

Abstract
Invasion, metastasis and therapy resistance are the major cause of cancer‐associated deaths, and the EMT‐inducing transcription factor ZEB1 is a crucial stimulator of these processes. While work on ZEB1 has mainly focused on its role as a transcriptional repressor, it can also act as a transcriptional activator. To further understand these two modes of action, we performed a genome‐wide ZEB1 binding study in triple‐negative breast cancer cells. We identified ZEB1 as a novel interactor of the AP‐1 factors FOSL1 and JUN and show that, together with the Hippo pathway effector YAP, they form a transactivation complex, predominantly activating tumour‐promoting genes, thereby synergising with its function as a repressor of epithelial genes. High expression of ZEB1, YAP, FOSL1 and JUN marks the aggressive claudin‐low subtype of breast cancer, indicating the translational relevance of our findings. Thus, our results link critical tumour‐promoting transcription factors: ZEB1, AP‐1 and Hippo pathway factors. Disturbing their molecular interaction may provide a promising treatment option for aggressive cancer types.
Keywords: AP‐1, breast cancer, epithelial to mesenchymal transition, ZEB1
Subject Categories: Cancer; Chromatin, Epigenetics, Genomics & Functional Genomics
Aggressive cellular states in human breast cancer are defined by context‐dependent interplay between transcriptional regulators ZEB1, AP‐1 and YAP.
Introduction
The transcription factor ZEB1 (zinc finger E‐box binding homeobox 1) is an activator of the embryonic epithelial to mesenchymal transition (EMT) programme, which, when hijacked by cancer cells, is considered a major driver of tumour progression (De Craene & Berx, 2013; Stemmler et al, 2019). Importantly, EMT is not a one‐way road but a highly dynamic and reversible process, providing cancer cells with the plasticity needed to cope with the different challenges on their way to distant metastasis formation (Chaffer et al, 2016). In addition to ZEB1, EMT can also be induced by other core EMT‐transcription factors (EMT‐TFs), namely ZEB2, TWIST1 and the two members of the Snail family, which most likely all have non‐redundant subfunctions (Stemmler et al, 2019; Yang et al, 2020). Notably, these core EMT‐TFs not only exert “classical” EMT properties, like loss of epithelial integrity and increased motility, but also confer other properties important for cancer progression, including stemness, survival and therapy resistance (Nieto et al, 2016).
The core EMT‐TF ZEB1 was shown to be particularly important for tumorigenicity and metastasis, by triggering the combined activation of cell motility and stemness properties (Vandewalle et al, 2009; Sanchez‐Tillo et al, 2011; Krebs et al, 2017). Its expression mediates aggressiveness, metastasis and therapy resistance in many different cancer types (Zhang et al, 2015; Stemmler et al, 2019). This includes epithelial cancers like breast and pancreatic cancer, but also non‐epithelial tumours, like glioblastoma (Karihtala et al, 2013; Siebzehnrubl et al, 2013; Bronsert et al, 2014; Kahlert et al, 2015; Lehmann et al, 2016; Krebs et al, 2017). In breast cancer, ZEB1 is highly expressed specifically in a fraction of the aggressive triple‐negative subtype (Karihtala et al, 2013; Lehmann et al, 2016).
ZEB1 can control target gene transcription by different modes of action. It binds to DNA via the two zinc finger clusters at its N‐ and C‐terminal ends, with each cluster binding to an individual E‐box motif (Remacle et al, 1999; Balestrieri et al, 2018). At least one E‐box needs to be a high‐affinity motif with the consensus sequence CAGGTG/A (Remacle et al, 1999). By recruiting additional cofactors, such as CtBP, ZEB1 represses the expression of epithelial genes (Postigo & Dean, 1999; Vandewalle et al, 2009). In addition to its role as a transcriptional repressor, ZEB1 has also been reported to act as a transcriptional activator (Gheldof et al, 2012). One important mechanism enabling its dual function as a transcriptional repressor and as an activator appears to be cooperativity with different transcription factor complexes (Gubelmann et al, 2014; Sanchez‐Tillo et al, 2015; Lehmann et al, 2016; Rosmaninho et al, 2018). We recently described that a selected set of tumour‐promoting target genes is activated by ZEB1 together with the Hippo pathway effectors YAP/TEAD (Lehmann et al, 2016). In a genome‐wide study using glioblastoma cell lines, ZEB1 was shown to cooperate with factors of the LEF/TCF family to activate expression of tumour‐promoting genes in brain cancer (Rosmaninho et al, 2018).
We here investigated transcriptional control by ZEB1 on a genome‐wide level in aggressive breast cancer cells to further dissect ZEB1 modes of action in a cancer of epithelial origin. In this context, we could not see any cooperation with LEF/TCF factors in this tumour type, but identified AP‐1 factors as novel partners of ZEB1 in activating cancer‐promoting genes together with YAP/TEAD.
Results
ZEB1 exhibits overlapping DNA binding with the AP‐1 factor JUN
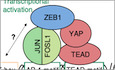
To gain genome‐wide insights into ZEB1 modes of action, we performed ChIP‐seq to analyse its binding pattern using the triple‐negative breast cancer cell line MDA‐MB‐231 as a model. We detected ZEB1 binding at 12,617 genomic sites (Fig EV1A), including promoter regions of many established ZEB1‐repressed target genes conferring an epithelial phenotype such as CDH1 or CRB3 (Fig EV1B; Aigner et al, 2007; Eger et al, 2005), as well as known ZEB1 activated genes such as the protumorigenic factors CTGF and CYR61 (Fig EV1C) (Lazarova et al, 2001; Lai et al, 2011; Lehmann et al, 2016). Furthermore, gene annotation of binding peaks in promoter regions and integration with gene expression data of ZEB1 knockdown compared to control MDA‐MB‐231 cells (Lehmann et al, 2016) corroborated both direct repressive and direct activating functions of ZEB1 (Fig EV1D and E). As expected, known motif discovery analysis retrieved the canonical ZEB1‐binding motif as the highest enriched motif in the ZEB1 peaks (Fig 1A). Sixty‐eight percent of the binding sites contained a high‐affinity ZEB1 motif (Fig 1A, Appendix Table S1), which was specifically enriched within 100 bp around the peak summit and as such located in a good position to mediate DNA binding of ZEB1 (Fig EV1F). In addition to this motif analysis, which relies on already identified DNA binding motifs, a de novo motif analysis was also performed to search for DNA motifs without relying on prior knowledge. This analysis identified a degenerated low affinity ZEB1 motif that was found at even higher frequency than the canonical ZEB1 motif (Appendix Table S2).
Figure EV1. ZEB1 ChIP‐seq in MDA‐MB‐231 cells.
- Heatmap representing the signal enrichment of ZEB1 ChIP‐seq and of control input DNA reads in 2 kb regions centred on the ZEB1 peak summits. Reads are clearly enriched in the peak regions in the ZEB1 ChIP‐seq but not in the control input DNA.
- Genome browser tracks of ZEB1 ChIP‐seq signal intensity showing specific enrichment at the promoter regions of known ZEB1‐repressed targets.
- Genome browser tracks of ZEB1 ChIP‐seq signal intensity showing specific enrichment at the promoter regions of known ZEB1‐activated targets.
- Distribution of ZEB1 peaks with respect to different genomic features. Promoters were defined as 1.5 kb upstream to 0.5 kb downstream of the TSS. Promoter peaks were annotated to their respective genes and integrated with transcriptome data of ZEB1 knockdown compared to control MDA‐MB‐231 cells to identify peaks associated with activated/repressed genes. Genes that were downregulated in the ZEB1 knockdown compared to control MDA‐MB‐231 cells were named “activated”, meaning activated in the presence of ZEB1. Genes that were upregulated after ZEB1 knockdown were called “repressed”, meaning repressed in the presence of ZEB1.
- Hypergeometric testing showed that ZEB1 promoter peaks are significantly associated with both, genes that are activated and genes that are repressed in the presence of ZEB1. Odds ratio (OR): measure of association between categorical variables with OR > 1 indicating a positive association. P‐value from Fisher's exact test.
- Enrichment profile of the consensus high‐affinity ZEB1 DNA binding motif in 600‐bp regions centred on the summits of ZEB1 peaks. The two grey dashed lines highlight the 200‐bp regions around the ZEB1 peak summits that were used for known and de novo HOMER motif analysis.
- Overlap between ZEB1 and YAP DNA binding sites in MDA‐MB‐231 cells. Sites are counted as overlapping when their peak summit positions are not more than 200 bp apart. P‐value from permutation test.
- Enrichment profile of YAP peak summit positions in 1 kb regions centred on the ZEB1 peak summits. The two grey dashed lines highlight the region of ± 200 bp around the ZEB1 peak summit positions where YAP peaks are counted as overlapping with ZEB1 peaks.
- Heatmap representing the signal enrichment of YAP ChIP‐seq reads in 2 kb regions centred on the ZEB1 peak summits.
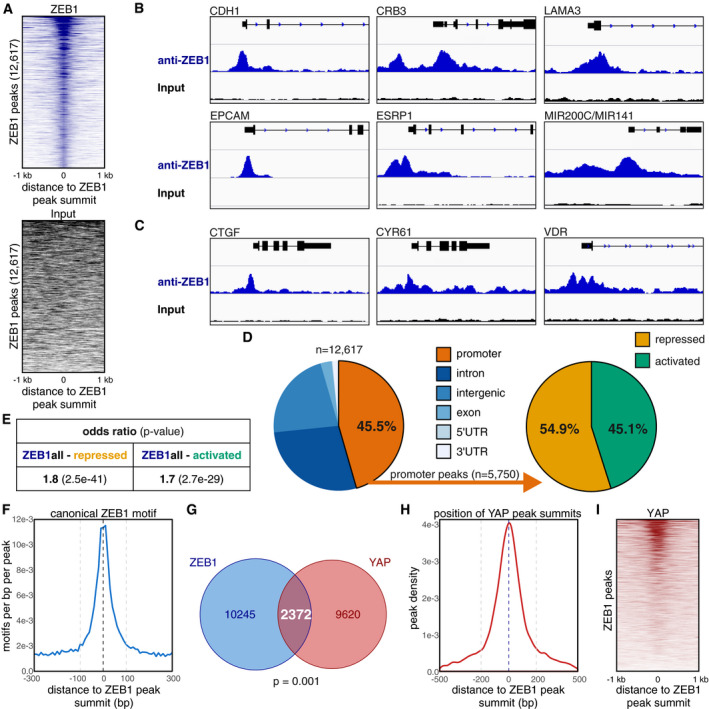
Figure 1. ZEB1 DNA binding sites overlap with YAP‐ and JUN‐binding sites.
- The three top enriched DNA binding motifs identified by HOMER known motif analysis on 200 bp regions centred on the ZEB1 peak summits. An extended list can be found in Appendix Table S1.
- Enrichment profiles of TEAD4 and AP‐1 consensus motifs in 600 bp regions centred on the ZEB1 peak summits. The two grey dashed lines highlight the 200 bp regions around the ZEB1 peak summits which were used for HOMER known motif analysis shown in (A).
- Overlap between ZEB1 and JUN DNA binding sites. Sites are counted as overlapping when their peak summit positions are not more than 200 bp apart. P‐value from permutation test.
- Enrichment profile of JUN peak summit positions in 1 kb regions centred on the ZEB1 peak summits. The two grey dashed lines highlight the region of ± 200 bp around the ZEB1 peak summit positions where JUN peaks are counted as overlapping with ZEB1 peaks.
- Heatmap representing the signal enrichment of JUN ChIP‐seq reads in 2 kb regions centred on the ZEB1 peak summits.
- Circle plot displaying the genome‐wide distribution of ZEB1 (outer middle circle, blue), JUN (inner middle circle, green) and YAP (inner circle, red) peaks across all chromosomes (outer circle). Peak density is visualised as a heatmap with more intense colours indicating a higher number of peaks in a specific location.
- ZEB1, JUN and YAP DNA binding overlaps at 1,993 genomic sites in MDA‐MB‐231 cells. Triple overlap is assumed when both JUN and YAP peak summit positions are within 200 bp of a ZEB1 peak summit position. P‐value from permutation test.
Strikingly, the second top enriched motif within the ZEB1 peaks was the consensus binding site for AP‐1 transcription factors. This motif was still present in more than 1/5 of all peaks (Fig 1A, Appendix Table S1), and similarly to the classical ZEB1‐binding motif, it was overrepresented within 100 bp around the peak summit (Fig 1B). This enrichment of AP‐1 motifs at the ZEB1 peak summit indicates joint binding of AP‐1 factors together with ZEB1 at the same genomic sites.
To investigate this hypothesis, we compared the ZEB1‐binding sites to a published ChIP‐seq data set of the AP‐1 factor JUN (also known as c‐JUN), also generated in MDA‐MB‐231 cells (Zanconato et al, 2015). More than 33% of the ZEB1 peaks overlap with sites of JUN‐binding (4,201 out of 12,617) (Fig 1C). Furthermore, JUN peak summits coincide with ZEB1 peak summits (Fig 1D) and JUN ChIP‐seq signal intensity increases around sites of ZEB1 binding (Fig 1E). These data suggest a cooperation between ZEB1 and the AP‐1 factor JUN.
The third top enriched motif was the TEAD‐binding motif, the main platform via which YAP binds to DNA (Zhao et al, 2008; Fig 1A, Appendix Table S1). The similar confined enrichment around the ZEB1 peak summit (Fig 1B) is consistent with the concept that ZEB1 can form a transcriptional activator complex together with the Hippo pathway effector YAP on genome‐wide level. Notably, in contrast to glioblastoma (Rosmaninho et al, 2018), we did not detect a significant enrichment of the LEF/TCF motif within the ZEB1 peaks in the breast cancer cells (Appendix Table S1).
To investigate whether ZEB1, AP‐1 and YAP bind all together or ZEB1 with either YAP or AP‐1 individually, we compared the DNA binding patterns of all three factors. To this end, we first verified the genome‐wide distribution of all three factors including a published data set of YAP binding peaks also generated by ChIP‐seq in MDA‐MB‐231 cells (Zanconato et al, 2015; Fig 1F). When we assessed the overlap of the ZEB1 and YAP peaks, we found that approximately 19% of the ZEB1‐binding sites overlap with YAP‐binding (2,372 out of 12,617) (Fig EV1G) and that YAP peak summits coincide with ZEB1 peak summits (Fig EV1H and I). Comparison among all three ChIP‐seq data sets showed that the largest fraction of common ZEB1‐YAP sites is additionally bound by JUN (approx. 84%; 1,993 out of 2,372) (Fig 1G). Moreover, approximately half of the common ZEB1‐JUN sites also showed YAP‐binding (1,993 out of 4,201). These findings suggest a genome‐wide cooperation of the EMT inducer ZEB1 with the Hippo effector YAP and the AP‐1 factor JUN in breast cancer. A cooperation of ZEB1 with LEF/TCF factors, as shown in glioblastoma, was not detected.
