

It has been hypothesized that the evolution of protein phosphoregulation drives phenotypic diversity across species of organisms, which impacts fitness during their evolution. However, there is a lack of information regarding linkage between the evolution of viral phosphoregulation and the phylogeny of virus species. In this study, we clarified the novel HSV-2 Us3 phosphoregulation of UL7 in infected cells, which is important for viral replication and pathogenicity in vivo. We also showed that the evolution of Us3 phosphoregulation of UL7 was linked to the phylogeny of viruses that are phylogenetically close to HSV-2 and to the phosphorylation requirements for the efficient in vivo viral fitness of HSV-2 and HSV-1, which are representative of viruses that have and have not evolved phosphoregulation, respectively. This study reports the first evidence showing that evolution of viral phosphoregulation coincides with phylogeny of virus species and supports the hypothesis regarding the evolution of viral phosphoregulation during viral evolution.
KEYWORDS: HSV, Us3 protein kinase, evolution, protein phosphorylation
ABSTRACT
Us3 proteins of herpes simplex virus 1 (HSV-1) and HSV-2 are multifunctional serine-threonine protein kinases. Here, we identified an HSV-2 tegument protein, UL7, as a novel physiological substrate of HSV-2 Us3. Mutations in HSV-2 UL7, which precluded Us3 phosphorylation of the viral protein, significantly reduced mortality, viral replication in the vagina, and development of vaginal disease in mice following vaginal infection. These results indicated that Us3 phosphorylation of UL7 in HSV-2 was required for efficient viral replication and pathogenicity in vivo. Of note, this phosphorylation was conserved in UL7 of chimpanzee herpesvirus (ChHV), which phylogenetically forms a monophyletic group with HSV-2 and the resurrected last common ancestral UL7 for HSV-2 and ChHV. In contrast, the phosphorylation was not conserved in UL7s of HSV-1, which belongs to a sister clade of the monophyletic group, the resurrected last common ancestor for HSV-1, HSV-2, and ChHV, and other members of the genus Simplexvirus that are phylogenetically close to these viruses. Thus, evolution of Us3 phosphorylation of UL7 coincided with the phylogeny of simplex viruses. Furthermore, artificially induced Us3 phosphorylation of UL7 in HSV-1, in contrast to phosphorylation in HSV-2, had no effect on viral replication and pathogenicity in mice. Our results suggest that HSV-2 and ChHV have acquired and maintained Us3 phosphoregulation of UL7 during their evolution because the phosphoregulation had an impact on viral fitness in vivo, whereas most other simplex viruses have not because the phosphorylation was not necessary for efficient fitness of the viruses in vivo.
IMPORTANCE It has been hypothesized that the evolution of protein phosphoregulation drives phenotypic diversity across species of organisms, which impacts fitness during their evolution. However, there is a lack of information regarding linkage between the evolution of viral phosphoregulation and the phylogeny of virus species. In this study, we clarified the novel HSV-2 Us3 phosphoregulation of UL7 in infected cells, which is important for viral replication and pathogenicity in vivo. We also showed that the evolution of Us3 phosphoregulation of UL7 was linked to the phylogeny of viruses that are phylogenetically close to HSV-2 and to the phosphorylation requirements for the efficient in vivo viral fitness of HSV-2 and HSV-1, which are representative of viruses that have and have not evolved phosphoregulation, respectively. This study reports the first evidence showing that evolution of viral phosphoregulation coincides with phylogeny of virus species and supports the hypothesis regarding the evolution of viral phosphoregulation during viral evolution.
INTRODUCTION
Protein phosphorylation is an effective and reversible posttranslational modification that controls most cellular machinery by regulating protein activity in eukaryotic cells (1). Accumulating evidence based on comprehensive analyses of protein phosphorylation in multiple species of organisms suggests that although functionally important phosphorylation sites (phosphosites) are under evolutionary constraint, they can diversify rapidly (2–4), which coincides with whole-genome duplication (4), a prominent factor of phenotypic diversity for organismal evolution (3, 4). These observations suggest that the evolution of protein phosphoregulation drives phenotypic diversity across species of organisms and impacts fitness during their evolution.
Because viruses depend on and hijack host cellular machinery for multiplication, most viruses that infect eukaryotic cells have evolved mechanisms that utilize protein phosphorylation to regulate their own viral proteins and to establish a cellular environment for efficient viral multiplication (5–8). Therefore, as hypothesized in organismal evolution, the evolution of phosphoregulation in virus species may be related to their phenotypic diversity and evolution. However, linkage between the evolution of phosphoregulation in virus species and their phenotypic diversity or phylogeny is unclear.
Herpesviruses, of the family Herpesviridae, are divided into three subfamilies: Alphaherpesvirinae, Betaherpesvirinae, and Gammaherpesvirinae. Herpes simplex virus 1 (HSV-1) and HSV-2 belong to the genus Simplexvirus in the Alphaherpesvirinae subfamily and cause various mucocutaneous and skin diseases in humans, including herpes labialis, genital herpes, herpetic whitlow, and keratitis, and life-threatening encephalitis (9). In contrast to most other viruses, herpesviruses encode a virus-specific protein kinase(s) (5, 7, 10). HSV-1 and HSV-2 encode serine/threonine Us3 protein kinases that are conserved in the Alphaherpesvirinae subfamily (5, 7, 11). HSV-1 Us3, the most commonly studied alphaherpesvirus Us3, performs multiple roles in viral replication by phosphorylating various viral and cellular proteins (7) and is critical for viral replication and pathogenicity in vivo (12, 13). However, Us3 phosphosites in HSV-1 proteins, whose phosphorylation impacts HSV-1 infection, are not always conserved in the corresponding HSV-2 proteins. For example, HSV-1 Us3 phosphosites in envelope glycoprotein B (gB) and Us3 (gB threonine at position 887 [Thr-887] and Us3 serine at position 147 [Ser-147]), whose phosphorylation promotes intracellular gB trafficking and Us3 catalytic activity, respectively, and which are required for efficient viral replication and pathogenicity in mice, are not conserved in HSV-2 gB and Us3 (12, 14–16). Thus, the acquisition of phosphosites in HSV proteins leads to phenotypic diversity of viral infections between HSV-1 and HSV-2 proteins. However, evolution of phosphoregulation in other herpesviruses related to HSVs has not been addressed; therefore, linkage between evolution of phosphoregulation among herpesvirus species and their phylogeny remains to be elucidated.
This study (i) identified and characterized the novel HSV-2 Us3 phosphoregulation of an HSV-2 protein not conserved in HSV-1 and (ii) investigated the linkage between the evolution of the identified phosphoregulation and the phylogeny of viruses phylogenetically close to HSVs.
RESULTS
Identification of HSV-2 UL7 as a novel substrate of HSV-2 Us3.
To identify Us3 target sites in HSV-2 proteins not conserved in the corresponding HSV-1 protein, we performed bioinformatic prediction of Us3 target sites in HSV-2 proteins using a consensus target sequence of Us3, RnX(pS/T)YY, where n is ≥2, X is any amino acid, pS/T is phosphorylated Ser or Thr, and Y is any amino acid except acidic residues. We determined this consensus target sequence based on earlier reports (17–19). We identified three putative Us3 target sites in HSV-2 UL7 (RRTpSSL), UL13 (RRRpSSPE), and UL47 (RRDpSAI), based on the following criteria: (i) that the three Us3 target sites were not conserved in HSV-1 UL7 (RQTSSL), UL13 (RRRASPE), and UL47 (DDDDEV) and (ii) that phosphorylation at the predicted phosphosites in the three Us3 target sites were detected in the phosphoproteomic analysis of HSV-2-infected U2OS cells reported previously (Table 1) (20). Among the three putative Us3 target sites in HSV-2 proteins, we focused on the site in HSV-2 UL7 (Fig. 1A).
TABLE 1.
Putative Us3-mediated phosphopeptides and phosphorylation sites identified by the previous MS/MS analysis
| Protein | Phosphorylation site(s) | Phosphopeptide sequencea | Mascot score |
|---|---|---|---|
| UL7 | Thr-288 | R(p)TSSLFYR | 43.41 |
| Ser-289 | RT(p)SSLFYR | 40.97 | |
| Ser-289 | T(p)SSLFYR | 33.83 | |
| UL13 | Ser-90 and Ser91 | RR(p)S(p)SPEAPGPAAK | 47.81 |
| Ser-90 | RS(p)SPEAPGPAAK | 44.24 | |
| UL47 | Thr-688 | RA(p)TGLGGPPRP | 33.81 |
| Ser-97 | RA(p)SFPRPR | 22.27 |
In peptide sequences, (p)S and (p)T indicate phosphorylated serine and threonine, respectively. For peptide identification, we conducted decoy database searching by Mascot and applied a filter for a false-positive rate of <1%.
FIG 1.

HSV-2 Us3-mediated phosphorylation of UL7 in cell cultures. (A) Amino acid sequence alignment of UL7 homologs from HSV-2 186, HSV-2 333, HSV-2 HG52, HSV-1(F), HSV-1(17), and HSV-1(KOS) strains. Amino acid sequences of HSV-2 UL7s, which match the Us3 consensus target sequence, are shaded. The putative Us3 target sequences of HSV-2 UL7s and the corresponding amino acid residues of HSV-1 UL7s are enclosed with a dotted square. The putative Us3 phosphosite, Ser-289, in HSV-2 UL7s, and corresponding serine residues of HSV-1 UL7s are boxed with a black square. At bottom, the conservation ratio has been deduced in the histogram. (B) HEK293FT cells were transfected with pcDNA-Myc-HSV-2-UL7, along with pFlag-HSV-2-Us3 or pFlag-HSV-2-Us3-K220M, harvested at 48 h postinfection, solubilized, separated in a Phos-tag(+) (top) or a Phos-tag(−) (middle and bottom) SDS-PAGE gel and analyzed by immunoblotting with antibodies to Myc (top and middle) or Flag (bottom). (C) The transfected HEK293FT cell lysates were untreated or treated with CIP overnight at 37°C and analyzed as described for panel B (top and middle). Digital images are representative of three independent experiments, and molecular mass markers are shown on the left (B and C).
To examine whether HSV-2 Us3 mediated phosphorylation of HSV-2 UL7, we performed phosphate affinity tag (Phos-tag) SDS-PAGE analysis, which visualizes the phosphorylation status of proteins as a distinct band shift (21). HSV-2 UL7 from HEK293FT cells cotransfected with plasmids expressing Myc-tagged HSV-2 UL7 (Myc-HSV-2 UL7) or Flag-tagged HSV-2 Us3 (Flag-HSV-2 Us3) was detected as three predominant bands with mobility differences in the Phos-tag(+) SDS-PAGE gel (Fig. 1B). After phosphatase treatment, the two slower-migrating (upper and middle) Myc-HSV-2 UL7 bands disappeared, and the amount of protein in the faster-migrating (lower) Myc-HSV-2 UL7 band was substantially increased in the Phos-tag(+) SDS-PAGE gel (Fig. 1C). Similarly, in cells cotransfected with plasmids expressing Myc-HSV-2 UL7 or a Flag-tagged HSV-2 Us3 kinase-dead mutant (Flag-HSV-2 Us3-K220M) in which the invariant lysine at Us3 residue 220 was replaced with methionine, the upper Myc-HSV-2 UL7 band was barely detectable, and the amounts of protein in the middle and lower Myc-HSV-2-UL7 bands were substantially decreased and increased, respectively, in the Phos-tag(+) SDS-PAGE gel (Fig. 1B). These results indicated that the two slower-migrating bands represented phosphorylated Myc-HSV-2 UL7 and that HSV-2 Us3 kinase mediated Myc-HSV-2 UL7 phosphorylation in these cotransfected cells. We should note that in cells cotransfected with plasmids expressing Myc-HSV-2 UL7 or Flag-HSV-2 Us3-K220M, the middle band was faint but consistently detected (Fig. 1B), suggesting that a cellular kinase(s) also mediated Myc-HSV-2 UL7 phosphorylation.
The putative Us3 target site identified in HSV-2 UL7 contained one threonine and two serines (Thr-288, Ser-289, and Ser-290) (Fig. 1A). To identify the minimal amino acids in these residues required for HSV-2 Us3-mediated phosphorylation of HSV-2 UL7, we constructed expression plasmids, with each expressing a series of mutant Myc-HSV-2 UL7s in which Thr-288, Ser-289, and/or Ser-290 was replaced with alanine(s). These mutants were tested by Phos-tag SDS-PAGE analyses. UL7 phosphorylation was determined as follows: (amount of protein in slower-migrating bands)/(amount of protein in slower migrating bands) + (amount of protein in faster-migrating band). The results were as follows. (i) The phosphorylation of HSV-2 UL7-TSS/AAA in which all threonine and serines were replaced with alanines (TSS/AAA) in the presence of Flag-HSV-2 Us3 was significantly lower than the level of wild-type HSV-2 UL7 phosphorylation but comparable to the level of phosphorylation of HSV-2 UL7-SS/AA, in which Ser-289 and Ser-290 were replaced with alanines (SS/AA) (Fig. 2A and D). (ii) The level of phosphorylation of HSV-2 UL7-S289A, in which Ser-289 was replaced with alanine (S289A) in the presence of Flag-HSV-2 Us3, was significantly lower than the level of wild-type HSV-2 UL7 phosphorylation but consistently higher than the level of HSV-2 UL7(SS/AA) phosphorylation (Fig. 2B and E). (iii) The phosphorylation level of HSV-2 UL7-T288A or UL7-S290A in which Thr-288 or Ser-290 was replaced with alanine (T288A or S290A), respectively, in the presence of Flag-HSV-2 Us3 was similar to the level of wild-type HSV-2 UL7 phosphorylation (Fig. 2B, C, E, and F). These results indicated that UL7 Ser-289 and Ser-290 were required for Us3-mediated phosphorylation of HSV-2 UL7 in cotransfected cells and suggested that Us3 phosphorylated UL7 at Ser-289 and/or Ser-290.
FIG 2.

Identification of phosphorylation site(s) of HSV-2 UL7 mediated by Us3 in cell-cultures. (A to C) HEK293 cells were transfected with pcDNA-Myc-HSV-2-UL7, pcDNA-Myc-HSV-2-UL7-TSS/AAA, pcDNA-Myc-HSV-2-UL7-SS/AA, pcDNA-Myc-HSV-2-UL7-T288A, pcDNA-Myc-HSV-2-UL7-S289A, or pcDNA-Myc-HSV-2-UL7-S290A and with pFlag-HSV-2-Us3 or pFlag-HSV-2-Us3-K220M using PEI (61). Then, the transfected cell lysates were analyzed as described in the legend of Fig. 1B. Digital images are representative of five (panels A and C) or seven (panel B) independent experiments, and molecular mass markers are shown on the left. (D to F) Quantitation of slower-migrating UL7 bands relative to the sum of the faster-migrating UL7 band and the slower-migrating UL7 bands shown in panels A to C. Each value is the mean ± standard errors of the mean of the results of five (panels A and C) or seven (panel D) independent experiments and is expressed relative to the mean value of the wild type normalized to 100. **, P < 0.01; ***, P < 0.001; ns, not significant by Tukey’s test (D to F).
HSV-2 Us3 directly phosphorylates HSV-2 UL7 in vitro.
