

Abstract
Shotgun proteomics using liquid chromatography coupled to mass spectrometry (LC-MS) is commonly used to identify peptides containing post-translational modifications. With the emergence of fast database search tools such as MSFragger, the approach of enlarging precursor mass tolerances during the search (termed ‘open search’) has been increasingly used for comprehensive characterization of post-translational and chemical modifications of protein samples. However, not all mass shifts detected using the open search strategy represent true modifications, as artifacts exist from sources such as unaccounted missed cleavages or peptide co-fragmentation (chimeric MS/MS spectra). Here we present Crystal-C, a computational tool that detects and removes such artifacts from open search results. Our analysis using Crystal-C shows that, in a typical shotgun proteomics dataset, the number of such observations is relatively small. Nevertheless, removing these artifacts helps to simplify the interpretation of the mass shift histograms, which in turn should improve the ability of open search-based tools to detect potentially interesting mass shifts for follow-up investigation.
INTRODUCTION
Protein post-translational modifications (PTMs) are crucial in many biological processes, including cellular regulation, signaling, and recognition1–7.Bottom-up proteomics, in which proteins are enzymatically digested and analyzed by liquid chromatography coupled to mass spectrometry (LC-MS), is widely used to uncover PTMs8–12. To identify PTMs from an LC-MS analysis, MS/MS spectra are searched against theoretical spectra generated from a target protein sequence database, resulting in thousands of peptide-spectrum matches (PSMs)13. In a traditional database search, the precursor mass tolerance is based on the mass accuracy of the analyzer (e.g. 20 ppm)14,15. Recently, an ‘open’ database search strategy has been proposed, where large precursor mass tolerances (e.g. 500 Da) are used to allow identification of peptides with a broad range of modifications16–21. In contrast to traditional searches, open searches use larger precursor mass tolerances to include more potential modified peptide candidates17. Consequently, large mass shifts between experimental and theoretical peptide masses naturally exist in open search results20.
The mass shifts identified using open search can be plotted as a histogram of delta mass values using tools such as DeltaMass22, and observed peaks in the mass shift histogram might correspond to true modifications (e.g., the delta mass value of ~79.96 implies a possible phosphorylation). However, observed mass shifts can also be potential artifacts. For example, if the database search only allowed fully digested peptides or only allowed up to one missed cleavage, an MS/MS spectrum belonging to a peptide with multiple missed cleavages may be assigned to the incomplete peptide sequence, resulting in mass shifts that correspond to the additional peptide sequence. Chimeric MS/MS spectra are also potential artifacts where two or more precursors in the isolation window are co-fragmented. Finally, incorrect determination of the precursor charge state may also introduce spurious mass shifts. While some PTM-centric open search tools have partially addressed these issues19,20, for accurate interpretation of open search results with MSFragger it is desirable to have an automated refinement method. Here we introduce Crystal-C, which detects and removes possible artifacts from open search results to enable focus on potential post-translation modifications.
METHODS
Experimental data.
A human HEK293 cell line data set from the PRIDE database (identifier: PXD001468)17 was used for performance evaluation. The data set was composed of 24 high-pH reversed phase fractions, each analyzed using a three-hour gradient on a Q-Exactive Orbitrap mass spectrometer.
Peptide identification using MS/MS database search.
All raw files were converted to the mzML file format using ProteoWizard23and searched against reviewed human protein sequences downloaded from UniProt (08/27/2019) with MSFragger version 2.118. Reversed protein sequences and contaminant sequences were added to the database by the Philosopher toolkit version 1.5.2 (https://philosopher.nesvilab.org/). For the closed search, lower and upper precursor mass tolerances were −20 and 20 ppm, respectively. For open searches, the lower and upper precursor mass tolerances were −150 and 500 Da, respectively. In both searches, only one missed cleavage was allowed. Cysteine carbamidomethylation was specified as a fixed modification, while methionine oxidation and N-terminal acetylation were specified as variable modifications. In order to have protein accession numbers of all proteins containing the identified peptide reported in pepXML file, MSFragger was run with report_alternative_proteins option set to 1. Default values were used for all remaining parameters.
Peptide validation and FDR filtering.
