The DICER1 protein is an RNase III enzyme that cleaves precursor miRNAs into mature miRNAs, their active form. miRNAs regulate mRNA expression. Loss-of-function germline mutations in the coding region of DICER1 are associated with tumor predisposition including increased risk of developing uncommon neoplasms such as PPB, ovarian SLCT, cystic nephroma, and Wilms tumor1,2. The history and/or the presence of these DICER1 related tumors should prompt germline DICER1 testing. DICER1 sequence analysis of tumors typically show a DICER1 loss of function mutation (commonly germline) and a somatic missense mutation affecting one of five codons in the RNase IIIb domain; so-called “hotspots,” on the other DICER1 allele. In a proportion of cases, the loss of function mutation and the RNase IIIb mutation will be limited to tumor cells only.7 In our case, molecular genetic testing of the tumor specimen performed due to the patient’s history of cystic nephroma revealed a somatic mutation of DICER1. The loss of function germ-line mutation was also detected.
Although DICER1 pathogenic variation has an autosomal dominant mode of inheritance, the variable penetrance of DICER1 germline mutations can also result in an unremarkable family history. In the present case, the family history was significant for Wilms’ tumor on her fathers’ side. Family members of the patient also underwent molecular genetic testing of DICER1 for the pathogenic variant identified in the proband, which showed a heterozygous pathogenic variant of DICER1 in her father, sister, paternal aunt and uncle who had a history of Wilms’ tumor as an infant. About 84% of DICER1 mutations are inherited from a parent. Individuals with a germline heterozygous mutation of DICER1 have a 50% chance of transmitting the mutated DICER1 allele to the offspring. As significant excesses in DICER1 related cancer risk has been recently reported among non-proband DICER1-carriers8, these family members were referred for genetic counseling, family education and imaging-based surveillance based on recommended strategies9. Family education regarding tumor risk and possible presenting symptoms is the cornerstone of early diagnosis. Briefly, current guidelines for individuals with germline pathogenic DICER1 variation suggest chest x-ray at birth and every 6 months until age 8 and annually until age 12 years with consideration of chest CT at 3–9 months of age and again at 2–3 years of age, prior to the peak diagnosis of pleuropulmonary blastoma. Guidelines for screening for renal tumors suggest abdominal ultrasound every 6 months until age 8 and every year until age 12. Pelvic ultrasound is suggested every 6 months for females due to risk of tumors of the gynecologic tract. Dilated eye exam is suggested annually from at least age 3 to 10 years. Thyroid nodules are common and thyroid cancer may occur, thus thyroid palpation is recommended at least annually with thyroid ultrasound suggested every 3 years.
In the female genital tract, RMS most commonly presents with a polypoid mass protruding from the vagina and/or cervix accompanied by vaginal bleeding10. However, in the present case, vulvar inspection and vaginoscopy showed no mass protruding from the vagina or the cervical canal. An irregular friable mass filling the endometrial cavity was detected by hysteroscopy. ERMS of the uterine cervix (cERMS), a generally uncommon site, is known to be associated with DICER1 mutations. The diagnosis of cERMS, irrespective of the age of onset, should be considered an indication for genetic testing for the DICER1 variation due to the high likelihood of the association5,6. Recently ERMS of the ovary has been also reported as a manifestation of the DICER1 syndrome3. RMS of the uterine corpus is more rare than of the cervix and our case suggests that ERMS of uterine corpus should be considered among the list of uncommon childhood tumors associated with the DICER1 variation.
The local treatment of ERMS of the uterine corpus is challenging, especially regarding conservative surgical treatment options other than hysterectomy and use of brachytherapy for local control10. Local resection with negative margins necessary to eliminate the need for radiotherapy can be achieved by hysterectomy in the RMS of the uterine corpus. In our case, chemotherapy was chosen as the initial treatment considering the possibility of conservative management in the absence of residual tumor following chemotherapy. After detection of microscopic residual tumor cells by second look hysteroscopy following seven cycles of chemotherapy, hysterectomy with bilateral pelvic lymphadenectomy was preferred for local control and also for the management of suspicious right external iliac lymph node detected during the initial staging work-up and disappeared following chemotherapy. The reasons for choosing surgical management were reducing the risk of recurrence and the possibility of avoiding radiotherapy and its possible long-term side effects such as vaginal stenosis, impaired sexual functions, infertility, gastrointestinal and urinary issues. When radiotherapy is indicated, brachytherapy should be considered for local control because it affects a smaller tissue volume than external-beam radiotherapy10.