To investigate whether HSV-2 Us3 directly phosphorylates UL7, we performed in vitro kinase assays using purified Us3 and UL7. We generated and purified wild-type HSV-2 Us3 and its kinase-dead mutant, each fused to glutathione S-transferase (GST) (GST-HSV-2 Us3 and GST-HSV-2 Us3-K220M, respectively), from Sf9 cells infected with recombinant baculoviruses expressing GST-HSV-2 Us3 or GST-HSV-2 Us3-K220M, respectively. Purified wild-type GST-HSV-2 Us3 was detected as one major band in a denaturing gel by silver staining, whereas purified GST-HSV-2 Us3-K220M was detected as two major bands, with the slower-migrating band predominating (Fig. 3A). These proteins reacted with anti-Us3 antiserum (Fig. 3B). We also generated and purified HSV-2 UL7 tagged with a Strep-tag (SE-HSV-2 UL7) and its mutant (SE-HSV-2 UL7-SS/AA) from HEK293FT cells transfected with a plasmid expressing SE-HSV-2 UL7 or SE-UL7-SS/AA and tested them as substrates using in vitro kinase assays with purified wild-type GST-HSV-2 Us3 and GST-HSV-2 Us3-K220M. SE-HSV-2 UL7, but not SE-HSV-2 UL7-SS/AA, was labeled with [γ-32P]ATP (Fig. 3D). SE-HSV-2 UL7 was not labeled by the kinase-dead mutant GST-HSV-2 Us3-K220M (Fig. 3D). SE-HSV-2 UL7 labeling by wild-type GST-HSV-2 Us3 was eliminated by phosphatase treatment (Fig. 3F). The expression of SE-HSV-2 UL7 and SE-HSV-2 UL7-SS/AA and identification of the SE-HSV-2 UL7-radiolabeled band were verified by Coomassie brilliant blue (CBB) staining (Fig. 3C and E). These results indicated that HSV-2 Us3 directly phosphorylated HSV-2 UL7 in vitro, which was dependent on HSV-2 UL7 Ser-289 and Ser-290, suggesting that HSV-2 Us3 directly phosphorylated HSV-2 UL7 at Ser-289 and/or Ser-290 in vitro.
FIG 3.
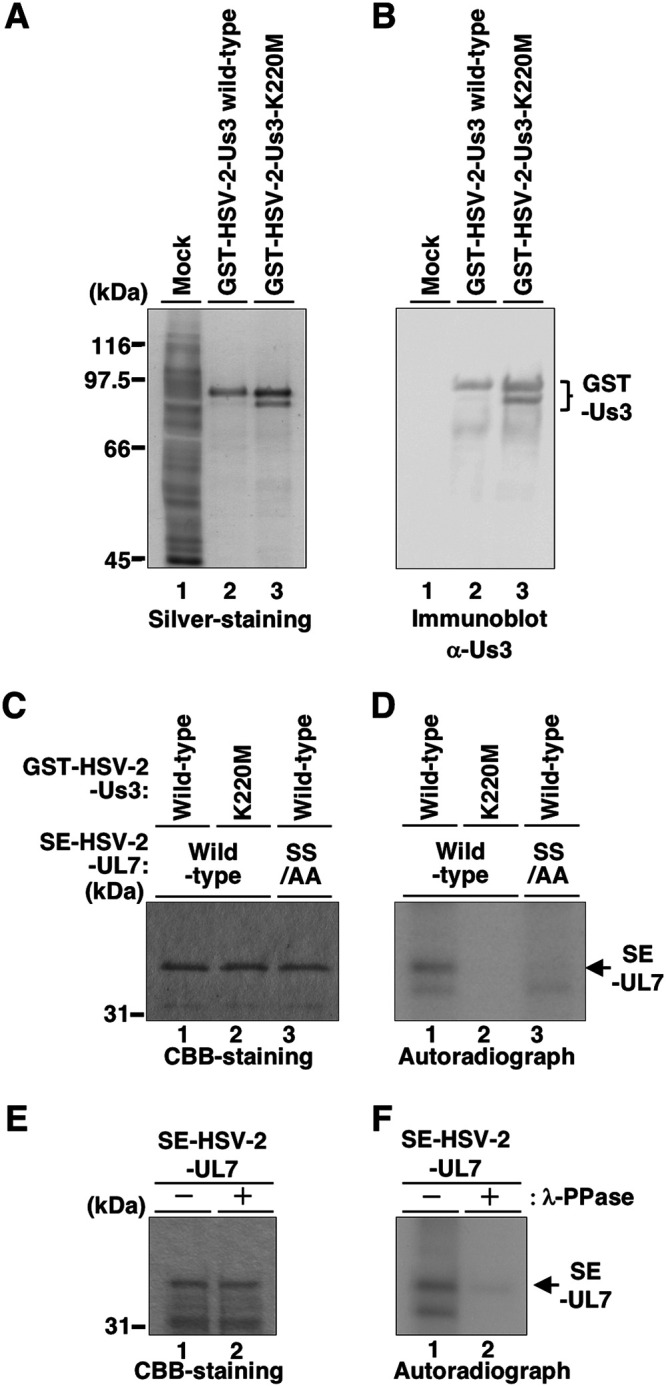
Purified HSV-2 Us3 directly phosphorylates purified UL7 in vitro. (A and B) GST-HSV-2-Us3 and GST-HSV-2-Us3-K220M were purified from Sf9 cells infected with the recombinant baculovirus Bac-GST-HSV-2-Us3 or Bac-GST-HSV-2-Us3-K220M, respectively. Then, mock-infected Sf9 cell lysate, purified GST-HSV-2-Us3, and GST-HSV-2-Us3-K220M were separated on denaturing gels and subjected to silver-staining (A) or transferred onto a nitrocellulose membrane and reacted with antibodies to Us3 (B). Digital images are representative of three independent experiments, and molecular mass markers are shown on the left. (C) SE-HSV-2-UL7 and SE-HSV-2-UL7-SS/AA were purified from HEK293FT cells transfected with pcDNA-SE-HSV-2-UL7 and pcDNA-SE-HSV-2-UL7-SS/AA, respectively. Purified SE-HSV-2-UL7 or SE-HSV-2-UL7-SS/AA in combination with purified GST-HSV-2-Us3 or GST-HSV-2-Us3-K220M was incubated in Us3 kinase buffer (50 mM Tris-HCl [pH 9.0], 20 mM MgCl2, 0.1% Nonidet P-40, and 1 mM dithiothreitol) containing 10 μM ATP, 10 μCi of [γ-32P]ATP for 30 min at 30°C, separated on a denaturing gel, and stained with CBB. (D) Autoradiograph of the gel shown in panel C. (E) Purified SE-HSV-2-UL7 labeled as described in panel C was then mock treated or treated with λ-PPase overnight at 30°C, separated on a denaturing gel, and stained with CBB. (F) Autoradiograph of the gel shown in panel E. Digital images are representative of two independent experiments, and molecular mass markers are shown on the left (panels C and E).
Construction of recombinant viruses using the newly generated HSV-2 BAC.
To examine the effects of HSV-2 UL7 and its phosphorylation by Us3 in the context of HSV-2 infection, we constructed a series of recombinant viruses using a self-excisable HSV-2 bacterial artificial chromosome (BAC) clone of strain 186 (pYEbac861Cre) generated in this study (Fig. 4). pYEbac861Cre was generated by inserting an expression cassette containing a gene encoding Cre recombinase into the intergenic site between the loxP site and the BAC sequence of HSV-2 BAC clone pYEbac861 (20) (Fig. 4). pYEbac861 is a derivative of HSV-2 BAC clone pYEbac356 (22) in which a spontaneous mutation in the UL13 locus of pYEbac356 was repaired (Fig. 4) (20). Recombinant viruses reconstituted from pYEbac861Cre and its derivatives were expected to excise BAC sequences via the functional Cre enzyme in HSV-2-infected cells, as described previously (22). This newly generated HSV-2 BAC (pYEbac861Cre) was characterized as follows. (i) When purified DNAs from strains YK356 (HSV-2/BAC) and YK785 (HSV-2/ΔBAC) reconstituted from pYEbac356 and pYEbac861Cre, respectively, were digested with EcoRI and analyzed by Southern blotting, the probe hybridized to fragment b plus c (7.4 kbp) in YK356 (HSV-2/BAC), whereas fragment b plus c was shifted to fragment b plus d (2.1 kbp) in YK785 (HSV-2/ΔBAC) as a result of the BAC excision (Fig. 5A and B). Therefore, the BAC vector sequence can be almost completely removed from the viral genome during the reconstitution of recombinant viruses in this system. (ii) Maintenance of OriL and OriS in YK785 (HSV-2/ΔBAC) was verified as described previously (22) (Fig. 5C) because the HSV-1 Ori sequences are unstable in bacteria (23). (iii) Expression levels of ICP27, UL50, UL34, and UL37 in Vero cells infected at a multiplicity of infection (MOI) of 3 with YK785 (HSV-2/ΔBAC) at 15 h postinfection were comparable to those of wild-type HSV-2 strain 186 (Fig. 5D). (iv) Growth curves for YK785 (HSV-2/ΔBAC) were similar to those of wild-type HSV-2 strain 186 in Vero cells infected at MOIs of 3 and 0.01 (Fig. 5E and F). (v) The 50% lethal dose (LD50) of YK785 (HSV-2/ΔBAC) for intracranial infection of mice was similar to that of wild-type HSV-2 186 (Table 2). (vi) Disease progression, survival, and vaginal viral titers of YK785 (HSV-2/ΔBAC) for vaginal infection of mice were similar to those of wild-type HSV-2 strain 186 (Fig. 5G to I). These results indicated that the YK785 (HSV-2/ΔBAC) virus reconstituted from pYEbac861Cre retained wild-type expression levels of viral proteins and replication kinetics in cell culture and maintained the virulence of wild-type HSV-2 strain 186 following intracranial and vaginal infection of mice.
FIG 4.

Strategy to construct a self-excisable HSV-2 186 BAC clone. Line 1, wild-type HSV-2 186 genome; line 2, intergenic region between HSV-2 186 UL50 and UL51 genes; lines 3 and 8, schematic diagrams of recombinant viruses YK351 and YK785, respectively; lines 4 and 5, schematic diagrams of E. coli plasmid pYEbac356 contained in DH10B and GS1783, respectively; line 6, schematic diagram of E. coli plasmid pYEbac861 contained in GS1783; line 7, schematic diagram of E. coli plasmid pYEbac861Cre contained in GS1783. To generate pYEbac861Cre, we modified the HSV-2 BAC clone pYEbac861 (20) by inserting an expression cassette containing a gene encoding Cre recombinase into the intergenic site between the loxP site and the BAC sequence. pYEbac861 is a derivative of an original HSV-2 BAC clone pYEbac356 in which a spontaneous mutation in the UL13 locus of pYEbac356 was repaired (20). Recombinant viruses reconstituted from pYEbac861Cre and its derivatives were expected to excise the BAC sequences through the functional Cre enzyme in HSV-2-infected cells as described previously (22).
FIG 5.

Characterization of a self-excisable HSV-2 186 BAC clone. (A) Schematic diagram of the genome structures of wild-type HSV-2 strain 186 and relevant domains of the recombinant viruses. Line 1, sequence arrangement of the wild-type HSV-2 186 genome; line 2, location of the EcoRI-HindIII fragment of pBS-UL51 used as a labeled probe in panel B; line 4, an enlarged portion of the EcoRI fragment including a portion of UL50 and UL51 genes in wild-type HSV-2 186; lines 3, 5 and 7, the fragment region (double-headed arrow); lines 6 and 8, summary of DNA fragments from recombinant viruses with or without the EGFP sequence, the BAC sequence, and poly(A) signals. (B) Southern blot analysis of DNA isolated from cells infected with wild-type HSV-2 186, YK356 (HSV-2 BAC), or YK785 (HSV-2 ΔBAC). Viral DNAs were digested with EcoRI, analyzed by electrophoresis on 0.8% agarose gels, and transferred to Hybond-N nylon membranes (GE Healthcare). The bands were hybridized with the 1,060-bp EcoRI-HindIII fragment of pBS-UL51 labeled with horseradish peroxidase using a direct nucleic acid labeling system (GE Healthcare) according to the manufacturer’s instructions. The letters on the right refer to the designations of the DNA fragments generated by restriction endonuclease EcoRI cleavage, which the probe hybridized. (C) A fragment containing OriL and OriS was amplified by PCR using primers 5′-CGAGGTGGGGGTTGTAGAAG-3′ and 5′-CTCTCCGATGGGGTCGAATG-3′ or 5′-CCTTCGCGATGCCGCCGCTGC-3′ and 5′-GCGATGCTAATGAGACCCTC-3′ from the HSV-2 186, YK356 (HSV-2 BAC), and YK785 (HSV-2 ΔBAC) genomes and subjected to agarose gel electrophoresis. (D) Vero cells were mock infected or infected with wild-type HSV-2 186, YK356 (HSV-2 BAC), or YK785 (HSV-2 ΔBAC) at an MOI of 3, harvested at 15 h postinfection, and then analyzed by immunoblotting with antibodies to ICP27, vdUTPase, UL34, UL37, and β-actin. IE, E, and L indicate immediate early, early, and late proteins, respectively. Digital images are representative of three independent experiments, and molecular size (panels B and C) and mass (panel D) markers are shown on the left. (E and F) Vero cells were infected at an MOI of 3 (E) or 0.01 (F) with wild-type HSV-2 186 or YK785 (HSV-2 ΔBAC). Total virus from cell culture supernatants and infected cells was harvested at the indicated times and assayed on Vero cells. Each value is the mean ± standard error of the mean of three independent experiments. (G) Twelve 6-week-old female ICR mice pretreated with medroxyprogesterone were infected vaginally with 0.75 × 104 PFU HSV-2 186 or YK785 (HSV-2 ΔBAC), and vaginal disease was scored every day for 26 days postinfection. Each data point is the mean ± the standard error of the mean of the scores. (H) The vaginal secretions of infected mice at 1 day postinfection in the experiment described in panel G were harvested, and virus titers were assayed on Vero cells. Each data point is the virus titer in the vaginal secretions of one mouse. The horizontal bars indicate the mean of each group. The dashed line indicates the limit of detection (2.5 PFU/ml). Viral titers below the limit of detection were assigned the value of the limit of detection and plotted on a graph, and statistical analysis was performed. (I) Survival of mice in the experiment described for panel G was monitored for 26 days postinfection. ns, not significant by a two-tailed Student's t test (E and F), Mann-Whitney U test (G and H), and log rank test (I).
TABLE 2.
LD50 values of wild-type HSV-2 186 and YK785 (HSV-2 ΔBAC) in mice following intracranial infection
| Virus | LD50 (PFU)a |
|---|---|
| Wild-type HSV-2 186 | 0.056 |
| YK785 (ΔBAC) | 0.092 |
Three-week-old female ICR mice were infected intracranially with serial 10-fold dilutions of each virus in groups of six per dilution and monitored for 21 days postinfection. LD50 values were determined by the Behrens-Karber method.
Using pYEbac861Cre, we constructed recombinant virus YK786 (HSV-2 UL7-SS/AA) encoding HSV-2 UL7-SS/AA, a UL7-null mutant virus YK787 (HSV-2ΔUL7), recombinant virus YK788 (HSV-2 UL7-repair) in which mutations in YK786 (HSV-2 UL7-SS/AA) and YK787 (HSV-2ΔUL7) were repaired, a HSV-2 Us3 kinase-dead mutant virus YK789 (HSV-2 Us3-K220M), and its repaired virus YK790 (HSV-2 Us3-KM-repair) (Fig. 6). YK787 (HSV-2 ΔUL7) was propagated on HSV-2 UL7-TetON-Vero cells in which the expression of UL7 was induced by addition of doxycycline (Fig. 7A).
FIG 6.

Schematic diagrams of recombinant HSV-2 or HSV-1 used in this study. (A) Schematic diagrams of the genome structure of wild-type HSV-2 186 and the recombinant viruses used in this study. Line 1, wild-type HSV-2 186 genome; line 2, domains of the UL6 to UL8 and Us2 to Us4 genes; lines 3 to 7, recombinant HSV-2 with mutations in the UL7 and/or Us3 genes. (B) Schematic diagrams of the genome structure of wild-type HSV-1(F) and the recombinant viruses used in this study. Line 1, wild-type HSV-1(F) genome; line 2, domains of the UL6 to UL8 and Us2 to Us4 genes; lines 3 to 6, recombinant HSV-1 with mutations in the UL7 and/or Us3 genes.
