

Precursor B cell acute lymphoblastic leukemia (B-ALL) is caused by genetic lesions in developing B cells that function as drivers for the accumulation of additional mutations in an evolutionary selection process. We investigated secondary drivers of leukemogenesis in a mouse model of B-ALL driven by PU.1/Spi-B deletion (Mb1-CreΔPB). Whole-exome-sequencing analysis revealed recurrent mutations in Jak3 (encoding Janus kinase 3), Jak1, and Ikzf3 (encoding Aiolos).
KEYWORDS: PU.1, Spi-B, ETS transcription factors, gene regulation, leukemia, reactive oxygen species, transcription factors
ABSTRACT
Precursor B cell acute lymphoblastic leukemia (B-ALL) is caused by genetic lesions in developing B cells that function as drivers for the accumulation of additional mutations in an evolutionary selection process. We investigated secondary drivers of leukemogenesis in a mouse model of B-ALL driven by PU.1/Spi-B deletion (Mb1-CreΔPB). Whole-exome-sequencing analysis revealed recurrent mutations in Jak3 (encoding Janus kinase 3), Jak1, and Ikzf3 (encoding Aiolos). Mutations with a high variant-allele frequency (VAF) were dominated by C→T transition mutations that were compatible with activation-induced cytidine deaminase, whereas the majority of mutations, with a low VAF, were dominated by C→A transversions associated with 8-oxoguanine DNA damage caused by reactive oxygen species (ROS). The Janus kinase (JAK) inhibitor ruxolitinib delayed leukemia onset, reduced ROS and ROS-induced gene expression signatures, and altered ROS-induced mutational signatures. These results reveal that JAK mutations can alter the course of leukemia clonal evolution through ROS-induced DNA damage.
INTRODUCTION
Acute lymphoblastic leukemia (ALL) is the most frequently occurring type of cancer in young children (1). Despite a high remission rate, ALL is still the leading cause of cancer-related deaths in children, and much needs to be done to reduce the long-term effects of chemotherapy toxicity (1). Of the many subtypes of ALL, 80% of pediatric ALLs are cancers of the B lymphocyte lineage (B-ALL) (1). Pre-B-ALL is associated with mutations or chromosomal translocations involving genes encoding transcription factors or proteins involved in B cell receptor (BCR) signaling (2). Pre-B-ALL cells are frequently addicted to Janus kinase signal transducer and activator of transcription (JAK-STAT) signaling pathways that are activated by interleukin-7 (IL-7) (3). Pre-B-ALLs, like all cancers, develop by an evolutionary process (4). Mutations that confer a survival or proliferation benefit to pro-B/pre-B cells, known as driver mutations, lead to the accumulation of secondary driver mutations and passenger mutations during leukemia evolution (5, 6). Whole-genome sequencing, whole-exome sequencing (WES), and targeted cancer gene sequencing have eliminated the bottleneck in identifying mutations that function as drivers of leukemogenesis (7). However, there is still a gap in utilizing biochemistry and mouse models to determine the molecular mechanisms of how identified mutations function as drivers of leukemogenesis. Animal models are critically important for understanding leukemia evolutionary biology (8).
Pre-BCR signals synergize with interleukin-7 receptor (IL-7R) signaling to drive proliferation of large pre-B cells (9). Expansion of pre-B cell numbers is accompanied by extensive changes to B cell metabolism, including high levels of reactive oxygen species (ROS) that play important roles as second messengers (10). Assembly of Ig light chains with Ig heavy chains forms a functional BCR that signals through Bruton tyrosine kinase (BTK) and B cell linker protein (BLNK) to inhibit IL-7R signaling and proliferation. All stages of B cell development are controlled by cell type-specific transcription factors, including early B cell factor (EBF), E2A, Pax5, Ikaros, Aiolos, PU.1, and Spi-B (11).
PU.1 (encoded by Spi1) and Spi-B (encoded by Spib) are transcription factors belonging to the E26 transformation-specific (ETS) family of proteins (12). PU.1 and Spi-B have similar DNA binding domains that interact with an overlapping set of DNA binding sites within the genome (the sequence AAGTGGAAGT [underlining indicates the core ETS binding site that is required for binding; nonunderlined sequence indicates nucleotides preferred but not required for binding]) (13). PU.1 is expressed in blood cells, beginning in hematopoietic stem cells, and has pioneer activity that can open sites of closed chromatin, allowing access to other factors (14). Spi-B is expressed specifically in the lymphocyte and plasmacytoid dendritic cell lineages (12). PU.1 and Spi-B are complementary transcriptional activators of genes involved in B cell signaling, including those for Bruton tyrosine kinase (Btk) (15) and B cell linker protein (Blnk) (16).
We previously described a mouse model in which a Spi1lox/lox allele (encoding PU.1) is deleted during B cell development under the control of the B cell-specific Mb1Cre gene. These mice are also germ line null for Spib (Spib−/−) (17). These mice, called Mb1-CreΔPB, to indicate that PU.1 and Spi-B are deleted in the B cell lineage, have a profound block to B cell development starting at the small pre-B cell stage, demonstrating a requirement for PU.1 and Spi-B in B cell development (17). Importantly, 100% of Mb1-CreΔPB mice develop B-ALL by 18 weeks of age (18). Previous work suggests that secondary driver mutations cooperate with PU.1/Spi-B deletion to induce leukemia (18). However, it is unclear whether secondary driver mutations are recurrent and, if so, what the mechanism(s) of mutagenesis is.
In this study, we investigated secondary drivers of leukemogenesis in the Mb1-CreΔPB mouse model using WES. We found that 5/8 leukemias had mutations in Jak3 (encoding Janus kinase 3), 2/8 had mutations in Jak1, and 3/8 had mutations in Ikzf3 (encoding the transcription factor Aiolos). Mutations with the highest variant allele frequency (VAF) were dominated by C→T transition mutations that were compatible with activation-induced cytidine deaminase (AID), whereas the majority of mutations, with the lowest VAF, were dominated by C→A transversions associated with ROS. Leukemia cells were dependent on high levels of ROS, driven by IL-7-dependent JAK-STAT signaling and altered antioxidant gene expression, which resulted in 8-oxoguanine (8-OxoG) DNA damage. The JAK inhibitor ruxolitinib inhibited leukemia cell growth, ROS production, and STAT5 phosphorylation in cultured leukemia cells. Rodent chow containing ruxolitinib increased survival and reduced tumor size in Mb1-CreΔPB mice. Gene expression analysis of leukemias from ruxolitinib-treated mice showed reduced ROS-induced gene expression, while WES analysis showed altered mutational signatures. These results reveal that JAK mutations can alter the course of leukemia clonal evolution through ROS-induced DNA damage.
RESULTS
WES of Mb1-CreΔPB leukemias reveals recurrent mutations in Jak1, Jak3, and Ikzf3.
Mb1+/Cre Spi1lox/lox Spib−/− mice (shortened to Mb1-CreΔPB mice) develop precursor B cell acute lymphoblastic leukemia (B-ALL) at 100% incidence with a median time to euthanasia of 18 weeks (18). Mb1-CreΔPB leukemias invariably express interleukin-7 receptor on their surface, have a pro-B/pre-B phenotype, and are either clonal or oligoclonal with respect to immunoglobulin (Ig) heavy chain and Igκ rearrangements (18). In this study, we sought to determine if there are recurrent secondary driver mutations associated with Mb1-CreΔPB leukemias. We performed whole-exome sequencing (WES) on genomic DNA prepared from the thymi of 8 leukemic mice, as well as control genomic DNA prepared from tail clips of the same mice (see Tables S1 and S2 in the supplemental material). All thymi from leukemic mice contained high frequencies of cells expressing the B cell-specific marker CD19. Single nucleotide variants (SNVs) for each of the 8 leukemias were determined by comparing leukemia and control sequences using three independent somatic-variant callers: Strelka, Varscan2, and FreeBayes (Data Set S1). Variable numbers of somatic variants were called using these three algorithms after applying a cutoff of >0.1 variant allele frequency (VAF), with Strelka showing the lowest sensitivity and FreeBayes showing the highest sensitivity (Table S2). SNVs were filtered further based on the prediction of high-impact functional missense, stop, start, and splice (MSSS) variants and mapping to genes. The average numbers of genes containing one or more variants were 75 using Strelka, 281 using VarScan, and 889 using FreeBayes. The average number of genes containing one or more variants, called by all three callers, was 20 per leukemia (Table S2).
The intersection of the genes impacted by the identified variants showed that 5/8 exomes had high-impact SNVs in Jak3, encoding Janus kinase 3, 2/8 exomes had high-impact SNVs in Jak1, encoding Janus kinase 1, and 3/8 exomes had high-impact SNVs in Ikzf3, encoding Aiolos, a zinc finger transcription factor highly related to Ikaros (IKZF1) (Fig. 1). No other genes had high-impact variants in more than one exome. All of these SNVs were present at high cancer cell frequency (CCF). A summary of the base changes, amino acid changes, and predicted impacts of identified variants in Jak3, Jak1, and Ikzf3 is shown in Table S3. All Jak3 and Jak1 mutants, except Jak3 T844M (encoding a change of T to M at position 844 of Janus kinase 3), encoded mutations located in the pseudokinase domain of these proteins, and their mutations were therefore predicted to function as activating mutations (19). Jak3 V670A, R653H, and T844M were previously shown to be activating mutations for IL-7-dependent signaling (18). In contrast, Ikzf3 R137* and H195Y encoded mutations located in zinc fingers 1 and 3, respectively (Table S3), and were therefore predicted to represent loss-of-function or dominant-negative mutations (20). In conclusion, recurrent mutations in Jak3 and Jak1 are activating mutations that likely act as secondary drivers of leukemogenesis by induction of the JAK-STAT signaling pathway.
FIG 1.

