

Significance Statement
Assessing a drug in a disease model more closely replicates the clinical situation if standard drugs are included in the study design. In a progressive-stage mouse model of obesity-related type 2 diabetes, bromoindirubin-3′-oxime (BIO) as an add-on to dual renin-angiotensin system (RAS)/sodium-glucose transporter (SGLT)-2 inhibition with metformin, ramipril, and empagliflozin showed remarkable effects. Quantitative end point analysis included the slope of measured GFR and filtration slit ultrastructure. Add-on BIO attenuated GFR decline by further reducing glomerulosclerosis, increasing podocyte numbers through sustaining specialization as well as inducing de novo differentiation from podocyte progenitors, and improving filtration slit density. The findings are a proof-of-concept for testing novel drugs for renoprotective effects beyond dual RAS/SGLT2 inhibition for diabetic kidney disease.
Keywords: glomerulosclerosis, diabetic nephropathy, translational research, diabetic kidney disease, regeneration
Abstract
Background
Progression of CKD in type 2 diabetes, despite dual inhibition of sodium-glucose transporter-2 and the renin-angiotensin system, remains a concern. Bromoindirubin-3′-oxime (BIO), previously reported to promote podocyte survival and regeneration, is a candidate additional drug to elicit renoprotective effects beyond therapy with metformin, ramipril, and empagliflozin (MRE). Evaluating a drug with standard therapeutics more closely mimics the clinical setting than evaluating the drug alone.
Methods
Uninephrectomized BKS-Lepr−/− (db/db) mice treated with or without MRE served as a model of progressive CKD in type 2 diabetes. Mice on or off MRE were randomized to only 4 weeks of add-on BIO or vehicle. The primary end point was slope of GFR (ΔGFR).
Results
Four weeks of MRE treatment alone did not affect ΔGFR, but significantly attenuated hyperglycemia, albuminuria, and glomerulosclerosis and increased podocyte filtration slit density, as assessed by STED super-resolution microscopy upon tissue clearing. BIO alone improved albuminuria, podocyte density in superficial and juxtamedullary nephrons, and podocyte filtration slit density. MRE+BIO combination therapy had additive protective effects on ΔGFR, glomerulosclerosis, podocyte density in juxtamedullary nephrons, and filtration slit density.
Conclusions
Add-on treatment with BIO for only 4 weeks attenuates progression of CKD beyond MRE therapy in mice with type 2 diabetes. Additional drug combinations may help to further delay ESKD in type 2 diabetes.
The epidemic of obesity and type 2 diabetes mellitus (T2DM) is a global health concern because diabetes complications such as CKD, blindness, foot ulcers, and cardiovascular disease imply disabling morbidity, mortality, and enormous health care costs.1,2 Dual inhibition of sodium-glucose transporter (SGLT)-2 and the renin-angiotensin system (RAS) in patients with T2DM can profoundly reduce cardiovascular events and the progression of CKD.3–7 Indeed, dual inhibition of SGLT2/RAS entirely abrogated any further decline of the GFR in patients with T2DM and early CKD, but only attenuated GFR decline in those with more advanced CKD.4,6 Thus, drugs that further attenuate or even reverse the residual GFR loss despite dual SGLT2/RAS inhibition are still needed for patients with T2DM.
Translational research activities for this prevalent segment of patients are challenging because most animal models mimic only very early phases of diabetic nephropathy. Animal models are selected for convenience, for their histomorphological lesions, or for albuminuria,8 but not for (1) CKD stage G2–4, (2) progressive GFR loss, or (3) obesity-related T2DM, patients who keep being recruited into clinical trials. A model addressing some of these issues and that can predict the outcome of clinical trials is on the basis of Lepr−/− (db/db) mice with obesity-related T2DM and surgical nephron reduction.9–13 However, any preclinical study omitting standard-of-care therapy ignores the fact that the standard of care may modulate the relative significance of a putative novel therapeutic target for disease progression. Thus, novel compounds should first demonstrate efficacy beyond therapy with antidiabetic drugs and dual RAS/SGLT2 inhibition in rodents before proceeding to clinical trials.
Podocyte loss is a critical pathomechanism in the progression of CKD in patients with diabetes because its consequences (macroproteinuria, glomerular scarring, and irreversible nephron loss) translate into GFR decline. The reasons for podocyte loss are manifold. RAS- and SGLT2-driven mechanisms imply increased shear stress to podocytes, and the robust nephroprotective effects of dual inhibition of RAS/SGLT2 ultimately prove the significance of podocyte stress in humans.14 A multitude of other molecular pathways affect podocyte malfunction and loss in diabetic kidney disease, e.g., disturbed insulin, Notch, or mammalian target of rapamycin signaling, altered proteostasis and autophagy, and glomerular inflammation, as well as bypassing cell cycle checkpoints toward mitotic catastrophe, to name just a few.15–19
Therefore, targeting podocyte malfunction and loss may be an appealing strategy as add-on treatment for patients with progressive diabetic kidney disease. Indeed, numerous studies have documented drug candidates with podocyte-protective effects in in vitro and in vivo models of podocyte injury, but rigorous testing of podocyte-directed therapies in a model mimicking closely the clinical scenario of residual progression of diabetic kidney disease beyond standard therapy is not yet available. Here, we focused on (2'Z,3′E)-6-bromoindirubin-3′-oxime (BIO), a compound modulating a number of the aforementioned molecular pathways,20 to endorse podocyte viability and function in vitro, and podocyte de novo formation and terminal differentiation in vivo.21–23 Altogether, we hypothesized that the particular renoprotective effects of BIO would attenuate CKD progression beyond metformin, ramipril, and empagliflozin (MRE) therapy in obese db/db mice with T2DM and progressive GFR decline.
CONCISE METHODS
Animals
Five-week-old BKS.Cg-m+/+Leprdb/BomTac mice were obtained from Taconic (Ry, Denmark). Diabetic mice homozygous for the loss-of-function mutation in the leptin receptor served as experimental group (db/db) and respective age-matched nondiabetic mice served as controls [wild-type (WT)]. Animals were housed in filter-top cages with a 12-hour dark/light cycle and unlimited access to food and water, and animal welfare was monitored throughout the duration of the study. Mice were terminated ahead of schedule when meeting predefined welfare scores, and were considered as dropouts in the survival analysis. The regional governmental authorities approved the experimental protocol on the basis of the European Union directive for the Protection of Animals Used for Scientific Purposes (2010/63/EU).