ZEB1 interacts with the AP‐1 factors JUN and FOSL1
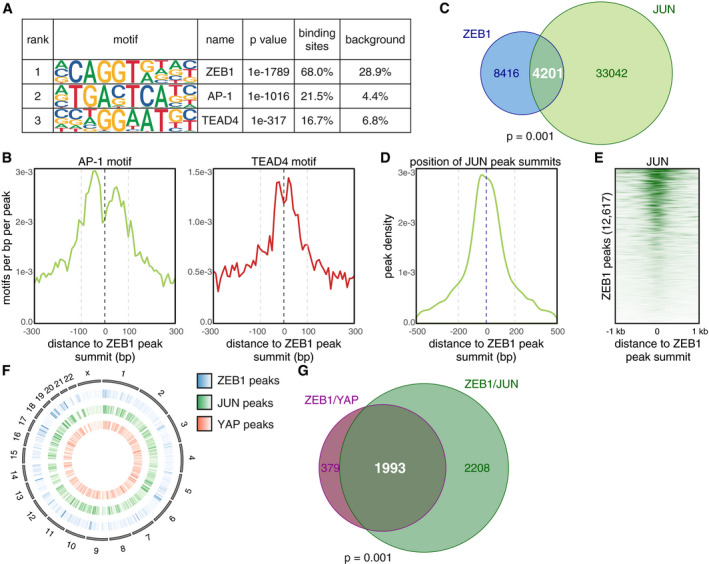
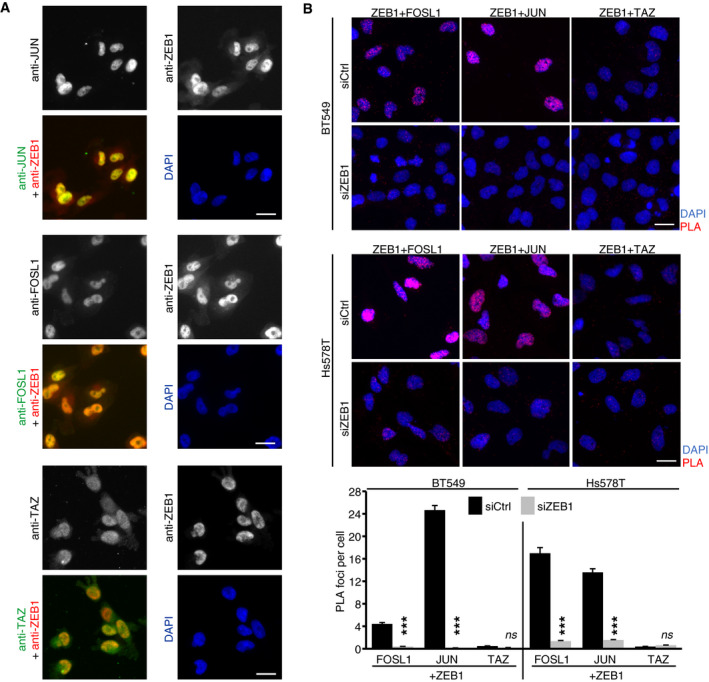
To identify whether ZEB1 and AP‐1 factors physically interact, we first tested if both ZEB1 and JUN are co‐expressed in MDA‐MB‐231 cells. We included the JUN co‐factor FOSL1 (also known as FRA‐1) in our analysis due to its known role in cancer progression and metastasis (Bakiri et al, 2015). Immunofluorescence labelling showed nuclear co‐localisation of ZEB1 with JUN and FOSL1 in MDA‐MB‐231 cells (Fig EV2A). An in situ proximity ligation assay for ZEB1 and the two AP‐1 factors further demonstrated that ZEB1 proteins are found in close proximity to JUN and FOSL1 proteins in the nuclei of MDA‐MB‐231 cells (Fig 2A), as well as in two additional basal type breast cancer cell lines, BT549 and Hs578T (Fig EV2B). As control, we used the Hippo pathway effector TAZ, for which we did not detect an interaction with ZEB1 in any of the three cell lines, although TAZ co‐localises with ZEB1 in the nucleus, as shown for MDA‐MB‐231 (Fig EV2A). We further tested whether ZEB1 interacts with JUN and FOSL1 by performing co‐immunoprecipitation (co‐IP) for all three factors and identified a physical interaction of endogenous ZEB1 with both JUN and FOSL1 (Fig 2B). Antibody and RNAi controls are shown in Appendix Fig S1A and B. Together, our results provide strong evidence that ZEB1 forms complexes with the AP‐1 factors JUN and FOSL1.
Figure EV2. Proximity ligation assay.
- Immunofluorescence staining of ZEB1, the AP‐1 factors JUN and FOSL1, and the Hippo pathway effector TAZ in MDA‐MB‐231 cells. All four proteins show a clear nuclear localisation. Scale bar = 20 μm.
- In situ proximity ligation assay (PLA) of ZEB1 with JUN, FOSL1 and TAZ shows close proximity of ZEB1 with JUN and FOSL1 but not TAZ in the nucleus of BT549 and Hs578T breast cancer cells indicated by red fluorescent dots. In the upper panel, representative microscopic images are shown. Scale bar = 20 μm. The lower panel shows a quantification of the PLA shown above. n = 2–3; shown is one representative experiment with means ± s.e.m. of at least 900 cells per condition; ***P ≤ 0.001; 2‐way ANOVA plus Tukey′s post‐test.
Figure 2. ZEB1 interacts with JUN and FOSL1.
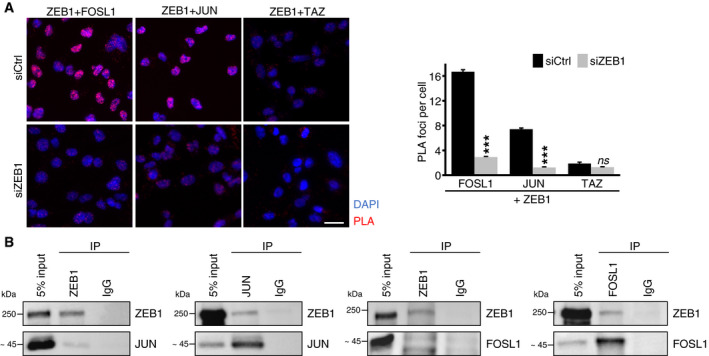
- In situ proximity ligation assay (PLA) of ZEB1 with JUN, FOSL1 and TAZ shows close proximity of ZEB1 with JUN and FOSL1 but not TAZ in the nucleus of MDA‐MB‐231 cells indicated by red fluorescent dots. As negative control, ZEB1 was transiently knocked down by siRNA. In the left panel, representative microscopic images are shown. Scale bar = 20 μm. Quantification of the PLA is shown on the right. n = 3; one representative experiment with means ± s.e.m. of at least 900 cells per condition is shown; ***P ≤ 0.001; 2‐way ANOVA plus Tukey′s post‐test.
- Co‐immunoprecipitation (Co‐IP) of endogenous ZEB1 and FOSL1 or JUN in MDA‐MB‐231 cells shows co‐precipitation of ZEB1 with FOSL1 and JUN.
ZEB1‐mediated transcriptional control has at least two modes of action
To further investigate whether cooperative binding of ZEB1 together with YAP and AP‐1 factors represents a specific mode of ZEB1‐mediated transcriptional control, we individually analysed two subsets of ZEB1‐binding sites in our ChIP‐seq data, hereafter called the ZEB1‐only and the ZEB1/YAP/JUN peaks. Importantly, ZEB1 binding to both subsets occurs in the same cell, here the aggressive, triple‐negative MDA‐MB‐231 cells. The ZEB1‐only peaks comprised a set of 5,963 locations where ZEB1 binds without any evidence for YAP‐ or JUN‐binding in close proximity. In contrast, at the 1,993 ZEB1/YAP/JUN sites, binding peaks of all three factors overlap pointing to the formation of putative ZEB1/YAP/AP‐1 complexes (Fig 3A). Peaks of both subsets were distributed over the whole genome (Fig 3B). However, examination of the specific genomic localisation of ZEB1‐only compared to ZEB1/YAP/JUN peaks revealed binding to different genomic elements. ZEB1‐only peaks were mostly located within 1 kb distance of a transcription start site (TSS) (Fig 3C). More specifically, 65.8% of the binding sites lay within promoter regions, defined as 1.5 kb upstream to 0.5 kb downstream of a TSS (Fig 3D). In contrast, ZEB1/YAP/JUN peaks were mostly located more distant to TSSs, with almost 40% of the peaks being 10–50 kb up‐ or downstream (Fig 3C). They were predominantly found in intronic and intergenic regions, with only 14.8% of peaks locating to promoters (Fig 3D). This analysis suggests that while ZEB1 without YAP and JUN preferentially controls transcription from promoter regions, ZEB1, YAP and JUN together act mainly from distal regulatory regions such as enhancers.
Figure 3. Characterisation of a ZEB1‐only and a ZEB1/YAP/JUN peak set.
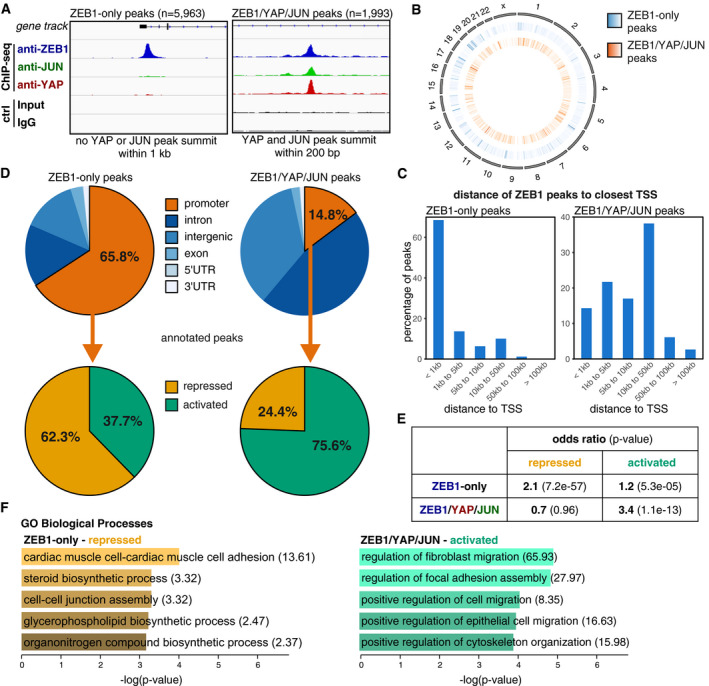
- Definition of two different ZEB1 peak subsets based on the presence or absence of overlapping YAP and JUN peaks. The two illustrative genome browser images show one example of a ZEB1‐only peak and one example of a ZEB1/YAP/JUN peak (at the EFEMP1 gene and within the SHROOM3 gene, respectively). Control (ctrl) is the input for ZEB1 and IgG for the YAP and JUN ChIP‐seqs. ChIP‐seqs and respective controls were scaled accordingly.
- Circle plot displaying the genome‐wide distribution of ZEB1‐only (middle circle, blue) and ZEB1/YAP/JUN peaks (inner circle, orange) across all chromosomes (outer circle). Peak density is visualised as a heatmap with more intense colours indicating higher accumulation of peaks at a specific location.
- Localisation of ZEB1 peak summits relative to the closest TSS.
- Distribution of ZEB1‐only and ZEB1/YAP/JUN peaks in respect to different genomic features. Promoters were defined as 1.5 kb upstream to 0.5 kb downstream of the TSS. Promoter peaks were annotated to their respective genes and integrated with transcriptome data of ZEB1 knockdown compared to control MDA‐MB‐231 cells.
- Hypergeometric testing showed that the association of ZEB1‐only promoter peaks with genes repressed in the presence of ZEB1 is stronger than with activated genes, while ZEB1/YAP/JUN promoter peaks are only associated with activated genes. Odds ratio (OR): measure of association between categorical variables with OR > 1 indicating a positive association. P‐value from Fisher's exact test.
- GO term analysis of the ZEB1‐only repressed and ZEB1/YAP/JUN activated gene sets using Enrichr. Odds ratios are given in brackets.
Next, we correlated the different modes of ZEB1‐binding with their effects on target gene expression. We here focused on genes exhibiting ZEB1‐only or ZEB1/YAP/JUN‐binding in their promoter region. Enhancer annotation to the respective target genes is currently being performed in an ongoing study. We assessed their differential expression levels in the aforementioned transcriptome analysis of ZEB1 knockdown compared to control MDA‐MB‐231 cells (Lehmann et al, 2016). Strikingly, over 60% of the genes with a ZEB1‐only peak at their promoter were repressed in the presence of ZEB1 (840 genes out of 1,348), whereas more than 75% of the genes with a ZEB1/YAP/JUN peak at their promoter were activated by ZEB1 (65 genes out of 86; Fig 3D). The association of ZEB1‐only peaks with gene repression and ZEB1/YAP/JUN peaks with gene activation was highly significant (Fig 3E), even with this small number of common ZEB1/YAP/JUN peaks in promoter regions. Analysis of the ZEB1‐only, repressed target gene set and the ZEB1/YAP/JUN, activated target gene set, revealed that repressed target genes are generally involved in epithelial biological processes like cell adhesion and cell–cell junction assembly, thus processes which need to be repressed during tumour progression. In contrast, activated target genes contribute to processes favouring tumour progression, like cell migration, invasion, metastasis and therapy resistance (Fig 3F, Table EV1). Together, these results suggest that ZEB1 without JUN and YAP acts predominantly as a transcriptional repressor, whereas in cooperation with YAP and AP‐1 factors, ZEB1 mainly acts as a transcriptional activator. Thus in the same (cancer) cell, ZEB1 can act in parallel as transcriptional repressor and activator on different, likely functional opposing sets of target genes, in summary favouring tumour progression.
ZEB1, YAP and AP‐1 factors synergistically induce target gene transcription
To verify that ZEB1, YAP and AP‐1 factors together form a transcriptional activator complex, we analysed the regulation of the above defined, activated gene set after extrinsic induction of EMT via TGFβ in MCF10A breast epithelial cells, which endogenously express YAP, JUN and FOSL1 (Appendix Fig S1D) and induce ZEB1 in response to TGFβ (Fig EV3A). As expected, TGFβ‐induced ZEB1 expression also activated expression of ZEB1/YAP/AP‐1 target genes but repressed the ZEB1‐only target CDH1 (Fig EV3A). Transient siRNA‐mediated knockdown of ZEB1 and to a lesser extent also FOSL1 and YAP inhibited TGFβ‐mediated activation (Fig 4A). Next, we performed luciferase reporter assays. We compared constructs harbouring different promoters and a known enhancer region (Liu et al, 2016), which exhibit binding of all three factors (Fig 4B), to a promoter construct of the epithelial polarity gene LLGL2, a known target gene repressed by ZEB1 (Spaderna et al, 2008), exhibiting ZEB1‐only binding (Fig 4C). Transient knockdown of ZEB1 in MDA‐MB‐231 cells, as well as in BT549 and Hs578T, led to a substantial reduction in luciferase activity in cells transfected with the constructs harbouring ZEB1/YAP/JUN‐binding, indicating an activating effect of ZEB1 on these constructs. In contrast, ZEB1 knockdown resulted in increased luciferase activity of the ZEB1‐only LLGL2 promoter, verifying the repressing function of ZEB1 in this context (Fig 4D, Fig EV3B and Appendix Fig S1C). Overexpression of combinations of the different factors in luminal‐type MCF7 cells, which lack endogenous expression of ZEB1, YAP, JUN and FOSL1, showed the highest synergistic activation of both the ANKRD1 and DOCK9 reporters when all four factors were overexpressed together (Fig 4E, Appendix Fig S1D and E). Together, these results suggest that ZEB1, YAP and AP‐1 factors synergistically induce target gene transcription.