PSMs from MSFragger were processed using PeptideProphet24,25 via the Philosopher toolkit. In both closed and open searches, expectation values, decoy probabilities, and semi-parametric modelling options were used. Accurate mass model binning and ppm mass error were used for closed searches, while a model mass width of 1000 and conservative level of −2 were used for open search. Both closed and open searches were processed using Philosopher filtering to achieve 1% PSM- and peptide- level FDR. The Philosopher ‘mapmods’ option was used for mapping mass shifts in open search results to UniMod26 annotations.
RESULTS AND DISCUSSION
The overview of the method is shown in Figure 1. Crystal-C performs three steps to sequentially check and remove potential artifacts from open search results: (1) peptides with missed cleavages, (2) semi-enzymatic peptides, and (3) chimeric spectra. Raw spectral files (in mzXML, mzML, or Thermo raw file format), open search results (in pepXML format), and the protein database used in the search (in standard FASTA file format) are taken as input. The delta mass value is defined as the identified peptide mass subtracted from the precursor neutral mass of a PSM.
Figure 1.
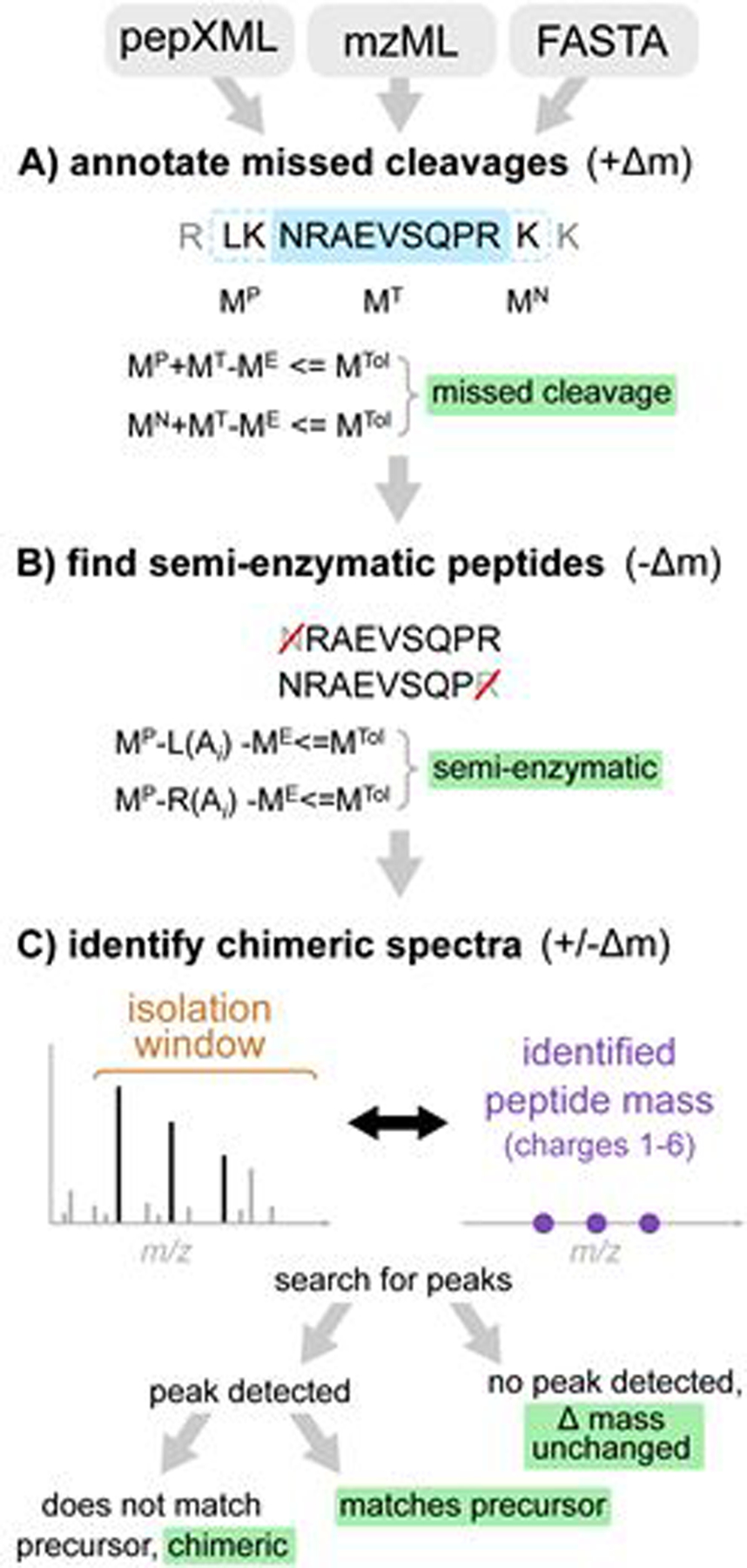
The workflow of Crystal-C as applied to each PSM from open search results. A) Find potential missed cleavage sites by searching the previous and next fully-enzymatic peptides of the identified peptide, where MTol is the mass tolerance (20 ppm by default), ME is the precursor neutral mass, MT is the identified peptide mass, and MP and MN are the previous and next adjacent fully enzymatic peptide masses, respectively. B) Check whether the PSM is semi-enzymatic by deleting one amino acid from the left or right side of the identified peptide sequence at a time and calculating the mass difference between ME and the remaining peptide sequence. If the mass difference is smaller than MTol, the remaining peptide sequence is regarded as semi-enzymatic. C) Find chimeric MS/MS spectra. Crystal-C searches for peaks from the identified peptide within the isolation window by comparing theoretical isotopic clusters (purple)to the MS1 spectrum. If a peak matching one of the theoretical isotope clusters is found in the isolation window and does not belong to the precursor, the PSM is considered chimeric.
