

Abstract
Anti–tumor necrosis factor (anti-TNF) therapy resistance is a major clinical challenge in inflammatory bowel disease (IBD), partly due to insufficient understanding of disease-site, protein-level mechanisms. Although proteomics data from IBD mouse models exist, data and phenotype discrepancies contribute to confounding translation from preclinical animal models of disease to clinical cohorts. We developed an approach called translatable components regression (TransComp-R) to overcome interspecies and trans-omic discrepancies between mouse models and human subjects. TransComp-R combines mouse proteomic data with patient pretreatment transcriptomic data to identify molecular features discernable in the mouse data that are predictive of patient response to therapy. Interrogating the TransComp-R models revealed activated integrin pathway signaling in anti-TNF–resistant colonic Crohn’s disease (cCD) and ulcerative colitis (UC) patients. As a step toward validation, we performed single-cell RNA sequencing (scRNA-seq) on biopsies from a cCD patient and analyzed publicly available immune cell proteomics data to characterize the immune and intestinal cell types contributing to anti-TNF resistance. We found that ITGA1 was expressed in T cells and that interactions between these cells and intestinal cell types were associated with resistance to anti-TNF therapy. We experimentally showed that the α1 integrin subunit mediated the effectiveness of anti-TNF therapy in human immune cells. Thus, TransComp-R identified an integrin signaling mechanism with potential therapeutic implications for overcoming anti-TNF therapy resistance. We suggest that TransComp-R is a generalizable framework for addressing species, molecular, and phenotypic discrepancies between model systems and patients to deliver translationally relevant biological insights.
One-sentence summary:
A platform for comparing trans-omics data sets between IBD mouse models and human patients reveals therapeutically relevant targets.
Editor’s summary:
Found in translation
An ongoing challenge for the development of new therapeutics is the difficulty in translating findings from preclinical animal models to human subjects, more so when different types of data (proteomics versus transcriptomics) are compared. Brubaker et al. developed a method to project transcriptomic data from patients with inflammatory bowel disease (IBD) into a principal components analysis of mouse proteomics data to investigate resistance to the anti-TNF antibody infliximab. This analysis, which suggested that integrin signaling contributed to resistance, was validated in experiments showing that inhibiting α1 integrin subunit signaling enhanced the ability of infliximab to suppress proinflammatory cytokine release from immune cells. These data suggest that this approach for comparing model system and patient data might reveal therapeutically relevant targets for other diseases.
Introduction
Crohn’s disease (CD) and ulcerative colitis (UC) are chronic inflammatory bowel diseases (IBDs) with an increasing global prevalence (1). Anti–tumor necrosis factor (anti-TNF) therapeutics, including infliximab and adalimumab (ADA), have emerged as remission-inducing therapies that are effective in 30 to 50% of patients, with up to 30% of these patients eventually developing a secondary nonresponse (2, 3). This high rate of nonresponse to anti-TNF therapy has motivated several studies examining the transcriptomic determinants of resistance (2, 4–9). These transcriptomic characterizations of anti-TNF resistance have yet to translate into effective strategies for overcoming resistance, potentially due to the lack of functional proteomic characterization of infliximab-resistant IBD. Whereas some mouse proteomics studies have measured thousands of proteins by mass spectrometry and provided a detailed view of signaling in inflamed and uninflamed conditions (10, 11), these did not include therapeutic stimuli, making it challenging to generalize these murine signaling characterizations to clinical therapeutic resistance.
Interspecies translational systems biology uses computational systems models to better translate biological insights from in vitro and nonhuman in vivo experimental models to the human disease context. Several studies have applied statistical and machine learning models to infer human disease biology from model systems (12–17), but a limitation of these methods was the need for comparable molecular data types and similar phenotypes between model systems and humans. Therefore, these methods are not appropriate for translating the IBD mouse model proteomic characterizations to understand infliximab resistance in patients. If the challenges of interspecies, trans-omic, inter-phenotypic translation could be overcome, then the available mouse proteomics data could provide valuable insights into the signaling networks associated with infliximab nonresponse in IBD.
Here, we developed a method to translate proteomics from IBD mouse models to patients, which we call translatable components regression (TransComp-R). TransComp-R projected human IBD transcriptomic data into a mouse proteomics principal component analysis (PCA) model and performed principal component (PC) regression against the human infliximab response phenotypes to identify the most translatable mouse PCs. Analysis of the proteins that defined separation along these latent variables identified activated integrin signaling, in both UC and CD, that separated infliximab responders and non-responders before treatment. Because IBD is a complex disorder that affects both host tissue and immune cell signaling, we obtained colonic biopsies from a CD patient to perform single-cell RNA-sequencing (scRNA-seq) and analyzed publicly available, proteomics data from sorted immune cells to identify the cell types and intercellular signaling pathways associated with the identified infliximab resistance signatures. The signaling network identified by TransComp-R and scRNA-seq described interactions between ITGA1+ T cells and colonic cell types. Further characterization of the intercellular signaling network between immune and colonic cell types implicated a collagen-binding integrin network associated with infliximab resistance in disease-relevant cell types. We then experimentally confirmed that inhibition of the α1 integrin subunit enhanced the cytokine-suppressive effects of infliximab in immune cells as assessed by Luminex analysis. The results of TransComp-R and our confirmatory analyses and experiments suggest that inhibition of the α1 integrin subunit may enhance the responsiveness of IBD patients to anti-TNF therapy.
Results
The molecular characteristics of infliximab resistance are tissue-specific
We analyzed publicly available gene expression data of colon and ileum biopsies from CD and UC patients, before the start of treatment with infliximab, to identify resistance signatures in each disease and tissue [responder (R) versus nonresponder (NR)] (4, 18). Differential expression analysis [Wilcoxon Mann-Whitney (WMW), false discovery rate (FDR) q < 0.25] identified several infliximab response–associated, differentially expressed genes (DEGs) in colonic Crohn’s disease (cCD, 2214 genes) and UC (996 genes) but found no substantial DEGs in ileal Crohn’s disease (iCD) patients (Fig. 1, A to C).
Fig 1. Tissue-specific signatures of infliximab resistance.
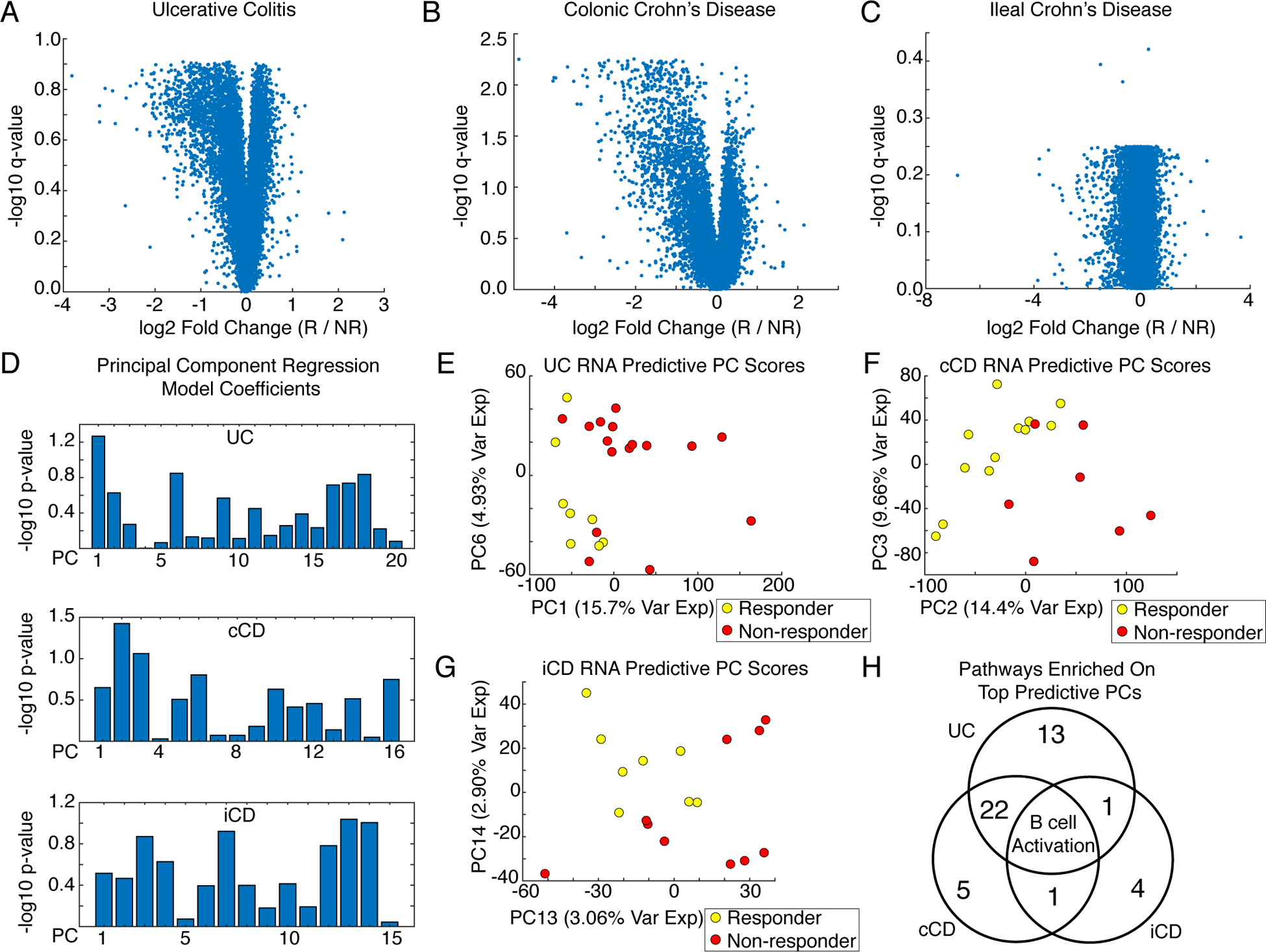
(A to C) Volcano plots of differential gene expression analysis (Wilcoxon Mann-Whitney, Benjamini Hochberg False Discovery Rate) from intestinal biopsies from patients with (A) Ulcerative colitis (UC, N = 24 patients), (B) colonic Crohn’s disease (cCD, N = 19 patients), and (C) ileal Crohn’s disease (iCD, N = 18 patients) comparing infliximab responders (R) and nonresponders (NR). Data were jittered for visualization. (D) PC regression coefficients predicting infliximab response from PCs explaining 95% variance in the UC, cCD, and iCD transcriptomic data. (E) Scores plot for the most predictive RNA PCs from the UC biopsies. (F) Scores plot for the most predictive RNA PCs in the cCD biopsies. (G) Scores plot for the most predictive RNA PCs in the iCD biopsies. (H) Comparison of pathways enriched for the top predictive PCs.