Study Design
The study design is described in detail in Supplemental Table 1. To achieve nephron reduction mimicking the aging- and injury-related nephron loss from clinical patients, animals underwent uninephrectomy (db/db 1K, n=43; WT, n=23) (Figure 1A). Surgery was performed at 6 weeks of age as described previously.13 All mice received a sodium-deficient diet (sodium <0.03%, E15430–244, ssniff Spezialdiäten GmbH, Soest, Germany) right upon surgery (T0) and all along the experiment as an attempt to further enhance glomerular hyperfiltration-related CKD progression as described (Figure 1A).24 At the age of 20 weeks (T12 of experiment), some db/db 1K mice were sacrificed for baseline histologic analysis (n=6). The rest of the animals in the db/db 1K group were maintained for further experiments (n=37). Group size calculation was on the basis of GFR as a primary end point and quantitative assumptions obtained from our previous studies.10,25,26 At this point, db/db 1K and WT mice were allocated by stratified randomization to MRE therapy, BIO treatment, or no treatment (groups and group sizes are detailed in Figure 2A, Supplemental Table 1). MRE therapy included 1500 mg/kg of metformin, 6 mg/kg of ramipril and 480 mg/kg of empagliflozin 12.5%. BIO (2 µmol/kg) was administered subcutaneously 5 days/wk. Additionally, a subgroup of animals treated with MRE was randomly selected for add-on treatment with BIO (Figure 5A, Supplemental Table 1). Kidney tissue was harvested for the evaluation of histopathology and gene expression at the end of the experiment (week 16). BIO’s safety analysis included histopathology and gene expression studies on the heart, pancreas, sciatic nerve, liver, skeletal muscle, and epididymal white, inguinal white, and brown adipose tissue upon collecting tissue samples at the end of the experiment. The procedures are described in detail in the Supplemental Methods.
Figure 1.
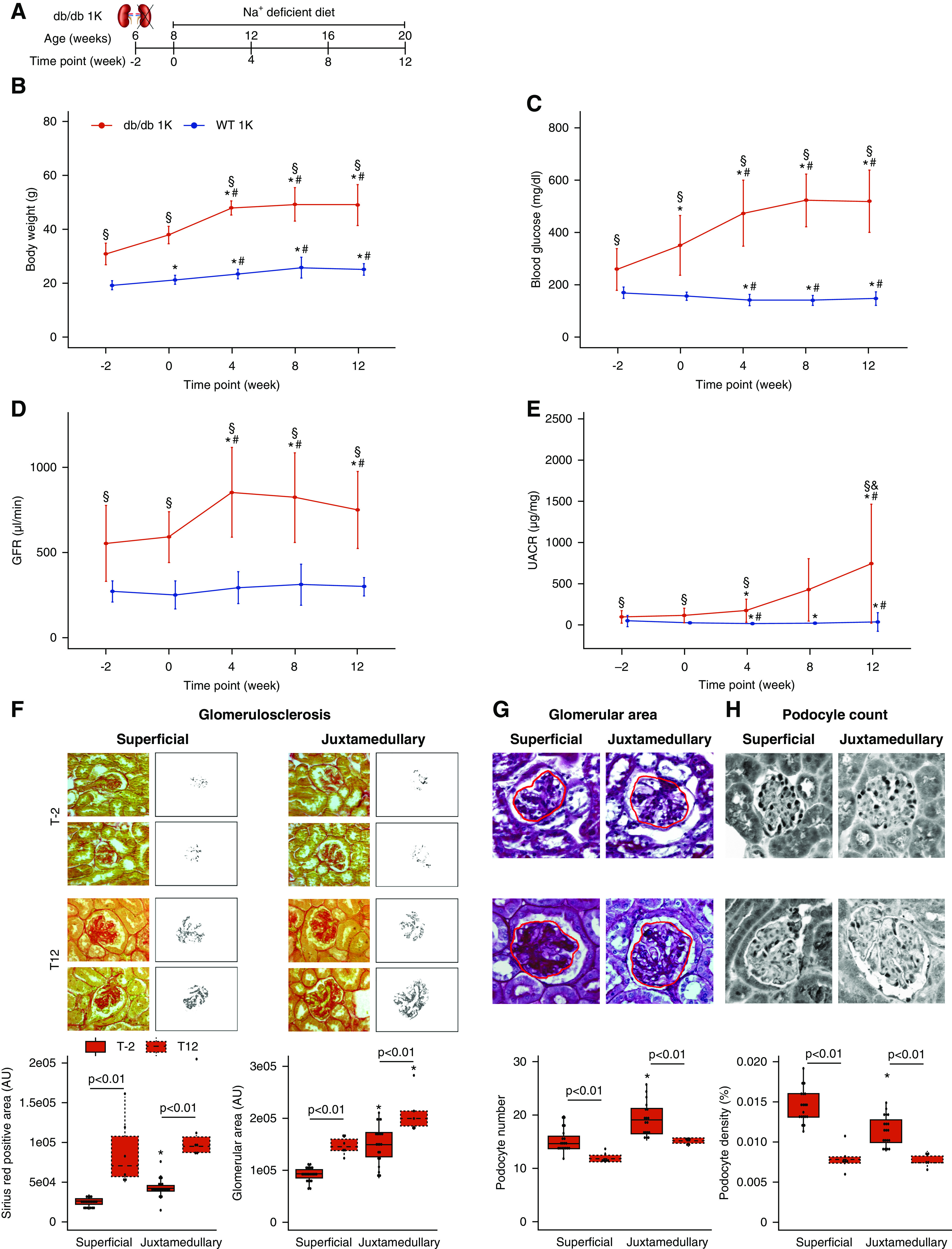
Establishment of a model of progressive glomerulosclerosis in male db/db mice with type 2 diabetes. (A) Schematic representation of surgeries in male BKS db/db. Mice underwent uninephrectomy of the right kidney at 6 weeks of age and were fed a sodium-deficient diet until 20 weeks of age (time point 12). (B) Body weight evolution, expressed as mean±SD (db/db 1K (n=34) and WT 1K (n=23)). (C) Blood glucose evolution, expressed as mean±SD (db/db 1K (n=34) and WT 1K (n=23)). (D) GFR evolution, expressed as mean±SD (db/db 1K (n=34) and WT 1K (n=23)). (E) UACR evolution, expressed as mean±SD (db/db 1K (n=26) and WT 1K (n=18)). (F) Glomerulosclerosis in db/db 1K mice, measured as Picro-Sirius Red0positive area (AU). Magnification, ×40. (G) Glomerular area assessment in db/db 1K mice, measured as the glomerular tuft area (AU) in PAS sections. Magnification, ×40. (H) Podocyte number assessment in WT-1 sections from db/db 1K mice. Podocyte density was calculated as the number of podocytes by glomerular tuft area section. Magnification, ×40. For all histologic analyses (F–H), the average number of glomeruli quantified at each time point is specified in Supplemental Table 2. *P value≤0.05 versus time point−2; #P value≤0.05 versus time point 0; §P value≤0.05 versus WT 1K. Statistical analyses performed are summarized in Supplemental Table 4. AU, arbitrary units.
Figure 2.