FIG 7.

HSV-2 Us3-mediated phosphorylation of UL7 in infected cells. (A) UL7-TetON-Vero cells were generated to support reconstitution and replication of YK787 (HSV-2 ΔUL7). UL7-TetON-Vero cells were mock treated or treated with doxycycline (DOX) (1 mg/ml), harvested at 24 h posttreatment, and analyzed by immunoblotting with antibodies to UL7 and β-actin. (B and C) Vero cells were mock infected or infected with wild-type HSV-2 186, YK789 (HSV-2 Us3-K220M), YK790 (HSV-2 Us3-KM-repair), YK786 (HSV-2 UL7-SS/AA), or YK788 (HSV-2 UL7-repair) at an MOI of 3, harvested at 8 h postinfection, solubilized, separated in Phos-tag(+) (top) or Phos-tag(−) (bottom) SDS-PAGE gels, and analyzed by immunoblotting with antibodies to UL7. (D) Vero cells were infected with wild-type HSV-2 186, harvested at 8 h postinfection, and lysed. The lysates were mock treated or treated with CIP overnight at 37°C and analyzed as described for panels B and C. (E and F) Vero cells were mock infected or infected with wild-type HSV-2 186, YK789 (HSV-2 Us3-K220M), or YK790 (HSV-2 Us3-KM-repair) at an MOI of 3, harvested at 8 h postinfection, and analyzed by immunoblotting with antibodies to UL7, Us3, UL37, and β-actin. (G and H) Vero cells were mock infected or infected with wild-type HSV-2 186, YK786 (HSV-2 UL7-SS/AA), YK787 (HSV-2 ΔUL7), or YK788 (HSV-2 UL7-repair) at an MOI of 3, harvested at 8 h postinfection, and analyzed as described for panels E and F. Digital images are representative of three independent experiments, and molecular mass markers are shown on the left (panels A, B, and E to H).
Us3-dependent phosphorylation of UL7 in HSV-2-infected cells.
To examine the phosphorylation of UL7 by Us3 in HSV-2-infected cells, Vero cells infected with wild-type HSV-2 186, YK789 (HSV-2 Us3-K220M), YK790 (HSV-2 Us3-KM-repair), YK786 (HSV-2 UL7-SS/AA), or YK788 (HSV-2 UL7-repair) at an MOI of 3 for 8 h were subjected to Phos-tag SDS-PAGE analyses. HSV-2 UL7 from cells infected with wild-type HSV-2 186, YK790 (HSV-2 Us3-KM-repair), or YK788 (HSV-2 UL7-repair) was detected as two predominant bands with mobility differences in Phos-tag(+) SDS-PAGE gels (Fig. 7B and C). After phosphatase treatment of lysate from wild-type HSV-2 186-infected cells, the slower-migrating HSV-2 UL7 band disappeared (Fig. 7D). Similarly, the slower-migrating band was not or only barely detectable with HSV-2 UL7 from cells infected with YK786 (HSV-2 UL7-SS/AA) or YK789 (HSV-2 Us3-K220M) (Fig. 7B and C). The HSV-2 Us3 kinase-dead mutation had no effect on the accumulation of UL7 (Fig. 7E) and Us3 (Fig. 7F) in HSV-2-infected cells. Similarly, the SS/AA mutation in HSV-2 UL7 had no effect on the accumulation of UL7 (Fig. 7G) and UL7 (Fig. 7H) in these infected cells. These results indicated that HSV-2 Us3 mediated UL7 phosphorylation in infected cells, which required UL7 Ser-289 and/or Ser-290, suggesting that HSV-2 Us3 phosphorylated UL7 at Ser-289 and/or Ser-290 in infected cells.
Effects of Us3 phosphorylation of HSV-2 UL7 on HSV-2 infection in vitro and in vivo.
To investigate the role of Us3 phosphorylation of HSV-2 UL7 in viral replication in cell cultures, Vero cells were infected with wild-type HSV-2 186, YK787 (HSV-2ΔUL7), YK786, (HSV-2 UL7-SS/AA), or YK788 (HSV-2 UL7-repair) at an MOI of 3 or 0.01, and viral titers were assayed at 18 or 36 h. Progeny virus yields of YK787 (HSV-2ΔUL7) were significantly lower than those of wild-type HSV-2 186 or YK788 (HSV-2-repair) at MOIs of 3 and 0.01 (Fig. 8A and B), indicating that HSV-1 UL7 was required for efficient viral replication in cell cultures, as reported for HSV-1 UL7 (24–27). In contrast, progeny virus yields of YK786 (HSV-2 UL7-SS/AA) were similar to those of wild-type HSV-2 186 or YK788 (HSV-2-repair) at MOIs of 3 and 0.01 (Fig. 8A and B).
FIG 8.

HSV-2 Us3 phosphorylation of UL7 contributes to viral replication and pathogenicity in mice following vaginal infection. (A and B) Vero cells were infected at an MOI of 3 (A) or 0.01 (B) with wild-type HSV-2 186, YK786 (HSV-2 UL7-SS/AA), YK787 (HSV-2 ΔUL7), or YK788 (HSV-2 UL7-repair). Total virus from cell culture supernatants and infected cells was harvested at 18 h (A) and 36 h (B) postinfection and assayed on UL7-TetON-Vero cells. Each value is the mean ± standard error of the mean of the results of three independent experiments. (C) Sixteen 6-week-old female ICR mice pretreated with medroxyprogesterone were infected vaginally with 0.75 × 104 PFU YK786 (HSV-2 UL7-SS/AA) or YK788 (HSV-2 UL7-repair) and scored for vaginal disease every day for 26 days postinfection. Each data point is the mean ± standard error of the mean of the scores. (D) The vaginal secretions of infected mice at 1 day postinfection in the experiment described in panel C were harvested, and virus titers were assayed on Vero cells. Each data point is the virus titer in the vaginal secretions of one mouse. The horizontal bars indicate the mean for each group. The dashed line indicates the limit of detection (2.5 PFU/ml). Viral titers below the limit of detection were assigned the value of the limit of detection and plotted on a graph, and statistical analysis was performed. (E) Survival of mice in the experiment described in panel C was monitored for 26 days postinfection. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, not significant by Tukey’s test (A and B), Mann-Whitney U test (C and D), and log rank test (E).
To examine the role of HSV-2 Us3 phosphorylation of UL7 in viral replication and pathogenesis in vivo, 6-week-old female ICR mice pretreated with medroxyprogesterone were infected vaginally with YK786 (HSV-2 UL7-SS/AA) or YK788 (HSV-2 UL7-repair). Survival of infected mice was monitored for 26 days, virus titers in vaginal secretions were determined at 1 day postinfection, and disease progression was observed at 1 to 26 days postinfection. SS/AA mutations in HSV-2 UL7 significantly reduced mortality of infected mice, development of vaginal disease at 6 to 26 days postinfection, and viral replication in the vagina at 1 day postinfection (Fig. 8C to E).
Taken together, these results suggest that although HSV-2 Us3 phosphorylation of UL7 had no effect on viral replication in cell culture, it was required for efficient HSV-2 replication and pathogenicity in vivo.
Effects of artificial Us3 phosphoregulation of UL7 in HSV-1 on viral infection.
Arg-287 in Us3 target sequences in HSV-2 UL7 was substituted for glutamine in HSV-1 UL7 (Fig. 1A). Because the corresponding amino acid sequences in HSV-1 UL7 did not match the Us3 target consensus sequence, HSV-1 UL7 may not be phosphorylated by Us3 in infected cells. In fact, HSV-1 UL7 from Vero cells infected with wild-type HSV-1(F) was detected as a single band in Phos-tag(+) SDS-PAGE gels (Fig. 9A), unlike HSV-2 UL7 from cells infected with wild-type HSV-2 strain 186 (Fig. 7B and C). We investigated the effects of artificial creation of the Us3 target site in HSV-1 UL7 in infected cells. We constructed recombinant virus YK791 (HSV-1 UL7-Q287R) in which HSV-1 UL7 Gln-287 was replaced with arginine (Q287R), its repaired virus YK792 (HSV-1 UL7-QR-repair), recombinant virus YK793 (HSV-1 UL7-QSS/RAA) encoding a mutant UL7 carrying the Q287R mutation and alanine substitutions in Ser-289 and Ser-290 (SS/AA), and recombinant virus YK794 (HSV-1 UL7-Q287R/Us3-K220M) encoding the UL7-Q287R mutant and enzymatically inactive Us3-K220M mutant (Fig. 6). In agreement with observations for HSV-2 UL7 from wild-type HSV-2 186-infected cells (Fig. 7B to D), HSV-1 UL7-Q287R from Vero cells infected with YK791 (HSV-1 UL7-Q287R) was detected as two predominant bands with mobility differences in Phos-tag(+) SDS-PAGE gels (Fig. 9A and B). The slower-migrating band disappeared after phosphatase treatment with lysate from infected cells (Fig. 9C), and the slower-migrating band of HSV-1 UL7 from cells infected with YK793 (HSV-1 UL7-QSS/RAA), YK794 (HSV-1 UL7-Q287R/Us3-K220M), or YK792 (HSV-1 UL7-QR-repair) was not detectable (Fig. 9A and B). Mutation(s) in each of these recombinant viruses had no effect on UL7 and Us3 protein accumulation in HSV-1-infected cells (Fig. 9D and E). These results indicated that HSV-1 Us3 did not phosphorylate UL7 in infected cells and that phosphorylation of the HSV-1 UL7-Q287R mutant was mediated by Us3 in infected cells, which required UL7 Ser-289 and/or Ser-290. This suggested that HSV-1 Us3 phosphorylated the UL7-Q287R mutant at Ser-289 and/or Ser-290 in infected cells.
FIG 9.

Effects of an artificial Us3 target site in HSV-1 UL7 on its phosphorylation in infected cells. (A and B) Vero cells were mock infected or infected with wild-type HSV-1(F), YK791 (HSV-1 UL7-Q287R), YK793 (HSV-1 UL7-QSS/RAA), YK794 (HSV-1 UL7-Q287R/Us3-K220M), or YK792 (HSV-1 UL7-QR-repair) at an MOI of 3, harvested at 8 h postinfection, solubilized, separated in Phos-tag(+) (top) or Phos-tag(−) (bottom) SDS-PAGE gels, and analyzed by immunoblotting with antibodies to UL7. (C) Vero cells were infected with YK791 (HSV-1 UL7-Q287R), harvested at 8 h postinfection, and lysed. The lysates were mock treated or treated with CIP overnight at 37°C and analyzed as described in panels A and B. (D and E) Vero cells were mock infected or infected with wild-type HSV-1(F), YK791 (HSV-1 UL7-Q287R), YK793 (HSV-1 UL7-QSS/RAA), YK794 (HSV-1 UL7-Q287R/Us3-K220M), or YK792 (HSV-1 UL7-QR-repair) at an MOI of 3, harvested at 8 h postinfection, and analyzed by immunoblotting with antibodies to Us3, gB, and β-actin. Digital images are representative of three independent experiments and molecular mass markers are shown on the left (panels A, D, and E).
Next, we examined effects of the Q287R mutation in HSV-1 UL7 on progeny virus yields in Vero cells and viral replication and pathogenicity in mice following vaginal infection. The Q287R mutation in HSV-1 UL7 had no effect on progeny virus yields in Vero cells at MOIs of 3 and 0.01, on mortality and development of vaginal disease in infected mice, and on viral replication in the vagina (Fig. 10A to G). Taken together, these results indicate that, unlike Us3 phosphorylation of UL7 in HSV-2, phosphorylation in HSV-1 by artificial creation of the Us3 target site in HSV-1 UL7 conferred no regulatory effects on viral replication and pathogenicity in vivo.
FIG 10.

Effects of an artificial Us3 target site in HSV-1 UL7 on viral infection in vitro and in vivo. (A to D) Vero cells were infected at an MOI of 3 (A and C) or 0.01 (B and D) with wild-type HSV-1(F), YK791 (HSV-1 UL7-Q287R), YK793 (HSV-1 UL7-QSS/RAA), YK794 (HSV-1 UL7-Q287R/Us3-K220M), or YK792 (HSV-1 UL7-QR-repair). Total virus from cell culture supernatants and infected cells was harvested at 18 h (A and C) and 36 h (B and D) postinfection and assayed on Vero cells. Each value is the mean ± standard error of the mean of three independent experiments. (E) Thirty-one 5-week-old female ICR mice pretreated with medroxyprogesterone were infected vaginally with 0.5 ×107 PFU YK791 (HSV-1 UL7-Q287R) or YK792 (HSV-1 UL7-QR-repair) and scored for vaginal disease every day for 26 days postinfection. Each data point is the mean ± standard error of the mean of the scores. (F) The vaginal secretions of infected mice at 1 day postinfection in the experiment described in panel E were harvested, and virus titers were assayed on Vero cells. Each data point is the virus titer in the vaginal secretions of one mouse. The horizontal bars indicate the mean of each group. (G) Survival of mice in the experiment described for panel E was monitored for 26 days postinfection. n.s., not significant by Tukey’s test (A to D), Mann-Whitney U test (E and F) and log rank test (G).
Evolution of Us3 phosphorylation of UL7 in simplex viruses.
To investigate linkage between evolution of Us3 phosphorylation of UL7 and phylogeny of simplex viruses, we performed phylogenetic analyses based on available whole-genome sequences of simplex viruses (Table 3). In agreement with earlier phylogenetic analyses of simplex viruses using parts of their viral genome sequences (28), HSV-2 and chimpanzee herpesvirus (ChHV) formed a monophyletic group, and HSV-1 belonged to a sister clade of the monophyletic group (Fig. 11A). Alignment of amino acid sequences of these simplex virus UL7 proteins showed that the Us3 target site in HSV-2 UL7 was completely conserved in the UL7 of ChHV (Fig. 11B). Next, we investigated whether ChHV Us3 mediated phosphorylation of UL7 via the putative Us3 target site. HEK293FT cells cotransfected with a plasmid expressing enhanced green fluorescent protein (EGFP) fused to wild-type ChHV UL7 (EGFP-ChHV UL7) or its mutant EGFP-ChHV UL7-SS/AA in which Ser-290 and Ser-291 in ChHV UL7 were replaced with alanines in combination with a plasmid expressing Flag-tagged wild-type ChHV Us3 (Flag-ChHV Us3) or its kinase-dead mutant Flag-ChHV Us3-K220M were analyzed on Phos-tag(+) SDS-PAGE gels. Ser-290 and Ser-291 in ChHV UL7 correspond to Ser-289 and Ser-290 in HSV-2 UL7 (Fig. 11B). EGFP-ChHV UL7 from cells cotransfected with plasmids expressing EGFP-ChHV UL7 or Flag-ChHV-Us3 was detected as three predominant bands with distinct mobility differences by Phos-tag(+) SDS-PAGE gel (Fig. 11C). After phosphatase treatment with lysate from cotransfected cells, the two slower-migrating bands were barely detectable, and the amount of protein in the faster-migrating band increased (Fig. 11D). Similarly, the two slower-migrating bands disappeared, and the amount of protein in the faster-migrating band increased in cells cotransfected with plasmids expressing EGFP-ChHV UL7 or Flag-ChHV Us3-K220M and in cells cotransfected with plasmids expressing EGFP-ChHV UL7-SS/AA or Flag-ChHV Us3 (Fig. 11C). Therefore, ChHV Us3-mediated phosphorylation of UL7 required Ser-290 and/or Ser-291, suggesting that ChHV Us3 phosphorylated UL7 at Ser-290 and/or Ser-291.
