

HCMV is a major global health concern, and antiviral chemotherapy remains problematic due to toxicity of available compounds and the emergence of drug-resistant viruses. Thus, an HCMV vaccine represents a priority for both governmental and pharmaceutical research programs. A major obstacle for the development of a vaccine is a lack of knowledge of the nature and specificities of protective immune responses that should be induced by such a vaccine. Glycoprotein B of HCMV is an important target for neutralizing antibodies and, hence, is often included as a component of intervention strategies. By generation of fusion-active gB chimeras, we were able to identify target structures of neutralizing antibodies that potently block gB-induced membrane fusion. This experimental system provides an approach to screen for antibodies that interfere with gB’s fusogenic activity. In summary, our data will likely contribute to both rational vaccine design and the development of antibody-based therapies against HCMV.
KEYWORDS: herpesvirus, antibodies, cell-cell fusion, glycoprotein B, human cytomegalovirus
ABSTRACT
Human cytomegalovirus (HCMV) is a ubiquitous pathogen that can cause severe clinical disease in allograft recipients and infants infected in utero. Virus-neutralizing antibodies defined in vitro have been proposed to confer protection against HCMV infection, and the virion envelope glycoprotein B (gB) serves as a major target of neutralizing antibodies. The viral fusion protein gB is nonfusogenic on its own and requires glycoproteins H (gH) and L (gL) for membrane fusion, which is in contrast to requirements of related class III fusion proteins, including vesicular stomatitis virus glycoprotein G (VSV-G) or baculovirus gp64. To explore requirements for gB’s fusion activity, we generated a set of chimeras composed of gB and VSV-G or gp64, respectively. These gB chimeras were intrinsically fusion active and led to the formation of multinucleated cell syncytia when expressed in the absence of other viral proteins. Utilizing a panel of virus-neutralizing gB-specific monoclonal antibodies (MAbs), we could demonstrate that syncytium formation of the fusogenic gB/VSV-G chimera can be significantly inhibited by only a subset of neutralizing MAbs which target antigenic domain 5 (AD-5) of gB. This observation argues for differential modes of action of neutralizing anti-gB MAbs and suggests that blocking the membrane fusion function of gB could be one mechanism of antibody-mediated virus neutralization. In addition, our data have important implications for the further understanding of the conformation of gB that promotes membrane fusion as well as the identification of structures in AD-5 that could be targeted by antibodies to block this early step in HCMV infection.
IMPORTANCE HCMV is a major global health concern, and antiviral chemotherapy remains problematic due to toxicity of available compounds and the emergence of drug-resistant viruses. Thus, an HCMV vaccine represents a priority for both governmental and pharmaceutical research programs. A major obstacle for the development of a vaccine is a lack of knowledge of the nature and specificities of protective immune responses that should be induced by such a vaccine. Glycoprotein B of HCMV is an important target for neutralizing antibodies and, hence, is often included as a component of intervention strategies. By generation of fusion-active gB chimeras, we were able to identify target structures of neutralizing antibodies that potently block gB-induced membrane fusion. This experimental system provides an approach to screen for antibodies that interfere with gB’s fusogenic activity. In summary, our data will likely contribute to both rational vaccine design and the development of antibody-based therapies against HCMV.
INTRODUCTION
Human cytomegalovirus (HCMV) is a ubiquitous betaherpesvirus that can cause severe disease in immunocompromised patients and newborns infected in utero. HCMV is the most frequent cause of congenital viral infection and affects 1 out of every 150 live-born infants worldwide (1). It is the leading infectious cause of childhood sensorineural hearing loss and an important cause of neurodevelopmental disorders (2). In addition, HCMV is a major cause of morbidity and mortality in recipients of solid-organ or stem cell transplants in both the early and late transplant periods and is thought to contribute to graft dysfunction leading to graft loss late after transplantation, contributing to an overall decrease in long-term survival of transplant recipients (3, 4). Current drug treatment options are limited, and their use is restricted by their toxicity and the development of drug-resistant virus isolates (5). Therefore, prophylactic vaccination has been argued to be the preferred approach for prevention of HCMV infection and disease (6).
The most successful HCMV vaccine tested to date, a recombinant monomeric glycoprotein B (gB) subunit vaccine adjuvanted with MF59, showed 50% efficacy in prevention of HCMV acquisition in a phase 2 trial in postpartum women (7). Furthermore, this vaccine was associated with reduced HCMV viremia and clinical indications for antiviral treatment in transplant recipients (8). Possible reasons proposed for the partial protection observed in clinical trials included a failure of the gB/MF59 vaccine to induce a potent and durable neutralizing antibody response (9–11). In addition, immunization with the gB/MF59 vaccine was reported to result in an antibody profile quite distinct from that during natural HCMV infection (9, 10).
As the primary viral fusion protein, HCMV gB is essential for entry into all cell types. It is also a highly immunogenic protein as gB-specific antibodies can be detected in all naturally infected individuals (12). A major fraction of neutralizing antibodies in sera from HCMV-seropositive donors was directed against gB, and the overall neutralizing capacity of immune serum correlated with the anti-gB antibody titer when it was assayed on fibroblasts (13, 14). As a result of these early findings, gB has long been considered the dominant protein for the induction of neutralizing antibodies; however, components of the pentameric complex gH/gL and UL128-UL131A have more recently received considerable attention as potential targets of clinically relevant virus-neutralizing antibodies (15). Of note, the neutralizing activity of antibodies against the pentameric complex, specifically those against proteins encoded by UL128-UL131A, has been shown to be limited to endothelial, epithelial, and dendritic cells since these monoclonal antibodies (MAbs) have limited activity on fibroblasts or trophoblasts, cells which are HCMV targets in vertical transmission (15–17). Critical to the discussion of the role of HCMV-neutralizing antibodies in the control of HCMV infection is the caveat that HCMV neutralization remains an in vitro determination of the capacity of antibodies to reduce replicating virus infectivity, usually measured by the inhibition of virus entry. Thus, multiple mechanisms could account for in vitro-defined HCMV-neutralizing antibody activity. However, even with this limitation of current knowledge, anti-gB MAbs show potent and robust neutralization independent of the target cell type, and, thus, gB remains an attractive target for inclusion in an HCMV vaccine. Consistent with these arguments, a number of studies using the guinea pig model demonstrated that congenital infection and mortality in pups could be reduced by gB recombinant protein subunit vaccination or by transfer of guinea pig CMV gB immune sera (18–20). Similarly, passive transfer of monoclonal antibodies reactive with murine CMV (MCMV) gB protected immunocompromised mice from MCMV infection (21). Last, recent findings indicate that immunization of mice with gB in a conformation more native than the soluble recombinant gB, as used in human vaccination trials, induced markedly higher titers of serum neutralizing antibodies (22, 23). Interestingly, virus-like particles (VLPs) that were used in the study by Kirchmeier et al. to induce HCMV-neutralizing antibodies were produced by coexpression of murine leukemia virus Gag and a chimeric gB/vesicular stomatitis virus glycoprotein G (VSV-G) protein that was noted to induce syncytium formation (23).
Glycoprotein gB is a homotrimeric type I viral envelope protein consisting of four distinct structural regions: a large ectodomain, a hydrophobic membrane-proximal region (MPR), a transmembrane domain (TM), and a C-terminal cytoplasmic domain (CTD). Additionally, five antigenic domains (AD) targeted by gB-specific antibodies have been identified, which, with the exception of AD-3 (linear epitope located within the CTD of gB), can induce neutralizing antibodies during infection (24). Of note, all neutralizing anti-gB MAbs have similar activities and neutralize 50% of input virus at concentrations in the nanomolar range in fibroblasts and epithelial/endothelial cells. Since the structure of gB is similar to that of the VSV-G protein, HCMV gB is regarded as a type III fusion protein (25). However, unlike VSV-G, HCMV gB requires additional proteins for membrane fusion and viral entry as its fusogenic activity is thought to be triggered by complexes containing glycoproteins gH/gL (26). The gB-gH/gL complex has been shown to form the minimal or core fusion machinery for herpesviruses (27).
At present, precise knowledge about the function and mode of action of anti-gB MAbs is lacking. One potential mechanism of neutralization by anti-gB MAbs could be inhibition of different steps of the fusion, a process that could be envisaged to block early steps in infectivity. One possible mechanism of anti-gB MAbs could be inhibition of the interaction between gB and the gH/gL complexes which would prevent formation of the minimal fusion machinery and activation of gB (26). Furthermore, analogous to other type III viral fusion proteins, such as VSV-G or baculovirus gp64 (25), herpesviral gBs are hypothesized to undergo refolding during fusion. Antibodies that inhibit the conformational rearrangement of gB from a putative fusion-competent prefusion conformation to the postfusion form could be another potential mode of action of virus-neutralizing anti-gB MAbs, a mechanism of virus neutralization proposed for antibodies reactive with other viruses, such influenza A virus (28, 29). Finally, antibody targeting of the fusogenic residues located within the hydrophobic fusion loops which are required for the fusogenic activity of gB is also conceivable because such a mechanism has been recently reported for the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) spike protein (30, 31). Differentiating between these possibilities with respect to antibody recognition and function would be important for identifying targets for the development of potent antiviral antibodies and small-molecule inhibitors but could also be helpful for a better understanding of the fusion process and the structural determinants of the prefusion conformation of gB.
In the current study, we engineered a number of gB mutant molecules with a goal to obtain an intrinsically fusion-active version of gB. Only proteins containing the TM domain and/or CTD of VSV-G or baculovirus gp64 fused to the ectodomain of gB showed fusogenic activity, a finding as noted above that was previously reported in a study using a gB/VSV-G chimera (23). In contrast to expression of fusion-inactive native gB, expression of these chimeras induced large multinucleated cell syncytia in different cell types. By performing cell fusion assays, we found that syncytium formation of gB/VSV-G could be potently blocked by MAbs specifically targeting the antigenic domain 5 (AD-5) of gB. In contrast, neutralizing MAbs targeting other domains of gB were significantly less effective in blocking fusion. These data provide new insight into the mechanism of HCMV neutralization by anti-gB antibodies and strongly argue for differential mechanisms of neutralizing anti-gB MAbs, including the inhibition of viral membrane fusion. In addition, this cell fusion assay will permit selective screening of MAbs and polyvalent antisera that can efficiently interfere with gB-mediated fusion and potentially lead to the development of more effective HCMV immunotherapeutics.
RESULTS
Generation of a fusion-competent HCMV gB.