We assessed pathway-level dysregulation between infliximab responders and nonresponders by performing PANTHER pathway enrichment analysis on genes significantly loaded on human RNA PCs predictive of infliximab response (19–21). This analysis also provided a computational control for TransComp-R. Predictive human PCs were identified by PC regression using PCs explaining 95% of the variance in the gene expression data. We defined significantly loaded genes as those in the top 10% absolute value loadings on a PC. No models were significantly predictive, however, patients separated by infliximab response along the most predictive PCs, and significant genes on these PCs were enriched for candidate pathways (Fig. 1, D to H, table S1). The implicated PCs were often lower rank PCs that insufficiently explained overall transcriptional variation but were more predictive than the typically studied PC1 and PC2. UC and cCD shared 22 resistance pathways, including several inflammatory signaling pathways, T cell activation, integrin signaling, vascular endothelial growth factor (VEGF), and wingless-type MMTV integration site family (WNT) signaling. B cell activation was associated with infliximab resistance in UC, cCD, and iCD.
Translatable components regression analysis of mouse data predicts signaling in patients
Although transcriptomics can identify pathways associated with infliximab resistance, gene signatures alone are an incomplete characterization of signaling (5, 6, 22). Previous proteomic studies in IBD have principally been serum-based, and not direct measurements of signaling at the site of disease (23–25). We previously obtained proteomic measurements from two IBD mouse models, the adoptive T cell transfer (TCT) and TNFΔARE (TNF-ARE) mice, in inflamed and uninflamed conditions (10, 11). No therapeutic stimuli were studied in these mice, potentially limiting the relevance of these datasets for human infliximab resistance. We developed a data-driven, systems-modeling framework called TransComp-R to translate mouse proteomics to understand infliximab resistance in IBD patients (Fig. 2). TransComp-R trains a PCA model using mouse proteins whose coding genes are homologs with infliximab response–associated human genes and projects the RNA samples from IBD patients into the mouse proteomics PC space. Classically, new samples are projected into a PCA model by normalizing the new data with the scaling factors from the training data and then multiplying by the PCA eigenvectors. This is not appropriate for interspecies, trans-omic projections because scaling factors cannot be assumed to be comparable between different species, sequencing platforms, or molecular data types. TransComp-R modifies this procedure by projecting in terms of relative human differences along mouse PCs, rather than absolute differences. This is accomplished by multiplying the normalized human matrix by the mouse PC eigenvectors. Once projected, a PC regression model is trained to relate human scores on mouse PCs to the human infliximab response, identifying predictive proteomic PCs and dysregulated proteins. The projection of human samples into the mouse PC space is a form of computational reverse-translation, and we can regard the mouse proteomic PCs that predicted the human clinical associations as the most humanized translatable components of the mouse (Fig. 2).
Fig. 2. TransComp-R methodology.
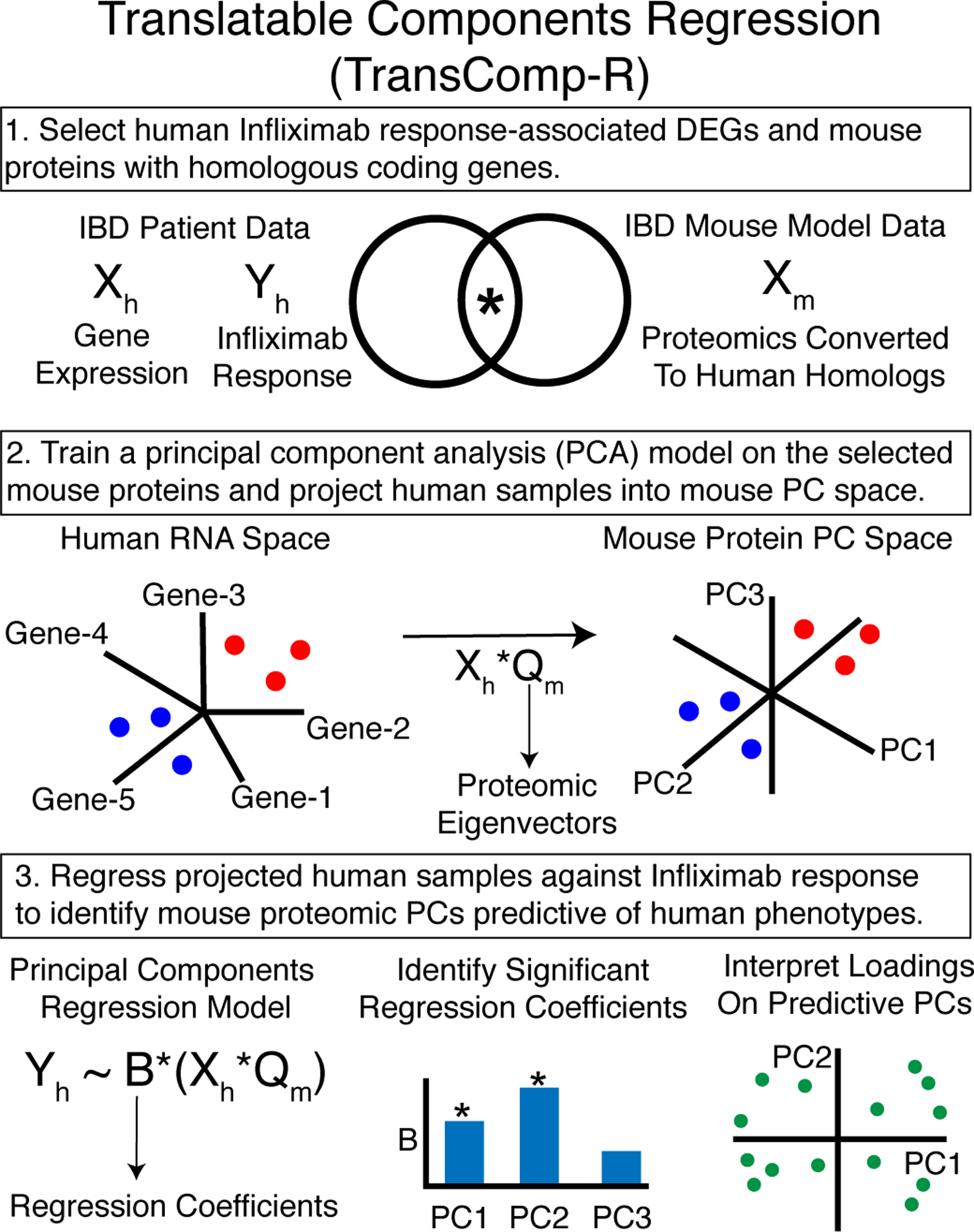
The various steps involved in TransComp-R are described.