Figure EV3. Common ZEB1/YAP/JUN target gene activation depends on EMT/ZEB1.
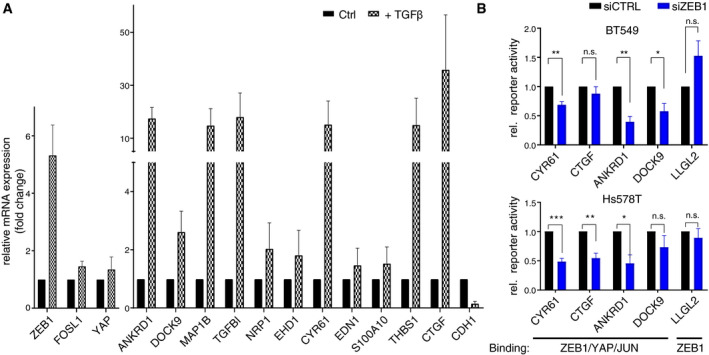
- qRT–PCR of ZEB1, FOSL1 and YAP, 11 ZEB1/YAP/JUN‐activated target genes and one ZEB1‐only repressed target gene, CDH1, in MCF10A cells after 7 days of TGFβ treatment compared to control treated cells. Both, control and TGFß‐treated cells were treated with control siRNA for 72 h before harvest. n = 3; shown is mean ± s.e.m.
- Luciferase reporter assay in BT549 and Hs578T cells comparing control and transient siRNA‐mediated knockdown of ZEB1. Reporter constructs of regulatory regions of genes shown in Fig 4 (B) and (C). n = 3; shown is mean ± s.e.m.; *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; unpaired two‐tailed Student's t‐test.
Figure 4. ZEB1, YAP and AP‐1 factors together activate target gene transcription.
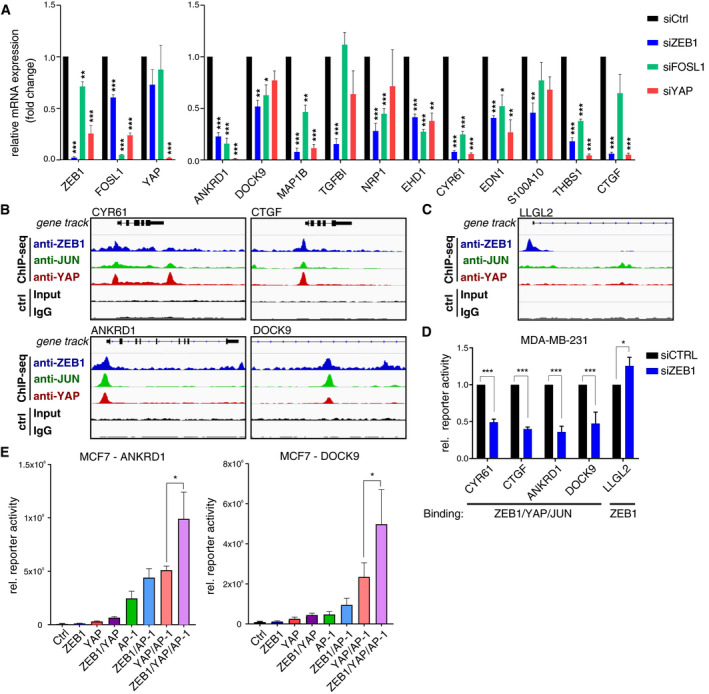
- qRT–PCR in TGFß‐treated MCF10A cells after siRNA‐mediated knockdown of ZEB1, FOSL1 or YAP compared to control knockdown cells. Knockdown efficiency of all three factors is shown on the left. The right panel shows their effects on 11 ZEB1/YAP/AP‐1‐activated target genes. n = 3; shown is mean ± s.e.m.; *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001; unpaired two‐tailed Student's t‐test.
- Overlap of ZEB1, YAP and JUN peaks at the promoter of CYR61, CTGF and ANKRD1 and in a known enhancer region of the DOCK9 gene.
- ZEB1‐only peak at the promoter of LLGL2, a well‐known ZEB1‐repressed target.
- Luciferase reporter assay in MDA‐MB‐231 cells comparing control and transient siRNA‐mediated knockdown of ZEB1. Reporter constructs of regulatory regions of genes shown in (B) and (C). n = 3–4; shown is mean ± s.e.m.; *P ≤ 0.05; ***P ≤ 0.001., unpaired two‐tailed Student's t‐test.
- Luciferase reporter assays with the ANKRD1 promoter and DOCK9 enhancer constructs in MCF7 cells upon overexpression of ZEB1, YAP and JUN/FOSL1 (AP‐1) or different combinations of the factors. n = 5 (ANKRD1) and n = 3 (DOCK9); shown is mean ± s.e.m.; *P ≤ 0.05; ratio paired t‐test. For reporter assays, firefly luciferase activity was normalised to co‐transfected Renilla luciferase.
Transcriptional activation does not require binding to ZEB1 high‐affinity motifs
When carefully analysing the DNA binding motifs present in ZEB1‐only vs. ZEB1/YAP/JUN peaks, we realised a striking difference. In the vast majority of ZEB1‐only peaks, both known and de novo motif analysis detected a very high enrichment of the canonical high‐affinity ZEB1‐binding motif (Z‐box) with the sequence CAGGTG/A. As expected, TEAD‐ and AP‐1‐binding motifs were only found in a low number of these sites (Fig 5A, Appendix Tables S3–S5). In contrast, in the ZEB1/YAP/JUN peaks, the top scoring motifs were the AP‐1 and TEAD consensus motifs, while the ZEB1‐binding motif exhibited a much lower enrichment, only 13% above background levels (Fig 5A). As direct binding of ZEB1 to DNA has been suggested to occur via its two zinc finger clusters to two separate Z‐boxes (Remacle et al, 1999), we inspected the multiplicity of the ZEB1 motif, i.e. how many sites were present within one peak, in ZEB1‐only compared to ZEB1/YAP/JUN peaks. Nearly 32% of the ZEB1‐only peaks contained at least two ZEB1‐binding motifs (1,903 of 5,963), while only about 5% of the ZEB1/YAP/JUN peaks harboured two or more of these sites (104 of 1,993). This observation indicates that most ZEB1‐binding motifs found in the ZEB1/YAP/JUN peaks do not recruit ZEB1 by themselves (or only inefficiently). Thus, we hypothesised that the recruitment of ZEB1 to DNA in complex with YAP and AP‐1 factors differs from classical ZEB1 DNA recruitment. In this context, ZEB1 does not require direct binding to its high‐affinity consensus motifs, but is recruited indirectly through YAP and AP‐1 proteins binding to their consensus DNA binding motifs.
Figure 5. The canonical high‐affinity ZEB1 motif is dispensable for the formation of a ZEB1 activator complex.
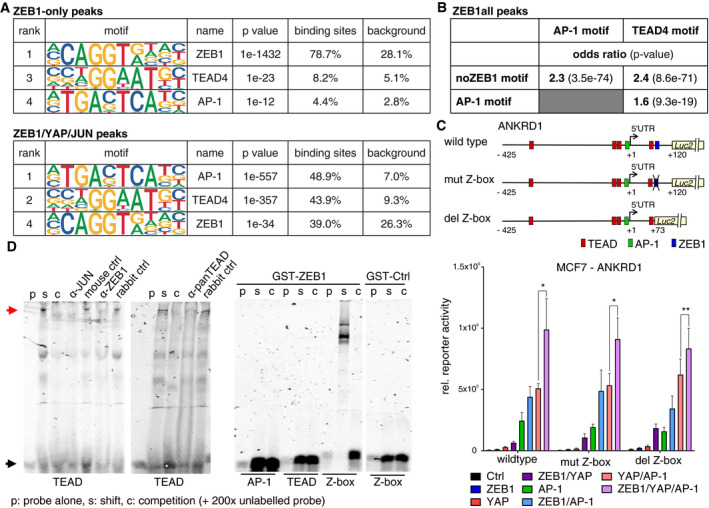
- Enrichment of the ZEB1, AP‐1 and TEAD4 DNA binding motifs is shown, as identified by HOMER known motif analysis on 200 bp regions centred on different subsets of ZEB1 peak summits. An extended list of the identified motifs can be found in Appendix Table S3 and S4.
- Hypergeometric testing showed that while there is a significant association between the AP‐1 and TEAD4 motif within ZEB1 peaks, both motifs are associated with the absence of the ZEB1 consensus motif (Z‐box). Odds ratio (OR): measure of association between categorical variables with OR > 1 indicating a positive association. P‐value from Fisher's exact test.
- The upper panel shows a schematic representation of the ANKRD1 luciferase reporter constructs. The lower panel shows luciferase reporter assays with these different ANKRD1 promoter constructs in MCF7 cells upon overexpression of ZEB1, YAP and JUN/FOSL1 or different combinations of the factors. n = 4–5; shown is mean ± s.e.m.; *P ≤ 0.05; **P ≤ 0.01; ratio paired t‐test. Firefly luciferase activity was normalised to co‐transfected Renilla luciferase. The wild‐type assay corresponds to the assay shown in Fig 4E.
- Left panel: EMSA using nuclear extracts from MDA‐MB‐231 and labelled TEAD binding side as probe (black arrow). Competition of specific binding complex (read arrow) with cold probe (C), anti‐JUN, ‐panTEAD, and ‐ZEB1 antisera. Right panel: recombinant GST‐ZEB1, but not GST‐Ctrl, only binds to Z‐box.
To investigate this hypothesis further, we tested for the statistical association between ZEB1, TEAD and AP‐1 consensus motifs in all ZEB1 peaks. Interestingly, there was no significant association of the ZEB1‐ with TEAD‐ or AP‐1‐binding motifs. On the contrary, there was a significant strong association between the absence of the ZEB1‐binding motif and the presence of an AP‐1‐ or TEAD‐binding motif (Fig 5B). On the other hand, there was a significant strong association between the presence of a TEAD‐binding motif and an AP‐1‐binding motif (Fig 5B). This analysis hints at a reduced importance of ZEB1‐binding motifs for recruitment of ZEB1 to sites where TEAD‐ and AP‐1 consensus motifs are present.
To further analyse the relevance of Z‐boxes for ZEB1/YAP/AP‐1 activator complex formation, we performed luciferase reporter assays with different ANKRD1 promoter constructs. Wild‐type sequences were compared to constructs in which the ZEB1‐binding motif was either mutated (mut Z‐box) or deleted (del Z‐box; Fig 5C). Neither deletion nor mutation of the ZEB1 motif resulted in altered luciferase activity after overexpression of different combinations of ZEB1, YAP, FOSL1 and JUN factors. This is consistent with our hypothesis that direct ZEB1 binding to Z‐boxes is not required in the activating context (Fig 5C). Additionally, by employing electrophoretic mobility shift assays we could confirm cooperative DNA binding of ZEB1, AP‐1 and YAP to a consensus binding site of TEAD (the DNA binding partner of YAP) under native conditions. Antibodies against each of the three factors specifically interfered with binding of a high molecular weight complex to the TEAD‐binding motif (Fig 5D). Importantly, direct DNA binding of recombinant ZEB1 via its C‐terminal zinc finger domain occurred only at the double Z‐box motif but not at the TEAD or the AP‐1 motif (Fig 5D). Taken together, these results support the hypothesis that direct binding of ZEB1 to Z‐boxes is not required for transcriptional activation by ZEB1/YAP/AP‐1 complexes.
Joint binding of ZEB1, YAP and JUN is associated with active genomic elements
We have shown here that ZEB1‐only binding preferentially represses target gene transcription, while joint binding of ZEB1, YAP and JUN together preferentially leads to gene activation (Fig 3D). We made use of the annotation of binding peaks to their respective genes and therefore only focused on peaks in promoter regions. To generate a genome‐wide picture of the chromatin status of ZEB1 bound regions, we performed ATAC‐seq, a method used to identify open chromatin. Chromatin accessibility can be used as a surrogate marker for the activity of genomic elements with active regions being more accessible than repressed regions. In order to assess how ZEB1 affects chromatin status, we performed ATAC‐seq on wild‐type, control and ZEB1 knockdown MDA‐MB‐231 cells. We took the regions that showed a ZEB1‐related gain or loss of accessibility in the ATAC‐seq, and overlaid our ChIP‐Seq data. Motif analysis showed that ZEB1 peaks within repressed regions contained the high‐affinity ZEB1 consensus motif more frequently compared to those in active genomic regions as identified by motif analysis. On the other hand, ZEB1 peaks in active genomic regions showed a substantial increase in the presence of TEAD and AP‐1 consensus motifs (Fig 6A, Appendix Tables S6–S8). This motif distribution supports a prominent role of AP‐1 and YAP/TEAD factors in ZEB1‐mediated gene activation and furthermore reflects the motif distribution seen in ZEB1‐only compared to ZEB1/YAP/JUN peaks.
Figure 6. Differential association of ZEB1‐only and ZEB1/YAP/JUN peaks with active and repressed genomic elements.
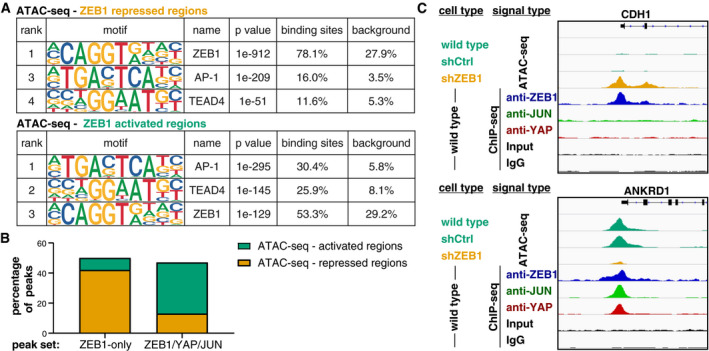
- Enrichment of the ZEB1, AP‐1 and TEAD4 DNA binding motifs identified by HOMER known motif analysis on 200 bp regions centred on ZEB1 peak summits associated with repressed or activated genomic regions identified by ATAC‐seq. An extended list of the identified motifs can be found in Appendix Tables S6 and S7.
- Comparison of ZEB1 ChIP‐seq peaks with ZEB1‐dependent changes in chromatin activation status identified by ATAC‐seq on control and ZEB1 knockdown MDA‐MB‐231 cells. Percentage of ZEB1‐only (n = 5,963) and ZEB1/YAP/JUN peaks (n = 1,993) falling into ZEB1‐repressed or ZEB1‐activated genomic regions are shown.
- Genome browser tracks of ZEB1, YAP and JUN ChIP‐seq signal intensity in wild‐type cells overlaid to ATAC‐seq signal intensity in wild‐type, control (shCtrl) and ZEB1 (shZEB1) knockdown MDA‐MB‐231 cells. One known ZEB1‐repressed target (CDH1) and one activated target (ANKRD1) are shown.