TABLE 3.
Strains and accession numbers of simplex viruses used for phylogenetic analyses
| Virus (designation) | Strain | GenBank accession no. |
|---|---|---|
| Herpes simplex virus 1 (HSV-1) | F | GU734771.1 |
| Herpes simplex virus 2 (HSV-2) | HG52 | NC_001798.2 |
| Chimpanzee herpes virus (ChHV) | 105640 | JQ360576.1 |
| Spider monkey herpesvirus (HVA-1) | Lennette | NC_034446.1 |
| Simian agent 8 (SA-8) | B264 | NC_006560.1 |
| B virus (Herpes B) | E2490 | NC_004812.1 |
| Leporid herpesvirus 4 (LHV-4) | LHV4012612 | NC_029311.1 |
| Parma wallaby herpesvirus (PWHV) | MaHV1.3076/08 | NC_029132.1 |
| Herpesvirus papio 2 (HVP-2) | X313 | NC_007653.1 |
| Marmoset herpesvirus (HVS-1) | MV 5-4 | NC_014567.1 |
FIG 11.

Phylogeny of simplexviruses and Us3 phosphorylation of UL7 in ChHV. (A) Phylogenetic tree of evolutionary relationships of taxa based on whole-genome sequences of viruses belonging to the genus Simplexvirus listed in Table 3. The evolutionary history was inferred using the neighbor-joining method (66). An optimal tree with a branch length summation of 1.42462506 is shown. The percentages of replicate trees in which the associated taxa clustered together in the bootstrap test (500 replicates) are shown next to the branches (69). The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The evolutionary distances were computed using the p-distance method (67) and are in units of the number of base differences per site. The rate variation among sites was modeled with a gamma distribution (shape parameter = 1). The analysis involved 10 nucleotide sequences. All positions containing gaps and missing data were eliminated. There were 70,936 positions in the final data set. Evolutionary analyses were conducted in MEGA7 (67). (B) Amino acid sequence alignment of UL7 homologs of HSV-2, ChHV, HSV-1, herpes B, SA-8, PaHV-2, LeHV-4, PWHV, HVA-1, and HVS-1. Amino acid sequences of HSV-2 and ChHV UL7 that matched the Us3 consensus target sequence are shaded. These Us3 target sequences and the corresponding residues of other viruses are enclosed with a dotted square. Phosphosites of UL7 in HSV-2 186 and the corresponding residues of other viruses are boxed with a black square. At the bottom, the conservation ratio has been deduced in a histogram. (C) HEK293FT cells were transfected with pEGFP-ChHV-UL7o or pEGFP-ChHV-UL7o-SS/AA, along with pFlag-ChHV-Us3o or pFlag-ChHV-Us3o-K220M (kinase-dead control), harvested at 48 h posttransfection, solubilized, separated in a Phos-tag(+) (top) or a Phos-tag(−) (middle and bottom) SDS-PAGE gel, and analyzed by immunoblotting with antibodies to GFP and Flag. (D) HEK293FT cells were transfected with pEGFP-ChHV-UL7o, along with pFlag-ChHV-Us3o, harvested at 48 h posttransfection, and lysed. The lysates were mock treated or treated with CIP overnight at 37°C and analyzed as described in panel C. Digital images are representative of three independent experiments, and molecular mass markers are shown on the left (C and D).
Sequences of UL7s in simplex viruses other than HSV-2 and ChHV corresponding to the Us3 target sites in HSV-2 and ChHV did not match the Us3 target consensus sequence (Fig. 11B). However, Us3 target specificity was similar to the specificities of the cellular protein kinases Akt and PKA (6, 29–31), and the corresponding sequences in marmoset herpesvirus (HVS-1) (KTRGESLF) matched the consensus target sequence of Akt [(R/K)XR/KXX(pS/T)] (Fig. 11B). Among simplex viruses tested, HVS-1 was the most phylogenetically distinct from HSV-2; therefore, we did not investigate whether HVS-1 Us3 phosphorylated UL7. These results suggested that Us3 phosphorylation of UL7 was not conserved in simplex viruses other than HSV-2 and ChHV and possibly HVS-1. Thus, evolution of Us3 phosphorylation of UL7 was linked to the phylogeny of simplex viruses that are physiologically close to HSV-2 and ChHV. It appears that viruses in the monophyletic group consisting of HSV-2 and ChHV acquired this phosphoregulation during their evolution.
To test this, we used maximum likelihood to reconstruct the sequence of the last common ancestor of UL7 for HSV-1, HSV-2, and ChHV (Anc-A UL7) (Fig. 12A and Table 4) and that of the last common ancestor of UL7 for HSV-2 and ChHV (Anc-B UL7) (Fig. 12A and Table 4). Anc-B UL7 contained the putative Us3 target site whose amino acid sequences were identical to those in UL7 of HSV-2 and ChHV (Fig. 12B), whereas corresponding amino acid sequences in Anc-A UL7 were identical to those in HSV-1 UL7 (Fig. 12B). These results suggested that Us3 phosphoregulation of UL7 was not conserved in Anc-A UL7, similar to results for HSV-1 UL7. Phos-tag(+) SDS-PAGE analyses similar to those of UL7 of ChHV (Fig. 11) showed that HSV-2 Us3 mediated phosphorylation of Anc-B UL7 and that this phosphorylation required Ser-288 and/or Ser-289, similar to findings for the UL7s of HSV-2 and ChHV. However, the viral kinase could not phosphorylate Anc-A UL7 unless a Us3 target site was artificially created by replacing Gln-286 with arginine, similar to UL7 of HSV-1 (Fig. 12C to F). Similar results were obtained with Phos-tag(+) SDS-PAGE analyses using Us3s of HSV-1 and ChHV (data not shown), suggesting that Us3 phosphorylation of UL7 was conserved in Anc-B UL7 but not in Anc-A UL7, in agreement with our hypothesis that the monophyletic group consisting of HSV-2 and ChHV acquired Us3 phosphoregulation of UL7 during evolution.
FIG 12.

Us3 phosphorylation of UL7 ancestors. (A) Phylogenetic tree based on UL7 nucleotide sequences using a maximum-likelihood estimation (70). The analysis involved the nucleotide sequences of 10 simplexvirus UL7 homologs listed in Table 3. Evolutionary analyses and inference of ancestral sequences were conducted in MEGA7 (67), which uses a maximum-likelihood approach to estimate ancestral character states and the phylogeny. A closed circle indicates the last common ancestral UL7 for HSV-1, HSV-2, and ChHV UL7s, termed Anc-A-UL7, whereas an open circle indicates the last common ancestral UL7 for HSV-2 and ChHV, termed Anc-B-UL7. (B) Amino acid sequence alignment of UL7s of HSV-2, ChHV, Anc-A, and Anc-B. Amino acid sequences of HSV-2, ChHV, and Anc-B UL7s that matched the Us3 consensus target sequence are shaded. These Us3 target sequences and the corresponding residues of HSV-1 and Anc-A are enclosed with a dotted square. Phosphosites of UL7 in HSV-2 and the corresponding residues of other viruses are boxed with a black square. At the bottom, the conservation ratio has been deduced in a histogram. (C and D) HEK293FT cells were transfected with pEGFP-Anc-A-UL7o, pEGFP-Anc-Ao-UL7-Q286R, or pEGFP-Anc-Ao-UL7-QSS/RAA, along with pFlag-HSV-2-Us3 or pFlag-HSV-2-Us3-K220M, harvested at 48 h posttransfection, solubilized, separated in a Phos-tag(+) (top) or a Phos-tag(−) (middle and bottom) SDS-PAGE gel, and analyzed by immunoblotting with antibodies to GFP or Flag. (D) HEK293FT cells were transfected with pEGFP-Anc-Ao-UL7, along with pFlag-HSV-2-Us3, harvested at 48 h posttransfection, and lysed. The lysates were mock treated or treated with CIP overnight at 37°C and analyzed as described in panel C. (E and F) HEK293FT cells were transfected with pEGFP-Anc-Bo-UL7 or pEGFP-Anc-Bo-UL7-SS/AA, along with pFlag-HSV-2-Us3 or pFlag-HSV-2-Us3-K220M, harvested at 48 h posttransfection, and assayed as described in panel C. (F) HEK293FT cells were transfected with pEGFP-Anc-Bo-UL7, along with pFlag-HSV-2-Us3, harvested at 48 h posttransfection, and assayed as described for panel D. Digital images are representative of three independent experiments, and molecular mass markers are shown on the left (C to F).
TABLE 4.
Summary of UL7 sequences inferred by bioinformatics analyses
| UL7 ancestor | Amino acid sequencea |
|---|---|
| AncA-UL7 | MAAPTPDDEGTAAAILKQAIAGDRSLVEVAEAISHQALLRMACEVRQVSDRQPRFTATSVVRVDVTPGCRLRFVLDGSPDDAYVASEDYFKRCCGQSTYRGFAVAVLTANEDHVHSLAVPPLVLLHRLSLFRPRDLRDFELACLLMYLENCPRSHATPSTFVKVSAWLGVVGRRTSPFERVRCLLLRSCHWILNTLMFMVHVKPFDDEFVLPHWYMARYLLANNPPPVLSALFCATPSSAFRLPGPIPRFDCVAYNPAGVMGSCWASEDLRAALVYWWLSETPKrqtsslfYQFC |
| AncB-UL7 | MADPTPADEGTAAAILKQAIAGDRSLVEVAEGISNQALLRMACEVRQVSDRQPRFTATSVVRVDVTPGGRLRFVLDGSSDDAYVASEDYFKRCGGQPTYRGFAVVVLTANEDHVHSLAVPPLVLLHRLSLFRPTDLRDFELVCLLMYLENCPRSHATPSLFVKVSAWLGVVARRASPFERVRCLLLRSCHWILNTLMCMAGVKPFDDELVLPHWYMAHYLLANNPPPVLSALFCATPSSAFQLPGPIPRMDCVAYNPAGVMGSCWKSKDLRSALVYWWLSGNPKRRTSSLFYQFC |
Us3 phosphorylation consensus sequences of AncA- and AncB-UL7 are underlined. Homologous residues of AncA-UL7 are in lowercase.
Evolution of Us3 phosphorylation of gB and Us3 itself in simplex viruses.
To further examine linkage between the evolution of phosphoregulation in simplex viruses and their phylogeny, we focused on HSV-1 Us3 phosphoregulation on gB Thr-887 and Us3 Ser-147 (14, 32), which are not conserved in HSV-2. Sequences in Us3 and gB homologs of other simplex viruses corresponding to Us3 target sites in HSV-1 gB and Us3 were missing or did not match the Us3 consensus target sequence (Fig. 13A and B). However, sequences in gB homologs of ChHV (RNKTRTNY) and HVS-1 (LRKRNTAR) matched consensus target sequences of Akt [(R/K)XR/KXX(pS/T)] and PKA [(R/K)R/KX(pS/T)], respectively. To examine whether ChHV Us3 mediated phosphorylation of gB via RNKTRTNY sequences, HEK293FT cells were cotransfected with a plasmid expressing EGFP tagged with a carboxyl-terminal domain of ChHV gB containing RNKTRTNY sequences (EGFP-ChHV gB769–903), EGFP tagged with a carboxyl-terminal domain of wild-type HSV-1 gB containing the Us3 target site (EGFP-HSV-1 gB770–904), or its mutant EGFP-HSV-1 gB770–904T887A in which the Us3 phosphorylation site in EGFP-HSV-1 gB770–904 was replaced with alanine in combination with that expressing Flag-HSV-1 Us3 or Flag-ChHV Us3. Transfected cells were lysed, immunoprecipitated with anti-GFP antibody, and subjected to immunoblotting with anti-phospho-AKT substrate antibody 110B7E, which recognizes proteins containing a phosphorylated serine or threonine and arginine at residue position −3 (RXXS or RXXT) and therefore should detect Us3 phosphorylation on the putative phosphorylation site in ChHV gB (Thr-886) and Us3 phosphorylation on HSV-1 gB Thr-887. The antibody recognized immunoprecipitated EGFP-HSV-1 gB770–904 but not EGFP-HSV-1 gB770–904T887A by immunoblotting (Fig. 14). In contrast, the antibody did not recognize immunoprecipitated EGFP-ChHV gB769–903 (Fig. 14A). We confirmed the anti-phospho-Akt substrate antibody-detected phosphorylation of EGFP-ChHV gB769–903 when ChHV gB Thr-886 was phosphorylated, based on the observation that the antibody functioned in an enzyme-linked immunosorbent assay (ELISA) with synthetic phosphopeptides corresponding to HSV-1 gB residues 881 to 893 or ChHV gB residues 880 to 892 in which Thr-887 or Thr-886 was artificially phosphorylated, respectively, but did not recognize the unphosphorylated ChHV gB peptide (Fig. 14B). These results indicated that ChHV Us3 did not phosphorylate ChHV gB via the sequence corresponding to the Us3 target site in HSV-1 gB. As with the HVS-1 Us3 phosphorylation of UL7, we did not investigate whether HVS-1 Us3 phosphorylated gB via the sequence described above.
FIG 13.

Evolution of the Us3 phosphorylation of UL7, gB, and Us3. (A and B) Amino acid sequence alignments of gB (A) and Us3 (B) homologs of simplex viruses. Amino acid sequences that matched the Us3 consensus target sequence are shaded. The Us3 target sequence of HSV-1 and the corresponding residues of other viruses are enclosed with a dotted square. Phosphosites of gB and Us3 in HSV-1 and the corresponding residues of other viruses are boxed with a black square. At the bottom, the conservation ratio has been deduced in a histogram. (C) Phylogenetic tree based on whole-genome sequences of simplex viruses listed in Table 3. Conservation of Us3 phosphorylation of UL7, gB, and Us3 itself based on panels A and B and on Fig. 11B is also indicated.
FIG 14.

Us3 phosphorylation of HSV-1 and ChHV gB in cell-cultures. (A) HEK293FT cells were transfected with pEGFP-HSV-1-gB770–904, pEGFP-HSV-1-gB770–904-T887A, or pEGFP-ChHV-gBo769–903, along with pFlag-HSV-1-Us3 or pFlag-ChHV-Us3o, harvested at 48 h posttransfection, immunoprecipitated (IP) with anti-GFP antibody (α-GFP), and analyzed by immunoblotting with anti-phospho-Akt substrate, anti-GFP, and anti-Flag antibodies. (B) Synthesized peptides shown in Table 10 were used to coat ELISA plates, and the reactivity of anti-phospho-Akt substrate antibody to each peptide was analyzed by ELISA. Each value is the mean ± standard error of the mean of the results of three independent experiments.
These results suggested that Us3 phosphorylation of Us3 itself was not conserved in simplex viruses other than HSV-1 and that Us3 phosphorylation of gB was not conserved in simplex viruses other than HSV-1 and possibly HVS-1. Thus, evolution of Us3 phosphoregulation of gB and Us3 itself was linked to phylogeny of simplex viruses, which are phylogenetically close to HSV-1, in agreement with evolution of Us3 phosphoregulation of UL7.
