

Abstract
Mutations in LAMA2 gene, encoding merosin, are generally responsible of a severe congenital-onset muscular dystrophy (CMD type 1A) characterized by severe weakness, merosin absence at muscle analysis and white matter alterations at brain Magnetic Resonance Imaging (MRI). Recently, LAMA2 mutations have been acknowledged as responsible of LGMD R23, despite only few cases with slowly progressive adult-onset and partial merosin deficiency have been reported. We describe 5 independent Italian subjects presenting with progressive limb girdle muscular weakness, brain white matter abnormalities, merosin deficiency and LAMA2 gene mutations. We detected 7 different mutations, 6 of which are new. All patients showed normal psicomotor development and slowly progressive weakness with onset spanning from childhood to forties. Creatin-kinase levels were moderately elevated. One patient showed dilated cardiomyopathy. Muscle MRI allowed to evaluate the degree and pattern of muscular involvement in all patients. Brain MRI was fundamental in order to address and/or support the molecular diagnosis, showing typical widespread white matter hyperintensity in T2-weighted sequences. Interestingly these alterations were associated with central nervous system involvement in 3 patients who presented epilepsy and migraine. Muscle biopsy commonly but not necessarily revealed dystrophic features. Western-blot was usually more accurate than immunohystochemical analysis in detecting merosin deficiency. The description of these cases further enlarges the clinical spectrum of LAMA2-related disorders. Moreover, it supports the inclusion of LGMD R23 in the new classification of LGMD. The central nervous system involvement was fundamental to address the diagnosis and should be always included in the diagnostic work-up of undiagnosed LGMD.
Key words: limb girdle muscular dystrophy, merosin, LAMA2 gene, brain MRI, muscle MRI, leukoencephalopathy
Introduction
Laminin-2 (also called merosin), a heterotrimeric protein composed of three different subunits (α2β1γ1), is a major component of the basement membrane of skeletal muscle fibres, Schwann cells in peripheral nerves, brain capillaries and submandibular glands 1. It belongs to a large family of glycoproteins involved in extracellular matrix architecture, cell adhesion and differentiation and mediates the attachment, migration, and organization of cells into tissues during embryonic development by interacting with other extracellular matrix components 1,2. Mutations in LAMA2 gene (chromosome 6q22.33), which encodes alpha 2 chain, have been so far identified as the cause of two different clinical phenotypes: an early-onset congenital muscular dystrophy (CMD), also known as CMD1A (congenital muscular dystrophy 1A), and a milder late-onset limb-girdle type muscular dystrophy (LGMD) phenotype.
CMD1A is one of the most frequent forms of classic CMD in Western countries. This form, commonly associated with complete absence of laminin-α2, is characterized by severe muscle weakness and hypotonia at birth or in the first six months of life, delayed motor development, inability to walk unsupported, joint contractures, high creatine-kinase (CK) levels and widespread white matter abnormalities on brain magnetic resonance imaging (MRI) 2-4.
In 1998 Bushby et al. described for the first time a case with adulthood onset and mutations in LAMA2 gene 5. These forms are generally characterized by slowly progressive proximal muscular weakness, white matter lesions on MRI and partial protein loss al muscle analysis 6-8.
Recently the 229th ENMC International Workshop revised LGMD nomenclature and classification, providing a precise definition of LGMD and including LAMA2 gene as causative of LGMD R23 9.
A potential correlation between the degree of merosin expression, the genotype and the clinical features has been described 10. In particular, mutations that lead to partial laminin-α2 deficiency are generally associated to milder phenotypes, which range from mild forms of CMD to adult-onset presentations. Mild CMD forms present a benign evolution and slow course, but are always characterized by delay in motor milestones acquisition 11-14, while the adult-onset LGMD forms generally do not show symptoms at birth. The adult-onset cases are generally due to missense or in-frame splice-site mutations while CMD cases are more frequently associated to nonsense or out-of-frame mutations.
While the prevalence of MDC1A is estimated to be 0.6/100,000 in Northern England (around 10-30% of all the MDC) 14-15, the prevalence of LGMD due to LAMA2 mutations is unknown. However it is believed to be very rare and since now only few cases with adult LGMD presentation have been reported 6-8,17-22.
Here we describe a small group of Italian subjects carrying homozygous or compound heterozygous mutations in LAMA2 gene, who developed mild and slowly progressive muscular weakness with limb girdle muscular dystrophy phenotype.
The description of these cases, which include patients with late onset seizures, very late disease onset as far as marginal muscle involvement, contributes to enlarge the spectrum of LAMA2 gene disorders. In many of these cases the incidental finding of brain white matter abnormalities was fundamental in order to direct the diagnosis.
Materials and methods
Clinical evaluation
Patients were selected from a cohort of adult LGMD patients evaluated at IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico of Milan and at Department of Neurosciences of Padua. All patients underwent a systematic clinical characterization, including comprehensive neurological, cardiac (electrocardiogram and echocardiogram) and respiratory (spirometry) assessments. Data about familiar and personal history, in particular about age of onset and progression of symptoms, were collected. Muscle strength was evaluated using the Medical Research Council (MRC) Scale. Electromyography (EMG) was performed at the beginning of the disease. The patients were also studied with both Brain and Muscle Magnetic Resonance Imaging (MRI). In selected cases further studies such as electroencephalography were performed.
Written informed consent was obtained (and preserved in the original form) from all subjects or their caregivers when primary diagnostic procedures were performed, with explicit consent to future uses for research purpose, according to the Declaration of Helsinki.
Brain and muscle imaging
Brain imaging data were obtained in all patients by nuclear Magnetic Resonance Imaging (MRI), A 1.5T scanner was used and spin echo (SE), turbo spin echo (TSE) and fluid attenuated inversion recovery (FLAIR) sequences with T1 and T2 weighted images in the three orthogonal planes were acquired.
Muscle MRI was performed using a 1.5T scanner, axial TSE T1 weighted- images and axial short tau inversion (STIR) T2-weighted images were acquired. Both proximal upper and lower limbs’ muscles were studied.
Muscle tissue analysis
All probands underwent muscle biopsy after giving written informed consent, according to Institutional guidelines. Morphological examination was performed according to standard procedures 23.
The proteins involved in LGMDs were studied using immunohistochemical (IHC) analyses with monoclonal antibodies directed against dystrophin (Novocastra, Newcastle upon Tyne, UK), sarcoglycans (Novocastra, Newcastle upon Tyne, UK), alpha-dystroglycan (a-DG) (Upstate Biotechnology, Lake Placid, NY), and caveolin-3 (Transduction Laboratories, Lexington, KY) as previously described. Calpain-3 and dysferlin expression were analyzed through Western blot (WB) analysis.
Immunostaining of laminin α2 was performed using two commercially available antibodies directed against the 80-kDa carboxyl-terminus (MAb1922; Chemicon, Temecula, California) and against the amino-terminus (NCL-MER3, Novocastra, Newcastle Upon Tyne, UK). Laminin-α2 immunoblot was performed using the MAb1922 antibody (Chemicon) 24.
Molecular analysis
Genomic DNA was extracted from peripheral blood samples according to standard procedures (Flexi Gene DNA Handbook, Qiagen). Molecular analysis of LAMA2 gene was performed through the amplification by Polimerase Chain reaction (PCR) and direct sequencing (ABI Prism 3100 Genetic Analyzer, Applied Biosystems, Foster City, CA) of all the exons and the adjacent intron regions. Primer sequences and PCR conditions are available upon request. Mutations were named according to the Leiden Muscular Dystrophy database (www.dmd.nl). For cDNA numbering, +1 corresponds to the A of the ATG translation initiation codon in the reference sequence.