We thus tested if the difference in motif content between ZEB1‐repressed and ZEB1‐activated genomic regions corresponds to a different distribution of ZEB1‐only compared to ZEB1/YAP/JUN peaks in regard to these regions. We found that more than fivefold more ZEB1‐only peaks localised to repressed genomic regions than to activated ones (2,529 compared to 473). Vice versa more than 2.5‐fold more ZEB1/YAP/JUN peaks fell into activated than into repressed regions (687 compared to 265; Fig 6B). Figure 6C shows one prototype ZEB1‐repressed gene (CDH1) and one prototype ZEB1‐activated gene (ANKRD1). As one would expect, the ZEB1‐only peak at the CDH1 promoter is associated with a condensed (repressed) chromatin conformation, while the ZEB1/YAP/JUN peaks at the ANKRD1 promoter are associated with open (active) chromatin. Together, these observations further support our finding that ZEB1 acts in concert with TEAD/YAP and AP‐1 factors to activate gene expression.
ZEB1 and AP‐1 mark aggressive breast cancer types
To investigate the translational and clinical relevance of a functional ZEB1/YAP/AP‐1 interaction, we analysed human breast cancer data sets. Using RNA expression data from the cancer cell line encyclopaedia (CCLE; Barretina et al, 2012; Cancer Cell Line Encyclopedia Consortium & Genomics of Drug Sensitivity in Cancer Consortium, 2015), we saw that expression of ZEB1 is highly correlated with FOSL1 expression and to a lesser extent with YAP and JUN expression (Fig 7A). Classification of the cell lines according to the different molecular subtypes of breast cancer (Parker et al, 2009; Prat et al, 2010) revealed that the aggressive claudin‐low subtype (a subfraction of triple‐negative breast cancer), known to harbour EMT features (Prat et al, 2010), is specifically marked by high expression of ZEB1, FOSL1 or YAP (Fig 7A and B). Of note, cell lines that lack expression of even only one of these three factors then belong to other subtypes. Indeed, breast cancer cell lines of the basal subtype displayed an elevated expression of YAP and FOSL1 but not of ZEB1 and almost all cell lines of the luminal A, luminal B and Her2+ subtypes lack expression of both ZEB1 and FOSL1 (Fig 7B). Importantly, the top ranked common activated target genes (Table EV1) are also highly expressed in the claudin‐low subset, whereas, as expected, epithelial ZEB1‐only repressed target genes behave the other way round, meaning they show lowest expression in the ZEB1 high claudin‐low subset of cell lines (Fig 7B, Appendix Table S9). These findings strongly support the biological relevance of our findings in the MDA‐MB‐231 cells for breast cancer cells of the claudin‐low subtype. Notably, although both claudin‐low breast cancer cells and glioblastoma cells express high levels of ZEB1, LEF1 levels in breast cancer cells are much lower than in glioblastoma cell lines, corroborating an important role of LEF1 in glioblastoma but not claudin‐low breast cancer (Fig EV4A).
Figure 7. ZEB1, FOSL1 and their common target genes are specifically enriched in breast cancer cell lines of the claudin‐low subtype.
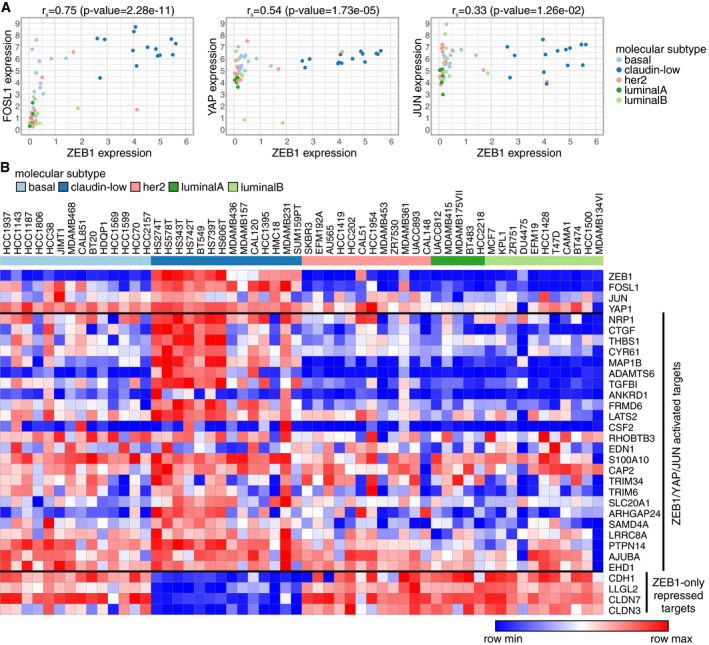
- Correlation of ZEB1 mRNA expression with the expression of YAP, FOSL1 and JUN across all breast cancer cell lines included in the cancer cell line encyclopaedia (CCLE). r s: Spearman's rank correlation coefficient.
- Heatmap of RNA‐seq expression data of all CCLE breast cancer cell lines.
Figure EV4. Clinical relevance in breast cancer.
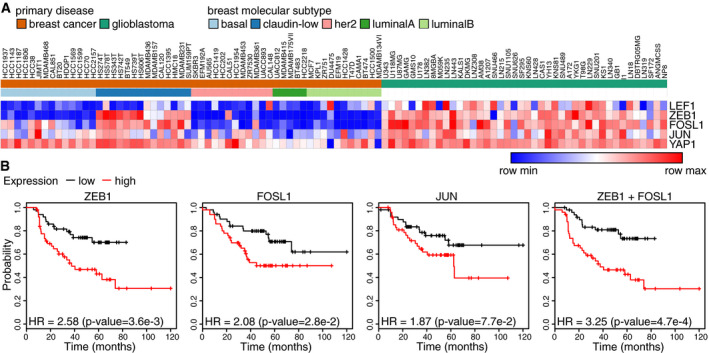
- Heatmap of RNA‐seq expression data of all CCLE breast cancer and glioblastoma cell lines.
- Kaplan–Meier plots from survival analyses of human ER /PR breast cancers showing relapse‐free survival after chemotherapy based on the expression of the indicated genes or gene combinations. HR: hazard ratio. P‐value from log‐rank test.
Further, we performed survival analysis using the KM‐plotter data analysis platform (Gyorffy et al, 2010) and focused on human breast cancers conventionally classified as hormone receptor negative (ER‐, PR‐), which include the most aggressive subtypes. In this context, high expression of ZEB1 was strongly associated with reduced distant metastasis‐free survival (DMFS), the major cause of poor clinical outcome (Fig 8A). Expression of the AP‐1 factors FOSL1 and JUN alone had no significant effect on DMFS in this specific subset of breast cancer patients. A combined expression of ZEB1 and FOSL1 however showed the strongest correlation with worse DMFS, in accordance with their combined expression in cell lines of the highly aggressive claudin‐low subtype (Fig 8A). Expression of each of the three transcription factors was also significantly associated with poor and particularly a combined expression of ZEB1 and FOSL1 with worst relapse‐free survival after chemotherapy (Fig EV4B), pointing to a potential therapy resistance phenotype. To discriminate between ZEB1 expression in tumour cells and tumour stromal cells, we immunohistochemically analysed protein expression of ZEB1, YAP and FOSL1 in human breast cancer specimen (Fig 8B). As previously shown for ZEB1 and YAP (Lehmann et al, 2016), nuclear expression of ZEB1 and FOSL1 in cancer cells was also highly correlated. More specifically, FOSL1 positive tumours had 16 times higher odds of being also ZEB1 positive (Fig 8B). When analysing mRNA expression levels of a core set of tumour‐promoting common ZEB1/YAP/AP‐1‐activated target genes in human breast cancers, molecular subtype analyses revealed that the claudin‐low tumours cluster in the group with high expression of this target set (Fig 8C left), whereas tumours with low expression of these genes are mainly of luminal type (Fig 8C right). Notably, expression of ZEB1, YAP and JUN correlates with that of their target genes. Irrespective of the subtype, we again detected a correlated expression of ZEB1 and common ZEB1/YAP/AP‐1 target genes that was independent of the subtype (Fig 8D, Appendix Table S10). Moreover, survival analysis of this breast cancer data set demonstrated that tumours with high expression of the selected core set of common ZEB1/YAP/AP‐1 target genes shown in Fig 8C correlated significantly with a worse relapse‐free (Fig 8E) and distant metastasis‐free survival (Fig 8F), which is in line with the results in hormone receptor negative (ER‐, PR‐) tumours shown in Fig 8A. Together, these analyses show that ZEB1 together with its binding partners YAP and AP‐1 as well as high expression of their common activated target genes marks aggressive breast cancers.
Figure 8. Clinical relevance of ZEB1, FOSL1 and their common activated target genes in breast cancer.
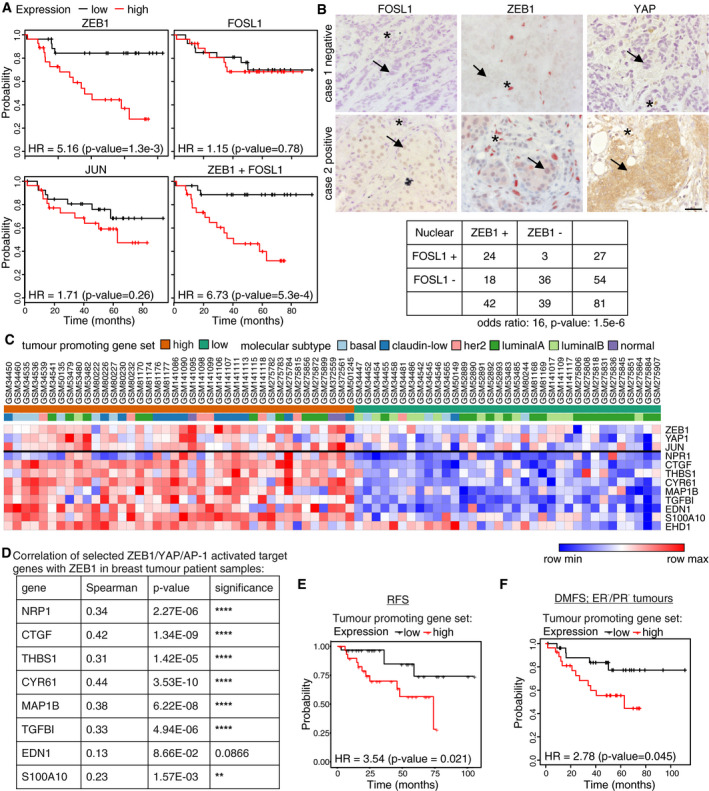
- Kaplan–Meier plots from survival analyses of human ER−/PR− breast cancers showing distant metastasis‐free survival (DMFS) based on the expression of the indicated genes or gene combinations. HR: hazard ratio. P‐value from log‐rank test.
- Hypergeometric testing showed a significant association between nuclear ZEB1 and FOSL1 expression on the protein level as determined by immunohistochemistry on 81 human breast cancer samples. The upper panel shows staining of a serial section of a negative and a positive case. Note that stromal cells (asterisks) are negative for FOSL1, and partially positive for ZEB1 and YAP, and that expression of ZEB1 in tumour cells (arrows) of positive cases is weaker than in stromal cells. Odds ratio (OR): measure of association between categorical variables with OR > 1 indicating a positive association. P‐value from Fisher's exact test. Scale bar = 20 μm.
- Heatmap of mRNA expression data of ZEB1, YAP, JUN and nine selected activated ZEB1/YAP/AP‐1 target genes in breast cancer patient samples (GSE18229). Genes were selected for known roles in tumour‐promoting properties like cell migration. Tumours in which ≥ 6 out of nine genes of the selected ZEB1/YAP/AP‐1 target gene set were expressed higher than the 60th percentile of expression were defined as “tumour‐promoting gene set high”; when expression of ≥ 6 out of the nine genes was below the 40th percentile, tumours were defined as “tumour‐promoting gene set low”. Tumours were annotated according to their molecular subtype.
- Correlation of ZEB1 mRNA expression with selected activated ZEB1/YAP/AP‐1 target genes which have known functions in cell migration in breast cancer patient samples (GSE18229). P‐value computed via the asymptotic t approximation.
- Kaplan–Meier plots from survival analyses of human breast cancers patient samples (GSE18229) showing relapse‐free survival (RFS) based on the combined expression of the nine selected ZEB1/YAP/AP‐1‐activated targets shown in (C). HR: hazard ratio. P‐value from log‐rank test.
- Kaplan–Meier plots from survival analyses of human ER‐/PR‐ breast cancers showing distant metastasis‐free survival (DMFS) based on the combined expression of the nine selected ZEB1/YAP/AP‐1‐activated targets shown in (C). HR: hazard ratio. P‐value from log‐rank test.
Discussion
Here, we present a genome‐wide study of ZEB1‐dependent transcriptional control in triple‐negative breast cancer. We find evidence that, in addition to its well characterised repressive function via direct binding to its canonical E‐box motif, ZEB1 is recruited to DNA by binding to AP‐1 and YAP factors. In cooperation with these factors, ZEB1 forms an activator complex potentiating the expression of tumour‐promoting target genes (Fig 9). Our results shed light on a direct functional link between important tumour‐promoting transcription factors: ZEB1, Hippo pathway and AP‐1‐factors.
Figure 9. Model of dual modes of transcriptional regulation by ZEB1.
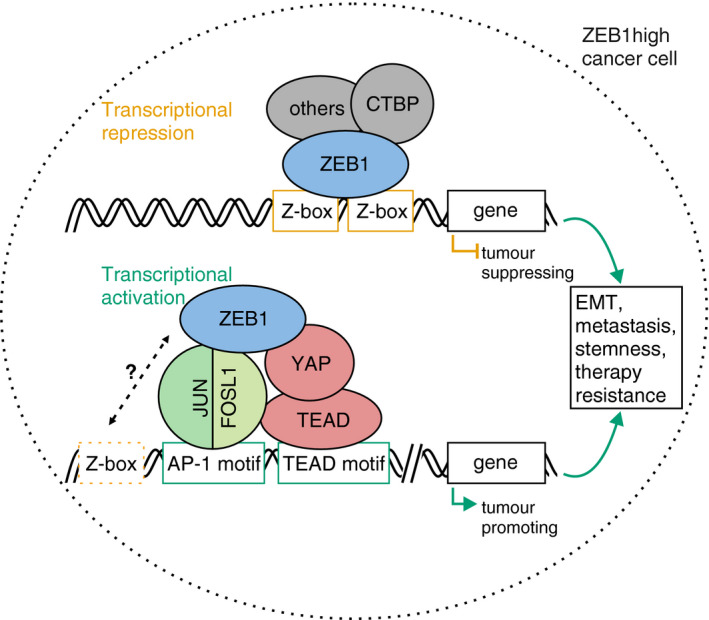
In order to act as a transcriptional repressor of epithelial differentiation genes, ZEB1 binds to DNA via a dual E‐box motif and recruits co‐repressors like CTBP. Within the same cell, recruitment of ZEB1 by YAP and the AP‐1 factors FOSL1 and JUN turns ZEB1 into a transcriptional activator of tumour‐promoting genes. This activator complex which we propose here can form independently of the presence of high‐affinity ZEB1 motifs and likely ZEB1 is indirectly recruited to DNA by AP‐1 and YAP/TEAD. It can however not be excluded that degenerated ZEB1 motifs support complex formation in a native chromatin context. While ZEB1 acts as a transcriptional repressor mainly from promoter regions, the ZEB1/YAP/AP‐1 complex preferentially locates to distal regulatory regions. Together, both modes of ZEB1 action exerted in the same cell contribute to cancer aggressiveness. Orange box: ZEB1‐specific E‐box motif. Dashed orange box: degenerated ZEB1 motifs. Green boxes: AP‐1 and TEAD consensus motifs.