This case is also unique with regard to her individual history and family history. She had cystic nephroma as an infant and underwent unilateral nephrectomy with no additional treatment. Her paternal uncle had a history of Wilms’ tumor as a young child. Cystic nephroma and Wilms’ tumor are associated with DICER1 mutations. In fact, when she presented with RMS, her history of cystic nephroma alerted us to the possibility of an underlying germline DICER1 mutation, however, at the time of their initial diagnosis DICER1 was a less recognized condition. Even now, there is a need to increase awareness about DICER1-associated tumors especially given the implications for individual and familial surveillance.
In conclusion, this report highlights the need to test for germline DICER1 mutation in the presence of the characteristic neoplasms such as cystic nephroma and/or ERMS of the gynecologic tract. The detection of a DICER1 mutation in an individual or family members is important to facilitate surveillance for related tumors, so that they may be detected at the earliest possible stage, potentially increasing survival and decreasing risks of late effects including risks to fertility. This case also shows the relationship between DICER1 mutation and embryonal rhabdomyosarcoma of the uterine corpus, expanding the spectrum of DICER1-associated tumors.
ACKNOWLEDGEMENT:
The authors thank Prof Emin Darendeliler from the Department of Radiation Oncology, Istanbul University, Oncology Institute, Prof. Carola Arndt from the Department of Pediatric Hematology-Oncology, Mayo Clinic for their helpful discussions in the treatment decisions; Anne K. Harris from the Children’s Hospitals and Clinics of Minnesota, and the International Pleuropulmonary Blastoma/ DICER1 Registry and International Ovarian and Testicular Stromal Tumor (OTST) Registry, Minneapolis, MN, Amanda Field, from the ResourcePath Laboratory, Sterling, VA for their valuable help in the DICER1 genetic testing. Ashley Hill and KrisAnn Schultz are supported by National Institutes of Health grant R01CA143167.
Footnotes
Declaration of interest: None
REFERENCES
- 1.Hill DA, Ivanovich J, Priest JR, Gurnett CA, Dehner LP, Desruisseau D, Jarzembowski JA, Wikenheiser-Brokamp KA, Suarez BK, Whelan AJ, Williams G, Bracamontes D, Messinger Y, Goodfellow PJ. DICER1 mutations in familial pleuropulmonary blastoma. Science. 2009. August 21;325(5943):965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stewart CJ, Charles A, Foulkes WD. Gynecologic Manifestations of the DICER1 Syndrome. Surg Pathol Clin. 2016;9(2):227–41. [DOI] [PubMed] [Google Scholar]
- 3.de Kock L, Druker H, Weber E, et al. Ovarian embryonal rhabdomyosarcoma is a rare manifestation of the DICER1 syndrome. Hum Pathol. 2015;46(6):917–22. [DOI] [PubMed] [Google Scholar]
- 4.Kirsch CH1, Goodman M, Esiashvili N. Outcome of female pediatric patients diagnosed with genital tract rhabdomyosarcoma based on analysis of cases registered in SEER database between 1973 and 2006. Am J Clin Oncol. 2014;37(1):47–50. [DOI] [PubMed] [Google Scholar]
- 5.Dehner LP, Jarzembowski JA, Hill DA. Embryonal rhabdomyosarcoma of the uterine cervix: a report of 14 cases and a discussion of its unusual clinicopathological associations. Mod Pathol 2012;25:602–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Doros L, Yang J, Dehner L, et al. DICER1 mutations in embryonal rhabdomyosarcomas from children with and without familial PPB-tumor predisposition syndrome. Pediatr Blood Cancer. 2012;59:558–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brenneman M, Field A, Yang J et al. Temporal order of RNase IIIb and loss-of-function mutations during development determines phenotype in pleuropulmonary blastoma / DICER1 syndrome: a unique variant of the two-hit tumor suppression model. Version 2. F1000Res. 2015;4:214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stewart DR, Best AF, Williams GM, et al. Neoplasm Risk Among Individuals With a Pathogenic Germline Variant in DICER1. Clin Oncol. 2019;37(8):668–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schultz KAP, Williams GM, Kamihara J, et al. DICER1 and Associated Conditions: Identification of At-risk Individuals and Recommended Surveillance Strategies. Clin Cancer Res. 2018;24(10):2251–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Minard-Colin V, Walterhouse D, Bisogno G, et al. Localized vaginal/uterine rhabdomyosarcoma-results of a pooled analysis from four international cooperative groups. Pediatr Blood Cancer. 2018;65(9):e27096. [DOI] [PMC free article] [PubMed] [Google Scholar]