TransComp-R identifies activated integrin signaling in infliximab-resistant IBD
We applied TransComp-R to identify proteomic signatures of infliximab response by projecting pre-infliximab treatment transcriptomics from cCD, UC, and iCD patients into mouse proteomic PCA models. Because different mouse models may describe different aspects of human disease, we used both the TCT and TNF-ARE mice to train separate TransComp-R models (10, 11). Infliximab response–associated genes were selected using an FDR threshold of q < 0.25 for UC and cCD. Because no genes met this threshold for iCD, we used the full dataset to train TransComp-R models. As a control, we trained PC regression models on the human transcriptomics data using the same genes selected for the TransComp-R models. Proteins significantly loaded on predictive PCs (top 10% absolute value loadings) were interpreted using PANTHER pathway enrichment analysis (19–21).
The cCD-TCT TransComp-R model was built using 335 proteins with homologous human DEGs. Although the human data alone did not produce a predictive model a TransComp-R model significantly predicted the infliximab response. The most predictive TCT PCs were PC5 and PC6 (5.85% total variance explained) (Fig. 3A, table S2). TCT mouse PC5 proteins were enriched for adenine and hypoxanthine salvage, coenzyme A biosynthesis, and vitamin D metabolism pathways, whereas TCT mouse PC6 proteins were enriched for adenine and hypoxanthine salvage and Alzheimer disease–presenilin pathways (table S3). The identification by TransComp-R of increased vitamin D metabolism in responders, a known biomarker of infliximab response, provides a positive control that the method can detect known response mechanisms (26, 27).
Fig. 3. TransComp-R of cCD and UC infliximab response against the IBD mouse models.
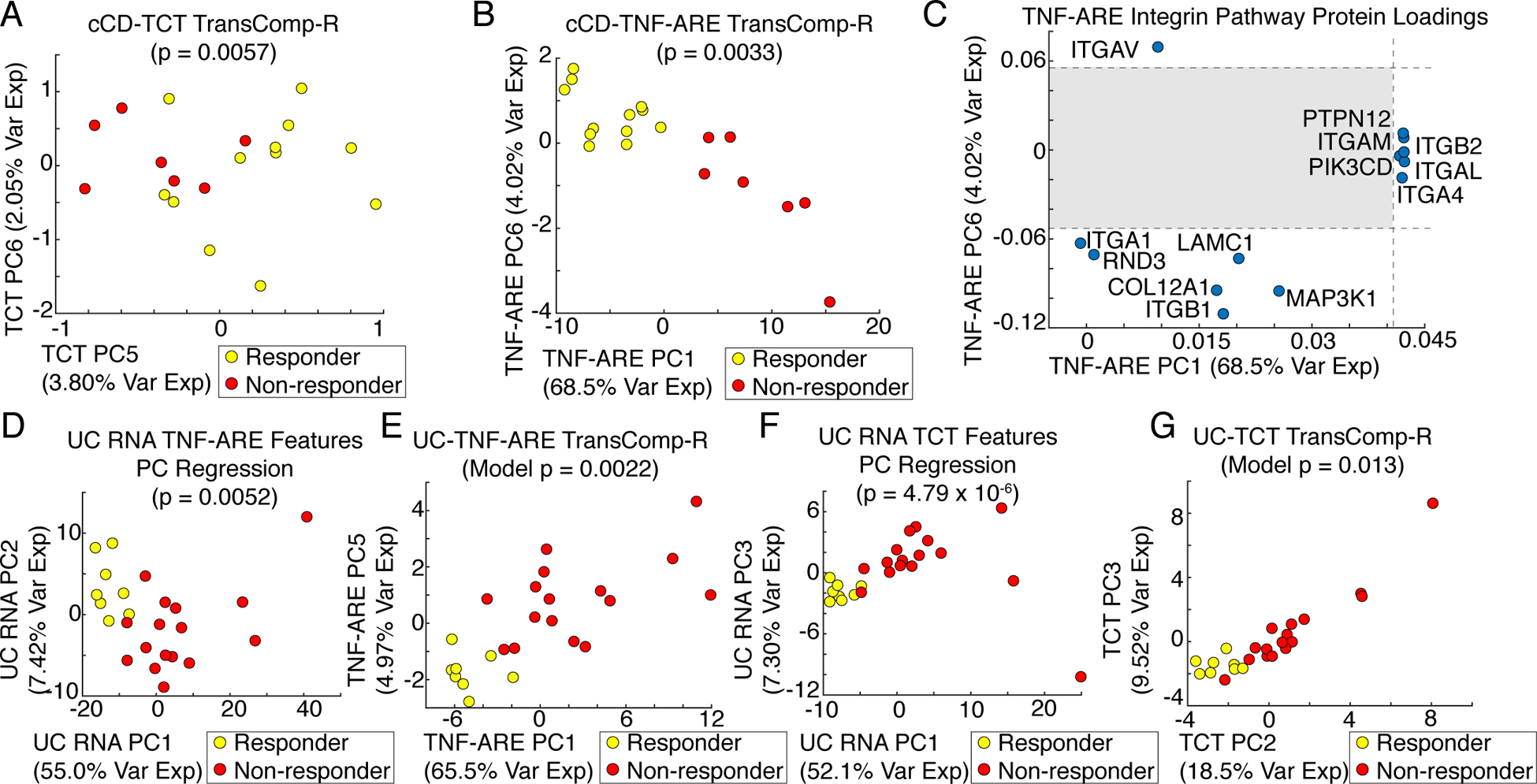
(A) Scores plot of the cCD-TCT mouse TransComp-R model of 335 infliximab resistance–associated, differentially expressed orthologous proteins (N = 19 patients). (B) Scores plot of the cCD-TNF-ARE mouse TransComp-R model of 810 infliximab resistance–associated, differentially expressed orthologous proteins (N = 19 patients). (C) Significant integrin pathway protein loadings on TNF-ARE mouse PC1 and PC6 with the non-significant region shaded (N = 8 mice). (D) Scores plot of the human UC principal component regression model of 368 infliximab resistance–associated, differentially expressed TNF-ARE mouse orthologous genes (N = 24 patients). (E) Scores plot of the UC-TNF-ARE mouse TransComp-R model of 368 infliximab resistance–associated differentially expressed orthologous proteins (N = 24 patients). (F) Scores plot of the human UC principal component regression model of 144 infliximab resistance–associated, differentially expressed TCT mouse orthologous genes (N = 24 patients). (G) Scores plot of the UC-TCT mouse TransComp-R model of 144 infliximab resistance–associated, differentially expressed orthologous proteins (N = 24 patients).
For the cCD-TNF-ARE TransComp-R model, 810 DEGs had homologous proteins in the TNF-ARE mouse data and these failed to train a predictive model with the cCD RNA data. The cCD-TNF-ARE TransComp-R model was predictive of the infliximab response, with the most predictive TNF-ARE PCs being PC1 and PC6 (72.5% total variance explained) (Fig. 3B, table S2). The proteins significantly loaded on TNF-ARE PC1 were enriched for 12 pathways, including B cell activation, T cell activation, interleukin signaling, inflammation medicated by chemokine and cytokine signaling, and integrin signaling (table S3). Integrin signaling was the most significantly enriched pathway on PC6, but the enrichment was driven by a different set of proteins than those on PC1. We plotted the variable loadings driving integrin pathway enrichment on PC1 and PC6 and found that the significant integrin-encoding genes driving enrichment on PC1 were associated with immune cell function (ITGA4, ITGAL, ITGAM) whereas those on PC6 encode collagen-binding integrins (ITGA1 and ITGB1) (Fig. 3C). On both PCs, a greater abundance of these integrins was associated with infliximab nonresponders, suggesting that multiple dimensions of activated integrin signaling are associated with resistance to infliximab.