The pathogenic nature of the mutations was confirmed by screening of 160 control healthy subjects and the parental origin of each mutation was assessed trough analysis of parental genomic DNA when available. Furthermore, the amino acid conservation was confirmed by comparison with the sequence in different species. In few patients, mRNA was isolated from muscle tissue with Eurozol. Then cDNA was produced through reverse transcription polymerase chain reaction (RT-PCR) using the Ready-To-Go RT-PCR kit (Amersham Pharmacia) and analysed by mRNA amplification and sequencing.
In one patient the diagnosis was achieved with Next Generation Sequencing (NGS) techniques and confirmed by Sanger sequencing. DNA samples underwent Motor HaloPlex Target Enrichment protocol, analyzing the coding regions of 89 genes; libraries were run on a HiSeq instrument and analyzed through a custom pipeline of bioinformatics analyses 25.
Results
We selected a cohort of 5 patients clinically diagnosed as LGMD and carrying mutations in LAMA2 gene. We identified 7 different mutation, 6 of which are novel. The molecular, clinical and bioptical features of the patients are summarised in Table I, Table II and Table III.
Table I.
Molecular data showing the two pathogenic mutations and their proteic effect for each patient. Most of the mutations were not previously described in literature.
| Pt | Nucleotide 1 | Protein 1 | Exon 1 | Ref. | Nucleotide 2 | Protein 2 | Exon 2 | Ref. |
|---|---|---|---|---|---|---|---|---|
| I | c.6742delC | p.Leu2248TrpfsX23 | 48 | New | c.8544C > G | p.His2848Gln | 60 | New |
| II.1 | c.4005T > C | p.Lys1469Arg | 30 | New | c.4005T > C | p.Lys1469Arg | 30 | New |
| III | c.2750 + 2 insT | p.Glu892Ala del 893_917 | IVS19 | New | c.2750 + 2 insT | p.Glu892Ala del 893_917 | IVS19 | New |
| IV | c.752T > C | p.Leu25Pro | 5 | New | c.7586_7589dup | p.Phe2531X | 55 | New |
| V | c.752T > C | p.Leu25Pro | 5 | New | c.7147C > T | p.Arg2383X | 50 | [35] |
Table II.
Clinical data about the patients described.
| Pt | I | II.1 | III | IV | V |
|---|---|---|---|---|---|
| Sex | M | F | M | M | F |
| Age (yrs) | 62 | 51 | 75 | 41 | 52 |
| Onset (age) | 28 y | Childhood | 40s | Childhood | Childhood |
| Symptoms at onset | Proximal weakness | Proximal weakness | Fatigability and distal weakness | Proximal weakness | Proximal weakness |
| Phenotype | LGMD | LGMD | LGMD | LGMD | LGMD |
| CK (UI/L) | 454-1000 | 300-400 | 500-800 | 677-1640 | 180-680 |
| Loss of ambulation | No (59 y) | No (51 y) | No (75y) | No (41y) | no (52y) |
| Joint retractions | No | Mild TT | No | TT and elbows | TT |
| Sural hypertrophy | Yes | Yes | Yes | No | No |
| Cardio-pulmonary involvement | BBD | PSVT | Dilated cardiomyopathy, atrial flutter | None | Mild MIP and MEP reduction |
| Muscle MRI | Severe asymmetric fibro-fatty replacement biceps brachii, triceps brachii, thigh. Selectively sparing of deltoid,sartorius, gracilis, rectus femoris and short head of the biceps femoris | Fatty substitution in trapezius, supraspinatus, subscapularis, infraspinatus and pectoral thigh and leg muscles with relative sparing of the sartorius, gracilis, short head of the biceps femoral and tibialis posterior muscles | Diffuse fibro-fatty substitution of the gluteal muscles, of the posterior thigh, adductor magnus and of the quadriceps, sparing only the rectus femoris. The shoulder muscles were mild affected | NA | Moderate fatty substitution of scapular girdle muscles, severe substitution of pelvic girdle and thigh muscles, with sparing of sartorius, tensor fasciae latae, obturator and iliopsoas, gracilis, biceps femoris and adductor longus on the right side (52 years) |
| Brain MRI | WMA (U fibres sparing) | WMA | WMA | WMA | WMA |
| CNS | None | None | Migraine, MCI, polyneuropathy | Epilepsy | Epilepsy |
| EMG | Neurogenic | NA | Mixed signs | Myogenic | Myogenic |
| Other features | Asymmetric weakness | Myalgia | Distal legs involvement | Generalized muscle atrophy | Hyperlordosis, tiptoe gait |
WMA: white matter abnormalities; MCI: mild cognitive impairment
Table III.
Bioptical data of the patient described.
| Patient | I | II.1 | III | IV | V | |||
|---|---|---|---|---|---|---|---|---|
| Age at muscle biopsy (yrs) | 40 | 45 | ND | 67 | 75 | ND | 11 | 30 |
| Biopsy pattern | Dystrophic | Myopathic | Dystrophic | Dystrophic | Dystrophic | ND | Myopathic | ND |
| Merosin WB (residual %) | ND | ND | 30% | 60% | 60.00% | ND | ND | 20% |
| Merosin IHC 300 kDa | Partial reduction | Partial reduction | ND | Normal/mild reduction | ND | ND | ND | Mild reduction |
| Merosin IHC 80 kDa | Partial reduction | Partial reduction | ND | Normal/mild reduction | Partial reduction | ND | ND | Mild reduction |
Patient I
Patient I was an Italian 30-year-old man without family history for neuromuscular disorders. He presented to medical attention with a LGMD phenotype characterized by progressive limb girdle weakness, predominantly involving lower limbs, and moderate CK levels increase (2-5x; range 454-1000 UI/L). He was the third son of non-consanguineous parents; his parents and his daughter did not show any muscular involvement and had normal CK levels. The patient was born after a normal pregnancy and had a normal psycho-motor development. He presented the first symptoms at 28 years of age when he started to complain difficulty in climbing stairs. The disease had a slowly progressive course with worsening of lower limbs weakness and asymmetrical involvement of proximal upper limb muscles starting from the age of 31 years. Electromyography showed neurogenic alterations without myopathic signs. Clinical evaluation at 59 years of age showed severe proximal atrophy (more pronounced at thigh and brachial biceps) and proximal lower limb weakness associated with bilateral calf hypertrophy. Weakness was asymmetric and involved predominantly the quadriceps (MRC 4+/5) and ileopsoas (MRC 4/5). At upper limb, brachial biceps was affected as well (MRC 3.5/5). Tendon reflexes were reduced at lower limbs. There was no distal, facial and bulbar involvement. Scoliosis, tendon retractions, cramps and myalgia were absent. The patient had a waddling hyperlordotic gate, possible on tiptoes but not on heels, and was able to walk for 400 meters at 6 minute-walking test. He raised from a chair pushing himself with upper limbs and was able to climb stairs with bilateral support. The patient lost the ability to raise from floor without external support at 55 years of age. Electrocardiogram and echocardiogram showed a normal cardiac function (ejection fraction 55%) and a right bundle branch block. Respiratory evaluation (spirometry and nocturnal saturation) did not reveal pulmonary involvement. The patient did not show cognitive impairment.
The patient underwent two muscle biopsies respectively at the age of 40 and 45 years. The first biopsy, performed on quadriceps muscle, showed a dystrophic pattern with connective tissue substitution. The second biopsy, obtained from deltoid muscle, showed mild fiber size variability, nuclear centralization and fiber splittings, without necrosis or inflammation. Furthermore the connective tissue was normally represented (Fig. 1). At IHC analysis, dystrophin, sarcoglycans, alpha-dystroglycan, caveolin-3 and emerin showed a normal signal. WB study revealed a normal signal of dysferlin and alpha-dystroglycan and a mild reduction of calpain-3 without mutations in the corresponding gene.
