

Abstract
Possible involvement of complement (C) systems in the pathogenesis of traumatic brain injury (TBI) was investigated by quantifying Cproteins in plasma astrocyte-derived exosomes (ADEs) of subjects with sports-related TBI (sTBI) and TBI in military veterans (mtTBI) without cognitive impairment. All sTBI subjects (n = 24) had mild injuries, whereas eight of the mtTBI subjects had moderate, and 17 had mild injuries. Plasma levels of ADEs were decreased after acute sTBI and returned to normal within months. Cprotein levels in ADEs were from 12- to 35-fold higher than the corresponding levels in neuron-derived exosomes. CD81 exosome marker-normalized ADE levels of classical pathway C4b, alternative pathway factor D and Bb, lectin pathway mannose-binding lectin (MBL), and shared neurotoxic effectors C3b and C5b-9 terminal C complex were significantly higher and those of C regulatory proteins CR1 and CD59 were lower in the first week of acute sTBI (n = 12) than in controls (n = 12). Most C abnormalities were no longer detected in chronic sTBI at 3–12 months after acute sTBI, except for elevated levels of factor D, Bb, and MBL. In contrast, significant elevations of ADE levels of C4b, factor D, Bb, MBL, C3b and C5b-9 terminal C complex, and depressions of CR1 and CD59 relative to those of controls were observed after 1–4 years in early chronic mtTBI (n = 10) and persisted for decades except for normalization of Bb, MBL, and CD59 in late chronic mtTBI (n = 15). Complement inhibitors may be useful therapeutically in acute TBI and post-concussion syndrome.
Keywords: extracellular vesicles, neurodegeneration, neuroinflammation
1 |. INTRODUCTION
Astrocytes are abundant glial cells in the human central nervous system (CNS) that normally support the neurons through promotion of development, nutrition, survival, dendrite outgrowth, and synapse formation.1–3 CNS responses to neuroinflammatory and neurodegenerative diseases include elevation in the total number of astrocytes and their increased differentiation into A1-type reactive astrocytes.4–6 An ensemble of immune cytokines are prominent inducers of A1-type astrocytes.7–9 A1-type astrocytes exhibit increased expression of pro-inflammatory pathways as well as neuron-directed toxic activities that damage synapses early and later destroy the neurons.2,6,10,11
Studies of postmortem brain tissues from patients with neurodegenerative diseases have delineated some putatively neurotoxic factors in A1-type astrocytes, but it is not clear which are involved in pathogenesis. Approximately 60% of glial fibrillary acidic protein-positive A1-type astrocytes in the prefrontal cortex of patients with Alzheimer’s disease (AD) contain prominently elevated levels of intact complement component 3 (C3) and potentially neurotoxic C3 fragments.4,6 Evidence of the possible pathogenic involvement of complement systems in AD had been presented, but it was not clear that astrocytes are the principal source of these complement mediators.4,12–14 Finding that reactive astrocytes and complement may protect neurons from proteinopathic factors in some animal models of AD further complicated interpretation of the role of complement in pathogenesis.15,16
Characterization of astrocyte-derived exosomes (ADEs) enriched from human plasma by sequential precipitation and immunochemical absorption showed much higher levels of the astrocyte biomarkers glutamine synthetase and glial fibrillary acidic protein than in neuron-derived exosomes (NDEs) enriched from the same plasmas.17 In contrast, NDEs had much higher levels than ADEs of the neuronal markers neurofilament light chain and neuron-specific enolase.17 Initial analyses of ADEs showed higher levels of β-site amyloid precursor protein-cleaving enzyme 1 and soluble amyloid precursor protein β of the Aβ42 peptide-generating system than in NDEs.17
Quantification of plasma ADE levels of complement proteins provided evidence for increased activation of the classical pathway and alternative amplification loop, but not the lectin pathway, in astrocytes of patients with AD compared to controls.18 Further, ADE content of the complement-regulatory membrane proteins CD59, CD46, decay-accelerating factor, and complement receptor type 1 (CR1), but not of fluid-phase factor I, were significantly lower in AD than controls.18 These data suggested a neurotoxic opsonic role for C3b and the possibility of direct neuronal membrane attack by C5b-9 terminal complement complex (TCC) in AD. Such complement abnormalities were detected in the phase of mild cognitive impairment several years preceding dementia and in clinically evident mild AD, but not in preclinical AD 5–12 years before memory loss.18,19
Less is known of the activation and pathogenic involvement of complement systems in TBI than in degenerative neurological diseases. Nonetheless, immunochemical quantification of complement proteins in frontal and temporal lobe tissues of TBI patients and mouse models of TBI showed that levels of C1q, C3b, C3d, C5b-9 TCC, and mannose-binding lectin (MBL) of the lectin complement pathway were elevated relative to those of controls.20–22 CSF levels of soluble C5b-9 TCC were elevated for up to 10 days after TBI and were highest in those with altered blood-brain barrier function.23 The effects of individual complement protein deficiencies and selective complement inhibitors on different stages of TBI were assessed in mouse models.24 These results suggested that C5b-9 TCC was most critical in mediating acute neural deficits, whereas C3b contributed to the secondary phase of injury by mediating progressive degeneration of dendritic and synaptic structures.20 The present study was designed to identify abnormalities in plasma ADE levels of complement mediators and inhibitors for groups of patients with TBI of different severity and at different times after TBI. The subject groups include patients sustaining sports-related TBI (sTBI) and military veterans suffering TBI (mtTBI) to further elucidate possible complement protein therapeutic targets in both settings.
