

Significance Statement
Extracellular matrix (ECM) replaces glomerular capillaries in FSGS. To evaluate differences in ECM remodeling between collapsing FSGS (cFSGS) and FSGS not otherwise specified (FSGS-NOS), we performed a proteomic analysis of glomerular ECM composition using human biopsies. Abundance of 58 ECM proteins differed: 41 were more abundant in cFSGS and 17 in FSGS-NOS. Increased abundance and coexpression of cathepsin B, cathepsin C, and annexin A3 characterized cells infiltrating glomerular tufts in cFSGS. These cells expressed markers of activated parietal epithelial cells, but not markers of podocytes. This work demonstrates multiple mechanisms of how dysregulated ECM remodeling underlies focal sclerosis. The work supports the important role of parietal epithelial cells in disease histopathology and identifies them as a possible therapeutic target, particularly for cFSGS.
Keywords: focal segmental glomerulosclerosis, extracellular matrix, collapsing FSGS, glomerular epithelial cells, cathepsin
Visual Abstract

Abstract
Background
The mechanisms leading to extracellular matrix (ECM) replacement of areas of glomerular capillaries in histologic variants of FSGS are unknown. This study used proteomics to test the hypothesis that glomerular ECM composition in collapsing FSGS (cFSGS) differs from that of other variants.
Methods
ECM proteins in glomeruli from biopsy specimens of patients with FSGS not otherwise specified (FSGS-NOS) or cFSGS and from normal controls were distinguished and quantified using mass spectrometry, verified and localized using immunohistochemistry (IHC) and confocal microscopy, and assessed for gene expression. The analysis also quantified urinary excretion of ECM proteins and peptides.
Results
Of 58 ECM proteins that differed in abundance between cFSGS and FSGS-NOS, 41 were more abundant in cFSGS and 17 in FSGS-NOS. IHC showed that glomerular tuft staining for cathepsin B, cathepsin C, and annexin A3 in cFSGS was significantly greater than in other FSGS variants, in minimal change disease, or in membranous nephropathy. Annexin A3 colocalized with cathepsin B and C, claudin-1, phosphorylated ERK1/2, and CD44, but not with synaptopodin, in parietal epithelial cells (PECs) infiltrating cFSGS glomeruli. Transcripts for cathepsins B and C were increased in FSGS glomeruli compared with normal controls, and urinary excretion of both cathepsins was significantly greater in cFSGS compared with FSGS-NOS. Urinary excretion of ECM-derived peptides was enhanced in cFSGS, although in silico analysis did not identify enhanced excretion of peptides derived from cathepsin B or C.
Conclusions
ECM differences suggest that glomerular sclerosis in cFSGS differs from that in other FSGS variants. Infiltration of activated PECs may disrupt ECM remodeling in cFSGS. These cells and their cathepsins may be therapeutic targets.
FSGS is the most common glomerular disorder leading to ESKD in North America.1,2 FSGS consists of a spectrum of histopathology with multiple causes, various presentations, and different outcomes and responses to therapy. The Columbia classification defines five patterns of histopathologic changes in FSGS: collapsing variant (collapse of the glomerular tuft with epithelial cell hyperplasia), tip variant (lesions at the urinary pole), perihilar variant (lesions at the vascular pole), cellular variant (endocapillary hypercellularity), and FSGS not otherwise specified (FSGS-NOS) for lesions that do not meet the criteria for the other four classes.3 The collapsing FSGS (cFSGS) variant defines a group of patients with more severe nephrotic syndrome, lower eGFR at presentation, and a higher likelihood of progression to ESKD.4–7 Additionally, cFSGS is associated with proliferation of parietal epithelial cells (PECs) and altered podocyte differentiation.8–10 The unique histologic and clinical characteristics suggest the pathophysiology of cFSGS differs from other FSGS variants.
A histopathologic finding common to all variants of FSGS is focal and segmental deposition of new extracellular matrix (ECM) that obliterates glomerular capillaries. ECM is a complex molecular structure that provides a physical scaffold for all tissues and regulates cell and tissue physiology. ECM composition is tissue specific and undergoes continuous remodeling.11–15 Proteomic studies by our group16 and by Lennon et al.17 identified about 250 proteins that comprise the normal glomerular ECM. Although alterations in glomerular ECM components occur in FSGS,9,18–21 a comprehensive analysis of ECM composition and differences among FSGS variants has not been performed. Thus, the contribution of abnormal ECM composition to development and progression of FSGS remains unclear. Based on the unique characteristics of cFSGS, we postulated that glomerular ECM composition in cFSGS differs from the other variants, and those differences may contribute to the unique characteristics of cFSGS. To address this hypothesis, proteomic and transcriptomic analysis of glomeruli isolated from human biopsies was performed.
Methods
Study Approval
The University of Louisville Human Studies Committee approved sample collection and use of de-identified samples provided by coauthors’ institutions. All study subjects provided informed consent before sample collection, except where samples were deemed exempt. All studies were conducted in compliance with the Declaration of Helsinki.
Tissue Collection
De-identified, formalin-fixed, paraffin-embedded renal tissue was provided by collaborating pathologists after confirmation of the diagnosis based on the Columbia classification criteria.3 Kidney tissue was cut into 10-μm sections on polyethylene terephthalate membrane frame slides and stained with Mayer hematoxylin following Leica LMD6500 Laser Microdissection System staining protocol. Laser capture microdissection of glomeruli was performed such that the Bowman’s capsule was excluded from samples (Supplemental Figure 1). Between 38 and 124 glomerular sections from 12 to 26 glomeruli for each subject were collected into tubes containing 20 µl of storage buffer (10 mM HEPES pH 7.0, 0.67 mM EDTA, 1× Halt Protease and Phosphatase Inhibitor [78442; Thermo Fisher Scientific, Waltham, MA]) and stored at −80°C.
Urine Collection
Single void urine samples were centrifuged at 1200 × g for 15 minutes at 4°C to remove cellular debris. Supernatants were aliquoted into 15 ml vials and stored at −80°C until used for sample preparation. Urine was diluted with an equal volume of 0.5% trifluoroacetic acid and then transferred to a Sartorius Vivaspin-2 concentrator (Vivaproducts, Littleton, MA) with a 5000 Da mol wt cutoff Hydrosart membrane. The Vivaspin-2 was spun at 3220 × g for 15 minutes at 4°C in an Eppendorf 5810R centrifuge. Peptides were concentrated and desalted using 1 ml Oasis HLB (Waters Corporation, Milford, MA) solid phase extraction cartridges as previously described.22 The ultrafiltrate was rinsed three times using 10 mM HEPES/0.5 mM EDTA, pH 7.6, and recovered by pipetting. The peptide and protein sample concentrations were determined by determined using a μBCA kit (Pierce, Rockford, IL).
ECM Enrichment and Protein Extraction
Protein extraction was performed using a two-step procedure previously reported.16 Isolated glomerular sections were de-cellularized with 25 mM ammonium hydroxide/0.5% Triton X-100 plus HALT Protease and Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific) followed by protein extraction of residual ECM-enriched pellet using an acid-labile surfactant (0.1% ProteaseMAX Surfactant in 0.05 M ammonium bicarbonate) with heating for 5 minutes at 95°C. The isolated proteins were digested overnight at 37°C with 20 ng sequencing-grade modified trypsin in 100 µl of 1 M urea with 0.1 M Tris hydrochloride, pH 8.5. The digests were filtered using the YM-10 (10,000 Da mol wt cutoff) device. The filtered and digested samples were desalted and concentrated using a reversed phase C18 PROTO Ultra MicroSpin desalting column (Nest Group, Southborough, MA). The recovered peptides were lyophilized and resuspended in 2% acetonitrile/0.1% formic acid before quantification using a NanoDrop 2000 (Thermo Fisher Scientific) at 205 nm.
Proteomic Liquid Chromatography Mass Spectrometry Data Collection
Glomerular ECM-Enriched Samples
Methods for the analysis of ECM-enriched, Triton X-100–insoluble pellet digests using liquid chromatography (LC) mass spectrometry (LCMS) were conducted as previously described.16 Peptide values were estimated using absorbance measurements at 205 nm by NanoDrop 2000. Peptide samples were separated with a 170 minute LC gradient using a Proxeon EASY-nLC 1000 (Thermo Fisher Scientific) ultra-high-performance LC (UHPLC) system and Dionex (Sunnyvale, CA) Acclaim PepMap 100 75 µm×2 cm, nanoViper (C18, 3 µm, 100 Å) trap, and a Dionex Acclaim PepMap RSLC 50 µm×15 cm, nanoViper (C18, 2 µm, 100 Å) separating column. The eluate was introduced via a Nanospray Flex source (Thermo Fisher Scientific) into an Orbitrap Elite electron-transfer dissociation mass spectrometer (Thermo Fisher). Data were acquired using an Nth Order Double Play (Xcalibur version 2.2; Thermo Fisher Scientific) with Fourier transform mass spectrometry (FTMS) MS1 scans (240,000 resolution) collected from 300 to 2000 m/z and ion trap mass spectrometry MS2 scans collected on up to 20 peaks having a minimum signal threshold of 5000 counts from the MS1 scan event.
Urine Peptidomics
Urine peptide samples were diluted to a final common concentration of 0.2 µg/µl in 2% acetonitrile/0.1% formic acid and loaded into autosampler vials before transfer to an Easy-nLC 1000 System. Peptides (500 ng) were separated using a 12 cm, 360 µm OD×100 µm ID fused silica tip packed with Aeris Peptide 3.6 µm XB-C18 100 Å material (Phenomenex, Torrance, CA) and high-resolution, label-free LCMS data collected as described above.
Urine Proteomics
Urine protein samples (50 µg) were reduced with dithiothreitol, alkylated using iodoacetamide, and digested using 2 µg mass spectrometry–grade trypsin/Lys-C protease mixture (Promega, Madison, WI). Protein digests were quantified by A205 nm measurements on a NanoDrop 2000 and 25 µg of each sample was used for labeling with one isobaric tagging reagent from a Tandem Mass Tag (TMT) 10-plex kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. A pooled internal standard comprising 5 µg admixtures from all sample digests was used as a reference normalization of TMT reporter ions between separate LCMS experiments. TMT10-plex–labeled admixtures of patient urine protein digests were separated using high pH reversed-phase chromatography with fraction concatenation using an Ultimate 3000SD UHPLC system (Thermo Fisher Scientific) with an XBridge BEH C18 130 Å, 5 µm 3×150 mm column (Waters Corporation) and 5%–90% acetonitrile/ammonium formate, pH 10 gradient. The concatenated samples were lyophilized and resuspended in 50 µl 2% acetonitrile/0.01% formic acid. The TMT-labeled samples were loaded into the autosampler of an EASY-nLC 1000 UHPLC system (Thermo Fisher Scientific) and separated using a 50 cm Acclaim PepMap RSLC column (Thermo Fisher Scientific) before transfer by Nanospray Flex source (Thermo Fisher Scientific) into an Orbitrap Elite electron-transfer dissociation mass spectrometer (Thermo Fisher Scientific). An Nth Order Double Play (Xcalibur version 2.2) was used to collect Orbitrap MS1 scans (60,000 resolution) for the range 300–2000 m/z and FTMS MS2 scans (higher-energy collisional dissociation activation; 60,000 resolution) on up to ten peaks that had a minimum signal threshold of 5000 counts from scan event one.