The two AP‐1 components FOSL1 and JUN, which we identified as ZEB1 binding partners in breast cancer, are known to be important players in tumour progression (Eferl & Wagner, 2003; Lopez‐Bergami et al, 2010). Particularly, FOSL1 has been heavily implicated in the regulation of EMT and metastasis (Shin et al, 2010; Caramel et al, 2013; Desmet et al, 2013; Bakiri et al, 2015) and also in this context has been shown to work in concert with JUN (Tam et al, 2013; Zhao et al, 2014a; Zhang et al, 2016). Our group has recently demonstrated that ZEB1 can directly interact with YAP forming an activator complex to induce the expression of a selected target gene set (Lehmann et al, 2016). The in‐depth genome‐wide analysis in breast cancer presented here corroborates the existence of such a complex on genome‐wide level. Furthermore, our results identify the AP‐1 factors JUN and FOSL1 as novel players in ZEB1‐mediated gene activation together with YAP/TEAD. Interestingly, a recent study performed in normal murine mammary gland epithelial cells bioinformatically predicted all three complex components, ZEB1, TEAD and AP‐1 factors, as major signalling hubs in EMT (Meyer‐Schaller et al, 2019). Moreover, a likely functional interaction of all three factors is also described in melanoma cells (Verfaillie et al, 2015).
ZEB1 has predominantly been described as a transcriptional repressor. Thorough analysis of the distribution of ZEB1 binding sites in respect to genomic elements revealed a differential localisation of ZEB1‐only sites (potentially repressive) compared to sites where ZEB1 binds in cooperation with YAP and AP‐1 factors (potentially activating). While ZEB1 represses target genes mainly from their promoter region, the ZEB1/YAP/AP‐1 activator complex appears to be mainly located in potential enhancers. This is in line with the reported localisation of YAP/AP‐1 complexes in enhancers (Zanconato et al, 2015) and may explain why direct ZEB1 activating functions have largely been neglected in previous studies. Accumulating evidence points towards an important role of enhancers in cancer (Sur & Taipale, 2016; Ge et al, 2017; Chen et al, 2018), in particular during the metastatic transition, which is a highly dynamic and plastic process, accompanied by massive enhancer reprogramming (McDonald et al, 2017; Roe et al, 2017).
Analyses of human breast cancers indicate translational and clinical relevance of our findings. Particularly, co‐expression of ZEB1 and FOSL1 in breast cancer cells marks the highly aggressive claudin‐low subtype, and importantly, the lack of one of the two factors is enough to classify them to another clinically established subtype (Fig 7A and B). In patient samples from hormone receptor negative breast cancers, which are in general associated with poor clinical outcome (Foulkes et al, 2010), high expression of ZEB1 predicts an additional increased risk for distant metastasis and therapy relapse, possibly by selecting for the claudin‐low subtype. This correlation is even stronger when combining ZEB1 and FOSL1 expression, fitting to their combined expression in cell lines of this subtype. High expression of AP‐1 factors alone does not significantly correlate with worse distant metastasis‐free survival (DMFS) in hormone receptor negative breast cancer. This might be due to the fact that FOSL1 shows elevated expression also in other hormone receptor‐negative subtypes, notably the basal subtype, which however do not express ZEB1. These data indicate that the functional interaction of ZEB1 and AP‐1 identified at genome‐wide level is also of translational relevance for the clinical behaviour of breast cancer, particularly for the metastatic risk. Such a tumour‐ and progression‐promoting function is also supported by the functional annotation of the identified ZEB1/YAP/AP‐1‐activated target factors, namely their role in cell migration, invasion, dissemination, metastasis, survival and therapy resistance (see Table EV1). These results are in line with a recent study investigating genome‐wide ZEB1 target genes in breast cancer, which also associated ZEB1 with activation of survival, therapy resistance and proliferation, mechanisms not directly linked to “classical” EMT features (Maturi et al, 2018).
In agreement with observations of a context‐ and tissue‐specific action mode of EMT‐TFs (summarised in Stemmler et al, 2019), our data are in contrast to data of a genome‐wide study in glioblastoma, which demonstrated a functional interaction of ZEB1 and LEF/TCF factors in activating cancer‐promoting genes in this non‐epithelial cancer type (Rosmaninho et al, 2018). Of note, we did not detect significant enrichment of the LEF/TCF motif in our breast cancer data set. Comparison of both complementary studies indicates context‐dependent action modes of EMT‐TFs. While we identified the same mechanism of ZEB1‐mediated gene repression through direct binding to a double Z‐box motif, its mode of transcriptional activation was different comparing breast cancer and glioblastoma. Notably, in both tumour types transcriptional activation does not depend on the presence of high‐affinity ZEB1 motifs, but is largely mediated via binding of ZEB1 to additional activator complexes, which however are tumour context‐dependent. In this case, the lower expression of LEF/TCF factors in breast cancer cells compared to glioblastoma cells (Fig EV4A) might explain the differences in the selection of ZEB1 binding partners.
In summary, our results add a new level of understanding to the transcriptional regulation by the cancer‐promoting factor ZEB1. We corroborate the observation that ZEB1 has different modes of action. By directly binding to two Z‐box motifs, ZEB1 represses the expression of genes suppressing tumour progression. By interacting with YAP and AP‐1 factors, ZEB1 activates the expression of tumour‐promoting gene sets (Fig 9). The presence of these factors together marks the most aggressive subtypes of breast cancer. Targeting ZEB1 activating function by disturbing its interaction with YAP or AP‐1 factors indicates potential novel treatment options for patients in a progressed disease state.
Materials and Methods
Cell culture
MDA‐MB‐231, MCF7, BT549, Hs578T and MCF10A cells were purchased from the American Type Culture Collection (ATCC). They were cultured under standard conditions in DMEM (Gibco) supplemented with 10% foetal bovine serum (Gibco) and regularly tested for mycoplasma contamination. For the stably transduced MDA‐MB‐231 shGFP and shZEB1 cell lines (Spaderna et al, 2008), cell culture medium was additionally supplemented with 1.0 μg ml−1 puromycin. MCF10A cells were cultured in DMEM/F12 (Invitrogen) containing 5% horse serum (Life Technologies), 20 ng ml−1 EGF (R&D Systems), 0.5 mg ml−1 hydrocortisone (Sigma), 0.1 mg ml−1 cholera toxin (Sigma), 10 mg ml−1 insulin (Invitrogen) and 10 mM HEPES (Thermo). For TGFß1 treatment, MCF10A medium was supplemented daily with 5 ng ml−1 TGFß1 (PeproTech) for 7 days. As TGFß1 was dissolved in a citric acid solution, the medium of control cells was supplemented with citric acid to concentration of 500 nM.
Plasmids
Generation of the CTGF and CYR61 luciferase reporter plasmids was previously described (Lehmann et al, 2016). The ANKRD1 luciferase reporter plasmids (wild type, del Z‐box) were generated by amplifying the ANKRD1 promoter (−425 to +120 and +73, respectively, rel. to TSS) from genomic DNA by PCR. The restriction sites XhoI and HindIII were incorporated into the primers, and the amplicon was inserted into pGL4.10 (E6651, Promega). Mutation of the Z‐Box was introduced by QuickchangeII site‐directed mutagenesis (Ambion) of the ANKRD1 wild‐type promoter construct using the complementary primer pair as shown in Appendix Table S11. For the DOCK9 luciferase reporter vector, the chromosomal region chr13:99,719,488–99,719,923 (hg19) was amplified from BAC DNA (RPCIB753F20107Q, Source Bioscience). Restriction enzyme sites XhoI and HindIII were incorporated into the primers, and the amplicon was inserted into pGL4.23 (E8411, Promega). Generation of the LLGL2 luciferase reporter plasmid was previously described (Spaderna et al, 2008). For generation of the pCIneo‐JUN‐FLAG and pCIneo‐FOSL1‐FLAG expression vectors, JUN or FOSL1 cDNA sequence was amplified by PCR from MSCV‐IP N‐HAonly FOSL1 (Addgene plasmid #34897) and MSCV‐IP N‐HAonly JUN (Addgene plasmid #34898), respectively. Both were a gift from Peter M. Howley (White et al, 2012). Restriction enzyme sites (EcoRI/XbaI) and c‐terminal FLAG‐tag were incorporated into the primers. The PCR products were inserted into pCIneo (Promega). pCIneo‐hZEB1 was a gift from Michel M. Sanders (University of Minnesota, Minneapolis, US), and pcDNA6‐V5‐YAP1 was a gift from Gerd Walz (University Hospital Freiburg, Freiburg, Germany).
ChIP (Chromatin‐immunoprecipitation)‐seq
MDA‐MB‐231 cells were washed twice with PBS (Ca+/Mg+) and crosslinked in 1.5 mM EGS (dissolved first in DMSO to 25 mM, then in PBS (Ca+/Mg+)) for 30 min at room temperature (RT). After additional washing with PBS (Ca+/Mg+), cells were further crosslinked with 1% formaldehyde in PBS (Ca+/Mg+) at RT for 10 min. Crosslinking was stopped by adding glycine to a final concentration of 0.125 M for 5 min at RT. Cells were washed twice with cold PBS, scraped off and centrifuged (300 g, 5 min, 4°C). Cells were washed with cold PBS and centrifuged again. Cells were resuspended in NP‐40 lysis buffer (0.5% NP‐40, 85 mM KCL, 5 mM HEPES (pH 7.9)) plus protease inhibitors (25xComplete, Roche) and incubated on ice for 10 min. Subsequently, cells were disrupted by Dounce homogenisation. Cells were centrifuged (2,400 g, 5 min, 4°C), and the pellet was resuspended in nuclei lysis buffer (50 mM Tris–HCl (pH 8.0), 10 mM EDTA, 1% SDS and protease inhibitors) and incubated on ice for 10 min. Aliquots of 200–300 μl (in 1.5 ml Eppis) were sonificated with a Diagenode Bioruptor to a fragment size of 300–500 bp. The mixture was centrifuged (16,000 g, 10 min, 4°C), and the supernatant was collected. Chromatin concentration was adjusted to 1 μg μl−1 and diluted 1:1 with IP‐buffer (20 mM HEPES (pH 8.0), 0.2 M NaCl, 2 mM EDTA, 0.1% sodium deoxycholate, 1% Triton X‐100, 1 mg ml−1 BSA, protease inhibitors). Chromatin was precleared by adding Dynabeads (Invitrogen Protein A and G Dynabeads, cat n. 100‐02D and 100‐04D, 1:1 ratio) and rocking at 4°C for 1.5 h. Afterwards, beads were captured. Immunoprecipitation was performed with 100 μg of chromatin in a volume of 1 ml diluted with IP‐buffer. 5 μg ZEB1 antibody (Santa Cruz, sc‐25388x) was added, and IP reaction was incubated rocking at 4°C overnight. Fifty microlitre of beads was added to each IP reaction that was then incubated rocking at 4°C for 2–3 h. Beads were captured and washed 5× with washing buffer (1 M HEPES (pH 7.9), 0.5 M EDTA, 10% NP‐40, 10% sodium deoxycholate, 8 M LiCl) and 1× with TE. Immune complexes were eluted by adding 100 μl elution buffer (0.1 M NaHCO3, 1% SDS) to the beads containing 250 μg ml−1 RNase A and 500 μg ml−1 proteinase K. Reaction was incubated for 1 h at 37°C and 65°C overnight. DNA was extracted using QIAquick columns (MinElute, 15 μl). Several IPs were pooled for sequencing.
Library preparation was performed with the NEBNext® Ultra DNA Library Prep Kit according to the manufacturer's instructions, and 50 or 100 bp paired‐end sequencing was conducted on an Illumina HighSeq2500.
ATAC (Assay for transposable‐accessible chromatin)‐seq
Chromatin accessibility mapping was performed using the ATAC‐seq method as previously described (Corces et al, 2017), with minor adaptations. Briefly, in each experiment ~50,000 sorted cells were pelleted by centrifuging for 10 min at 4°C at 500 g. After centrifugation, the pellet was carefully lysed in 50 μl resuspension buffer supplemented with NP‐40 (Sigma), Tween‐20 and Digitonin (10 mM Tris–HCl pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.1% NP‐40, 0.1% Tween‐20, 0.01% Digitonin), and incubated for 3 min on ice. Then, 1 ml of ice‐cold resuspension buffer supplemented with 0.1% Tween‐20 was added, and the sample was centrifuged at 4°C at 500 g for 10 min. The supernatant was discarded, and the cell pellet was carefully resuspended in the transposition reaction (25 μl 2× TD buffer (Illumina), 2.5 μl TDE1 (Illumina), 16.5 μl PBS, 5 μl nuclease‐free water, 0.5 μl 1% Digitonin (Promega), 0.5 μl 10% Tween‐20 (Sigma)) for 30 min at 37°C on a shaker at 1,000 rpm. Following DNA purification with the Clean and Concentrator‐5 kit (Zymo) eluting in 23 μl, 2 μl of the eluted DNA was used in a quantitative 10 μl PCR (1.25 μM forward and reverse custom Nextera primers (Corces et al, 2017), 1× SYBR green final concentration) to estimate the optimum number of amplification cycles with the following programme 72°C 5 min; 98°C 30 s; 25 cycles: 98°C 10 s, 63°C 30 s, 72°C 1 min; the final amplification of the library was carried out using the same PCR programme and the number of cycles according to the Cq value of the qPCR. Library amplification using custom Nextera primers was followed by SPRI size selection with AmpureXP beads to exclude fragments larger than 1,200 bp. DNA concentration was measured with a Qubit fluorometer (Life Technologies). The libraries were sequenced as 126 bp paired‐end reads using an Illumina HighSeq2500.
Luciferase reporter assay
Cells were seeded in 24‐well plates in triplicates. The next day, they were transfected with the FuGENE HD transfection reagent (Promega, E2311) according to the manufacturer's instructions. Thereby, 100 ng of firefly luciferase reporter vector and 30 ng of pRL‐TK Renilla luciferase control reporter vector (Promega, E2241) and the following amounts of expression vectors were transfected: ZEB1 (100 ng), YAP, FOSL1, JUN (25 ng). Transfection reactions were adjusted to equal amounts with the corresponding empty control vectors. For reporter assays after transient knockdown, cells were transfected with siRNA 1 day after seeding. Transfection of reporter constructs followed the next day. In all cases, cells were harvested after 72 h and lysed in passive lysis buffer (Promega, E1941). Luciferase activity was measured using the Dual‐Luciferase Reporter Assay system according to Hampf and Gossen (2006) using a CentroXS³ LB 960 Luminometer (Berthold). Values of the firefly luciferase were normalised to their corresponding Renilla values, serving as a transfection control.
siRNA transfection
For transient knockdown, MDA‐MB‐231, BT549, Hs578T or MCF10A cells were transfected with siRNAs using the Lipofectamine RNAiMAX transfection reagent (Life Technologies, 13778‐075). Transfection was performed according to the manufacturer's protocol for 72 h. The final amount of siRNA was adjusted to a final concentration of 5 pmol per 24 well and 80 pmol per six well. siRNAs were obtained from Ambion (Silencer Select siRNAs) with the following sequences: hs ZEB1, 5′‐GGUAGAUGGUAAUGUAAUATT‐3′ (s229971); hs FOSL1, 5′‐CCAUCUGCAAAAUCCCGGAtt‐3′ (s15585); hs siJUN, 5′‐GGCACAGCUUAAACAGAAAtt‐3′ (s7658); hs siYAP, 5′‐AGAGAUACUUCUUCUUAAAUCAtt‐3′; Silencer Select Negative Control #1 siRNA (4390844).