Venn diagrams showing overlap in gene variants between exome sequences. Numbers outside colored shapes indicate sequenced leukemia exomes. Numbers inside colored shapes indicate numbers of variants called by three variant callers.
Mutational signature analysis reveals distinct patterns of DNA damage.
Analysis of whole-exome and whole-genome sequences from thousands of human cancers revealed at least 30 distinct mutational signatures (21). To determine mutational signatures in our 8 WES sequences of Mb1-CreΔPB mouse leukemia, we used the deconstructSigs R package that identifies mutational signatures in exome sequences based on comparison to human COSMIC (Catalogue Of Somatic Mutations In Cancer) version 2 mutational signatures (22). DeconstructSigs analysis of output from Strelka (Fig. S1) or VarScan2 (not shown) showed that the two most common mutational signatures identified were signatures 18 and 24 (Fig. 2A). In contrast, analysis of output from FreeBayes showed mutational signatures 3 and 4 only (not shown). Mutational signatures 3 and 4 are flat signatures with no particular enrichment in mutation type (21). Therefore, we speculate that this result is due to the high number of SNVs called by FreeBayes relative to the numbers called by Strelka and Varscan2 (Table S2). Mutational signatures 18 and 24, discovered from both Strelka and Varscan2 data, are characterized by a high frequency of C→A transversions (Fig. S1) (7, 23). C→A transversions are thought to be caused by high levels of reactive oxygen species (ROS) causing oxidation of guanine, resulting in 8-oxoguanine (8-OxoG) mispairing with adenine following one round of replication (24). C→T transition mutations are thought to be induced primarily by APOBEC family enzymes, including activation-induced cytidine deaminase (AID) (25). To gain insight into the mechanism(s) of mutagenesis in the Mb1-CreΔPB mouse model, SNVs called by Strelka were placed into bins based on VAF to determine the frequency of C→A transversion relative to C→T transition mutations. This analysis showed that SNVs with VAFs of >0.3 had higher frequencies of C→T transitions relative to their frequencies of C→A transversions (Fig. 2B). This result suggests different mutational processes for high-VAF mutations and low-VAF mutations.
FIG 2.

Evidence for distinct mutational processes. (A) Frequencies of mutational signatures. The pie chart shows frequencies of top mutational signatures for each of 8 leukemias analyzed by the indicated mutation caller. (B) Frequencies (percentages) of C→A transversions compared to C→T transitions in each of 6 VAF bins. (C) The ratios of C→A transversions to C→T transitions are lower at high VAF (0.334 to 0.5) than at low VAF (0.167 to 0.334) (n = 8; unpaired t test; *, P < 0.01). (D) Variants are enriched for WRC (A/T, A/G, C) motifs at high VAF frequency compared to their enrichment at low VAF frequency (repeated-measures one-way analysis of variance [ANOVA]; *, P < 0.05; **, P < 0.01; ****, P < 0.0001).
Libraries from leukemias 853, 854, and 857 were prepared separately from those from leukemias 406, 968, 973, 856, and 932 and exhibited higher ratios of C→A transversions to C→T transitions (Fig. S1). It has previously been noted that preparation of genomic DNA libraries by high-energy sonication can result in the presence of an 8-oxoguanine library preparation artifact, albeit at a VAF of <0.1 (23). To exclude potential artifacts, we reanalyzed our data by excluding SNVs at a VAF of <0.167. We also used the Broad Institute d-ToxoG tool to remove predicted 8-OxoG artifacts. SNVs were then placed into two bins, those at 0.167 to 0.334 VAF and those at 0.334 to 0.5 VAF, and the ratios of C→A transversions to C→T transitions were determined for the 8 leukemia exomes. Analysis showed that the ratios of C→A transversions to C→T transitions were higher at VAFs of <0.334 than at VAFs of >0.334 (Fig. 2C). We noted that, of the mutations in Ikzf3, Jak1, and Jak3 described in Table S3, most of which are at high VAF and CCF, 7/10 were C→T transitions, 2 were T→C or T→G, and 1 was a C→A transversion. Taken together, these results suggest the presence of two distinct mutational processes in Mb1-CreΔPB mice, one occurring early in leukemia development and leading to C→T transitions at high VAF and the second occurring later in leukemia development and leading to C→A transversions at low VAF.
AID is an APOBEC family member expressed exclusively in the B cell lineage that is implicated in mutagenesis in B cell leukemia and lymphoma (25). AID induces C→T transition mutations with preference for WRC (A/T, A/G, C) sequences (26). To determine whether the high-VAF (>0.334) mutations called by Strelka were compatible with mutagenesis induced by AID, the sequence context was examined. Analysis showed that WRC trinucleotide motifs were significantly enriched for high-VAF variants relative to their enrichment for low-VAF variants (Fig. 2C). To determine if the 7 C→T mutations described in Table S3 were compatible with an AID-induced signature, the sequence context was examined. Three of 7 C→T mutations were in a CAC trinucleotide sequence, while 3/7 were in an AGC trinucleotide sequence and 1/7 was in a TAC trinucleotide sequence. Therefore, the sequence context of high-VAF variants, including Jak3, Jak1, and Ikzf3 C→T transition mutations, is compatible with the preferred recognition sequence for AID.
High levels of ROS in leukemias from PU.1, Spi-B-deficient mice.
The high frequency of C→A transversion mutations in Mb1-CreΔPB leukemias suggested 8-oxoguanine DNA damage due to reactive oxygen species (ROS) (27). To determine if the gene expression profile of leukemia cells showed evidence of ROS, 100-bp paired-end RNA-seq was performed on the eight Mb1-CreΔPB leukemia samples summarized in Table S1. Paired-end reads were analyzed using Cufflinks, and normalized gene expression was determined as fragments per kilobase of transcript per million mapped reads (FPKM). The levels of gene expression from leukemia samples were compared to the gene expression from nonleukemic PU.1/Spi-B-deficient pro-B cells (17) using gene set enrichment analysis (GSEA) (28). GSEA analysis revealed positive enrichment in leukemia cells for gene ontology (GO) gene sets “GO response to ROS” (191 genes) (Fig. 3A) and “GO response to oxidative stress” (352 genes) (Fig. 3B). GSEA revealed negative enrichment for “GO positive regulation of ROS” (86 genes) (Fig. 3C). These results suggest an impact of ROS on gene expression in Mb1-CreΔPB leukemias.
FIG 3.

Gene expression analysis of leukemias reveals altered responses to reactive oxygen species. (A to C) Gene set enrichment analysis revealed positive association with “GO response to ROS” (191 genes) (A) and “GO response to oxidative stress” (352 genes) (B) and negative association with “GO positive regulation of ROS” (86 genes) (C). FDR, false discovery rate. (D) Heat map of antioxidant gene expression in PU.1-induced i660BM cells (Spi1-induced 1 to 3), uninduced i660BM cells (Control 1 to 3), and individual leukemia samples. Gene names and functions are indicated on the right. Red indicates increased expression; blue indicates decreased expression. (E) ROS levels detected by H2DCFDA staining and flow cytometry in cultured wild-type pro-B cells (WT3 Pro-B) and leukemia 973 cells (973). Numbers indicate mean fluorescence intensities (MFI).
Published literature was examined to prioritize genes that have a role in producing or clearing/scavenging ROS. No prooxidant genes were eligible due to undetectable expression. However, 29 antioxidant genes were expressed and showed substantial differences, with most showing downregulation in leukemia cells (Fig. 3D). Genes for catalase (Cat), glutathione peroxidase (Gpx1), and peroxiredoxin (Prdx4 and Prdx5) enzymes, important for the control of intracellular hydrogen peroxide (29), were among the most highly downregulated genes in Mb1-CreΔPB leukemia cells compared to their expression in preleukemic cells (Fig. 3D). Overall, these results suggest that antioxidant responses are reduced in Mb1-CreΔPB leukemias relative to their levels in control cells.
In order to determine whether ROS was increased in leukemia cells relative to the amount in control cells, cell lines were generated from leukemias by culturing in IL-7-conditioned medium. Cell line 973 was chosen as the representative cell line since it grew robustly in culture and had a Jak1 mutation, identified using whole-exome sequencing (Fig. 1; Table S3). Cell line 973 leukemia cells and wild-type (WT) fetal-liver-derived pro-B cells were incubated with 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA), a cell-permeant dye that becomes brightly fluorescent upon oxidization. 973 cells had high levels of H2DCFDA staining relative to the staining in WT pro-B cells that expressed PU.1 and Spi-B (Fig. 3E). To determine if PU.1 plays a role in control of ROS in preleukemic cells, bone marrow cells from young (6- to 8-week-old) ΔB (Spib−/−) or Mb1-CreΔPB mice were placed in culture with 5% IL-7-conditioned medium for 2 weeks to generate proliferating pro-B/pre-B cells and then stained with H2DCFDA. Flow cytometric analysis showed that Mb1-CreΔPB pro-B/pre-B cells had increased ROS compared to the amount in ΔB cells (Fig. S2). Taken together, these results suggest high levels of ROS in both preleukemic and leukemia cells from Mb1-CreΔPB mice.
Detection of 8-oxoguanine DNA damage in Mb1-CreΔPB leukemia cells.
To determine if there was 8-oxoguanine DNA damage in Mb1-CreΔPB leukemias, thymic leukemias were freshly isolated, placed in short-term culture (5 to 7 days) to select for nonapoptotic cells, and then cytospun onto glass slides, followed by paraformaldehyde fixation and staining with anti-8-oxoguanine antibody and 4′,6-diamidino-2-phenylindole (DAPI) (Fig. 4A). Wild-type fetal-liver-derived pro-B cells were used as controls. 8-Oxoguanine-stained leukemia cell nuclei had a median fluorescence intensity (MFI) that was 27.9-fold greater than the MFI of cells stained with secondary antibody only (Fig. 4B). The median fluorescence of wild-type cells was 9.3-fold greater than that of cells stained with secondary antibody only (Fig. 4B). 8-Oxoguanine-stained leukemia cells had 3.4-fold higher levels of fluorescence than 8-OxoG stained wild-type cells (Fig. 4B). We conclude that Mb1-CreΔPB leukemia cells had increased levels of 8-oxoguanine DNA damage compared to the levels in wild-type pro-B cells. These results suggest that Mb1-CreΔPB leukemia cells have increased levels of ongoing DNA damage as a consequence of high ROS levels.