Mass Spectrometry Data Analysis
Tissue Proteomics
The individual cFSGS, FSGS-NOS, or normal control (NC) LCMS data sets were analyzed by Proteome Discoverer version 1.4.1.14 using Mascot version 2.5.1 and SequestHT searches with the December 18, 2019 version of the UniprotKB Homo sapiens reviewed reference proteome canonical and isoform sequences. Search parameters included: variable methionine or proline oxidation (+16 Da), fixed cysteine carbamidomethylation (+57 Da), up to two missed tryptic cleavages, 50 ppm precursor error for MS1 Orbitrap FTMS data, 1 Da error for MS2 data sets, and minimum peptide length of four amino acids. To estimate the false discovery rate (FDR), a Target Decoy PSM Validator node was included in the Proteome Discoverer workflow. The resulting .msf files from Proteome Discoverer were loaded into Scaffold Q+S version 4.4.5 for comparative proteomics. Scaffold was used to calculate the FDR (strict, 1% FDR; relaxed, 5% FDR) using the Peptide and Protein Prophet algorithms. The Scaffold Local FDR algorithm was enabled during loading to accommodate instrumental mass measurement drift in Orbitrap data. Final peptide level information used to build quantitative results were filtered by a 2 ppm precursor mass accuracy. Accepted protein-level data were annotated with human gene ontology information from the Gene Ontology Annotations Database (geneontology.org). Protein intensity-based absolute quantification (iBAQ) scores were exported as Excel files for statistical analysis. As described previously,23 missing values were imputed and data normalized before statistical testing by ANOVA analysis (one-way ANOVA in R version 3.5.0, using the R script aov) with Benjamini–Hochberg correction for multiple comparisons and post hoc t tests. Missing values were imputed by singular value decomposition using “impute.MAR” function of the R package “imputeLCMD.” iBAQ scores were normalized by variance stabilizing normalization by applying the “justvsn” function available in R package “vsn.”
Urine Peptidomics
Peptidomic data sets were recalibrated postacquisition (RecalOffline, Xcalibur version 2.2) to the +2 charge state of the uromodulin peptide SVIDQSRVLNLGPITR, a urinary peptide consistently identified at high confidence in all samples. The recalibrated files were researched in Peaks Studio X (Bioinformatics Solutions Inc., Waterloo, ON, Canada) using the Denovo, PeaksDB, PeaksPTM, and Label-Free Q algorithms and the August 8, 2018 version of the UniProtKB-reviewed reference proteome canonical and isoform H. sapiens sequences (proteome identifier, UP000005640). Search parameters included: variable methionine oxidation (+15.9949 Da), no enzyme specified, 15 ppm precursor error for MS1 Orbitrap FTMS data, 0.5 Da error for MS2 data sets, and common post-translational modifications in the PeaksPTM algorithm. The peptide, feature, and protein-peptide comma separated values files were exported from the Label-Free Q result into Microsoft Excel 2016 and normalized to urine protein concentration. The statistical testing for significant, differentially abundant urinary peptides was determined using the one-way ANOVA in R (version 3.5.0) using the R-script aov. A P value <0.05 was considered significant.
Urine Proteomics
Proteomic data files were searched using Mascot version 2.5.1 and SequestHT against the August 8, 2018 version of the UniprotKB-reviewed reference proteome canonical and isoform H. sapiens sequences (proteome identifier, UP000005640) using Proteome Discoverer version 2.2.0.388 (Thermo Fisher Scientific). Search parameters included: variable methionine oxidation (+15.9949 Da) and protein N-terminal acetylation (+42.0106 Da), fixed cysteine carbamidomethylation (+57.0215 Da) and TMT6-plex modification (+229.16293 at any N-terminal and K), up to two missed tryptic cleavages, 10 ppm precursor error for MS1 Orbitrap FTMS data, 0.02 Da error for MS2 data sets, and minimum peptide length of six amino acids. A proteins text file was exported from Proteome Discoverer, data were normalized to urine protein concentration, and statistical tests were done in Microsoft Excel 2016 and R version 3.5.0.
The statistical testing for significant, differentially abundant proteins within glomerular tissue or urine was performed using the Kruskal–Wallis ANOVA (H test) with post hoc correction for multiple comparisons as provided within Scaffold Q+S. The statistical testing for significant, differentially abundant urinary peptides was performed using PeaksX data set normalized to urine protein concentration in Microsoft Excel 2016 by one-way ANOVA in R (version 3.5.0) using the R-script aov. A P value <0.05 was considered significant.
Identification of Candidate Proteases
Urine peptides assigned with high confidence (PeaksX criteria) were exported into comma separated values files for upload and analysis by the Proteasix (http://www.proteasix.org/) algorithm using the “peptide-centric” prediction tool based on curated, known, and observed cleavage events enabling assignment of protease identities from the MEROPS database.24
Immunohistochemistry
Immunohistochemistry (IHC) of paraffin-embedded renal biopsy sections was performed as previously described.25 Antibodies to identify glomerular proteins were chosen based on validation by the Human Proteome Atlas Project (https://www.proteinatlas.org).26 Primary antibodies to the following proteins were used: cathepsin C (3 µg/ml, catalog AF1071; R&D Systems, Minneapolis, MN), annexin A3 (1:2000, catalog HPA013398; Sigma, St. Louis, MO), cathepsin B (1:2000, catalog 31817; Cell Signaling, Danvers, MA), CD44 (1:100, Cell Signaling 16728; Abcam, Cambridge, MA), and phosphorylated ERK (pERK; 1:100, catalog 7383; Santa Cruz Biotechnology, Dallas, TX). Sections were incubated with primary antibodies overnight at 4°C. All sections, including negative controls, were then incubated with biotinylated secondary antibodies (Vector Labs) for 30 minutes, followed by incubation in avidin/biotin enzyme complex (Vectastain Elite ABC Kit, Vector Labs) for 30 minutes. Proteins were detected after color development using 3,3′-diaminobenzidine as substrate (Vector Labs). Digital images were acquired with a Q Color 5 camera attached to an Olympus BX51 microscope using Image-Pro software. Cathepsin C and annexin A3 glomerular tuft immunostaining was quantified with Image-Pro 6.2 software (Media Cybernetics, Silver Spring, MD) on images captured with a 40× objective. The percentage of immunostaining in each glomerular tuft was determined by the ratio of immunostained area to total glomerular tuft surface area.
Matrisome Identification within Proteomic Data Sets
Identification of ECM proteins was performed by comparing proteins expressed to the combination of the Matrisome database (http://matrisomeproject.mit.edu/analytical-tools/matrisome-annotator/)27 and previously published glomerular ECM databases.16,17
Confocal Microscopy
Renal biopsy sections were cleared of paraffin, rehydrated, and subjected to antigen retrieval and blocking with 5% horse serum as described above for IHC. Biopsy sections were incubated simultaneously with anti–cathepsin C and anti–annexin A3 antibodies or anti-synaptopodin (1:100, catalog sc21537; Santa Cruz Biotechnology) and anti–annexin A3 antibodies, at the antibody dilutions described above, overnight at 4°C. Sections were incubated with anti-rabbit Alexa Fluor 488 for detection of annexin A3, followed by anti-goat Alexa Fluor 546 to detect cathepsin C or synaptopodin. For costaining of annexin A3 with claudin-1 (1:250, catalog 211737; Abcam) or cathepsin B, sections were first incubated with anti–annexin A3 overnight at 4°C, followed by incubation with anti-rabbit Alexa Fluor 488. Sections were again blocked with 5% horse serum, followed by incubation with anti–claudin-1 antibody or anti–cathepsin B, overnight at 4°C. Claudin-1 or cathepsin B were detected with anti-rabbit Alexa Fluor 546. Nuclei were stained with 4′,6-diamidino-2-phenylindole. Images were acquired using an Olympus Fluoview FV-1000 confocal coupled to an Olympus 1×81 inverted microscope, a PlanApoN 60× objective, and FV-10 ASW 2.1 software. A multichannel scanning configuration with sequential line scanning was set up for acquisition of 4′,6-diamidino-2-phenylindole, AF488, and AF546. The optimal brightness setting for each channel was configured by determining the HV setting yielding maximal intensity without saturation. Each of the settings was tested against negative control sections to ensure exclusion of nonspecific emission.
Glomerular Transcriptome
Quantitative microarray (European Renal cDNA Bank; ERCB) and RNA-sequencing (RNA-seq; Nephrotic Syndrome Research Network, NEPTUNE) expression data from available ERCB data sets were accessed through the Nephroseq platform and from NEPTUNE data sets accessed through an Ancillary Studies award.28,29 In brief, the ERCB data were developed using RNA extracted from microdissected glomeruli to generate cDNA libraries that were sequenced by the University of Michigan DNA Sequencing Core Facility on a HiSeq 4000. Quality control was performed to examine read quality, duplication rates, GC content, adapter content, and nucleotide distribution using the FastQC toolkit. Reads were aligned to the GRCh38 genome with the STAR aligner (2.5.2), and transcript-level quantification was performed using Kallisto.30 NEPTUNE RNA-seq ENST data were converted to gene names. The ERCB and NEPTUNE data were filtered for glomerular transcripts by matching with the gene name of the identified regulated matrisome proteomic data. The differences in transcript expression means were compared by t test without multiple comparisons test corrections. A P value <0.05 was considered significant.
Statistical Analysis
Differences in glomerular tissue proteomes and transcriptomes and urine proteome and peptidome were determined by ANOVA and post hoc t tests. Significance for P values and multiple comparisons–adjusted P values (q values) by Benjamini–Hochberg correction were a priori set at <0.05. Statistical significance estimates and fold-change values were used to identify candidates for IHC or confocal microscopy tissue confirmation studies. Area under the curve (AUC) of the receiver operator characteristic plots were used to determine the classification model performance of glomerular cathepsin C and annexin A3 staining to accurately classify patient biopsy specimen diagnoses.
Results
Glomerular ECM in FSGS
To identify glomerular ECM proteins, enrichment of glomerular sections was performed by sequential extraction with ammonium hydroxide/Triton X-100 to disrupt cells followed by protein extraction with ProteaseMAX (Promega).16 Preliminary studies showed this procedure increased the number and quantity of ECM proteins identified from patients without renal disease (NC) and FSGS-NOS glomerular sections (Supplemental Figure 2). Glomerular ECM proteins were identified from 38 to 124 glomerular sections from each of seven patients diagnosed with cFSGS, six with FSGS-NOS, and two NCs.16 The number of glomeruli per patient from which sections were obtained was 19±2 (mean±SEM; range, 12–26). A total of 1402 unique proteins were identified by combining results of all 15 patients, of which 1148 were identified from cFSGS, 1104 from FSGS-NOS, and 900 from NCs (Supplemental Table 1). ECM proteins were identified from a combined list of the Matrisome database27 and ECM proteins previously identified from human glomeruli.16,17 A total of 166 ECM proteins were identified (Supplemental Table 2), of which 158 ECM proteins were identified from cFSGS glomeruli, 136 from FSGS-NOS, and 109 from NCs. The relative abundance of 32 basement membrane matrisome proteins, 33 structural matrisome proteins, and 101 matrisome-associated proteins, based on established categories,27,31 is shown in Supplemental Table 3. Differences in abundance of individual proteins were analyzed by ANOVA with post hoc determination of q value by Benjamini–Hochberg analysis and by the fold differences based on iBAQ between NCs and cFSGS or FSGS-NOS. A total of 79 ECM proteins demonstrated a significant difference in abundance between FSGS and NCs. An additional 25 ECM proteins demonstrated a fivefold or greater difference in abundance between FSGS and NCs in the absence of statistical significance. Of those 104 proteins, 85 showed enhanced abundance in FSGS, of which 31 were increased in both cFSGS and FSGS-NOS, 37 in cFSGS only, and 17 in FSGS-NOS only. A total of 19 proteins showed reduced abundance in FSGS, five in both cFSGS and FSGS-NOS, five in cFSGS, and nine in FSGS-NOS.
To identify ECM proteins with differential abundance between cFSGS and FSGS-NOS, three separate screens were applied to the 104 ECM proteins with altered abundance in FSGS. First, iBAQ parameters from high-resolution proteomic data sets were analyzed by ANOVA followed by a post hoc Benjamini–Hochberg test (q value).23 A total of 48 ECM proteins were significantly different (q≤0.05), of which abundance was greater in cFSGS for 35 proteins and in FSGS-NOS for 13 proteins. A volcano plot of that analysis is shown in Figure 1. Second, four ECM proteins analyzed by ANOVA without correction for multiple comparisons showed statistically significant differences between cFSGS and FSGS-NOS. Finally, six glomerular ECM proteins demonstrated a threefold or greater difference in protein abundance, determined by the average of iBAQ scores. Table 1 lists those 58 ECM proteins, of which 41 were more abundant in cFSGS and 17 were more abundant in FSGS-NOS.
Figure 1.