There were 368 UC infliximab response–associated DEGs with homologous proteins in the TNF-ARE mouse data. These genes resulted in a predictive model using UC RNA PCs to predict the infliximab response, with the most predictive UC RNA PCs being PC1 and PC2 (62.4% total variance explained) (Fig. 3D, table S2). The significant proteins on UC PC1 were enriched for 20 pathways, among them integrin signaling and T cell activation (table S3). The gene encoding the immune cell trafficking–associated α4 integrin subunit (ITGA4) was among the genes driving enrichment of the integrin pathway, consistent with the finding from the cCD-TNF-ARE TransComp-R model. The UC-TNF-ARE TransComp-R model was slightly more predictive of infliximab response than was the human UC RNA model, with the most predictive TNF-ARE PCs being PC1 and PC5 (70.5% total variance explained) (Fig. 3D, table S2). The significant proteins on these PCs were enriched for six pathways, all of which were identified by the UC PC regression model except for the cadherin signaling pathway (table S3). Both the UC-TNF-ARE TransComp-R and UC RNA models identified the ITGA4 gene, indicating that mouse α4 integrin subunit activity may be translationally predictive of patient infliximab resistance.
There were 144 DEGs with homologous proteins in the TCT mouse data. These genes resulted in a predictive model using human UC RNA PCs to predict infliximab response, with the most predictive PCs being PC1 and PC3 (59.4% total variance explained) (Fig. 3E, table S2). The significant genes on both UC RNA PCs were enriched for eight pathways, with both PCs enriched for integrin signaling involving ILK, ACTN1, CAV1, and FYN (table S3). The UC-TCT TransComp-R model was slightly less predictive of infliximab response, with the most predictive TCT PCs being PC2 and PC3 (27.0% total variance explained) (Fig. 3E, table S2). Of the nine pathways enriched on the TCT PCs, four were also enriched on the predictive UC RNA PCs (table S3). These pathways included the hypoxia response to HIF activation, p53 pathway by glucose deprivation, p53 pathway feedback loops 2, and the phosphoinositide 3-kinase (PI3K) pathway, indicating that these pathways have both transcriptomic and proteomic relevance to infliximab resistance.
For iCD, neither the human RNA PCs nor the TransComp-R models were significantly predictive of infliximab response regardless of which mouse models were used, suggesting that there must be a signal in the human data being projected for TransComp-R to provide a predictive model. Differences in the coverage and depth of protein homologs for human genes did not appear to be a substantial factor in TransComp-R performance, with the lower coverage TCT data training predictive TransComp-R models in the UC and cCD cases. Although mouse PC1 and PC2 explained a greater proportion of proteomic variance, TransComp-R revealed that the lower-rank PCs were often more predictive of the human therapeutic response (fig. S1, table S2). Whereas the initial mouse PCA models separated mice along PCs describing inflammation and inter-mouse variability, TransComp-R detected the proteomic signal predictive of therapeutic response in humans, a signal not necessarily related to differences between mice. Comparison of PC regression and TransComp-R model P values indicates that in all cases except the UC-TCT TransComp-R model, TransComp-R better separated patients by infliximab response with mouse proteomic PCs than models built with human transcriptomic PCs. This is a strength of the TransComp-R framework: the ability to identify cross-species proteomic signatures that better predict the infliximab response than training comparable models on human transcriptomic data using the same features.
Interrogating cell type–specific integrin signaling signatures of infliximab resistance in cCD
The integrin signaling pathway was significantly activated in infliximab-resistant UC and cCD patients, with different subsets of integrins and integrin-associated proteins implicated on predictive PCs (Fig. 3, tables S2 and S3). In both disease subtypes, immune cell trafficking integrins were identified as being increased in resistant patients in both human RNA PC regression and mouse protein TransComp-R models. Immune cell trafficking integrin signaling has previously been associated with IBD pathobiology and resistance to anti-TNF therapeutics (9, 28–30). TransComp-R also identified a collagen-binding integrin signature increased in infliximab-resistant cCD, an uncharacterized mechanism of resistance to infliximab that we investigated further. TransComp-R was performed on samples containing a mixture of colonic and immune cells, making it challenging to associate resistance signaling with particular cell types. To develop therapeutic strategies to overcome therapeutic resistance, it is necessary to verify the activity of the collagen-binding integrin signaling network in cCD patients and to characterize the cell types responsible. To address these questions, we performed single-cell RNA sequencing (scRNA-seq) on two biopsies from a cCD patient and analyzed a publicly available proteomics dataset of 28 sorted immune cell types (ImmProt) (31).
We mined ImmProt for cell types expressing the cCD integrin signaling network proteins that contribute to infliximab resistance (Fig. 3) (31). Clustering of protein copy numbers revealed cell-specific expression of certain key proteins, including specialization of MAP3K1 to neutrophils, ITGA1 (the α1 integrin subunit) to activated natural killer (NK) cells, and ITGB1 (the β1 integrin subunit) in macrophages and NK cells (Fig. 4A). Whereas broad involvement of macrophages is a hallmark of CD, the specificity of MAP3K1 to neutrophils suggests that this cell type may be a key player in infliximab resistance. Furthermore, the potential for immune cell populations to secrete ligands of integrin α1β1 suggests a range of possible interactions between immune and colonic cell types that may facilitate infliximab resistance in cCD.
Fig. 4. Cell type–specific signaling characterization of the collagen-binding integrin signatures associated with anti-TNF resistance.
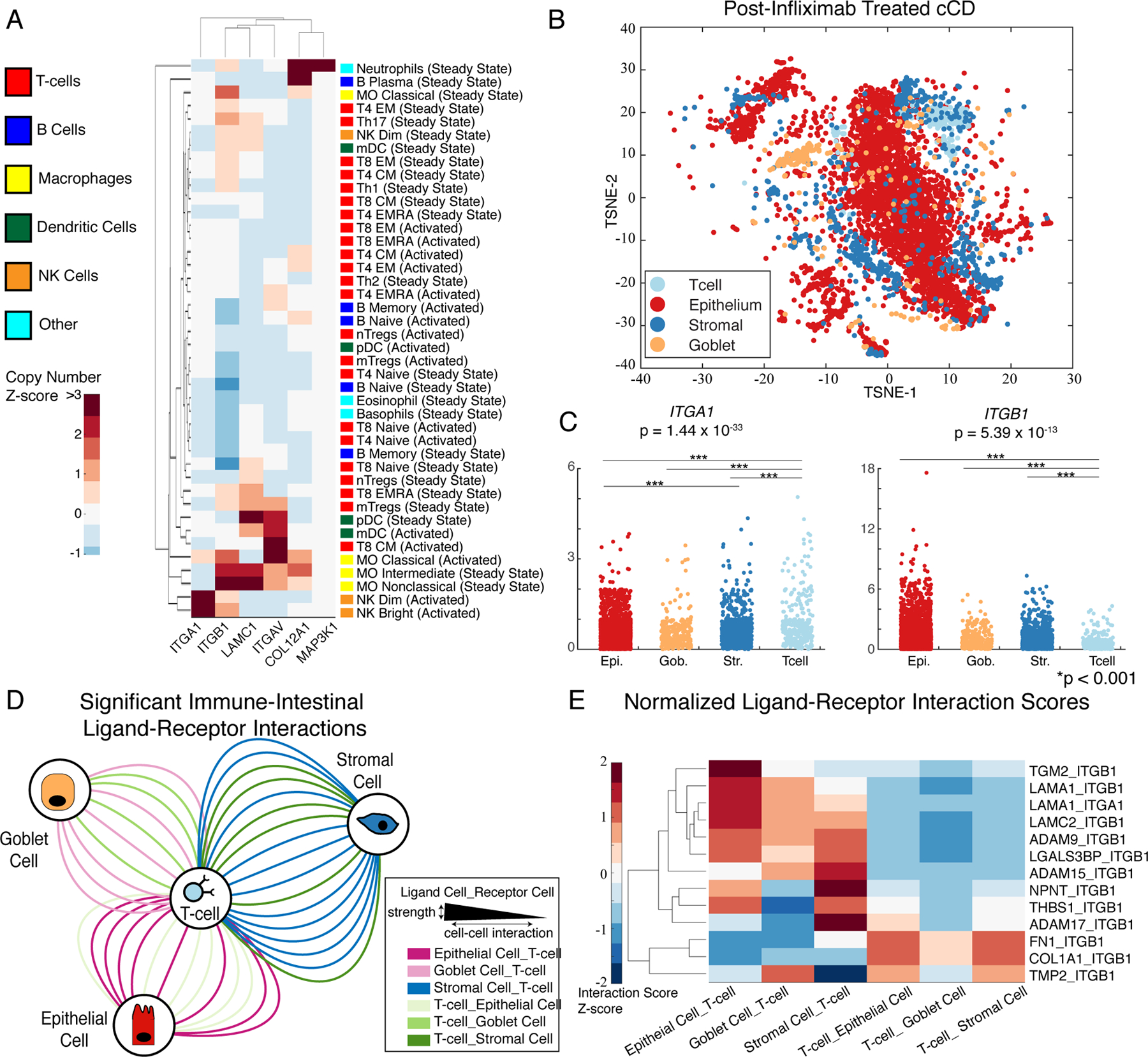
(A) Copy numbers of the indicated proteins from FACS-sorted immune cell populations (N = 3 or 4 donors). (B) TSNE of colonic CD biopsies (N = 5195 cells). (C) Differential expression of the genes ITGA1 and ITGB1 (which encode collagen-binding integrins) in the indicated cell populations [Kruskal-Wallis test; Epi, epithelial; Gob, goblet; Str, stromal (N = 5195 cells)]. (D) Network of significant ligand-receptor (LR) scores between intestinal and immune cell types containing at least one infliximab resistance signature gene (N = 5195 cells). Cell types are connected by edges indicating that a top-ranked (top 10% scores) LR interaction was present, whereas edge thickness indicates interaction strength. (E) Normalized LR scores within each pair of cell-cell interactions (N = 5195 cells).