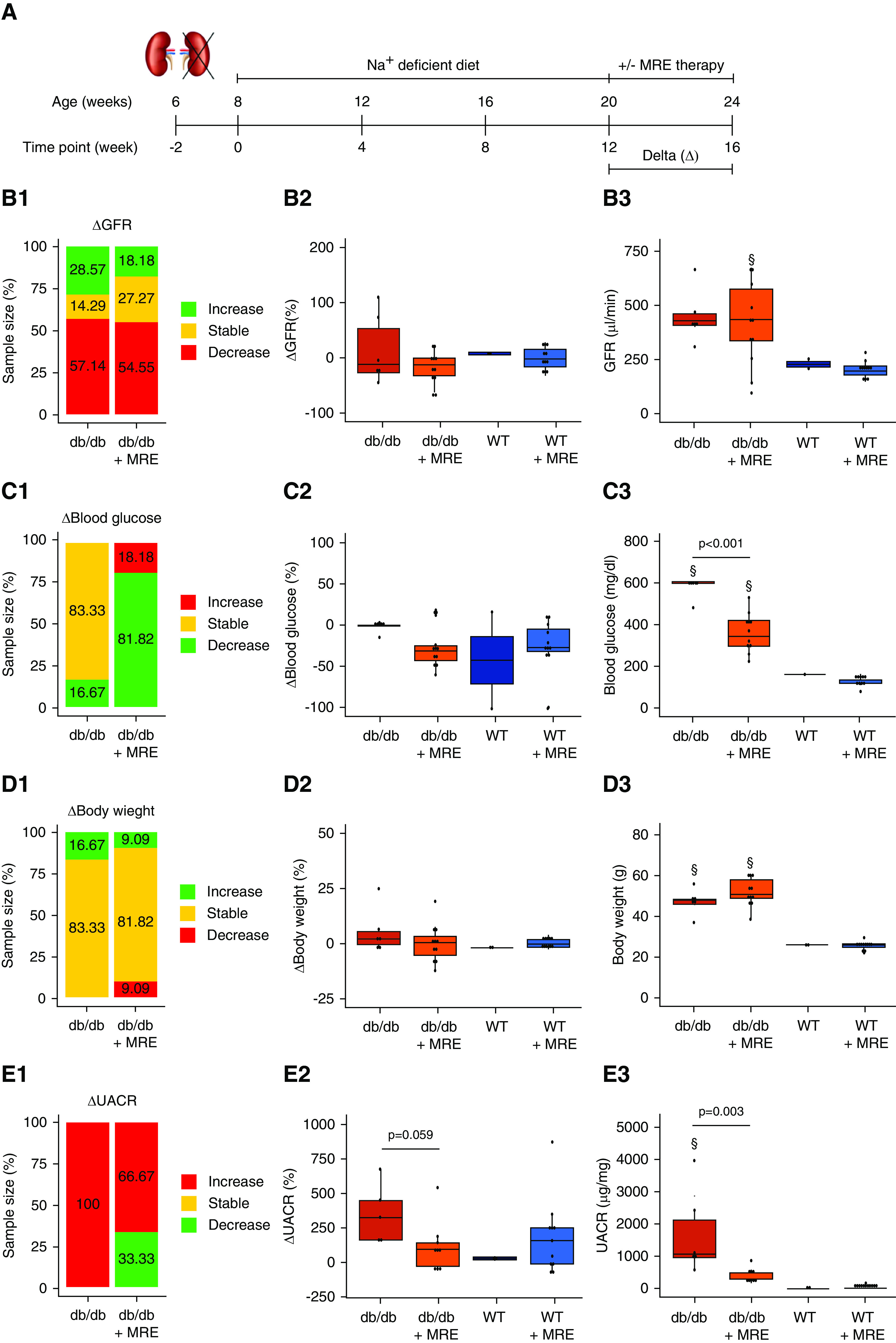
Effect of MRE therapy on primary and secondary outcomes. (A) Schematic representation of the experiment to assess the effect of MRE therapy versus no therapy. Uninephrectomized mice were treated with a sodium-deficient diet or MRE therapy (included in the sodium-deficient diet) from weeks 12 to 16. (B) GFR assessment. (B1) ΔGFR classification, expressed as the variation in GFR from weeks 12 to 16. Mice were classified as having an increase in GFR (>10%, green), a stable GFR (±10%, yellow), or a decrease (<10%, red) between the two time points indicated. The percentage of mice in each of these groups is specified in the stacked bar graph. (B2) ΔGFR values, expressed as the percentage of variation in GFR from weeks 12 to 16. (B3) GFR at end point (week 16). (C) Blood glucose assessment. (C1) Δ Blood glucose classification, expressed as the variation in blood glucose levels from weeks 12 to 16. Mice were classified as having an increase in blood glucose (>10%, red), stable levels (±10%, yellow), or a decrease (<10%, green) between the two time points indicated. The percentage of mice in each of these groups is specified in the stacked bar graph. (C2) Δ Blood glucose values, expressed as the percentages of variation in blood glucose from weeks 12 to 16. (C3) Blood glucose levels at end point (week 16). (D) Body weight assessment. (D1) Δ Body weight classification, expressed as the variation in body weight from weeks 12 to 16. Mice were classified as having an increase in body weight (>10%, green), stable weight (±10%, yellow), or a decrease (<10%, red) between the two time points indicated. The percentage of mice in each of these groups is specified in the stacked bar graph. (D2) Δ Body weight values, expressed as the percentage of variation in body weight from weeks 12 to 16. (D3) Body weight at end point (week 16). (E) UACR assessment. (E1) ΔUACR classification, expressed as the variation in UACR from weeks 12 to 16. Mice were classified as having an increase in UACR (>10%, red), stable levels (±10%, yellow), or a decrease (<10%, green) between the two time points indicated. The percentage of mice in each of these groups is specified in the stacked bar graph. (E2) ΔUACR values, expressed as the percentage of variation in UACR from weeks 12 to 16. (E3) UACR levels at end point (week 16). §P value≤0.05 versus respective WT group. Statistical analyses performed are summarized in Supplemental Table 4.
Figure 5.