Figure 1.
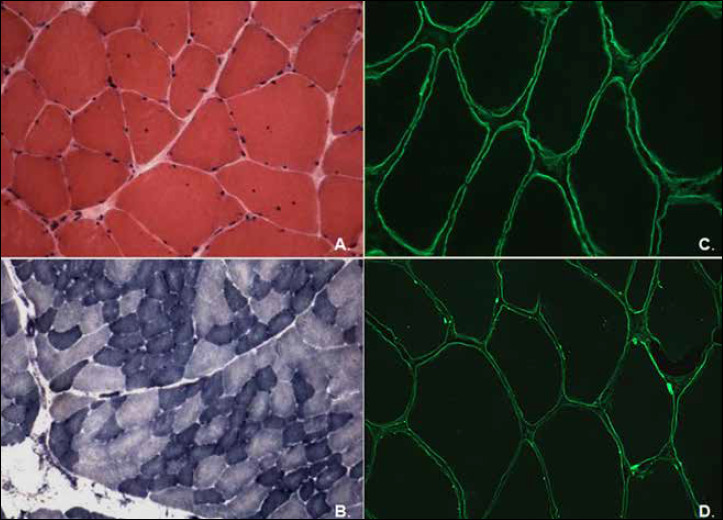
Muscle biopsy Patient I. (A) Haematoxylin and eosin (H&E), 40x: few fibres with internal nuclei and fibre splittings; (B) Reduced nicotinamide adenine dinucleotide dehydrogenase- tetrazolium reductase (NADH-TR), 25x, showing mild variation in fibre size. Immunolabelling of laminin alfa2 to 80 kDa fragment in a control muscle (C) and in our case (D), showing a partial protein expression in some areas of the sarcolemma (arrows). (Immunolabelling with amino-terminus antibody is not shown).
At the age of 57 years, after two transient episodes of tinnitus and blurred vision, the patient underwent brain MRI which showed radiological signs of leukoencephalopathy with bilateral, symmetric and diffuse T2 hyperintensity of peri- and sovra-ventricular white matter, extending to the subcortical regions and sparing U fibres. Posterior limb of internal capsule was only mildly involved. No signs of atrophy was found and the corpus callosum and posterior fossa structures were regular (Fig. 2). Eye examination was also normal.
Figure 2.
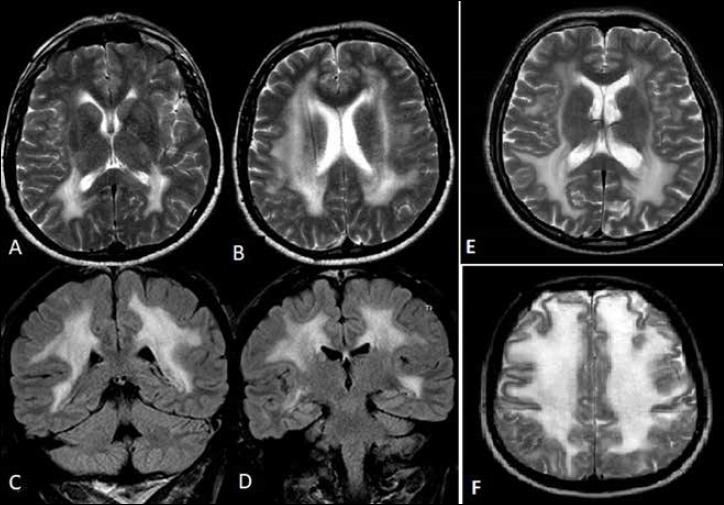
Brain MRI showing diffuse abnormal high signal intensity involving peri and sovra-ventricular white matter in axial T2 images (patient I: A-B; patient III: E; patient II: F) and coronal T2 FLAIR images (patient I: C-D).
According to the brain MRI finding, IHC and WB of merosin were performed. At IHC merosin labelling showed a partial reduction with both antibodies in few muscle fibers (Fig. 1), while the WB analysis showed a severe reduction of the protein expression (Fig. 3).
Figure 3.
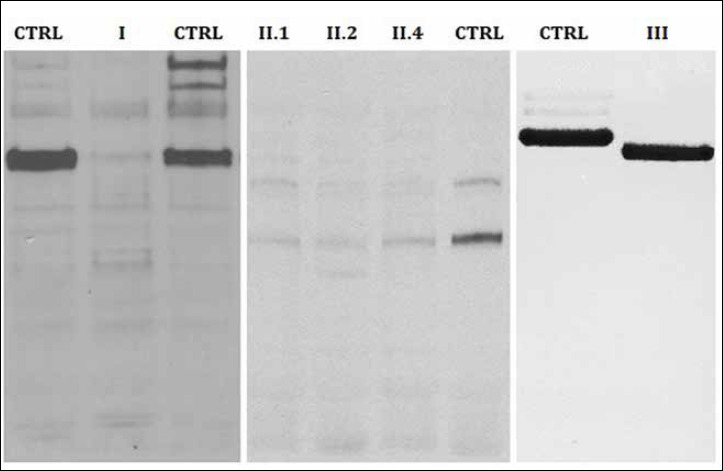
Western-blot analysis. Merosin Western-blot analysis showing a severe deficiency with monoclonal MAb1922 antibody.
Muscle MRI, performed at 59 years of age, revealed a significant muscular impairment at both arms and thighs. In particular the most involved muscle of the upper limbs was the biceps brachii, followed by the triceps brachii, while deltoid was selectively spared. The muscles of the thigh showed atrophy and an high signal in T1-weighted images, which suggested fibro-fatty replacement. Some muscles were selectively spared, in particular the sartorius, the rectus femoris and the short head of the biceps femoris. The right thigh was more impaired than the controlateral one, where also the semitendinosus muscle was partially spared. Atrophy and substitution were detected also in the medial portion of gastrocnemius. No inflammatory signs were found on STIR T2 images (Fig. 4).
Figure 4.
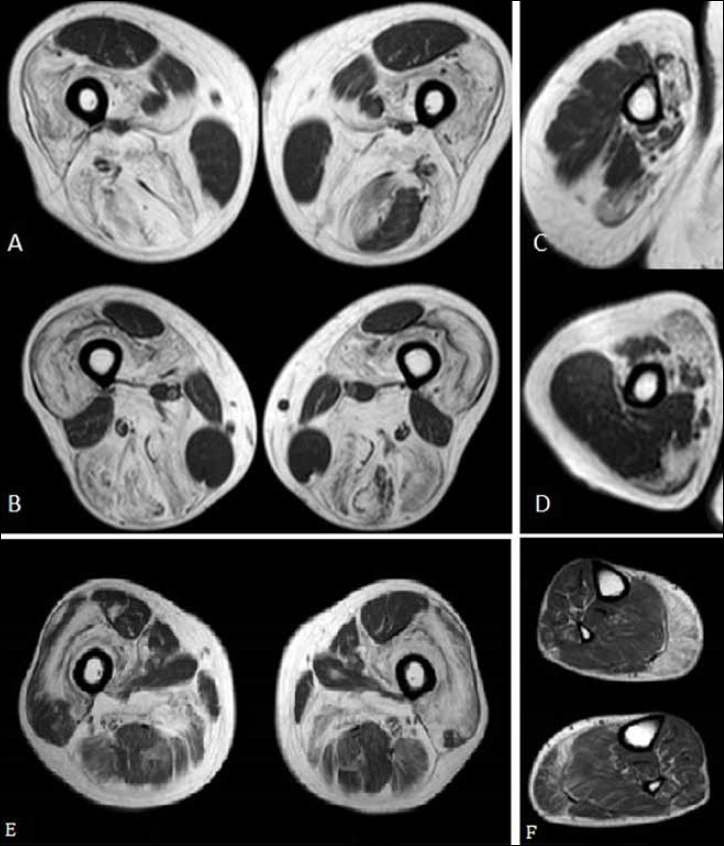
Muscle MRI. Axial TSE T1 images of the thighs (A-B) and of the right arm (C-D) of patient I, showing a diffuse atrophy and hyperintensity signal, as for fatty degeneration; some muscles were selectively spared, in particular the deltoid, the sartorius, the gracilis, the rectus femoris and the short head of the biceps femoris; the left thigh semitendinosus muscle was partially spared too. Axial T1 images of the thighs (E) and legs (F) of patient III, showing diffuse fibro-fatty substitution of the gluteal muscles, of the posterior thigh, adductor magnus and of the quadriceps; milder connective substitution in the medial gastrocnemious bilaterally.