2 |. MATERIALS AND METHODS
2.1 |. Patient evaluation and experimental design
For sTBI, participants were students from the University of Denver or from the University of California, San Francisco (Table 1). All sustained mild TBIs in the course of playing high-impact sports. Acute TBI was defined by the standard NCAA clinical criteria for diagnosis and included 12 participants who donated plasma for this study within 7 days of an episode of sTBI.25,26 Half of the subjects with acute sTBI had suffered one (1/12) or two (5/12) mild TBIs prior to the episode of this study, but all had occurred at least 10 months before the study episode (Table 1). Chronic sTBI was defined as having had at least two TBIs, the most recent of which was no less than 3 months and no more than 12 months before donation of blood for this study (Table 1). Only two of 12 controls for sTBI had one mild TBI at least 5 years prior to participation in this study. On entry into the study, all sTBI participants underwent a cognitive test battery, including Mini-Mental State Examination (MMSE) and other measures of memory, and balance testing.26
TABLE 1.
Demographics and TBI history of participants
| Age (mean ± SEM) | Sex (female/male) | Total TBIs-subjects | ||
|---|---|---|---|---|
| A. sTBIa | Interval between last TBI and blood donation (mean ± SEM) | |||
| Control (n = 12) | 21.6 ± 0.68 | 6/6 | None | – |
| Acute TBI (n = 12) | 21.3 ± 0.40 | 6/6 | 1–5 | ≤1 week |
| 2–1 | ||||
| Chronic TBI (n = 12) | 20.1 ± 0.48 | 6/6 | 2–6 | 6.58 ± 0.89 months |
| 3–3 | ||||
| 4–3 | ||||
| B. mtTBIb | Months between last TBI and blood donation (mean ± SEM) | |||
| Chronic control | ||||
| Early (n = 5) | 38.0 ± 3.89 | 1/4 | None | – |
| Late (n = 5) | 74.6 ± 1.81 | 2/3 | None | – |
| Chronic TBI | ||||
| Early (n = 10) | 38.4 ± 2.44 | 3/7 | 1–7 | 28.2 ± 4.74 |
| 2–3 | ||||
| Late (n = 15) | 77.0 ± 1.86 | 2/13 | 1–4 | 561 ± 58.4 |
| 2–5 | ||||
| 3–3 | ||||
| 4–3 |
Abbreviations: mtTBI, military-inflicted TBI; sTBI, sports-related TBI.
Acute sTBI participants had no previous episodes of TBI (6/12) or up to two previous episodes at least 12 months before the study episode, whereas chronic sTBI participants had at least two previous episodes with the most recent being no more than 12 months before the study episode.
Early chronic mtTBI encompasses participants studied 1–4 years after TBI and late chronic mtTBI those studied ≥12 years after TBI.
All mtTBI participants were military veterans residing at the Veterans Home of California in Yountville, CA or in communities in Northern California. Military veteran participants were recruited for the study through flyers, social events, and word of mouth. The total cohort of 35 had an age range of 30–79 years for controls (n = 10) and 29–93 years for mtTBI subjects (n = 25). Early chronic mtTBI subjects (n = 10) donated blood for this study 1–4 years after their TBI and late chronic mtTBI subjects (n = 15) donated blood 12 years or more after their TBI (Table 1). mtTBI was defined initially as a self-reported head injury resulting in documented medical attention that included a neurological examination. Then the Ohio State University TBI Identification Method, a structured clinical interview recommended by the National Institute of Neurological Disorders and Stroke as a Common Data Element for the retrospective assessment of lifetime TBI in clinical research, was used to assess mtTBI history and determine the severity of mtTBI.27 The severity of mtTBI was mild for 17 mtTBI subjects and moderate for eight mtTBI subjects. No mtTBI subject had cognitive impairment as assessed by the MMSE and detailed neuropsychological testing, as described.28,29 As norms are less established for very old participants, cognitive impairment for the elderly was defined operationally using a composite Z-score, as described.29,30
One investigator (E.J.G.) isolated ADEs and NDEs from all plasmas together by the same procedures and completed ELISAs without knowledge of clinical data. The protocol and procedures of this study received prior approval by the Institutional Review Boards of the University of Denver, University of California, San Francisco and the Veterans Administration Medical Center of San Francisco. Informed Consent was obtained from each participant.
2.2 |. Blood sampling of patients and control participants
Ten milliliters of venous blood were drawn by syringe into 0.5 mL of saline with EDTA and centrifuged for 15 minutes at 2500 × g. Plasma samples were stored in 0.25 mL aliquots at −80°C.