Volcano plot shows significant differences in glomerular ECM protein expression between cFSGS and FSGS-NOS. Of 166 proteins, abundance of 42 proteins differed by ANOVA analysis with Benjamini–Hochberg correction for multiple comparisons (adjusted P<0.05). The y axis is defined as the −log10 multiple comparisons adjusted P value (F adj P-value) and the x axis defined as the log2 fold change (Log2 FC) of ECM protein abundance in isolated glomeruli from patients with cFSGS (n=7), compared with those with FSGS-NOS (n=6). Red symbols denote increased abundance in cFSGS for 32 proteins. Blue symbols denote increased abundance in FSGS-NOS for ten proteins. Symbols denoting differentially abundant proteins are annotated by gene name.
Table 1.
ECM proteins with differential abundance between cFSGS and FSGS-NOS
| Protein Name | Gene Name | Log2 Fold Change | P Value | q Value | Category | Division |
|---|---|---|---|---|---|---|
| Proteins with greater abundance in cFSGS | ||||||
| Annexin A3 | ANXA3 | 3.04 | 1.96e−07 | 1.69e−06 | ECM-affiliated proteins | Matrisome associated |
| Apolipoprotein C3 | APOC3 | >5.0 | 1.92e−11 | 5.65e−10 | ECM-affiliated proteins | Matrisome associated |
| Azurocidin | AZU1 | >5.0 | 3.81e−08 | 3.92e−07 | ECM regulators | Matrisome associated |
| Complement C1q A chain | C1QA | >5.0 | 4.55e−05 | 2.81e−04 | ECM-affiliated proteins | Matrisome associated |
| Complement C1q B chain | C1QB | >5.0 | 7.11e−09 | 8.09e−08 | ECM-affiliated proteins | Matrisome associated |
| Complement C1q C chain | C1QC | 3.20 | 7.58e−04 | 3.09e−03 | ECM-affiliated proteins | Matrisome associated |
| Collagen α-1 (XII) chain | COL12A1 | 2.31 | 0.0443 | 0.1115 | Collagens | Structural matrisome |
| Collagen α-1 (XXI) chain | COL21A1 | 2.91 | 4.04e−03 | 0.017 | Collagens | Structural matrisome |
| Collagen α-1 (VII) chain | COL7A1 | 3.30 | 0.0462 | 0.1149 | Collagens | Basement membrane |
| Cysteine-rich transmembrane BMP regulator 1 | CRIM1 | 4.58 | 8.05e−12 | 3.23e−10 | ECM glycoproteins | Structural matrisome |
| Chondroitin sulfate proteoglycan 4 | CSPG4 | 1.13 | 6.56e−04 | 2.74e−03 | ECM-affiliated proteins | Matrisome associated |
| Cathepsin B | CTSB | 3.09 | 0.3219 | 0.4759 | ECM regulators | Matrisome associated |
| Cathepsin C | CTSC | 3.61 | 4.21e−07 | 3.41e−06 | ECM regulators | Matrisome associated |
| Cathepsin G | CTSG | 5.67 | 1.52e−07 | 1.36e−06 | ECM regulators | Matrisome associated |
| Cathepsin L | CTSL | 4.96 | 5.06e−08 | ECM regulators | Matrisome associated | |
| ECM protein 1 | ECM1 | 4.10 | 4.31e−05 | 2.24e−04 | ECM glycoproteins | Structural matrisome |
| Neutrophil elastase | ELANE | 4.55 | 4.61e−09 | 5.71e−08 | ECM regulators | Matrisome associated |
| Coagulation factor XII | F12 | 4.15 | 4.21e−05 | 2.21e−04 | ECM regulators | Matrisome associated |
| Fibrillin 2 | FBN2 | 2.26 | 2.09e−07 | 1.76e−06 | ECM glycoproteins | Structural matrisome |
| Fibrinogen-like protein 1 | FGL1 | 4.25 | 1.69e−09 | 3.35e−08 | ECM glycoproteins | Structural matrisome |
| Hydroxysteroid 17-β dehydrogenase 12 | HSD17B12 | 3.09 | 1.12e−04 | 6.69e−04 | ECM-affiliated proteins | Matrisome associated |
| Insulin-like growth factor binding protein acid-labile subunit | IGFALS | >5.0 | 0.1133 | 0.2317 | ECM glycoproteins | Structural matrisome |
| Insulin-like growth factor binding protein 7 | IGFBP7 | 2.77 | 2.71e−09 | 3.64e−08 | ECM glycoproteins | Structural matrisome |
| Inter-α-trypsin inhibitor heavy chain 5 | ITIH5 | 2.85 | 1.09e−03 | 4.25e−03 | ECM regulators | Matrisome associated |
| Kininogen 1 | KNG1 | 1.63 | 5.74e−05 | 2.91e−04 | ECM regulators | Matrisome associated |
| Laminin subunit α 3 | LAMA3 | 2.57 | 2.42e−03 | 0.011 | ECM glycoproteins | Basement membrane |
| Laminin subunit β 1 | LAMB1 | 1.85 | 3.78e−03 | 0.013 | ECM glycoproteins | Basement membrane |
| Galectin 3 | LGALS3 | 2.53 | 4.76e−09 | 5.74e−08 | ECM-affiliated proteins | Matrisome associated |
| Galectin 7 | LGALS7 | 3.67 | 2.62e−06 | 1.72e−05 | ECM-affiliated proteins | Matrisome associated |
| Lectin, mannose binding 1 | LMAN1 | 2.26 | 3.98e−08 | 4.00e−07 | ECM-affiliated proteins | Matrisome associated |
| Matrix metalloproteinase 9 | MMP9 | >5.0 | 0.0978 | 0.2216 | ECM regulators | Matrisome associated |
| Myeloperoxidase | MPO | 3.25 | 1.77e−03 | 6.53e−03 | ECM regulators | Matrisome associated |
| Procollagen-lysine,2-oxoglutarate 5-dioxygenase 3 | PLOD3 | 2.54 | 0.0115 | 0.0353 | ECM regulators | Matrisome associated |
| Proteoglycan 3 | PRG3 | >5.0 | 9.77e−12 | 3.39e−10 | Proteoglycans | Structural matrisome |
| Serine protease 23 | PRSS23 | 4.22 | 6.04e−07 | 4.58e−06 | ECM regulators | Matrisome associated |
| S100 calcium binding protein A8 | S100A8 | 2.18 | 0.0492 | 0.121 | Secreted factors | Matrisome associated |
| S100 calcium binding protein A9 | S100A9 | >5.0 | 2.31e−07 | 1.93e−06 | Secreted factors | Matrisome associated |
| Serpin family D member 1 | SERPIND1 | 4.12 | 3.88e−08 | 3.96e−07 | ECM regulators | Matrisome associated |
| Transthyretin | TTR | 2.88 | 2.18e−04 | 1.20e−03 | ECM regulators | Matrisome associated |
| Serpin family F member 2 | SERPINF2 | 4.18 | 6.47e−09 | 7.65e−08 | ECM regulators | Matrisome associated |
| Uromodulin | UMOD | 4.66 | 5.75e−12 | 2.48e−10 | ECM glycoproteins | Structural matrisome |
| Proteins with greater abundance in FSGS-NOS | ||||||
| Biglycan | BGN | 2.98 | 2.12e−09 | 2.97e−08 | Proteoglycans | Structural matrisome |
| Collagen α-1 (I) chain | COL1A1 | 3.71 | 4.08e−03 | 0.0139 | Collagens | Structural matrisome |
| Collagen α-2 (I) chain | COL1A2 | 2.66 | 4.24e−03 | 0.0143 | Collagens | Structural matrisome |
| Collagen α-1 (XV) chain | COL15A1 | 3.28 | 0.2057 | 0.3533 | Collagens | Basement membrane |
| Collagen α-1 (III) chain | COL3A1 | 1.70 | 0.4772 | 0.6235 | Collagens | Basement membrane |
| Cystatin-C | CST3 | 4.37 | 1.05e−08 | 1.57e−07 | ECM regulators | Matrisome associated |
| Fibulin 5 | FBLN5 | 0.76 | 0.0327 | 0.0859 | ECM glycoproteins | Structural matrisome |
| Filaggrin family member 2 | FLG2 | 1.22 | 0.0161 | 0.0475 | Secreted factors | Matrisome associated |
| HtrA serine peptidase 1 | HTRA1 | 4.10 | 3.58e−06 | 2.29e−05 | ECM regulators | Matrisome associated |
| Inhibin β E chain | INHBE | 4.32 | 6.17e−11 | 2.22e−09 | Secreted factors | Matrisome associated |
| Inter-α-trypsin inhibitor heavy chain H4 | ITIH4 | 1.68 | 3.03e−03 | 0.0131 | ECM regulators | Matrisome associated |
| Lipoprotein(a) | LPA | 1.86 | 1.81e−04 | 8.30e−04 | ECM regulators | Matrisome associated |
| Peroxidasin | PXDN | 3.07 | 5.59e−03 | 0.0227 | ECM glycoproteins | Structural matrisome |
| Serum amyloid A1 | SAA1 | 5.65 | 1.01e−05 | 5.97e−05 | ECM regulators | Matrisome associated |
| Serpin family B member 12 | SERPINB12 | 2.18 | 1.22e−03 | 5.90e−03 | ECM regulators | Matrisome associated |
| Serpin family G member 1 | SERPING1 | 3.34 | 0.8085 | 0.8878 | ECM regulators | Matrisome associated |
| von Willebrand factor | VWF | 1.92 | 0.0118 | 0.0362 | ECM glycoproteins | Structural matrisome |
Validation of ECM Protein Abundance Differences
To validate differences in ECM protein abundance, IHC was performed on a separate cohort of human biopsies diagnosed as cFSGS, FSGS-NOS, and NC for 15 of 41 ECM proteins more abundant in cFSGS: annexin A3, azurocidin, cathepsin B, cathepsin C, cathepsin G, neutrophil elastase, coagulation factor XII, IGF binding protein acid-labile subunit, IGF binding protein 7, kininogen 1, myeloperoxidase, lectin mannose binding 1, proteoglycan 3, serpin D1, and transthyretin. A visually obvious increase in expression and difference in staining pattern for annexin A3, cathepsin C, and cathepsin B were observed (Figure 2). All three proteins stained PECs with normal morphology lining Bowman’s capsule in all three groups of patients. Cells lining Bowman’s capsule with an abnormal morphology characterized by cytoplasmic hyperplasia and enlarged nuclei typical of activated PECs19,32 also stained for the three proteins in glomeruli from patients with cFSGS and FSGS-NOS (Figure 2, D and E). Obvious staining in and around cells infiltrating glomerular tufts was seen in about half of cFSGS glomeruli (Figure 2, J–O). Examples of staining for the other proteins is shown in Supplemental Figure 3.
Figure 2.