DISCUSSION
HSV-1 UL7, a structural component of the virion tegument, a proteinaceous layer located between the nucleocapsid and envelope, is conserved in all herpesviruses. HSV-1 UL7 and its homolog of a porcine alphaherpesvirus, pseudorabies virus, are important for viral replication and cell-to-cell spread in cell culture (24, 25, 33), viral replication, and pathogenicity in vivo (27, 34) and are involved in final envelopment in conjunction with another tegument protein, UL51 (26). This effect on final envelopment was also shown for a UL7 homolog of a human betaherpesvirus, human cytomegalovirus (35). In contrast, HSV-2 UL7 has not been characterized, and the regulatory mechanism of UL7 in infected cells has not been investigated in any herpesviruses to date. Here, we report that HSV-2 UL7 was regulated by Us3 phosphorylation in infected cells and that this phosphoregulation was important for viral replication and pathogenicity in vivo. This is the first report to identify a physiological HSV-2 Us3 substrate in infected cells. Similar to Us3 phosphoregulation of UL7 in HSV-2 clarified in this study, HSV-1 Us3 was reported to phosphorylate another major tegument protein, UL47 (36), which promoted viral replication and pathogenicity in vivo without affecting viral replication in cell cultures. HSV-1 UL47 is involved in HSV-1 morphogenesis (37). These observations suggested that optimal regulation of tegument proteins by Us3 phosphorylation during virion morphogenesis is critical for HSV infection in vivo but not for HSV replication in cell culture.
Divergent regulation of gene expression, protein-protein interactions, and protein posttranslational regulation are thought to drive phenotypic variation across species of organisms (4, 38–40) and possibly across virus species as well during their evolution. Here, we focused on the role of one of these mechanisms, that of viral protein phosphoregulation in viral evolution. The evolution of protein phosphoregulation might contribute to phenotypic diversity and the evolution of species. Thus, by analyzing resurrected ancestral proteins, evolution of an essential function of a transcription factor, CCAAT/enhancer binding protein-β (C/EBP-β) that induces prolactin during pregnancy in placental animals, was shown to coincide with reorganization of multiple phosphosites in the transcription factor during evolution from nonplacental to placental animals (40). Similarly, we examined the acquisition of Us3 phosphoregulation of UL7 in simplex viruses and the reconstituted last common ancestors for HSV-1, HSV-2, and ChHV. Evolution of Us3 phosphoregulation of UL7 was linked to phylogeny of simplex viruses other than HVS-1, the most phylogenetically distinct from HSV-2 (Fig. 13C). To the best of our knowledge, this is the first evidence showing that evolution of phosphoregulation of a viral protein in a virus species coincides with its phylogeny. Acquisition of the Us3 phosphoregulation of UL7 in HSV-2, ChHV, and the common ancestors of HSV-2 and ChHV might impact the fitness of these viruses, whereas acquisition of phosphoregulation in HSV-1, the common ancestor of HSV-2, ChHV, and HSV-1 and most other simplex viruses had little effect. Therefore, during evolution, ChHV and HSV-2 maintained Us3 phosphoregulation of UL7, but HSV-1 and most other simplex viruses did not if they acquired phosphoregulation. Supporting this hypothesis, we experimentally showed that Us3 phosphoregulation of UL7 in HSV-2 was required for efficient viral replication and pathogenicity in vivo, whereas artificial Us3 phosphorylation of UL7 in HSV-1 had little effect. Similarly, Us3 phosphoregulation of Us3 Ser-147 and gB Thr-887 in HSV-1, which phylogenetically forms a monophyletic group, was not conserved in Us3 and gB homologs of simplex viruses other than HVS-1 (Fig. 13C). There are other instances of the evolution of Us3 phosphoregulation linked to phylogeny of simplex viruses, suggesting that HSV-1 acquired Us3 phosphoregulation of gB and Us3 itself during its evolution. Taken together, as hypothesized in organismal evolution (40), these observations suggest that evolution of phosphoregulation of viral proteins may drive phenotypic variation across virus species during their evolution; however, further studies on evolutional and phylogenetic linkage to viral phosphoregulation in other virus species and on how variations in viral phosphoregulation translate into phenotypic changes with a measurable impact on viral fitness are required. These studies may be accelerated by the increased body of data on phosphoproteomes in various virus infections (41–43). Of note, significant numbers of phosphosites are thought to be nonfunctional. Therefore, the studies require not only identification of viral phosphosites comprehensively but also their functional in-depth analyses as conducted in this study.
MATERIALS AND METHODS
Cells and viruses.
Vero, rabbit skin, HEK293FT, Plat-GP, and Sf9 cells, as well as wild-type HSV-1(F) and HSV-2 strain 186, were described previously (44–47). Recombinant virus YK356 (HSV-2 BAC) reconstituted from pYEbac356 containing an HSV-2 strain 186 sequence with a BAC sequence between UL50 and UL51, was described previously (22).
Construction of a self-excisable HSV-2 186 BAC.
To construct Escherichia coli GS1783 containing a self-excisable HSV-2 BAC plasmid (pYEbac861Cre) (Fig. 4), a two-step Red-mediated mutagenesis procedure was performed as described previously (32, 48), except that the primers, plasmids, and Escherichia coli strains listed in Table 5 were used.
TABLE 5.
Oligonucleotide sequences, plasmids, and E. coli GS1873 strains for the construction of recombinant viruses
| Recombinant virus | Oligonucleotide sequence (5′–3′) | Plasmid DNA template (reference) | E. coli GS1873 straina (source or reference) |
|---|---|---|---|
| YK785 (HSV-2 ΔBAC) | TAGTGAACCGTCAGATCCGCTAGCGCTACCGGTCGCCACCATGCCCAAGAAGAAGAGGAAGGTGTCCA | pcDNA-CREin-KanS (50) | GS1783/pYEbac861 (20) |
| GTAAAACCTCTACAAATGTGGTATGGCTGATTATGATCAGCTAATCGCCATCTTCCAGCA | |||
| YK786 (HSV-2 UL7-SS/AA) | GGTGTATTGGTGGCTTTCGGGGAGCCCCAAACGACGGACCGCGGCGCTTTTCTATCGGTTTTGCAGGATGACGACGATAAGTAGGG | pEP-KanS (48) | GS1783/pYEbac861Cre (this study) |
| ACGTTTATTTTCCGGAGTTAGCAAAACCGATAGAAAAGCGCCGCGGTCCGTCGTTTGGGGCTCCCAACCAATTAACCAATTCTGATTAG | |||
| YK787 (HSV-2 ΔUL7) | GTCGAACGCAGGGATGGCCGACCCCACGCCCGCCGATGAGCTTTCGGGGAGCCCCAAACGAGGATGACGACGATAAGTAGGG | pEP-KanS (48) | Containing the YK786 genome (this study) |
| AGAAAAGCGACGAGGTCCGTCGTTTGGGGCTCCCCGAAAGCTCATCGGCGGGCGTGGGGTCAACCAATTAACCAATTCTGATTAG | |||
| YK789 (HSV-2 Us3-K220M) | TGATAGCAGCCACCCGAACTACCCTCATCGGGTAATCGTCATGGCGGGGTGGTACGCCAGAGGATGACGACGATAAGTAGGG | pEP-KanS (48) | GS1783/pYEbac861Cre (this study) |
| GCCGCGCCTCGTGGCTCGTGCTGGCGTACCACCCCGCCATGACGATTACCCGATGAGGGTCAACCAATTAACCAATTCTGATTAG | |||
| YK790 (HSV-2 Us3-KM-repair) | TGATAGCAGCCACCCGAACTACCCTCATCGGGTAATCGTCAAGGCGGGGTGGTACGCCAGAGGATGACGACGATAAGTAGGG | pEP-KanS (48) | Containing the YK789 genome (this study) |
| GCCGCGCCTCGTGGCTCGTGCTGGCGTACCACCCCGCCTTGACGATTACCCGATGAGGGTCAACCAATTAACCAATCTGATTAG | |||
| YK791 (HSV-1 UL7-Q287R) | CCTCTGGTCTATTGGTGGCTTTCGGAGACCCCAAAACGACGGACGTCGTCGCTGTTTTATCAGGATGACGACGATAAGTAGGG | pEP-KanS (48) | GS1783/pYEbac8102Cre (50) |
| TCCTAAAATTCAACAAAACTGATAAAACAGCGACGACGTCCGTCGTTTTGGGGTCTCCGAACAACCAATTAACCAATTCTGATTAG | |||
| YK792 (HSV-1 UL7-QR-repair) | CCTCTGGTCTATTGGTGGCTTTCGGAGACCCCAAAACGACAGACGTCGTCGCTGTTTTATCAGGATGACGACGATAAGTAGGG | pEP-KanS (48) | Containing the YK791 genome (this study) |
| TCCTAAAATTCAACAAAACTGATAAAACAGCGACGACGTCTGTCGTTTTGGGGTCTCCGAACAACCAATTAACCAATTCTGATTAG | |||
| YK793 (HSV-1 UL7-QSS/RAA) | CCTCTGGTCTATTGGTGGCTTTCGGAGACCCCAAAACGACGGACGGCGGCGCTGTTTTATCAGTTTTGTAGGATGACGACGATAAGTAGGG | pEP-KanS (48) | Containing the YK791 genome (this study) |
| GGTTTATTTCCTAAAATTCAACAAAACTGATAAAACAGCGCCGCCGTCCGTCGTTTTGGGGTCTCCGAACAACCAATTAACCAATTCTGATTAG | |||
| YK794 (HSV-1 UL7-Q287R/Us3-K220M) | GACAGCAGCCACCCAGATTACCCCCAACGGGTAATCGTGATGGCGGGGTGGTACACGAGCACAGGATGACGACGATAAGTAGGG | pEP-KanS (48) | Containing the YK791 genome (this study) |
| CAGTCGCGCCTCGTGGCTCGTGCTCGTGTACCACCCCGCCATCACGATTACCCGTTGGGGCAACCAATTAACCAATTCTGATTAG |
Containing HSV-BAC.
Plasmids.
pcDNA-Myc or pcDNA-SE was constructed by annealing oligonucleotides (Table 6) and cloning it into pcDNA3.1(+) (Thermo Fisher Scientific). Based on the genome information of ChHV (Table 3) and inferred ancestral sequences of UL7 (AncA- or AncB-UL7) (Table 4), pUC57-ChHV-Us3o, pUC57-ChHV-UL7o, pUC57-ChHV-gBCTDo, pUC57-AncA-UL7o, and pUC57-AncB-UL7o (Table 7) were synthesized by GenScript. These sequences were codon-optimized variants. pFLAG-ChHV-Us3o, pcDNA-Myc-ChHV-UL7o, pEGFP-ChHV-gBo769–903, pEGFP-AncA-UL7o, and pEGFP-AncB-UL7o were constructed by subcloning the DNA fragments into the recipient plasmids listed in Table 8, in frame with the tag sequence. pEGFP-HSV-1-gB770–904 was constructed by PCR amplifying HSV-1 gB (codons 770 to 904) using the primers and DNA template listed in Table 9 and cloning it into pEGFP-C2 (Clontech). Other plasmids were constructed by standard PCR procedures using the primers, DNA templates, and parental plasmids listed in Table 9.
TABLE 6.
Oligonucleotide sequences and parental plasmids for modification by annealed oligonucleotide cloning
| Parental plasmida | Oligonucleotide sequence (5′–3′) |
|---|---|
| pcDNA-Myc | AGCTTGCCACCATGGAGCAGAAGCTGATCTCAGAGGAGGACCTGG |
| GATCCCAGGTCCTCCTCTGAGATCAGCTTCTGCTCCATGGTGGCA | |
| pcDNA-SE | AGCTTACCATGGCTAGCTGGAGCCACCCGCAGTTCGAGAAAGGTGGAGGTGCCCGAGGTGGATCGGGAGGTGGATCGTGGAGCCACCCGCAGTTCGAAAAAGGAGGTTCAGAGAATTTGTATTTTCAGGGTG |
| AATTCACCCTGAAAATACAAATTCTCTGAACCTCCTTTTTCGAACTGCGGGTGGCTCCACGATCCACCTCCCGATCCACCTCGGGCACCTCCACCTTTCTCGAACTGCGGGTGGCTCCAGCTAGCCATGGTA-3′ |
The recipient plasmid was pcDNA3.1(+) (Thermo Fisher Scientific).
TABLE 7.