2.3 |. Enrichment of plasma ADEs for enzyme-linked assay quantification of proteins
Aliquots of 0.25 mL plasma were incubated with 0.1 mL thromboplastin D (Thermo Fisher Scientific, Waltham, MA), followed by addition of 0.15 mL of calcium- and magnesiumfree Dulbecco’s balanced salt solution (DBS) with protease inhibitor cocktail (Roche, Indianapolis, IN) and phosphatase inhibitor cocktail (Thermo Fisher Scientific; DBS++). After centrifugation at 3000 × g for 30 minutes at 4°C, total exosomes were harvested from resultant supernatants by precipitation with 126 μL per tube of ExoQuick (System Biosciences, Mountain View, CA) and centrifugation at 1500 × g for 30 minutes at 4°C. To enrich ADEs, total exosomes were resuspended in 0.35 mL of DBS and incubated for 60 minutes at room temperature with 2.0 μg of mouse anti-human glutamine aspartate transporter (ACSA-1) biotinylated antibody (Miltenyi Biotec, Auburn, CA) in 50 μL of 3% bovine serum albumin (BSA; 1:3.33 dilution of Blocker BSA 10% solution in DBS; Thermo Fisher Scientific) per tube with mixing, followed by addition of 10 μL of streptavidin-agarose Ultralink resin (Thermo Fisher Scientific) in 40 μL of 3% BSA and incubation for 30 minutes at room temperature with mixing.17 After centrifugation at 800 X g for 10 minutes at 4°C and removal of the supernatant, each pellet was suspended in 100 μL of cold 0.05M glycine-HCl (pH 3.0) by gentle mixing for 10 seconds and centrifuged at 4000 X g for 10 minutes, all at 4°C. Supernatants then were transferred to clean tubes containing 25 μL of 10% BSA and 10 μL of 1M Tris-HCl (pH 8.0) and mixed gently. An aliquot of 5 μL was removed from each tube for ADE counts before addition of 370 μL of mammalian protein extraction reagent (M-PER, Thermo Fisher Scientific). Resultant 0.5 mL lysates of ADEs were stored at −80°C. NDEs were prepared as described.31
For counting of exosomes, each suspension was diluted 1:50 in PBS. The mean diameter (nanometers) and concentration (particles per milliliter) of exosomes in each suspension were determined by nanoparticle tracking analysis (NTA) using the Nanosight NS500 system with a G532nm laser module and NTA 3.1 nanoparticle tracking software (Malvern Instruments, Malvern, United Kingdom). Camera settings were as follows: gain 366; shutter 31.48; and frame rate 24.9825 frames/s. Brownian motion was captured by performing 5 repeated 60 s video recordings.
ADE and NDE proteins were quantified by enzyme-linked immunosorbent assay (ELISA) kits for human tetraspanning exosome marker CD81, complement fragment C4b (ARP American Research Products, Waltham, MA; Cusabio Technology, College Park, MD), glutamine synthetase, CR1, factor I (ARP American Research Products; Cloud-Clone Corp, Katy, TX), glial fibrillary acidic protein (EMD Millipore, Billerica, MA), complement fragment C3b, complement factor B (Abcam, Cambridge, MA), Bb fragment of complement factor B (Quidel-Microvue, San Diego, CA), TCC C5b-9 (Aviva Systems, San Diego, CA), CD59, MBL (Ray Biotech, Norcross, GA), and complement factor D (Thermo Fisher Scientific-Invitrogen, LaFayette, CO).
The mean value for all determinations of CD81 in each assay group was set at 1.00, and relative values of CD81 for each sample were used to normalize their recovery.
2.4 |. Statistical analyses
The Shapiro-Wilks test showed that data in all 54 sets, except three, were distributed normally. Statistical significance of differences between means for TBI groups and normal controls were determined with an unpaired Student t test, including a Bonferroni correction. For the three non-normally distributed sets, significance was determined by a Mann-Whitney U test.
3 |. RESULTS
The ages and sex distribution were the same for participants in the control, acute, and chronic sTBI groups (Table 1). Subsequently determined ADE data were indistinguishable for the early and late chronic mtTBI control groups, so results for these 10 subjects were considered together as one control group for both the early and late mtTBI subjects. Sex distribution was the same for the control and early chronic mtTBI groups, but men were overrepresented in the late chronic mtTBI group (Table 1). No subject in any group had cognitive impairment.
As was found in a previous study of extracellular vesicle complement proteins,18 their levels for the same six subjects in an acute phase of sTBI were significantly higher in ADEs than NDEs (Table 2). Plasma levels of NDEs had been shown to be decreased acutely after TBI.32 In the current study, plasma levels of ADEs determined by counts and CD81 content were significantly lower for the acute sTBI group, but not any of the chronic TBI groups, relative to those of corresponding controls (Table 3). Therefore, all values for ADE complement proteins in Figures 1 and 2 were normalized for CD81 content.