It has been reported that HCMV gB when expressed by itself is nonfusogenic but when expressed together with gH/gL can induce cell-cell fusion (27). We explored different possibilities to generate an intrinsically fusion-active gB. Structural analyses of herpes simplex virus 1 (HSV-1) gB have proposed that the MPR, TM, and CTD of gB form a uniquely folded pedestal beneath the ectodomain and that the CTD acts as a clamp that controls the fusogenic activity of gB’s ectodomain across the membrane (32, 33). Therefore, in an initial attempt to generate a fusogenic form of gB, we concentrated on the CTD and produced a number of C-terminal mutants of HCMV gB, which are depicted in Fig. 1. Within its carboxy-terminal tail, gB contains a number of motifs which are putatively involved in endocytosis, e.g., tyrosine- and dileucine-containing sorting motifs at positions 845/894 and 883/884, respectively. Similar intracellular trafficking signals in the CTD of HSV gB have been shown to promote fusion activity of HSV gB (34–36). In contrast to fusogenic phenotypes of HSV gB following mutations in the CTD, mutation of the predicted sorting motifs (gB-mutEndo, Y845A LL883/884AA Y894A) failed to produce a fusogenic form of HCMV gB as the phenotype of this mutant was similar to that of wild-type (wt) gB (Fig. 1A, frames a to d). We next replaced the TM and CTD of HCMV gB with that of a hyperfusogenic HSV-1 gB variant harboring the LL871/872AA mutation in the cytoplasmic tail (35, 36). This HCMV-HSV gB chimera was also fusion inactive (Fig. 1A, frames e and f), suggesting that additional regulatory mechanisms other than those provided by the cytosolic domains of alpha- and betaherpesvirus fusogens are required for generation of a fusogenic form of HCMV gB.
FIG 1.

gB/gp64 and gB/VSV-G chimeras are capable of forming cell syncytia. (A and B) HEK293T cells transiently expressing native gB, the CTD mutants gB-mutEndo and gB-HSVmutLL, the gB/gp64 chimera gB703/461gp64, and the gB-VSV-G chimeras gB771/483VSV-G, gB750/463VSV-G, and gB749/468VSV-G (alternatively termed gB/VSV-G). For indirect immunofluorescence analysis, cells were stained with anti-gB MAb 27-287, and nuclei were visualized by counterstaining with DAPI. The schematic representations on the right-hand side illustrate the domain architecture of native HCMV gB and the respective mutants. The individual protein domains are displayed in different colors; signal peptide (SP), membrane proximal region (MPR), transmembrane (TM) domain, and C-terminal domain (CTD/CT) are indicated. (A) gB-mutEndo harbors the endocytosis motif mutations Y845A LL883/884AA Y894A in the CTD. The gB-HSVmutLL chimera contains the TM and CTD of HSV-1 including the LL871/872AA mutation of hyperfusogenic HSV-1 gB. (B) Domains derived from gp64 and VSV-G are displayed in blue and magenta, respectively. Numbers indicate the first and last amino acids of gB (black), gp64 (blue), and VSV-G (magenta), respectively. (C) Box plot of the number of nuclei per syncytium as counted upon transfection of HEK293T cells with the individual gB constructs indicated in panel B and in Fig. 2B. At about 2 days posttransfection, cells were fixed and stained for expression of the gB/chimera. At least 50 gB-positive cells/syncytium were counted for each construct.
Since none of these C-terminal mutations increased the fusogenic activity of gB in our assay, we next constructed a set of chimeric proteins consisting of gB and the structurally related class III viral fusion protein G of vesicular stomatitis virus (VSV-G) and glycoprotein 64 (gp64) of baculovirus. These chimeras contained either the ectodomain and TM domain of gB fused to the CTD of VSV-G (gB771/483VSV-G) or only the ectodomain of gB fused to the TM domain and CTD of VSV-G or gp64 with various portions of the MPR of VSV-G (gB750/463VSV-G and gB749/468VSV-G) or gp64 (gB703/461gp64), respectively (Fig. 1B, schematic representation). In contrast to wt gB with native TM and CTD, which predominantly showed single-positive cells (Fig. 1B, frames a and b), the gB/gp64 and gB/VSV-G chimeric constructs were able to form multinucleated cell syncytia (Fig. 1B, frames c to k), indicating that these chimeras adopt a protein conformation which is capable of inducing the membrane fusion process. To unambiguously confirm this observation, we quantified the nuclei per syncytium formed by HEK293T cells that were transfected with our set of constructs (indicated in Fig. 1B and 2B). As illustrated in Fig. 1C, the highest numbers of nuclei per syncytium were detectable with gB749/468VSV-G (Fig. 1B, frames i and k), which was therefore used for all further experiments and is hereafter referred to as gB/VSV-G.
FIG 2.

Fusion of gB/VSV-G is mediated by the ectodomain of gB. HEK293T cells were transiently transfected with native gH or a gH/VSV-G chimera (A) and the gB/VSV-G chimera gB749/468VSV-G or a fusion loop mutant thereof (gB/VSV-G FLmut) (B). For indirect immunofluorescence analysis, cells were stained with the anti-gH MAb SA4 (A) and the anti-gB MAb 27-287 (B). Cell nuclei were visualized by counterstaining with DAPI. In the linear representations of the gH and gB constructs on the right-hand side, protein domains are defined as described in the legend to Fig. 1. In the gH/VSV-G chimera the TM and CTD of gH were replaced by those of VSV-G (amino acids 468 to 511). To eliminate gB fusion activity, the fusion loop mutant gB/VSV-G FLmut was generated by replacing hydrophobic residues crucial for fusion with more hydrophilic ones (YIY155-157GHR and W240A).
Fusogenic activity of gB/VSV-G is conferred by the ectodomain of gB.
Recent findings have provided evidence for a crucial role of the C terminus of VSV-G in the fusion process (37). Therefore, we set out to define which region of the gB/VSV-G chimera was responsible for driving membrane fusion. In analogy to gB/VSV-G, we also fused the TM domain and CTD of VSV-G to the ectodomain of another type I HCMV glycoprotein, glycoprotein H (gH) (Fig. 2A, schematic representation), which together with gL and gB forms the minimal or core fusion machinery (27). Similar to native gH (Fig. 2A, frames a and b), no syncytium formation was detectable in the case of gH/VSV-G (Fig. 2A, frames c and d), indicating that attaching VSV-G to any nonfusogenic membrane protein is not sufficient to render a membrane protein fusogenic.
The viral fusogen, gB, contains two hydrophobic fusion loops which have been shown to be indispensable for gB’s fusogenic activity in cell-cell fusion assays (31). In particular, residues YIY155–157 and W240 have been shown to be crucial for mediating fusion as exchanging these hydrophobic residues for their less hydrophobic HSV-1 gB counterpart (GHR and A) has been shown to result in fusion-inactive HCMV gB (31). If fusion activity of gB/VSV-G is mediated by the fusion loops within the ectodomain of gB, then exchange of the crucial residues should render the protein nonfusogenic. Mutation of the fusion loops within the gB/VSV-G chimera (gB/VSV-G FLmut) (Fig. 2B, schematic representation) resulted in a chimeric gB that could no longer induce syncytia, in contrast to the cell fusion activity observed following expression of gB/VSV-G (Fig. 1C and 2B, compare frames a and b to frames c and d). This result indicated that the fusion activity of gB/VSV-G requires the native sequence of the fusion loops of gB.
gB/VSV-G induces cell-cell fusion in multiple cell types.
In order to determine whether the fusion capacity of gB/VSV-G was cell type independent, we stably expressed gB in various cells of human (HEK293, adult retinal pigmented epithelium [ARPE-19], RPE, and human foreskin fibroblast [HFF] cells) and murine (ST2) origin via lentivirus transduction (Fig. 3). Addition of doxycycline allowed control of the initiation of gB/VSV-G expression and subsequently the fusion process. While expression of native gB again resulted in only single-positive cells (Fig. 3A to E, frames a and b for HEK293, ARPE-19, RPE, ST2, and HFF, respectively), the chimeric gB/VSV-G was able to induce the formation of large multinucleated syncytia that consistently contained >10 nuclei in all cell types tested except HFF cells (Fig. 3A to D, frames c and d for HEK293, ARPE-19, RPE, and ST2 cells, respectively). Even in the case of murine ST2 cells, large syncytia were clearly visible (Fig. 3D, frames c and d), indicating that the fusion activity of gB/VSV-G is not restricted to cells of human origin but occurs in a species-independent manner. Interestingly, no syncytium formation was detectable following gB/VSV-G expression in HFF cells (Fig. 3E, frames c and d). This finding was in agreement with results reported by Vanarsdall et al., who also observed the lack of cell-cell fusion in normal human fibroblasts upon coexpression of the minimal fusion machinery gB/gH/gL, a result that was in contrast to results with other cell types (27).
FIG 3.

gB/VSV-G forms syncytia in various cell types. (A to E) Syncytium formation was analyzed in the indicated cell types of human (A, B, C, and E) and murine (D) origin stably expressing gB or gB/VSV-G by lentiviral transduction. The expression of gB and gB/VSV-G was induced by treatment with 500 ng/ml doxycycline. At 48 h after doxycycline induction, cells were stained with the anti-gB MAb 27-287 recognizing the gp58 part of gB. Cell nuclei were counterstained with DAPI.
gB/VSV-G expression in cis is sufficient for cell-cell fusion.
To further characterize the determinants of gB/VSV-G-mediated membrane fusion, we tested whether this process requires expression of gB/VSV-G in cis or in trans (Fig. 4). For this assay, we generated bidirectional lentiviral expression constructs, which allowed coexpression of fluorescent marker proteins such as enhanced green fluorescent protein (EGFP) or mCherry (mCh) together with gB or gB/VSV-G (Fig. 4A). Following lentiviral transduction, we obtained ARPE-19 cells, which expressed EGFP/mCherry either alone or in combination with gB or gB/VSV-G (gB+EGFP, gB+mCh, gB/VSV-G+EGFP, or gB/VSV-G+mCh). To assay for fusion in cis, we mixed cells expressing mCherry and gBs (gB+mCh and gB/VSV-G+mCh) with cells expressing EGFP only (Fig. 4B and C, frames a to f). For detection of fusion in trans, the gB+mCh- and gB/VSV-G+mCh-expressing cells were cocultivated with gB+EGFP- or gB/VSV-G+EGFP-expressing cells (Fig. 4B and C, frames g to m). The detection of a yellow signal in the merged channel was considered indicative of a cell-cell fusion event. As expected, in the case of wt gB, no yellow staining was detectable irrespective of whether gB+mCh-expressing cells were mixed with EGFP- or gB+EGFP-expressing cells (Fig. 4B, merge1). By costaining gB with MAb 27-287 (Fig. 4B, frames d and k), we confirmed that the gB+mCh and gB+EGFP cells expressed gB (Fig. 4B, merge2). This result further confirmed previous findings that native gB when expressed in the absence of other viral proteins was nonfusogenic. In contrast to this result, yellow syncytia were readily detectable in the case of the gB/VSV-G-expressing samples (Fig. 4C, merge1). Interestingly, a yellow signal was detected following cocultivation of gB/VSV-G+mCh- with EGFP-expressing cells only (Fig. 4C, frame e). This result indicated that gB/VSV-G has the capacity to fuse with gB-negative cells, as was also evident from the merge2 image of Fig. 4C, where only the mCherry-expressing and not the EGFP-expressing cells were positive for gB staining (Fig. 4C, frame f). Thus, these data demonstrated that expression in cis is sufficient for gB/VSV-G-mediated membrane fusion.
FIG 4.