FIG 4.
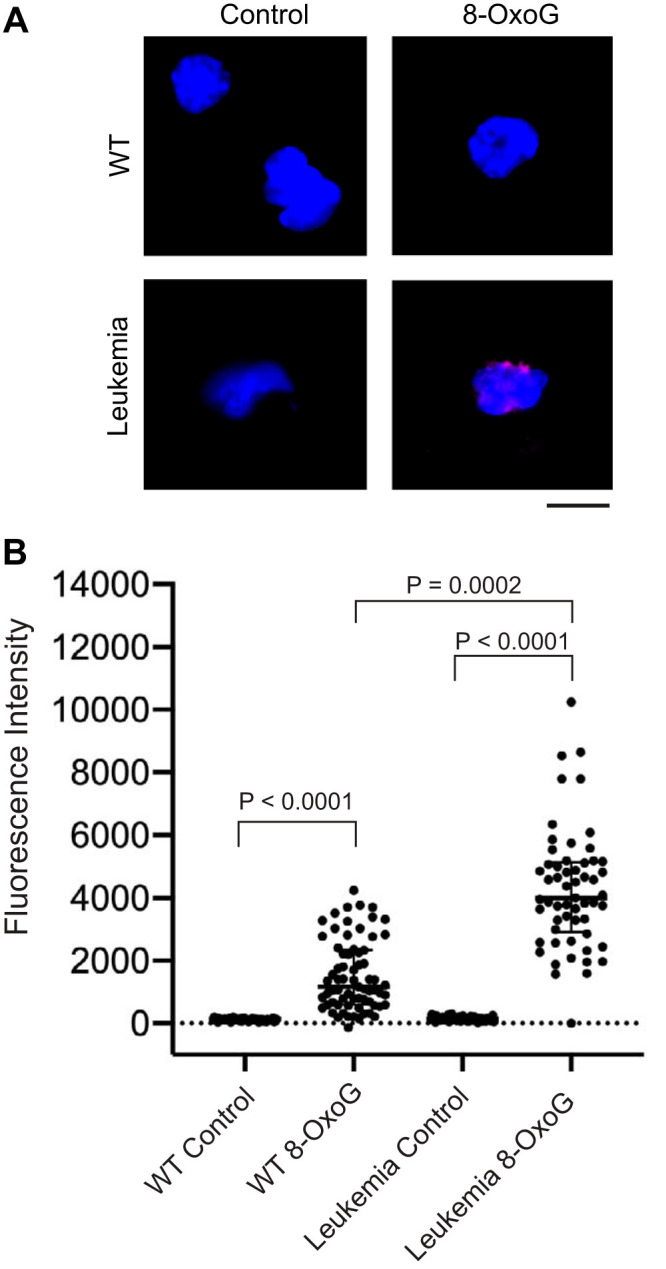
8-Oxoguanine staining in wild-type fetal-liver-derived pro-B cells compared to short-term-cultured leukemia cells. (A) Images of representative nuclei. Shown are overlaid images of DAPI-stained intact nuclei (blue) and anti-8-OxoG antibody (red) from pro-B cells (WT) and leukemia cells (Leukemia). Staining was performed as described in Materials and Methods. The scale bar indicates 10 μm. (B) Quantification of 8-OxoG staining in intact nuclei. Dots indicate nuclei of individual cells visualized from wild-type pro-B cells (WT; control = 58 nuclei, 8-OxoG = 69 nuclei) or short-term-cultured Mb1-CreΔPB leukemia cells (Leukemia; control = 56 nuclei, 8-OxoG = 57 nuclei). Cells were stained with secondary antibody only (Control) or with anti-8-OxoG antibody and secondary antibody (8-OxoG). Statistics were determined by Kruskal-Wallis with post hoc Dunn’s test.
Requirement of high ROS for proliferation and gene expression of Mb1-CreΔPB leukemia cells.
ROS has been shown to promote proliferation and survival of leukemia cells (27, 29). A number of cellular signaling pathways, including the JAK-STAT pathway downstream from IL-7R, generate ROS and are also activated by ROS in a positive feedback loop (10, 30). To determine if Mb1-CreΔPB leukemia cells require ROS for survival, cell line 973 cells were cultured with the antioxidant N-acetylcysteine (NAC) (30) and proliferation was determined using MTT [3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide] assays and confirmed by cell counting, since the MTT assay can be sensitive to ROS. Concentrations of NAC above 1 μM inhibited proliferation of 973 cells as determined by MTT assay (Fig. 5A). To determine if interleukin-7 signaling plays a role in ROS generation, we performed cell counting assays to identify a concentration of IL-7 at which cells survived but did not proliferate (Fig. 5B). Consequently, 0.05% conditioned medium was selected as a low IL-7 concentration and 5% conditioned medium was selected as a high IL-7 concentration (Fig. 5B). To increase ROS, 973 cells were cultured with hydrogen peroxide (H2O2), a form of ROS that serves as a second messenger for cellular signaling pathways (10). Under culture conditions using 5% IL-7-conditioned medium, the addition of H2O2 reduced cell counts (Fig. 5C). Interestingly, under culture conditions with 0.05% IL-7, the addition of H2O2 at concentrations up to 50 μM stimulated proliferation of 973 cells (Fig. 5D). Taken together, these results suggest that 973 cells require a specific level of ROS, above or below which proliferation is reduced.
FIG 5.

Effects of antioxidant N-acetylcysteine and prooxidant hydrogen peroxide on cell proliferation and gene expression of cultured leukemia cells. (A) N-Acetylcysteine (NAC) treatment reduces cell counts of cultured 973 cells in a dose-dependent manner. MTT assays were performed, and the absorbance was determined. Zero indicates vehicle control. OD, optical density. (B) Dose-response of cultured pro-B cells to interleukin-7. % IL CM, percentage of IL-7 in conditioned medium. (C) Hydrogen peroxide (H2O2) reduces cell counts of cultured 973 cells in a dose-dependent manner. Experiment was performed using 5% IL-7-conditioned medium. (D) Under low-IL-7 conditions, H2O2 increases cell counts of cultured 973 cells in a dose-dependent manner. The experiment was performed using 0.5% IL-7-conditioned medium. P value was determined by ordinary one-way ANOVA of biological replicate (n = 3) experiments. (E, F) Gene expression analysis. RT-qPCR was performed to determine relative fold changes in the indicated mRNA transcripts after 24 h of culture with NAC (D) or H2O2 (E). Statistical analysis was performed using one-sample t and Wilcoxon tests (n = 5 biological replicate experiments). Error bars indicate standard errors of the means. *, P < 0.05; **, P < 0.01; ***; P < 0.001, ****, P < 0.0001.
Next, we determined the effects of half-maximal doses of NAC (2.5 μM) and H2O2 (50 μM) (determined from the results shown in Fig. 5A to D) on gene expression in 973 cells. Neil1 encodes a DNA glycosylase that is important for repair of guanine oxidation products, whose transcription is activated by DNA damage and ROS (31). We found that Neil1 was downregulated by NAC and upregulated by H2O2, indicating that this gene is activated by ROS (Fig. 5E and F). Rad51 encodes a key protein involved in DNA repair and is transcriptionally upregulated in response to DNA damage (24). Rad51 was downregulated by NAC and upregulated by H2O2, suggesting that this gene responds to ROS-induced DNA damage (Fig. 5E and F). Sod2 encodes superoxide dismutase 2, a mitochondrion-located enzyme whose function is to catalyze the dismutation of superoxide (O2−) into molecular oxygen and H2O2 (29). Sod2 was not significantly affected by NAC treatment but was strongly downregulated by H2O2 (Fig. 5E and F). Gpx1 encodes glutathione peroxidase 1, the function of which is to catalyze a reaction between glutathione and ROS to oxidize glutathione disulfide, reducing oxidative stress within the cell (29). Gpx1 was downregulated in response to NAC and upregulated in response to H2O2 (Fig. 5E and F). Finally, we examined changes in the expression of Cat, encoding catalase, which catalyzes dismutation of H2O2 to water and oxygen (29). Cat expression was downregulated by NAC and upregulated 100-fold in response to H2O2 (Fig. 5E and F). In summary, in response to upregulation of ROS by H2O2 treatment in cultured leukemia cells, Neil1, Rad51, Gpx1, and Cat were upregulated, while these genes were downregulated in response to treatment with NAC. Downregulation of antioxidant gene expression by NAC suggests that this compound functions directly as an antioxidant, resulting in reduced requirement for antioxidant gene expression. In contrast, H2O2 was capable of stimulating antioxidant gene expression in 973 cells. These results show that gene expression is dynamically regulated by ROS in cultured leukemia cells.
Regulation of proliferation and gene expression of Mb1-CreΔPB leukemia cells by Necrox-5 and ML334.
Necrox-5 is a small molecule that specifically targets the mitochondria and was reported to inhibit calcium flux and have antioxidant activity (32, 33). Necrox-5 significantly inhibited the proliferation of 973 cells at concentrations above 2 μM as determined by MTT assay (Fig. S3A). Unexpectedly, at 3 μM (half maximal), Necrox-5 induced increases in antioxidant gene expression that were similar to those induced by H2O2 (Fig. S3B). Necrox-5 also resulted in reduced levels of ROS as determined by H2DCFDA staining (Fig. S3C). These results suggest that Necrox-5 interferes with mitochondrial signaling to the antioxidant gene response.