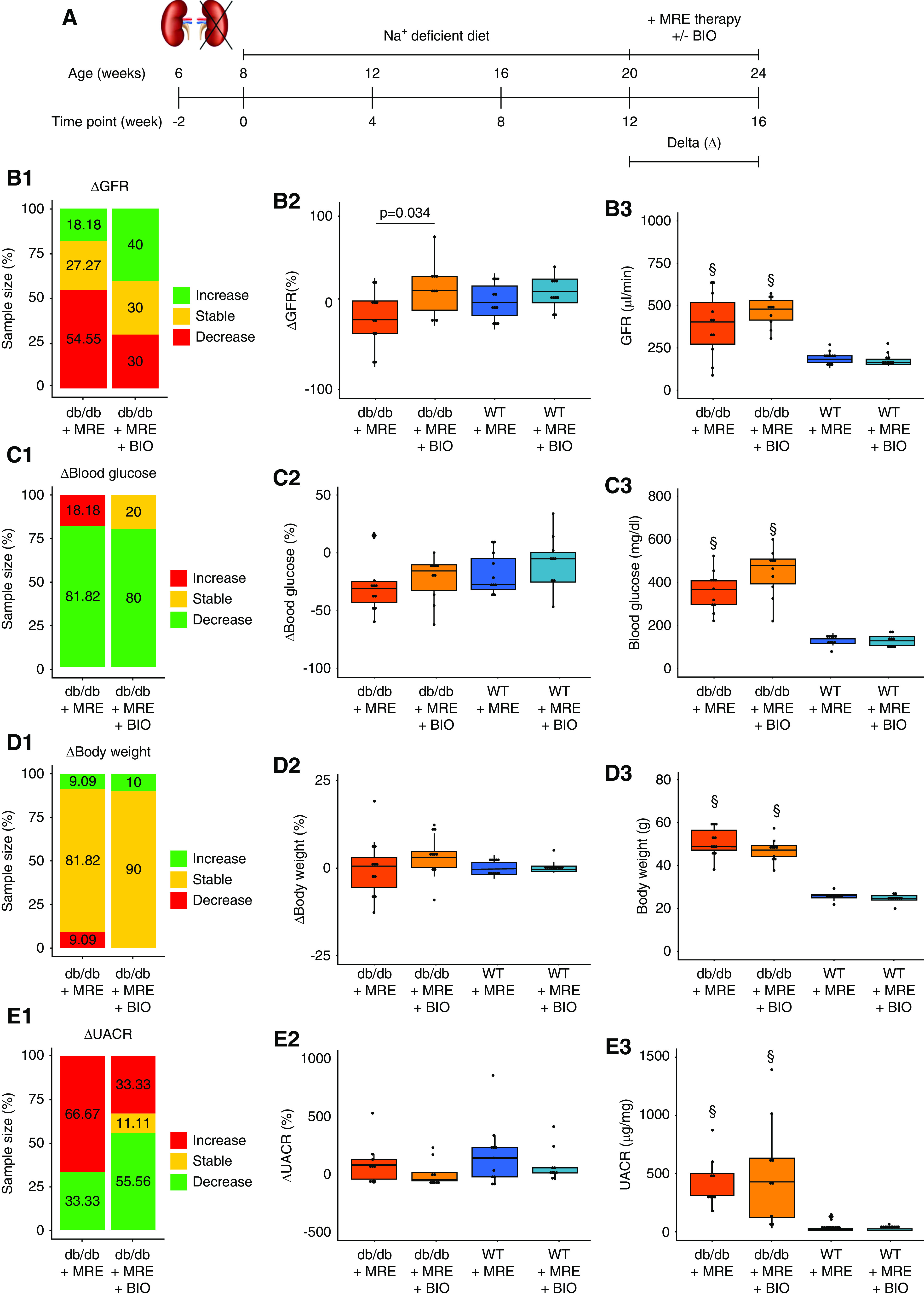
Effect of combined MRE+BIO therapy on primary and secondary outcomes. (A) Schematic representation of the experiment to assess the effect of combined MRE+BIO therapy versus MRE therapy alone. Uninephrectomized mice were treated with MRE therapy (included in the sodium-deficient diet) or MRE+BIO (2 µmol/kg, subcutaneously) from weeks 12 to 16. (B) GFR assessment. (B1) ΔGFR classification, expressed as the variation in GFR from weeks 12 to 16. Mice were classified as having an increase in GFR (>10%, green), a stable GFR (±10%, yellow), or a decrease (<10%, red) between the two time points indicated. The percentage of mice in each of these groups is specified in the stacked bar graph. (B2) ΔGFR values, expressed as the percentage of variation in GFR from weeks 12 to 16. (B3) GFR at end point (week 16). (C) Blood glucose assessment. (C1) Δ Blood glucose classification, expressed as the variation in blood glucose levels from weeks 12 to 16. Mice were classified as having an increase in blood glucose (>10%, red), stable levels (±10%, yellow), or a decrease (<10%, green) between the two time points indicated. The percentage of mice in each of these groups are specified in the stacked bar graph. (C2) Δ Blood glucose values, expressed as the percentage of variation in blood glucose from weeks 12 to 16. (C3) Blood glucose levels at end point (week 16). (D) Body weight assessment. (D1) Δ Body weight classification, expressed as the variation in body weight from weeks 12 to 16. Mice were classified as having an increase in body weight (>10%, green), stable weight (±10%, yellow), or a decrease (<10%, red) between the two time points indicated. The percentage of mice in each of these groups is specified in the stacked bar graph. (D2) Δ Body weight values, expressed as the percentage of variation in body weight from weeks 12 to 16. (D3) Body weight at end point (week 16). (E) UACR assessment. (E1) ΔUACR classification, expressed as the variation in UACR from weeks 12 to 16. Mice were classified as having an increase in UACR (>10%, red), stable levels (±10%, yellow), or a decrease (<10%, green) between the two time points indicated. The percentage of mice in each of these groups is specified in the stacked bar graph. (E2) ΔUACR values, expressed as the percentage of variation in UACR from weeks 12 to 16. (E3) UACR levels at end point (week 16). §P value≤0.05 versus respective WT group. Statistical analyses performed are summarized in Supplemental Table 4.
Primary End Point
The primary end point was the slope of GFR (ΔGFR). For GFR measurement, mice were anesthetized with isoflurane, and a miniaturized imager device built from two light-emitting diodes, a photodiode, and a battery (MediBeacon, Mannheim, Germany) was mounted via double-sided adhesive tape onto each shaved animal’s neck. Animals were conscious in a single cage during the recording (approximately 1.5 hour). Before the intravenous injection of 150 mg/kg FITC-Sinistrin (MediBeacon, Mannheim, Germany), the skin’s background signal was recorded for 5 minutes. After removing the imager device, the data were analyzed using Mannheim Pharma and Diagnostics Lab Software (MediBeacon, Mannheim, Germany). The GFR (μl/min) was calculated from the decrease of fluorescence intensity over time (i.e., plasma t1/2 of FITC-Sinistrin) using a two-compartment model, the body weight of the mouse, and an empirical conversion factor.27 For the assessment of CKD progression, ΔGFR, calculated as the percentage of variation during the treatment period (week 12 to week 16), was analyzed. In addition, the variation in GFR from week 12 to week 16 was classified as increase (>10%), stable (±10%), or decrease (<10%). The number of mice in each group was quantified and expressed as a percentage.
Secondary End Points
Biochemical Parameters
Body weight and blood glucose levels, monitored with an Accu-check sensor (Roche, Manheim, Germany), were analyzed every 4 weeks. Urine samples were collected every 4 weeks for the estimation of creatinine (DiaSys Diagnostic Systems, Holzheim, Germany) and albumin (Bethyl Laboratories, Montgomery, MD). For the assessment of CKD progression, we calculated Δ urinary albumin-creatinine ratio (ΔUACR) as the percentage of variation during the treatment period (weeks 12–16). In addition, the variation in UACR from weeks 12 to 16 was classified as increase (>10%), stable (±10%), or decrease (<10%). The number of mice in each group were quantified and expressed as a percentage.