gB/VSV-G can induce fusion in cis and in trans. (A) Linear schematic representation of the lentiviral constructs having bidirectional promoters used for generating ARPE-19 cells expressing EGFP or mCherry alone as well as in combination with gB (gB+mCh and gB+EGFP) and gB/VSV-G (gB/VSV-G+mCh and gB/VSV-G+EGFP), respectively. Expression from the tetracycline-inducible bidirectional promoter (pTRE3Gbi) is controlled by addition of doxycycline to the cell culture medium. Abbreviations: LTR, long terminal repeat; Ψ, psi packaging signal; RRE, Rev response element; cPPT, central polypurine tract; SV40pA, simian virus 40 polyadenylation signal; mCMV, minimal cytomegalovirus promoter; MCS, multiple-cloning site; BGHpA, bovine growth hormone polyadenylation signal; UBC, ubiquitin c promoter; rtTA3, reverse tetracycline-regulated transactivator gene; IRES, internal ribosome entry site; neo, neomycin resistance gene; WPRE, woodchuck hepatitis virus posttranscriptional regulatory element. (B and C) Lentivirally transduced ARPE-19 cells coding for gB and mCherry (gB+mCh) or gB/VSV-G and mCherry (gB/VSV-G+mCh), as indicated, were cocultivated with ARPE-19 cells encoding either EGFP (EGFP), gB and EGFP (gB+EGFP), or gB/VSV-G and EGFP (gB/VSV-G+EGFP), as indicated. At 48 h postinduction of transgene expression (EGFP, mCherry, gB, and gB/VSV-G) by addition of 500 ng/ml doxycycline, the cells were costained for gB (MAb 27-287). Merge1, overlay of mCherry and EGFP images; merge2, overlay of mCherry, EGFP, and gB images.
Syncytium formation of gB/VSV-G can be blocked by gB-specific MAbs.
Since cell fusion induced by gB/VSV-G was dependent on only the ectodomain of gB (Fig. 2), we addressed the question of whether this process could be inhibited by gB-specific MAbs (Fig. 5). Initially, we tested a set of diverse anti-gB MAbs targeting different antigenic domains (AD) of gB (MAbs 1G2, SM5-1, C23, SM10, and 27-287). Following transfection of HEK293T cells with the gB/VSV-G construct, the cells were incubated with 25 μg/ml of the individual gB MAbs. Interestingly, we found that compared to untreated cells (mock) in which gB/VSV-G expression again led to large multinucleated cell formations (Fig. 5, frames a and b), the sizes of gB/VSV-G-formed syncytia as estimated from the number of nuclei contained in individual syncytia were variable depending on the MAb that was added to the culture (Fig. 5, frames c to m). While most of the MAbs displayed only a moderate effect on syncytium formation (Fig. 5, frames e and f for MAb SM5-1, frames g and h for C23, and frames l and m for 27-287), incubation with MAb 1G2 or SM10 almost completely inhibited cell-cell fusion, as evidenced by the finding of almost entirely single nuclei containing positive cells (Fig. 5, frames c and d for 1G2 and frames i and k for SM10). This result indicated that MAbs directed against the ectodomain of gB could inhibit gB/VSV-G-mediated fusion, albeit with markedly different efficiencies.
FIG 5.
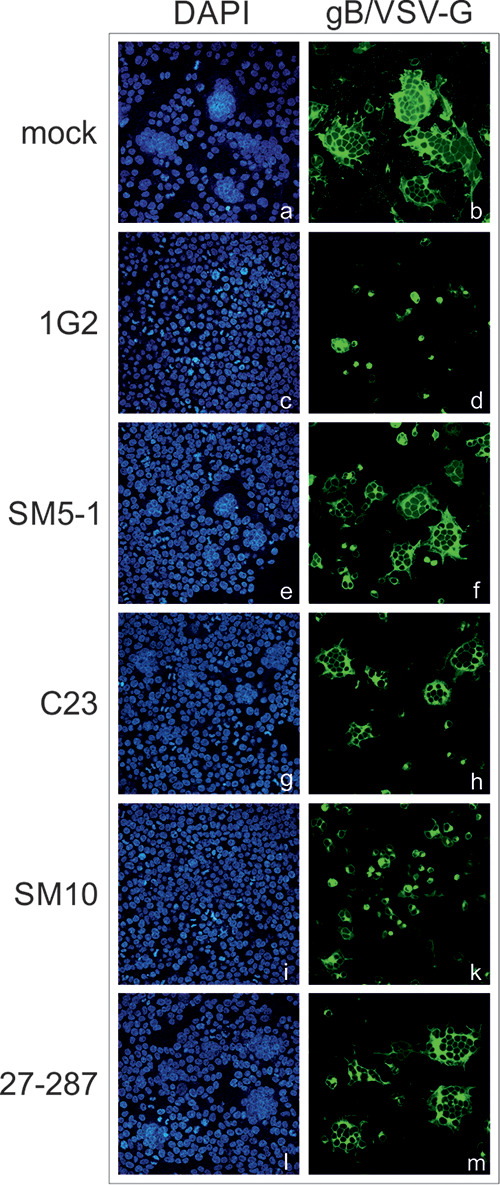
Effects of anti-gB MAbs on gB/VSV-G-induced syncytium formation. HEK293T cells were transiently transfected with the gB/VSV-G expression construct. At 4 h posttransfection, the cells were either mock treated (no antibody) or incubated with 25 μg/ml of the individual anti-gB MAbs (1G2, SM5-1, C23, SM10, and 27-287) as indicated. For indirect immunofluorescence analysis, cells were stained with the anti-gB MAb 27-287. Cell nuclei were visualized by DAPI staining.
AD-5-specific gB MAbs efficiently prevent syncytium formation.
In order to quantify the observed syncytium-inhibitory capacity of MAbs, we established a cell fusion assay which utilized automated quantification of syncytium formation, thus removing potential observer bias in quantification of syncytia (Fig. 6). For direct visualization and counting of syncytia, we generated EGFP-tagged versions of gB (gB-EGFP) and gB/VSV-G (gB/VSV-G-EGFP) (Fig. 6A, schematic representation). Initially, we confirmed that tagging the C terminus of the gB/VSV-G protein with EGFP did not influence its fusogenic activity. In contrast to cells transfected with fusion-negative gB-EGFP (Fig. 6A, frames a and b), large multinucleated syncytia were readily detectable following gB/VSV-G-EGFP expression (Fig. 6A, frames c and d). For quantitation of the cell fusion inhibition assay, we transfected HEK293T cells with the gB-EGFP and gB/VSV-G-EGFP constructs and at 4 h posttransfection added a set of anti-gB MAbs targeting the different AD of gB which have also been shown to exhibit different virus-neutralizing antibody activities (AD-1, SDZ 89-104, 27-39, and 27- 287; AD-2, C23; AD-4, SM5-1; AD-5, 1G2, SM10, and 2C2). As controls, we included the human CMV hyperimmune globulin (HIG) Cytotect as well as two anti-gH MAbs (SA4 and 14-4b), which should serve as controls and have no effect on gB/VSV-G-induced cell fusion. Untreated cells (no antibody) served as a negative control. At 48 h posttransfection, EGFP signals were documented with a Fluorospot reader as shown in Fig. 6B. Syncytia were then defined as fluorescent spots exceeding the size detected for nonfusogenic gB-EGFP and estimated to contain >3 nuclei (compare Fig. 1B and C) and quantified as described in Materials and Methods. The results of at least three independent experiments were combined and are summarized in Fig. 6C. As expected, the anti-gH MAbs SA4 and 14-4b failed to prevent syncytial cell formation (Fig. 6C). Interestingly, the polyclonal serum Cytotect at concentrations of 150 μg/ml was also not active in this assay (Fig. 6C). In contrast, the other anti-gB MAbs, with the exception of MAbs SDZ 89-104 and 27-39, significantly reduced the number of syncytia (Fig. 6C). Of note, the MAbs 1G2, SM10, and 2C2 were highly active and almost completely inhibited cell fusion (Fig. 6C). In addition, titration experiments demonstrated that 10 μg/ml of a potent fusion blocker like 1G2 was sufficient to nearly completely inhibit cell fusion and that even 2.5 μg/ml of 1G2 resulted in over 50% inhibition (Fig. 6D). Combining different antibodies failed to identify a combination of antibodies that could efficiently inhibit syncytium formation, and all remained less effective than 1G2 treatment alone (Fig. 6E). Interestingly, as illustrated in Fig. 6F, the most potent inhibitors of fusion, 1G2, SM10, and 2C2, which are highlighted by a red box, all target the same structural domain of gB (domain I) as they are all AD-5-specific MAbs (24). Thus, our data demonstrated that gB-mediated membrane fusion can efficiently be inhibited by MAbs directed against the AD-5 of gB and suggested that this cell fusion assay could aid in the identification of MAbs which specifically target AD-5 and, in turn, the gB fusion process.
FIG 6.

Identification of anti-gB MAbs that potently block membrane fusion. (A) Immunofluorescence analysis of HEK293T cells transiently expressing gB-EGFP or gB/VSV-G-EGFP. The gB proteins were visualized by their EGFP moiety. Cell nuclei were stained with DAPI. (B) Automated quantification of the syncytium formation. Representative cell fusion assay images taken by a Fluorospot reader are shown. HEK293T cells in 24-well plates transiently expressing gB-EGFP or gB/VSV-G-EGFP were left without (w/o) treatment or treated with 25 μg/ml of the indicated gB-specific MAb (27-287, C23, SM5-1, or 1G2), and images were recorded 48 h following transfection. The numbers in the left corner of the images represent the syncytium counts of each well as calculated by the ImmunoSpot, version 6.0.0.2, software, which marked counted spots by yellow edging (see magnification of untreated cell samples). (C) Cell fusion assay determining the capacity of gB-specific MAbs to prevent gB/VSV-G-induced syncytium formation. HEK293T cells in 24-well plates were transfected with the gB/VSV-G-EGFP expression construct. At 4 h posttransfection, the cultures were washed, and fresh medium (no antibody) or medium containing 25 μg/ml of the anti-gB (SDZ 89-104, 27-39, 27-287, C23, SM5-1, 1G2, SM10, or 2C2) and anti-gH (SA4 and 14-4b) MAbs as well as 150 μg/ml of the HIG Cytotect was added. Images were recorded 48 h after transfection by a Fluorospot reader, and syncytium counts were measured as specified in Materials and Methods using ImmunoSpot, version 6.0.0.2, software. Values were derived minimally from biological triplicates and represent mean values ± standard deviations. Syncytium counts were calculated relative to the level of the mock control (no antibody), which was set to 100%. Statistical analysis was performed by ordinary one-way analysis of variance (ANOVA). n.s., not significant; **, P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤ 0.0001. (D and E) Cell fusion assays with decreasing amounts of the AD-5-specific MAb 1G2 (D) or antibody combinations (E) as indicated. The total amount of IgG that was added to the cells was 25 μg/ml in all combinations. Shown are representative results from three independent experiments. Syncytium counts were calculated relative to the level of the mock control (no antibody), which was set to 100%. (F) Ribbon diagram of a gB monomer, with the structural domains I to V colored as in the linear representation of gB in panel A. The characterized antigenic domains AD-1 to AD-5 of gB are indicated, and the anti-gB MAbs applied in the cell fusion assay shown in panel B are grouped according to their respective target structures. The potent fusion blockers (1G2, SM10, and 2C2), which all bind to AD-5, are highlighted by a red box.