High levels of ROS induce dissociation of Keap1 from Nrf2, followed by translocation of Nrf2 to the nucleus to activate antioxidant gene expression (10). The experimental compound ML334 activates antioxidant responses by inducing Keap1-Nrf2 dissociation (34). ML334 reduced cell counts of cultured 973 cells in a dose-dependent manner (Fig. S3D). At 50 μM, ML334 resulted in upregulation of antioxidant gene expression, including Sod2 (Fig. S3E). Taken together with the results presented above, these experiments demonstrate that Mb1-CreΔPB leukemia cells require ROS for proliferation and/or survival and that reduction of ROS by treatment with NAC, Necrox-5, or ML334 results in altered antioxidant gene expression.
Ruxolitinib inhibits proliferation and alters mutational signatures of Mb1-CreΔPB leukemia cells.
JAK-STAT signaling increases cellular ROS, and increased levels of cellular ROS provide positive feedback to augment JAK-STAT signaling (27, 30). Ruxolitinib (Jakafi, INC424) is a JAK inhibitor that is effective against human and murine JAK1 to -3 (JAK1–3) (3, 35). Since leukemias in Mb1-CreΔPB mice are IL-7 dependent (18, 36) and IL-7 signals primarily through JAK1 and -3 (19), we set out to determine whether proliferation of Mb1-CreΔPB leukemia cells could be inhibited by ruxolitinib. Proliferation of 973 cells in culture was efficiently inhibited by ruxolitinib as determined by cell counting (Fig. 6A) or MTT assay (Fig. 6B), with half-maximal activity at 75 nM. Culture of 973 cells with 75 nM ruxolitinib resulted in reduced STAT5 phosphorylation (Fig. 6C), as well as reduced ROS as determined by H2DCFDA staining (Fig. 6D). Furthermore, treatment of 973 cells with 75 nM ruxolitinib resulted in upregulated antioxidant gene expression (Fig. 6E). To determine if reduced ROS might be responsible for reduced proliferation, we asked whether the addition of H2O2 could rescue proliferation of ruxolitinib-treated 973 cells. The addition of 50 μM H2O2 partly rescued cell counts of 973 cells treated with ruxolitinib (Fig. 6F). These results indicate that inhibition of JAK-STAT signaling downstream from IL-7 signaling by ruxolitinib resulted in reduced ROS.
FIG 6.

Effects of ruxolitinib on cell proliferation, gene expression, and reactive oxygen species. (A, B) Ruxolitinib treatment reduces cell counts of cultured 973 cells in a dose-dependent manner. Cell-counting assays (A) or MTT assays (B) were performed. A concentration of 0 indicates the vehicle control. (B) Absorbance. The P value was determined by one-way ANOVA of biological replicate (n = 3) experiments. (C) Reduced STAT5 phosphorylation in ruxolitinib-treated 973 cells. Anti-phosphorylated-STAT5 antibody (P-Stat5) or total STAT5 immunoblotting was performed on 973 cells treated with 75 nM ruxolitinib for 24 h. (D) Reduced ROS in 973 cells treated for 24 h with ruxolitinib, as determined by H2DCFDA staining. Numbers indicate mean fluorescence intensities. (E) Gene expression analysis. RT-qPCR was performed to determine relative fold changes in the indicated mRNA transcripts after 24 h of culture with ruxolitinib. Statistical analysis was performed by one-sample t and Wilcoxon tests (n = 5 biological replicate experiments). Error bars indicate standard errors of the means. *, P < 0.05; **, P < 0.01; ns, not significant. (F) H2O2 partially rescues suppressed 973 cell counts induced by ruxolitinib. 973 cells were cultured for 3 days with 75 nM ruxolitinib with or without 50 μM H2O2.
Based on the recurrence of Jak1/-3 mutations and the ROS gene signature in Mb1-CreΔPB leukemias, we hypothesized that ruxolitinib would delay leukemia incidence in Mb1-CreΔPB mice. To test this idea, Mb1-CreΔPB mice were fed ruxolitinib chow or control rodent chow between 4 and 8 weeks of age and then switched to regular chow and monitored until signs of illness appeared (Fig. 7A, top). Ruxolitinib-treated and control mice consumed on average 6.9 g and 7.1 g of chow per day, respectively. Four of 8 ruxolitinib-treated Mb1-CreΔPB mice developed B-ALL with delayed time to euthanasia, while 4/8 Mb1-CreΔPB mice did not develop signs of leukemia before the experiment was terminated at 58 days (Fig. 7A, bottom). Ruxolitinib-treated Mb1-CreΔPB mice had reduced thymus weight compared to that of control mice (Fig. 7B). Despite reduced cellularity, 7/8 mice had extensive CD19+ leukemia cell infiltration in the thymus at the time of euthanasia, while 1 mouse had a few infiltrating CD19+ leukemia cells in the thymus. These results showed that ruxolitinib treatment between 4 and 8 weeks of age significantly delayed time to euthanasia in Mb1-CreΔPB mice.
FIG 7.

Effects of ruxolitinib on Mb1-CreΔPB mouse survival, leukemia gene expression, and mutational signatures. (A) Bottom, survival of Mb1-CreΔPB mice after being fed control (n = 5) or ruxolitinib (n = 8) mouse chow for 4 weeks. Top, schematic of ruxolitinib mouse chow experiment. Statistical significance of the Kaplan-Meier curve was determined using the log rank (Mantel-Cox) test. (B) Reduced thymus weights at euthanasia in Mb1-CreΔPB mice fed with control or ruxolitinib (Ruxo) chow between 4 and 8 weeks of age. Horizontal bars and error bars represent mean values ± standard deviations. Statistical significance was determined using an unpaired t test (n = 5 control mice, n = 8 ruxolitinib-treated mice). (C) Heat map of gene expression in Mb1-CreΔPB mice fed with control or ruxolitinib chow between 4 and 8 weeks of age. The heat map shows FPKM of genes found to be differentially expressed by DESeq2 (adjusted P value, <0.05). (D) Gene set enrichment analysis of RNA-seq analysis using the antioxidant gene set shown in Fig. 3D. (E) Reduced frequencies of Jak1–3 variants in leukemias from mice fed with ruxolitinib chow between 4 and 8 weeks of age compared to the frequencies in leukemias from mice fed with control chow. Shown is a stacked bar graph showing the added frequencies of mice with Jak1–3 variants detected by three independent variant callers. (F, G) Heat maps of mutational signatures in leukemias from Mb1-CreΔPB mice fed with control or ruxolitinib chow between 4 and 8 weeks of age. Colors indicate weights of mutational signatures called by Strelka (F) or Varscan2 (G) for WES results of leukemias.
To determine if delayed leukemia development was associated with altered ROS and DNA damage, we performed transcriptome sequencing (RNA-seq) on leukemia cells from four control and five ruxolitinib-treated mice, as shown in Fig. 7A (summarized in Table S4). Normalized gene expression was determined as FPKM using the Cufflinks suite, and differential gene expression was determined using DESeq2 (37). More genes were downregulated than upregulated in ruxolitinib-treated leukemias, with highly pro-B-/pre-B-specific genes like VpreB1, Igll1, and Cd79b being among the most highly downregulated genes (Fig. 7C). Antioxidant and electron transport chain genes were also downregulated in ruxolitinib-treated leukemias (Fig. 7C). Furthermore, GSEA analysis using the antioxidant gene expression set shown in Fig. 3D showed highly significant downregulation of antioxidant gene expression (Fig. 7D). These results suggest that ruxolitinib reduced ROS in leukemia cells, resulting in downregulation of antioxidant gene expression.
Finally, WES was performed on leukemia cells from four control and seven ruxolitinib-treated mice, as shown in Fig. 7A and summarized in Table S4. SNVs were called using Strelka, Varscan2, and FreeBayes variant callers as described above. No variants were identified that were in common between all leukemias and all three callers. The frequencies of Jak1, Jak2, and Jak3 variants were reduced in leukemias from ruxolitinib-treated mice compared to their frequencies in control mice, as would be expected if ruxolitinib preferentially targets leukemic clones with Jak1–3 driver mutations (Fig. 7E). As previously noted, FreeBayes called variants with relaxed stringency compared to the reporting by Strelka and Varscan2 (Fig. 7E). Next, mutational signature analysis was performed on variants identified by Strelka (Fig. 7F) and Varscan2 (Fig. 7G). As noted above and shown in Fig. 2A, ROS-associated mutation signatures 18 and 24 were among the most heavily weighted mutational signatures in control leukemias (Fig. 7F and G). For leukemias with delayed onset from mice fed from 4 to 8 weeks with ruxolitinib chow, there was increased variability in signatures 18 and 24, accompanied by increases in frequencies of calling and weights of mutational signatures 10, 15, 20, and 29 for Strelka (Fig. 7F; Fig. S4A) and of mutation signatures 3, 5, 14, 20, and 29 for Varscan2 (Fig. 7G). For variants with a VAF of >0.3, which were expected to have arisen earlier during clonal evolution, there was an even more pronounced spreading of mutational signatures than for variants with a VAF of <0.3 (Fig. S4B and C). In summary, these data indicate that feeding with ruxolitinib chow for 4 to 8 weeks resulted in delayed onset of leukemia, and the leukemias that developed had not only an altered gene expression signature but also an altered mutational signature. These results suggest that Jak mutations affect the clonal evolution of leukemia by leading to additional DNA damage through mechanisms that may include ROS.