TABLE 2.
Differences in levels of complement proteins in ADEs and NDEs of acute sTBI
| C4b | Factor D | Bb | C3b | C5b-9 TCC | |
|---|---|---|---|---|---|
| ADEs | 124 208 ± 7469 | 25 341 ± 5116 | 606 352 ± 128 438 | 23 798 ± 3916 | 687 ± 96.1 |
| NDEs | 9769 ± 732 | 960 ± 68.6 | 17 130 ± 2120 | 1310 ± 53.6 | 19.4 ± 2.05 |
Note: All values are mean pg/mL ± SEM for plasmas of six sTBI subjects in the acute phase after injury. Differences all are significant at P < .001.
TABLE 3.
Acute decrease in plasma ADE concentration after TBI
| A. sTBI | Control | Acute | Chronic |
|---|---|---|---|
| CD81 (pg/mL) | 1250 ± 36.7 | 959 ± 86.7* | 1311 ± 61.3 |
| Counts (×109/mL) | 56.0 ± 1.75 | 43.8 ± 1.78** | 56.5 ± 1.41 |
| B. mtTBI | Control | Early chronic | Late chronic |
| CD81 (pg/mL) | 1658 ± 264 | 1160 ± 38.6 | 1282 ± 42.2 |
| Counts (×109/mL) | 60.7 ± 1.33 | 56.5 ± 1.33 | 58.7 ± 1.33 |
Note: Unpaired t test comparison to control value with *P < .01 and **P < .001.
Abbreviations: mtTBI, military-inflicted TBI; sTBI, sports-related TBI.
FIGURE 1.
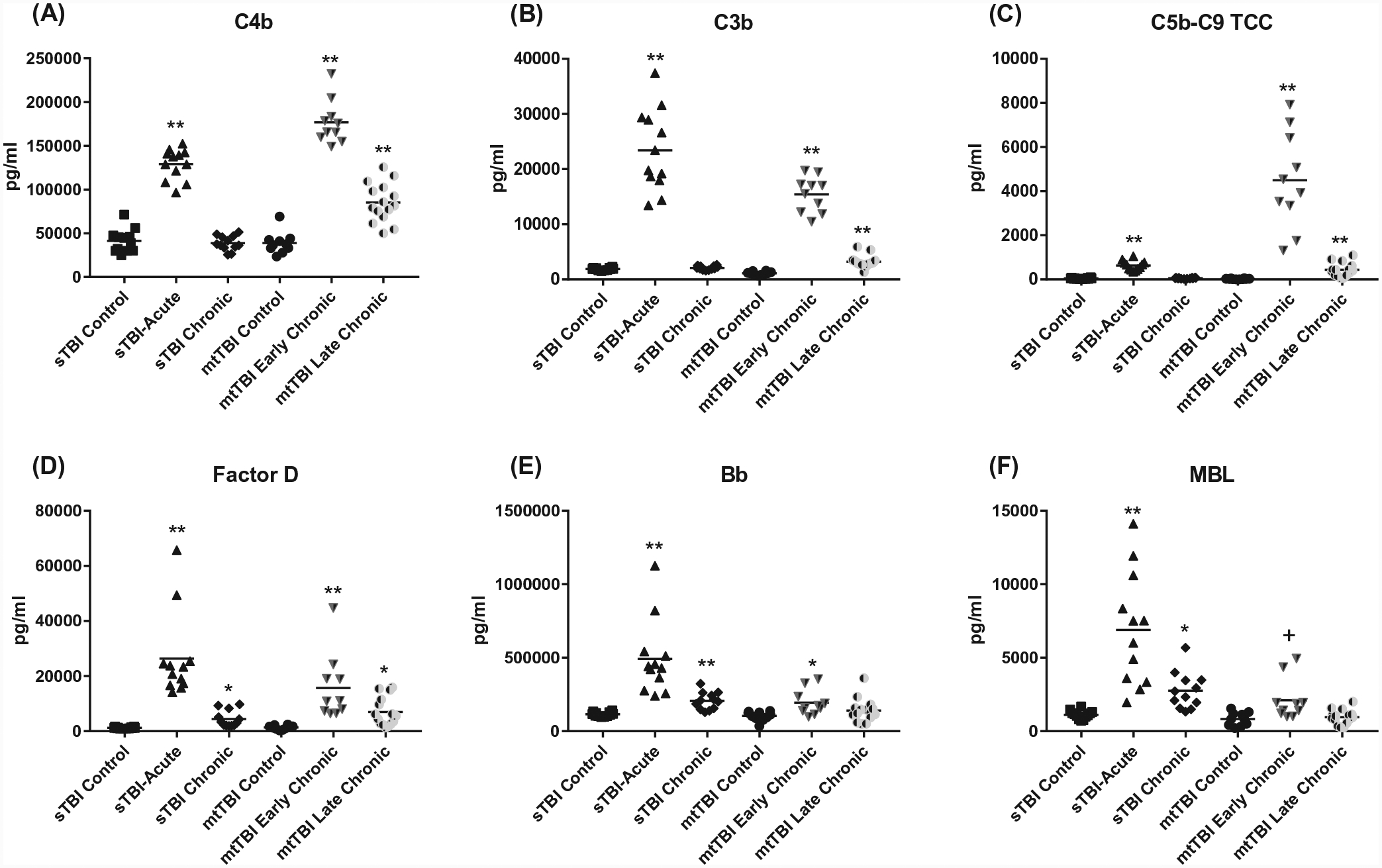
ADE levels of complement effector proteins in sTBI and mtTBI subject groups. Each point represents the value for a control or TBI participant and the horizontal line in point clusters is the mean level for that group. Mean ± SEM for sTBI control, sTBI acute, sTBI chronic, mtTBI control, mtTBI early chronic, and mtTBI late chronic participant values, respectively, are 741 ± 79.6, 501 ± 21.6, 887 ± 74.0, 841 ± 54.9, 483 ± 90.6, and 586 ± 45.9 pg/mL for C4b (A), 1889 ± 65.4, 23 414 ± 2139, 2091 ± 101, 1107 ± 90.6, 15 420 ± 1022, and 3207 ± 289 pg/mL for C3b (B), 45.2 ± 7.25, 629 ± 62.2, 53.4 ± 6.46, 24.3 ± 5.29, 4493 ± 690, and 436 ± 80.5 pg/mL for C5b-9 TCC (C), 741 ± 79.6, 501 ± 21.6, 887 ± 74.0, 841 ± 54.9, 483 ± 90.6, and 586 ± 45.9 pg/mL for factor D (D), 115 423 ± 4298, 491 384 ± 73 188, 206 574 ± 17 089, 103 866 ± 9960, 194 506 ± 27 240, and 140 820 ± 20 312 pg/mL for Bb (E), and 1121 ± 82.4, 6899 ± 1112, 2752 ± 363, 829 ± 144, 2109 ± 440, and 955 ± 127 pg/mL for MBL (F). The significance of differences shown between values for controls and TBI subjects were calculated by an unpaired Student’s t test; +P < .05, *P < .01, **P < .0001
FIGURE 2.
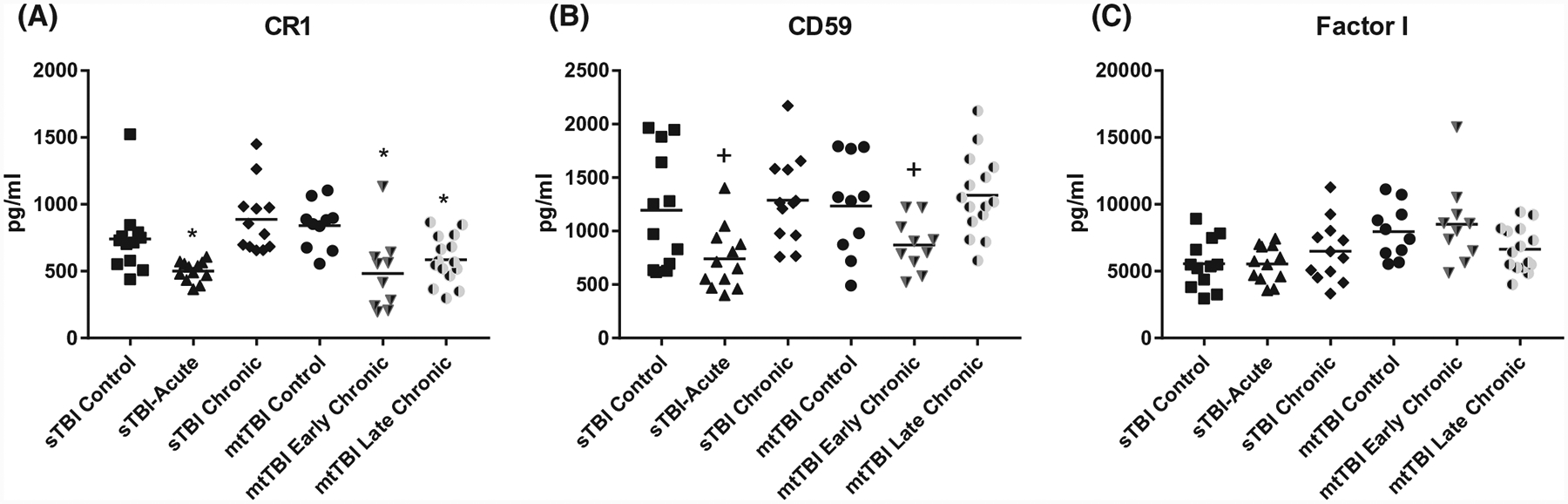