Inhibition of gB/VSV-G fusion does not correlate with neutralization activity of gB MAbs.
Next, we determined if there was a correlation between MAb neutralizing capacity and inhibition of cell fusion. Initially, the relative neutralization activity of the gB MAbs used in the fusion assay was determined in fibroblasts (HFF) and epithelial cells (ARPE-19) by calculating the concentration of IgG needed to achieve 50% inhibition of viral infection (IC50) (Fig. 7A). Except for 27-287 and 27-39 which had IC50 values of >20 μg/ml and therefore were defined as nonneutralizing (nnt), all other anti-gB MAbs shared similar neutralization capacities in both cell types (Fig. 7A). These findings were consistent with previous reports that have described these MAbs in greater detail (24, 38–40). Interestingly, these MAbs were not equally effective in the inhibition of cell fusion induced by the gB/VSV-G chimeric protein (Fig. 6B and C), thus providing evidence that there was a lack of correlation between the virus neutralization activity of anti-gB MAbs and their inhibitory activities in gB-driven cell fusion in our assay. This interpretation was further supported by correlation analyses, as shown in Fig. 7B and C, where the IC50 values of these MAbs were plotted against the calculated syncytium counts of Fig. 6C. These findings clearly demonstrated that potent virus-neutralizing MAbs such as the AD-2-specific MAb C23 or the AD-4-specific MAb SM5-1 had only moderate inhibitory activity in the gB-mediated cell-cell fusion assay but had values identical to or even far lower IC50 than the values of the potent AD-5-specific MAbs (1G2, SM10, and 2C2) that efficiently blocked cell-cell fusion (Fig. 7B and C). Together, these results demonstrated that inhibition of gB fusion was dependent on the gB domain that was targeted by the antibody rather than on neutralization activity of gB MAbs. Perhaps more importantly, these findings provide initial evidence for differential mechanisms of action of anti-gB MAbs in the early events of virus infection and argue that inhibition of gB-mediated fusion could represent a potential correlate of protective antibody responses to HCMV.
FIG 7.
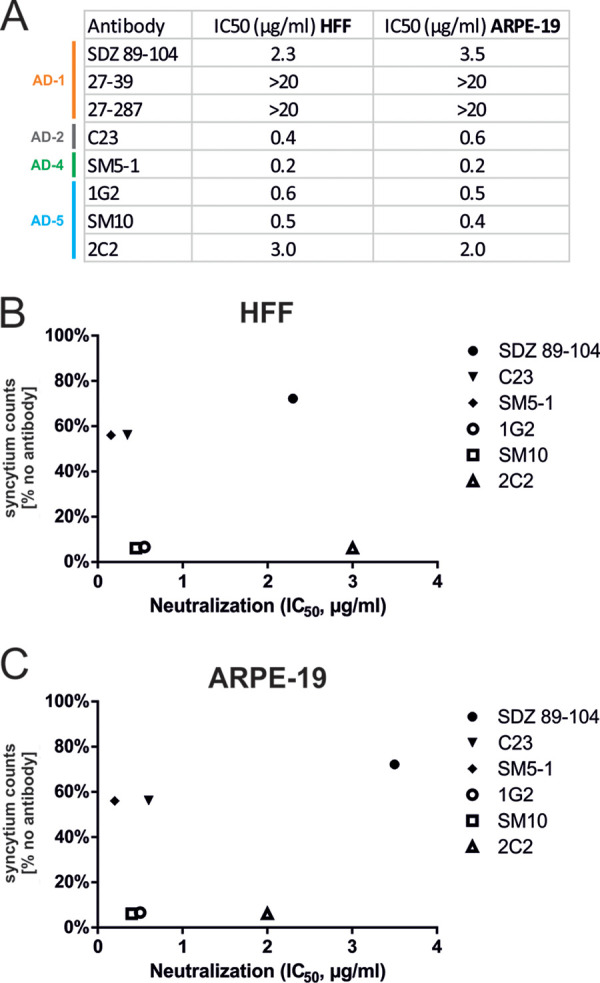
Neutralization activity of gB MAbs is not a direct correlate for cell fusion inhibition. (A) Neutralizing activities of respective anti-gB MAbs in HFF and ARPE-19 cells as indicated. The antibodies were incubated with the luciferase-expressing HCMV AD169 or TB40/E strain. HFF (AD169) or ARPE-19 cells (TB40/E) were infected with the antibody-virus mixture, and luciferase activity was measured 48 h later. The IC50 was then determined. (B and C) Correlation analyses of the capacity of anti-gB MAbs to block fusion in relation to their neutralization activities. For each antibody, the calculated syncytium counts of Fig. 6C, which serve as a measure for cell fusion inhibition and were determined relative to the no-antibody control (set to 100%), were plotted against the IC50s for neutralization in HFF or ARPE-19 cells.
Capacity of human polyclonal antibodies to block gB/VSV-G fusion.
Finally, we determined whether polyclonal antibodies purified from plasma samples of HCMV-positive donors could block gB’s fusion activity and thereby prevent the formation of gB/VSV-G-induced syncytia in our cell fusion assay (Fig. 8). For this, we used lentivirally transduced HEK293 cells expressing a tetracycline (Tet)-regulated fluorescently labeled gB/VSV-G-EGFP that upon doxycycline induction formed large syncytia. We applied antibody preparations at 800 μg/ml from 40 individual HCMV-positive blood donors, which were characterized previously as elite neutralizers that were reported to have high levels of virus-neutralizing antibodies as measured by the capacity to limit infectivity in vitro of cell free virus (41). In contrast to the efficient fusion inhibitory activity of 1G2 (Fig. 8, red dot), these IgG preparations from elite neutralizers (Fig. 8, black dots) proved to be rather ineffective in inhibiting gB’s cell fusion, displaying activity in this assay that was similar to that of the HCMV HIG Cytotect (Fig. 8, green dot). Some IgG preparations even failed completely to prevent syncytium formation of gB/VSV-G (Fig. 8, black dots ≥100%). Thus, we concluded from these data that anti-AD5-specific antibodies directly targeting the process of gB-driven fusion seem to be underrepresented in sera of HCMV-positive individuals that have been selected secondary to high levels of HCMV-neutralizing antibodies (41).
FIG 8.
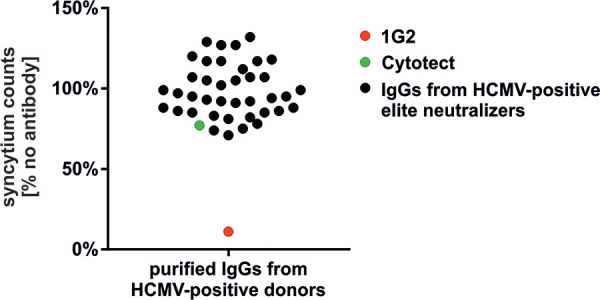
Human polyclonal antibodies fail to efficiently block gB/VSV-G-mediated fusion. Purified IgGs from plasma samples derived from 40 individual HCMV-positive elite neutralizers were tested in the cell fusion assay for their ability to prevent gB/VSV-G-induced syncytium formation. Lentivirally transduced HEK293 cells in 96-well plates were treated with 100 ng/ml of doxycycline to induce expression of fluorescently tagged gB/VSV-G-EGFP. In parallel, 25 μg of 1G2 and 800 μg/ml of either the individual elite neutralizer IgG preparations or the HCMV HIG Cytotect were added to the medium. Untreated cells (no antibody) served as the mock control. Images were recorded 24 h after addition of the IgG preparations by a Fluorospot reader, and syncytium counts were measured as specified in Materials and Methods using ImmunoSpot, version 6.0.0.2, software. Values were derived from biological triplicates. Syncytium counts were calculated relative to the level of the mock control (no antibody), which was set to 100%.
DISCUSSION
Similar to other fusogenic virion envelope proteins, HCMV gB plays an essential role in early steps of virus infection, the cell-to-cell spread of intracellular virions, and the fusion of infected cells leading to the formation of multinucleated cells that have been observed in tissues of HCMV-infected individuals and in vitro following inoculation of viral stocks with very high titers. Because gB has been shown to be essential for virus infectivity and spread and because of its immunodominance in the induction of adaptive immune responses following HCMV infection, gB has been viewed as an essential component of prophylactic vaccines. Several candidate vaccines that have been evaluated in early clinical trials have included gB (42–44). The initial phase II studies with a recombinant gB/MF59 subunit vaccine were encouraging but showed only partial protection (7, 8). The analysis of adaptive immune responses induced by the vaccine also demonstrated that a better understanding of the immune response elicited by gB will be needed to generate vaccines that will elicit improved protective antibody responses (7–11, 45). Moreover, these studies also demonstrated that antiviral activities in addition to virus-neutralizing antibodies (MAbs) as assayed in conventional in vitro assays potentially contribute to protective responses. To date, there is limited progress in the definition of immune correlates of protection from HCMV infections, particularly for correlates of protective antiviral antibodies. Perhaps one major factor for this lack of understanding is the identification and application of relevant in vitro surrogate assays that will allow the discovery of correlates of protective antibodies. Neutralization assays have been used for decades, yet the potential complexity and redundancy of mechanisms of in vitro antibody-mediated neutralization of HCMV infectivity will require deconvolution and identification of specific mechanisms that can then be translated into protective responses in vivo. Similarly, the development of therapeutic MAbs for treatment of HCMV infections will also require an increased level of understanding of the mechanisms of antiviral antibodies in order to generate and to identify antibodies with therapeutic activity. The identification of immunological correlates of protection from in vitro studies has been hampered by the lack of a definitive understanding of the characteristics of protective anti-gB antibodies, including antibody recognition of structures present in the native or prefusion conformation of the gB trimer that are required for the fusion activity associated with virion gB. These studies are further complicated by the requirement that the conserved heterodimeric gH/gL complex must be coexpressed as only in the presence of the minimal or core fusion machinery consisting of gH/gL and trimeric forms of gB can gB adopt a fusion-active conformation (27). Thus, dissecting mechanisms of fusion as well as of fusion inhibition by MAbs and polyvalent immune sera will require a considerably better understanding of the structure of the fusion complex.