Molecular analysis showed the presence of two compound heterozygous mutation in LAMA2 gene: the frame-shift c.6742delC (p.Leu2248TrpfsX23) in exon 48 and the missense mutation c.8544C > G (p.His2848Gln) in exon 60. Both mutations are novel. Both parents as far as the daughter of the patient carried the mutation in a heterozygous state.
Patient II
Subject II.1 is a 51-years-old woman presenting for progressive muscular weakness involving predominantly limb girdle muscles. She started to complain muscular weakness during childhood with difficulties in running. The disease showed a progressive course during the following years. The patient lost the ability of rising from supine position at 10 years of age and of climbing stairs at 30 years of age. Myalgias were reported from adolescence. The neurological examination at 50 years of age showed symmetrical limb girdle weakness, sural pseudo-hypertrophy, bilateral mild Achilles tendon thightness, lumbar hyperlordosis and waddling gait. Weakness mainly involved lower limbs muscles, in particular gluteus (MRC 2/5), ileopsoas (MRC 4/5), quadriceps (MRC 4/5) and tibialis anterior (MRC 4/5). Deltoid (MRC 4/5) was the only muscle involved at upper limbs. The patient was able to raise from a chair only with external support. Cranial nerves and cognitive functions were normal. No episodes of loss of consciousness were reported. CK levels were mildly elevated (300-400 U/l). Pulmonary function was normal. The patient was affected by paroxysmal supraventricular tachycardia since the age of 45 years old and was treated with beta-blocker therapy.
Her parents were not apparently consanguineous but carried the same last name and were born in the same town. The patient had two unaffected children, but five relatives on her mother’s side presented muscular involvement (Suppl. Fig. 1), suggesting a pseudo-dominant pattern of inheritance. The patient’s mother (Subject II.2) showed proximal muscular weakness and lost independent ambulation at 55 years of age. Three of her brothers and one sisters also showed neurmuscolar involvement. In particular, one aunt (Subject II.5) lost ambulation at 60 years of age. Another uncle (Subject II.3) showed sural pseudohypertrophy since adolescence and lower limbs weakness since adulthood. In the following years he developed atrophy in gluteus and thigh muscles, waddling hyperlordotic gait and upper limb involvement with deltoid pseudo-hypertrophy and right fingers common extensor atrophy. His electromyography displayed myopathic signs. He died at 47 years of age in a road accident. Another aunt (Subject II.4) presented slowly progressive weakness in pelvic girdle muscles since childhood, associated with axial muscle involvement, sural pseudo-hypertrophy, lower limb areflexia, myopathic signs at EMG and CK increase (400 UI/L). At 71 years of age she required support for walking. Moreover, the maternal grandfather (Subject II.6) was also affected by an unspecified neuromuscular disease.
Suppl. Figure 1.
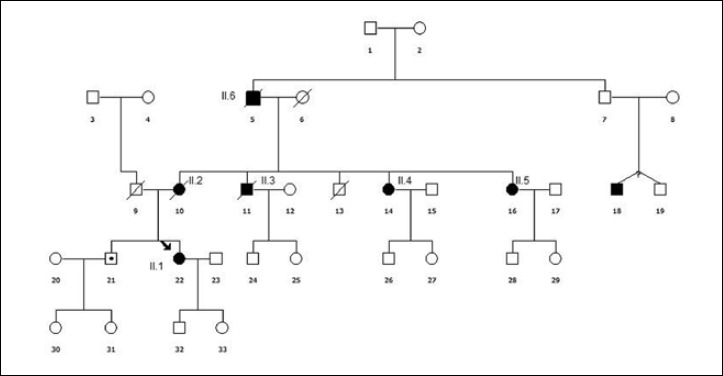
Pedigree of patient II suggesting a pseudo-dominant inheritance.
Muscle biopsy was performed in our proband and in subjects II.2 and II.4. The proband showed a dystrophic pattern, subject II.2 neurogenic alterations and subject II.4 a myopathic pattern. In the proband immunohystochemical analysis for caveolin-3, alpha-sarcoglycan, gamma-sarcoglycan, alpha-dystroglycan, dysferlin and dystrophin were normal, while Calpain-3 (94 kD) was mildly reduced.
NGS analysis revealed an homozygous missense mutation (c.4405T > C; p.Lys1469Arg) in exon 30 of LAMA2 gene. This mutation was previously described in compound heterozygosis in a patient with adulthood onset of mild proximal myopathy, polyneuropathy, dilated cardiomyopathy with conduction defects, histological features similar to Inclusion Body Myositis and partial laminin alpha-2 reduction 17. This homozygous mutation was confirmed by Sanger sequencing both in the patient and in subject II.4. Unfortunately it was not possible to confirm the segregation in the other members of the family; however Western blot analysis was performed in subject II.1 II.2 and II.4 demonstrating severe merosin deficiency (30% of residual protein) in all of them (Fig. 3).
Brain MRI was performed in subject II.1 and revealed widespread periventricular and subcortical white matter hyperintensity in T2-weighted sequences and a reduction of ventricular system dimensions and cortical sulci, suggestive for white matter edema (Fig. 2). Signal abnormalities were found also in the corticospinal tract and to a lesser extent in the splenium of corpus callosum, posterior portion of the thalamus and dentate nucleus (basal ganglia were spared) 25-28.
Muscle MRI, performed at 50 years of age, detected almost complete fatty substitution in thigh and leg muscles with relative sparing of the sartorius, gracilis, short head of the biceps femoral and tibialis posterior muscles. In the upper limbs the trapezius, supraspinatus, subscapularis, infraspinatus and pectoral muscles were mainly involved; milder signs of fibro-fatty infiltration were present also in deltoid muscles.
Patient III
Patient III is a 75-year-old male, born from consanguineous parents (first cousins). He suffered from migraine since adolescence, almost unresponsive to common headache drugs. Family history was positive for dementia (the mother and two maternal aunts affected by). None of the other family members had neuromuscular disease history. The patient started complaining of mild exercise intolerance in his fourties. Although he referred a worsening in fatigability, the muscular involvement remained almost stable over the years. At last evaluation he presents just a mild muscular weakness in distal legs associated with a mild calf hypertrophy. He always had high serum CK levels (500-800 U/l). The EMG showed a mixed neurogenic and mild myogenic pattern. EMG also documented a chronic polyneuropathy even if the patient has never complained neuropathic symptoms. Over the years the patient developed a dilated cardiomyopathy, recently worsened by atrial flutter episodes due to an abnormal ventricular pre-excitation, treated with catheter ablation when he was 73.
In his sixties a brain MRI, performed during the migraine follow-up, showed diffuse white matter lesions, spreading over the years to almost the whole brain white matter (Fig. 2), without clinical significant correlation. MMSE was of 26/30 and psychometric tests were globally normal. Because of headache, family history of dementia and MRI findings, a gene analysis for CADASIL was performed but turned out normal.