Summary of constructed plasmids and synthesized plasmids
| Constructed plasmid | Synthesized DNA sequencea |
|---|---|
| pUC57-ChHV-Us3o | AAGCTTGCATGTAGGAAGTTCTGTCGAGTGTATGGAAGACCCGATAAAAGGCAGGAGGCCAGCGTCCCACCCGAGACCAAAACCGCCCCAGTCTTCCCTAGCGGCACCTTTTACACACCTGCAGAGGATGCATATCTGGCATCTGGCTCTCCTGAGACAATCCACCCAAGCCGGCCCCCTTCCCCTGGAGAGGCAGCACGGCTGAGACAGCTGCAGGAGATCCTGGCCCAGATGCACTCCGATGAGGACTACCCTATCGTGGATGCAGCAGGAGCAGAGGAGGGCGGCGAGGCAGACGATGACGCCCCAGATGACGTGGCCTACCCCGATGACTATGCCGAGGGCAGGTTCCTGTCTATGGTGAGCACAGCCCCTCTGCCAGGCGACGGCGGACACCTGCCCGTGCCATGCAGAGCAGCACACTCTGATGTGAGAACCTGTGACTCCACAAAAGTGGGCGCCACCGGCTTTACACCAGAGGAGCTGGATACCATGGACCGGGAGGCCCTGAGAGCCATCTCTAGGGGCTGCAAGCCACCCAGCACACTGGCCAAGCTGGTGACAGGCCTGGGCTTCACAATTCACGGCGCCCTGATCCCTGGATCTGAGGGCTGCGTGTTCGATAGCTCCCACCCTGACTACCCACAGAGAGTGATCGTGAAGGCCGGCTGGTATGCCAGCACCTCCCACGAGGCCAGACTGCTGCGGAGACTGGATCACCCAGCCATCCTGCCTCTGCTGGACCTGCACGTGGTGAGCGGAGTGACCTGCCTGGTGCTGCCCAAGTACCAGTCCGATCTGTACACATATCTGTCTAAGAGGCCAAGCCCCCTGGGACACCTGCAGATTGCAGCCGTGTCCAGACAGCTGCTGTCTGCCATCGATTATGTGCACAGGGAGGGCATCATCCACCGCGACATCAAGACCGAGAACATCTTCATCAATACACCTGAGAACATCTGTCTGGGCGATTTTGGAGCAGCATGCTTTGTGCGGGGATGTAGATCTAGCCCATTTCAGTACGGCATCGCCGGCACCATCGACACAAATGCCCCCGAGGTGCTGGCCGGCGATCCTTATACCCAGGTCATCGACATCTGGTCTGCCGGCCTGGTCATCTTCGAGACAGCCGTGCACAACGCCTCCCTGTTTTCTGCCCCCAGGGATCCTGAGAGGCGCCCATGCGACTCCCAGATCGCCAGGATCATCCGCCAGGCCCAGGTGCACGTGGATGAGTTCCCTACCCACGCAGAGTCCAGACTGACCGCACACTATCGGAGCCGGGCAGCAGGAAACAATAGACCCGCCTGGACCCGGCCTGCCTGGACAAGATACTATAAGATCCACACCGATGTGGAGTATCTGATCTGTAAGGCCCTGACATTTGACGCCGCTCTGAGACCTTCCGCCGCCGAACTGCTGAGACTGCCACTGTTTCATCCAAAGTGAGAATTC |
| pUC57-ChHV-Us7o | GAATTCGGCCGACCCAACCCCCGCAGATGAAGGGACAGCAGCCGCTACAATCCTGAAACAGGCAATCGCCGGAGACAGATCACTGGTGGAGGTGGCCGAGGGCATCTCCAATCAGGCCCTGCTGAGAATGGCATGTGAGGTGCGGCAGGTGTCTGATAGACAGCCACGCTTCACCGCCACAAGCGTGGTGAGGGTGGACGTGACACCTGGCGGCAGGCTGAGATTTGTGCTGGATGGCAGCTCCGACGATGCCTACGTGGCCTCTGAGGACTATTTCAAGAGATGTGGCGGACAGCCTACCTACAGGGGCTTTGCCGTGGTGGTGCTGACAGCCAACGAAGACCACGTGCACAGCCTGGCAGTGCCCCCTCTGGTGCTGCTGCACAGACTGTCCCTGTTCAGACCAACCGACCTGAGGGATTTTGAGCTGGTGTGCCTGCTGATGTATCTGGAGAATTGTCCAAAGAGCCACGCCACACCCTCCCTGTTCGTGAAGGTGTCCGCCTGGCTGGGAGTGGTGGCAAGAAGAGCATCTCCCTTTGCCCGGGTGAGATGCCTGCTGCTGCGGTCTTGTCACTGGATCCTGAACACCCTGATGTGCATGGCCGGCATCAAGCCTTTCGACGATGGCCTGGTGCTGCCACACTGGTACATGGCCCACTATCTGCTGGCCAACAATCCACCCCCTGTGCTGAGCGCCCTGTTCTGTGCCACACCACAGTCTAGCGCCTTTCAGCTGCCAGGCCCCATCCCTCGCATGGATTGCGTGGCCTACAATCCCGCCGGCGTGATGGGCTCCTGTTGGAAGTCTAAGGACCTGCGGAGCGCCCTGGTGTATTGGTGGCTGAGCGGCAATAGGAAGCGGCGGACATCATCCCTGTTTTACCAGTTTTGTTAAGATATC |
| pUC57-ChHV-gBo769–903 | AAGCTTGAATTCTTTATGTCTAACCCCTTCGGGGCTCTGGCCGTGGGACTGCTGGTGCTGGCCGGGCTGGCTGCCGCCTTCTTTGCCTTTCGCTATGTGCTGCGGCTGCAGAGCAACCCAATGAAGGCCCTGTACCCCCTGACCACAAAGGAGCTGAAGAATCCCGACCCTCAGGGAGTCGGAGGAGAGGGAGAGGAGGGAGGAGGATTCGATGAGGCAAAGCTGGCCGAGGCCCAGGAGATGATCAGGTATATGGCCCTGGTGTCTGCCATGGAGCGCACCGAGCACAAGGCCAAGAAGAAGGGCACAAGCGCCCTGCTGAGCTCCAAGGTGACCAACATGGTGCTGCGGAAGAGAAATAAAACTCGGTATTCCCCACTGCATAATGAAGATGAGGCCGGAGATGAGGACGAACTGTGAGTCGACAAGCTT |
| pUC57-AncA-UL7o | AAGCTTGAATTCGCCGCCCCAACCCCAGACGATGAGGGAACAGCAGCAGCAATCCTGAAGCAGGCAATCGCAGGCGATAGGTCCCTGGTGGAGGTGGCCGAGGCCATCTCTCACCAGGCCCTGCTGAGGATGGCATGCGAGGTGCGCCAGGTGAGCGACAGGCAGCCAAGATTCACCGCCACATCCGTGGTGCGCGTGGACGTGACCCCAGGATGTAGGCTGCGCTTTGTGCTGGATGGCTCTCCCGACGATGCCTACGTGGCCAGCGAGGACTATTTCAAGAGGTGCTGTGGCCAGTCTACCTACAGGGGATTTGCAGTGGCCGTGCTGACAGCCAACGAGGATCACGTGCACTCCCTGGCAGTGCCACCTCTGGTGCTGCTGCACAGGCTGTCTCTGTTCCGGCCCAGAGACCTGAGAGATTTTGAGCTGGCCTGCCTGCTGATGTATCTGGAGAATTGTCCTCGGAGCCACGCCACCCCATCCACATTCGTGAAGGTGTCTGCCTGGCTGGGAGTGGTGGGCCGGAGAACCAGCCCTTTTGAGAGGGTGCGCTGCCTGCTGCTGCGGAGCTGTCACTGGATCCTAAACACACTGATGTTCATGGTGCACGTGAAGCCCTTCGACGATGAGTTTGTGCTGCCTCACTGGTACATGGCCAGATATCTGCTGGCCAACAATCCACCACCTGTGCTGTCCGCCCTGTTCTGCGCAACACCTAGCTCCGCCTTTCGGCTGCCTGGCCCAATCCCCAGATTTGATTGCGTGGCCTACAATCCAGCAGGCGTGATGGGCTCCTGTTGGGCATCTGAGGACCTGAGGGCCGCCCTGGTGTACTGGTGGCTGTCCGAGACCCCTAAGCGCCAGACATCTAGCCTGTTCTATCAGTTTTGTTGACTCGAGAAGCTT |
| pUC57-AncB-UL7o | AAGCTTGAATTCGCCGACCCAACCCCAGCAGATGAGGGAACAGCAGCAGCAATCCTGAAGCAGGCAATCGCAGGCGATCGGAGCCTGGTGGAGGTGGCAGAGGGAATCTCCAACCAGGCCCTGCTGAGAATGGCCTGCGAGGTGAGGCAGGTGTCTGACCGGCAGCCAAGATTCACCGCCACAAGCGTGGTGAGAGTGGACGTGACCCCAGGAGGCCGGCTGAGATTTGTGCTGGATGGCAGCTCCGACGATGCCTACGTGGCCAGCGAGGACTACTTCAAGCGGTGCGGAGGACAGCCAACCTACAGGGGCTTTGCCGTGGTGGTGCTGACAGCCAACGAGGATCACGTGCACAGCCTGGCAGTGCCACCTCTGGTGCTGCTGCACAGGCTGTCCCTGTTCCGCCCTACCGACCTGCGGGATTTTGAGCTGGTGTGCCTGCTGATGTATCTGGAGAATTGTCCTCGCTCTCACGCCACACCAAGCCTGTTCGTGAAGGTGTCCGCCTGGCTGGGAGTGGTGGCACGGAGAGCCTCTCCATTTGAGAGGGTGCGCTGCCTGCTGCTGCGGTCTTGTCACTGGATCCTAAATACCCTGATGTGCATGGCCGGCGTGAAGCCCTTCGACGATGAGCTGGTGCTGCCTCACTGGTACATGGCCCACTATCTGCTGGCCAACAATCCACCCCCTGTGCTGTCCGCCCTGTTCTGTGCCACACCTTCTAGCGCCTTTCAGCTGCCTGGCCCAATCCCCAGAATGGATTGCGTGGCCTACAACCCAGCCGGCGTGATGGGCAGCTGTTGGAAGTCCAAGGACCTGAGGTCCGCCCTGGTGTACTGGTGGCTGTCTGGCAATCCTAAGAGGCGCACATCCTCTCTGTTCTATCAGTTTTGCTGACTCGAGAAGCTT |
Restriction enzyme sites used for cloning are underlined. Open reading frame sequences are shown in black. Restriction enzyme sites not used for cloning are shown in italic font.
TABLE 8.
DNA fragments and parental plasmids for the construction of plasmids
| Constructed plasmid | DNA fragment from parental plasmid (size) | Recipient plasmid (source) |
|---|---|---|
| pFlag-ChHV-Us3o | EcoRI-HindIII fragment from pUC57-ChHV-Us3o (1,449 bp) | pFlag-CMV-2 (Sigma-Aldrich) |
| pcDNA-Myc-ChHV-UL7o | EcoRI-EcoRV fragment from pUC57-ChHV-UL7o (900 bp) | pcDNA-Myc (this study) |
| pEGFP-ChHV-gBo769–903 | EcoRI-SalI fragment from pUC57-ChHV-gBCTDo (414 bp) | pEGFP-C2 (Clontech) |
| pEGFP-AncA-UL7o | EcoRI-XhoI fragment from pUC57-AncA-UL7o (891 bp) | pEGFP-C2 (Clontech) |
| pEGFP-AncB-UL7o | EcoRI-XhoI fragment from pUC57-AncA-UL7o (891 bp) | pEGFP-C2 (Clontech) |
| pEGFP-ChHV-UL7o | EcoRI-XhoI fragment from pcDNA-SE-ChHV-UL7 (933 bp) | pEGFP-C2 (Clontech) |
| pEGFP-ChHV-UL7o-SS/AA | EcoRI-XhoI fragment from pcDNA-SE-ChHV-UL7o-SS/AA (921 bp) | pEGFP-C2 (Clontech) |
| pcDNA-SE-HSV-2-UL7o-SS/AA | BamHI-XhoI fragment of pcDNA-Myc-HSV-2-UL7-SS/AA (320 bp) | pcDNA-SE-HSV-2-UL7 (this study) |
TABLE 9.
Oligonucleotide sequences and DNA templates for the construction of plasmids
| Constructed plasmid | Oligonucleotide sequence (5′–3′) | PCR DNA template (reference or source) | Recipient plasmid (source) |
|---|---|---|---|
| pBS-UL7 | GATAAGCTTGATATCGAATTCACACCGCGTCCGTCAAGGTGC | pYEbac861Cre (this study) | pBluescript II KS(+) (Stratagene) |
| CGCTCTAGAACTAGTGGATCGACGGCGCGGTTTTGTTTCTC | |||
| pBS-UL51 | GCGAATTCGTCCGTTCTGTCGTCGCTAT | Wild-type HSV-2 186 genome | pBluescript II KS(+) (Stratagene) |
| GCAAGCTTGGTACGCGTCGTAGCTGAAT | |||
| pRetroX-TRE3G-UL7 | GCGCGGCCGCGCCACCATGGCCGCCGCGACGGCCGAC | Wild-type HSV-2 186 genome | pRetroX-TRE3G (Clontech) |
| GCGCGGCCGCTTAGCAAAACCGATAGAAAAGC | |||
| pAcGHLT-HSV-2-Us3 | GCGCGGCCGCTCACTTAGGGTGGAATAGCG | YK813 (HSV-2 Us3-repair) genome (22) | pAcGHLT-B (Pharmingen) |
| GCGAATTCTGGCCTGTCGTAAGTTCTGTGG | |||
| pAcGHLT-HSV-2-Us3-K220M | GCGCGGCCGCTCACTTAGGGTGGAATAGCG | YK811 (HSV-2 Us3-K220M) genome (22) | pAcGHLT-B (Pharmingen) |
| GCGAATTCTGGCCTGTCGTAAGTTCTGTGG | |||
| pFlag-HSV-1-Us3 | GCGAATTCAGCCTGTCGTAAGTTTTGTCG | pBS-Us3 (60) | pFlag-CMV-2 (Sigma-Aldrich) |
| GCGATATCTCATTTCTGTTGAAACAGCG | |||
| pFlag-HSV-1-Us3-K220M | GCGAATTCAGCCTGTCGTAAGTTTTGTCG | pBS-Us3-K220M (60) | pFlag-CMV-2 (Sigma-Aldrich) |
| GCGATATCTCATTTCTGTTGAAACAGCG | |||
| pFlag-HSV-2-Us3 | GCGAATTCGGCCTGTCGTAAGTTCTGTGG | pBS-HSV-2-Us3 (58) | pFlag-CMV-2 (Sigma-Aldrich) |
| GCGATATCTCACTTAGGGTGGAATAGCG | |||
| pFlag-HSV-2-Us3-K220M | GCGAATTCAGCCTGTCGTAAGTTCTGTGG | pAcGHLT-HSV-2-Us3-K220M (this study) | pFlag-CMV-2 (Sigma-Aldrich) |
| GCGATATCTCACTTAGGGTGGAATAGCG | |||
| pFlag-ChHV-Us3o-K220M | |||
| 1st PCR-A | GCAAGCTTGCATGTAGGAAGTT | pUC57-ChHV-Us3o (this study) | pFlag-CMV-2 (Sigma-Aldrich) |
| TGCTGGCATACCAGCCGGCCATCACGATCACTCTCTGTGGG | |||
| 1st PCR-B | CCCACAGAGAGTGATCGTGATGGCCGGCTGGTATGCCAGCA | ||
| GCGAATTCTCACTTTGGATGAA | |||
| 2nd PCR | GCAAGCTTGCATGTAGGAAGTT | Mixture of 1st PCR fragments | |
| GCGAATTCTCACTTTGGATGAA | |||
| pMAL-HSV-2-UL7 | GCGTCGACATGGCCGACCCCACGCCCGC | pYEbac356 (22) | pMAL-c (New England BioLabs) |
| GCAAGCTTTTAGCAAAACCGATAGAAAAG | |||
| pMAL-HSV-2-UL34 | GCGAATTCATGGCGGGGATGGGGAAGCCCTAC | pYEbac356 (22) | pMAL-c (New England BioLabs) |
| GCGTCGACTCATAGGCGCGCGCCAACCGCC | |||
| pcDNA-Myc-HSV-2-UL34 | GCGAATTCTGCGGGGATGGGGAAGCCCTA | pMAL-HSV-2-UL34 (this study) | pcDNA-Myc (this study) |
| GCGTCGACTCATAGGCGCGCGCCAACCGCC | |||
| pcDNA-SE-HSV-2-UL7 | GCGATATCAAATGGCCGACCCCACGCCCGCCG | pMAL-HSV-2-UL7 (this study) | pcDNA-SE (this study) |
| GCCTCGAGTTAGCAAAACCGATAGAAAAG | |||
| pcDNA-Myc-HSV-2-UL7 | GCGATATCGCCACCATGGAGCAGAAGCTGATCTCAGAGGAGGACCTGGCCGACCCCACGCCCGCCGA | pYEbac356 (22) | pcDNA3.1(+) (Thermo Fisher Scientific) |
| GCCTCGAGTTAGCAAAACCGATAGAAAAG | |||
| pcDNA-SE-ChHV-UL7o | GCGAATTCGCCGACCCAACCCCCGCAGA | pcDNA-Myc-ChHV-UL7o (this study) | pcDNA-SE (this study) |
| GCGATATCTTAACAAAACTGGTAAAACAGG | |||
| pEGFP-HSV-1-gB770–904 | GCGAATTCTTCATGTCCAACCCCTTTGG | Wild-type HSV-1(F) genome | pEGFP-C2 (Clontech) |
| GCGGATCCTCACAKUGGTCGTCCTCGTCGG | |||
| pcDNA-Myc-HSV-2-UL7-T288A | GCGATATCGCCACCATGGAGCAGAAGCTGATCTCAGAGGAGGACCTGGCCGACCCCACGCCCGCCGA | pYEbac356 (22) | pcDNA3.1(+) (Thermo Fisher Scientific) |
| GCCTCGAGTTAGCAAAACCGATAGAAAAGCGACGAGGCCCGTCG | |||
| pcDNA-Myc-HSV-2-UL7-S289A | GCGATATCGCCACCATGGAGCAGAAGCTGATCTCAGAGGAGGACCTGGCCGACCCCACGCCCGCCGA | pYEbac356 (22) | pcDNA3.1(+) (Thermo Fisher Scientific) |
| GCCTCGAGTTAGCAAAACCGATAGAAAAGCGACGCGGTCCG | |||
| pcDNA-Myc-HSV-2-UL7-S290A | GCGATATCGCCACCATGGAGCAGAAGCTGATCTCAGAGGAGGACCTGGCCGACCCCACGCCCGCCGA | pYEbac356 (22) | pcDNA3.1(+) (Thermo Fisher Scientific) |
| GCCTCGAGTTAGCAAAACCGATAGAAAAGCGCCGAGGTCCGTCG | |||
| pcDNA-Myc-HSV-2-UL7-SS/AA | GCGATATCGCCACCATGGAGCAGAAGCTGATCTCAGAGGAGGACCTGGCCGACCCCACGCCCGCCGA | pYEbac356 (22) | pcDNA3.1(+) (Thermo Fisher Scientific) |
| GCCTCGAGTTAGCAAAACCGATAGAAAAGCGCCGCGGTCCGTCG | |||
| pcDNA-Myc-HSV-2-UL7-TSS/AAA | GCGATATCGCCACCATGGAGCAGAAGCTGATCTCAGAGGAGGACCTGGCCGACCCCACGCCCGCCGA | pYEbac356 (22) | pcDNA3.1(+) (Thermo Fisher Scientific) |
| GCCTCGAGTTAGCAAAACCGATAGAAAAGCGCCGCGGCCCGTCG | |||
| pcDNA-SE-ChHV-UL7o-SS/AA | GCGAATTCGCCGACCCAACCCCCGCAGA | pcDNA-Myc-ChHV-UL7o (this study) | pcDNA-SE (This study) |
| GCGATATCTTAACAAAACTGGTAAAACAGGGCTGCTGTCCGCCGCTTCCTATTGC | |||
| pEGFP-AncA-UL7o-Q286R | ATGCAAGCTTGAATTCGCCG | pEGFP-AncA-UL7o (this study) | pEGFP-C2 (Clontech) |
| GCCTCGAGTCAACAAAACTGATAGAACAGGCTAGATGTTCGGCGCTTAGGGGTCTCGGAC | |||
| pEGFP-AncA-UL7o-QSS/RAA | ATGCAAGCTTGAATTCGCCG | pEGFP-AncA-UL7o (this study) | pEGFP-C2 (Clontech) |
| GCCTCGAGTCAACAAAACTGATAGAACAGGGCAGCTGTTCGGCGCTTAGGGGTCTCGGAC | |||
| pEGFP-AncB-UL7o-SS/AA | ATGCAAGCTTGAATTCGCCG | pEGFP-AncB-UL7o (this study) | pEGFP-C2 (Clontech) |
| GCCTCGAGTCAGCAAAACTGATAGAACAGAGCGGCTGTGCGCCTCTTAGGATTGC | |||
| pEGFP-HSV-1-gB770–904-T887A | |||
| 1st PCR-A | GCGAATTCTTCATGTCCAACCCCTTTGG | Wild-type HSV-1(F) genome | pEGFP-C2 (Clontech) |
| GGGAACTTGGGTGTAGTTGGCGTTGCGGCGCTTGCGCATGA | |||
| 1st PCR-B | TCATGCGCAAGCGCCGCAACGCCAACTACACCCAAGTTCCC | ||
| GCGGATCCTCACAGGTCGTCCTCGTCGG | |||
| 2nd PCR | GCGAATTCTTCATGTCCAACCCCTTTGG | Mixture of 1st PCR fragments | |
| GCGGATCCTCACAGGTCGTCCTCGTCGG |
Establishment of stable Vero cells with tetracycline-inducible UL7 expression.