Having shown that the infliximab resistance–associated integrin signaling network proteins were expressed in immune cells, we profiled this network in the physiological cCD context of mixed immune and colonic cell types. We analyzed the post-infliximab-treated cCD samples from bulk gene expression data to see whether dysregulation in collagen-binding integrin signaling persisted after treatment. Both before and after treatment, the infliximab resistance–associated integrin pathway genes were more highly expressed in infliximab-resistant patients relative to infliximab-sensitive patients (fig. S2). Activity of the genes in this signature after treatment suggests that the collagen-binding integrin signature is a durable feature of infliximab-resistant cCD. We could therefore compare the integrin signaling network from TransComp-R to scRNA-seq data from left and right colonic biopsies obtained from a CD patient after anti-TNF treatment to characterize the cell types and intercellular integrin signaling network.
Cells from left and right colonic biopsies of a cCD patient after anti-TNF treatment were merged to generate a compendium of 5195 cells for analysis. We applied a Gaussian Mixture Model (GMM)–based approach to classify cell types using a set of marker genes curated from the literature (table S4) (32–34). The GMM identified four distinct cell types, including an epithelial cell population, goblet cells, stromal cells, and T cells in each biopsy, which we visualized by t-distributed stochastic neighbor embedding (TSNE) (table S5, Fig. 4B). Although it is possible that other sub-populations of cell types were present within the epithelial category, the accuracy of GMM classification degraded on our data when we tried to predict additional cell types.
We performed differential expression analysis (Kruskal-Wallis test) on the infliximab-resistance signature genes to characterize cell type–specific signaling of the collagen-binding integrin signaling network (table S6, Fig. 4C). In the merged, single-cell dataset, four genes were differentially expressed between cell types, including ITGA1, ITGAV, ITGB1, and RND3. ITGB1 was more highly expressed in intestinal cell types than in T cells and was expressed to a similar extent as that of ITGA1 in T cells. ITGA1 was significantly overexpressed in T cells relative to its expression in intestinal cell types, suggesting that the collagen-binding integrin α1β1 primarily functions through immune cell interactions with intestinal cell types.
Having identified a population of T cells expressing the genes (ITGA1 and ITGB1) encoding the integrin α1β1 as potential mediators of infliximab resistance, we characterized intercellular communication with a scoring algorithm that uses gene expression data to infer potential ligand-receptor (LR) interactions between cells (32). We scored 2567 LR interactions and identified 41 highly ranked interactions (top 10% of all interaction scores) implicating the integrin α1β1 (inferred by expression of ITGA1 or ITGB1). We visualized these interactions between cell types in a network using Cytoscape, with nodes for each cell type and edges for LR interactions, colored by LR directionality (Fig. 4D) (35).
Interactions through the integrin α1β1 were highly prevalent between ITGA1-positive T cells and colonic cell types. The LR interaction inference model showed that ITGA1+ T cells might interact with goblet, epithelial, and stromal cells through intestinal cell type secretion of the laminin, LAMA1, which would interact with the integrin α1β1 (Fig. 4E). Interactions between T cells and stromal cells accounted for the highest number of significant interactions and the highest inferred interaction scores (Fig. 4D). Although some colonic cell types expressed ITGB1 and would be expected to interact with T cells through T cell–secreted ligands, the highest scoring cell-cell interactions between colonic cells and T cells occurred when the ligands were associated with colonic cells and the integrin α1β1 components were associated with T cells (Fig. 4E). However, these interactions were not inferred from proteomics, and do not account for the localization of potentially interacting cells, necessitating further experimental characterization of integrin α1 subunit-mediated signaling.
Activated collagen-binding integrin signaling contributes to infliximab resistance
Having shown by immune cell proteomics and scRNA-seq that the collagen-binding integrin α1β1 was active in cCD patients, we sought to support our hypothesis that activated integrin α1 subunit signaling contributes to resistance to anti-TNF therapy. We treated peripheral blood mononuclear cells from four independent donors with anti-ITGA1, anti-TNFα, and the combination of both antibodies, and then measured cytokine responses at 2, 6, and 10 hours after treatment to assess the role of ITGA1+ immune cells in mediating the anti-inflammatory effects of anti-TNF therapy (Fig. 5, Table S7). Inhibiting ITGA1 alone did not induce significant changes in a panel of 27 cytokines at any time points (Kruskal-Wallis, P < 0.05). Treatment with anti-TNFα antibody alone induced significant inhibition of the production of six cytokines at different times (TNFα, IL-10, IL1-RA, G-CSF, MCP1, and IL-15). In contrast, the combined inhibition of ITGA1 and TNFα induced significant inhibition of between 11 and 26 cytokines over time (Fig. 5). This finding suggests that activated integrin α1 subunit signaling in immune cells contributes to resistance to anti-TNF therapy and that inhibiting integrin α1 enhances the anti-inflammatory effects of anti-TNF therapy.
Fig. 5. Luminex cytokine profiling of PBMCs treated with anti-ITGA1, anti-TNFα, or both.
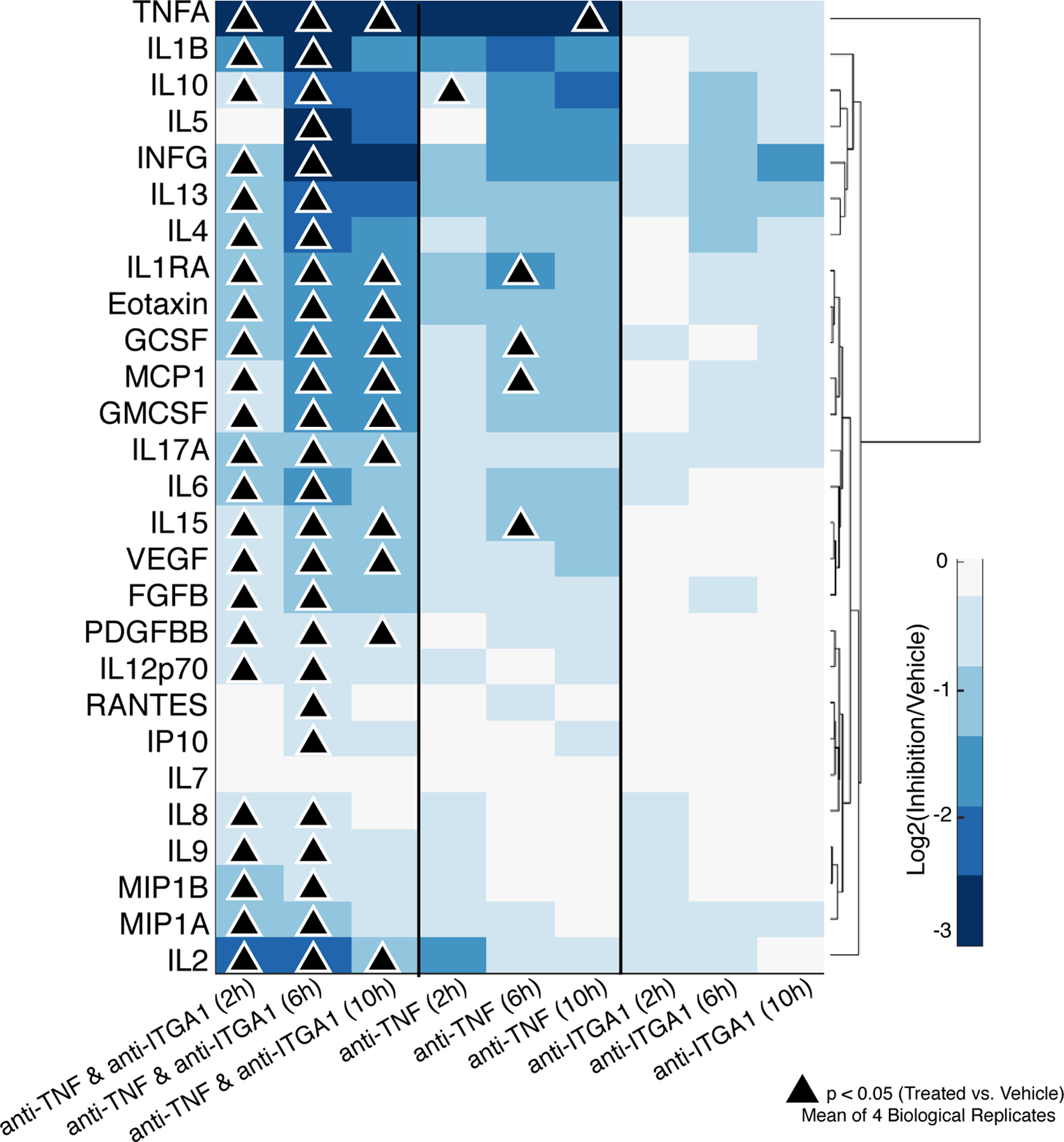
Values indicate a mean of donors (N = 4 biological replicates). P values were determined by Kruskal-Wallis and Wilcoxon Mann-Whitney.