The patient underwent to muscle biopsies respectively at the age of 65 and 73 years (Fig. 5). The first biopsy showed some scattered COX negative fibres leading to a mitochondrial disease suspect. The immunofluorescence study for dystrophin, caveolin-3 resulted normal. Based on the suspicion of mitochondrial pathology, mtDNA long time PCR on muscle tissue was performed showing multiple deletions. Several nuclear genes involved in multiple deletions were screened (POLG1, ANT1, POLG2, TYMP and PEO1) but turned out negative.
Figure 5.
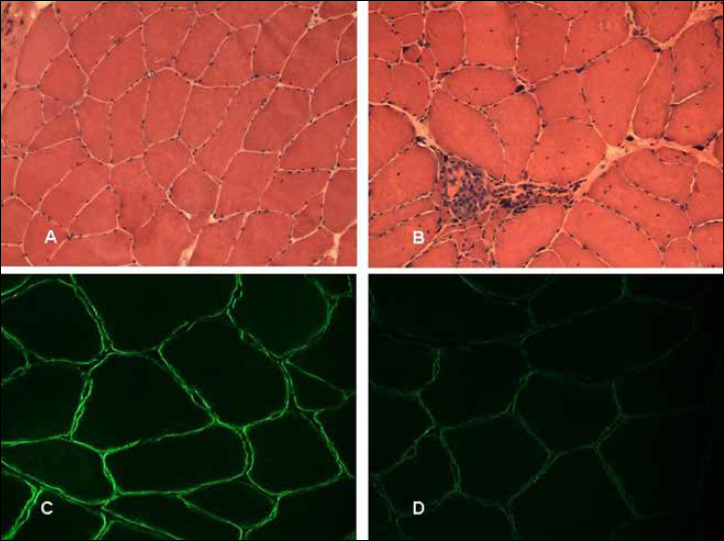
Muscle biopsy Patient III. Hematoxilin and Eosin stain on the the first muscle biopsy (A) and on the second (B). Scattered hypotrophic fibers in (A). Dystrophic pattern showing necrosis, nuclei centralizations and increase in connective tissue (B). Immunofluorescence staining (Merosin antibody) on the first muscle biopsy (C) and on the second (D). Original Magnification 40X (Merosin Laminin Alpha2). Normal Merosin binding (C) and scattered mild Merosin reduction of binding (D).
Eight years later, a second muscle biopsy and a muscle MRI were performed. The MRI showed a diffuse fibro-fatty substitution of the glutei, the posterior thigh, adductor magnus and quadriceps, sparing only the rectus femoris. A diffuse but milder connective substitution was also present in lower legs muscles, particularly in the medial gastrocnemious twins. The shoulder muscles were mild affected. No inflammation was detected (Fig. 4). The biopsy showed the presence of diffuse fibre atrophy and several necrotic fibres with a mild increase in muscular connective tissue. A few COX negative fibres were still present. At immunofluorescence, a mild, diffuse reduction of merosin alpha 2 came out (Fig. 5). Western Blot analysis of merosin on muscles shown a reduction of 40% in both the muscle biopsies (Fig. 3).
According to the MRI and bioptical results LAMA2 gene was analyzed showing the presence of the following homozygous new mutation: c.2750+2 insT (IVS19), bringing to an in frame deletion of 75 nucleotides (c.2675-2749 del 75nt) as demonstrated by cDNA analysis. cDNA electrophoresis showed a band shorter than normal control, which corresponds with the production of a shorter protein product compared to the usual laminin size (p.Glu892Ala del893_917). The healthy sister of the patient resulted heterozygotic carrier of the mutation.
Patient IV
Patient IV is a 42-year-old man with progressive muscular weakness starting during childhood. The parents described him as clumsy. He showed difficulties in running since 18 years of age and in climbing stairs since 30 years of age. He underwent surgery for tibio-tarsal retraction. He did not show cognitive involvement but presented since 35 years of age partial seizures. Cardiac and respiratory involvement were absent. Last neurological examination showed waddling gait, proximal weakness (2~4/5 at MRC evaluation) and elbow tendon retractions. CK levels were mildly increased (1640-677 U/L) and muscle biopsy showed mild connective tissue increase and fibre I atrophy. Merosin was not tested. Brain MRI showed widespread white matter alteration.
Molecular analysis of LAMA2 gene revealed the presence of the following heterozygous mutations c.752T > C (p.Leu251Pro) in exon 5 and c.7586_7589dup (p.Phe2531X) in exon 55, confirming the diagnosis.
Patient V
Patient V is a 53-year-old patient with negative family history. She came to medical attention at 30 years of age for progressive muscular weakness, with difficulties in climbing stairs and in rising from floor since 45 years of age. Retrospectively she presented motor delay with achievement of independent ambulation at 2 years of age and difficulties in running. At last evaluation neurological examination showed waddling gait, proximal weakness at four limbs (4/5 at MRC scale), tibio-tarsal retractions. CK were mildly elevated (180-680 U/L). Muscle biopsies showed myopathic signs. The patient did not show cognitive impairment. At 26 years of age she presented generalized seizures (both absences and tonic-clonic episodes).. Brain MRI showed widespread alterations of brain white matter.
Molecular analysis showed the following compound heterozygous mutations in LAMA2 gene: c.752T > C (p.Leu25Pro) in exon 5 and c.7147C > T (p.Arg2383X) in exon 50; both mutations were not previously described.
Review of the literature
We performed a wide PubMed search of the literature concerning LGMD-like presentation associated to mutation in LAMA2 gene. We used the most commonly used entries and selected only papers written in English and reporting a complete molecular characterization.
We selected 14 papers 6-8,17-22,30-34. One paper 30 was excluded because it described three siblings with adulthood onset but predominant distal phenotype.
The clinical and molecular characteristics of the patients described in the remaining 13 papers are summarized in Table IV.
Table IV.
Review of the literature. LGMD patients with LAMA2 gene mutations described in literature: clinical and molecular characteristics.
| Clinical aspects | ||||||||
|---|---|---|---|---|---|---|---|---|
| Pt | Sex | Onset (age) | Ambulation (age) | Brain MRI | CNS/PNS | CK | Other features | Ref |
| 1 | M | 12 yrs | 16 m | WMC | None | 2417 | Three sister affected | [16] |
| 2 | M | < 29 yrs | 18 m | WMC | IQ 85 | 250-1236 | Rimmed vacuoles; dilated cardiomyopathy, arrhytmias | [17,29] |
| 3.1 | M | 15 yrs | Normal | WMC, cerebellar hypotrophy | Demyelinating neuropathy | 4x | Severe contractures; mild respiratory involvement | [18,33] |
| 3.2 | F | Childhood | Hip dislocation | WMC | None | 2x | Contractures | [18,33] |
| 4 | M | 4 yrs | 18 m | WMC | None | NA | Contractures | [32] |
| 5 | M | Childhood | 18 m | WMC | Epilepsy, sensory-motor neuropathy | 1429 | Contractures | [31] |
| 6 | M | 14 mo* | 12 m | WMC | Low normal IQ, epilepsy | 655 | - | [6] |
| 7.1 | M | 23 yrs | Normal | WMC | Epilepsu ? | 309 | - | [6] |
| 7.2 | F | 40 yrs | 2 yrs | WMC | Mild executive function deficit, epilepsy ? | 405 | - | [6] |
| 8 | M | 10 yrs | Normal | WMC | Epilepsy | 1053 | - | [6] |
| 9 | M | 59 yrs | Normal | WMC | Mild executive funcion deficit, trigeminal neuralgia | 859 | - | [6] |
| 10.1 | F | 30 yrs | Normal | Subcortical and deep WMC | Epilepsy | 280 | Occasional rimmed vacuoles | [7] |
| 10.2 | M | NA | Normal | WMC | None | NA | Rimmed vacuoles | [7] |
| 11 | F | 5 yrs | 12 m | Deep parietal WMC | Sensorimotor neuropathy | 653 | - | [19] |
| 12 | M | 56 yrs | Normal | WMC | None | 1171 | - | [8] |
| 13 | F | 1 yr | Delayed | WMC | None | 2148 | Contractures | [8] |
| 14 | M | 10 yrs | Normal | WMC | Epilepsy | 1053 | - | [8] |
| 15.1 | M | 8 yrs | 16 m | WMC | None | 398-2103 | Hyperreflexia | [21] |
| 15.2 | F | < 3 yrs | 14 m | WMC | None | 4100 | - | [21] |
| 16.1 | M | Childhood | Delayed | WMC, globi pallidi involvement | None | 400 | Contractures, rigid spine, dilated cardiomyopathy, atrial fibrillation | [20] |
| 16.2 | M | 7 yrs | Normal | ND | Sensorimotor neuropathy | 400 | Contracture, rigid spine | [20] |
* no progression until 17 yrs; WMC: white matter changes
Overall 21 cases with LGMD presentation have been described since now. Age of onset spanned from 14 months to 59 years of age. However the patient with onset at 14 months complained waddling gait and frequent falls, but achieved independent ambulation at 12 months of age and did not show progression of weakness until 17 years of age. Only two patients showed mild delay in motor milestones, while all the other subjects achieved independent ambulation and had normal milestones. CK levels were moderately increased (average 1032 +/-926 UI/L, range 280-4100 UI/L). All patients were still ambulant at last evaluation (age range 11-65 years).