Glomerular Size
We harvested and fixed kidneys in 4% formalin in PBS and embedded them in paraffin. Three-micrometer sections were stained with periodic acid–Schiff (PAS) reagent to quantify glomerular size. Briefly, randomly selected 40× images containing superficial or juxtamedullary glomeruli were captured for each kidney. We specify the average number of images acquired per section for each time point in Supplemental Table 2. Quantification of glomerular tuft area was performed for each glomerular type by two observers in a blinded fashion using Image J (1.51u) software.
Glomerulosclerosis
We used Picro-Sirius Red (0.1% collagen; Sigma-Aldrich, St Louis, MO) stain to quantify diffused glomerulosclerosis in 40× images containing superficial and juxtamedullary glomeruli (Supplemental Table 2). Subsequently, images were posterized using GIMP software (2.8.22) to isolate the red color, which stains collagen deposition. Percentage of glomerulosclerosis in each glomerulus was quantified on the modified images using Image J by two observers in a blinded fashion.
Podocyte Number and Density
Three-micrometer sections were stained with Wilms tumor 1 (WT-1) (1:200; Santa Cruz Biotechnology, Dallas, TX) for identification of podocytes. Images at 40× magnification containing superficial and juxtamedullary glomeruli were captured (Supplemental Table 2), and WT-1-positive cells were quantified independently in each glomerular type. Podocyte density was calculated as the number of podocytes per respective glomerular tuft area, and expressed as percentage and by using the method published previously by Venkatareddy et al.28 (described in detail in Supplemental Figure 1 and the Supplemental Methods).
Quantification of Filtration Slit Density
For quantification of filtration slit density as a marker of podocyte tertiary foot process coverage, optical clearing of the kidneys was performed. Briefly, kidneys were dissected and immediately incubated at 4°C in hydrogel solution (4% v/v acrylamide, 40.025% v/v bisacrylamide, 0.25% w/v VA-044 initiator, 4% PFA, and 1× PBS) for 1 day. The gel was polymerized at 37°C for 3 hours, and the presence of oxygen was minimized by filling tubes to the top with hydrogel solution. Samples were removed from the hydrogel solution, immersed in clearing solution (200 mmol/l boric acid and 4% SDS, pH 8.5), and incubated for 1 day. Kidneys were cut into 0.5-mm thick slices using a Vibratome and incubated at 50°C for 5 days with clearing solution changed every day. Before immunolabeling, samples were incubated in PBST (0.1% Triton-X in 1× PBS) for 1 day. During immunolabeling, PBST was used as diluent in all steps. Samples were incubated with primary antibody for 24 hours at 37°C and then washed in PBST for 8 hours at 37°C, followed by secondary antibody incubation for 24 hours at 37°C and washing for 8 hours at 37°C before mounting. To stain for podocin, a rabbit anti-podocin primary antibody (P0372; 1:50; Sigma-Aldrich) and a goat anti-rabbit Alexa Fluor-532 secondary antibody (1:100; Life Technologies, Monza, Italy) were used. Samples were immersed in 40% (w/v) fructose with 0.5% (v/v) 1-thioglycerol for 2 hours and then transferred to 80% (w/w) fructose with 0.5% (v/v) 1-thioglycerol for 24 hours. Samples were mounted on microscope slides with cavities (1320002; Marienfeld, Germany) before imaging. Stimulated Emission Depletion (STED) microscopy xyz images were acquired in bidirectional mode with a Leica SP8 STED 3× confocal microscope system. Alexa Fluor 532-secondary antibody was excited with a 532-nm tuned white light laser (WLL) and emission collected from 540 to 580 nm. A 660-nm pulsed-depletion laser was used with a gating between 0.7 and 6 ns. Images were acquired with a Leica HC PL APO CS2 100×/1.40 oil STED White objective. Collected images were deconvolved with Huygens Professional software. For the quantification of filtration slit coverage, z stacks of podocin signal were acquired (at least 5-µm thick). All of the images of each z stack were merged, and the foot process length and the area of the field were manually dissected and measured with Image J. The total length of the foot processes was divided by the total area of interest. This was carried out for five randomly selected areas for each glomerulus of at least five glomeruli per mouse. Identifying glomeruli as superficial or juxtamedullary was not possible with that method. To generate three-dimensional (3D) reconstructions of glomeruli, we stained for nephrin. A sheep anti-nephrin primary antibody (AF4269, 1:50; R&D Systems) and a donkey anti-sheep Alexa Fluor-Tetramethylrhodamine secondary antibody (1:80; Life Technologies) were used. Z-series stacks were obtained from 80-μm kidney slices. Images were collected at 1-μm intervals by using a Leica SP8 confocal microscope and deconvolved with Huygens Professional software. We used image processing software from Leica Microsystems “Leica Application Suite X” for 3D reconstruction.
Tubular Size
We used Picro-Sirius Red stain to quantify tubular areas in 20× images. All of the histologic structures other than proximal tubules were removed from the images and images were posterized using GIMP (2.8.22) software to isolate the yellow color, which represented the tubular area, and remove the red color staining collagen. The number of tubules per section was then quantified and divided by the tubular area of a section to quantify the average tubular size. Quantification was performed in a blinded fashion using Image J software.
Open Capillary Analysis
To quantify glomerular damage, we identified open capillaries in superficial and juxtamedullary glomeruli, and quantified them at 40× magnification in ≥20 glomeruli/animal.
Single-Cell RNA Sequencing Analysis of Gsk-3β
A single-cell RNA sequencing data set from Fu et al.29 was queried for the expression of GSK-3β in glomerular cells of control and streptozotocin-induced diabetic eNOS−/− mice.
Cell Culture
Glomeruli isolation and in vitro analysis are described in the Supplemental Methods section.
Safety Analysis
The need for termination ahead of schedule on the basis of welfare scoring was assessed as a marker of survival throughout the experiment as part of the safety analysis of all treatments (Supplemental Figure 2). A detailed description of the methods applied to assess safety in nonrenal tissue is provided in the Supplemental Methods section.
Statistical Analysis
Data are presented as mean with SD or as boxplot statistics. Before statistical analysis, data were analyzed for normal distribution by checking the distribution of the data against the expected normal distribution with a quantile-quantile (Q-Q) plot and confirmation with the Shapiro–Wilk test. We tested normally distributed data for statistically significant differences via ANOVA and post hoc Tukey’s correction was used for multiple comparisons. Non-normally distributed data were compared using Wilcoxon signed-rank testing or Kruskal–Wallis testing with post hoc Dunn’s test correction for multiple comparisons (Supplemental Table 4). Survival was plotted on Kaplan–Meier curves and comparisons between groups were evaluated using log-rank tests. A value of P<0.05 was considered to indicate statistical significance. All statistical analyses were performed with R (3.5.3).
RESULTS
Hyperfiltration, Increase in Nephron Dimensions, and Progressive Glomerulosclerosis in db/db Mice
Uninephrectomized db/db mice (db/db 1K) were already heavier at baseline and their body weights increased much faster as compared with uninephrectomized nondiabetic mice (WT 1K) (Figure 1B). These db/db 1K mice displayed increased hyperglycemia and GFR throughout the study (Figure 1, C and D). UACR started to increase after surgery, probably as a result of increased hyperfiltration in the remnant nephrons, and remained significantly higher than in WT controls at week 12 (Figure 1E). In db/db 1K mice, no losses occurred in the following 2 weeks after surgery, but around 30% of the db/db 1K mice included in the experiment were lost ahead of schedule before week 12, as compared with 10% of WT 1K mice (Supplemental Figure 2A).