ADE levels of complement regulatory proteins in sTBI and mtTBI subject groups. Each point represents the value for a control or TBI participant and the horizontal line in point clusters is the mean level for that group. Mean ± SEM for sTBI control, sTBI acute, sTBI chronic, mtTBI control, mtTBI early chronic, and mtTBI late chronic participant values, respectively, are 741 ± 79.6, 501 ± 21.6, 887 ± 74.0, 841 ± 54.9, 483 ± 90.6, and 586 ± 45.9 pg/mL for CR1 (A), 1196 ± 157, 740 ± 84.5, 1289 ± 118, 1233 ± 147, 870 ± 76.0, and 1334 ± 97.0 pg/mL for CD59 (B), and 5566 ± 538, 5545 ± 388, 6492 ± 666, 7957 ± 633, 8509 ± 967, and 6640 ± 430 pg/mL for factor I (C). The significance of differences shown between values for controls and TBI subjects were calculated by an unpaired Student’s t test; +P < .05, *P < .01
In the acute phase of sTBI, within 1 week of injury, CD81-normalized ADE levels of complement proteins C4b, factor D, Bb, and MBL of all three pathways as well as shared effector proteins C3b and C5b-9 TCC were significantly higher than those of controls (Figure 1). Concurrently, those of the membrane-associated complement regulatory proteins CR1 and CD59 (Figure 2), but not fluid-phase factor I, were significantly lower than those of controls. In the chronic phase sTBI group, from 3 months to 1 year after TBI, CD81-normalized ADE levels of alternative pathway factors D and Bb as well as lectin pathway MBL, but not of classical pathway C4b or the effector proteins C3b and C5b-9 TCC, were significantly higher than those of controls (Figure 1). This suggests distinctively persistent, but limited, activation of the alternative and lectin pathways in sTBI. None of the CD81-normalized ADE levels of any of the complement regulatory proteins were lower in chronic sTBI than controls (Figure 2).
No plasmas were available from subjects with acute mtTBI. In early chronic mtTBI, 1–4 years after injury, ADE levels of complement proteins in all three pathways and the shared effector proteins C3b and C5b-9 TCC all were significantly higher (Figure 1) and of the membrane-associated complement regulatory proteins CR1 and CD59, but not factor I, were significantly lower than in controls (Figure 2). In late chronic mtTBI, 12 or more years after injury, ADE levels of complement proteins C4b, factor D, and the shared effector proteins C3b and C5b-9 TCC, but not of Bb or MBL, were significantly higher (Figure 1) and levels of the membrane-associated complement regulatory protein CR1, but not CD59 or factor I, were significantly lower than in controls (Figure 2). Tentative assembly of a time-course for generation of the neurotoxic complement factors C3b and C5b-9 TCC by one or more pathways following a TBI suggests initiation as soon as days after injury and possible persistence for years after TBI depending on the nature and severity of the injury.