DISCUSSION
In this study, we investigated secondary drivers of leukemogenesis in the Mb1-CreΔPB mouse model using whole-exome sequencing (WES). WES analysis revealed that 5/8 leukemias had mutations in Jak3 (encoding Janus kinase 3), 2/8 had mutations in Jak1, and 3/8 had mutations in Ikzf3 (encoding the transcription factor Aiolos). Mutations with the highest VAFs were dominated by C→T transition mutations, whereas the majority of mutations with the lowest VAFs were dominated by C→A transversions. Leukemia cells were dependent on high levels of ROS driven by IL-7-dependent JAK-STAT signaling. Leukemia cells had altered antioxidant gene expression and 8-OxoG DNA damage. The JAK inhibitor ruxolitinib inhibited leukemia cell growth, ROS production, and STAT5 phosphorylation in cultured leukemia cells. Rodent chow containing ruxolitinib increased the survival time of Mb1-CreΔPB mice, resulting in a gene expression signature indicating lower levels of ROS and a mutational signature indicating altered ROS-induced DNA damage. Taken together, these data suggest that Jak1/-3 mutations lead to altered IL-7-dependent proliferation driven by ROS, cooperating with JAK/STAT signaling, and followed by 8-OxoG-induced accumulation of additional mutations to drive disease evolution.
Mutations in JAK3 and JAK1 are recurrent in human leukemia and frequent in Ph-like human leukemia (3, 38). The Jak3 and Jak1 mutations identified in our mouse model correspond to mutations observed in human leukemia. The human equivalent of Jak3 R653H (JAK3 R657Q) and V670A (JAK3 V674F) was identified as an activating mutation in leukemia (39). The human equivalent of Jak3 A568V (JAK3 A572V) was identified as an activating mutation in T cell leukemia (40). Jak1 V657F corresponds to human JAK1 V658F, which has been shown to be a frequent activating human driver mutation in ALL (41). The human equivalent of Jak1 F837V (JAK1 F838V) was identified in one case of T cell lymphoma (42). Mutations in IKZF3 (AIOLOS) are recurrent in human hypodiploid ALL and frequently occur in zinc finger domains (43). The Ikzf3 H195Y mutation is predicted to disrupt zinc finger 3, leading to a dominant-negative protein similar to that resulting from the human IKZF3 H163Y mutation (44). Therefore, the Mb1-CreΔPB mouse represents a powerful model for leukemogenesis in which each mouse independently evolves its own leukemia through the development of secondary driver mutations corresponding to mutations observed in human leukemia.
Previous studies have shown that many types of human cancer, including human leukemia, have levels of ROS that are higher than in normal cells (45). These high levels of ROS are generated either from the electron transport chain or from prooxidant enzymes, including the NOX family (46). High ROS levels increase cellular signaling pathways, including JAK signaling, by diverse mechanisms, including inactivation of protein tyrosine kinases and PTEN, leading to further increases in cellular metabolism and ROS (10, 29). However, the danger of sustained high levels of ROS is cellular damage, including 8-OxoG DNA damage (45). JAK-STAT signaling has previously been implicated directly in the generation of high levels of ROS, associated with DNA damage and cancer (47).
Reduced levels of antioxidant genes have been observed previously in precursor B cell acute lymphoblastic leukemia (27). JAK3 activation of STAT5 has been shown to repress antioxidant gene expression, leading to increased ROS (48). Therefore, we speculate that the high levels of ROS observed in Mb1-CreΔPB leukemia cells are caused in part by deregulation of JAK signaling but also are due in part to STAT5-mediated repression of antioxidant gene expression (Fig. 3). In accord with this, ruxolitinib treatment increased antioxidant gene expression and reduced ROS (Fig. 7C and D). Treatment with the antioxidant NAC led to downregulation of antioxidant gene expression (Fig. 5D). Conversely, treatment with Necrox-5 or ML334 resulted in activation of antioxidant gene expression and reduced ROS (Fig. S3). Our observation that Necrox-5, a mitochondrion-targeted ROS scavenger and Ca2+ flux inhibitor, reduced ROS and upregulated antioxidant activity suggests that the mitochondrial electron transport chain is the major source of ROS in Mb1-CreΔPB leukemias. Cytoplasmic prooxidant enzymes were not significantly expressed in our cells, as described in Results and shown by the results in Fig. 3D. These experiments suggest that high levels of ROS and, consequently, 8-OxoG DNA damage are expected to be a hallmark of dysregulated JAK-STAT signaling in leukemia. In accord with this idea, mutational signature analysis of human pediatric cancers revealed that a substantial frequency of precursor B-ALLs have high frequencies of C→A transversions thought to be caused by 8-oxoguanine DNA damage (7).
Precursor B-ALL is thought to originate from large pre-B cells, and the phenotype and metabolic state of large pre-B cells is similar to that of B-ALL cells (6). Large pre-B cells have high levels of metabolic activity, as well as the highest levels of ROS of any B cell stage measured (49). It has recently been shown that transcription factors PAX5 and IKZF1 have gatekeeper functions for repression of cellular metabolism (50). Interestingly, PAX5 and IKZF1 are among the most frequently mutated genes in childhood pre-B-ALL (50, 51). Mutation of PAX5 and IKZF1 results in activation of cellular metabolism, which can cooperate with additional driver mutations to induce malignant transformation (50). We speculate that PU.1 and Spi-B also function as gatekeepers for cellular metabolism (see Fig. S2 in the supplemental material) and that mutations in Spi1/Spib cooperate strongly with Jak1/-3 mutations to activate metabolism, leading to ROS, further activation of JAK-STAT signaling, and DNA damage. Activated STAT5 downstream from JAK3 signaling would be expected to enforce signaling both by activation of cellular metabolism and by direct repression of antioxidant genes (48).
SPI1 and SPIB mutations have been identified in human leukemia (52, 53). SPI1 and SPIB are transcriptionally repressed by ETV6-RUNX1 and ETO-RUNX1 translocation products in human B-ALL and acute myeloid leukemia (AML) (54, 55). Importantly, ETV6-RUNX1 (t12;21) leukemias represent nearly 25% of childhood leukemias (2). It was recently shown that B cells from mice lacking the transcription factors PU.1 and interferon (IFN) regulatory factor 8 (IRF8) have increased mRNA transcript levels of Aicda, encoding AID, suggesting that AID expression is constrained by PU.1 and IRF8 (56). In another study, it was shown that B cells from mice lacking both PU.1 and Spi-B had robust upregulation of Aicda (57). These studies suggest that AID expression is constrained by PU.1 and Spi-B interacting with IRF4/-8, although the mechanism of this has not been investigated. Deregulated expression of Aicda represents a potential mechanism for induction of secondary driver mutations in the Mb1-CreΔPB mouse model.
The successful implementation of clinically approved JAK inhibitors (ruxolitinib, tofacitinib, baracitinib, and itacitinib) demonstrates that inhibition of JAK-STAT signaling is beneficial in a wide variety of cancers and chronic inflammatory and autoimmune diseases (58). It will be useful to explore other JAK inhibitors in our system, particularly tofacitinib, since it is a dual JAK1/-3 inhibitor, whereas JAK2 might be largely irrelevant in the Mb1-CreΔPB mouse model. Combination therapy using JAK inhibitors and ROS scavengers may also be effective, as demonstrated in other systems. It will also be necessary to test the efficacy of JAK inhibitors after disease onset, since ruxolitinib may not be as effective in this context (58). The development of covalent kinase inhibitors is a dynamic area of research that is expected to continue producing new clinically relevant drugs (59).
Overall, this work suggests that activating JAK mutations, which are frequent in human leukemia, are expected to lead to increased ROS and 8-oxoguanine DNA damage, which will result in additional secondary driver mutations that will affect the clonal evolution of the disease. JAK inhibitors may be effective at targeting leukemia by breaking the positive feedback cycle of JAK-STAT signaling, ROS production, and 8-OxoG DNA damage that leads to clonal evolution of the disease.
MATERIALS AND METHODS
Animal care.
C57BL/6 mice were purchased from Charles River Laboratories (Saint-Constant, QC, Canada). Mb1+/Cre Spi1lox/lox Spib−/− mice (shortened to Mb1-CreΔPB mice) were generated by mating Mb1+/+ Spi1lox/lox Spib−/− female mice to Mb1+/Cre Spi1lox/lox Spib−/− male mice and genotyped as described previously (18). Four-week-old preleukemic littermate Spib/Spi1-deficient mice were fed 10 g of ruxolitinib chow (2,000 mg/kg of body weight; provided by Novartis) or control chow for 30 days, after which all mice were switched to regular chow. Upon signs of illness indicated by lethargy, dyspnea, and piloerection, mice were euthanized in fulfilment of ethical standards provided by the Western University Council on Animal Care.
WES.
DNAs from leukemias and matched tails were extracted using the Wizard genomic DNA purification kit (Promega, Madison, WI). WES was performed by the Genome Quebec Innovation Centre using the SureSelectXT mouse all exon kit (Agilent Technologies, Mississauga, Canada) for exome target capture, the SureSelectXT target enrichment system to produce the paired-end DNA libraries, and the Illumina HiSeq4000 (Illumina, San Diego, CA) for exome sequencing. The BAM files generated had adaptor sequences trimmed using TrimGalore! and aligned to the reference mm10 genome using Bowtie2. Processed files were analyzed using mutation callers Strelka (60), VarScan2 (61), and FreeBayes (62) with the standard settings. Annotation was performed on these files using SnpEffect (63). Mutational signatures were identified using deconstructSigs (22). GetFlanks was used to obtain the 15 bases preceding the C→T mutation. Cancer cell frequency (CCF) was calculated according to the formula CCF = VAF × (1/purity)[CN × purity + 2(1 − purity)], where CN stands for cell number. Venn diagrams were prepared using the Van de Peer laboratory tool available at http://bioinformatics.psb.ugent.be/beg/tools/venn-diagrams.
RNA sequencing.