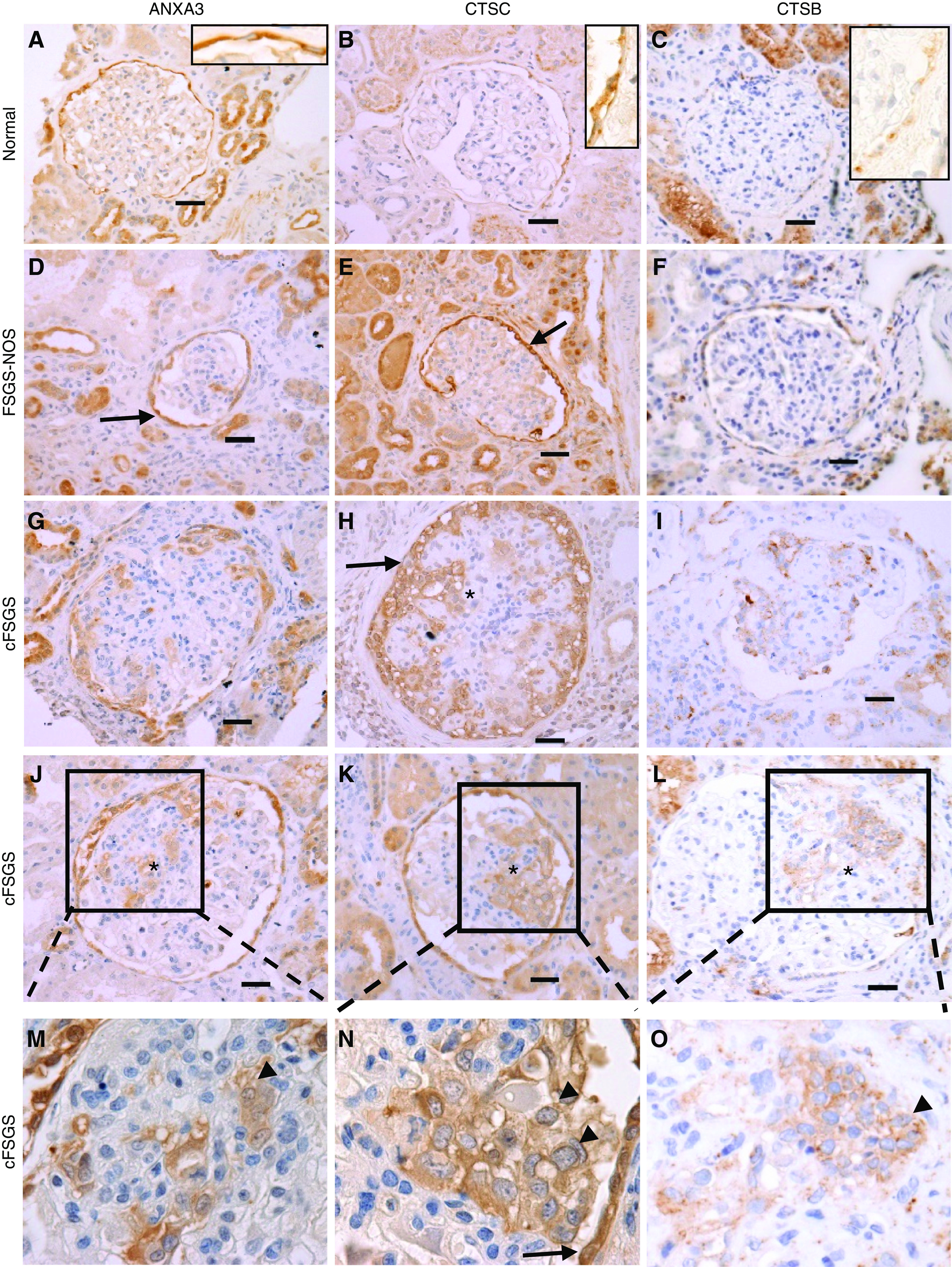
Glomerular tufts stain for annexin A3, cathepsin B, and cathepsin C in cFSGS. Glomerular expression of annexin A3, cathepsin C, and cathepsin B was visualized by IHC. Representative photomicrographs of glomeruli are shown for protein staining in (A–C) NCs, (D–F) FSGS-NOS, and (G–O) cFSGS. Arrows identify PECs along Bowman’s capsule showing hypertrophy and enlarged nuclei. (M–O) Show enlargements of the boxes in (J–L) showing infiltration of glomerular tufts by stained cells (arrowheads). The asterisk marks areas of glomerular tuft staining. Scale bars, 30 μm. Original magnification, 40×.
The staining patterns suggested cathepsin B and cathepsin C were expressed by normal PECs, and their expression was increased in activated PECs. Colocalization of cathepsin C and cathepsin B with annexin A3 was confirmed by confocal microscopy (Figure 3). That colocalization occurred in cells with normal and abnormal morphology lining Bowman’s capsule and in cells within glomerular tufts. To confirm that annexin A3–stained cells expressed markers of PECs, and not podocytes, colocalization of annexin A3 with claudin-1 and synaptopodin was examined. Annexin A3 and claudin-1 extensively colocalized in cells lining Bowman’s capsule and infiltrating glomeruli in cFSGS (Figure 4A). Syndaptopodin did not colocalize to annexin A3–stained cells, whether lining Bowman’s capsule or infiltrating glomeruli (Figure 4B). Taken together, the staining patterns suggest that cells infiltrating glomeruli in cFSGS represent PECs.
Figure 3.

Annexin A3 co-loclaizes with cathepsin C and cathepsin B in FSGS glomeruli. Sections of renal biopsies from patients with (A–C) cFSGS, (D–F) FSGS-NOS, and (G–I) NCs were stained for annexin A3, cathepsin C or cathepsin B, and nuclei. The panels show photomicrographs of single plane confocal images of staining for (A, D, and G) annexin A3 only, for (B, E, and H) cathepsin C or cathepsin B only, and (C, F, and I) merged images. The merged images demonstrate cathepsin C and cathepsin B colocalization with annexin A3. Scale bars, 30 μm. Original magnification, 60×.
Figure 4.

Annexin A3 colocalizes with claudin-1, but not synaptopodin in cFSGS. (A) Sections of renal biopsies from patients with (A–C) cFSGS, (D–F) FSGS-NOS, and (G–I) NCs immunostained for annexin A3, claudin-1, and nuclei stained with 4′,6-diamidino-2-phenylindole. The panels show staining for (A, D, and G) annexin A3 only, for (B, E, and H) claudin-1 only, and (C, F, and I) merged images. The merged images show that annexin A3 and claudin-1 colocalize. Photomicrographs are 1-µm, single-plane images acquired by confocal microscopy. Original magnification of all images, 60×. Scale bars, 30 µm. (B) Sections of a renal biopsy from a patient with cFSGS immunostained for annexin A3, synaptopodin, and nuclei stained with 4′,6-diamidino-2-phenylindole. The panels show staining for (A, D, and G) annexin A3 only, for (B, E, and H) synaptopodin only, and (C, F, and I) merged images, in three different glomeruli from the same patient. The merged images show that annexin A3 and synaptopodin do not colocalize. Photomicrographs are 1-µm, single-plane images acquired by confocal microscopy. Original magnification of all images, 60×. Scale bars, 30 µm.
In addition to altered morphology, PEC activation is associated with ERK1/2-dependent expression of CD44.9,18–20,33,34 To determine if annexin A3–positive cells with abnormal morphology expressed activation markers, IHC for annexin A3, CD44, and pERK was performed on serial cFSGS biopsy sections. Figure 5, A–C, shows that many annexin A3–positive cells with abnormal morphology stained for CD44 and pERK. Additionally, annexin A3–positive cells infiltrating glomerular tufts stained for CD44 (Figure 5, D–I).
Figure 5.
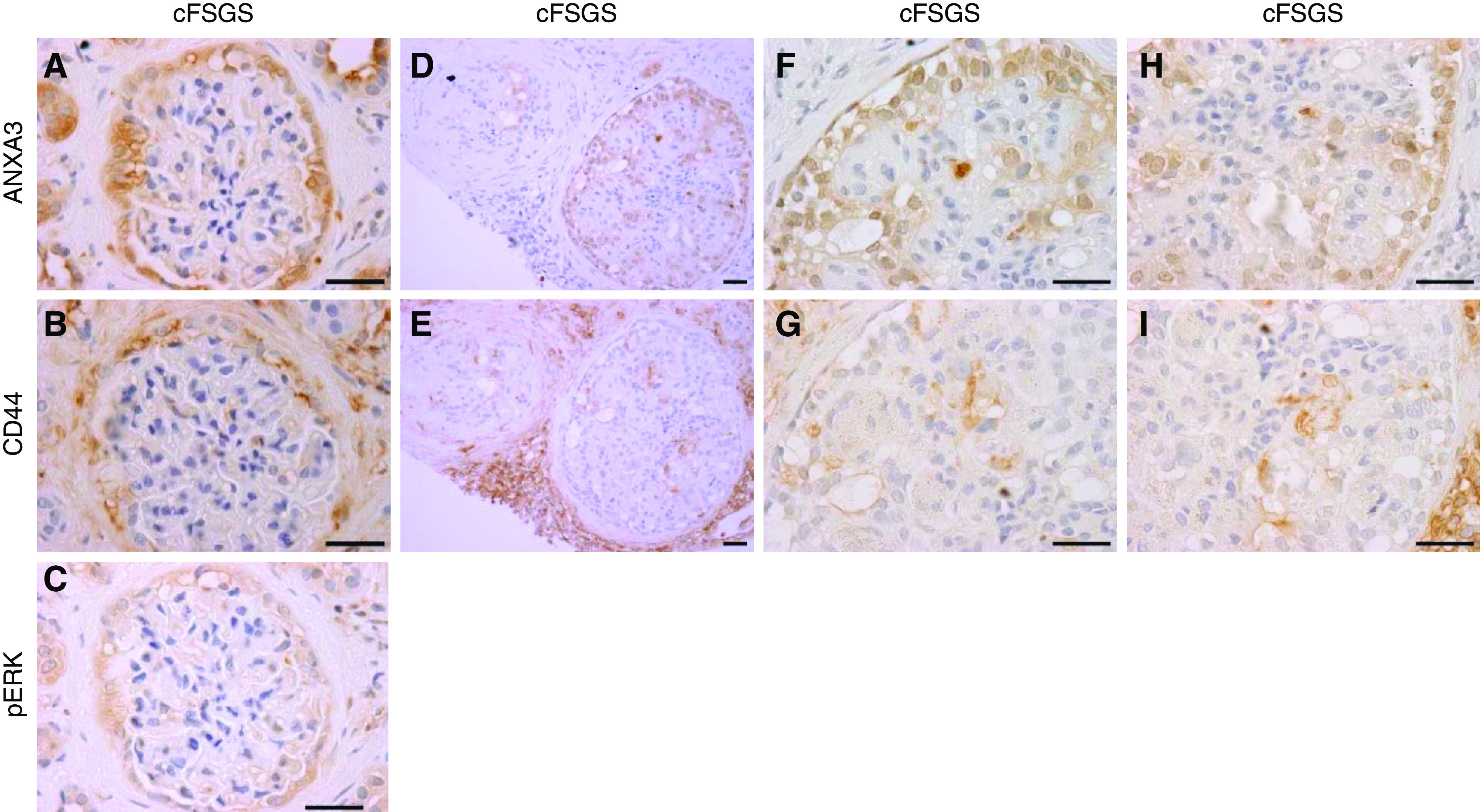
Annexin A3–stained PECs express CD44 and pERK in cFSGS. Photomicrographs show IHC staining patterns for (A) annexin A3, (B) CD44, and (C) pERK in serial sections of a cFSGS glomerulus. PECs showed hypertrophy/hyperplasia, enlarged nuclei, and adhesions to glomerular tufts. Annexin A3, CD44, and pERK showed the same staining pattern. (D and E) Serial sections of a single cFSGS glomerulus with annexin A3–stained PECs infiltrating the glomerular tuft, demonstrating CD44 staining of plasma membranes (E). (F–I) Enlarged portions of the same glomerulus as in (D and E). Scale bars, 30 µm. Original magnification, 100× for (A–C) and (F–I), and 40× for (D and E).
Cathepsin C and Annexin A3 Expression in Proteinuric Glomerular Diseases
Due to limited human kidney biopsy tissue, comparison of protein expression in proteinuric glomerular diseases focused on cathepsin C and annexin A3. IHC was performed on 64 kidney biopsy specimens containing at least four nonsclerotic glomeruli diagnosed with various histologic classes of FSGS, minimal change disease (MCD), membranous nephropathy (MN), or NC. The number of glomeruli evaluated per patient was 12±7 (mean±SD; range, 4–35). Figure 6 illustrates the percentage of glomeruli with staining of normal PECs, staining of PECs with abnormal morphology, and staining of cells infiltrating glomerular tufts for individual patients with each glomerular disease; Figure 7 shows representative photomicrographs. The numeric data are shown in Supplemental Table 4. Biopsies from all patients showed annexin A3 and cathepsin C staining of morphologically normal PECs lining Bowman’s capsule in >80% of glomeruli. Staining for both cathepsin C and annexin A3 of PECs with hyperplastic morphology lining Bowman’s capsule occurred in 35%–65% of glomeruli from patients with glomerular disease and in 10%–25% of glomeruli obtained from patients without known renal disease.
Figure 6.