In order to overcome these limitations and to study the fusion activity of gB in the absence of an interaction with gH/gL and, thus, a trimolecular complex, we attempted to generate a gB variant which was intrinsically fusion active. Initially we attempted to make gB fusogenic by introducing mutations in the putative endocytosis motifs of the cytoplasmic tail of HCMV gB according to a previously described approach that resulted in the generation of a hyperfusogenic HSV-1 gB variant (LL871/872) (35, 36). However, neither mutation of the predicted HCMV gB sorting motifs (gB-mutEndo, Y845A LL883/884AA Y894A) nor exchange of the TM and CTD of HCMV gB with that of the hyperfusogenic HSV-1 LL871/872 gB variant (gB-HSVmutLL) resulted in a fusogenic form of HCMV gB (Fig. 1A). Since the CTD is generally believed to negatively regulate the fusion activity of herpesviral gB proteins (32), we next generated various chimeras in which we replaced parts of the MPR, the TM, and/or CTD of HCMV gB with those of the structurally related class III fusogens VSV-G and baculovirus gp64, respectively. In contrast to native gB, we observed widespread syncytium formation indicative of membrane fusion in cells expressing these chimeras. Among the different chimeras tested, gB749/468VSV-G (alternatively termed gB/VSV-G) had the most pronounced effect, leading to the formation of large multinucleated syncytia (Fig. 1B), as also observed during generation of gB pseudotypes (23). The CTD of gB, which is part of the pedestal that forms underneath the ectodomain, has been suggested to act as a clamp that stabilizes gB’s ectodomain in its prefusion conformation. Replacing the large CTD of gB with the unstructured and short cytoplasmic tail of gp64 or VSV-G most likely uncoupled the ectodomain from this stabilizing structure (32, 33). Therefore, we propose that gB in the gB/gp64 and gB/VSV-G chimeras adopted a fusion-competent conformation that mediated fusion and syncytium formation with gB-negative cells, including several human cell types and cells of murine origin (Fig. 3 and 4). Furthermore, our findings indicated that the fusion activity of gB/VSV-G was conferred by the ectodomain of gB as mutations of the hydrophobic residues within the fusion loops of gB749/468VSV-G (gB/VSV-G) eliminated syncytium formation and, perhaps more interesting, that anti-gB-specific MAbs reactive with specific domains within the ectodomain inhibited the gB/VSV-G-mediated fusion process (Fig. 2 and 5).
To quantify the capacity of anti-gB MAbs to inhibit gB/VSV-G-mediated membrane fusion, we used automated quantification of syncytium formation. This assay allowed us to quantify anti-gB MAb activity independent of any observer bias that can confound the results from studies that rely on manual detection of syncytia. The results revealed that the anti-gB MAbs which potently blocked fusion of gB/VSV-G shared unique target specificities. At present, five antigenic domains (AD) have been identified on HCMV gB, with AD-1, -2, -4, and -5 inducing antibodies with various levels of virus-neutralizing activity (46). Interestingly, we found that only AD-5-specific MAbs (1G2, SM10, and 2C2) could prevent cell-cell fusion of gB/VSV-G while MAbs targeting the remaining antigenic domains exhibited significantly less activity (Fig. 6). Since most of these MAbs demonstrated comparable in vitro virus neutralization capacities in the nanomolar range in different cell types (Fig. 7), these results indicated that inhibition of gB-mediated fusion was dependent on the specific domain targeted by the MAb that could not be defined by the virus-neutralizing activity of anti-gB MAbs when assayed in vitro. By implication, this observation argues that anti-gB antibody-mediated virus neutralization as measured in vitro almost certainly includes multiple mechanisms leading to the reduction of infectivity, as has been reported in other viruses (47).
The capacity of MAbs directed at domain I of gB, i.e., AD-5, to inhibit cell fusion is perhaps not surprising as this region of gB has been shown to harbor the predicted fusion loops and often has been referred to as the fusion domain of herpesviral gB. Yet, to our knowledge, none of the domain I-specific monoclonal antibodies described for HCMV or other herpesviruses has been shown to target the hydrophobic fusion loops directly, including the antibodies used in the current study, indicating that they block fusion via a different mechanism (38). Binding of antibodies to AD-5 could induce long-range conformational effects that could in turn modulate the fusogenic activity of the fusion loops, as has been hypothesized for gB domain I-specific neutralizing antibodies directed at HSV-1 (48). Another potential mode of action of AD-5 MAbs could be the restriction of conformational changes which gB undergoes during the fusion process. Analogous to other type III viral fusion proteins such as VSV-G or baculovirus gp64, herpesviral gBs have also been hypothesized to refold during fusion. In fact, for VSV-G, for which post- and prefusion structures are available, a space-consuming conformational change seems plausible (49). Thus, these required conformational rearrangements in gB from a fusion-active prefusion form to the postfusion form could be blocked by MAbs targeting AD-5. Such molecular events that are targeted by anti-AD-5 MAbs remain an important objective of further studies as results from such studies could help identify prefusion conformations of gB as well define targets for the development of potent antiviral antibodies. Last, the availability of an intrinsically fusogenic gB variant that did not require the gH/gL complex for activation to a fusogenic form allowed us to postulate that AD-5-specific MAbs could inhibit gB-mediated fusion without targeting a gB-gH/gL interaction. In addition, our data indicated that neutralizing MAbs targeting antigenic domains of gB other than AD-5 block HCMV infection by different modes of action, perhaps by inhibition of protein-protein interactions of the minimal fusion machinery gB-gH/gL. Such a mechanism of action in vivo could be limited by sequence variations in either gB or gH/gL and result in decreased antiviral activity. In contrast, antibodies targeting domains that have an essential role in gB-mediated fusion could be expected to be conserved among different viral isolates and, thus, are less likely to be affected by sequence variations within gB and/or gH/gL. These possibilities further support the concept that anti-gB antibodies can affect virus neutralization by different mechanisms, thus introducing additional complexity into our understanding of the contribution of anti-gB antibodies to immune correlates of protection. It is of importance that several human polyclonal antibody preparations lacked the ability to efficiently block gB’s fusion activity in our assay (Fig. 8). Not only the HCMV HIG Cytotect but also antibody preparations from elite donors which were selected due to their extraordinary neutralization capacity (41) were ineffective in preventing gB-mediated fusion. This suggests that AD-5-specific MAbs targeting the direct fusion process of gB are likely to be underrepresented in sera from healthy HCMV-infected individuals. This is an interesting observation, given the fact that the potent fusion blockers 1G2, SM10, and 2C2 were isolated from three different donors and thus do not seem to represent MAbs with extremely rare or exceptional target specificities (24).
In summary, we have demonstrated that AD-5-specific gB antibodies can directly target the gB-mediated fusion process, as evidenced by their capacity to inhibit gB-mediated generation of syncytia that resemble syncytia of cytomegalic cells associated with CMV infection in vivo. Such molecular events that are targeted by anti-AD-5 MAbs remain an important objective of further studies as results from such studies could help identify prefusion conformations of gB as well as define targets for the development of potent antiviral antibodies. Thus, the induction of antibodies specific for AD-5 should be considered at least one potential measure of an effective prophylactic vaccine, and the development of antibodies targeting this antigenic site could translate into powerful therapeutic agents for passive immunotherapy to modify HCMV infection in populations at risk for HCMV disease. The fact that we are able to differentiate neutralizing gB MAbs into those with fusion inhibiting activities and those without this activity opens up new possibilities to combine therapeutic anti-gB antibodies with different modes of action. In this regard, the established cell fusion assay would seem to represent a valuable tool for the screening and identification of antibodies with defined functional specificities.
MATERIALS AND METHODS
Oligonucleotides and expression constructs.
The oligonucleotide primers used for this study were purchased from Biomers GmbH (Ulm, Germany) and are listed in Table 1. The expression plasmids coding for gB (codon optimized for eukaryotic gene expression) of HCMV strains AD169 (pcADgBcoop) and TB40/E (pcTB40gBcoop), the gB sorting motif mutant gB-mutEndo (Y845A LL883/884AA Y894A), the gB-HSV-1 fusion protein gB-HSVmutLL (LL871/872AA), the chimeras composed of vesicular stomatitis virus G protein fused to gB of AD169 (pcADgB749-G) or TB40/E (pcTB40gB750-G), Autographa californica nucleopolyhedrovirus gp64 fused to gB of TB40/E (gB703/461gp64), and the gH/VSV-G fusion protein (pcUL75/VSV-G) were obtained by cloning of the respective nucleotide sequences synthesized by GeneArt (Thermo Fisher Scientific, Waltham, MA), Integrated DNA Technologies (IDT, Coralville, IA), or Synbio Technologies (Monmouth Junction, NJ) into a pcDNA3.1 expression plasmid. The gB-VSV-G fusion proteins gB771/483VSV-G and gB750/463VSV-G were generated by overlap extension PCR using plasmids pc58 (pcDNA3-based vector coding for AD169 gB) and pLP/VSV-G (Invitrogen/Thermo Fisher Scientific, Waltham, MA) as templates. For amplification of gB771/483VSV-G, the primer pairs gB685-EcoV/gB771-VSV-G CT3 and gB771-VSV-G CT5/VSV-G-G511-Xho3 were used, while gB750/463VSV-G was obtained with the primer pairs gB685-EcoV/gB750-VSV-G TM3 and gB750-VSV-G TM5/VSV-G511-Xho3. The respective PCR products were cloned into pc58. For generation of the fusion loop mutant gB/VSV-G FLmut (residues YIY155–157 and W240 replaced with GRH and A), the respective mutations were introduced by insertion of the corresponding sequence of pcDomI+II-FLmut (38) into pcADgB749-G by EcoNI and SgrAI digestion. The resulting construct was named pMN146. Expression plasmids coding for EGFP-tagged gB and gB/VSV-G (gB-EGFP and gB/VSV-G-EGFP) were generated by insertion of an EGFP coding sequence, amplified by PCR from pEGFP-N1 (TaKaRa Clontech, Saint-Germaine-en-Laye, France) with primers EGFP-Not5 and EGFPXba3, into plasmid pc58, giving rise to pc58EGFP. In a next step, the EGFP nucleotide sequence was transferred from pc58EGFP into pcTB40gBcoop via EcoRI and XbaI digestion and into pcTB40gB750-G via EcoRI and NotI digestion, resulting in plasmids pcTB40gBEGFP and pcTB40gB/VSV-G-EGFP, respectively. The vector coding for wt gH (pRc/CMV-UL75) was described elsewhere (50).
TABLE 1.