Missed cleavage sites.
For each peptide identified with a large positive delta mass, Crystal-C checks whether the precursor neutral mass matches the same peptide but with additional residues due to a missed enzymatic cleavage. The protein sequence associated with the PSM is digested in silico to generate the two possible peptides that correspond to the identified peptide with a missed cleavage on either side. If either of these missed-cleavage peptides matches the precursor neutral mass (within the user-defined tolerance, 20 ppm by default), Crystal-C replaces the original peptide identification with the missed-cleavage peptide and updates the delta mass value in the pepXML file. If both missed cleavage peptides match within the tolerance, then the match with the smallest mass shift is used. When a PSM maps to multiple proteins (including alternative proteins) in the sequence database, all sequences are considered.
Semi-Enzymatic Peptides.
Another possible artifact in open search results is semi-enzymatic peptides, which are peptides that do not conform to the enzymatic cleavage rule (e.g. trypsin cleavage rule trypsin was used) at one of the termini. Such peptides may be observed due to non-tryptic proteolytic cleavage prior to mass spectrometry analysis or cleavage caused by in-source fragmentation. Crystal-C checks whether each PSM with a large negative delta mass is semi-enzymatic by sequentially removing amino acids from one end of the identified peptide to generate subsequences. If the mass of the subsequence matches the precursor neutral mass within the user-defined tolerance (default 20 ppm), the original peptide sequence and delta mass are replaced by the subsequence and the newly calculated delta mass, respectively. This procedure is performed from both left to right and right to left on the peptide, and if two subsequences match the precursor mass, the match with the smaller delta mass is selected.
Chimeric MS/MS spectra.