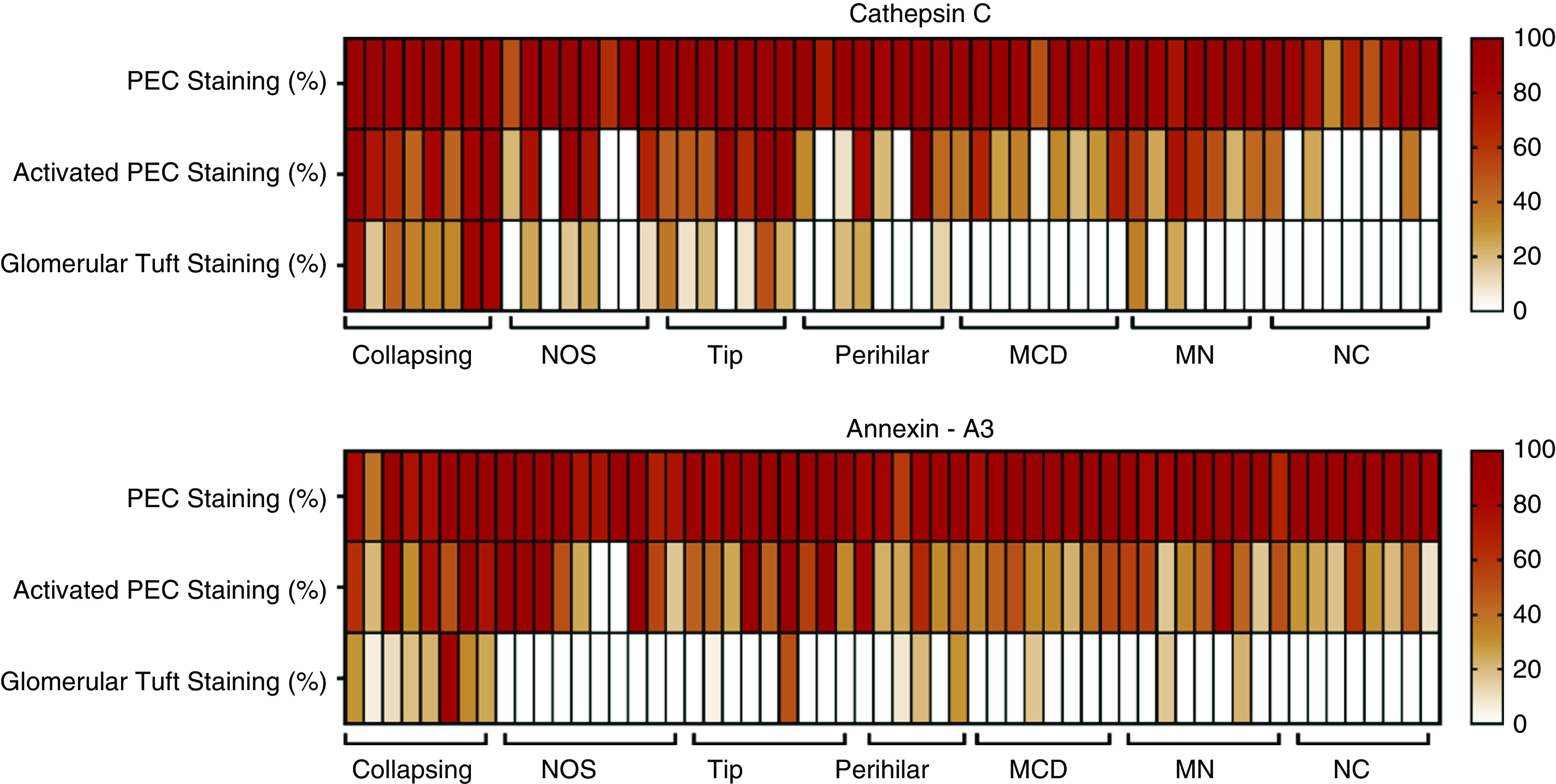
Enhanced annexin A3 and cathepsin C staining in glomerular tufts in cFSGS, compared to other proteinuric glomerular diseases. Heat maps show the percentage of glomeruli with annexin A3 and cathepsin C staining of PECs with normal or hyperplastic morphology and cells infiltrating glomerular tufts for all patients with FSGS variants, MCD, MN, or no glomerular disease (NC).
Figure 7.


Glomerular tuft staining for annexin A3 and cathepsin C is prominent in cFSGS, compared to other proteinuria glomerular diseases. Representative images of IHC staining for annexin A3 and cathepsin C in biopsies from (A and B) NCs, (C and D) cFSGS, (E and F) FSGS-NOS, (G–J) FSGS tip lesion, (K and L) FSGS perihilar lesion, (M and N) MCD, and (O and P) MN. Scale bars, 30 μm. Original magnification, 40×.
All patients with cFSGS exhibited the presence of cells staining for cathepsin C and annexin A3 within glomerular tufts in 51%±10% of glomeruli (mean±SEM; range, 17%–85%). As shown in Figure 7 and Supplemental Table 4, staining for cathepsin C and annexin A3 covered a large portion of glomerular surface area in affected cFSGS glomeruli. Glomerular tuft staining for cathepsin C and annexin A3 in biopsies from other classes of FSGS was much less common and covered a smaller percentage of surface area. Fewer than 20% of glomeruli demonstrated cathepsin C or annexin A3 staining within glomerular tufts of biopsies from patients with FSGS-NOS and perihilar or tip lesions. In the tip lesion, glomerular tuft staining occurred in areas of adhesion at the urinary pole (Figure 7, G–J) and positive glomerular staining in perihilar FSGS was observed near the vascular pole (Figure 7, K and L). Minimal tuft staining was rarely seen in patients diagnosed with MN and MCD and was absent in NCs.
By regression analysis, both the percentage of glomeruli per biopsy with tuft staining for cathepsin C and annexin A3 and the percentage of glomerular surface area staining for cathepsin C were significantly different between cFSGS and other histologic classes of FSGS (P<0.000015). Classification of biopsy glomerular tuft staining by receiver operator characteristic curves for percentage of glomeruli stained separated cFSGS from the other histologic FSGS variants and from all proteinuric glomerular diseases studied (Figure 8). The AUC for percentage of glomeruli stained was 0.916±0.054 (P=0.0006) for cathepsin C (Figure 8A) and 0.918±0.049 (P=0.0005) for annexin A3 (Figure 8B) when cFSGS was compared with other FSGS variants. These classifications were optimal at 29% of glomerular tufts stained for cathepsin C with a resulting sensitivity of diagnosing cFSGS of 87.5% and a specificity to exclude other diseases of 91.3%. For annexin A3, 10% of glomerular tuft staining provided a sensitivity of 87.5% and a specificity of 88%. When compared with FSGS variants, MCD, and MN, the AUC for cathepsin C was 0.947±0.032 (P=0.0001) with a sensitivity of 87.5% and a specificity of 93.8%. For annexin A3, AUC was 0.940±0.031 (P=0.0001) with a sensitivity of 87.5% and a specificity of 88%. Thus, detection of glomerular tuft infiltration by cells containing markers for PECs may improve the sensitivity of biopsy diagnosis of cFSGS.
Figure 8.

Receiver operating characteristic (ROC) curves showing glomerular tuft staining for cathepsin C and annexin A3 predict cFSGS. ROC plots showing predictive capability of the percentage of glomeruli with tuft staining for cathepsin C or annexin A3. (A) The ROC curve for the percentage of glomeruli with cathepsin C staining of glomerular tufts comparing cFSGS to all other variants of FSGS. (B) The ROC curve for percentage of glomeruli with annexin A3 staining of glomerular tufts comparing cFSGS to all other variants of FSGS. (C) The ROC curve for the percentage of glomeruli with cathepsin C staining comparing cFSGS with other FSGS variants, MCD, and MN. (D) The ROC curve for annexin A3 staining comparing cFSGS with other FSGS variants, MCD, and MN. The AUC analysis and P value are in the box accompanying each panel. Patients with cFSGS had a mean±SEM serum creatinine of 3.81±0.67 mg/dl and mean±SEM urine protein excretion of 9.54±1.97 mg/24 h. Patients with the other variants had a mean±SEM serum creatinine of 1.82±0.24 mg/dl and mean±SEM urine protein excretion of 8.1±1.95 gm/24 h.
Evidence for Differences in Synthesis and Degradation of ECM Proteins in FSGS
The differential abundance of glomerular ECM proteins between cFSGS and FSGS-NOS could be due to differences in matrix production and/or degradation. To define the basis for differential ECM protein abundance, gene expression, urine excretion of intact proteins, and urine excretion of peptides from differentially abundant ECM proteins were compared among patients with cFSGS, FSGS-NOS, and NCs. Table 2 shows differences in gene expression between FSGS and NC and between cFSGS and FSGS-NOS for ECM proteins identified as differentially abundant by mass spectrometry. Glomerular gene expression comparing FSGS with NC was performed for 32 of the 58 ECM proteins with differential abundance between cFSGS and FSGS-NOS (Table 2). Transcripts for 16 of 32 genes examined showed a significantly greater expression in FSGS glomeruli, including CTSC.
Table 2.
Transcript analysis for ECM proteins with differential abundance between cFSGS and FSGS-NOS
| Protein Name | Gene Name | Log2 Fold Change (FSGS to NC) | q Value | Log2 Fold Change (cFSGS to FSGS- NOS) | P Value |
|---|---|---|---|---|---|
| Proteins with greater abundance in cFSGS | |||||
| Annexin A3 | ANXA3 | 1.13 | 0.070 | 0.80 | 0.068 |
| Apolipoprotein C3 | APOC3 | 0.61 | 0.030 | −0.72 | 0.091 |
| Azurocidin | AZU1 | 1.03 | 0.380 | 0.122 | 0.852 |
| Complement C1q A chain | C1QA | ND | 0.30 | 0.473 | |
| Complement C1q B chain | C1QB | ND | 0.28 | 0.556 | |
| Complement C1q C chain | C1QC | ND | 0.40 | 0.089 | |
| Collagen α-1 (XII) chain | COL12A1 | ND | 0.40 | 0.034 | |
| Collagen α-1 (XXI) chain | COL21A1 | ND | −0.19 | 0.527 | |
| Collagen α-1(VII) chain | COL7A1 | 1.28 | 0.001 | 0.07 | 0.505 |
| Cysteine-rich transmembrane BMP regulator 1 | CRIM1 | 0.86 | 0.001 | −0.10 | 0.524 |
| Chondroitin sulfate proteoglycan 4 | CSPG4 | 1.07 | 0.080 | 0.00 | 1.000 |
| Cathepsin B | CTSB | ND | 0.54 | 0.043 | |
| Cathepsin C | CTSC | 1.5 | 0.001 | 0.54 | 0.252 |
| Cathepsin G | CTSG | 0.96 | 0.190 | 0.29 | 0.722 |
| Cathepsin L | CTSL | ND | −0.20 | 0.166 | |
| ECM protein 1 | ECM1 | 2.67 | 0.001 | −0.10 | 0.834 |
| Neutrophil elastase | ELANE | ND | 0.17 | 0.577 | |
| Coagulation factor XI | F12 | 0.94 | 0.090 | −0.06 | 0.858 |
| Fibrillin 2 | FBN2 | 1.35 | 0.001 | 0.26 | 0.373 |
| Fibrinogen-like protein 1 | FGL1 | ND | ND | ||
| Hydroxysteroid 17-β dehydrogenase 12 | HSD17B12 | ND | 0.40 | 0.047 | |
| Insulin-like growth factor binding protein acid labile | IGFALS | 0.92 | 0.030 | 0.07 | 0.704 |
| Insulin-like growth factor binding protein 7 | IGFBP7 | 1.06 | 0.070 | −0.20 | 0.483 |
| Inter-α-trypsin inhibitor heavy chain 5 | ITIH5 | ND | −0.29 | 0.363 | |
| Kininogen 1 | KNG1 | 0.71 | 0.001 | 0.27 | 0.614 |
| Laminin subunit α 3 | LAMA3 | ND | 0.31 | 0.162 | |
| Laminin subunit β 1 | LAMB1 | 1.11 | 0.160 | 0.13 | 0.447 |
| Galectin 3 | LGALS3 | ND | 0.52 | 0.025 | |
| Galectin 7 | LGALS7 | ND | −0.39 | 0.924 | |
| Lectin, mannose binding 1 | LMAN1 | 0.88 | 0.010 | 0.00 | 1.000 |
| Matrix metalloproteinase 9 | MMP9 | ND | −0.10 | 0.799 | |
| Myeloperoxidase | MPO | ND | −0.08 | 0.880 | |
| Procollagen-lysine,2-oxoglutarate 5-dioxygenase 3 | PLOD3 | 1.13 | 0.001 | 0.84 | 0.090 |
| Proteoglycan 3 | PRG3 | 0.95 | 0.080 | ND | |
| Serine protease 23 | PRSS23 | 1.48 | 0.001 | 1.15 | 0.050 |
| S100 calcium binding protein | S100A8 | 0.91 | 0.320 | −0.35 | 0.412 |
| S100 calcium binding protein | S100A9 | 0.88 | 0.290 | −0.31 | 0.344 |
| Serpin family D member 1 | SERPIND1 | 1.13 | 0.060 | −0.35 | 0.224 |
| Transthyretin | TTR | ND | −0.25 | 0.619 | |
| Serpin family F member 2 | SERPINF2 | ND | 0.00 | 1.000 | |
| Uromodulin | UMOD | 0.69 | 0.001 | −0.50 | 0.510 |
| Proteins with greater abundance in FSGS-NOS | |||||
| Biglycan | BGN | 1.2 | 0.020 | −0.02 | 0.560 |
| Collagen α-1 (I) chain | COL1A1 | 1.49 | 0.001 | 1.80 | 0.004 |
| Collagen α-2 (I) chain | COL1A2 | 2.82 | 0.001 | 0.70 | 0.160 |
| Collagen α-1 (XV) chain | COL15A1 | 1.94 | 0.001 | 0.09 | 0.824 |
| Collagen α-1 (III) chain | COL3A1 | ND | 0.20 | 0.640 | |
| Cystatin-C | CST3 | ND | 0.30 | 0.409 | |
| Fibulin 5 | FBLN5 | 0.95 | 0.230 | −0.30 | 0.389 |
| Filaggrin family member 2 | FLG2 | ND | 0.00 | 1.000 | |
| HtrA serine peptidase 1 | HTRA1 | 0.97 | 0.280 | −0.75 | 0.236 |
| Inhibin β E chain | INHBE | ND | 0.05 | 0.990 | |
| Inter-α-trypsin inhibitor heavy chain H4 | ITIH4 | ND | −0.53 | 0.642 | |
| Lipoprotein(a) | LPA | 0.93 | 0.070 | −0.36 | 0.465 |
| Peroxidasin | PXDN | ND | 1.00 | 0.015 | |
| Serum amyloid A1 | SAA1 | ND | −0.14 | 0.827 | |
| Serpin family B member 12 | SERPINB12 | ND | ND | ||
| Serpin family G member 1 | SERPING1 | 1.05 | 0.160 | 0.5 | 0.044 |
| von Willebrand factor | VWF | 1.00 | 0.280 | 0.10 | 0.680 |
ND, not determined.