Contractures were reported in 6 patients, associated to rigid spine in two cases. One patient showed mild respiratory involvement while cardiac involvement was reported in two cases. Brain white matter abnormalities were present in all patients and were generally severe confluent white matter lesions, with exception of one case who showed only subtle bilateral signal abnormalities on the deep parietal lobe 20. Cerebellar abnormalities were present in two subjects. Central nervous system involvement was variable and included epilepsy (3 patients), sporadic episodes of loss of consciousness (2 cases), mild mental retardation (2 subjects) and mild deficit of executive functions (2 patients). The remaining subjects had normal cognitive performances. Three patients showed peripheral neuropathy. Interestingly merosin staining generally showed partial reduction with exception of two cases which showed respectively absence at IHC study and severe reduction at WB analysis. In some cases merosin immunostaining revealed only a subtle decrease, in particular when antibody that recognizes the 80 kDa fragment was used, which could be erroneously interpreted as normal (Tab. III). Muscle biopsy showed a dystrophic pattern, rimmed vacuoles were present in three cases. As far as mutational analysis is concerned the majority of patients carried at least one missense mutation with the exception of the patient described by Di Blasi et al. which presented a nonsense mutation which however was associated with the skipping of the exon containing the mutation, thereby resulting in a restored open reading frame 19.
Discussion
Since now only 21 cases of patients with LAMA2 gene mutations associated with adult-onset phenotype have been reported worldwide (Tab. IV). These cases are generally associated with partial reduction of merosin, the presence of almost one in-frame mutation, moderate increase of CK levels.
In this paper we describe 5 new subjects with LAMA2 gene mutations associated with late onset phenotype characterized by mild, slowly evolving, proximal muscular involvement.
Mean age of onset was 23.2 ± 11.3 years; all patients showed normal milestones and achievement of independent ambulation; only one patient was described as clumsy during childhood, all patients were ambulant at last evaluation. In particular, patients I and III showed a very mild clinical phenotype, with the latest age of onset (respectively 62 and 75 years of age) described since now. Moreover patient III also showed a very mild muscular involvement. The other patients showed predominantly proximal muscle weakness, with distal involvement described only in one subject. CK were moderately elevated (640 ± 267.8 UI/L). Cardiac involvement was present in two patients who developed respectively paroxysmal supraventricular tachycardia and dilated cardiomiopathy associated to atrial flutter. Respiratory involvement was absent in all patients. Joint retraction and sural hypertrophy can be present. Electromyography showed a myopathic pattern occasionally associated to neurogenic signs (2 patients). Neurogenic signs were also detected in the biopsy of one patient (II.2). Peripheral neuropathy has been described in LAMA2-related diseases, mainly as a sensorimotor demyelinating polyneuropathy 14,32, with possible axonal involvement 20 due to abnormal myelinogenesis linked to the deficitary expression of merosin in the peripheral nerve Schwann cells. Two patients showed CNS involvement with epilepsy starting in adulthood, respectively at 26 and 35 years of age; seizures can be common in CMD but are rare in LGMD cases and generally start at younger age (Tab. IV). Migraine was present in patient III and never described before in association with this form of LGMD.
Brain MRI, which was abnormal in the majority of the patients, is fundamental to detect some specific aspects that can address the diagnosis. It should be performed even if central nervous system involvement is entirely subclinical. For example in patient I the molecular diagnosis was reached only after the accidental finding of white matter abnormalities at brain MRI, which addressed protein analysis and molecular investigation.
Muscle damage detected at muscle MRI is hard to compare with what is described in other patients because literature lacks MRI data in patients with partial merosin deficiency and LGMD phenotype. Only in a single report, a younger patients showed in her adolescence similar muscle MRI findings, although more severe that those found in the present series 20.
Our patients showed diffuse atrophy and fatty degeneration of thighs with selective sparing of the sartorius, the gracilis, the rectus femoris and the short head of the biceps femoris also at later stages. A diffuse but milder connective substitution was also present at lower legs, particularly in the medial gastrocnemious twins while upper limbs were less affected. No inflammatory signs were found.
Molecular analysis showed seven different mutations in LAMA2 gene, six of which are new. Merosin deficient patients with a LGMD phenotype carry at least one missense or in-frame mutation. Patient III carries the novel homozygous mutation IVS19+2 insT which affects splicing bringing to a 75 bp in-frame deletion in cDNA transcript (c.2675-2749 del 75nt). We did not notice any correlation between mutations and disease severity.
Muscle biopsy analysis showed a myopathic pattern. In patient III the availability of two different muscle samples, collected at 13 years of distance, allowed to study disease progression from a histological point of view. In this case, at disease onset, disease epiphenomena like fibres atrophy and subtle mitochondrial abnormalities may lead to misdiagnosis.
All patients showed partial reduction of merosin at protein analysis. Western-blot data are available only in three patients, among them the patient with milder reduction (60% of residual protein) showed a later onset than the two patients with more severe deficiency (30% of residual protein), but this data should be confirmed in larger samples.
Furthermore we noticed a considerable variability in merosin IHC and WB staining pattern depending on the antibody used. This variability has been previously described. Jones et al. analysed with two different antibodies 58 muscle biopsy samples demonstrating that 40% of them showed a differential staining 33. In particular the Chemicon antibody MAB1922, which was used in our patients, showed a milder degree of deficiency when compared to that observed with the Alexis MAB4H8-2. This significative difference between IHC and WB analysis results also by Di Blasi et al. 18. Interestingly also Bushby et al. 5 described a group of patients with a late onset myopathy and the unusual finding of a reduction of merosin on immunoblotting but not on IHC analysis. The presence of severe merosin reduction at WB but not at IHC in this group of patients and in our probands could be explained by different reactivity of the antibody, by the extensive processing of homogenization which is necessary for WB analysis or by a patient’s specific predisposition to merosin fragility.
Furthermore in some cases protein analysis could be misleading because, especially when only amino-terminus antibodies are used, it can underestimate merosin deficiency. WB analysis with antibodies directed against both the amino- and carboxyl-terminus of the protein must therefore be used when a LGMD associated to merosin deficiency is suspected.