Discussion
The challenge of interspecies translation becomes complicated by discrepancies in phenotypes and molecular data types between clinical cohorts and pre-clinical experimental systems. Here, we demonstrated the utility of the TransComp-R framework for translating proteomic dysregulation in IBD mouse models to a phenotypically and molecularly mismatched clinical cohort of patients to characterize resistance to anti-TNF therapy. We identified a signature for infliximab resistance that linked observations of immune trafficking integrins, laminins, and collagen-binding integrins in IBD to the clinical challenge of overcoming resistance to anti-TNF therapies. Our verification of a collagen-binding integrin anti-TNF therapy resistance network in immune cell proteomics, patient biopsies, and in vitro experiments suggests a larger role for ITGA1+ immune cell signaling in CD pathobiology and biologic therapy resistance.
Together, our results indicate an expanded role for collagen-binding integrin signaling in the clinical phenotype of infliximab-resistant cCD. In healthy colon, ITGA1 expression is localized to colonic crypts in (36), and inhibition of integrin α1β1 components is protective against colitis in the DSS and TNBS mouse models of IBD (37, 38). Previous studies examining tissue-independent markers of memory T cells have also implicated the α1 integrin subunit as a consistent surface marker of tissue-resident memory T cells (39). Our results suggest that collagen-binding integrin signaling associated with infliximab resistance is likely facilitated by a memory T cell population through interactions with colonic cell types. The combined evidence of our scRNA-seq data, ImmProt analysis, and in vitro experiments suggests that the immune cells expressing ITGA1 mediate an intercellular signaling network that facilitates CD disease progression and infliximab resistance.
Our results also indicate that MAP3K1 signaling may be playing a role in infliximab resistance as an intracellular mediator of the extracellular signaling cues from collagen-binding integrin signaling. Activation of α1-containing integrin on the surface of cells is capable of activating MAP3K1 (40–43). Further studies showed that MAP3K1 can activate RAF1, which is itself an activator of the extracellular signal–regulated kinase (ERK) and c-Jun N-terminal kinase (JNK) signaling cascades (44). A small clinical trial of a RAF1 inhibitor showed that targeting the JNK signaling cascade in macrophages could induce remission in cCD and that this could be achieved in infliximab nonresponsive patients (45–47). MAP3K1 also carries a cCD specific SNP, rs832582, which may predispose carriers to a more intense inflammatory response (48). A potential biomarker of infliximab resistance in cCD could therefore be MAP3K1 signaling in neutrophils, stromal cells, or both, as well as potentially the IBD risk SNP rs832582 for MAP3K1. The convergence of multiple signaling disruptions by collagen-binding integrins and MAP3K1 suggests that patients expressing these disease characteristics should be considered for a therapeutic course other than anti-TNF therapy.
A powerful computational feature of TransComp-R is that it identifies mouse proteomic PCs predictive of human phenotypes despite these PCs explaining little variation in the mouse proteomics data. TransComp-R accomplishes this by first assessing whether the projection of human samples into the mouse PC space can produce an overall significantly predictive regression model. If this condition is met, then the top-two most predictive PCs can be interpreted to identify the most translatable biology. In general, a predictive TransComp-R model is not guaranteed and, if produced, the top two PCs may not be individually significantly predictive. Despite this, using top candidate PCs from a predictive TransComp-R model can reveal translational insights that can be validated experimentally, as in the present study.
In standard PCA, latent variables are constructed to explain the variance in the training dataset (mouse proteomics), rather than the relationship of the PCs to a phenotype or to reflect variance of a secondary dataset (human transcriptomics). Because the mouse PCA model was built with data from mice with different phenotypes than the human CD dataset (mouse inflamed vs. uninflamed; human infliximab responder vs. nonresponder), it is not surprising that mouse PC1 and PC2 did not separate projected human phenotypes as well as did lower-ranked mouse PCs. This suggests that the most translatable biology may not be that which most immediately explicates the experimental groups, but instead indicates that a computational modeling approach such as TransComp-R can more insightfully recover translationally relevant biology. Further extensions of TransComp-R may be able to account for different splicing variants, isoforms, and protein posttranslational modifications. In particular, TransComp-R currently requires only using one-to-one mouse-human homologous proteins-genes. To better understand species-specific associations, it may be necessary to incorporate information about homologs that map to multiple genes and proteins across species. Overall, we believe that TransComp-R is widely applicable to challenges of translation in other disease contexts, model systems, and types of molecular data.
Materials and Methods
Analysis of human CD gene expression data
Colonic and ileal CD transcriptomic data were obtained from the gene expression omnibus (GEO), accession number GSE16879, using Bioconductor tools and normalized by the robust multichip average method (4, 18, 49, 50). Differential expression analysis was performed using the Wilcoxon Mann-Whitney test with Benjamini Hochberg False Discovery Rate (FDR) correction and significance defined by q < 0.25. PC regression was used on the entire gene expression dataset to identify human RNA PCs predictive of therapeutic response, with PANTHER pathway enrichment performed on highly loaded proteins on predictive PCs (19–21).
Analysis of mouse proteomics datasets
The TCT and TNF-ARE mouse proteomics datasets were obtained from two studies examining proteomic changes between inflamed and uninflamed mice (10, 11). The mouse protein identifiers were mapped to their coding genes and converted to human gene symbols using the Mouse Genome Informatics databases (51, 52). Only one-to-one mouse-human homologs were retained for the analysis.
Translatable components regression
When constructing a PCA model, it is often desirable to project observations from another dataset into that model to examine how the variability explained by the model relates to those new observations. This requires normalizing the new observation, usually by mean-centering and scaling by the standard deviation (SD) of the data used to train the PCA model. However, if the new observation is measured on a different sequencing platform, comes from a different species, or is of a different molecular data type, then this centering and scaling by training data factors is not well defined and may distort the projected observation. Therefore, cross-species, cross-omic, and cross-platform projections of biological datasets and observations should not be undertaken by the standard PCA projection method.
The primary component of a PCA model is identification of the eigenvectors of the covariance matrix of the training data, that is, the PCs that explain the greatest possible amount of variability in the training dataset. Although these vectors define a basis that has a particular interpretation for the training dataset, we can ask how the new observations project relative to this coordinate system. This is done by first internally normalizing the new observations by their own mean and SD, to define the relative spread of each variable, and then multiplying these normalized observations by the eigenvectors of the training dataset. Once projected, we performed PC regression of the projected data against any outcome or phenotypic variable of the new observations to identify the PCs of the training data that best explained the phenotype from the new observations. Here, we performed TransComp-R for a given mouse model and human IBD cohort pairing and assessed whether the model was significantly predictive of patient infliximab response based upon the regression P value. If the overall model was significant, then we interpreted the two most significant mouse PCs to identify proteins that were significantly loaded on the top two PCs. If the overall model P value was not significant, then we considered the model not to be predictive and we did not interpret the PCs.