To determine if there was gene expression differential abundance of ECM proteins between cFSGS and FSGS-NOS, RNA-seq–based transcriptomic data obtained from the NEPTUNE database (https://www.rarediseasesnetwork.org/cms/neptune) were analyzed for patients with cFSGS (n=11) and FSGS-NOS (n=21). Due to the small sample size, unadjusted P values were used to rank order transcript differential abundance between groups. Table 2 shows that, of the 38 genes observed for ECM proteins more abundant in cFSGS, five genes showed greater expression in cFSGS at the P<0.05 level, including CTSB. Of the 15 genes observed for ECM proteins more abundant in FSGS-NOS, three genes showed greater expression at the P<0.05 level.
As additional support for increased production of ECM proteins in cFSGS, quantitative urine excretion of ECM proteins was determined by mass spectrometry in three patients each with cFSGS and FSGS-NOS, and the relative abundances were compared. A total of 93 ECM proteins showed a significantly greater abundance in urine from patients with cFSGS, whereas 21 ECM proteins were significantly more abundant in urine from FSGS-NOS patients (Supplemental Table 5). Only eight of the 93 ECM proteins more abundant in cFSGS urine were also more abundant in glomeruli, including cathepsin B and cathepsin C. On the other hand, seven ECM proteins more abundant in cFSGS urine were present in greater abundance in FSGS-NOS glomeruli. Similarly, three of 21 ECM proteins more abundant in urine from patients with FSGS-NOS were present in greater abundance in cFSGS glomeruli. Thus, urine excretion of ECM proteins appears to be a poor predictor of glomerular ECM composition. Because both cathepsin B and C were present in tubular cells (Figure 3), enhanced urine excretion cannot be conclusively linked to glomerular abundance of those proteins.
To screen for degradation of ECM proteins by cathepsin B and C in cFSGS glomeruli, excretion of urine peptides of <5000 kDa was quantified for each of three patients with cFSGS, FSGS-NOS, and MCD. MCD was used as the control to account for filtered peptides due to increased glomerular basement membrane permeability and degradation of filtered serum proteins by tubule cells. A total of 902 peptides from 152 unique proteins was identified, 45 of which were ECM proteins represented by 365 peptides. Comparing the three patients with MCD to all six patients with FSGS, 34 peptides derived from 25 ECM proteins showed significantly enhanced abundance in urine from patients with FSGS (Supplemental Table 6). Of the 35 peptides with significantly different abundance between cFSGS and FSGS-NOS, all showed greater abundance in cFSGS. To examine the potential role of cathepsin B and cathepsin C in ECM degradation in cFSGS, protease prediction with Proteasix (www.proteasix.org) was performed for urine peptides from ECM proteins, only accepting proteases with experimentally observed cleavage sites. No peptides with cleavage sites for cathepsin C were identified. A total of eight peptides with cleavage sites for cathepsin B were identified, none of which showed differential urine excretion. Peptides with cathepsin cleavage sites and increased abundance in urine from patients with cFSGS were derived from cathepsin D or cathepsin L. A list of all protease groups predicted to be responsible for cleavage sites on urine ECM proteins is shown in Supplemental Table 7.
Discussion
We performed a proteomic and transcriptomic analysis of human ECM constituents to gain an understanding of the differences in ECM remodeling that distinguishes cFSGS from other FSGS variants. Significant differences were identified in cFSGS glomerular ECM composition. Proteomic analysis found 41 ECM proteins with increased abundance in cFSGS and 17 with increased abundance in FSGS-NOS. Those proteins included members of all categories of matrisome proteins, including basement membrane, structural, ECM-affiliated, and ECM regulators. IHC and confocal microscopy confirmed the enhanced expression and colocalization of three proteins—cathepsin B, cathepsin C, and annexin A3—in cells lining Bowman’s capsule and infiltrating capillary tufts in cFSGS. Those three proteins were not seen in normal glomerular tufts and were uncommon in glomerular tufts from other FSGS variants, MCD, and MN.
Although the origin of cells infiltrating glomerular tufts in cFSGS cannot be stated with certainty, substantial evidence indicates they are activated PECs. A number of reports indicated a contribution of PECs to the development of FSGS in humans and animal models, including their proliferation in cFSGS and increased synthesis of ECM.9,18,19,32,33 Consistent with a previous study of human FSGS,9 we found that infiltrating cells expressed markers of PECs (claudin-1 and annexin A3), whereas a marker of podocytes (synaptopodin) was absent. PEC activation depends on ERK1/2-mediated expression of CD44, an adhesion receptor for hyaluronic acid and other ECM proteins.18,19,32,33,35–37 Colocalization of annexin A3, CD44, and pERK1/2 in our study is also consistent with a PEC origin of cells infiltrating cFSGS glomeruli. Both cathepsin B and C were detected in PECs of normal glomeruli, and both proteases were present in infiltrating cells. Finally, lineage tracing studies in mice show PECs contribute to proliferating epithelial cells within glomeruli in mouse models of FSGS.18,38 The basis for PEC migration into glomerular tufts in cFSGS remains to be determined. Because CD44 is a receptor for a number of ECM components, it is possible that altered glomerular ECM composition in cFSGS contributes to PEC migration into glomeruli.
Migration of PECs into glomerular tufts introduced two proteases, cathepsin B and cathepsin C, not normally present. Our study suggests that PEC activation increases expression of both cathepsins. Glomerular transcript expression for cathepsin C was increased in FSGS, compared with NC. Transcript expression for cathepsin B was increased in cFSGS, compared with FSGS-NOS. IHC staining for both proteases was more intense in PECs with hyperplastic morphology. Additionally, urine proteomic analysis showed significantly greater urinary excretion of both cathepsins in patients with FSGS, compared with those with no renal disease. Those findings are consistent with single nephron proteomics performed in two mouse models of FSGS showing that cathepsin B was significantly increased in glomeruli.39 A potential role for cathepsin B in mediating FSGS was suggested by the resistance of cathepsin B knockout mice to nephrotoxic serum-induced proteinuria.39
Cathepsin B and C are members of a family of 11 cysteine proteases.40 Cathepsins are normally present in high concentrations in lysosomes, where they are crucial for protein processing and degradation.41 Cathepsins are also secreted into the extracellular space, where they participate in ECM remodeling, receptor and adhesion molecule shedding, activation of cytokines and growth factors, generation of bioactive fragments from ECM proteins, cleavage of cell junction proteins, and cell migration.12–14,41–43 Cathepsin B degrades a number of basement membrane proteins, including laminins, and degrades intact glomerular basement membrane.41,44 In silico protease prediction, however, failed to identify increased urine excretion of peptides generated by either cathepsin. Those findings suggest the possibility that increased glomerular expression of cathepsin B and C function differently from ECM remodeling in cFSGS. Several cathepsins, including cathepsin B, differentially regulate TGFβ signaling that is linked to ECM production. Altered cathepsin activity was observed in unilateral ureteral obstruction–mediated renal fibrosis, and inhibition of cathepsin activity altered the degree of fibrosis.45 As pharmacologic inhibitors of specific cathepsins are being developed,46 defining the role of cathepsins in cFSGS could provide new therapeutic approaches.
Only five of 38 transcripts from proteins with increased abundance in cFSGS showed increased expression. In addition to cathepsin B, a transcript for PRSS23 was among those increased. Two previous studies showed increased gene expression for PRSS23 in FSGS.47,48 Increased expression of PRSS23 in PECs was associated with proteinuria and loss of renal function in FSGS, lupus nephritis, diabetic nephropathy, and MN,49 and evidence from a mouse model of FSGS suggested a role for PRSS23 in the development of a migratory PEC phenotype.49 The absence of increased gene expression for the majority of differentially abundant ECM proteins suggests that transcriptional regulation of protein synthesis is not responsible for most differences in ECM protein abundance in FSGS. Differential ECM protein abundance could be due to altered degradation rates or to increased translational regulation of ECM synthesis, as has been described in idiopathic pulmonary fibrosis.50 Differences in urinary peptide excretion derived from ECM proteins in our study indicate ECM degradation is increased in cFSGS. Because the ECM peptides were not derived from the proteins showing differential abundance, it is possible those proteins are more resistant to proteolysis.
A number of limitations to our study exist. Heterogeneity of glomerular changes in FSGS limits identification of the focal and local changes in ECM composition. Because our proteomic approach combined sections from 12 to 26 glomeruli from each patient, our data present average differences in ECM composition. The application of proteomics to single glomeruli with defined histologic changes will improve detection of altered ECM protein expression. Confirmation studies that visualize the protein of interest are needed to determine the pattern of expression within abnormal glomeruli. It is possible that IHC validation of our proteomic findings from Triton X-100–insoluble pellets failed due to differences in sample handling. The inability to perform lineage tracing studies in humans resulted in using expression of markers presumed to be specific for PECs and podocytes to identify the cell type infiltrating glomeruli in cFSGS. Marker expression, coupled with previous animal studies, justifies the preliminary conclusion that infiltrating cells are primarily composed of PECs. The possibility exists, however, that dedifferentiation of podocytes results in altered expression of the markers used in this study. The use of urine protein and peptide excretion to reflect ECM expression and protease activity in glomeruli has a number of limitations. Blood could be the source of proteins and peptides in the urine due to altered glomerular filtration barrier in FSGS. That possibility was addressed by using urine from MCD as a control for urine peptide excretion. Although 114 ECM proteins showed differential urine excretion between cFSGS and FSGS-NOS, only 18 of those proteins were differentially abundant in glomeruli. Additionally, ten of those 18 ECM proteins showed increased urine excretion in the patient group with a relative decrease in glomerular abundance. The expression of cathepsin B and C by renal tubule cells provides another source for increased urine excretion of those proteases. We conclude that technical and biologic considerations limit the ability of urine excretion of ECM proteins to predict glomerular ECM composition. Similarly, urinary peptide excretion may not accurately reflect the level of glomerular ECM degradation due to peptide processing or reabsorption by renal tubules. The predictive algorithms for protease cleavage sites may miss noncanonical amino acid sequences identified by cathepsins, because candidate proteases were not identified for 40% of peptides with differential urine excretion.