RNA was prepared from leukemias using Qiagen RNeasy (Qiagen, Hilden, Germany). RNA samples were sequenced by the Genome Quebec Innovation Centre using the TruSeq stranded total RNA library preparation kit and the Illumina HiSeq4000 PE100 (Illumina, San Diego, CA). BAM files were trimmed using TrimGalore! and aligned to mm10 using TopHat2. Cufflinks was used to generate transcript counts, expressed as fragments per kilobase of transcript per million mapped reads (FPKM). DESeq2 differential gene expression analysis (37) and gene set enrichment analysis (GSEA) (28) were performed using standard settings. The eligibility of prooxidant and antioxidant genes for heat map analysis was determined based on genes that were expressed (FPKM > 0.5) and differed in expression by more than 1.3-fold (highest FPKM/lowest FPKM). Heat maps were generated using the heatmap function in RStudio 1.2.5.
Cell culture.
Thymus or bone marrow cells were cultured in Iscove’s modified Dulbecco’s medium (IMDM) with 10% fetal bovine serum (FBS), 1× penicillin–streptomycin–l-glutamine, 5 × 10−5 M β-mercaptoethanol, and IL-7-conditioned medium prepared as previously described (64). A cell line was established from leukemia 973 by passaging every 48 to 72 h. Fetal-liver-derived pro-B cells were cultured on ST2 stromal cells as previously described (64). Cell counting or MTT assays (Trevigen, Gaithersburg MD) were performed using 72 h of culture in various concentrations of N-acetylcysteine (MilliporeSigma, Oakville, ON, Canada), hydrogen peroxide (Fisher Scientific, Mississauga, ON, Canada), Necrox-5 (Bio-Techne, Oakville, ON, Canada), ML334 (Bio-Techne), or ruxolitinib (Abcam, Toronto, ON, Canada). For gene expression experiments, cells were cultured for 24 h before RNA was prepared using TRIzol (Thermo-Fisher Scientific). Primers for reverse transcriptase quantitative PCR (RT-qPCR) experiments are shown in Table S5 in the supplemental material. For immunoblot analysis, whole-cell lysates were generated using Laemmli buffer and probed with anti-STAT5 and anti-phosphorylated-STAT5 (anti-p-STAT5) antibodies (Cell Signaling Technology, Danvers, MA). Imaging was performed using a ChemiDoc XRS+ imager (Bio-Rad, Hercules, CA).
8-Oxoguanine DNA damage immunofluorescence staining.
Wild-type pro-B cells or leukemia cells from the enlarged thymus of moribund Mb1-CreΔPB mice were cultured for <2 weeks and then cytospun onto glass slides at 1 × 105 cells/slide for 5 min at 500 rpm. Air-dried slides were fixed with 4% paraformaldehyde (MilliporeSigma) in phosphate-buffered saline (PBS) for 30 min and then rinsed and treated at room temperature with 0.2 mg/ml RNase A (MilliporeSigma) for 1 h. Slides were rinsed with 0.3% Triton X-100 in PBS, incubated in 2 M HCl for 10 min, and rinsed with 50 mM Tris base for neutralization. Slides were blocked with 5% normal goat serum (Southern Biotech, Birmingham AL), Fc Block antibody (BD Biosciences, Mississauga, ON, Canada), and 0.1% Triton X-100 (MilliporeSigma). Slides were then rinsed and incubated with biotinylated anti-8-oxoguanine antibody (15A3; Abcam) for 1 h at room temperature, followed by incubation with Alexa Fluor 647-conjugated antibiotin antibody (Jackson ImmunoResearch, West Grove, PA) overnight at 4°C. Coverslips were placed over cells in DAPI-containing Fluoromount mounting medium (Thermo-Fisher Scientific, Mississauga ON, Canada). Imaging was performed with a Leica DMI6000B wide-field microscope at ×630 magnification. Image analysis was performed using ImageJ. DAPI fluorescence was used to delineate the boundaries of intact nuclei, and the average fluorescence intensity for Alexa Fluor 647 in the corresponding regions was quantified. The Iglewicz-and-Hoaglin modified Z score was used to identify and remove outliers, defined as nuclei with an average fluorescence intensity Z score of >3.5.
Statistical analysis.
All data reported in this article were graphed as mean values ± standard errors of the means (SEM). Statistical analyses were conducted with Prism 8.2.2 (GraphPad Software, La Jolla, CA) using tests indicated in the figure legends.
Reagent table.
A reagent table is provided as Table S6.
Data availability.
Data are available under BioProject accession number PRJNA588255 in the Sequence Read Archive.
Supplementary Material
ACKNOWLEDGMENTS
We thank Novartis Pharma AG (Basel, Switzerland) and Incyte corporation (Wilmington, DE) for providing ruxolitinib mouse chow and drug-free vehicle chow used in these studies. We acknowledge Aren Marshall and the laboratory of Fred Dick (Department of Biochemistry, Western University) for a protocol and advice with 8-OxoG staining, Charles Yin and Bryan Heit (Microbiology and Immunology, Western University) for assistance and advice with wide-field microscopy, Greg Dekaban (Microbiology and Immunology, Western University) for assistance with cytospinning, Parisa Shooshtari (Pathology and Laboratory Medicine, Western University) for advice on bioinformatics analysis, and Angelo Empleo and Devanshi Shukla for technical assistance with experiments.
This work was supported by the Leukemia and Lymphoma Society of Canada via grant number 569086 and by Canadian Institutes of Health Research via grant number 142250.
R.P.D. conceived the study. M.L., C.R.B., B.R.D.O., R.C., J.F., K.C., D.K., J.I., and D.M. performed experiments and analyzed the data. M.A. analyzed data. M.L. and R.P.D. wrote the manuscript. All authors had final approval of the submitted manuscript.
We declare no competing financial interests.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Iacobucci I, Mullighan CG. 2017. Genetic basis of acute lymphoblastic leukemia. J Clin Oncol 35:975–983. doi: 10.1200/JCO.2016.70.7836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roberts KG, Mullighan CG. 2015. Genomics in acute lymphoblastic leukaemia: insights and treatment implications. Nat Rev Clin Oncol 12:344–357. doi: 10.1038/nrclinonc.2015.38. [DOI] [PubMed] [Google Scholar]
- 3.Roberts KG, Yang Y-L, Payne-Turner D, Lin W, Files JK, Dickerson K, Gu Z, Taunton J, Janke LJ, Chen T, Loh ML, Hunger SP, Mullighan CG. 2017. Oncogenic role and therapeutic targeting of ABL-class and JAK-STAT activating kinase alterations in Ph-like ALL. Blood Adv 1:1657–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Greaves M, Maley CC. 2012. Clonal evolution in cancer. Nature 481:306–313. doi: 10.1038/nature10762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Swaminathan S, Klemm L, Park E, Papaemmanuil E, Ford A, Kweon SM, Trageser D, Hasselfeld B, Henke N, Mooster J, Geng H, Schwarz K, Kogan SC, Casellas R, Schatz DG, Lieber MR, Greaves MF, Muschen M. 2015. Mechanisms of clonal evolution in childhood acute lymphoblastic leukemia. Nat Immunol 16:766–774. doi: 10.1038/ni.3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DeKoter RP, Gibson PJ. 2016. B cell leukemia and lymphoma, p 279 In Ratcliffe MJH. (ed), Encyclopedia of immunobiology, Academic Press, Oxford, UK. [Google Scholar]
- 7.Ma X, Liu Y, Liu Y, Alexandrov LB, Edmonson MN, Gawad C, Zhou X, Li Y, Rusch MC, John E, Huether R, Gonzalez-Pena V, Wilkinson MR, Hermida LC, Davis S, Sioson E, Pounds S, Cao X, Ries RE, Wang Z, Chen X, Dong L, Diskin SJ, Smith MA, Auvi JMG, Meltzer PS, Lau CC, Perlman EJ, Maris JM, Meshinchi S, Hunger SP, Gerhard DS, Zhang J. 2018. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature 555:371–376. doi: 10.1038/nature25795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kohnken R, Porcu P, Mishra A. 2017. Overview of the use of murine models in leukemia and lymphoma research. Front Oncol 7:22. doi: 10.3389/fonc.2017.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clark MR, Mandal M, Ochiai K, Singh H. 2014. Orchestrating B cell lymphopoiesis through interplay of IL-7 receptor and pre-B cell receptor signalling. Nat Rev Immunol 14:69–80. doi: 10.1038/nri3570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ray PD, Huang BW, Tsuji Y. 2012. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal 24:981–990. doi: 10.1016/j.cellsig.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pang SH, Carotta S, Nutt SL. 2014. Transcriptional control of pre-B cell development and leukemia prevention. Curr Top Microbiol Immunol 381:189–213. doi: 10.1007/82_2014_377. [DOI] [PubMed] [Google Scholar]