Immunofluorescence and in situ proximity ligation assay
For immunofluorescence, cells were seeded on glass coverslips. If necessary, siRNA knockdown was performed after 24 h. Cells were washed with PBS once and fixed using 4% formaldehyde for 10 min at RT. Cells were washed again with PBS and incubated in permeabilisation solution (0.1% Triton X‐100 in PBS) for 10 min at RT. After washing with PBS, cells were incubated with blocking solution (3% BSA in PBS) for 30 min at RT. Primary antibodies against ZEB1 (1:300, Sigma‐Prestige, HPA027524), JUN (1:100, DB Biosciences, 610327), FOSL1 (1:100, Santa Cruz, sc‐28310) and TAZ (1:200, BD Biosciences, 560235) were diluted in blocking solution and incubated for 1 h at RT. Cells were washed twice and subsequently incubated with Alexa 488‐ and Alexa 594‐conjugated secondary antibodies (Life Technologies, A‐11037, A‐11029, A‐11034, 1:500 in blocking buffer) for 1 h at RT protected from the light. Cells were washed thrice and stained with 300 nM DAPI (Sigma‐Aldrich, D9542) for 2 min. Cells were again washed twice and mounted using Citifluor antifading solution (Science Services, CT17970‐100). Specimen were analysed using the Leica DM5500 B microscope, and images were processed using the Leica Application Suite X software. For Proximity ligation assay (PLA), cells were seeded, treated and stained with primary antibodies as for immunofluorescence except for permeabilisation in 0.4% Triton X‐100 in PBS (instead of 0.1%) and incubation with the primary antibody at 4°C overnight. PLA was then performed with the Duolink in situ Detection Reagents Orange (Merck, DUO92007) according to the manufacturer's protocol. Anti‐rabbit PLUS (Merck, DUO92002) and anti‐mouse MINUS (Merck, DUO92004) PLA probes were used. Images were captured by microscopy (Leica DM5500 B) using a 20× objective. Batch image analysis as well as merging DAPI and PLA channels for representative images was performed using CellProfiler software (Kamentsky et al, 2011).
Co‐immunoprecipitation assay
Co‐immunoprecipitation (Co‐IP) was performed using nuclear extracts from MDA‐MB‐231 cells. Cells were cultured on 15 cm dishes, rinsed once with PBS and harvested. Cells were first lysed in lysis buffer A (10 mM HEPES pH 7.9, 10 mM KCl, 0.1 mM EDTA, 1 mM DTT, 1x complete protease inhibitor (Roche)), by brief vortexing and incubation for 15 min on ice. NP‐40 was added to a final concentration of 0.5%, and it was vortexed for 10 sec followed by centrifugation (17,000 g, 4°C, 2 min). The pellet was washed twice with lysis buffer A, then resuspended in lysis buffer B (20 mM HEPES pH 7.9, 400 mM NaCl, 1 mM EDTA, 1 mM DTT, 1× complete protease inhibitor (Roche)). The suspension was incubated in a shaker (4°C, 1,000 rpm) for 30 min. The lysate was cleared by centrifugation (10 min, 4°C, 17,000 g). Equal amounts of protein were used for each individual IP. For the IP, first the required antibody was coupled to the beads (anti‐ZEB1 (Santa Cruz, sc‐25388x), anti‐FOSL1 (Santa Cruz, sc‐183), anti‐JUN (BD Biosciences, 610327)). Ten microlitre Dynabeads Protein G was washed twice in citrate–phosphate wash buffer (24.47 mM Citric acid, 51.68 mM Na2HPO4 (pH 5.0)) and then resuspended in 100 μl lysis buffer B containing 500 ng of the specific antibody. Samples were incubated for 40 min at RT on a rotating wheel. The antibody–Dynabeads complexes were washed twice with citrate–phosphate buffer. Nuclear extracts were diluted to a protein concentration of 2 μg μl−1 with buffer B, and 500 μg of protein was added to the beads. The mixture was incubated at 4°C for 1.5 h on a rotating wheel. Subsequently, the beads were washed thrice in IP washing buffer (lysis buffer B 1:5 in PBS). Immunoprecipitates were eluted with 1x Laemmli buffer (62.5 mM Tris–HCl pH 6.8, 2% (w/v) SDS, 0.2% (w/v) bromophenol blue, 10% (v/v) glycerol; diluted in buffer B) by incubation at 95°C for 5 min. Beads were removed, and ß‐mercaptoethanol was added to a final concentration of 10%. Samples were boiled again for 5 min at 95°C and submitted to Western blot analysis.
Quantitative RT–PCR
Total RNA was isolated using the RNeasy Plus Mini Kit (74136, Qiagen) following the manufacturer's instructions. Reverse transcription was subsequently performed using the Revert Aid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, K1622) according to the manufacturer's protocol. cDNA was amplified using gene specific primers, the Universal Probe Library (Roche, 04869877001) and the TaqMan Universal Master Mix (Applied Biosystems, 4440040) according to the manufacturer's instructions. Samples were run in triplicates in a LightCycler 480 (Roche) and normalised to GAPDH. For primer sequences, see Appendix Table S13.
Western blotting
For preparation of whole cell lysates, cells were washed with ice‐cold PBS and then lysed in lysis buffer (150 mM NaCl, 50 mM Tris–HCl pH 8.0, 0.5% Na‐Desoxycholate (w/v), 0.1% SDS (v/v), 1% NP‐40 (v/v), 1x complete protease inhibitor (Roche), 1 mM PMSF) for 30 min on ice. Lysates were cleared by centrifugation at 13,300 rpm and 4°C. Protein concentration was determined using the BCA assay (Thermo Scientific, 23225) in a flat‐bottom 96‐well plate according to the manufacturer's protocol. Proteins were separated by SDS–PAGE and transferred to a nitrocellulose membrane (GE Healthcare, 10600007) by wet blotting. The following antibodies were used for protein detection: ZEB1 (Sigma‐Prestige, HPA027524, 1:5,000 order 1:2,000), JUN (BD Biosciences, 610327, 1:1,000), FOSL1 (Santa Cruz, sc‐183, 1:1,000), YAP (Abcam, ab52771, 1:10,000), V5 (Invitrogen, 460705, 1:5,000), FLAG (Sigma, F7425, 1:5,000), β‐Actin (Sigma, A5441, 1:10,000). Protein bands were visualised using Western Lightning Plus‐ECL (PerkinElmer, NEL103001EA) according to the manufacturer's instructions and developed using a ChemiDoc MP Imaging System (Bio‐Rad).
Electrophoretic mobility shift assay (EMSA)
Electrophoretic mobility shift assays were performed as described (Brabletz et al, 1999). DNA sequences that were used as double‐stranded DNA oligonucleotides for probes and for competition are listed in Appendix Table S12. The probes were 5′‐CY 5.5‐labelled for direct in gel detection using a Chemidoc fluorescent imager (Bio‐Rad). The following antibodies were used for protein detection: ZEB1 (Novus, NBP1‐88845, 0.5 μg), JUN (BD Biosciences, 610327, 0.5 μg) and TEAD (Cell Signaling Technologies, 13295, 0.5 μg).
Immunohistochemistry
Immunohistochemistry for ZEB1 on 81 formalin‐fixed, paraffin‐embedded samples of different histological types of breast cancers from patients who underwent surgery was done as previously described (Wellner et al, 2009). Samples were retrieved from local archives, and usage was approved by the Ethics Committee of the University of Erlangen‐Nuremberg (no. 374–14 Bc). In brief, sections (4 mm) were deparaffinised, rehydrated and pretreated in a pressure cooker in 10 mM citrate buffer pH 6.0. They were then incubated with a polyclonal antiserum against ZEB1, diluted 1:800 (Sigma, #HPA027524) at 4°C overnight. Slides were washed three times with TBS/0.05% Tween‐20 and developed with the EnVision‐System (DAKO, #K4003, #K4001) and DAB colour reaction according to the manufacturer's protocol. Finally, they were counterstained with Mayer's haematoxylin, diluted 1:10 (Merck; #1.09249.0500) for 60–90 s. For detection of FOSL1 and YAP expression, samples were stained with mouse anti‐FOSL1 (Santa Cruz, sc‐28310, 1:100) and rabbit anti‐YAP (Proteintech #13584‐1‐AP, diluted 1:400).
Statistical analyses
All statistical analyses were performed using GraphPad Prism 8 software (GraphPad Software, Inc.) or the R statistical environment (R Core Team, 2018). In all assays, results are plotted as mean ± s.e.m. from biological replicates as indicated in the figure legends. Unpaired two‐tailed Student's t‐test was applied for the reporter assays and qRT–PCR analysis after siRNA‐mediated knockdown. For all other luciferase assays, a ratio paired t‐test was performed. For the PLA, at least 900 cells from 25 randomly chosen microscopic fields per condition were quantified for each replicate experiment and 2‐way ANOVA plus Tukey′s post‐test was applied. Correlation of mRNA‐mRNA pairs in cell lines was analysed calculating the Spearman's rank correlation coefficient. We followed common standards in the field and assumed data follow a normal distribution (although sample sizes are too small for this assumption to be tested), and significance was determined without assuming similar variance. Statistical significance of the association between gene expression in human breast tumours was assessed using Fisher's exact test. The log‐rank test was used to compare Kaplan–Meier survival curves. Statistical significance is presented as follows: *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001.
Data analysis
Preprocessing and analysis of ZEB1 ChIP‐seq data
Raw reads were quality filtered and adapter‐trimmed using cutadapt v.1.9.1 (Martin, 2011). Trimmed reads were subsequently aligned to the human reference genome (Ensembl GRCh37, primary assembly, release 85) using Bowtie2 (v. 2.2.9) (Langmead & Salzberg, 2012). Reads were aligned in paired‐end mode, with discordant or unpaired alignments discarded. In order to keep only uniquely mapping reads, reads were filtered for a MAPQ value of ≥ 30 using samtools. Redundant reads were removed using Picard's MarkDuplicates. Peaks were called using MACS2 (v. 2.1.0) (Zhang et al, 2008). As two biological replicates from two independent experiments were performed for each ChIP‐seq, the Irreproducible Discovery Rate (IDR) framework (Li et al, 2011) was used to assess the consistency of replicate experiments. Peaks were called separately on individual ChIP replicates but using merged input files. As recommended for IDR analysis, a relaxed p‐value threshold of 1e‐2 was set. The final list of peaks was selected with an IDR threshold of 0.05 in order to obtain a conservative set of peaks from merged replicates. Peaks overlapping blacklisted regions as defined by the ENCODE project (The Encode Project Consortium, 2012) were discarded. Unless otherwise specified, all further analysis was performed in the R (v. 3.4.0)/Bioconductor environment (Huber et al, 2015; R Core Team, 2018) with plots generated relying on the ggplot2 library (v. 3.0.0; Wickham, 2016).
Preprocessing and analysis of YAP/JUN publicly available ChIP‐seq data
ChIP‐seq data sets for YAP and JUN in MDA‐MB‐231 cells were downloaded from Gene Expression Omnibus under accession number GSE66081 (Zanconato et al, 2015). Raw data were analysed with the same pipeline as described above for ZEB1. As these data sets are single‐end sequencing data, analysis parameters were adjusted accordingly. Instead of input, the corresponding IgG control was used for each ChIP.
Preprocessing and analysis of ATAC‐seq data
ATAC‐seq reads were trimmed using Skewer (Jiang et al, 2014) and aligned to the hg19 assembly of the human genome using Bowtie2 (Langmead & Salzberg, 2012) with the very‐sensitive parameter and a maximum fragment length of 2,000. Duplicate and unpaired reads were removed using the sambamba (Tarasov et al, 2015) markdup command, and reads with mapping quality > 30 and alignment to the nuclear genome were kept. All downstream analyses were performed on these filtered reads. Peak calling was performed using HOMER (Heinz et al, 2010) with the following approach: two different peak sets were called, once with the options “‐style factor ‐fragmentLength 150 ‐size 250 ‐minDist 250 ‐L 4 ‐fdr 0.0001 ‐tbp 1″, to identify highly robust small regions of open chromatin, and in a second approach with the parameters “‐region ‐fragmentLength 150 ‐size 150 ‐minDist 350 ‐L 4 ‐fdr 0.0001 ‐tbp 1” to identify larger regions of open chromatin. The two peak sets were then intersected using the bedtools intersect command with the “‐u” option to create the peak set that was used for downstream analysis. For the comparative analysis, we created a consensus region set by merging the called peaks from all involved samples using bedtools, and we quantified the accessibility of each region in each sample by counting the number of reads from the filtered BAM file that overlapped each region. Peaks overlapping blacklisted features as defined by the ENCODE project (The Encode Project Consortium, 2012) were discarded. DESeq2 (Love et al, 2014) was used on the raw count values for each sample and regulatory element to identify differential chromatin accessibility between samples. Significant regions were defined as having an FDR‐corrected (Benjamini–Hochberg corrected) P‐value below 0.05, an absolute log2fold change above 1, and a mean accessibility equal to or greater than 10. UCSC genomic naming conventions were converted to Ensembl naming with CrossMap (Zhao et al, 2014b). ZEB1 peak summit positions located inside a differential ATAC‐seq region were extracted using bedtools intersect specifying –u (Quinlan & Hall, 2010; Quinlan, 2014).
Visualisation in the genome browser
For visualisation in the IGV browser (v. 2.4.8; Robinson et al, 2011; Thorvaldsdottir et al, 2013), replicates of processed ChIP‐seq and ATAC‐seq data were merged. Signalling tracks were generated using the bamCoverage command from deeptools (v. 2.5.3; Ramirez et al, 2014), and the number of reads per bin was normalised to reads per kilobase per million reads (RPKM). Encode blacklisted regions were excluded from the analysis.
Peak annotation
ChIP‐seq peaks were annotated to genes using the annotatePeakInBatch function from ChIPpeakAnno package (v. 3.10.1; Zhu et al, 2010; Zhu, 2013) setting the following options output=“overlapping”, FeatureLocForDistance=“TSS”, bindingRegion=c (‐1,500, 500). The gtf file from Ensembl GRCh37 v. 85 was used as annotation, by keeping only “transcript” features.
Gene expression analysis by Affymetrix microarrays
The gene expression data used here (Lehmann et al, 2016) is available at the ArrayExpress database with the identifier E‐MTAB‐3482. In brief, two stable MDA‐MB‐231 knockdown clones for ZEB1 (shZEB1) were compared to two control clones (shCtrl). Data were preprocessed using RMA, as implemented in the Bioconductor package affy (v. 1.54.0) (Gautier et al, 2004). Afterwards, differential expression analysis of ZEB1 knockdown samples versus control samples was performed relying on the limma package (v. 3.32.2; Ritchie et al, 2015; Phipson et al, 2016). P‐values were adjusted using the Benjamini–Hochberg correction; annotation of probe sets to genes relied on the annotation file provided by Affymetrix for HG‐U133_Plus_2 (v. 36).