In summary, a proteomic analysis of glomerular ECM composition shows distinct differences between cFSGS and FSGS-NOS, suggesting the mechanisms of abnormal ECM remodeling differ. Our data indicate that both enhanced ECM protein synthesis and altered degradation contribute to abnormal ECM remodeling in FSGS. Extensive glomerular tuft infiltration of cells with an activated PEC phenotype differentiates cFSGS from other FSGS variants and other proteinuric glomerular diseases. Those cells introduce a new set of proteases, cathepsins B and C, into glomeruli. Evidence from animal studies suggests those proteases contribute to development of FSGS. Whereas podocyte injury likely initiates FSGS, our data support the concept that PEC activation and migration determine disease severity. Targeting PECs for therapeutic intervention may alter disease progression, particularly in cFSGS. Multiple pathways leading to focal sclerotic lesions complicate identification of therapeutic targets for disruption of ECM deposition in FSGS. Our data point to cathepsin proteases as possible candidates for pharmacologic intervention.
Disclosures
D. Caster reports being site principal investigator for clinical trials from Aurinia, Calliditas, Mallinckrodt, and Retrophin and being on the advisory board for Retrophin. M. Kretzler reports nonfinancial support from the University of Michigan and has a patent PCT/EP2014/073413 “Biomarkers and methods for progression prediction for CKD” issued. J. Wetzels reports support and honoraria from Achillion, Chemocentryx, Shire, and Virfor Pharma. All remaining authors have nothing to disclose.
Funding
NEPTUNE is a part of the NIH Rare Disease Clinical Research Network, supported through a collaboration between the Office of Rare Diseases Research, National Center for Advancing Translational Sciences, and the NIDDK, grant U54‐DK‐083912. Additional funding and/or programmatic support for this project was also provided by the University of Michigan, NephCure Foundation, and the Halpin Foundation. This project was supported by NIH grant R01DK110077 to J. Klein; NIH grant R01DK091584 to M. Merchant; NIH grant 1K08DK102542 to D. Caster; ZonMw AGIKO grant 92003587 to I. Rood; the James Y. McCullough Award Fund to M Merchant, M. Barati, and K. McLeish; and the National Center for Advancing Translational Sciences grant UL1TR002240 for the Michigan Institute for Clinical and Health Research. L. Mariani reports grants from Boehringer Ingelheim, Complexa Inc., Goldfinch Bio, NIDDK, during the conduct of the study.
Supplementary Material
Acknowledgments
We would like to thank all members of the NEPTUNE consortium listed below.
From the NEPTUNE enrolling centers: J. Sedor (principal investigator), K. Dell (principal investigator), M. Schachere (study coordinator), and J. Negrey (study coordinator) at Cleveland Clinic (Cleveland, OH); K. Lemley (principal investigator) and S. Tang (study coordinator) at Children’s Hospital Los Angeles (Los Angeles, CA); T. Srivastava (principal investigator) and A. Garrett (study coordinator) at Children’s Mercy Hospital (Kansas City, MO); C. Sethna (principal investigator) and R. Odusayana (study coordinator) at Cohen Children’s Hospital (New Hyde Park, NY); G. Appel (principal investigator) and M. Toledo (study coordinator) at Columbia University (New York, NY); L. Barisoni (principal investigator) at Duke University (Durham, NC); L. Greenbaum (principal investigator), C. Wang (coinvestigator), and B. Lee (study coordinator) at Emory University (Atlanta, GA); S. Adler (principal investigator), C. Nast (principal investigator; Cedars-Sinai Medical Center, Los Angeles, CA), and J. LaPage (study coordinator) at Harbor-University of California Los Angeles Medical Center; A. Athavale (principal investigator) at John H. Stroger Jr. Hospital of Cook County (Chicago, IL); A. Neu (principal investigator) and S. Boynton (study coordinator) at Johns Hopkins Medicine (Baltimore, MD); F. Fervenza (principal investigator), M. Hogan (coinvestigator), J. Lieske (principal investigator), and V. Chernitskiy (study coordinator) at Mayo Clinic (Rochester, MN); F. Kaskel (principal investigator), N. Kumar (principal investigator), and P. Flynn (study coordinator) at Montefiore Medical Center (Bronx, NY); J. Kopp (principal investigator) and J. Blake (study coordinator) at National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) Intramural (Bethesda, MD); H. Trachtman (principal investigator), O. Zhdanova (coinvestigator), F. Modersitzki (study coordinator), and S. Vento (study coordinator) at New York University Medical Center (New York, NY); R. Lafayette (principal investigator) and K. Mehta (study coordinator) at Stanford University (Stanford, CA); C. Gadegbeku (principal investigator), D. Johnstone (coinvestigator), and S. Quinn-Boyle (study coordinator) at Temple University (Philadelphia, PA); D. Cattran (principal investigator), M. Hladunewich (coinvestigator), H. Reich (coinvestigator), P. Ling (study coordinator), and M. Romano (study coordinator) at University Health Network Toronto; A. Fornoni (principal investigator) and C. Bidot (study coordinator) at University of Miami (Miami, FL); M. Kretzler (principal investigator), D. Gipson (principal investigator), A. Williams (study coordinator), and J. LaVigne (study coordinator) at University of Michigan (Ann Arbor, MI); V. Derebail (principal investigator), K. Gibson (principal investigator), E. Cole (study coordinator), and S. Grubbs (study coordinator) at University of North Carolina (Chapel Hill, NC); L. Holzman (principal investigator), K. Meyers (coinvestigator), K. Kallem (study coordinator), and J. Lalli (study coordinator) at University of Pennsylvania (Philadelphia, PA); K. Sambandam (principal investigator), Z. Wang (study coordinator), and F. Chambers (study coordinator) at University of Texas Southwestern (Dallas, TX); A. Jefferson (principal investigator), S. Hingorani (coinvestigator), K. Tuttle (coinvestigator; Providence Medical Research Center, Spokane, WA), N. Johnson (study coordinator), S. Dismuke (study coordinator), and A. Cooper (study coordinator; Providence Medical Research Center, Spokane, WA) at University of Washington (Seattle, WA); and B. Freedman (principal investigator), J.J. Lin (coinvestigator), and S. Gray (study coordinator) at Wake Forest University Baptist Health (Winston-Salem, NC).
From the data analysis and coordinating center: M. Kretzler, L. Barisoni, C. Gadegbeku, B. Gillespie, D. Gipson, L. Holzman, L. Mariani, M. Sampson, P. Song, J. Troost, J. Zee, E. Herreshoff, S. Li, C. Lienczewski, T. Mainieri, M. Wladkowski, A. Williams, and D. Zinsser.
From the digital pathology committee: Carmen Avila-Casado (University Health Network Toronto), Serena Bagnasco (Johns Hopkins), Joseph Gaut (Washington University), Stephen Hewitt (National Cancer Institute), Jeff Hodgin (University of Michigan), Kevin Lemley (Children’s Hospital Los Angeles), Laura Mariani (University of Michigan), Matthew Palmer (University of Pennsylvania), Avi Rosenberg (NIDDK), Virginie Royal (Montreal), David Thomas (University of Miami), Jarcy Zee (Arbor Research), and cochairs Laura Barisoni (Duke University) and Cynthia Nast (Cedar Sinai).
From the NIDDK program office: C. Roy and S. Mendley.
From the National Center for Advancing Translational Sciences program office: T. Urv and P.J. Brooks.
Dr. Kenneth McLeish and Dr. Michael Merchant conceived and designed the study; Dr. Michelle Barati, Dr. Liliane Hobeika, Dr. Jeffrey Hodgin, Dr. Jon Bernard Klein, Dr. Matthias Kretzler, Ms. Ming Li, Dr. Laura Mariani, Dr. Michael Merchant, Dr. Johnathan Troost, and Mr. Daniel Wilkey performed and interpreted experiments; Dr. Susan Coventry, Dr. Jessica Hata, and Dr. Christopher Patrick Larsen evaluated and provided anonymous renal biopsy material; Dr. Dawn Caster, Dr. Jeroen Deegens, Dr. Ilse Rood, and Dr. Jack Wetzels collected and provided anonymous urine samples; Dr. Michael Brier performed statistical analyses; Dr. Michelle Barati and Dr. Michael Merchant prepared figures; Dr. Michelle Barati, Dr. Michael Merchant, and Dr. Kenneth McLeish wrote the manuscript; Dr. Michael Brier, Dr. Jon Bernard Klein, Dr. Matthias Kretzler, Dr. Liliane Hobeika, and Dr. Johnathan Troost assisted in editing the manuscript.
Dr. Dawn Caster reports grant support from Aurinia and Mallinckrodt; personal fees from Retrophin, outside the submitted work. Dr. Jeffrey Hodgin reports grant support from Complexa Inc., Goldfinch Bio, and Pfizer Inc., outside the submitted work. Dr. Matthias Kretzler reports grants from AstraZeneca, Boehringer Ingelheim, Certa, Chan Zuckerberg Initiative, Complexa Inc., Eli Lilly, Elpidera, European Union Innovative Medicine Initiative, Gilead, Goldfinch Bio, Janssen, JDRF, Merck, NovoNordisc, and Pfizer Inc., outside the submitted work. Dr. Laura Mariani reports grants from Pfizer Inc., and personal fees from Reata Pharmaceuticals, outside the submitted work. Dr. Jonathan P. Troost reports grant support from Complexa Inc., Goldfinch Bio, and Pfizer Inc.; other from General Electric; and other from Procter & Gamble, outside the submitted work. Dr. Jack Wetzels reports personal fees from Novartis; personal fees from Retrophin; and personal fees from Shire, outside the submitted work.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
Data Sharing Statement
Data files for acquired LCMS data (.RAW), for peak lists (.mgf), and compressed search results (.mzIdentML) were deposited in MassIVE (http://massive.ucsd.edu/) data repository (MassIVE identifier, MSV000079914) of the Center for Computational Mass Spectrometry at the University of California, San Diego and shared with the ProteomeXchange (www.proteomexchange.org). The data files for ERCB microarray data are available at Gene Expression Omnibus (GEO; www.ncbi.nlm.nih.gov/geo/) under GEO numbers GSE47183, GSE32591, and GSE104948.
Supplemental Material
This article contains the following supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2019070696/-/DCSupplemental.
Supplemental Figure 1. Laser capture of glomeruli excludes Bowman’s capsule.
Supplemental Figure 2. Comparison of ECM protein enrichment by extraction method.
Supplemental Figure 3. Immunohistochemistry of cFSGS, FSGS-NOS, and NC for selected glomerular ECM proteins.
Supplemental Table 1. All glomerular proteins identified from cFSGS, FSGS-NOS, and NC. (Mass spectrometry data contained in a supplemental spreadsheet.)
Supplemental Table 2. Gene names of glomerular ECM proteins identified in NC, FSGS-NOS, and cFSGS.
Supplemental Table 3. Relative abundance of glomerular ECM proteins.
Supplemental Table 4. Glomerular cathepsin and annexin A3 staining of various proteinuric glomerular diseases.
Supplemental Table 5. Comparison of urinary excretion of ECM proteins.
(Mass spectrometry data contained in a supplemental spreadsheet.)
Supplemental Table 6. Peptides derived from ECM proteins showing increased urine excretion. (Mass spectrometry data contained in a supplemental spreadsheet.)
Supplemental Table 7. Proteases associated with urine ECM peptides.