Immune cell proteomics analysis
Quantitative proteomics data from FACS-sorted cells were obtained from the study of Rieckmann et al. (31) and analyzed for protein copy numbers of significantly loaded integrin pathway proteins identified by TransComp-R (Fig. 5). Immune cell populations not expressing any protein from the network were excluded, together with proteins not measured in the dataset. Data were z-score–normalized by protein and clustered to identify groups of coregulated proteins expressed in similar immune cell types. Analysis was performed in MATLAB_R2018b.
Collection of patient samples
The study protocol was approved by the Institutional Review Board at Vanderbilt University Medical Center. Written informed consent was obtained for analysis of demographics, medication history, serum, and tissue biopsies obtained at the time of endoscopic procedures as part of routine clinical care to evaluate for disease activity and response to therapy in a patient with ileo-colonic CD. The patient underwent an overnight fast and received polyethylene glycol electrolyte solution for bowel preparation before colonoscopy. At the time of colonoscopy, biopsy specimens were obtained from the right and left colon (two bites in each location with large capacity biopsy forceps). The specimens were placed in a 1.5-ml Eppendorf tube with RPMI medium, placed on ice, and transported to the lab for further processing for scRNA-seq analysis.
Tissue processing
Biopsies were delivered from endoscopy in cold RPMI and were transferred to DPBS (without Ca or Mg) with 4 mM EDTA and 0.5mM DTT to chelate for 1 hour before being lightly triturated in DPBS. Tissues were then resuspended in DPBS containing cold-active protease (5 mg/ml, Sigma) with DNase (2.5 mg/ml, Sigma) and incubated for 20 min at 4 to 6˚C with a rocking motion. Trituration with a P1000 pipette needle was performed on the dissociated suspension to yield single cells, which were then filtered through a 35-μm mesh and washed into DPBS. Essentially, the entire specimen was dissociated and used for subsequent steps. Live cell density was counted based on the number of Trypan Blue–positive cells. Cells were adjusted to a density of 150,000 cells/ml and Optiprep was added to a final concentration of 16% immediately before encapsulation.
inDrop single-cell RNA-seq
Single-cell encapsulation of gut epithelial tissue was performed using the inDrop platform (1CellBio) with an in vitro transcription library preparation protocol, as previously described (53, 54). The inDrop platform uses CEL-Seq in preparation for sequencing and is summarized as follows: (i) reverse transcription (RT); (ii) ExoI nuclease digestion; (iii) SPRI purification (SPRIP); (iv) second-strand synthesis; (v) SPRIP; (vi) T7 in vitro transcription linear amplification; (vii) SPRIP; (viii) RNA fragmentation; (ix) SPRIP; (x) primer ligation, (xi) RT; and (xii) library enrichment PCR. The number of cells encapsulated was calculated by approximating the density of the single-cell suspension multiplied by the bead loading efficiency during the duration of encapsulation. Approximately 3000 cells for each sample entered the microfluidic chip. After library preparation, which was performed as described earlier, the samples were sequenced using Nextseq 500 (Illumina) with a 150-bp paired-end sequencing kit in a customized sequencing run. After sequencing, reads were filtered, sorted by their designated barcode, and aligned to the reference transcriptome using the InDrop pipeline (55, 56).
Single-cell filtering
scRNA-seq count data were filtered using several steps. First, the cumulative read inflection point was plotted. A cutoff of approximately 25 to 30% beyond the inflection point was used to exclude low-quality barcodes but retain cells with small library sizes. The filtered data were then normalized for library size, transformed, and visualized using t-SNE with 100 PCs. Density peak clustering was performed, and user-defined thresholds were set to obtain 10 to 20 clusters. Library size rank and combined mitochondrial gene expression were overlaid onto the t-SNE space, and low-quality cells were removed using these criteria. Canonical marker genes of cell types were also overlaid onto the t-SNE space to ensure that cell types of interest were not removed during filtering.
Cell type classification and ligand-receptor interaction scoring
Cell type markers were selected from previous single-cell analyses of colonic and intestinal tissue contexts and used to train a Gaussian mixture model (GMM) on the log-normalized expression data as previously described (32–34). Differential expression analysis was performed using the Kruskal-Wallis test (P < 0.05) on infliximab resistance marker genes. We then characterized the intercellular signaling network of ligand-receptor (LR) interactions between identified cell types by assigning a score based on the product of average receptor abundance in a cell type with the average ligand abundance in the interacting cell type as previously described (32). LR interaction scores in the top 10% of all interaction scores across cell types that contained at least one infliximab resistance signature gene were retained for downstream analysis and interpretation.
PBMC stimulation experiments
Peripheral blood mononuclear cells (PBMCs) were isolated from fresh whole blood (male) from human donors (Research Blood Components, Watertown MA.) and cryobanked in liquid nitrogen until use, as described previously (57). Frozen PBMCs were thawed, seeded at 2 × 106/ml in duplicate wells, and stimulated with PMA/ionomycin Cell Stimulation Cocktail (Thermofisher cat# 00-4970-93), in the presence or absence of anti-TNF (Bio-rad cat#MCA6090) with or without anti-ITGA1 (EMD Millipore cat# MAB1973, clone-FB12). Cells were cultured in RPMI, 10% heat-inactivated FBS, 2 mM Glutamax (Thermofisher cat#35050061). After incubation for 2, 6, or 10 hours, the cells were centrifuged and the supernatant retained for Luminex analysis. Conditioned medium was diluted three-fold in sample buffer + 2%(w/v) BSA (Bioplex 27-plex cat# MK500KCAF0Y) and assayed as previously described (57). Cytokine amounts were calculated with Bioplex Manager software v6.1.
Supplementary Material
Fig. S1. Comparison of high-rank and TransComp-R principal components.
Fig. S2. Comparison of integrin pathway genes before and after treatment with infliximab.
Table S3. TransComp-R and PC regression pathway enrichment.
Table S2. TransComp-R and PC regression coefficients.
Table S1. Human RNA pathway enrichment.
Table S4. Cell type classification markers.
Table S5. GMM training and cross-validation performance.
Table S6. Single-cell DEG results.
Table S7. Luminex data for anti-ITGA1 and anti-TNF stimulation experiments.
Acknowledgments:
The authors wish to thank J. Hambor for helpful discussions on the methods and manuscript.
Funding: D.K.B. received funding from the Research Beyond Borders SHINE (Strategic Hub for Innovation New Therapeutic Concept Exploration) program of Boehringer Ingelheim Pharmaceuticals. E.A.S. was funded by KL2TR002245 (National Center for Advancing Translational Sciences) and a DDRC Pilot and Feasibility Grant from the National Institute of Diabetes and Digestive and Kidney Diseases (P30DK058404). K.S.L. was funded by NIH grant R01DK103831. D.A.L. received funding from the US Army Research Office Institute for Collaborative Biotechnologies (Grant: W911NF-19-2-0026).
Footnotes
Competing interests: The authors declare that they have no competing interests.
Data and materials availability: Single-cell data were deposited in the Gene Expression Omnibus (GEO Accession Number GSE141553). The TransComp-R code has been deposited at Mathworks File Exchange (Number 77987). All other data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.