Integration of ChIP‐seq and gene expression data
Integration of the ZEB1 ChIP‐seq data and the microarray expression data was performed on the basis of the Ensembl gene IDs, respectively, associated with the ZEB1 peaks and the array probe sets. Only probes with an adjusted P‐value ≤ 0.01 were considered. Genes represented by both repressive and activating probes were filtered out. Significance of the association of different peak sets with activation or repression of gene expression was assessed with the GeneOverlap package (v. 1.12.0; Shen & Sinai, 2013). The list of genes with a significant expression change in the transcriptome data (adjusted P‐value of ≤ 0.01) was compared to the list of genes that have at least one associated probe in the microarray and a peak of the considered peak set (e.g. ZEB1all or ZEB1/YAP/JUN) assigned to them. The overlap of both lists was calculated using the newGeneOverlap function, and significance testing was performed with the testGeneOverlap function. Genome size was set to the number of genes on the microarray. P‐values from all performed tests were adjusted together for multiple testing with Benjamini–Hochberg correction.
Genomic distribution
The distance from the peak summit positions (calculated by MACS2) to the closest TSS was calculated using bedtools closest function (Quinlan & Hall, 2010; Quinlan, 2014). Upstream distances were reported as negative values. In case of ties, the upstream values were kept. The distribution of peaks according to different genomic features was calculated using a custom R script (R v. 3.5.1). Promoter regions were defined as 1.5 kb upstream to 0.5 kb downstream of a TSS as described before for peak annotation. All other types of genomic regions were assigned using the assignChromosomeRegion function from the ChIPpeakAnno package (v. 3.14.2; Zhu et al, 2010; Zhu, 2013) and the GenomicFeatures package (v. 1.32.2; Lawrence et al, 2013). Precedence was set to promoters, immediateDownstream, fiveUTRs, threeUTRs, exons and introns.
Motif discovery in ChIP‐seq peaks
Known and de novo motif analysis were performed with the findMotifsGenome function of the HOMER software (Heinz et al, 2010; v. 4.9.1) using Ensembl GRCh37 v. 85 as custom genome. Motif search was performed in 200‐bp regions centred on the peak summits. Motifs associated with the same TF were grouped together, and the one with the highest percentage detected in the target sequences was considered as representative motif for the TF. To ensure comparability, the motifs selected as representative using all ZEB1 peaks were used for all other peak subsets as well. To create motif density plots, the HOMER annotatePeaks function was used with bin size set to 10 bp. Motif logos in main figures were drawn with ggseqlogo library (v. 0.1; Wagih, 2017). Co‐occurrence of specific motifs in the ZEB1 peaks was identified using the annotatePeaks from HOMER specifying –nmotifs ‐matrix. Search parameters were set the same way as described above for the findMotifsGenome function. To test for the significant association of specific motifs, Fisher's exact test was used and the entire population was set to the number of ZEB1 peaks. P‐values from all performed tests were adjusted together for multiple testing with Benjamini–Hochberg correction.
Identification of overlapping peaks
To identify overlapping peaks and calculate the distance between their summits, the findOverlapsOfPeaks function of the ChIPpeakAnno package (v. 3.10.2; Zhu et al, 2010; Zhu, 2013) was used with connectedPeaks =“keepAll”. Peaks were considered as overlapping when their summit positions were not further than 200 bp apart. Scaled Venn diagrams were drawn with the VennDiagram package (v. 1.6.20; Chen, 2018). Significance of the overlap was assessed utilising the function peakPermTest of the ChIPpeakAnno package (v. 3.18.2; R v. 3.6.1), which estimates a p‐value relying on a permutation test. As pool of potential binding sites, transcription factor binding site clusters (V3) from ENCODE were utilised (http://hgdownload.cse.ucsc.edu/goldenPath/hg19/encodeDCC/wgEncodeRegTfbsClustered/wgEncodeRegTfbsClusteredV3.bed.gz) and the number of permutations was set to 1,000. Heatmaps to assess the signal enrichment of ChIP‐seq reads in specific genomic regions were created using the computeMatrix reference point and plotHeatmap functions from deeptools (v. 2.5.3; Ramirez et al, 2014).
Extraction of peak subsets
The ZEB1‐only peak set contains all those ZEB1 peaks that have no adjacent YAP or JUN peak summit within 1,000 bp up‐ or downstream of the ZEB1 peak summit. To select this peak set, the distance between ZEB1 peak summits and the next YAP or JUN peak summit was calculated with bedtools closest ‐D ref ‐t all (Quinlan & Hall, 2010; Quinlan, 2014). Afterwards, the overlap between ZEB1 peaks having, respectively, no YAP or no JUN peak within 1,000 bp was calculated by the subsetByOverlaps function from the GenomicRanges package (Lawrence et al, 2013). The ZEB1/YAP/JUN peak set contains all those ZEB1 peaks that have both a YAP and a JUN peak summit within 200 bp up‐ or downstream of the ZEB1 peak summit. Overlapping peaks were identified using the findOverlapsOfPeaks function from the ChIPpeakAnno package.
Gene ontology (GO) term analysis
Gene ontology term analysis was performed using the online tool Enrichr (https://amp.pharm.mssm.edu/Enrichr; Kuleshov et al, 2016). Gene lists were supplied in form of gene symbols, and the annotation GO Biological Process 2018 was used.
Circle plots
Circle plots were performed relying on the public Galaxy web platform https://usegalaxy.eu/ (Afgan et al, 2018). Plots were created using the Circos tool (Krzywinski et al, 2009). The karyotype file from Ensembl GRCh37 v. 85 was used. For peak files, peak numbers were counted in bins of 1 Mb and peak numbers per bin were visualised as a heat map with more intense colours indicating a higher number of peaks.
Analysis of breast cancer gene expression data
The expression data set from the cancer cell line encyclopaedia (CCLE; Barretina et al, 2012; Cancer Cell Line Encyclopedia Consortium, Genomics of Drug Sensitivity in Cancer Consortium, 2015) was downloaded from https://depmap.org/portal/download/, and molecular subtyping was performed using the genefu package (v. 2.12.0; R v. 3.5.1; Gendoo et al, 2016). The molecular.subtyping function was used with the pam50 (Parker et al, 2009) or claudinLow (Prat et al, 2010) classifier. The human breast cancer expression data set (Prat et al, 2010) was downloaded from Gene Expression Omnibus (accession number GSE18229) considering only the GPL1390 microarray platform. Expression values of probes mapping to the same gene were averaged and only breast tumour samples were considered, so that a total of 188 samples with molecular subtype classification according to Prat et al was analysed. Association between expression of different genes was calculated using Spearman's rank correlation and p‐value computed via the asymptotic t approximation. Expression heat maps were generated using the Morpheus software from the Broad Institute (Morpheus software).
Survival analysis
Survival analyses of the GSE18229‐ GPL1390 breast cancer data set were performed relying on the package survival (v. 3.1‐8; R v 3.6.1). For each considered gene, “high” and “low” samples were identified as those samples where the gene expression was respectively lower than the 40th or higher than the 60th percentile. For analysis of the activated tumour‐promoting gene set (comprising the genes NRP1, CTGF, THBS1, CYR61, MAP1B, TGFBI, S100A10, EDN1 and EHD1), the expression “high” group contains samples where at least six out of the nine genes in the set have “high” expression and analogously for the expression “low” group. Kaplan–Meier survival curves for the two groups were compared via the log‐rank test. For further meta‐analysis of published data sets for breast cancer, we used the KM‐plotter (http://kmplot.com/analysis; Gyorffy et al, 2010). Patient samples (ER‐/PR‐) were selected and grouped for “high” (higher than 75th percentile) or “low” (lower than 25th percentile) mRNA expression of the selected gene or gene set and compared. For analyses of ZEB1/FOSL1 combined, the “multigene classifier” mode was applied in which the mean expression of the probes associated with the two genes was used. For relapse‐free survival plus chemotherapy, the patient cohort was restricted to patients who had received either adjuvant or neoadjuvant chemotherapy. For analysis of the activated tumour‐promoting gene set in ER‐/PR‐ patient samples, the “multigene classifier” mode of the KM‐plotter was applied and mean expression associated with probes for the following genes – NRP1, CTGF, THBS1, CYR61, MAP1B, TGFBI, S100A10, EDN1 and EHD1 – was used. The following microarray probes were used for each gene: 212764_at for ZEB1, 204420_at for FOSL1, 201464_x_at for JUN, 212298_at for NRP1, 209101_at for CTGF, 201110_s_at for THBS1, 201289_at for CYR61, 212233_at for MAP1B, 201506_at for TGFBI, 200872_at for S100A10, 218995_s_at for EDN1 and 208112_x_at for EHD1.
Conflict of interest
The authors declare that they have no conflict of interest.
Author contributions
NF planned and carried out experiments and bioinformatics analyses and wrote the manuscript. FF planned, performed and supervised bioinformatics analyses and helped with writing the manuscript. SW and HS planned and performed the interaction studies. IF and KJ planned and performed target gene evaluation. K.G. planned and performed the ZEB1 ChIP‐seq. SL contributed to bioinformatics analyses of ChIP‐seq data. JK and DR planned and performed the ATAC‐seq. CS supervised and analysed the ATAC‐seq and gave critical input to the manuscript. UB was responsible for sequencing the ZEB1 ChIP‐seq. RLE gave critical input to scientific discussions and the manuscript. MPS supervised ChIP experiments and gave critical input to scientific discussions. TB planned and coordinated the project, analysed immunohistochemistry data and wrote the manuscript. SB planned and coordinated the project, designed and analysed the reporter assays and wrote the manuscript.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 2
Source Data for Figure 4
Source Data for Figure 5
Acknowledgements
We greatly thank E. Bauer and B. Schlund for expert technical assistance and Aline Bozec for fruitful scientific discussions. This work was supported by grants to T.B. from the German Research Foundation (SFB992/C06; BR 1399/9‐1; 1399/10‐1; 1399/13‐1) and from the IZKF Erlangen (F4‐46) and to S.B. from the German Research Foundation (BR 4145/1‐1; 4145/2‐1), to R.L.E. from FAU (ELAN: 29624020). Open access funding enabled and organized by Projekt DEAL.
The EMBO Journal (2020) 39: e103209
Contributor Information
Thomas Brabletz, Email: thomas.brabletz@fau.de.
Simone Brabletz, Email: simone.brabletz@fau.de.
Data availability
All sequencing data generated for this study have been deposited in the ArrayExpress database at EMBL‐EBI under accession number E‐MTAB‐8258 (ZEB1 ChIP‐seq data) and E‐MTAB‐8264 (ATAC‐seq data). Analysis scripts are available from the corresponding author on reasonable request.
References
- Afgan E, Baker D, Batut B, van den Beek M, Bouvier D, Cech M, Chilton J, Clements D, Coraor N, Gruning BA et al (2018) The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res 46: W537–W544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aigner K, Dampier B, Descovich L, Mikula M, Sultan A, Schreiber M, Mikulits W, Brabletz T, Strand D, Obrist P et al (2007) The transcription factor ZEB1 (deltaEF1) promotes tumour cell dedifferentiation by repressing master regulators of epithelial polarity. Oncogene 26: 6979–6988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakiri L, Macho‐Maschler S, Custic I, Niemiec J, Guio‐Carrion A, Hasenfuss SC, Eger A, Muller M, Beug H, Wagner EF (2015) Fra‐1/AP‐1 induces EMT in mammary epithelial cells by modulating Zeb1/2 and TGFbeta expression. Cell Death Differ 22: 336–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balestrieri C, Alfarano G, Milan M, Tosi V, Prosperini E, Nicoli P, Palamidessi A, Scita G, Diaferia GR, Natoli G (2018) Co‐optation of tandem DNA repeats for the maintenance of mesenchymal identity. Cell 173: 1150–1164 e14 [DOI] [PubMed] [Google Scholar]
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehar J, Kryukov GV, Sonkin D et al (2012) The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483: 603–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brabletz T, Jung A, Hlubek F, Lohberg C, Meiler J, Suchy U, Kirchner T (1999) Negative regulation of CD4 expression in T cells by the transcriptional repressor ZEB. Int Immunol 11: 1701–1708 [DOI] [PubMed] [Google Scholar]
- Bronsert P, Kohler I, Timme S, Kiefer S, Werner M, Schilling O, Vashist Y, Makowiec F, Brabletz T, Hopt UT et al (2014) Prognostic significance of Zinc finger E‐box binding homeobox 1 (ZEB1) expression in cancer cells and cancer‐associated fibroblasts in pancreatic head cancer. Surgery 156: 97–108 [DOI] [PubMed] [Google Scholar]
- Cancer Cell Line Encyclopedia Consortium, Genomics of Drug Sensitivity in Cancer Consortium (2015) Pharmacogenomic agreement between two cancer cell line data sets. Nature 528: 84–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caramel J, Papadogeorgakis E, Hill L, Browne GJ, Richard G, Wierinckx A, Saldanha G, Osborne J, Hutchinson P, Tse G et al (2013) A switch in the expression of embryonic EMT‐inducers drives the development of malignant melanoma. Cancer Cell 24: 466–480 [DOI] [PubMed] [Google Scholar]
- Chaffer CL, San Juan BP, Lim E, Weinberg RA (2016) EMT, cell plasticity and metastasis. Cancer Metastasis Rev 35: 645–654 [DOI] [PubMed] [Google Scholar]
- Chen H (2018) VennDiagram: Generate High‐Resolution Venn and Euler Plots. R package version 1620
- Chen H, Li C, Peng X, Zhou Z, Weinstein JN, Cancer Genome Atlas Research Network , Liang H (2018) A pan‐cancer analysis of enhancer expression in nearly 9000 patient samples. Cell 173: 386–399 e12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott‐Armstrong NA, Vesuna S, Satpathy AT, Rubin AJ, Montine KS, Wu B et al (2017) An improved ATAC‐seq protocol reduces background and enables interrogation of frozen tissues. Nat Methods 14: 959–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2018) R: a language and environment for statistical computing. Vienna: R Foundation for Statistical Computing; [Google Scholar]
- De Craene B, Berx G (2013) Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer 13: 97–110 [DOI] [PubMed] [Google Scholar]
- Desmet CJ, Gallenne T, Prieur A, Reyal F, Visser NL, Wittner BS, Smit MA, Geiger TR, Laoukili J, Iskit S et al (2013) Identification of a pharmacologically tractable Fra‐1/ADORA2B axis promoting breast cancer metastasis. Proc Natl Acad Sci USA 110: 5139–5144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eferl R, Wagner EF (2003) AP‐1: a double‐edged sword in tumorigenesis. Nat Rev Cancer 3: 859–868 [DOI] [PubMed] [Google Scholar]
- Eger A, Aigner K, Sonderegger S, Dampier B, Oehler S, Schreiber M, Berx G, Cano A, Beug H, Foisner R (2005) DeltaEF1 is a transcriptional repressor of E‐cadherin and regulates epithelial plasticity in breast cancer cells. Oncogene 24: 2375–2385 [DOI] [PubMed] [Google Scholar]
- Foulkes WD, Smith IE, Reis‐Filho JS (2010) Triple‐negative breast cancer. N Engl J Med 363: 1938–1948 [DOI] [PubMed] [Google Scholar]