References
- 1.Rosenberg AZ, Kopp JB: Focal segmental glomerulosclerosis. Clin J Am Soc Nephrol 12: 502–517, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sim JJ, Bhandari SK, Batech M, Hever A, Harrison TN, Shu YH, et al.: End-stage renal disease and mortality outcomes across different glomerulonephropathies in a large diverse US population. Mayo Clin Proc 93: 167–178, 2018. [DOI] [PubMed] [Google Scholar]
- 3.D’Agati VD, Fogo AB, Bruijn JA, Jennette JC: Pathologic classification of focal segmental glomerulosclerosis: A working proposal. Am J Kidney Dis 43: 368–382, 2004. [DOI] [PubMed] [Google Scholar]
- 4.Stokes MB, D’Agati VD: Morphologic variants of focal segmental glomerulosclerosis and their significance. Adv Chronic Kidney Dis 21: 400–407, 2014. [DOI] [PubMed] [Google Scholar]
- 5.Laurin L-P, Gasim AM, Derebail VK, McGregor JG, Kidd JM, Hogan SL, et al.: Renal survival in patients with collapsing compared with not otherwise specified FSGS. Clin J Am Soc Nephrol 11: 1752–1759, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Raja R, Nada R, Yadav AK, Kumar A, Goyal A, Kumar V, et al.: A prospective study of collapsing focal segmental glomerulosclerosis. Ren Fail 38: 894–898, 2016. [DOI] [PubMed] [Google Scholar]
- 7.Tsuchimoto A, Matsukuma Y, Ueki K, Tanaka S, Masutani K, Nakagawa K, et al.: Utility of Columbia classification in focal segmental glomerulosclerosis: Renal prognosis and treatment response among the pathological variants [published online ahead of print January 11, 2019]. Nephrol Dial Transplant [DOI] [PubMed] [Google Scholar]
- 8.Albaqumi M, Soos TJ, Barisoni L, Nelson PJ: Collapsing glomerulopathy. J Am Soc Nephrol 17: 2854–2863, 2006. [DOI] [PubMed] [Google Scholar]
- 9.Dijkman H, Smeets B, van der Laak J, Steenbergen E, Wetzels J: The parietal epithelial cell is crucially involved in human idiopathic focal segmental glomerulosclerosis. Kidney Int 68: 1562–1572, 2005. [DOI] [PubMed] [Google Scholar]
- 10.Dijkman HB, Weening JJ, Smeets B, Verrijp KC, van Kuppevelt TH, Assmann KK, et al.: Proliferating cells in HIV and pamidronate-associated collapsing focal segmental glomerulosclerosis are parietal epithelial cells. Kidney Int 70: 338–344, 2006. [DOI] [PubMed] [Google Scholar]
- 11.Theocharis AD, Skandalis SS, Gialeli C, Karamanos NK: Extracellular matrix structure. Adv Drug Deliv Rev 97: 4–27, 2016. [DOI] [PubMed] [Google Scholar]
- 12.Fonović M, Turk B: Cysteine cathepsins and extracellular matrix degradation. Biochim Biophys Acta 1840: 2560–2570, 2014. [DOI] [PubMed] [Google Scholar]
- 13.Menou A, Duitman J, Crestani B: The impaired proteases and anti-proteases balance in Idiopathic Pulmonary Fibrosis. Matrix Biol 68–69: 382–403, 2018. [DOI] [PubMed] [Google Scholar]
- 14.Ricard-Blum S, Vallet SD: Fragments generated upon extracellular matrix remodeling: Biological regulators and potential drugs. Matrix Biol 75–76: 170–189, 2019. [DOI] [PubMed] [Google Scholar]
- 15.Vizovišek M, Fonović M, Turk B: Cysteine cathepsins in extracellular matrix remodeling: Extracellular matrix degradation and beyond. Matrix Biol 75–76: 141–159, 2019. [DOI] [PubMed] [Google Scholar]
- 16.Hobeika L, Barati MT, Caster DJ, McLeish KR, Merchant ML: Characterization of glomerular extracellular matrix by proteomic analysis of laser-captured microdissected glomeruli. Kidney Int 91: 501–511, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lennon R, Byron A, Humphries JD, Randles MJ, Carisey A, Murphy S, et al.: Global analysis reveals the complexity of the human glomerular extracellular matrix. J Am Soc Nephrol 25: 939–951, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smeets B, Kuppe C, Sicking EM, Fuss A, Jirak P, van Kuppevelt TH, et al.: Parietal epithelial cells participate in the formation of sclerotic lesions in focal segmental glomerulosclerosis. J Am Soc Nephrol 22: 1262–1274, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kuppe C, Gröne HJ, Ostendorf T, van Kuppevelt TH, Boor P, Floege J, et al.: Common histological patterns in glomerular epithelial cells in secondary focal segmental glomerulosclerosis. Kidney Int 88: 990–998, 2015. [DOI] [PubMed] [Google Scholar]
- 20.Roeder SS, Barnes TJ, Lee JS, Kato I, Eng DG, Kaverina NV, et al.: Activated ERK1/2 increases CD44 in glomerular parietal epithelial cells leading to matrix expansion. Kidney Int 91: 896–913, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chan GC, Eng DG, Miner JH, Alpers CE, Hudkins K, Chang A, et al.: Differential expression of parietal epithelial cell and podocyte extracellular matrix proteins in focal segmental glomerulosclerosis and diabetic nephropathy. Am J Physiol Renal Physiol 317: F1680–F1694, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Merchant ML, Perkins BA, Boratyn GM, Ficociello LH, Wilkey DW, Barati MT, et al.: Urinary peptidome may predict renal function decline in type 1 diabetes and microalbuminuria. J Am Soc Nephrol 20: 2065–2074, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Srivastava S, Merchant M, Rai A, Rai SN: Standardizing proteomics workflow for liquid chromatography-mass spectrometry: Technical and statistical considerations. J Proteomics Bioinform 12: 48–55, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Klein J, Eales J, Zürbig P, Vlahou A, Mischak H, Stevens R: Proteasix: A tool for automated and large-scale prediction of proteases involved in naturally occurring peptide generation. Proteomics 13: 1077–1082, 2013. [DOI] [PubMed] [Google Scholar]
- 25.Barati MT, Powell DW, Kechavarzi BD, Isaacs SM, Zheng S, Epstein PN, et al.: Differential expression of endoplasmic reticulum stress-response proteins in different renal tubule subtypes of OVE26 diabetic mice. Cell Stress Chaperones 21: 155–166, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, et al.: Proteomics. Tissue-based map of the human proteome. Science 347: 1260419, 2015. [DOI] [PubMed] [Google Scholar]
- 27.Naba A, Clauser KR, Ding H, Whittaker CA, Carr SA, Hynes RO: The extracellular matrix: Tools and insights for the “omics” era. Matrix Biol 49: 10–24, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gadegbeku CA, Gipson DS, Holzman LB, Ojo AO, Song PX, Barisoni L, et al.: Design of the Nephrotic Syndrome Study Network (NEPTUNE) to evaluate primary glomerular nephropathy by a multidisciplinary approach. Kidney Int 83: 749–756, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ju W, Nair V, Smith S, Zhu L, Shedden K, Song PXK, et al. ; ERCB, C-PROBE, NEPTUNE, and PKU-IgAN Consortium : Tissue transcriptome-driven identification of epidermal growth factor as a chronic kidney disease biomarker. Sci Transl Med 7: 316ra193, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bray NL, Pimentel H, Melsted P, Pachter L: Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol 34: 525–527, 2016. [DOI] [PubMed] [Google Scholar]
- 31.Randles MJ, Humphries MJ, Lennon R: Proteomic definitions of basement membrane composition in health and disease. Matrix Biol 57–58: 12–28, 2017. [DOI] [PubMed] [Google Scholar]
- 32.Smeets B, Stucker F, Wetzels J, Brocheriou I, Ronco P, Gröne HJ, et al.: Detection of activated parietal epithelial cells on the glomerular tuft distinguishes early focal segmental glomerulosclerosis from minimal change disease. Am J Pathol 184: 3239–3248, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smeets B, Te Loeke NA, Dijkman HBPM, Steenbergen MLM, Lensen JFM, Begieneman MPV, et al.: The parietal epithelial cell: A key player in the pathogenesis of focal segmental glomerulosclerosis in Thy-1.1 transgenic mice. J Am Soc Nephrol 15: 928–939, 2004. [DOI] [PubMed] [Google Scholar]
- 34.Eymael J, Sharma S, Loeven MA, Wetzels JF, Mooren F, Florquin S, et al.: CD44 is required for the pathogenesis of experimental crescentic glomerulonephritis and collapsing focal segmental glomerulosclerosis. Kidney Int 93: 626–642, 2018. [DOI] [PubMed] [Google Scholar]
- 35.Roeder SS, Stefanska A, Eng DG, Kaverina N, Sunseri MW, McNicholas BA, et al.: Changes in glomerular parietal epithelial cells in mouse kidneys with advanced age. Am J Physiol Renal Physiol 309: F164–F178, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aruffo A, Stamenkovic I, Melnick M, Underhill CB, Seed B: CD44 is the principal cell surface receptor for hyaluronate. Cell 61: 1303–1313, 1990. [DOI] [PubMed] [Google Scholar]
- 37.Goodison S, Urquidi V, Tarin D: CD44 cell adhesion molecules. Mol Pathol 52: 189–196, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kuppe C, Leuchtle K, Wagner A, Kabgani N, Saritas T, Puelles VG, et al.: Novel parietal epithelial cell subpopulations contribute to focal segmental glomerulosclerosis and glomerular tip lesions. Kidney Int 96: 80–93, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Höhne M, Frese CK, Grahammer F, Dafinger C, Ciarimboli G, Butt L, et al.: Single-nephron proteomes connect morphology and function in proteinuric kidney disease. Kidney Int 93: 1308–1319, 2018. [DOI] [PubMed] [Google Scholar]
- 40.Rossi A, Deveraux Q, Turk B, Sali A: Comprehensive search for cysteine cathepsins in the human genome. Biol Chem 385: 363–372, 2004. [DOI] [PubMed] [Google Scholar]
- 41.Vidak E, Javoršek U, Vizovišek M, Turk B: Cysteine cathepsins and their extracellular Roles: Shaping the microenvironment. Cells 8: E264, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tong B, Wan B, Wei Z, Wang T, Zhao P, Dou Y, et al.: Role of cathepsin B in regulating migration and invasion of fibroblast-like synoviocytes into inflamed tissue from patients with rheumatoid arthritis. Clin Exp Immunol 177: 586–597, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wolters PJ, Laig-Webster M, Caughey GH: Dipeptidyl peptidase I cleaves matrix-associated proteins and is expressed mainly by mast cells in normal dog airways. Am J Respir Cell Mol Biol 22: 183–190, 2000. [DOI] [PubMed] [Google Scholar]
- 44.Baricos WH, Cortez SL, Le QC, Zhou YW, Dicarlo RM, O’Connor SE, et al.: Glomerular basement membrane degradation by endogenous cysteine proteinases in isolated rat glomeruli. Kidney Int 38: 395–401, 1990. [DOI] [PubMed] [Google Scholar]
- 45.Zhang X, Zhou Y, Yu X, Huang Q, Fang W, Li J, et al.: Differential roles of cysteinyl cathepsins in TGF-β signaling and tissue fibrosis. iScience 19: 607–622, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Korkmaz B, Caughey GH, Chapple I, Gauthier F, Hirschfeld J, Jenne DE, et al.: Therapeutic targeting of cathepsin C: From pathophysiology to treatment. Pharmacol Ther 190: 202–236, 2018. [DOI] [PubMed] [Google Scholar]
- 47.Hodgin JB, Borczuk AC, Nasr SH, Markowitz GS, Nair V, Martini S, et al.: A molecular profile of focal segmental glomerulosclerosis from formalin-fixed, paraffin-embedded tissue. Am J Pathol 177: 1674–1686, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bennett MR, Czech KA, Arend LJ, Witte DP, Devarajan P, Potter SS: Laser capture microdissection-microarray analysis of focal segmental glomerulosclerosis glomeruli. Nephron, Exp Nephrol 107: e30–e40, 2007. [DOI] [PubMed] [Google Scholar]
- 49.Harder JL, Menon R, Otto EA, Zhou J, Eddy S, Wys NL, et al. ; European Renal cDNA Bank (ERCB); Nephrotic Syndrome Study Network (NEPTUNE) : Organoid single cell profiling identifies a transcriptional signature of glomerular disease. JCI Insight 4: e122697, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Parker MW, Rossi D, Peterson M, Smith K, Sikström K, White ES, et al.: Fibrotic extracellular matrix activates a profibrotic positive feedback loop. J Clin Invest 124: 1622–1635, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.