Oligonucleotides used for plasmid construction
| Name | Sequence |
|---|---|
| gB685-EcoV | GATTGAATTCAACTCGTACAAG |
| gB771-VSV-G CT3 | CTTTAATTTAATGCAAAGATGGATACCAACTCGATAGATCAAATAAGTGATAATGACTACGGC |
| gB771-VSV-G CT5 | GCCGTAGTCATTATCACTTATTTGATCTATCGAGTTGGTATCCATCTTTGCATTAAATTAAAG |
| VSV-G511-Xho3 | GATTGCTCGAGCTTACTTTCCAAGTC |
| gB750-VSV-G TM3 | TATGATAAAGAAAAGAGGCAATAGAGCTGGGGTTTTTGAGGAAGGTGGCAACGCCTTC |
| gB750-VSV-G TM5 | GAAGGCGTTGCCACCTTCCTCAAAAACCCCAGCTCTATTGCCTCTTTTTTCTTTATCATA |
| EGFP-Not5 | GATTGCGGCCGCCATGGTGAGCAAGGGCGAG |
| EGFPXba3 | ATGATCTAGAGTCGCGGCCGCTTTAC |
| c-CRS-mut | GCGTGTACGGTGGGAGGCCTATATAAGCAGAGCCTAGGTAGGGAGAAGTCAGATCGCCTGGAGACGCC |
| nc-CRS-mut | GGCGTCTCCAGGCGATCTGACTTCTCCCTACCTAGGCTCTGCTTATATAGGCCTCCCACCGTACACGC |
| 5′-BGHpA-Xba,SexA1,Asc,Nsi,Pme | GCATTCTAGAACCAGGTAGGCGCGCCTAATGCATAGTTTAAACTAGGCCCTATTCTATAGTGTCACCTAAATGCTAGAGC |
| 3′-BGHpA-XcmI | GAATCCAGATCTTGGGTGGGATACCCCCTAGAGCCCCAGC |
| 5′-pInd-ADcoop/TB40gBcoop-AscI | CATAGGCGCGCCATGGAAAGCCGGATCTGGTGCC |
| 3′-pInd-AD/TBgBcoop-PmeI | CATAGTTTAAACTCACACGTTCTCTTCCTCGTCG |
| 3′-pInd-TB/ADgB751-G-PmeI | CATAGTTTAAACTTACTTGCCCAGCCGGTTC |
| 3′-EGFP-PmeI | CATAGTTTAAACTTACTTGTACAGCTCGTC |
| 5′-BglII-SV40pA-pTREbi | GCATAGATCTCACCTGACGTCGGCAGTGAAAAAAATGC |
| 3′-Asc1,SexA1-pTRE3Gbi | GCATGGCGCGCCTACCTGGTGGGCCCCGGTGTCGACTTTACGAGG |
The lentiviral constructs for doxycycline-inducible expression of gB and gB/VSV-G (corresponding to TB40/E-based gB750/468VSV-G) were generated using a modified pInducer20-based vector (51). First, the cis repression signal (CRS) of the HCMV major immediate early promoter (MIEP) was mutated by site-directed mutagenesis using primers c-CRS-mut and nc-CRS-mut in order to prevent IE2p86-based autorepression and downregulation of transgene expression following HCMV infection (52). In addition, a bovine growth hormone polyadenylation signal (BGH-pA) as well as a multiple-cloning site was inserted by cloning of a PCR product obtained with primers 5′-BGHpA-Xba,SexA1,Asc,Nsi,Pme and 3′-BGHpA-XcmI as well as pHM971 (53) as the template, resulting in plasmid pMN1. Finally, PCR-amplified TB40/E gB was introduced, which was obtained using primers 5′-pInd-ADcoop/TB40gBcoop-AscI and 3′-pInd-AD/TBgBcoop-PmeI together with pcTB40gBcoop as the template, yielding plasmid pMN4. For introduction of gB/VSV-G, gB750/468VSV-G was amplified by PCR with primers 5′-pInd-ADcoop/TB40gBcoop-AscI and 3′-pInd-TB/ADgB751-G-PmeI using pcTB40gB750-G as the template, which resulted in plasmid pMN5. In addition, fluorescently labeled gB-EGFP and gB/VSV-G-EGFP were introduced into pMN1 by PCR amplification using primers 5′-pInd-ADcoop/TB40gBcoop-AscI and 3′-EGFP-PmeI as well as plasmid pcTB40gBEGFP or pcTB40gB/VSV-G-EGFP as templates, resulting in plasmids pMN67 and pMN68, respectively.
In order to generate bidirectional doxycycline-inducible vectors with fluorescent marker genes, first pTRE3G-Bi-mCherry (pMN50) was generated via digestion of plasmid pTRE3G-bi-mCherry-PIK3CA (kindly provided by A. Rajput, Albuquerque, NM) with BamHI and BglII, followed by religation. For pTRE3G-Bi-EGFP (pMN51), mCherry from pMN50 was cut out via EcoRI and XbaI and replaced by EGFP excised with the same enzymes from pEGFP-N1. Lentiviral control vectors encoding the bidirectional doxycycline-responsive promoter and mCherry or EGFP but no additional transgene were generated by insertion of the BglII+AscI-digested PCR-product of templates pMN50/pMN51 amplified with primers 5′-BglII-SV40pA-pTREbi and 3′-Asc1,SexA1-pTRE3Gbi into the BamHI and AscI sites of pMN1, thereby generating pMN59 (mCherry+empty) and pMN60 (EGFP+empty), respectively. In analogy, the monodirectional TRE2 promoters from pMN4 and pMN5 were removed via BamHI and AscI digestion and subsequently replaced by the BglII- and AscI-digested PCR product of templates pMN50 and pMN51 amplified with primers 3′-Asc1,SexA1-pTRE3Gbi and 5′-BglII-SV40pA-pTREbi. This procedure resulted in the bidirectional doxycycline-inducible plasmids coexpressing mCherry and gB (pMN62) or mCherry and gB/VSV-G (pMN63). The corresponding bidirectional doxycycline-inducible vectors expressing EGFP instead of mCherry are named pMN65 and pMN66, respectively.
Cells and viruses.
Human embryonic kidney cells (HEK293 and HEK293T), primary human foreskin fibroblasts (HFF), human retinal pigmented epithelial cells (ARPE-19 and RPE), and murine stromal cells (ST2) were cultured in Dulbecco's modified Eagle’s medium (DMEM) (Gibco/Thermo Fisher Scientific, Waltham, MA) supplemented with 10% fetal calf serum (FCS) (Sigma-Aldrich, St. Louis, MO), glutamine (100 μg/ml), and gentamicin (350 μg/ml).
Luciferase-expressing HCMV AD169 (24) and TB40/E (54) cells were propagated in human fetal lung fibroblasts (MRC-5), and viral titers were determined by titration of virus stocks in MRC-5 cells, using an indirect immunofluorescence assay with the MAb p63-27, directed against the HCMV immediate early 1 (IE1) protein (55), or by measurement of luciferase activity.
Antibodies.
The following antibodies were used: gB AD-1-specific human MAbs SDZ 89-104 (56), 27-39 (57), 27-287 (58); gB AD-2-specific human MAb C23 (Ti23) (59); gB AD-4-specific human MAb SM5-1 (24); gB AD-5-specific human MAbs 1G2, SM10, and 2C2 (38); the mouse anti-gH MAbs AP86-SA4, referred to as SA4 (60), and 14-4b (61). The human CMV hyperimmune globulin Cytotect was purchased from Biotest (Dreieich, Germany). The Alexa Fluor 488-, 555-, and 647-conjugated secondary antibodies for indirect immunofluorescence experiments were purchased from Molecular Probes (Karlsruhe, Germany). Polyclonal anti-HCMV antibodies were purified via protein A chromatography from plasma samples from HCMV-seropositive blood donors provided by the German Red Cross Blood-Transfusion Service, Baden-Württemberg and Hessen, with informed consent according to human experimentation guidelines (Ethical Board of Ulm University vote number 53/14) as previously described (41, 62).
Generation of gB-expressing cells by lentivirus transduction.
For the generation of gB-, gB/VSV-G-, gB-EGFP-, and gB/VSV-G-EGFP-expressing HFF, HEK293, HEK293T, or ST2 cells, replication-deficient lentiviruses were generated by using pINDUCER20-based expression vectors. HEK293, ARPE-19, and RPE cells stably expressing EGFP/mCherry alone or in combination with gB or gB/VSV-G were generated by using pINDUCER20-based lentiviral vectors harboring bidirectional promoters derived from pTRE3G-Bi-EGFP/pTRE3G-Bi-mCherry (kindly provided by A. Rajput, Albuquerque, NM). For this purpose, HEK293T cells were seeded into 10-cm dishes (5 × 106 cells) cotransfected with the lentiviral expression vectors, together with packaging plasmids psPAX2 and pLP/VSV-G, by using Lipofectamine 2000 reagent (Invitrogen, Karlsruhe, Germany). Viral supernatants were harvested at 48 h posttransfection, cleared by centrifugation, filtered, and stored at −80°C. The respective cells were incubated for 24 h with lentiviral supernatants in the presence of 7.5 μg/ml Polybrene (Merck Chemicals GmbH, Darmstadt, Germany). Stably transduced cell populations were selected by the addition of 500 μg/ml Geneticin to the cell culture medium. Transgene expression was induced by addition of 500 ng/ml doxycycline to the cell culture medium.
Indirect immunofluorescence analysis.
For indirect immunofluorescence analysis, lentivirally transduced cells or transfected HEK293T cells (calcium phosphate precipitation) were grown on coverslips. At 48 h posttransfection or doxycycline-induced transgene expression, the cells were washed three times with phosphate-buffered saline (PBS), followed by fixation with 3% paraformaldehyde for 10 min at room temperature. Then, the cells were permeabilized with PBS–0.2% Triton X-100 on ice for 20 min, followed by incubation with the respective primary or secondary antibodies for 30 min at 37°C. Finally, the cells were mounted by using Vectashield mounting medium plus 4′,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories, Burlingame, CA). The samples were examined by using a Leica TCS SP5 confocal microscope with a 488-nm, 543-nm, or 633-nm laser line, scanning each channel separately under image capture conditions that eliminated channel overlap. The images were then exported, processed with Adobe Photoshop CS5, and assembled by using CorelDraw X6. For quantification of nuclei per syncytium, HEK293T cells were fixed about 36 h after transfection with the respective gB/chimera and incubated with the anti-gB antibody 27-287.
Cell fusion assay.
HEK293T cells seeded in 24-well plates (2 × 105 cells/well) were transfected with the gB/EGFP and gB/VSV-G-EGFP expression constructs by calcium phosphate precipitation. At 4 h posttransfection, the cell culture medium was replaced with medium containing 25 μg/ml of the respective anti-gB and anti-gH MAbs or 150 μg of the HCMV hyperimmune globulin Cytotect. After 48 h, cells were fixed with 3% paraformaldehyde, and plates were imaged with a CTL Immunospot S6 analyzer (Cellular Technology Limited, Bonn, Germany). Then, syncytium formation was quantified using ImmunoSpot, version 6.0.0.2, software (Cellular Technology Limited, Bonn, Germany). Syncytia were defined as EGFP signals being larger than the fluorescent spots detected for nonfusogenic gB-EGFP (spot sizes of >0.012 mm2).
Virus neutralization assay.
HFF or ARPE-19 cells were seeded in 96-well plates (1 × 104 cells/well). Luciferase-expressing HCMV AD169 (infection of HFF cells) and TB40/E (infection of ARPE-19 cells) cells were preincubated with serial log2 dilutions of anti-gB MAbs for 1 h at 37°C, and the mixtures were added to the cells for 4 h. The inoculum was replaced with fresh medium, and the cells were incubated at 37°C for 48 h. Cells were lysed using 100 μl of Glo lysis buffer (Promega) per well. Thirty microliters of each cell lysate was transferred to white 96-well LIA plates (Costar), and 50 μl of assay buffer (15 mM KH2PO4, 25 mM glycylglycine, 15 mM MgSO4, 4 mM EGTA, 5 mM ATP, 1 mM dithiothreitol [DTT]) was added to each well. Luciferase activity was measured by injection of 50 μl of d-luciferin (PJK GmbH) solution (25 mM glycylglycine, 15 mM MgSO4, 4 mM EGTA, 2 mM DTT, and 0.05 mM d-luciferin) per well, and detection of chemiluminescence was performed by use of an Orion microplate luminometer (Berthold Technologies). The data were plotted as percent neutralization versus that in control wells, to which no antibody was added, and the 50% inhibitory concentrations (IC50s) were determined.