- 12.Su GH, Ip HS, Cobb BS, Lu MM, Chen HM, Simon MC. 1996. The Ets protein Spi-B is expressed exclusively in B cells and T cells during development. J Exp Med 184:203–214. doi: 10.1084/jem.184.1.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Solomon LA, Li SKH, Piskorz J, Xu LS, DeKoter RP. 2015. Genome-wide comparison of PU.1 and Spi-B binding sites in a mouse B lymphoma cell line. BMC Genomics 16:76. doi: 10.1186/s12864-015-1303-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iwasaki H, Somoza C, Shigematsu H, Duprez EA, Iwasaki-Arai J, Mizuno S, Arinobu Y, Geary K, Zhang P, Dayaram T, Fenyus ML, Elf S, Chan S, Kastner P, Huettner CS, Murray R, Tenen DG, Akashi K. 2005. Distinctive and indispensable roles of PU.1 in maintenance of hematopoietic stem cells and their differentiation. Blood 106:1590–1600. doi: 10.1182/blood-2005-03-0860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Christie DA, Xu LS, Turkistany SA, Solomon LA, Li SK, Yim E, Welch I, Bell GI, Hess DA, DeKoter RP. 2015. PU.1 opposes IL-7-dependent proliferation of developing B cells with involvement of the direct target gene Bruton tyrosine kinase. J Immunol 194:595–605. doi: 10.4049/jimmunol.1401569. [DOI] [PubMed] [Google Scholar]
- 16.Xu LS, Sokalski KM, Hotke K, Christie DA, Zarnett O, Piskorz J, Thillainadesan G, Torchia J, DeKoter RP. 2012. Regulation of B cell linker protein transcription by PU.1 and Spi-B in murine B cell acute lymphoblastic leukemia. J Immunol 189:3347–3354. doi: 10.4049/jimmunol.1201267. [DOI] [PubMed] [Google Scholar]
- 17.Batista CR, Li SKH, Xu LS, Solomon LA, DeKoter RP. 2017. PU.1 regulates Ig light chain transcription and rearrangement in pre-B cells during B cell development. J Immunol 198:1565–1574. doi: 10.4049/jimmunol.1601709. [DOI] [PubMed] [Google Scholar]
- 18.Batista CR, Lim M, Laramee A-S, Abu-Sardanah F, Xu LS, Hossain R, Bell GI, Hess DA, DeKoter RP. 2018. Driver mutations in Janus kinases in a mouse model of B-cell leukemia induced by deletion of PU.1 and Spi-B. Blood Adv 2:2798–2810. doi: 10.1182/bloodadvances.2018019950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morris R, Kershaw NJ, Babon JJ. 2018. The molecular details of cytokine signaling via the JAK/STAT pathway. Protein Sci 27:1984–2009. doi: 10.1002/pro.3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rebollo A, Schmitt C. 2003. Ikaros, Aiolos and Helios: transcription regulators and lymphoid malignancies. Immunol Cell Biol 81:171–175. doi: 10.1046/j.1440-1711.2003.01159.x. [DOI] [PubMed] [Google Scholar]
- 21.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale AL, Boyault S, Burkhardt B, Butler AP, Caldas C, Davies HR, Desmedt C, Eils R, Eyfjord JE, Foekens JA, Greaves M, Hosoda F, Hutter B, Ilicic T, Imbeaud S, Imielinski M, Jager N, Jones DT, Jones D, Knappskog S, Kool M, Lakhani SR, Lopez-Otin C, Martin S, Munshi NC, Nakamura H, Northcott PA, Pajic M, Papaemmanuil E, Paradiso A, Pearson JV, Puente XS, Raine K, Ramakrishna M, Richardson AL, Richter J, Rosenstiel P, Schlesner M, Schumacher TN, Span PN, Teague JW, et al. 2013. Signatures of mutational processes in human cancer. Nature 500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rosenthal R, McGranahan N, Herrero J, Taylor BS, Swanton C. 2016. deconstructSigs: delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol 17:31. doi: 10.1186/s13059-016-0893-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pugh TJ, Morozova O, Attiyeh EF, Asgharzadeh S, Wei JS, Auclair D, Carter SL, Cibulskis K, Hanna M, Kiezun A, Kim J, Lawrence MS, Lichenstein L, McKenna A, Pedamallu CS, Ramos AH, Shefler E, Sivachenko A, Sougnez C, Stewart C, Ally A, Birol I, Chiu R, Corbett RD, Hirst M, Jackman SD, Kamoh B, Khodabakshi AH, Krzywinski M, Lo A, Moore RA, Mungall KL, Qian J, Tam A, Thiessen N, Zhao Y, Cole KA, Diamond M, Diskin SJ, Mosse YP, Wood AC, Ji L, Sposto R, Badgett T, London WB, Moyer Y, Gastier-Foster JM, Smith MA, Guidry Auvil JM, Gerhard DS, et al. 2013. The genetic landscape of high-risk neuroblastoma. Nat Genet 45:279–284. doi: 10.1038/ng.2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kino K, Hirao-Suzuki M, Morikawa M, Sakaga A, Miyazawa H. 2017. Generation, repair and replication of guanine oxidation products. Genes Environ 39:21. doi: 10.1186/s41021-017-0081-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Casellas R, Basu U, Yewdell WT, Chaudhuri J, Robbiani DF, Di Noia JM. 2016. Mutations, kataegis and translocations in B cells: understanding AID promiscuous activity. Nat Rev Immunol 16:164–176. doi: 10.1038/nri.2016.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Larijani M, Frieder D, Basit W, Martin A. 2005. The mutation spectrum of purified AID is similar to the mutability index in Ramos cells and in ung(-/-)msh2(-/-) mice. Immunogenetics 56:840–845. doi: 10.1007/s00251-004-0748-0. [DOI] [PubMed] [Google Scholar]
- 27.Prieto-Bermejo R, Romo-González M, Pérez-Fernández A, Ijurko C, Hernández-Hernández Á. 2018. Reactive oxygen species in haematopoiesis: leukaemic cells take a walk on the wild side. J Exp Clin Cancer Res 37:125. doi: 10.1186/s13046-018-0797-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Irwin ME, Rivera-Del Valle N, Chandra J. 2013. Redox control of leukemia: from molecular mechanisms to therapeutic opportunities. Antioxid Redox Signal 18:1349–1383. doi: 10.1089/ars.2011.4258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Iiyama M, Kakihana K, Kurosu T, Miura O. 2006. Reactive oxygen species generated by hematopoietic cytokines play roles in activation of receptor-mediated signaling and in cell cycle progression. Cell Signal 18:174–182. doi: 10.1016/j.cellsig.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 31.Das A, Hazra TK, Boldogh I, Mitra S, Bhakat KK. 2005. Induction of the human oxidized base-specific DNA glycosylase NEIL1 by reactive oxygen species. J Biol Chem 280:35272–35280. doi: 10.1074/jbc.M505526200. [DOI] [PubMed] [Google Scholar]
- 32.Park J-H, Kim HK, Jung H, Kim KH, Kang MS, Hong JH, Yu BC, Park S, Seo S-K, Choi IW, Kim SH, Kim N, Han J, Park SG. 2017. NecroX-5 prevents breast cancer metastasis by AKT inhibition via reducing intracellular calcium levels. Int J Oncol 50:185–192. doi: 10.3892/ijo.2016.3789. [DOI] [PubMed] [Google Scholar]
- 33.Kim HJ, Koo SY, Ahn B-H, Park O, Park DH, Seo DO, Won JH, Yim HJ, Kwak H-S, Park HS, Chung CW, Oh YL, Kim SH. 2010. NecroX as a novel class of mitochondrial reactive oxygen species and ONOO(-) scavenger. Arch Pharm Res 33:1813–1823. doi: 10.1007/s12272-010-1114-4. [DOI] [PubMed] [Google Scholar]
- 34.Hu L, Magesh S, Chen L, Wang L, Lewis TA, Chen Y, Khodier C, Inoyama D, Beamer LJ, Emge TJ, Shen J, Kerrigan JE, Kong A-N, Dandapani S, Palmer M, Schreiber SL, Munoz B. 2013. Discovery of a small-molecule inhibitor and cellular probe of Keap1-Nrf2 protein-protein interaction. Bioorg Med Chem Lett 23:3039–3043. doi: 10.1016/j.bmcl.2013.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fridman JS, Scherle PA, Collins R, Burn TC, Li Y, Li J, Covington MB, Thomas B, Collier P, Favata MF, Wen X, Shi J, McGee R, Haley PJ, Shepard S, Rodgers JD, Yeleswaram S, Hollis G, Newton RC, Metcalf B, Friedman SM, Vaddi K. 2010. Selective inhibition of JAK1 and JAK2 is efficacious in rodent models of arthritis: preclinical characterization of INCB028050. J Immunol 184:5298–5307. doi: 10.4049/jimmunol.0902819. [DOI] [PubMed] [Google Scholar]
- 36.Sokalski KM, Li SK, Welch I, Cadieux-Pitre HA, Gruca MR, DeKoter RP. 2011. Deletion of genes encoding PU.1 and Spi-B in B cells impairs differentiation and induces pre-B cell acute lymphoblastic leukemia. Blood 118:2801–2808. doi: 10.1182/blood-2011-02-335539. [DOI] [PubMed] [Google Scholar]