References and Notes
- 1.Ng SC et al. , Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population-based studies. Lancet 390, 2769–2778 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Schmitt H et al. , Expansion of IL-23 receptor bearing TNFR2+ T cells is associated with molecular resistance to anti-TNF therapy in Crohn’s disease. Gut (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ben-Horin S, Kopylov U, Chowers Y, Optimizing anti-TNF treatments in inflammatory bowel disease. Autoimmun Rev 13, 24–30 (2014). [DOI] [PubMed] [Google Scholar]
- 4.Arijs I et al. , Mucosal gene expression of antimicrobial peptides in inflammatory bowel disease before and after first infliximab treatment. PLoS One 4, e7984 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Halloran B et al. , Molecular patterns in human ulcerative colitis and correlation with response to infliximab. Inflamm Bowel Dis 20, 2353–2363 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mesko B et al. , Peripheral blood derived gene panels predict response to infliximab in rheumatoid arthritis and Crohn’s disease. Genome Med 5, 59 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gaujoux R et al. , Cell-centred meta-analysis reveals baseline predictors of anti-TNFalpha non-response in biopsy and blood of patients with IBD. Gut (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peters LA et al. , A functional genomics predictive network model identifies regulators of inflammatory bowel disease. Nat Genet 49, 1437–1449 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arijs I et al. , Effect of vedolizumab (anti-alpha4beta7-integrin) therapy on histological healing and mucosal gene expression in patients with UC. Gut 67, 43–52 (2018). [DOI] [PubMed] [Google Scholar]
- 10.Lyons J et al. , Integrated in vivo multiomics analysis identifies p21-activated kinase signaling as a driver of colitis. Sci Signal 11 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Strasser SD et al. , Substrate-based kinase activity inference identifies MK2 as driver of colitis. Integr Biol (Camb) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brubaker DK, Proctor EA, Haigis KM, Lauffenburger DA, Computational translation of genomic responses from experimental model systems to humans. PLoS Comput Biol 15, e1006286 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brubaker DK et al. , Proteogenomic Network Analysis of Context-Specific KRAS Signaling in Mouse-to-Human Cross-Species Translation. Cell Syst 9, 258–270 e256 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Normand R et al. , Found In Translation: a machine learning model for mouse-to-human inference. Nat Methods 15, 1067–1073 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Poussin C et al. , The species translation challenge-a systems biology perspective on human and rat bronchial epithelial cells. Sci Data 1, 140009 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Seok J, Evidence-based translation for the genomic responses of murine models for the study of human immunity. PLoS One 10, e0118017 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brubaker DK, Lauffenburger DA, Translating preclinical models to humans. Science 367, 742–743 (2020). [DOI] [PubMed] [Google Scholar]
- 18.Edgar R, Domrachev M, Lash AE, Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 30, 207–210 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thomas PD et al. , PANTHER: a library of protein families and subfamilies indexed by function. Genome Res 13, 2129–2141 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ashburner M et al. , Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25, 25–29 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gene Ontology C, Gene Ontology Consortium: going forward. Nucleic Acids Res 43, D1049–1056 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arijs I et al. , Mucosal gene signatures to predict response to infliximab in patients with ulcerative colitis. Gut 58, 1612–1619 (2009). [DOI] [PubMed] [Google Scholar]
- 23.Drobin K et al. , Targeted Analysis of Serum Proteins Encoded at Known Inflammatory Bowel Disease Risk Loci. Inflamm Bowel Dis 25, 306–316 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meuwis MA et al. , Proteomics for prediction and characterization of response to infliximab in Crohn’s disease: a pilot study. Clin Biochem 41, 960–967 (2008). [DOI] [PubMed] [Google Scholar]
- 25.Townsend P et al. , Serum Proteome Profiles in Stricturing Crohn’s Disease: A Pilot Study. Inflamm Bowel Dis 21, 1935–1941 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Abreu MT et al. , Measurement of vitamin D levels in inflammatory bowel disease patients reveals a subset of Crohn’s disease patients with elevated 1,25-dihydroxyvitamin D and low bone mineral density. Gut 53, 1129–1136 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reich KM, Fedorak RN, Madsen K, Kroeker KI, Role of Vitamin D in Infliximab-induced Remission in Adult Patients with Crohn’s Disease. Inflamm Bowel Dis 22, 92–99 (2016). [DOI] [PubMed] [Google Scholar]
- 28.Banse C, Armengol-Debeir L, Vittecoq O, Effectiveness of vedolizumab for Crohn’s disease with spondyloarthritis in fail with two TNF blocking agents. Joint Bone Spine (2017). [DOI] [PubMed] [Google Scholar]
- 29.Barre A, Colombel JF, Ungaro R, Review article: predictors of response to vedolizumab and ustekinumab in inflammatory bowel disease. Aliment Pharmacol Ther 47, 896–905 (2018). [DOI] [PubMed] [Google Scholar]
- 30.Fuchs F et al. , Clinical Response to Vedolizumab in Ulcerative Colitis Patients Is Associated with Changes in Integrin Expression Profiles. Front Immunol 8, 764 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rieckmann JC et al. , Social network architecture of human immune cells unveiled by quantitative proteomics. Nat Immunol 18, 583–593 (2017). [DOI] [PubMed] [Google Scholar]
- 32.Kumar MP et al. , Analysis of Single-Cell RNA-Seq Identifies Cell-Cell Communication Associated with Tumor Characteristics. Cell Rep 25, 1458–1468 e1454 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kinchen J et al. , Structural Remodeling of the Human Colonic Mesenchyme in Inflammatory Bowel Disease. Cell 175, 372–386 e317 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Parikh K et al. , Colonic epithelial cell diversity in health and inflammatory bowel disease. Nature 567, 49–55 (2019). [DOI] [PubMed] [Google Scholar]
- 35.Shannon P et al. , Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13, 2498–2504 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boudjadi S, Carrier JC, Groulx JF, Beaulieu JF, Integrin alpha1beta1 expression is controlled by c-MYC in colorectal cancer cells. Oncogene 35, 1671–1678 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Krieglstein CF et al. , Collagen-binding integrin alpha1beta1 regulates intestinal inflammation in experimental colitis. J Clin Invest 110, 1773–1782 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fiorucci S et al. , Importance of innate immunity and collagen binding integrin alpha1beta1 in TNBS-induced colitis. Immunity 17, 769–780 (2002). [DOI] [PubMed] [Google Scholar]
- 39.Kumar BV et al. , Human Tissue-Resident Memory T Cells Are Defined by Core Transcriptional and Functional Signatures in Lymphoid and Mucosal Sites. Cell Rep 20, 2921–2934 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Szklarczyk D et al. , STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res 43, D447–452 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kanehisa M, Goto S, KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28, 27–30 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K, KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res 45, D353–D361 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kanehisa M, Sato Y, Furumichi M, Morishima K, Tanabe M, New approach for understanding genome variations in KEGG. Nucleic Acids Res 47, D590–D595 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Karandikar M, Xu S, Cobb MH, MEKK1 binds raf-1 and the ERK2 cascade components. J Biol Chem 275, 40120–40127 (2000). [DOI] [PubMed] [Google Scholar]
- 45.Lowenberg M et al. , Specific inhibition of c-Raf activity by semapimod induces clinical remission in severe Crohn’s disease. J Immunol 175, 2293–2300 (2005). [DOI] [PubMed] [Google Scholar]
- 46.Hommes D et al. , Inhibition of stress-activated MAP kinases induces clinical improvement in moderate to severe Crohn’s disease. Gastroenterology 122, 7–14 (2002). [DOI] [PubMed] [Google Scholar]
- 47.Waetzig GH, Rosenstiel P, Nikolaus S, Seegert D, Schreiber S, Differential p38 mitogen-activated protein kinase target phosphorylation in responders and nonresponders to infliximab. Gastroenterology 125, 633–634; author reply 635–636 (2003). [DOI] [PubMed] [Google Scholar]
- 48.Morrell ED et al. , Genetic Variation in MAP3K1 Associates with Ventilator-Free Days in Acute Respiratory Distress Syndrome. Am J Respir Cell Mol Biol 58, 117–125 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gentleman RC et al. , Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 5, R80 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Irizarry RA et al. , Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4, 249–264 (2003). [DOI] [PubMed] [Google Scholar]
- 51.Blake JA et al. , Mouse Genome Database (MGD)-2017: community knowledge resource for the laboratory mouse. Nucleic Acids Res 45, D723–D729 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Eppig JT et al. , Mouse Genome Informatics (MGI): reflecting on 25 years. Mamm Genome 26, 272–284 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Herring CA et al. , Unsupervised Trajectory Analysis of Single-Cell RNA-Seq and Imaging Data Reveals Alternative Tuft Cell Origins in the Gut. Cell Syst 6, 37–51 e39 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu Q et al. , Quantitative assessment of cell population diversity in single-cell landscapes. PLoS Biol 16, e2006687 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Klein AM et al. , Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 161, 1187–1201 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zilionis R et al. , Single-cell barcoding and sequencing using droplet microfluidics. Nat Protoc 12, 44–73 (2017). [DOI] [PubMed] [Google Scholar]
- 57.Schrier SB, Hill AS, Plana D, Lauffenburger DA, Synergistic Communication between CD4+ T Cells and Monocytes Impacts the Cytokine Environment. Scientific Reports 6, 34942 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Comparison of high-rank and TransComp-R principal components.
Fig. S2. Comparison of integrin pathway genes before and after treatment with infliximab.
Table S3. TransComp-R and PC regression pathway enrichment.
Table S2. TransComp-R and PC regression coefficients.
Table S1. Human RNA pathway enrichment.
Table S4. Cell type classification markers.
Table S5. GMM training and cross-validation performance.
Table S6. Single-cell DEG results.
Table S7. Luminex data for anti-ITGA1 and anti-TNF stimulation experiments.