In this experimental setting, we assessed histologic parameters in superficial and juxtamedullary nephrons in db/db 1K mice at baseline (T−2) and at week 12. As expected, at week 12 we observed a significant increase in Picro-Sirius red-positive mesangial area, a surrogate parameter for glomerulosclerosis in both types of glomeruli (Figure 1F). Similarly, glomerular tuft areas in PAS sections as well as the areas of tubular cross sections were increased as a structural consequence of persistent single nephron hyperfiltration (Figure 1G). Podocyte numbers and densities, expressed as podocyte numbers by glomerular tuft areas, were significantly reduced in both superficial and juxtamedullary nephrons (Figure 1H, Supplemental Table 5). Thus, db/db 1K mice display typical functional and structural features of progressive CKD in obesity-related type 2 diabetes.
Nephroprotective Effects of MRE
To test for any renoprotective effects of MRE in this setting, db/db 1K and WT 1K mice were randomized to treatment with MRE (db/db/WT+MRE) or no treatment (db/db/WT) from week 12 to week 16 (Figure 2A).
Primary Outcome
Starting from the state of diabetes-related hyperfiltration at week 12, more than one half of the untreated db/db 1K mice experienced a decline in GFR from week 12 to week 16, whereas around 14% maintained GFR and 29% increased it. Treatment with MRE for 4 weeks had no significant effect on ΔGFR or GFR in absolute numbers in db/db or WT mice (Figure 2B).
Secondary Outcomes
As secondary outcomes, we assessed blood glucose, body weight, albuminuria, and histologic parameters at the end of the study. Untreated db/db 1K mice displayed hyperglycemia at levels around four-times higher than those of WT mice and, as expected, the two antidiabetic drugs among the MRE combination significantly reduced blood glucose levels down to levels around two-times higher than those of WT mice (Figure 2C). In WT 1K mice there was no such effect (Figure 2C). In contrast, 4 weeks of MRE did not affect the profound obesity of db/db or WT 1K mice (Figure 2D). MRE therapy decreased albuminuria in around 33% of db/db 1K mice, whereas all nontreated db/db 1K mice increased the levels of albuminuria within the study period (Figure 2E). Thus, MRE therapy significantly reduced albuminuria (Figure 2E). In db/db 1K mice, MRE significantly reduced diffuse glomerulosclerosis in superficial as well as juxtamedullary nephrons (Figure 3A). Four weeks of MRE therapy did not significantly alter the dimensions of glomeruli and tubules, although we noted a nonsignificant trend toward smaller tubule dimensions. (Figure 3, B and C). When assessing podocyte loss/recovery, we occasionally noted podocytes detaching from the glomerular tuft of cortical as well as juxtamedullary nephrons in untreated db/db 1K mice WT-1+, a phenomenon not seen with any of the therapies (Supplemental Figure 3). Indeed, MRE therapy significantly increased podocyte numbers and podocyte densities in both types of nephrons in db/db 1K mice (Figure 3, D and E, Supplemental Table 5). Interestingly, MRE treatment restored podocyte numbers in superficial nephrons to the level of db/db mice before uninephrectomy at T−2 (Figure 3D), whereas juxtamedullary nephrons remained with less podocytes upon MRE treatment (Figure 3E). However, podocyte densities in both types of nephrons remained way below those of young db/db mice before uninephrectomy (T−2) (Figure 3E). In relation to podocyte quantification, filtration slit density was assessed as a marker of ultrastructural podocyte injury by STED super-resolution microscopy upon tissue clearing. MRE significantly improved filtration slit density along the glomerular filtration barrier as compared with nontreated db/db 1K mice, but not up to the level of WT mice (Figure 3F). This was also evident from 3D reconstructions of confocal microscopy images of entire glomeruli stained for nephrin of mice of all three groups that depicted the effect of MRE therapy on type 2 diabetes-related loss of podocytes and foot process effacement in podocytes surrounding these gaps (Figure 4, Supplemental Figure 4).
Figure 3.
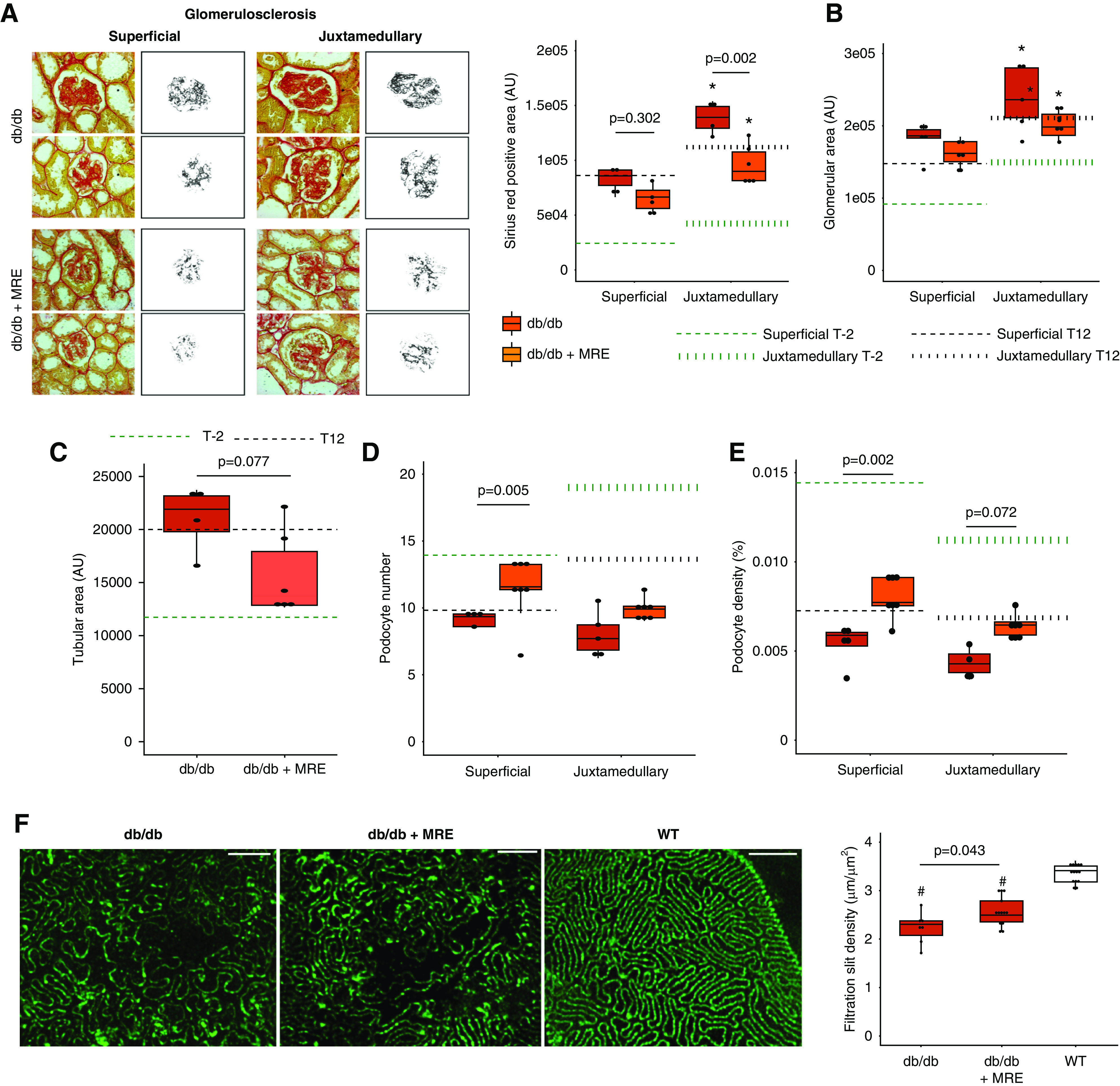
Effect of MRE therapy on histologic outcomes at end point. (A) Glomerulosclerosis, measured as Picro-Sirius Red-positive area (AU). Magnification, ×40. (B) Glomerular area, measured as the glomerular tuft area (AU) in PAS sections. (C) Tubular size, assessed in Picro-Sirius Red sections. (D) Podocyte number, assessed in WT-1 sections. (E) Podocyte density, calculated as the number of podocytes per glomerular tuft area. In (A–D), dotted green lines represent the levels at baseline (week −2) from each histologic assessment. Dotted black lines represent the levels before starting the treatment (week 12). In addition, both superficial and juxtamedullary nephrons were assessed independently, and the average numbers of glomeruli quantified are summarized in Supplemental Table 2. (F) Filtration slit density. Density of filtration slit was assessed as a marker of terminal podocyte differentiation by using STED-super-resolution microscopy upon tissue clearing. The quantification was carried out for five randomly selected areas for each glomerulus of at least five glomeruli per mouse. Bars, 2 µm. Magnification, ×100. *P value≤0.05 versus respective superficial glomeruli. #P value≤0.05 versus WT. Statistical analyses performed are summarized in Supplemental Table 4. AU, arbitrary units.