Vero cells were transduced with supernatants of Plat-GP cells cotransfected with pMDG (49) and pRetroX-Tet3G (TaKaRa), selected with 1 mg/ml G418 (Wako), and further transduced with supernatants of Plat-GP cells cotransfected with pMDG and pRetroX-TRE3G-UL7. For tetracycline-inducible (TetON) UL7 expression, after double selection with 5 μg/ml puromycin (Sigma) and 1 mg/ml G418, resistant cells were cloned from a single colony and designated HSV-2 UL7-TetON-Vero cells.
Mutagenesis of viral genomes and generation of recombinant HSV-2 and -1.
All recombinant viruses (Fig. 6), except YK788 (HSV-2 UL7-repair), were generated by two-step Red-mediated mutagenesis as described previously (32, 48), except that the primers, plasmids, and E. coli GS1873 strains containing HSV-2-BAC or HSV-1 BAC listed in Table 5 were used. Recombinant virus YK788 (HSV-2 UL7-repair) (Fig. 6), in which the YK787 (HSV-2 ΔUL7) mutation was repaired, was generated by cotransfection of rabbit skin cells with the YK787 (HSV-2 ΔUL7) genome and pBS-UL7, as described previously (50, 51). After the third round of plaque purification, restoration of original sequences was confirmed by sequencing (51). In experiments in which YK787 (HSV-2 ΔUL7) was used, viruses were propagated and assayed in HSV-2 UL7-TetON-Vero cells.
Antibodies.
Commercial antibodies used in this study were mouse monoclonal antibodies to Flag (M2; Sigma), β-actin (AC15; Sigma), gB (H1817; Virusys), ICP27 (8.F.137B; Abcam), and Myc (PL14; MBL), rabbit monoclonal antibody to phospho-Akt substrate [RXX(S*/T*)] (110B7E; Cell Signaling Technology), and rat monoclonal antibody to GFP (FM264; BioLegend). Rabbit polyclonal antibodies to UL7, UL34, UL37, UL50, and Us3 were described previously (32, 52–55).
Southern blotting, immunoblotting and immunoprecipitation.
Southern blotting, immunoblotting, and immunoprecipitation were performed as described previously (50, 56, 57).
Phos-tag SDS-PAGE.
Phos-tag SDS-PAGE analyses were performed as described previously (54, 58, 59), except that denaturing gels containing 57 μM MnCl2 and 29 μM Phos-tag acrylamide (Wako) were used for untagged and myc-tagged UL7 or 71 μM MnCl2 and 36 μM Phos-tag acrylamide were used for EGFP-tagged UL7.
Generation of recombinant baculoviruses.
BAC-GST-HSV-2-Us3 or BAC-GST-HSV-2-Us3-K220M was generated as described previously (45, 60), except that pAcGHL-HSV-2-Us3, pAcGHL-HSV-2-Us3(K220M), and BacPAK6 viral DNA were used (TaKaRa).
Production and purification of GST and MBP fusion proteins.
GST-HSV-2-Us3 and GST-HSV-2-Us3-K220M were purified using glutathione-Sepharose resin (GE Healthcare) from Sf9 cells infected with BAC-GST-HSV-2-Us3 or BAC-GST-HSV-2-Us3-K220M as described previously (60). MBP-HSV-2-UL34 (where MBP is maltose binding protein) and MBP-lacZ were expressed in E. coli BL21 Star(DE3) transformed with pMAL-HSV-2-UL34 and pMAL-c and purified using amylose resin (New England BioLabs) as described previously (45).
Production and purification of Strep-tagged proteins in HEK293FT cells.
HEK293FT cells transfected with pcDNA-SE-HSV-2-UL7 and pcDNA-SE-HSV-2-UL7-SS/AA using polyethylenimine (PEI) (61) were harvested at 48 h posttransfection and lysed in radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1% Nonidet P-40 [NP-40], 0.5% deoxycholate, 0.1% sodium dodecyl sulfate) containing a protease inhibitor cocktail (Nacalai Tesque). Supernatants obtained after centrifugation of lysates were precleared by incubation with protein A beads (GE Healthcare) at 4°C for 30 min and then reacted with StrepTactin beads (IBA) for 2 h. StrepTactin beads were collected by brief centrifugation and washed twice with high-salt buffer (1 M NaCl, 10 mM Tris-HCl [pH 8.0], 0.2% NP-40), once with low-salt buffer (0.1 M NaCl, 10 mM Tris-HCl [pH 8.0], 0.2% NP-40), six times with RIPA buffer, and four times with Us3 kinase buffer (50 mM Tris-HCl [pH 9.0], 20 mM MgCl2, 0.1% NP-40, 1 mM dithiothreitol).
In vitro kinase assay.
MBP fusion or Strep-tagged proteins captured on amylose or StrepTactin beads, respectively, were used as substrates for in vitro kinase assays with GST-HSV-2-Us3 and GST-HSV-2-Us3-K220M, as described previously (32, 60).
Phosphatase treatment.
Phosphorylated proteins were treated with λ-protein phosphatase (λ-PPase) or alkaline phosphatase (calf intestinal phosphatase [CIP]) (New England Bio-Labs) as described previously (32, 60).
Animal studies.
Three-week-old female ICR mice (Charles River) were infected intracranially with 10-fold serial dilutions of each virus as described previously (22, 62) and were monitored for survival for 21 days postinfection. LD50 values were calculated by the Behrens-Karber method (63). In other studies, 6- or 5-week-old female ICR mice (Charles River) pretreated with 1.67 mg of medroxyprogesterone (OK Injection; ANB Laboratories) were infected intravaginally with 0.75 × 104 PFU of HSV-2 or 0.5 × 107 PFU of HSV-1 viruses as described previously (20, 62). Mice were monitored for survival for 26 days postinfection. Scoring of vaginal disease severity and virus titers in vaginal secretions of mice were determined as described previously (20, 64). All animal experiments were carried out in accordance with guidelines for proper conduct of animal experiments of the Science Council of Japan. The protocol was approved by the Institutional Animal Care and Use Committee (IACUC) of the Institute of Medical Science, University of Tokyo (IACUC protocol approvals 19–26, PA11-81, and PA16-69).
Phylogenetic analysis.
A phylogenetic tree of 10 nucleotide sequences of the viral genome belonging to simplex viruses, listed in Table 3, was constructed as follows. All positions containing gaps and missing data were eliminated. There were 70,936 positions in the final data set. Multiple sequence alignments of these nucleotide sequences were constructed using MAFFT (65). Evolutionary history was inferred using the neighbor-joining method (66). Evolutionary analyses were conducted in MEGA7 (67). Ancestral sequences of UL7 genes were reconstructed based on phylogenetic analysis of 10 nucleotide sequences of UL7 gene homologs belonging to simplex viruses using MUSCLE (68) implemented in MEGA7 (67). Ancestral sequences were inferred with MEGA7 (67), which uses maximum likelihood to estimate ancestral character states and phylogeny.
ELISA.
Peptides listed in Table 10 were purchased from Toray Research Center, Inc., and coupled to ELISA plates with a peptide coating kit (TaKaRa) according to the manufacturer’s instructions. After anti-phospho-Akt substrate antibody (Cell Signaling Technology) incubation, anti-rabbit IgG horseradish peroxidase (HRP)-linked F(ab’)2 fragment donkey (GE Healthcare) and TMB (3,3',5,5'-tetramethylbenzidine) solution (eBioscience) were added, and signaling was quantified using an EnSpire plate reader (Perkin Elmer).
TABLE 10.
Summary of synthesized peptides
| Peptide | Peptide sequencea |
|---|---|
| Phospho-HSV-1 gB881–893 | MRKRRN(p)TNYTQVP |
| Phospho-ChHV gB880–892 | LRKRNK(p)TRYSPLH |
| ChHV gB880–892 | LRKRNKTRYSPLH |
In peptide sequences, (p)T indicates phosphorylated threonine.
ACKNOWLEDGMENTS
We thank Risa Abe, Keiko Sato, and Tohru Ikegami for their excellent technical assistance.
This study was supported by Grants for Scientific Research from the Japan Society for the Promotion of Science, Grants for Scientific Research on Innovative Areas from the Ministry of Education, Culture, Science, Sports and Technology of Japan (16H06433, 16H06429, 16K21723, 18H04968, and 19H04826), contract research funds from the Program of Japan Initiative for Global Research Network on Infectious Diseases (J-GRID) (JP18fm0108006) and the Research Programs on Emerging and Reemerging Infectious Diseases (19fk018105h0001 and 20wm0125002h0001) from the Japan Agency for Medical Research and Development (AMED), a grant for the Joint Research Project of the Institute of Medical Science, the University of Tokyo, grants from the Takeda Science Foundation, the Uehara Memorial Foundation, the Naito Foundation, and the Waksman Foundation of Japan, and a GSK Japan Research Grant 2018.