PSMs with a delta mass outside the narrow mass tolerance range that are not classified as missed cleavage peptides or semi-enzymatic peptides may result from co-fragmentation events. Co-fragmentation occurs when two or more precursor ions are simultaneously isolated for MS/MS analysis, resulting in chimeric MS/MS spectra27. When a co-fragmented peptide (rather than the intended target) is identified as the top scoring peptide for the MS/MS spectrum, the precursor m/z value of the corresponding MS/MS scan would not match the m/z value of the identified co-fragmented peptide ion. Note that the resulting absolute delta mass value in such cases could be small (within several Da, corresponding to the isolation window of the instrument) or very large. This depends on whether the co-fragmented peptide ion has the charge state matching the value reported for the MS/MS scan. To determine whether a PSM resulted from a chimeric MS/MS spectrum, Crystal-C compares the identified peptide to the MS1 spectrum to find whether a possible co-isolated precursor (or simply an incorrect charge state assignment) can explain the observed delta mass. The identified peptide mass and a user-defined charge state range (1–6 by default)are used to calculate three theoretical isotopic m/z values (including the monoisotope, 1st isotope, and 2nd isotope) for each charge, generating 18 total isotopic m/z values (3 isotopes, 6 charge states for each). Next, Crystal-C searches for these 18 isotopic m/z values within the precursor isolation window to determine if there is a matching precursor (within a 20ppm default tolerance).If a match is found, Crystal-C checks whether this peak belongs to the isotopic cluster of the precursor within the tolerance. If the matched peak does not belong to the precursor, the PSM is considered to be a chimeric MS/MS spectrum as a peak from a different precursor was found in the isolation window. Crystal-C then updates the precursor neutral mass and the delta mass value according to the mass of the newly identified co-isolated precursor, as this precursor can completely explain the PSM. In this way, Crystal-C corrects delta masses that can be explained by co-isolation or incorrect charge state assignment.
Evaluation of Crystal-C.
To demonstrate the performance of the tool in detecting and removing artifacts, and to better understand their frequency in a typical dataset, we compared the open search results with and without Crystal-C using a HEK293 cell line data. Detailed data processing steps are described in the Method Section. Table 1 shows the number of PSMs and peptides identified by closed search, open search without Crystal-C, and open search with Crystal-C. Open search (with or without Crystal-C) identified significantly (>30%) more PSMs compared to closed search, in line with previous reports that a large proportion of unassigned spectra can be identified as modified peptides with the open search strategy7,8. Comparing open search results without and with Crystal-C, 566089 PSMs were commonly identified at 1% PSM and 1% peptide FDR. Among them, 74.8% had experimental masses that matched the identified peptide with an absolute delta mass value smaller than 20 ppm (including spectra where Crystal-C was able to detect the monoisotopic peak in the MS1 scans and correct the 12C/13C isotope error). These PSMs represent unmodified peptides, or peptides with a modification specified as a variable modification in the search (i.e. oxidized methionine or N-terminal acetylation) (Table 2, labeled as “no mass shift” category). Overall, the number of PSMs with unexplained mass shifts was greatly decreased after using Crystal-C. Within the commonly identified PSMs that had a mass shift outside of 20ppm, 1.1% and 0.3% were re-annotated by Crystal-C as peptides containing missed cleavages and semi-tryptic peptides, respectively. Only 363 PSMs (0.06%) have their precursor charge states corrected by Crystal-C. For 7.5% of PSMs, Crystal-C detected a peak in the MS1 spectrum that was within the instrument precursor isolation window that matched (within 20 ppm tolerance) the theoretical mass of the identified peptide. These cases can be explained as PSMs identified from chimeric MS/MS spectra, and they fall into two distinct groups (Figure 2): group A with a small delta mass value (mostly contained within the −4 to 4 Da region), and group B with delta mass values above 356 Da. The PSMs in group A are due to co-fragmentation of two precursors of the same charge state, whereas PSMs group B can be explained as identifications with the wrong initial charge state assignment. In addition to identifying and correcting chimeric spectra that manifested as small mass shifts, Crystal-C is able to fix large delta mass values that would otherwise have appeared to be chemical modifications. After the correction by Crystal-C, the delta masses of PSMs in both groups were adjusted to 0. These results demonstrate the ability of Crystal-C to correct artifacts from open search results.
Table 1.
The number of identified PSMs and peptides, filtered by both 1% PSM FDR and 1% peptide FDR levels. The total number of MS/MS scans in the dataset is 1121158.
| Closed | Open Search | |||
|---|---|---|---|---|
| Without Crystal-C | With Crystal-C | |||
| Min. PeptideProphet Probability at 1% FDR | PSM level | 0.3454 | 0.6095 | 0.6827 |
| Peptide level | 0.9376 | 0.9612 | 0.9428 | |
| Number | PSMs | 511225 | 585444 | 617719 |
| Peptides | 118804 | 119229 | 122652 | |
Table 2.