Statistical analysis.
Statistical analysis was performed by ordinary one-way analysis of variance (ANOVA) using GraphPad Prism (version 6; GraphPad Software, USA).
ACKNOWLEDGMENTS
We express sincere gratitude to Anna-Katharina Wiegers for her practical and conceptual contributions to this study. We thank Ashwani Rajput and Guanghua Wan (University of New Mexico, Albuquerque, NM) for generously providing the plasmid pTRE3G-bi-mCherry-PIK3CA. Antibody C23 (TI23) was a kind gift of Teijin Pharma Limited, Japan.
We thank Fondation Dormeur, Vaduz, for an equipment grant to purchase the CTL Immunospot S6 Ultimate UV Analyzer (Cellular Technology Limited, Bonn, Germany). This work was supported by grants from the NIH (grants 1R21AI126886-01 and 1R01AI089956-01 to W.J.B.) and the Else Kröner-Fresenius Foundation (grant 2016-A126 to A.K. and C.S.).
REFERENCES
- 1.Manicklal S, Emery VC, Lazzarotto T, Boppana SB, Gupta RK. 2013. The “silent” global burden of congenital cytomegalovirus. Clin Microbiol Rev 26:86–102. doi: 10.1128/CMR.00062-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dreher AM, Arora N, Fowler KB, Novak Z, Britt WJ, Boppana SB, Ross SA. 2014. Spectrum of disease and outcome in children with symptomatic congenital cytomegalovirus infection. J Pediatr 164:855–859. doi: 10.1016/j.jpeds.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ramanan P, Razonable RR. 2013. Cytomegalovirus infections in solid organ transplantation: a review. Infect Chemother 45:260–271. doi: 10.3947/ic.2013.45.3.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Teira P, Battiwalla M, Ramanathan M, Barrett AJ, Ahn KW, Chen M, Green JS, Saad A, Antin JH, Savani BN, Lazarus HM, Seftel M, Saber W, Marks D, Aljurf M, Norkin M, Wingard JR, Lindemans CA, Boeckh M, Riches ML, Auletta JJ. 2016. Early cytomegalovirus reactivation remains associated with increased transplant-related mortality in the current era: a CIBMTR analysis. Blood 127:2427–2438. doi: 10.1182/blood-2015-11-679639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chou S. 2015. Approach to drug-resistant cytomegalovirus in transplant recipients. Curr Opin Infect Dis 28:293–299. doi: 10.1097/QCO.0000000000000170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arvin AM, Fast P, Myers M, Plotkin S, Rabinovich R, National Vaccine Advisory Committee. 2004. Vaccine development to prevent cytomegalovirus disease: report from the National Vaccine Advisory Committee. Clin Infect Dis 39:233–239. doi: 10.1086/421999. [DOI] [PubMed] [Google Scholar]
- 7.Pass RF, Zhang C, Evans A, Simpson T, Andrews W, Huang ML, Corey L, Hill J, Davis E, Flanigan C, Cloud G. 2009. Vaccine prevention of maternal cytomegalovirus infection. N Engl J Med 360:1191–1199. doi: 10.1056/NEJMoa0804749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Griffiths PD, Stanton A, McCarrell E, Smith C, Osman M, Harber M, Davenport A, Jones G, Wheeler DC, O'Beirne J, Thorburn D, Patch D, Atkinson CE, Pichon S, Sweny P, Lanzman M, Woodford E, Rothwell E, Old N, Kinyanjui R, Haque T, Atabani S, Luck S, Prideaux S, Milne RS, Emery VC, Burroughs AK. 2011. Cytomegalovirus glycoprotein-B vaccine with MF59 adjuvant in transplant recipients: a phase 2 randomised placebo-controlled trial. Lancet 377:1256–1263. doi: 10.1016/S0140-6736(11)60136-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nelson CS, Huffman T, Jenks JA, Cisneros de la Rosa E, Xie G, Vandergrift N, Pass RF, Pollara J, Permar SR. 2018. HCMV glycoprotein B subunit vaccine efficacy mediated by nonneutralizing antibody effector functions. Proc Natl Acad Sci U S A 115:6267–6272. doi: 10.1073/pnas.1800177115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baraniak I, Kropff B, Ambrose L, McIntosh M, McLean GR, Pichon S, Atkinson C, Milne RSB, Mach M, Griffiths PD, Reeves MB. 2018. Protection from cytomegalovirus viremia following glycoprotein B vaccination is not dependent on neutralizing antibodies. Proc Natl Acad Sci U S A 115:6273–6278. doi: 10.1073/pnas.1800224115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cui X, Meza BP, Adler SP, McVoy MA. 2008. Cytomegalovirus vaccines fail to induce epithelial entry neutralizing antibodies comparable to natural infection. Vaccine 26:5760–5766. doi: 10.1016/j.vaccine.2008.07.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schoppel K, Kropff B, Schmidt C, Vornhagen R, Mach M. 1997. The humoral immune response against human cytomegalovirus is characterized by a delayed synthesis of glycoprotein-specific antibodies. J Infect Dis 175:533–544. doi: 10.1093/infdis/175.3.533. [DOI] [PubMed] [Google Scholar]
- 13.Britt WJ, Vugler L, Butfiloski EJ, Stephens EB. 1990. Cell surface expression of human cytomegalovirus (HCMV) gp55-116 (gB): use of HCMV-recombinant vaccinia virus-infected cells in analysis of the human neutralizing antibody response. J Virol 64:1079–1085. doi: 10.1128/JVI.64.3.1079-1085.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marshall GS, Rabalais GP, Stout GG, Waldeyer SL. 1992. Antibodies to recombinant-derived glycoprotein B after natural human cytomegalovirus infection correlate with neutralizing activity. J Infect Dis 165:381–384. doi: 10.1093/infdis/165.2.381. [DOI] [PubMed] [Google Scholar]
- 15.Macagno A, Bernasconi NL, Vanzetta F, Dander E, Sarasini A, Revello MG, Gerna G, Sallusto F, Lanzavecchia A. 2010. Isolation of human monoclonal antibodies that potently neutralize human cytomegalovirus infection by targeting different epitopes on the gH/gL/UL128-131A complex. J Virol 84:1005–1013. doi: 10.1128/JVI.01809-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zydek M, Petitt M, Fang-Hoover J, Adler B, Kauvar LM, Pereira L, Tabata T. 2014. HCMV infection of human trophoblast progenitor cells of the placenta is neutralized by a human monoclonal antibody to glycoprotein B and not by antibodies to the pentamer complex. Viruses 6:1346–1364. doi: 10.3390/v6031346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cui X, Freed DC, Wang D, Qiu P, Li F, Fu TM, Kauvar LM, McVoy MA. 2017. Impact of antibodies and strain polymorphisms on cytomegalovirus entry and spread in fibroblasts and epithelial cells. J Virol 91:e01650-16. doi: 10.1128/JVI.01650-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cardin RD, Bravo FJ, Pullum DA, Orlinger K, Watson EM, Aspoeck A, Fuhrmann G, Guirakhoo F, Monath T, Bernstein DI. 2016. Replication-defective lymphocytic choriomeningitis virus vectors expressing guinea pig cytomegalovirus gB and pp65 homologs are protective against congenital guinea pig cytomegalovirus infection. Vaccine 34:1993–1999. doi: 10.1016/j.vaccine.2016.03.005. [DOI] [PubMed] [Google Scholar]
- 19.Schleiss MR, Choi KY, Anderson J, Mash JG, Wettendorff M, Mossman S, Van Damme M. 2014. Glycoprotein B (gB) vaccines adjuvanted with AS01 or AS02 protect female guinea pigs against cytomegalovirus (CMV) viremia and offspring mortality in a CMV-challenge model. Vaccine 32:2756–2762. doi: 10.1016/j.vaccine.2013.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chatterjee A, Harrison CJ, Britt WJ, Bewtra C. 2001. Modification of maternal and congenital cytomegalovirus infection by anti-glycoprotein b antibody transfer in guinea pigs. J Infect Dis 183:1547–1553. doi: 10.1086/320714. [DOI] [PubMed] [Google Scholar]
- 21.Bootz A, Karbach A, Spindler J, Kropff B, Reuter N, Sticht H, Winkler TH, Britt WJ, Mach M. 2017. Protective capacity of neutralizing and non-neutralizing antibodies against glycoprotein B of cytomegalovirus. PLoS Pathog 13:e1006601. doi: 10.1371/journal.ppat.1006601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cui X, Cao Z, Wang S, Lee RB, Wang X, Murata H, Adler SP, McVoy MA, Snapper CM. 2018. Novel trimeric human cytomegalovirus glycoprotein B elicits a high-titer neutralizing antibody response. Vaccine 36:5580–5590. doi: 10.1016/j.vaccine.2018.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kirchmeier M, Fluckiger AC, Soare C, Bozic J, Ontsouka B, Ahmed T, Diress A, Pereira L, Schodel F, Plotkin S, Dalba C, Klatzmann D, Anderson DE. 2014. Enveloped virus-like particle expression of human cytomegalovirus glycoprotein B antigen induces antibodies with potent and broad neutralizing activity. Clin Vaccine Immunol 21:174–180. doi: 10.1128/CVI.00662-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Potzsch S, Spindler N, Wiegers AK, Fisch T, Rucker P, Sticht H, Grieb N, Baroti T, Weisel F, Stamminger T, Martin-Parras L, Mach M, Winkler TH. 2011. B cell repertoire analysis identifies new antigenic domains on glycoprotein B of human cytomegalovirus which are target of neutralizing antibodies. PLoS Pathog 7:e1002172. doi: 10.1371/journal.ppat.1002172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baquero E, Albertini AA, Gaudin Y. 2015. Recent mechanistic and structural insights on class III viral fusion glycoproteins. Curr Opin Struct Biol 33:52–60. doi: 10.1016/j.sbi.2015.07.011. [DOI] [PubMed] [Google Scholar]