Conclusions
Since now only few cases of LGMD with LAMA2 gene mutations have been described. LGMD R23 is a rare cause of LGMD, although this entity might be under-recognized. This could obviously be due to its rarity, but also to the fact that some contributory examinations, such as brain MRI, are not routinely performed in LGMD patients.
Overall the description of our cases further enlarges the spectrum of clinical phenotypes associated to mutations in LAMA2 gene. In particular our case sample includes patients with very late age of onset and very mild muscular involvement. Furthermore, the description of muscle MRI in our sample expands the knowledge about muscle imaging in this form of LGMD. WB analysis and brain MRI should be useful in order to suspect this form of LGMD also in pauci-symptomatic patients.
Figures and tables
Table IV.
Review of the literature. LGMD patients with LAMA2 gene mutations described in literature: clinical and molecular characteristics.
| Bioptical and molecular aspects | ||||
|---|---|---|---|---|
| Pt | Merosin ICH 300/80 kDa | Merosin WB | Molecular analysis | Ref |
| 1 | Severe / mild reduction | NA | Linkage analysis | [16] |
| 2 | Partial / partial reduction | Severe reduction | c.4405T > C (p.Cys1469Arg); c.4645C > T (p.Arg1549X) | [17,29] |
| 3.1 | Moderate / moderate reduction | Partial reduction | c.2230C > T (p.Arg744X + skipping of exon 15) in homozygosis | [18,33] |
| 3.2 | Slight / slight reduction | Partial reduction | c.2230C > T (p.Arg744X + skipping of exon 15) in homozygosis | [18,33] |
| 4 | Absent / absent | Absent | NA | [32] |
| 5 | Moderate / moderate reduction | NA | c.1847G > A (p.Gly600Arg) in homozygosis | [31] |
| 6 | Severe / mild reduction | 50% reduction | c.850G > A homozygous (p.Gly284Arg) | [6] |
| 7.1 | Severe / mild reduction | 65% reduction | c.728T > C (p.Leu243Pro); c.4860+2T > G_4860+3insGCC (p.Phe1573_Lys1620delinsSerfsX49) |
[6] |
| 7.2 | ND | ND | c.728T > C (p.Leu243Pro); c.4860+2T > G_4860+3insGCC (p.Phe1573_Lys1620delinsSerfsX49) |
[6] |
| 8 | ND / mild reduction | 50% reduction | c.397-1_397-15del (p.Val133_Gln135delinsArgX5); c.7431A > T (p.Arg2477Ser) |
[6] |
| 9 | NA | Absent | c.454T > G homozygous (p.Trp152Gly) | [6] |
| 10.1 | Mild reduction / normal | NA | c.2749+1G > A (Spl?); c.1177T > G (p.Cys393Gly) |
[7] |
| 10.2 | NA | NA | c.2749+1G > A (Spl?); c.1177T > G (p.Cys393Gly) |
[7] |
| 11 | Subtle / subtle reduction | Normal | c.391C > T (p.Gln131X); c.4487C > T (p.Ala496Val); (associated c.595T > A p.Cys199Ser exon 4) | [19] |
| 12 | Residual / NA | 1/3 residual | NA | [8] |
| 13 | Residual /NA | Absent | c.3758T > GT (?) (p.Leu1253Arg); c.35T > G (p.Leu12Arg) | [8] |
| 14 | Residual / NA | 1/3 residual | NA | [8] |
| 15.1 | NA / normal | NA | c.1358G > C (p.Cys453Ser); deletion exon 36-65 | [21] |
| 15.2 | NA | NA | c.1358G > C (p.Cys453Ser); deletion exon 36-65 | [21] |
| 16.1 | NA | NA | c.4533delT (p.Gly1512fsX); c.611C > T (p.Ser204Phe) | [20] |
| 16.2 | NA | NA | c.4533delT (p.Gly1512fsX); c.611C > T (p.Ser204Phe) | [20] |
Acknowledgements
This research received funding support from Telethon Grant GUP10006. Telethon Genetic Biobanks Network GTB07001E was the source of the DNA used in this study.
We thank Associazione Amici del Centro Dino Ferrari for their support.
References
- 1.Hohenester E, Yurchenco PD. Laminins in basement membrane assembly. Cell Adh Migr 2013;7:56-63. https://doi.org/10.4161/cam.21831 10.4161/cam.21831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yurchenco PD. Basement membranes: cell scaffoldings and signaling platforms. Cold Spring Harb Perspect Biol 2011;3 https://doi.org/10.1101/cshperspect.a004911 10.1101/cshperspect.a004911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de los A Beytía M, Dekomien G, Hoffjan S, et al. High creatine kinase levels and white matter changes: clinical and genetic spectrum of congenital muscular dystrophies with laminin alpha-2 deficiency. Mol Cell Probes 2014;28:118-22. https://doi.org/10.1016/j.mcp.2013.11.002 10.1016/j.mcp.2013.11.002 [DOI] [PubMed] [Google Scholar]
- 4.Tomé FM, Evangelista T, Leclerc A, et al. Congenital muscular dystrophy with merosin deficiency. C R Acad Sci III, Sci Vie 1994;317:351-7. [PubMed] [Google Scholar]
- 5.Bushby K, Anderson LV, Pollitt C, et al. Abnormal merosin in adults. A new form of late onset muscular dystrophy not linked to chromosome 6q2. Brain 1998;121:581-8. https://doi.org/10.1093/brain/121.4.581 10.1093/brain/121.4.581 [DOI] [PubMed] [Google Scholar]
- 6.Gavassini BF, Carboni N, Nielsen JE, et al. Clinical and molecular characterization of limb-girdle muscular dystrophy due to LAMA2 mutations. Muscle Nerve 2011;44:703-9. https://doi.org/10.1002/mus.22132 10.1002/mus.22132 [DOI] [PubMed] [Google Scholar]
- 7.Rajakulendran S, Parton M, Holton JL, et al. Clinical and pathological heterogeneity in late-onset partial merosin deficiency. Muscle Nerve 2011;44:590-3. https://doi.org/10.1002/mus.22196 10.1002/mus.22196 [DOI] [PubMed] [Google Scholar]
- 8.Løkken N, Born AP, Duno M, et al. LAMA2-related myopathy: frequency among congenital and limb-girdle muscular dystrophies. Muscle Nerve 2015;52:547-53. https://doi.org/10.1002/mus.24588 10.1002/mus.24588 [DOI] [PubMed] [Google Scholar]
- 9.Straub V, Murphy A, Udd B, et al. 229th ENMC international workshop: limb girdle muscular dystrophies – Nomenclature and reformed classification Naarden, the Netherlands, 17-19 March 2017 Neuromuscul Disord 2018;28:702-10. https://doi.org/10.1016/j.nmd.2018.05.007 10.1016/j.nmd.2018.05.007 [DOI] [PubMed] [Google Scholar]
- 10.Geranmayeh F, Clement E, Feng LH, et al. Genotype-phenotype correlation in a large population of muscular dystrophy patients with LAMA2 mutations. Neuromuscul Disord 2010;20:241-50. https://doi.org/10.1016/j.nmd.2010.02.001 10.1016/j.nmd.2010.02.001 [DOI] [PubMed] [Google Scholar]
- 11.Naom I, D’Alessandro M, Sewry CA, et al. Laminin alpha 2-chain gene mutations in two siblings presenting with limb-girdle muscular dystrophy. Neuromuscul Disord 1998;8:495-501. https://doi.org/10.1016/s0960-8966(98)00065-0 10.1016/s0960-8966(98)00065-0 [DOI] [PubMed] [Google Scholar]
- 12.Naom I, D’alessandro M, Sewry CA, et al. Mutations in the laminin alpha2-chain gene in two children with early-onset muscular dystrophy. Brain 2000;123(Pt 1):31-41. https://doi.org/10.1093/brain/123.1.31 10.1093/brain/123.1.31 [DOI] [PubMed] [Google Scholar]