The maximal increases in C3b and C5b-9 TCC observed in acute sTBI were no longer sustained months later in chronic sTBI, along with subsidence of the acute increases in C4b, factor D, Bb, and MBL (Figure 1). For early chronic mtTBI with post-TBI intervals similar to chronic sTBI, the ADE levels of C3b, C5b-9 TCC, C4b, factor D, Bb, and MBL all were at their highest point for the mtTBI set and C3b, C5b-9 TCC, C4b, and factor D remained elevated in late chronic mtTBI. All episodes of sTBI were mild, whereas some episodes of mtTBI were moderate. When subjects with early chronic mtTBI were evaluated in relation to severity of TBI, those with moderate TBI (n = 4) had significantly higher mean ± SEM levels of ADE C3b and C5b-C9 at 18 482 ± 784 pg/mL and 3295 ± 790 pg/mL (P = .0072 and .0157), respectively, than those with mild TBI (n = 6) at 13 463 ± 1005 pg/mL and 1318 ± 134 pg/mL. There were no other correlations between the clinical severity of mtTBI and the level of complement effector proteins in mtTBI in this limited set of study subjects.
4 |. DISCUSSION
The elevated ADE levels of complement component C4b of the classical pathway and factors D and Bb of the alternative pathway in acute sTBI relative to controls resemble in magnitude those observed in AD (Figure 1).18,19 However, a distinctive increase in ADE levels of MBL in the same subjects relative to those of controls suggests recruitment of the lectin complement pathway that was not seen in Alzheimer’s disease. Thus in acute sTBI, all three complement pathways appear to contribute rapidly to the increased ADE levels of C3b and C5b-9 TCC that are capable of damaging synapses and injuring neurons.
The ADE levels of C4b, factor D, Bb, and MBL, and the resultant effector components C3b and C5b-9 TCC all are elevated significantly within days and probably hours in acute sTBI relative to controls (Figure 1). Similarly, ADE levels of the complement regulatory membrane proteins CR1 and CD59 decline within days after TBI (Figure 2). In contrast, the time-courses of correction of ADE levels of complement proteins to normal in TBI is extremely variable, presumably in relation to the type, severity, and other characteristics of the injury. Return of ADE levels of C4b, C3b, C5b-9 TCC, CR1, and CD59, but not factor D, Bb, or MBL, to normal was complete in sTBI within months. In contrast, return of ADE levels of Bb, MBL, and CD59 to those of controls in mtTBI required many years and normalization of ADE levels of C4b, C3b, C5b-9 TCC, and CR1 was not complete decades after injury. This may explain why many individuals with blast injuries have persistent post-concussive symptoms of a wide range from disordered balance to psychiatric issues for many years.