- 26.Vanarsdall AL, Johnson DC. 2012. Human cytomegalovirus entry into cells. Curr Opin Virol 2:37–42. doi: 10.1016/j.coviro.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vanarsdall AL, Ryckman BJ, Chase MC, Johnson DC. 2008. Human cytomegalovirus glycoproteins gB and gH/gL mediate epithelial cell-cell fusion when expressed either in cis or in trans. J Virol 82:11837–11850. doi: 10.1128/JVI.01623-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kallewaard NL, Corti D, Collins PJ, Neu U, McAuliffe JM, Benjamin E, Wachter-Rosati L, Palmer-Hill FJ, Yuan AQ, Walker PA, Vorlaender MK, Bianchi S, Guarino B, De Marco A, Vanzetta F, Agatic G, Foglierini M, Pinna D, Fernandez-Rodriguez B, Fruehwirth A, Silacci C, Ogrodowicz RW, Martin SR, Sallusto F, Suzich JA, Lanzavecchia A, Zhu Q, Gamblin SJ, Skehel JJ. 2016. Structure and function analysis of an antibody recognizing all influenza A subtypes. Cell 166:596–608. doi: 10.1016/j.cell.2016.05.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ekiert DC, Bhabha G, Elsliger MA, Friesen RH, Jongeneelen M, Throsby M, Goudsmit J, Wilson IA. 2009. Antibody recognition of a highly conserved influenza virus epitope. Science 324:246–251. doi: 10.1126/science.1171491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Poh CM, Carissimo G, Wang B, Amrun SN, Lee CY, Chee RS, Fong SW, Yeo NK, Lee WH, Torres-Ruesta A, Leo YS, Chen MI, Tan SY, Chai LYA, Kalimuddin S, Kheng SSG, Thien SY, Young BE, Lye DC, Hanson BJ, Wang CI, Renia L, Ng L. 2020. Two linear epitopes on the SARS-CoV-2 spike protein that elicit neutralising antibodies in COVID-19 patients. Nat Commun 11:2806. doi: 10.1038/s41467-020-16638-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sharma S, Wisner TW, Johnson DC, Heldwein EE. 2013. HCMV gB shares structural and functional properties with gB proteins from other herpesviruses. Virology 435:239–249. doi: 10.1016/j.virol.2012.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cooper RS, Heldwein EE. 2015. Herpesvirus gB: a finely tuned fusion machine. Viruses 7:6552–6569. doi: 10.3390/v7122957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cooper RS, Georgieva ER, Borbat PP, Freed JH, Heldwein EE. 2018. Structural basis for membrane anchoring and fusion regulation of the herpes simplex virus fusogen gB. Nat Struct Mol Biol 25:416–424. doi: 10.1038/s41594-018-0060-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tugizov S, Maidji E, Xiao J, Pereira L. 1999. An acidic cluster in the cytosolic domain of human cytomegalovirus glycoprotein B is a signal for endocytosis from the plasma membrane. J Virol 73:8677–8688. doi: 10.1128/JVI.73.10.8677-8688.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beitia Ortiz de Zarate I, Cantero-Aguilar L, Longo M, Berlioz-Torrent C, Rozenberg F. 2007. Contribution of endocytic motifs in the cytoplasmic tail of herpes simplex virus type 1 glycoprotein B to virus replication and cell-cell fusion. J Virol 81:13889–13903. doi: 10.1128/JVI.01231-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Atanasiu D, Saw WT, Gallagher JR, Hannah BP, Matsuda Z, Whitbeck JC, Cohen GH, Eisenberg RJ. 2013. Dual split protein-based fusion assay reveals that mutations to herpes simplex virus (HSV) glycoprotein gB alter the kinetics of cell-cell fusion induced by HSV entry glycoproteins. J Virol 87:11332–11345. doi: 10.1128/JVI.01700-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ci Y, Yang Y, Xu C, Shi L. 2018. Vesicular stomatitis virus G protein transmembrane region is crucial for the hemi-fusion to full fusion transition. Sci Rep 8:10669. doi: 10.1038/s41598-018-28868-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wiegers AK, Sticht H, Winkler TH, Britt WJ, Mach M. 2015. Identification of a neutralizing epitope within antigenic domain 5 of glycoprotein B of human cytomegalovirus. J Virol 89:361–372. doi: 10.1128/JVI.02393-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kropff B, Burkhardt C, Schott J, Nentwich J, Fisch T, Britt W, Mach M. 2012. Glycoprotein N of human cytomegalovirus protects the virus from neutralizing antibodies. PLoS Pathog 8:e1002999. doi: 10.1371/journal.ppat.1002999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Utz U, Britt W, Vugler L, Mach M. 1989. Identification of a neutralizing epitope on glycoprotein gp58 of human cytomegalovirus. J Virol 63:1995–2001. doi: 10.1128/JVI.63.5.1995-2001.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Falk JJ, Winkelmann M, Stohr D, Alt M, Schrezenmeier H, Krawczyk A, Lotfi R, Sinzger C. 2018. Identification of elite neutralizers with broad and potent neutralizing activity against human cytomegalovirus (HCMV) in a population of HCMV-seropositive blood donors. J Infect Dis 218:876–885. doi: 10.1093/infdis/jiy229. [DOI] [PubMed] [Google Scholar]
- 42.Gerna G, Lilleri D. 2019. Human cytomegalovirus (HCMV) infection/re-infection: development of a protective HCMV vaccine. New Microbiol 42:1–20. [PubMed] [Google Scholar]
- 43.Schleiss MR, Permar SR, Plotkin SA. 2017. Progress toward Development of a Vaccine against Congenital Cytomegalovirus Infection. Clin Vaccine Immunol 24:e00268-17. doi: 10.1128/CVI.00268-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xia L, Su R, An Z, Fu TM, Luo W. 2018. Human cytomegalovirus vaccine development: immune responses to look into vaccine strategy. Hum Vaccin Immunother 14:292–303. doi: 10.1080/21645515.2017.1391433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bernstein DI, Munoz FM, Callahan ST, Rupp R, Wootton SH, Edwards KM, Turley CB, Stanberry LR, Patel SM, McNeal MM, Pichon S, Amegashie C, Bellamy AR. 2016. Safety and efficacy of a cytomegalovirus glycoprotein B (gB) vaccine in adolescent girls: a randomized clinical trial. Vaccine 34:313–319. doi: 10.1016/j.vaccine.2015.11.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mach MW, Spindler N, Winkler T. 2013. Protective humoral immunity, p 215–231. In Reddehase M. (ed), Cytomegaloviruses: from molecular pathogenesis to intervention. Caister Academic Press, Norfolk, United Kingdom. [Google Scholar]
- 47.Klasse PJ. 2014. Neutralization of virus infectivity by antibodies: old problems in new perspectives. Adv Biol 2014:1–24. doi: 10.1155/2014/157895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cairns TM, Fontana J, Huang ZY, Whitbeck JC, Atanasiu D, Rao S, Shelly SS, Lou H, Ponce de Leon M, Steven AC, Eisenberg RJ, Cohen GH. 2014. Mechanism of neutralization of herpes simplex virus by antibodies directed at the fusion domain of glycoprotein B. J Virol 88:2677–2689. doi: 10.1128/JVI.03200-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Backovic M, Jardetzky TS. 2011. Class III viral membrane fusion proteins. Adv Exp Med Biol 714:91–101. doi: 10.1007/978-94-007-0782-5_3. [DOI] [PubMed] [Google Scholar]
- 50.Reschke M, Revello MG, Percivalle E, Radsak K, Landini MP. 1999. Constitutive expression of human cytomegalovirus (HCMV) glycoprotein gpUL75 (gH) in astrocytoma cells: a study of the specific humoral immune response. Viral Immunol 12:249–262. doi: 10.1089/vim.1999.12.249. [DOI] [PubMed] [Google Scholar]
- 51.Meerbrey KL, Hu G, Kessler JD, Roarty K, Li MZ, Fang JE, Herschkowitz JI, Burrows AE, Ciccia A, Sun T, Schmitt EM, Bernardi RJ, Fu X, Bland CS, Cooper TA, Schiff R, Rosen JM, Westbrook TF, Elledge SJ. 2011. The pINDUCER lentiviral toolkit for inducible RNA interference in vitro and in vivo. Proc Natl Acad Sci U S A 108:3665–3670. doi: 10.1073/pnas.1019736108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Reuter N, Reichel A, Stilp AC, Scherer M, Stamminger T. 2018. SUMOylation of IE2p86 is required for efficient autorepression of the human cytomegalovirus major immediate-early promoter. J Gen Virol 99:369–378. doi: 10.1099/jgv.0.001021. [DOI] [PubMed] [Google Scholar]
- 53.Hofmann H, Floss S, Stamminger T. 2000. Covalent modification of the transactivator protein IE2-p86 of human cytomegalovirus by conjugation to the ubiquitin-homologous proteins SUMO-1 and hSMT3b. J Virol 74:2510–2524. doi: 10.1128/jvi.74.6.2510-2524.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Scrivano L, Sinzger C, Nitschko H, Koszinowski UH, Adler B. 2011. HCMV spread and cell tropism are determined by distinct virus populations. PLoS Pathog 7:e1001256. doi: 10.1371/journal.ppat.1001256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Andreoni M, Faircloth M, Vugler L, Britt WJ. 1989. A rapid microneutralization assay for the measurement of neutralizing antibody reactive with human cytomegalovirus. J Virol Methods 23:157–167. doi: 10.1016/0166-0934(89)90129-8. [DOI] [PubMed] [Google Scholar]
- 56.Wagner B, Kropff B, Kalbacher H, Britt W, Sundqvist VA, Ostberg L, Mach M. 1992. A continuous sequence of more than 70 amino acids is essential for antibody binding to the dominant antigenic site of glycoprotein gp58 of human cytomegalovirus. J Virol 66:5290–5297. doi: 10.1128/JVI.66.9.5290-5297.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Britt WJ, Vugler LG. 1992. Oligomerization of the human cytomegalovirus major envelope glycoprotein complex gB (gp55-116). J Virol 66:6747–6754. doi: 10.1128/JVI.66.11.6747-6754.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schoppel K, Hassfurther E, Britt W, Ohlin M, Borrebaeck CA, Mach M. 1996. Antibodies specific for the antigenic domain 1 of glycoprotein B (gpUL55) of human cytomegalovirus bind to different substructures. Virology 216:133–145. doi: 10.1006/viro.1996.0040. [DOI] [PubMed] [Google Scholar]
- 59.Meyer H, Masuho Y, Mach M. 1990. The gp116 of the gp58/116 complex of human cytomegalovirus represents the amino-terminal part of the precursor molecule and contains a neutralizing epitope. J Gen Virol 71:2443–2450. doi: 10.1099/0022-1317-71-10-2443. [DOI] [PubMed] [Google Scholar]
- 60.Urban M, Britt W, Mach M. 1992. The dominant linear neutralizing antibody-binding site of glycoprotein gp86 of human cytomegalovirus is strain specific. J Virol 66:1303–1311. doi: 10.1128/JVI.66.3.1303-1311.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Simpson JA, Chow JC, Baker J, Avdalovic N, Yuan S, Au D, Co MS, Vasquez M, Britt WJ, Coelingh KL. 1993. Neutralizing monoclonal antibodies that distinguish three antigenic sites on human cytomegalovirus glycoprotein H have conformationally distinct binding sites. J Virol 67:489–496. doi: 10.1128/JVI.67.1.489-496.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Krawczyk A, Arndt MAE, Grosse-Hovest L, Weichert W, Giebel B, Dittmer U, Hengel H, Jäger D, Schneweis KE, Eis-Hübinger AM, Roggendorf M, Krauss J. 2013. Overcoming drug-resistant herpes simplex virus (HSV) infection by a humanized antibody. Proc Natl Acad Sci U S A 110:6760–6765. doi: 10.1073/pnas.1220019110. [DOI] [PMC free article] [PubMed] [Google Scholar]