Figure 4.

Three-dimensional reconstruction of glomeruli stained for nephrin upon optical tissue clearing. Images show representative glomeruli of mice from each group. Signal represents nephrin protein within the slit diaphragm. Z-series stacks were obtained from 80-μm kidney slices. Images were collected at 1-μm intervals. WT 1K mice show smaller glomeruli with an intact foot process coverage of the glomerular capillaries, i.e., the intact glomerular filtration barrier. Db/db 1K mice show large defects in coverage representing denudated areas (black areas) with podocyte loss (marked with an asterisk). Arrowheads indicate filtration slits between podocytes showing foot process effacement. MRE and BIO treatment reduced the area of denudation, and BIO especially reduced signs of foot process effacement. Bars, 20 µm. Magnification, ×40.
Safety Outcomes
All WT animals survived up to the end of the study, whereas two mice each in the db/db 1K and db/db 1K + MRE groups dropped out ahead of schedule. The comparable survival in both db/db groups does not raise MRE-related safety concerns, consistent with the human experience with these drugs (Supplemental Figure 2B).
Together, the data suggest that short-term MRE therapy does not (yet) improve GFR but elicits numerous other renoprotective effects on CKD in db/db mice consistent with respective clinical trials.
Glycogen Synthase Kinase-3β as a Therapeutic Target in Diabetic Kidney Disease
GSK-3β inhibition with BIO can attenuate albuminuria in db/db mice,23 but the mechanism of action and its efficacy beyond MRE therapy remain uncertain. According to the human protein atlas, GSK-3β RNA and protein are expressed at low levels in the healthy human kidney.30 To better understand the cell type-specific expression and the effect of diabetes on cell type-specific Gsk3β, mRNA expression was tested using the recent single-cell RNA sequencing analysis of glomerular cells from diabetic and nondiabetic mice.29 Gsk3β transcripts were present in all glomerular cells (Supplemental Figure 5A). Moreover, we did not observe a significant difference in its expression between diabetic and control mice (Supplemental Figure 5, B and C), indicating that the constitutive expression of GSK-3β is not much affected by diabetes at the single-cell level. Nevertheless, treating db/db 1K mice on a low-salt diet with BIO from weeks 12 to 16 validated the previously reported effect on albuminuria and revealed significant renoprotective effects on podocyte densities in cortical and juxtamedullary nephrons, and on podocyte filtration slit density (Supplemental Tables 5 and 6). 3D reconstruction of entire glomeruli revealed smaller areas of podocyte denudation and better foot process preservation in the surrounding areas (Figure 4). In addition, survival was slightly better without BIO therapy (Supplemental Figure 2C). Thus, GSK-3β may be a valuable therapeutic target to attenuate CKD progression beyond MRE therapy in type 2 diabetes.
Additive Renoprotective Effects of MRE and BIO Therapy
Finally, we tested the therapeutic effects of BIO added to MRE therapy in db/db and WT 1K mice (Figure 5A).
Primary Outcome
Only 30% of the mice treated with the combined therapy showed a decline of GFR, compared with >50% in the MRE alone group (Figure 5B). This resulted in a significant reduction of ΔGFR after the 4-week treatment period with add-on BIO (Figure 5B). This implies a protective effect on progressive GFR decline as compared with treatment with MRE alone.
Secondary Outcomes
BIO added to MRE had no effects on blood glucose levels and body weight (Figure 5, C and D), and did not further decrease albuminuria (Figure 5E). However, the MRE+BIO combination increased the percentage of mice decreasing albuminuria as compared with MRE therapy alone (Figure 5E). The MRE+BIO combination had no effect on the aforementioned parameters in WT mice (Figure 5, B–E). Regarding glomerulosclerosis, the MRE+BIO combination showed significant protection beyond MRE in juxtamedullary nephrons (Figure 6A) without affecting the dimensions of glomeruli or tubuli (Figure 6, B and C), but increased the number of open glomerular capillaries that may have contributed to a better GFR (Supplemental Figure 6). In the same nephrons, the MRE+BIO combination also increased podocyte numbers up to the levels of young db/db mice before uninephrectomy (T−2) and significantly increased podocyte density beyond MRE, although densities still remained below those of young mice (Figure 6, D and E). Consistent with these findings, BIO significantly further improved filtration slit density at the glomerular filtration barrier, and almost completely restored the tuft structure and the glomerular filtration barrier, as noted from the 3D reconstructions of nephrin-stained glomeruli upon optical tissue clearing (Figure 7).
Figure 6.