PSMs commonly identified by both closed and open search, as categorized by Crystal-C.
| Category | Number | % |
|---|---|---|
| No mass shift | 423411 | 74.8 |
| Missed cleavage peptides | 6301 | 1.1 |
| Semi-tryptic peptides | 1468 | 0.3 |
| Chimeric spectra | 42370 | 7.5 |
| Remaining with mass shift | 92539 | 16.3 |
Figure 2.
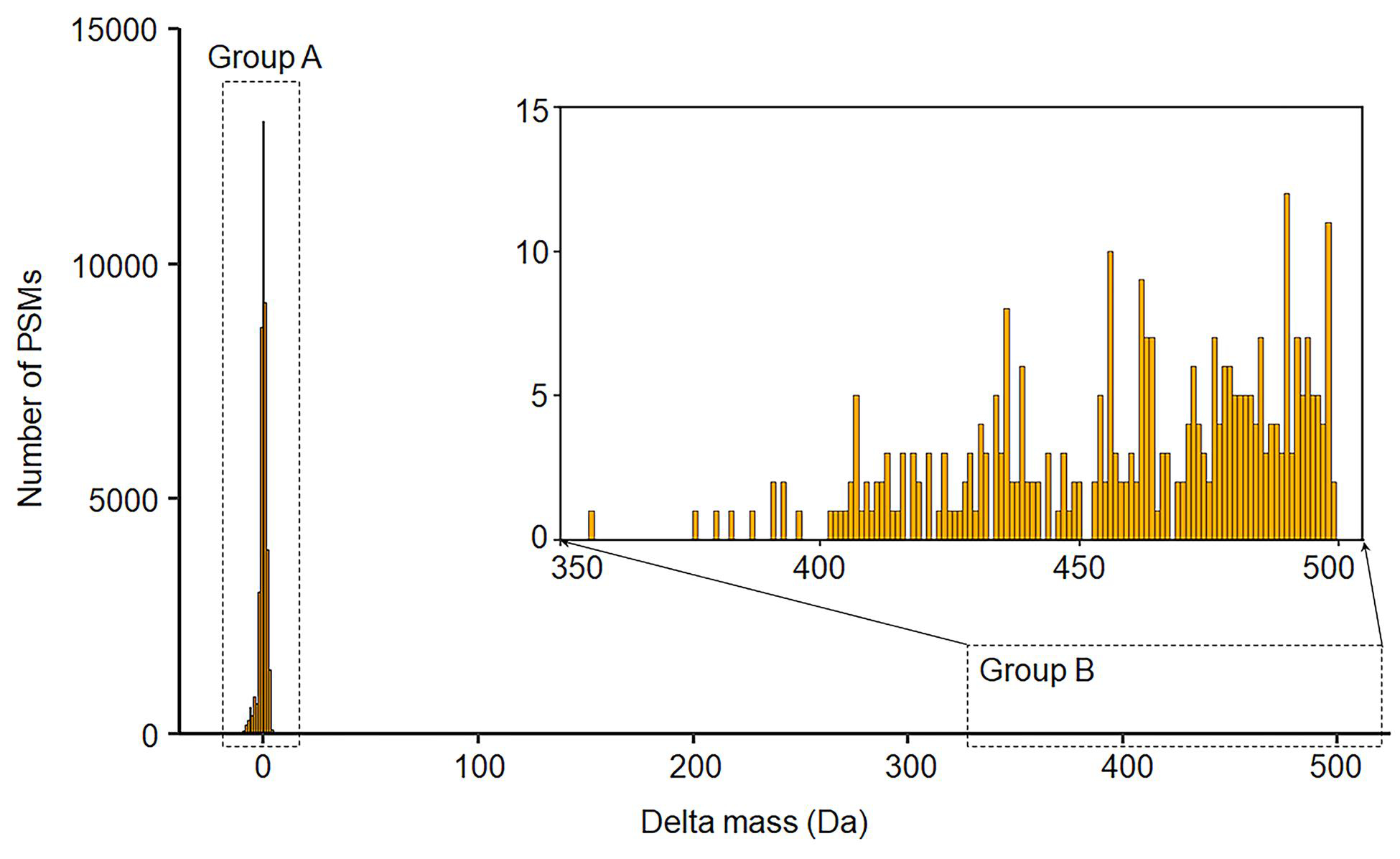
The delta mass shift of the 42370 PSMs annotated as chimeric spectra by Crystal-C. Note that the delta masses of these PSMs are found between-12.84 and 3.99 (group A), and from 356.05 to 499.34 (group B).