The time of recruitment of ADE neuronally toxic effector products of the classical and alternative pathways of complement in AD is late relative to that of elevations of NDE levels of β-amyloid peptides and a range of forms of P-tau in the decades long course of disease.18 Elevated plasma NDE levels of β-amyloid peptides and P-tau moieties, as well as diverse constituents of damaged neurons and their synapses, are detected up to 15 years prior to memory loss and other clinical evidence of cognitive decline.33 ADE levels of complement regulatory membrane proteins were significantly reduced up to 12 years before clinical evidence of cognitive losses in AD, but ADE levels of neurotoxic complement proteins first became abnormally high late in the course at about 5 years before cognitive loss in AD and about 3 years before conversion of MCI to Alzheimer’s dementia.18,19
The present results have several implications for potential therapeutic applications of complement pathway inhibitors in TBI and AD. Generation of the effector proteins C3b and C5b-9 TCC involve two or more pathways in both diseases. Thus alleviation of complement effects would require targeting of multiple shared elements, such as the C3 convertase C4bC2b of the classical and lectin pathways and the C3 convertase C3bBb of the alternative pathway. In contrast, inhibition of binding of C3b to C3 convertase to prevent formation of a C5 convertase or suppression of C5 convertase activity could substantially decrease generation of C5b-9 TCC in both diseases. The second relevant point is the time of pathogenic involvement of complement mediators. In TBI, inhibitors would likely need to be administered immediately and for months to be optimally effective. In AD, any benefits will require treatment only in the late phases after recruitment of complement pathways. Overall, the results demonstrate that ADE complement and other protein cargoes should be meaningfully considered when evaluating biomarkers of TBI and neurodegenerative diseases as their levels are significantly higher than those of NDEs from the same patients (Table 2).17,18
ACKNOWLEDGMENTS
The authors are grateful to Judith H. Goetzl (JHSF, CJL) for expert preparation of the illustrations. The results reported are from studies supported by the Department of Defense [WB1XWH-14-2-0137] (K.Y., C.B.P.), an intramural pilot grant from DU (A.L., K.G., B.D., A.-C.G.) and the Intramural Research Program of the National Institutes on Aging (M.M., D.K., D.T., N.G.).
Funding information
U.S. Department of Defense (DOD), Grant/Award Number: WB1XWH-14-2-0137; University of Denver (DU), Grant/Award Number: 999; HHS | NIH | National Institute on Aging (NIA)
Abbreviations:
- AD
Alzheimer’s disease
- ADE
astrocyte-derived exosome
- C
complement
- DBS
Dulbecco’s balanced salt solution
- MBL
mannose-binding lectin
- MMSE
mini-mental state examination
- mtTBI
TBI in military veterans
- NDE
neuron-derived exosome
- TBI
traumatic brain injury
- sTBI
sports-related TBI
Footnotes
CONFLICT OF INTEREST
Edward J. Goetzl has filed a USA Patent application for the ADE methodology described, but other authors have no conflicts to report.
REFERENCES
- 1.Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol. 2010;119:7–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zamanian JL, Xu L, Foo LC, et al. Genomic analysis of reactive astrogliosis. J Neurosci. 2012;32:6391–6410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anderson MA, Burda JE, Ren Y, et al. Astrocyte scar formation aids central nervous system axon regeneration. Nature. 2016;532:195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ben Haim L, Carrillo-de Sauvage MA, Ceyzeriat K, Escartin C. Elusive roles for reactive astrocytes in neurodegenerative diseases. Front Cell Neurosci. 2015;9:278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goetzl EJ, Miller BL. Multicellular hypothesis for the pathogenesis of Alzheimer’s disease. Faseb J. 2017;31:1792–1795. [DOI] [PubMed] [Google Scholar]
- 6.Liddelow SA, Barres BA. Reactive astrocytes: production, function, and therapeutic potential. Immunity. 2017;46:957–967. [DOI] [PubMed] [Google Scholar]
- 7.Choi SS, Lee HJ, Lim I, Satoh J, Kim SU. Human astrocytes: secretome profiles of cytokines and chemokines. PLoS ONE. 2014;9:e92325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crotti A, Ransohoff RM. Microglial physiology and pathophysiology: insights from genome-wide transcriptional profiling. Immunity. 2016;44:505–515. [DOI] [PubMed] [Google Scholar]
- 9.Liddelow SA, Guttenplan KA, Clarke LE, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541:481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sofroniew MV. Multiple roles for astrocytes as effectors of cytokines and inflammatory mediators. Neuroscientist. 2014;20:160–172. [DOI] [PubMed] [Google Scholar]