- Gautier L, Cope L, Bolstad BM, Irizarry RA (2004) affy–analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 20: 307–315 [DOI] [PubMed] [Google Scholar]
- Ge Y, Gomez NC, Adam RC, Nikolova M, Yang H, Verma A, Lu CP, Polak L, Yuan S, Elemento O et al (2017) Stem cell lineage infidelity drives wound repair and cancer. Cell 169: 636–650 e14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gendoo DM, Ratanasirigulchai N, Schroder MS, Pare L, Parker JS, Prat A, Haibe‐Kains B (2016) Genefu: an R/Bioconductor package for computation of gene expression‐based signatures in breast cancer. Bioinformatics 32: 1097–1099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gheldof A, Hulpiau P, van Roy F, De Craene B, Berx G (2012) Evolutionary functional analysis and molecular regulation of the ZEB transcription factors. Cell Mol Life Sci 69: 2527–2541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubelmann C, Schwalie PC, Raghav SK, Roder E, Delessa T, Kiehlmann E, Waszak SM, Corsinotti A, Udin G, Holcombe W et al (2014) Identification of the transcription factor ZEB1 as a central component of the adipogenic gene regulatory network. Elife 3: e03346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyorffy B, Lanczky A, Eklund AC, Denkert C, Budczies J, Li Q, Szallasi Z (2010) An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res Treat 123: 725–731 [DOI] [PubMed] [Google Scholar]
- Hampf M, Gossen M (2006) A protocol for combined Photinus and Renilla luciferase quantification compatible with protein assays. Anal Biochem 356: 94–99 [DOI] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK (2010) Simple combinations of lineage‐determining transcription factors prime cis‐regulatory elements required for macrophage and B cell identities. Mol Cell 38: 576–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber W, Carey VJ, Gentleman R, Anders S, Carlson M, Carvalho BS, Bravo HC, Davis S, Gatto L, Girke T et al (2015) Orchestrating high‐throughput genomic analysis with Bioconductor. Nat Methods 12: 115–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Lei R, Ding SW, Zhu S (2014) Skewer: a fast and accurate adapter trimmer for next‐generation sequencing paired‐end reads. BMC Bioinformatics 15: 182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahlert UD, Suwala AK, Raabe EH, Siebzehnrubl FA, Suarez MJ, Orr BA, Bar EE, Maciaczyk J, Eberhart CG (2015) ZEB1 promotes invasion in human fetal neural stem cells and hypoxic glioma neurospheres. Brain Pathol 25: 724–732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamentsky L, Jones TR, Fraser A, Bray MA, Logan DJ, Madden KL, Ljosa V, Rueden C, Eliceiri KW, Carpenter AE (2011) Improved structure, function and compatibility for Cell Profiler: modular high‐throughput image analysis software. Bioinformatics 27: 1179–1180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karihtala P, Auvinen P, Kauppila S, Haapasaari KM, Jukkola‐Vuorinen A, Soini Y (2013) Vimentin, zeb1 and Sip1 are up‐regulated in triple‐negative and basal‐like breast cancers: association with an aggressive tumour phenotype. Breast Cancer Res Treat 138: 81–90 [DOI] [PubMed] [Google Scholar]
- Krebs AM, Mitschke J, Losada ML, Schmalhofer O, Boerries M, Busch H, Boettcher M, Mougiakakos D, Reichardt W, Bronsert P et al (2017) The EMT‐activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat Cell Biol 19: 518–529 [DOI] [PubMed] [Google Scholar]
- Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA (2009) Circos: an information aesthetic for comparative genomics. Genome Res 19: 1639–1645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A et al (2016) Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 44: W90–W97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai D, Ho KC, Hao Y, Yang X (2011) Taxol resistance in breast cancer cells is mediated by the hippo pathway component TAZ and its downstream transcriptional targets Cyr61 and CTGF. Cancer Res 71: 2728–2738 [DOI] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL (2012) Fast gapped‐read alignment with Bowtie 2. Nat Methods 9: 357–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M, Huber W, Pages H, Aboyoun P, Carlson M, Gentleman R, Morgan MT, Carey VJ (2013) Software for computing and annotating genomic ranges. PLoS Comput Biol 9: e1003118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarova DL, Bordonaro M, Sartorelli AC (2001) Transcriptional regulation of the vitamin D(3) receptor gene by ZEB. Cell Growth Differ 12: 319–326 [PubMed] [Google Scholar]
- Lehmann W, Mossmann D, Kleemann J, Mock K, Meisinger C, Brummer T, Herr R, Brabletz S, Stemmler MP, Brabletz T (2016) ZEB1 turns into a transcriptional activator by interacting with YAP1 in aggressive cancer types. Nat Commun 7: 10498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Brown JB, Huang H, Bickel PJ (2011) Measuring reproducibility of high‐throughput experiments. The Annals of Applied Statistics 5: 1752–1779 [Google Scholar]
- Liu X, Li H, Rajurkar M, Li Q, Cotton JL, Ou J, Zhu LJ, Goel HL, Mercurio AM, Park JS et al (2016) Tead and AP1 coordinate transcription and motility. Cell Rep 14: 1169–1180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez‐Bergami P, Lau E, Ronai Z (2010) Emerging roles of ATF2 and the dynamic AP1 network in cancer. Nat Rev Cancer 10: 65–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 15: 550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M (2011) Cutadapt removes adapter sequences from high‐throughput sequencing reads. EMBnet Journal 17: 10–12 [Google Scholar]
- Maturi V, Enroth S, Heldin CH, Moustakas A (2018) Genome‐wide binding of transcription factor ZEB1 in triple‐negative breast cancer cells. J Cell Physiol 233: 7113–7127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald OG, Li X, Saunders T, Tryggvadottir R, Mentch SJ, Warmoes MO, Word AE, Carrer A, Salz TH, Natsume S et al (2017) Epigenomic reprogramming during pancreatic cancer progression links anabolic glucose metabolism to distant metastasis. Nat Genet 49: 367–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer‐Schaller N, Cardner M, Diepenbruck M, Saxena M, Tiede S, Lüönd F, Ivanek R, Beerenwinkel N, Christofori G (2019) A hierarchical regulatory landscape during the multiple stages of EMT. Dev Cell 48: 539–553.e6 [DOI] [PubMed] [Google Scholar]
- Morpheus software Morpheus software https://software.broadinstitute.org/morpheus.
- Nieto MA, Huang Ruby Y‐J, Jackson Rebecca A, Thiery Jean P (2016) EMT: 2016. Cell 166: 21–45 [DOI] [PubMed] [Google Scholar]
- Parker JS, Mullins M, Cheang MC, Leung S, Voduc D, Vickery T, Davies S, Fauron C, He X, Hu Z et al (2009) Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol 27: 1160–1167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phipson B, Lee S, Majewski IJ, Alexander WS, Smyth GK (2016) Robust hyperparameter estimation protects against hypervariable genes and improves power to detect differential expression. Ann Appl Stat 10: 946–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postigo AA, Dean DC (1999) ZEB represses transcription through interaction with the corepressor CtBP. Proc Natl Acad Sci USA 96: 6683–6688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prat A, Parker JS, Karginova O, Fan C, Livasy C, Herschkowitz JI, He X, Perou CM (2010) Phenotypic and molecular characterization of the claudin‐low intrinsic subtype of breast cancer. Breast Cancer Res 12: R68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR (2014) BEDTools: The Swiss‐Army tool for genome feature analysis. Curr Protoc Bioinformatics 47: 11 12 1–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez F, Dundar F, Diehl S, Gruning BA, Manke T (2014) deepTools: a flexible platform for exploring deep‐sequencing data. Nucleic Acids Res 42: W187–W191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remacle JE, Kraft H, Lerchner W, Wuytens G, Collart C, Verschueren K, Smith JC, Huylebroeck D (1999) New mode of DNA binding of multi‐zinc finger transcription factors: deltaEF1 family members bind with two hands to two target sites. EMBO J 18: 5073–5084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK (2015) limma powers differential expression analyses for RNA‐sequencing and microarray studies. Nucleic Acids Res 43: e47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP (2011) Integrative genomics viewer. Nat Biotechnol 29: 24–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roe JS, Hwang CI, Somerville TDD, Milazzo JP, Lee EJ, Da Silva B, Maiorino L, Tiriac H, Young CM, Miyabayashi K et al (2017) Enhancer reprogramming promotes pancreatic cancer metastasis. Cell 170: 875–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosmaninho P, Mukusch S, Piscopo V, Teixeira V, Raposo AA, Warta R, Bennewitz R, Tang Y, Herold‐Mende C, Stifani S et al (2018) Zeb1 potentiates genome‐wide gene transcription with Lef1 to promote glioblastoma cell invasion. EMBO J [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez‐Tillo E, de Barrios O, Valls E, Darling DS, Castells A, Postigo A (2015) ZEB1 and TCF4 reciprocally modulate their transcriptional activities to regulate Wnt target gene expression. Oncogene 34: 5760–5770 [DOI] [PubMed] [Google Scholar]
- Sanchez‐Tillo E, Siles L, de Barrios O, Cuatrecasas M, Vaquero EC, Castells A, Postigo A (2011) Expanding roles of ZEB factors in tumorigenesis and tumor progression. Am J Cancer Res 1: 897–912 [PMC free article] [PubMed] [Google Scholar]
- Shen L, Sinai M (2013) GeneOverlap: Test and visualize gene overlaps. R package version 1120
- Shin S, Dimitri CA, Yoon SO, Dowdle W, Blenis J (2010) ERK2 but not ERK1 induces epithelial‐to‐mesenchymal transformation via DEF motif‐dependent signaling events. Mol Cell 38: 114–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siebzehnrubl FA, Silver DJ, Tugertimur B, Deleyrolle LP, Siebzehnrubl D, Sarkisian MR, Devers KG, Yachnis AT, Kupper MD, Neal D et al (2013) The ZEB1 pathway links glioblastoma initiation, invasion and chemoresistance. EMBO Mol Med 5: 1196–1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaderna S, Schmalhofer O, Wahlbuhl M, Dimmler A, Bauer K, Sultan A, Hlubek F, Jung A, Strand D, Eger A et al (2008) The transcriptional repressor ZEB1 promotes metastasis and loss of cell polarity in cancer. Cancer Res 68: 537–544 [DOI] [PubMed] [Google Scholar]
- Stemmler MP, Eccles RL, Brabletz S, Brabletz T (2019) Non‐redundant functions of EMT transcription factors. Nat Cell Biol 21: 102–112 [DOI] [PubMed] [Google Scholar]
- Sur I, Taipale J (2016) The role of enhancers in cancer. Nat Rev Cancer 16: 483–493 [DOI] [PubMed] [Google Scholar]
- Tam WL, Lu H, Buikhuisen J, Soh BS, Lim E, Reinhardt F, Wu ZJ, Krall JA, Bierie B, Guo W et al (2013) Protein kinase C alpha is a central signaling node and therapeutic target for breast cancer stem cells. Cancer Cell 24: 347–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarasov A, Vilella AJ, Cuppen E, Nijman IJ, Prins P (2015) Sambamba: fast processing of NGS alignment formats. Bioinformatics 31: 2032–2034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Encode Project Consortium (2012) An integrated encyclopedia of DNA elements in the human genome. Nature 489: 57–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorvaldsdottir H, Robinson JT, Mesirov JP (2013) Integrative Genomics Viewer (IGV): high‐performance genomics data visualization and exploration. Brief Bioinform 14: 178–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandewalle C, Van Roy F, Berx G (2009) The role of the ZEB family of transcription factors in development and disease. Cell Mol Life Sci 66: 773–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verfaillie A, Imrichova H, Atak ZK, Dewaele M, Rambow F, Hulselmans G, Christiaens V, Svetlichnyy D, Luciani F, Van den Mooter L et al (2015) Decoding the regulatory landscape of melanoma reveals TEADS as regulators of the invasive cell state. Nat Commun 6: 6683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagih O (2017) ggseqlogo: a versatile R package for drawing sequence logos. Bioinformatics 33: 3645–3647 [DOI] [PubMed] [Google Scholar]
- Wellner U, Schubert J, Burk UC, Schmalhofer O, Zhu F, Sonntag A, Waldvogel B, Vannier C, Darling D, Zur Hausen A et al (2009) The EMT‐activator ZEB1 promotes tumorigenicity by repressing stemness‐inhibiting microRNAs. Nat Cell Biol 11: 1487–1495 [DOI] [PubMed] [Google Scholar]
- White EA, Sowa ME, Tan MJ, Jeudy S, Hayes SD, Santha S, Munger K, Harper JW, Howley PM (2012) Systematic identification of interactions between host cell proteins and E7 oncoproteins from diverse human papillomaviruses. Proc Natl Acad Sci USA 109: E260–E267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H (2016) ggplot2: elegant graphics for data analysis. New York, NY: Springer‐Verlag; [Google Scholar]
- Yang J, Antin P, Berx G, Blanpain C, Brabletz T, Bronner M, Campbell K, Cano A, Casanova J, Christofori G et al (2020) Guidelines and definitions for research on epithelial‐mesenchymal transition. Nat Rev Mol Cell Biol 21: 341–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanconato F, Forcato M, Battilana G, Azzolin L, Quaranta E, Bodega B, Rosato A, Bicciato S, Cordenonsi M, Piccolo S (2015) Genome‐wide association between YAP/TAZ/TEAD and AP‐1 at enhancers drives oncogenic growth. Nat Cell Biol 17: 1218–1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Sun Y, Ma L (2015) ZEB1: at the crossroads of epithelial‐mesenchymal transition, metastasis and therapy resistance. Cell Cycle 14: 481–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Wu J, Luo S, Lechler T, Zhang JY (2016) FRA1 promotes squamous cell carcinoma growth and metastasis through distinct AKT and c‐Jun dependent mechanisms. Oncotarget 7: 34371–34383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W et al (2008) Model‐based analysis of ChIP‐Seq (MACS). Genome Biol 9: R137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Ye X, Yu J, Li L, Li W, Li S, Yu J, Lin JD, Wang CY, Chinnaiyan AM et al (2008) TEAD mediates YAP‐dependent gene induction and growth control. Genes Dev 22: 1962–1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Qiao Y, Jonsson P, Wang J, Xu L, Rouhi P, Sinha I, Cao Y, Williams C, Dahlman‐Wright K (2014a) Genome‐wide profiling of AP‐1‐regulated transcription provides insights into the invasiveness of triple‐negative breast cancer. Cancer Res 74: 3983–3994 [DOI] [PubMed] [Google Scholar]
- Zhao H, Sun Z, Wang J, Huang H, Kocher JP, Wang L (2014b) CrossMap: a versatile tool for coordinate conversion between genome assemblies. Bioinformatics 30: 1006–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu LJ (2013) Integrative analysis of ChIP‐chip and ChIP‐seq dataset. Methods Mol Biol 1067: 105–124 [DOI] [PubMed] [Google Scholar]
- Zhu LJ, Gazin C, Lawson ND, Pages H, Lin SM, Lapointe DS, Green MR (2010) ChIPpeakAnno: a Bioconductor package to annotate ChIP‐seq and ChIP‐chip data. BMC Bioinformatics 11: 237 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Table EV1
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 2
Source Data for Figure 4
Source Data for Figure 5
Data Availability Statement
All sequencing data generated for this study have been deposited in the ArrayExpress database at EMBL‐EBI under accession number E‐MTAB‐8258 (ZEB1 ChIP‐seq data) and E‐MTAB‐8264 (ATAC‐seq data). Analysis scripts are available from the corresponding author on reasonable request.