REFERENCES
- 1.Sugiyama N, Ishihama Y. 2016. Large-scale profiling of protein kinases for cellular signaling studies by mass spectrometry and other techniques. J Pharm Biomed Anal 130:264–272. doi: 10.1016/j.jpba.2016.05.046. [DOI] [PubMed] [Google Scholar]
- 2.Gnad F, Forner F, Zielinska DF, Birney E, Gunawardena J, Mann M. 2010. Evolutionary constraints of phosphorylation in eukaryotes, prokaryotes, and mitochondria. Mol Cell Proteomics 9:2642–2653. doi: 10.1074/mcp.M110.001594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beltrao P, Trinidad JC, Fiedler D, Roguev A, Lim WA, Shokat KM, Burlingame AL, Krogan NJ. 2009. Evolution of phosphoregulation: comparison of phosphorylation patterns across yeast species. PLoS Biol 7:e1000134. doi: 10.1371/journal.pbio.1000134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Studer RA, Rodriguez-Mias RA, Haas KM, Hsu JI, Viéitez C, Solé C, Swaney DL, Stanford LB, Liachko I, Böttcher R, Dunham MJ, de Nadal E, Posas F, Beltrao P, Villén J. 2016. Evolution of protein phosphorylation across 18 fungal species. Science 354:229–232. doi: 10.1126/science.aaf2144. [DOI] [PubMed] [Google Scholar]
- 5.Jacob T, Van den Broeke C, Favoreel HW. 2011. Viral serine/threonine protein kinases. J Virol 85:1158–1173. doi: 10.1128/JVI.01369-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rubio RM, Mohr I. 2019. Inhibition of ULK1 and Beclin1 by an alpha-herpesvirus Akt-like Ser/Thr kinase limits autophagy to stimulate virus replication. Proc Natl Acad Sci U S A 116:26941–26950. doi: 10.1073/pnas.1915139116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kato A, Kawaguchi Y. 2018. Us3 protein kinase encoded by HSV: the precise function and mechanism on viral life cycle. Adv Exp Med Biol 1045:45–62. doi: 10.1007/978-981-10-7230-7_3. [DOI] [PubMed] [Google Scholar]
- 8.Arii J, Goto H, Suenaga T, Oyama M, Kozuka-Hata H, Imai T, Minowa A, Akashi H, Arase H, Kawaoka Y, Kawaguchi Y. 2010. Non-muscle myosin IIA is a functional entry receptor for herpes simplex virus-1. Nature 467:859–862. doi: 10.1038/nature09420. [DOI] [PubMed] [Google Scholar]
- 9.Roizman B, Knipe DM, Whitley RJ. 2013. Herpes simplex viruses, p 1823–1897. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed Lippincott-Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 10.Erazo A, Kinchington PR. 2010. Varicella-zoster virus open reading frame 66 protein kinase and its relationship to alphaherpesvirus US3 kinases. Curr Top Microbiol Immunol 342:79–98. doi: 10.1007/82_2009_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gao J, Finnen RL, Sherry MR, Le Sage V, Banfield BW. 2020. Differentiating the roles of UL16, UL21 and Us3 in the nuclear egress of herpes simplex virus capsids. J Virol 94:e00738-20. doi: 10.1128/JVI.00738-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sagou K, Imai T, Sagara H, Uema M, Kawaguchi Y. 2009. Regulation of the catalytic activity of herpes simplex virus 1 protein kinase Us3 by autophosphorylation and its role in pathogenesis. J Virol 83:5773–5783. doi: 10.1128/JVI.00103-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meignier B, Longnecker R, Mavromara-Nazos P, Sears AE, Roizman B. 1988. Virulence of and establishment of latency by genetically engineered deletion mutants of herpes simplex virus 1. Virology 162:251–254. doi: 10.1016/0042-6822(88)90417-5. [DOI] [PubMed] [Google Scholar]
- 14.Kato A, Arii J, Shiratori I, Akashi H, Arase H, Kawaguchi Y. 2009. Herpes simplex virus 1 protein kinase Us3 phosphorylates viral envelope glycoprotein B and regulates its expression on the cell surface. J Virol 83:250–261. doi: 10.1128/JVI.01451-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Imai T, Arii J, Minowa A, Kakimoto A, Koyanagi N, Kato A, Kawaguchi Y. 2011. Role of the herpes simplex virus 1 Us3 kinase phosphorylation site and endocytosis motifs in the intracellular transport and neurovirulence of envelope glycoprotein B. J Virol 85:5003–5015. doi: 10.1128/JVI.02314-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Imai T, Sagou K, Arii J, Kawaguchi Y. 2010. Effects of phosphorylation of herpes simplex virus 1 envelope glycoprotein B by Us3 kinase in vivo and in vitro. J Virol 84:153–162. doi: 10.1128/JVI.01447-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Purves FC, Deana AD, Marchiori F, Leader DP, Pinna LA. 1986. The substrate specificity of the protein kinase induced in cells infected with herpesviruses: studies with synthetic substrates [corrected] indicate structural requirements distinct from other protein kinases. Biochim Biophys Acta 889:208–215. doi: 10.1016/0167-4889(86)90106-0. [DOI] [PubMed] [Google Scholar]
- 18.Leader DP. 1993. Viral protein kinases and protein phosphatases. Pharmacol Ther 59:343–389. doi: 10.1016/0163-7258(93)90075-o. [DOI] [PubMed] [Google Scholar]
- 19.Leader DP, Deana AD, Marchiori F, Purves FC, Pinna LA. 1991. Further definition of the substrate specificity of the alpha-herpesvirus protein kinase and comparison with protein kinases A and C. Biochim Biophys Acta 1091:426–431. doi: 10.1016/0167-4889(91)90210-O. [DOI] [PubMed] [Google Scholar]
- 20.Koyanagi N, Kato A, Takeshima K, Maruzuru Y, Kozuka-Hata H, Oyama M, Arii J, Kawaguchi Y. 2018. Regulation of herpes simplex virus 2 protein kinase UL13 by phosphorylation and its role in viral pathogenesis. J Virol 92:e00807-18. doi: 10.1128/JVI.00807-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kinoshita E, Kinoshita-Kikuta E, Takiyama K, Koike T. 2006. Phosphate-binding tag, a new tool to visualize phosphorylated proteins. Mol Cell Proteomics 5:749–757. doi: 10.1074/mcp.T500024-MCP200. [DOI] [PubMed] [Google Scholar]
- 22.Morimoto T, Arii J, Tanaka M, Sata T, Akashi H, Yamada M, Nishiyama Y, Uema M, Kawaguchi Y. 2009. Differences in the regulatory and functional effects of the Us3 protein kinase activities of herpes simplex virus 1 and 2. J Virol 83:11624–11634. doi: 10.1128/JVI.00993-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cunningham C, Davison AJ. 1993. A cosmid-based system for constructing mutants of herpes simplex virus type 1. Virology 197:116–124. doi: 10.1006/viro.1993.1572. [DOI] [PubMed] [Google Scholar]
- 24.Tanaka M, Sata T, Kawaguchi Y. 2008. The product of the herpes simplex virus 1 UL7 gene interacts with a mitochondrial protein, adenine nucleotide translocator 2. Virol J 5:125. doi: 10.1186/1743-422X-5-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feutz E, McLeland-Wieser H, Ma J, Roller RJ. 2019. Functional interactions between herpes simplex virus pUL51, pUL7 and gE reveal cell-specific mechanisms for epithelial cell-to-cell spread. Virology 537:84–96. doi: 10.1016/j.virol.2019.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Albecka A, Owen DJ, Ivanova L, Brun J, Liman R, Davies L, Ahmed MF, Colaco S, Hollinshead M, Graham SC, Crump CM. 2017. Dual function of the pUL7-pUL51 tegument protein complex in herpes simplex virus 1 infection. J Virol 91:e02196-16. doi: 10.1128/JVI.02196-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu X, Fan S, Zhou J, Zhang Y, Che Y, Cai H, Wang L, Guo L, Liu L, Li Q. 2016. The mutated tegument protein UL7 attenuates the virulence of herpes simplex virus 1 by reducing the modulation of alpha-4 gene transcription. Virol J 13:152. doi: 10.1186/s12985-016-0600-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wertheim JO, Smith MD, Smith DM, Scheffler K, Kosakovsky Pond SL. 2014. Evolutionary origins of human herpes simplex viruses 1 and 2. Mol Biol Evol 31:2356–2364. doi: 10.1093/molbev/msu185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Benetti L, Roizman B. 2004. Herpes simplex virus protein kinase US3 activates and functionally overlaps protein kinase A to block apoptosis. Proc Natl Acad Sci U S A 101:9411–9416. doi: 10.1073/pnas.0403160101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chuluunbaatar U, Roller R, Feldman ME, Brown S, Shokat KM, Mohr I. 2010. Constitutive mTORC1 activation by a herpesvirus Akt surrogate stimulates mRNA translation and viral replication. Genes Dev 24:2627–2639. doi: 10.1101/gad.1978310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Naghavi MH, Gundersen GG, Walsh D. 2013. Plus-end tracking proteins, CLASPs, and a viral Akt mimic regulate herpesvirus-induced stable microtubule formation and virus spread. Proc Natl Acad Sci U S A 110:18268–18273. doi: 10.1073/pnas.1310760110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kato A, Tanaka M, Yamamoto M, Asai R, Sata T, Nishiyama Y, Kawaguchi Y. 2008. Identification of a physiological phosphorylation site of the herpes simplex virus 1-encoded protein kinase Us3 which regulates its optimal catalytic activity in vitro and influences its function in infected cells. J Virol 82:6172–6189. doi: 10.1128/JVI.00044-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fuchs W, Granzow H, Klopfleisch R, Klupp BG, Rosenkranz D, Mettenleiter TC. 2005. The UL7 gene of pseudorabies virus encodes a nonessential structural protein which is involved in virion formation and egress. J Virol 79:11291–11299. doi: 10.1128/JVI.79.17.11291-11299.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Klopfleisch R, Klupp BG, Fuchs W, Kopp M, Teifke JP, Mettenleiter TC. 2006. Influence of pseudorabies virus proteins on neuroinvasion and neurovirulence in mice. J Virol 80:5571–5576. doi: 10.1128/JVI.02589-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.MacManiman JD, Meuser A, Botto S, Smith PP, Liu F, Jarvis MA, Nelson JA, Caposio P. 2014. Human cytomegalovirus-encoded pUL7 is a novel CEACAM1-like molecule responsible for promotion of angiogenesis. mBio 5:e02035. doi: 10.1128/mBio.02035-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kato A, Liu Z, Minowa A, Imai T, Tanaka M, Sugimoto K, Nishiyama Y, Arii J, Kawaguchi Y. 2011. Herpes simplex virus 1 protein kinase Us3 and major tegument protein UL47 reciprocally regulate their subcellular localization in infected cells. J Virol 85:9599–9613. doi: 10.1128/JVI.00845-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu Z, Kato A, Shindo K, Noda T, Sagara H, Kawaoka Y, Arii J, Kawaguchi Y. 2014. Herpes simplex virus 1 UL47 interacts with viral nuclear egress factors UL31, UL34, and Us3 and regulates viral nuclear egress. J Virol 88:4657–4667. doi: 10.1128/JVI.00137-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lynch VJ, Tanzer A, Wang Y, Leung FC, Gellersen B, Emera D, Wagner GP. 2008. Adaptive changes in the transcription factor HoxA-11 are essential for the evolution of pregnancy in mammals. Proc Natl Acad Sci U S A 105:14928–14933. doi: 10.1073/pnas.0802355105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nnamani MC, Ganguly S, Erkenbrack EM, Lynch VJ, Mizoue LS, Tong Y, Darling HL, Fuxreiter M, Meiler J, Wagner GP. 2016. A derived allosteric switch underlies the evolution of conditional cooperativity between HOXA11 and FOXO1. Cell Rep 15:2097–2108. doi: 10.1016/j.celrep.2016.04.088. [DOI] [PubMed] [Google Scholar]
- 40.Lynch VJ, May G, Wagner GP. 2011. Regulatory evolution through divergence of a phosphoswitch in the transcription factor CEBPB. Nature 480:383–386. doi: 10.1038/nature10595. [DOI] [PubMed] [Google Scholar]
- 41.Hutchinson EC, Denham EM, Thomas B, Trudgian DC, Hester SS, Ridlova G, York A, Turrell L, Fodor E. 2012. Mapping the phosphoproteome of influenza A and B viruses by mass spectrometry. PLoS Pathog 8:e1002993. doi: 10.1371/journal.ppat.1002993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kulej K, Avgousti DC, Sidoli S, Herrmann C, Della Fera AN, Kim ET, Garcia BA, Weitzman MD. 2017. Time-resolved global and chromatin proteomics during herpes simplex virus type 1 (HSV-1) infection. Mol Cell Proteomics 16:S92–S107. doi: 10.1074/mcp.M116.065987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ivanov A, Ramanathan P, Parry C, Ilinykh PA, Lin X, Petukhov M, Obukhov Y, Ammosova T, Amarasinghe GK, Bukreyev A, Nekhai S. 2019. Global phosphoproteomic analysis of Ebola virions reveals a novel role for VP35 phosphorylation-dependent regulation of genome transcription. Cell Mol Life Sci 77:2579–2603. doi: 10.1007/s00018-019-03303-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maruzuru Y, Ichinohe T, Sato R, Miyake K, Okano T, Suzuki T, Koshiba T, Koyanagi N, Tsuda S, Watanabe M, Arii J, Kato A, Kawaguchi Y. 2018. Herpes simplex virus 1 VP22 inhibits AIM2-dependent inflammasome activation to enable efficient viral replication. Cell Host Microbe 23:254–265.e7. doi: 10.1016/j.chom.2017.12.014. [DOI] [PubMed] [Google Scholar]
- 45.Kawaguchi Y, Kato K, Tanaka M, Kanamori M, Nishiyama Y, Yamanashi Y. 2003. Conserved protein kinases encoded by herpesviruses and cellular protein kinase cdc2 target the same phosphorylation site in eukaryotic elongation factor 1δ. J Virol 77:2359–2368. doi: 10.1128/jvi.77.4.2359-2368.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Asano S, Honda T, Goshima F, Watanabe D, Miyake Y, Sugiura Y, Nishiyama Y. 1999. US3 protein kinase of herpes simplex virus type 2 plays a role in protecting corneal epithelial cells from apoptosis in infected mice. J Gen Virol 80:51–56. doi: 10.1099/0022-1317-80-1-51. [DOI] [PubMed] [Google Scholar]
- 47.Ejercito PM, Kieff ED, Roizman B. 1968. Characterization of herpes simplex virus strains differing in their effects on social behaviour of infected cells. J Gen Virol 2:357–364. doi: 10.1099/0022-1317-2-3-357. [DOI] [PubMed] [Google Scholar]
- 48.Tischer BK, von Einem J, Kaufer B, Osterrieder N. 2006. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 40:191–197. doi: 10.2144/000112096. [DOI] [PubMed] [Google Scholar]
- 49.Zufferey R, Nagy D, Mandel RJ, Naldini L, Trono D. 1997. Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat Biotechnol 15:871–875. doi: 10.1038/nbt0997-871. [DOI] [PubMed] [Google Scholar]
- 50.Kato A, Oda S, Watanabe M, Oyama M, Kozuka-Hata H, Koyanagi N, Maruzuru Y, Arii J, Kawaguchi Y. 2018. Roles of the Phosphorylation of Herpes Simplex Virus 1 UL51 at a Specific Site in Viral Replication and Pathogenicity. J Virol 92:e01035-18. doi: 10.1128/JVI.01035-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sugimoto K, Uema M, Sagara H, Tanaka M, Sata T, Hashimoto Y, Kawaguchi Y. 2008. Simultaneous tracking of capsid, tegument, and envelope protein localization in living cells infected with triply fluorescent herpes simplex virus 1. J Virol 82:5198–5211. doi: 10.1128/JVI.02681-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nozawa N, Daikoku T, Yamauchi Y, Takakuwa H, Goshima F, Yoshikawa T, Nishiyama Y. 2002. Identification and characterization of the UL7 gene product of herpes simplex virus type 2. Virus Genes 24:257–266. doi: 10.1023/a:1015332716927. [DOI] [PubMed] [Google Scholar]
- 53.Watanabe D, Ushijima Y, Goshima F, Takakuwa H, Tomita Y, Nishiyama Y. 2000. Identification of nuclear export signal in UL37 protein of herpes simplex virus type 2. Biochem Biophys Res Commun 276:1248–1254. doi: 10.1006/bbrc.2000.3600. [DOI] [PubMed] [Google Scholar]
- 54.Kato A, Tsuda S, Liu Z, Kozuka-Hata H, Oyama M, Kawaguchi Y. 2014. Herpes simplex virus 1 protein kinase Us3 phosphorylates viral dUTPase and regulates its catalytic activity in infected cells. J Virol 88:655–666. doi: 10.1128/JVI.02710-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shiba C, Daikoku T, Goshima F, Takakuwa H, Yamauchi Y, Koiwai O, Nishiyama Y. 2000. The UL34 gene product of herpes simplex virus type 2 is a tail-anchored type II membrane protein that is significant for virus envelopment. J Gen Virol 81:2397–2405. doi: 10.1099/0022-1317-81-10-2397. [DOI] [PubMed] [Google Scholar]
- 56.Kawaguchi Y, Van Sant C, Roizman B. 1997. Herpes simplex virus 1 alpha regulatory protein ICP0 interacts with and stabilizes the cell cycle regulator cyclin D3. J Virol 71:7328–7336. doi: 10.1128/JVI.71.10.7328-7336.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Arii J, Watanabe M, Maeda F, Tokai-Nishizumi N, Chihara T, Miura M, Maruzuru Y, Koyanagi N, Kato A, Kawaguchi Y. 2018. ESCRT-III mediates budding across the inner nuclear membrane and regulates its integrity. Nat Commun 9:3379. doi: 10.1038/s41467-018-05889-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shindo K, Kato A, Koyanagi N, Sagara H, Arii J, Kawaguchi Y. 2016. Characterization of a herpes simplex virus 1 (HSV-1) chimera in which the Us3 protein kinase gene is replaced with the HSV-2 Us3 gene. J Virol 90:457–473. doi: 10.1128/JVI.02376-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fujii H, Kato A, Mugitani M, Kashima Y, Oyama M, Kozuka-Hata H, Arii J, Kawaguchi Y. 2014. The UL12 protein of herpes simplex virus 1 is regulated by tyrosine phosphorylation. J Virol 88:10624–10634. doi: 10.1128/JVI.01634-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kato A, Yamamoto M, Ohno T, Kodaira H, Nishiyama Y, Kawaguchi Y. 2005. Identification of proteins phosphorylated directly by the Us3 protein kinase encoded by herpes simplex virus 1. J Virol 79:9325–9331. doi: 10.1128/JVI.79.14.9325-9331.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Boussif O, Lezoualc'h F, Zanta MA, Mergny MD, Scherman D, Demeneix B, Behr JP. 1995. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proc Natl Acad Sci U S A 92:7297–7301. doi: 10.1073/pnas.92.16.7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kato A, Shindo K, Maruzuru Y, Kawaguchi Y. 2014. Phosphorylation of a herpes simplex virus 1 dUTPase by a viral protein kinase, Us3, dictates viral pathogenicity in the central nervous system but not at the periphery. J Virol 88:2775–2785. doi: 10.1128/JVI.03300-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tanaka M, Kagawa H, Yamanashi Y, Sata T, Kawaguchi Y. 2003. Construction of an excisable bacterial artificial chromosome containing a full-length infectious clone of herpes simplex virus type 1: viruses reconstituted from the clone exhibit wild-type properties in vitro and in vivo. J Virol 77:1382–1391. doi: 10.1128/jvi.77.2.1382-1391.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Morrison LA, Da Costa XJ, Knipe DM. 1998. Influence of mucosal and parenteral immunization with a replication-defective mutant of HSV-2 on immune responses and protection from genital challenge. Virology 243:178–187. doi: 10.1006/viro.1998.9047. [DOI] [PubMed] [Google Scholar]
- 65.Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Saitou N, Nei M. 1987. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 67.Kumar S, Stecher G, Tamura K. 2016. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Felsenstein J. 1985. Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39:783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x. [DOI] [PubMed] [Google Scholar]
- 70.Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59:307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]