- 13.Hayashi YK, Ishihara T, Domen K, et al. A benign allelic form of laminin alpha 2 chain deficient muscular dystrophy. Lancet 1997;349:1147 https://doi.org/10.1016/S0140-6736(05)63023-1 10.1016/S0140-6736(05)63023-1 [DOI] [PubMed] [Google Scholar]
- 14.Deodato F, Sabatelli M, Ricci E, et al. Hypermyelinating neuropathy, mental retardation and epilepsy in a case of merosin deficiency. Neuromuscul Disord 2002;12:392-8. https://doi.org/10.1016/s0960-8966(01)00312-1 10.1016/s0960-8966(01)00312-1 [DOI] [PubMed] [Google Scholar]
- 15.Clement EM, Feng L, Mein R, et al. Relative frequency of congenital muscular dystrophy subtypes: analysis of the UK diagnostic service 2001-2008. Neuromuscul Disord 2012;22:522-7. https://doi.org/10.1016/j.nmd.2012.01.010 10.1016/j.nmd.2012.01.010 [DOI] [PubMed] [Google Scholar]
- 16.Norwood FLM, Harling C, Chinnery PF, et al. Prevalence of genetic muscle disease in Northern England: in-depth analysis of a muscle clinic population. Brain 2009;132:3175-86. https://doi.org/10.1093/brain/awp236 10.1093/brain/awp236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tan E, Topaloglu H, Sewry C, et al. Late onset muscular dystrophy with cerebral white matter changes due to partial merosin deficiency. Neuromuscul Disord 1997;7:85-9. https://doi.org/10.1016/s0960-8966(96)00421-x 10.1016/s0960-8966(96)00421-x [DOI] [PubMed] [Google Scholar]
- 18.Di Blasi C, Mora M, Pareyson D, et al. Partial laminin alpha2 chain deficiency in a patient with myopathy resembling inclusion body myositis. Ann Neurol 2000;47:811-6. https://doi.org/10.1002/1531-8249(200006)47:6<811 [DOI] [PubMed] [Google Scholar]
- 19.Di Blasi C, He Y, Morandi L, et al. Mild muscular dystrophy due to a nonsense mutation in the LAMA2 gene resulting in exon skipping. Brain 2001;124:698-704. https://doi.org/10.1093/brain/124.4.698 10.1093/brain/124.4.698 [DOI] [PubMed] [Google Scholar]
- 20.Chan SHS, Foley AR, Phadke R, et al. Limb girdle muscular dystrophy due to LAMA2 mutations: diagnostic difficulties due to associated peripheral neuropathy. Neuromuscul Disord 2014;24:677-83. https://doi.org/10.1016/j.nmd.2014.05.008 10.1016/j.nmd.2014.05.008 [DOI] [PubMed] [Google Scholar]
- 21.Harris E, McEntagart M, Topf A, et al. Clinical and neuroimaging findings in two brothers with limb girdle muscular dystrophy due to LAMA2 mutations. Neuromuscul Disord 2017;27:170-4. https://doi.org/10.1016/j.nmd.2016.10.009 10.1016/j.nmd.2016.10.009 [DOI] [PubMed] [Google Scholar]
- 22.Ding J, Zhao D, Du R, et al. Clinical and molecular genetic analysis of a family with late-onset LAMA2-related muscular dystrophy. Brain Dev 2016;38:242-9. https://doi.org/10.1016/j.braindev.2015.08.005 10.1016/j.braindev.2015.08.005 [DOI] [PubMed] [Google Scholar]
- 23.Dubowitz V, Sewry C, Oldfors A. Muscle biopsy: a practical approach, 4th Ed. New York: Elsevier; 2014. [Google Scholar]
- 24.Pegoraro E, Marks H, Garcia CA, et al. Laminin alpha2 muscular dystrophy: genotype/phenotype studies of 22 patients. Neurology 1998;51:101-10. https://doi.org/10.1212/wnl.51.1.101 10.1212/wnl.51.1.101 [DOI] [PubMed] [Google Scholar]
- 25.Savarese M, Di Fruscio G, Mutarelli M, et al. MotorPlex provides accurate variant detection across large muscle genes both in single myopathic patients and in pools of DNA samples. Acta Neuropathol Commun 2014;2 https://doi.org/10.1186/s40478-014-0100-3 10.1186/s40478-014-0100-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Philpot J, Topaloglu H, Pennock J, et al. Familial concordance of brain magnetic resonance imaging changes in congenital muscular dystrophy. Neuromuscul Disord 1995;5:227-31. https://doi.org/10.1016/0960-8966(94)00047-d 10.1016/0960-8966(94)00047-d [DOI] [PubMed] [Google Scholar]
- 27.Farina L, Morandi L, Milanesi I, et al. Congenital muscular dystrophy with merosin deficiency: MRI findings in five patients. Neuroradiology 1998;40:807-11. https://doi.org/10.1007/s002340050689 10.1007/s002340050689 [DOI] [PubMed] [Google Scholar]
- 28.Lamer S, Carlier RY, Pinard JM, et al. Congenital muscular dystrophy: use of brain MR imaging findings to predict merosin deficiency. Radiology 1998;206:811-6. https://doi.org/10.1148/radiology.206.3.9494506 10.1148/radiology.206.3.9494506 [DOI] [PubMed] [Google Scholar]
- 29.van der Knaap MS, Smit LM, Barth PG, et al. Magnetic resonance imaging in classification of congenital muscular dystrophies with brain abnormalities. Ann Neurol 1997;42:50-9. https://doi.org/10.1002/ana.410420110 10.1002/ana.410420110 [DOI] [PubMed] [Google Scholar]
- 30.Carboni N, Marrosu G, Porcu M, et al. Dilated cardiomyopathy with conduction defects in a patient with partial merosin deficiency due to mutations in the laminin-α2-chain gene: a chance association or a novel phenotype? Muscle Nerve 2011;44:826-8. https://doi.org/10.1002/mus.22228 10.1002/mus.22228 [DOI] [PubMed] [Google Scholar]
- 31.Kevelam SH, van Engelen BGM, van Berkel CGM, et al. LAMA2 mutations in adult-onset muscular dystrophy with leukoencephalopathy. Muscle Nerve 2014;49:616-7. https://doi.org/10.1002/mus.24147 10.1002/mus.24147 [DOI] [PubMed] [Google Scholar]
- 32.Di Muzio A, De Angelis MV, Di Fulvio P, et al. Dysmyelinating sensory-motor neuropathy in merosin-deficient congenital muscular dystrophy. Muscle Nerve 2003;27:500-6. https://doi.org/10.1002/mus.10326 10.1002/mus.10326 [DOI] [PubMed] [Google Scholar]
- 33.Jones KJ, Morgan G, Johnston H, et al. The expanding phenotype of laminin 2 chain (merosin) abnormalities: case series and review. 9. https://doi.org/10.1136/jmg.38.10.649 10.1136/jmg.38.10.649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morandi L, Blasi CD, Farina L, et al. Clinical correlations in 16 patients with total or partial laminin alfa 2 deficiency characterized using antibodies against 2 fragments of the protein. Arch Neurol 1999;56:7 https://doi.org/10.1001/archneur.56.2.209 10.1001/archneur.56.2.209 [DOI] [PubMed] [Google Scholar]
- 35.Pegoraro E, Fanin M, Trevisan CP, et al. A novel laminin alpha2 isoform in severe laminin alpha2 deficient congenital muscular dystrophy. Neurology 2000;55:1128-34. https://doi.org/10.1212/wnl.55.8.1128 10.1212/wnl.55.8.1128 [DOI] [PubMed] [Google Scholar]