- 11.Ceyzeriat K, Abjean L, Carrillo-de Sauvage MA, Ben Haim L, Escartin C. The complex STATes of astrocyte reactivity: howare they controlled by the JAK-STAT3 pathway? Neuroscience. 2016;330:205–218. [DOI] [PubMed] [Google Scholar]
- 12.Stevens B, Allen NJ, Vazquez LE, et al. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131:1164–1178. [DOI] [PubMed] [Google Scholar]
- 13.Lian H, Yang L, Cole A, et al. NFkappaB-activated astroglial release of complement C3 compromises neuronal morphology and function associated with Alzheimer’s disease. Neuron. 2015;85:101–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hong S, Beja-Glasser VF, Nfonoyim BM, et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. 2016;352:712–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kraft AW, Hu X, Yoon H, et al. Attenuating astrocyte activation accelerates plaque pathogenesis in APP/PS1 mice. Faseb J. 2013;27:187–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heppner FL, Ransohoff RM, Becher B. Immune attack: the role of inflammation in Alzheimer disease. Nat Rev Neurosci. 2015;16:358–372. [DOI] [PubMed] [Google Scholar]
- 17.Goetzl EJ, Mustapic M, Kapogiannis D, et al. Cargo proteins of plasma astrocyte-derived exosomes in Alzheimer’s disease. Faseb J. 2016;30:3853–3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goetzl EJ, Schwartz JB, Abner EL, Jicha GA, Kapogiannis D. High complement levels in astrocyte-derived exosomes of Alzheimer disease. Ann Neurol. 2018;83:544–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Winston CN, Goetzl EJ, Schwartz JB, Elahi FM, Rissman RA. Complement protein levels in plasma astrocyte-derived exosomes are abnormal in conversion from mild cognitive impairment to Alzheimer’s disease dementia. Alzheimers Dement. 2019; 11 : 61–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alawieh A, Langley EF, Weber S, Adkins D, Tomlinson S. Identifying the role of complement in triggering neuroinflammation after traumatic brain injury. J Neurosci. 2018;38:2519–2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hammad A, Westacott L, Zaben M. The role of the complement system in traumatic brain injury: a review. J Neuroinflammation. 2018;15:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.De Blasio D, Fumagalli S, Orsini F, et al. Human brain trauma severity is associated with lectin complement pathway activation. J Cereb Blood Flow Metab. 2019;39:794–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stahel PF, Morganti-Kossmann MC, Perez D, et al. Intrathecal levels of complement-derived soluble membrane attack complex (sC5b-9) correlate with blood-brain barrier dysfunction in patients with traumatic brain injury. J Neurotrauma. 2001;18:773–781. [DOI] [PubMed] [Google Scholar]
- 24.Carpanini SM, Torvell M, Morgan BP. Therapeutic inhibition of the complement system in diseases of the central nervous system. Front Immunol. 2019;10:362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ruff RM, Iverson GL, Barth JT, et al. Recommendations for diagnosing a mild traumatic brain injury: a National Academy of Neuropsychology education paper. Arch Clin Neuropsychol. 2009;24:3–10. [DOI] [PubMed] [Google Scholar]
- 26.Maroon JC, Lovell MR, Norwig J, Podell K, Powell JW, Hartl R. Cerebral concussion in athletes: evaluation and neuropsychological testing. Neurosurgery. 2000;47:659–669; discussion 669–672. [DOI] [PubMed] [Google Scholar]
- 27.Corrigan JD, Bogner J. Initial reliability and validity of the Ohio State University TBI identification method. J Head Trauma Rehabil. 2007;22:318–329. [DOI] [PubMed] [Google Scholar]
- 28.Ivnik RJ, Smith GE, Petersen RC, Boeve BF, Kokmen E, Tangalos EG. Diagnostic accuracy of four approaches to interpreting neuropsychological test data. Neuropsychology. 2000;14:163–177. [DOI] [PubMed] [Google Scholar]
- 29.Goetzl EJ, Peltz CB, Mustapic M, Kapogiannis D, Yaffe KC. Neuron-derived plasma exosome proteins after remote traumatic brain injury. J Neurotrauma. 2020; 37:382–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harris ME, Ivnik RJ, Smith GE. Mayo’s older Americans normative studies: expanded AVLT recognition trial norms for ages 57 to 98. J Clin Exp Neuropsychol. 2002;24:214–220. [DOI] [PubMed] [Google Scholar]
- 31.Goetzl EJ, Kapogiannis D, Schwartz JB, et al. Decreased synaptic proteins in neuronal exosomes of frontotemporal dementia and Alzheimer’s disease. Faseb J. 2016;30:4141–4148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goetzl EJ, Elahi FM, Mustapic M, et al. Altered levels of plasma neuron-derived exosomes and their cargo proteins characterize acute and chronic mild traumatic brain injury. Faseb J. 2019;33:5082–5088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fiandaca MS, Kapogiannis D, Mapstone M, et al. Identification of preclinical Alzheimer’s disease by a profile of pathogenic proteins in neurally derived blood exosomes: a case-control study. Alzheimers Dement. 2015;11(6):600–607.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]