- 37.Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jain N, Roberts KG, Jabbour E, Patel K, Eterovic AK, Chen K, Zweidler-McKay P, Lu X, Fawcett G, Wang SA, Konoplev S, Harvey RC, Chen I-M, Payne-Turner D, Valentine M, Thomas D, Garcia-Manero G, Ravandi F, Cortes J, Kornblau S, O’Brien S, Pierce S, Jorgensen J, Shaw KRM, Willman CL, Mullighan CG, Kantarjian H, Konopleva M. 2017. Ph-like acute lymphoblastic leukemia: a high-risk subtype in adults. Blood 129:572–581. doi: 10.1182/blood-2016-07-726588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Losdyck E, Hornakova T, Springuel L, Degryse S, Gielen O, Cools J, Constantinescu SN, Flex E, Tartaglia M, Renauld J-C, Knoops L. 2015. Distinct acute lymphoblastic leukemia (ALL)-associated Janus kinase 3 (JAK3) mutants exhibit different cytokine-receptor requirements and JAK inhibitor specificities. J Biol Chem 290:29022–29034. doi: 10.1074/jbc.M115.670224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Elliott NE, Cleveland SM, Grann V, Janik J, Waldmann TA, Dave UP. 2011. FERM domain mutations induce gain of function in JAK3 in adult T-cell leukemia/lymphoma. Blood 118:3911–3921. doi: 10.1182/blood-2010-12-319467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hornakova T, Staerk J, Royer Y, Flex E, Tartaglia M, Constantinescu SN, Knoops L, Renauld J-C. 2009. Acute lymphoblastic leukemia-associated JAK1 mutants activate the Janus kinase/STAT pathway via interleukin-9 receptor alpha homodimers. J Biol Chem 284:6773–6781. doi: 10.1074/jbc.M807531200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roberti A, Dobay MP, Bisig B, Vallois D, Boechat C, Lanitis E, Bouchindhomme B, Parrens M-C, Bossard C, Quintanilla-Martinez L, Missiaglia E, Gaulard P, de Leval L. 2016. Type II enteropathy-associated T-cell lymphoma features a unique genomic profile with highly recurrent SETD2 alterations. Nat Commun 7:12602. doi: 10.1038/ncomms12602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Holmfeldt L, Wei L, Diaz-Flores E, Walsh M, Zhang J, Ding L, Payne-Turner D, Churchman M, Andersson A, Chen S-C, McCastlain K, Becksfort J, Ma J, Wu G, Patel SN, Heatley SL, Phillips LA, Song G, Easton J, Parker M, Chen X, Rusch M, Boggs K, Vadodaria B, Hedlund E, Drenberg C, Baker S, Pei D, Cheng C, Huether R, Lu C, Fulton RS, Fulton LL, Tabib Y, Dooling DJ, Ochoa K, Minden M, Lewis ID, To LB, Marlton P, Roberts AW, Raca G, Stock W, Neale G, Drexler HG, Dickins RA, Ellison DW, Shurtleff SA, Pui C-H, Ribeiro RC, et al. 2013. The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat Genet 45:242–252. doi: 10.1038/ng.2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Churchman ML, Qian M, Te Kronnie G, Zhang R, Yang W, Zhang H, Lana T, Tedrick P, Baskin R, Verbist K, Peters JL, Devidas M, Larsen E, Moore IM, Gu Z, Qu C, Yoshihara H, Porter SN, Pruett-Miller SM, Wu G, Raetz E, Martin PL, Bowman WP, Winick N, Mardis E, Fulton R, Stanulla M, Evans WE, Relling MV, Pui C-H, Hunger SP, Loh ML, Handgretinger R, Nichols KE, Yang JJ, Mullighan CG. 2018. Germline genetic IKZF1 variation and predisposition to childhood acute lymphoblastic leukemia. Cancer Cell 33:937–948.e8. doi: 10.1016/j.ccell.2018.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gorrini C, Harris IS, Mak TW. 2013. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov 12:931–947. doi: 10.1038/nrd4002. [DOI] [PubMed] [Google Scholar]
- 46.Jayavelu AK, Müller JP, Bauer R, Böhmer S-A, Lässig J, Cerny-Reiterer S, Sperr WR, Valent P, Maurer B, Moriggl R, Schröder K, Shah AM, Fischer M, Scholl S, Barth J, Oellerich T, Berg T, Serve H, Frey S, Fischer T, Heidel FH, Böhmer F-D. 2016. NOX4-driven ROS formation mediates PTP inactivation and cell transformation in FLT3ITD-positive AML cells. Leukemia 30:473–483. doi: 10.1038/leu.2015.234. [DOI] [PubMed] [Google Scholar]
- 47.Themanns M, Mueller KM, Kessler SM, Golob-Schwarzl N, Mohr T, Kaltenecker D, Bourgeais J, Paier-Pourani J, Friedbichler K, Schneller D, Schlederer M, Zebedin-Brandl E, Terracciano LM, Han X, Kenner L, Wagner K-U, Mikulits W, Kozlov AV, Heim MH, Gouilleux F, Haybaeck J, Moriggl R. 2016. Hepatic deletion of Janus kinase 2 counteracts oxidative stress in mice. Sci Rep 6:34719. doi: 10.1038/srep34719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bourgeais J, Ishac N, Medrzycki M, Brachet-Botineau M, Desbourdes L, Gouilleux-Gruart V, Pecnard E, Rouleux-Bonnin F, Gyan E, Domenech J, Mazurier F, Moriggl R, Bunting KD, Herault O, Gouilleux F. 2017. Oncogenic STAT5 signaling promotes oxidative stress in chronic myeloid leukemia cells by repressing antioxidant defenses. Oncotarget 8:41876–41889. doi: 10.18632/oncotarget.11480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stein M, Dutting S, Mougiakakos D, Bosl M, Fritsch K, Reimer D, Urbanczyk S, Steinmetz T, Schuh W, Bozec A, Winkler TH, Jack H-M, Mielenz D. 2017. A defined metabolic state in pre B cells governs B-cell development and is counterbalanced by Swiprosin-2/EFhd1. Cell Death Differ 24:1239–1252. doi: 10.1038/cdd.2017.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chan LN, Chen Z, Braas D, Lee J-W, Xiao G, Geng H, Cosgun KN, Hurtz C, Shojaee S, Cazzaniga V, Schjerven H, Ernst T, Hochhaus A, Kornblau SM, Konopleva M, Pufall MA, Cazzaniga G, Liu GJ, Milne TA, Koeffler HP, Ross TS, Sánchez-García I, Borkhardt A, Yamamoto KR, Dickins RA, Graeber TG, Müschen M. 2017. Metabolic gatekeeper function of B-lymphoid transcription factors. Nature 542:479–483. doi: 10.1038/nature21076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vitanza NA, Zaky W, Blum R, Meyer JA, Wang J, Bhatla T, Morrison DJ, Raetz EA, Carroll WL. 2014. Ikaros deletions in BCR-ABL-negative childhood acute lymphoblastic leukemia are associated with a distinct gene expression signature but do not result in intrinsic chemoresistance. Pediatr Blood Cancer 61:1779–1785. doi: 10.1002/pbc.25119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mullighan CG, Goorha S, Radtke I, Miller CB, Coustan-Smith E, Dalton JD, Girtman K, Mathew S, Ma J, Pounds SB, Su X, Pui C-H, Relling MV, Evans WE, Shurtleff SA, Downing JR. 2007. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature 446:758–764. doi: 10.1038/nature05690. [DOI] [PubMed] [Google Scholar]
- 53.Mueller BU, Pabst T, Osato M, Asou N, Johansen LM, Minden MD, Behre G, Hiddemann W, Ito Y, Tenen DG. 2003. Heterozygous PU.1 mutations are associated with acute myeloid leukemia. Blood 101:2074. doi: 10.1182/blood-2002-12-3903. [DOI] [PubMed] [Google Scholar]
- 54.Pang SH, Minnich M, Gangatirkar P, Zheng Z, Ebert A, Song G, Dickins RA, Corcoran LM, Mullighan CG, Busslinger M, Huntington ND, Nutt SL, Carotta S. 2016. PU.1 cooperates with IRF4 and IRF8 to suppress pre-B-cell leukemia. Leukemia 30:1375–1387. doi: 10.1038/leu.2016.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vangala RK, Heiss-Neumann MS, Rangatia JS, Singh SM, Schoch C, Tenen DG, Hiddemann W, Behre G. 2003. The myeloid master regulator transcription factor PU.1 is inactivated by AML1-ETO in t(8;21) myeloid leukemia. Blood 101:270–277. doi: 10.1182/blood-2002-04-1288. [DOI] [PubMed] [Google Scholar]
- 56.Wang H, Jain S, Li P, Lin J-X, Oh J, Qi C, Gao Y, Sun J, Sakai T, Naghashfar Z, Abbasi S, Kovalchuk AL, Bolland S, Nutt SL, Leonard WJ, Morse HC III. 2019. Transcription factors IRF8 and PU.1 are required for follicular B cell development and BCL6-driven germinal center responses. Proc Natl Acad Sci U S A 116:9511–9520. doi: 10.1073/pnas.1901258116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Willis SN, Tellier J, Liao Y, Trezise S, Light A, O’Donnell K, Garrett-Sinha LA, Shi W, Tarlinton DM, Nutt SL. 2017. Environmental sensing by mature B cells is controlled by the transcription factors PU.1 and SpiB. Nat Commun 8:1426. doi: 10.1038/s41467-017-01605-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim S-K, Knight DA, Jones LR, Vervoort S, Ng AP, Seymour JF, Bradner JE, Waibel M, Kats L, Johnstone RW. 2018. JAK2 is dispensable for maintenance of JAK2 mutant B-cell acute lymphoblastic leukemias. Genes Dev 32:849–864. doi: 10.1101/gad.307504.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Abdeldayem A, Raouf YS, Constantinescu SN, Moriggl R, Gunning PT. 2020. Advances in covalent kinase inhibitors. Chem Soc Rev 49:2617–2687. doi: 10.1039/c9cs00720b. [DOI] [PubMed] [Google Scholar]
- 60.Saunders CT, Wong WSW, Swamy S, Becq J, Murray LJ, Cheetham RK. 2012. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics 28:1811–1817. doi: 10.1093/bioinformatics/bts271. [DOI] [PubMed] [Google Scholar]
- 61.Koboldt DC, Larson DE, Wilson RK. 2013. Using VarScan 2 for germline variant calling and somatic mutation detection. Curr Protoc Bioinform 44:15.4.1–15.4.17. doi: 10.1002/0471250953.bi1504s44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Garrison E, Marth G. 2012. Haplotype-based variant detection from short read sequencing. arXiv 1207.3907 [q-bio.GN]. https://arxiv.org/abs/1207.3907.
- 63.Reumers J, Schymkowitz J, Ferkinghoff-Borg J, Stricher F, Serrano L, Rousseau F. 2005. SNPeffect: a database mapping molecular phenotypic effects of human non-synonymous coding SNPs. Nucleic Acids Res 33:D527–D532. doi: 10.1093/nar/gki086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schweitzer BL, DeKoter RP. 2004. Analysis of gene expression and Ig transcription in PU.1/Spi-B-deficient progenitor B cell lines. J Immunol 172:144–154. doi: 10.4049/jimmunol.172.1.144. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are available under BioProject accession number PRJNA588255 in the Sequence Read Archive.