CONCLUSION
The identification of PTMs is critical to the understanding of complex cell processes, but presents a challenge to LC-MS proteomics methods28. Open database searching offers a straightforward approach to discover PTMs on proteolytic peptides. However, observed mass shifts can be difficult to interpret, as some may be attributed to sample preparation (missed proteolytic cleavages, non-enzymatic cleavages) or data acquisition and processing (chimeric spectra, incorrect charge state determination) rather than chemical post-translational modification. To improve the ability of open search methods to discover real PTMs, we developed Crystal-C to detect and remove possible artifacts from open search results. While Crystal-C does not attempt to re-search and re-score PSMs following artifact removal, the tool aids discovery of real mass shifts in the data. Re-annotating search results with corrected mass shifts not only helps uncover true modifications, but also allows users to monitor the quality of the samples. For example, one can quickly evaluate digestion efficiency from the number of unexpected missed cleavages and semi-tryptic peptides reported by Crystal-C. Importantly, even after Crystal-C analysis, there remains a significant number of mass-shifted PSMs. Understanding the nature of these modified peptides (biological modifications, sample handling artifacts, amino acid variants, etc.) represents another significant challenge that will be addressed with downstream computational tools currently under development in our lab.
Crystal-C is implemented in Java 8 and can be used in both Windows and Linux operating systems. It is currently compatible with conventional LC-MS/MS data, and can be used with the commonly used proteolytic enzymes (including Trypsin and Lys-C). The software tool is freely available for download (https://github.com/nesvilab/Crystal-C), and is included in the graphical user interface FragPipe (https://github.com/nesvilab/FragPipe).
ACKNOWLEDGMENT
This work was supported in part by NIH Grants R01-GM-094231, U24-CA210967, and U2C-ES030164. We thank all Nesvizhskii lab members for useful discussion, and users of Crystal-C for their valuable feedback.
REFERENCES
- 1.Doerig C, Rayner JC, Scherf A & Tobin AB Post-translational protein modifications in malaria parasites. Nat Rev Microbiol 13, 160–172, doi: 10.1038/nrmicro3402 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Benjdia A, Guillot A, Ruffie P, Leprince J & Berteau O Post-translational modification of ribosomally synthesized peptides by a radical SAM epimerase in Bacillus subtilis. Nat Chem 9, 698–707, doi: 10.1038/nchem.2714 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Riley NM & Coon JJ Phosphoproteomics in the Age of Rapid and Deep Proteome Profiling. Anal Chem 88, 74–94, doi: 10.1021/acs.analchem.5b04123 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pagel O, Loroch S, Sickmann A & Zahedi RP Current strategies and findings in clinically relevant post-translational modification-specific proteomics. Expert Rev Proteomic 12, 235–253, doi: 10.1586/14789450.2015.1042867 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang XY & Qian KV Protein O-GlcNAcylation: emerging mechanisms and functions. Nat Rev Mol Cell Bio 18, 452–465, doi: 10.1038/nrm.2017.22 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang HZ et al. iPTMnet: an integrated resource for protein post-translational modification network discovery. Nucleic Acids Res 46, D542–D550, doi: 10.1093/nar/gkx1104 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu HD et al. PTMD: A Database of Human Disease-associated Post-translational Modifications. Genom Proteom Bioinf 16, 244–251, doi: 10.1016/j.gpb.2018.06.004 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aebersold R & Mann M Mass-spectrometric exploration of proteome structure and function. Nature 537, 347–355, doi: 10.1038/nature19949 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Stepanova S & Kasicka V Recent developments and applications of capillary and microchip electrophoresis in proteomic and peptidomic analyses. J Sep Sci 39, 198–211, doi: 10.1002/jssc.201500973 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Ortea I, O’Connor G & Maquet A Review on proteomics for food authentication. J Proteomics 147, 212–225, doi: 10.1016/j.jprot.2016.06.033 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Misra BB Updates on resources, software tools, and databases for plant proteomics in 2016–2017. Electrophoresis 39, 1543–1557, doi: 10.1002/elps.201700401 (2018). [DOI] [PubMed] [Google Scholar]
- 12.Carrera M, Canas B & Gallardo JM Advanced proteomics and systems biology applied to study food allergy. Curr Opin Food Sci 22, 9–16, doi: 10.1016/j.cofs.2017.12.001 (2018). [DOI] [Google Scholar]
- 13.Na S & Paek E Software eyes for protein post-translational modifications. Mass Spectrom Rev 34, 133–147, doi: 10.1002/mas.21425 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Eng JK, Searle BC, Clauser KR & Tabb DL A face in the crowd: recognizing peptides through database search. Mol Cell Proteomics 10, R111 009522, doi: 10.1074/mcp.R111.009522 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sinitcyn P, Rudolph JD & Cox J Computational Methods for Understanding Mass Spectrometry-Based Shotgun Proteomics Data. Annu Rev Biomed Da S 1, 207–234, doi: 10.1146/annurev-biodatasci-080917-013516 (2018). [DOI] [Google Scholar]
- 16.Chi H et al. Comprehensive identification of peptides in tandem mass spectra using an efficient open search engine. Nat Biotechnol, doi: 10.1038/nbt.4236 (2018). [DOI] [PubMed] [Google Scholar]
- 17.Chick JM et al. A mass-tolerant database search identifies a large proportion of unassigned spectra in shotgun proteomics as modified peptides. Nat Biotechnol 33, 743–749, doi: 10.1038/nbt.3267 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kong AT, Leprevost FV, Avtonomov DM, Mellacheruvu D & Nesvizhskii AI MSFragger: ultrafast and comprehensive peptide identification in mass spectrometry-based proteomics. Nat Methods 14, 513–520, doi: 10.1038/nmeth.4256 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Na S, Kim J & Paek E MODplus: Robust and Unrestrictive Identification of Post-Translational Modifications Using Mass Spectrometry. Anal Chem 91, 11324–11333, doi: 10.1021/acs.analchem.9b02445 (2019). [DOI] [PubMed] [Google Scholar]
- 20.Solntsev SK, Shortreed MR, Frey BL & Smith LM Enhanced Global Post-translational Modification Discovery with MetaMorpheus. Journal of proteome research 17, 1844–1851, doi: 10.1021/acs.jproteome.7b00873 (2018). [DOI] [PubMed] [Google Scholar]
- 21.Tsur D, Tanner S, Zandi E, Bafna V & Pevzner PA Identification of post-translational modifications via blind search of mass-spectra. Proc IEEE Comput Syst Bioinform Conf, 157–166 (2005). [DOI] [PubMed] [Google Scholar]
- 22.Avtonomov DM, Kong A & Nesvizhskii AI DeltaMass: Automated Detection and Visualization of Mass Shifts in Proteomic Open-Search Results. Journal of proteome research 18, 715–720, doi: 10.1021/acs.jproteome.8b00728 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kessner D, Chambers M, Burke R, Agus D & Mallick P ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics 24, 2534–2536, doi: 10.1093/bioinformatics/btn323 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Keller A, Nesvizhskii AI, Kolker E & Aebersold R Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem 74, 5383–5392 (2002). [DOI] [PubMed] [Google Scholar]
- 25.Ma K, Vitek O & Nesvizhskii AI A statistical model-building perspective to identification of MS/MS spectra with PeptideProphet. BMC Bioinformatics 13 Suppl 16, S1, doi: 10.1186/1471-2105-13-S16-S1 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Creasy DM & Cottrell JS Unimod: Protein modifications for mass spectrometry. Proteomics 4, 1534–1536, doi: 10.1002/pmic.200300744 (2004). [DOI] [PubMed] [Google Scholar]
- 27.Houel S et al. Quantifying the impact of chimera MS/MS spectra on peptide identification in large-scale proteomics studies. Journal of proteome research 9, 4152–4160, doi: 10.1021/pr1003856 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yakubu RR, Weiss LM & Silmon de Monerri NC Post-translational modifications as key regulators of apicomplexan biology: insights from proteome-wide studies. Mol Microbiol 107, 1–23, doi: 10.1111/mmi.13867 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]