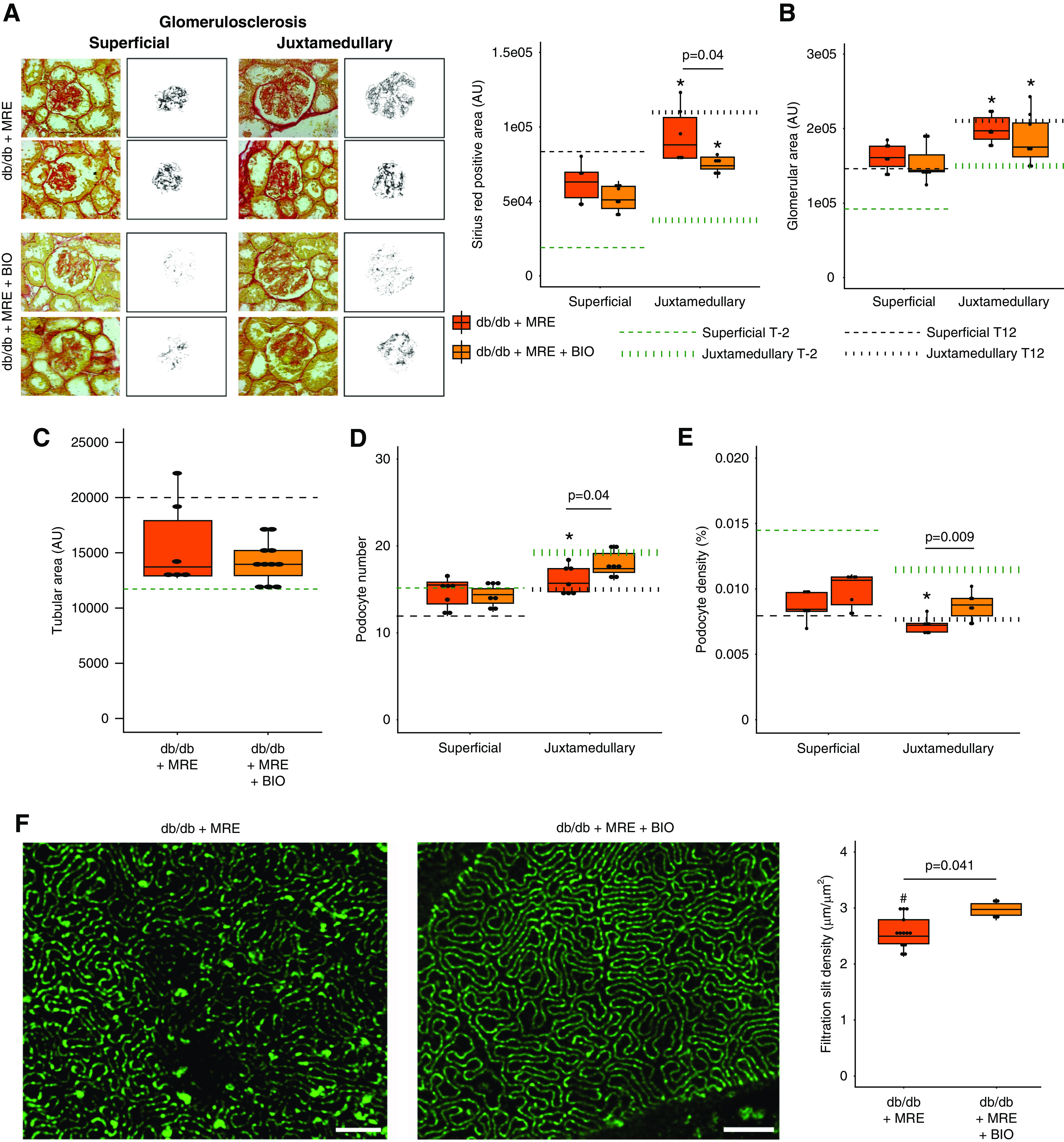
Effect of combined MRE+BIO therapy on histologic outcomes at end point. (A) Glomerulosclerosis, measured as Picro-Sirius Red-positive area (AU). Magnification, ×40. (B) Glomerular area, measured as the glomerular tuft area (AU) in PAS sections. (C) Tubular size, assessed in Picro-Sirius Red sections. (D) Podocyte number, assessed in WT-1 sections. (E) Podocyte density, calculated as the number of podocytes per glomerular tuft area. In (A–D), dotted green lines represent the levels at baseline (week −2) from each histologic assessment. Dotted black lines represent the levels before starting the treatment (week 12). In addition, both superficial and juxtamedullary nephrons were assessed independently, and the average number of glomeruli quantified are summarized in Supplemental Table 2. (F) Filtration slit density. Density of filtration slit was assessed as a marker of terminal podocyte differentiation by using STED-super resolution microscopy upon tissue clearing. The quantification was carried out for five randomly selected areas for each glomerulus of at least five glomeruli per mouse. Bars, 2 µm. Magnification, ×100. *P value≤0.05 versus respective superficial glomeruli. Statistical analyses performed are summarized in Supplemental Table 4. AU, arbitrary units.
Figure 7.

Three-dimensional reconstruction of glomeruli stained for nephrin upon optical tissue clearing. Images show representative glomeruli of mice from each group. Signal represents nephrin protein within the slit diaphragm. Z-series stacks were obtained from 80-μm kidney slices. Images were collected at 1-μm intervals. Db/db 1K mice treated with MRE show residual defects in coverage representing denudated areas (black areas) with podocyte loss (marked with an asterisk). Arrowheads indicate filtration slits between podocytes showing foot process effacement. Dual MRE+BIO treatment improves these residual defects. Bars, 20 µm. Magnification, ×40.
Safety Outcomes
BIO had a small but statistically significant negative effect on survival as compared with the respective nondiabetic control group (Supplemental Figure 2D). With both treatments, premature dropouts were associated to urinary tract infections (Supplemental Figure 2E). An extended safety analysis of numerous nonrenal organs did not reveal any safety concerns regarding the MRE+BIO combination in comparison with MRE alone (Supplemental Table 7).
Effects of BIO on Mouse Glomeruli in vitro
BIO is known to have direct effects on cultured glomerular cells but we questioned its effects on entire mouse glomeruli in vitro.21–23 Glomeruli could be obtained from mouse kidneys at high purity (Supplemental Figure 7A). The isolation procedure itself was associated with marked downregulation of the constitutive mRNA expression of the slit membrane protein nephrin, the structural protein α-SMA, and the cell cycle regulator Cyclin D1, but not of IL-6, which is not expressed inside glomeruli under basal conditions (Supplemental Figure 7, B–E). Glucose exposure increased IL-6 mRNA levels but none of the other transcripts. BIO induced a significant upregulation of nephrin but did not affect the other mRNAs (Supplemental Figure 7, B–E). Thus, BIO is a specific inductor of nephrin in mouse glomeruli in nondiabetic as well as diabetic conditions.
Discussion
We had hypothesized that BIO would attenuate CKD progression beyond MRE therapy in obese db/db mice with T2DM. Our results confirm this concept in terms of protection from glomerulosclerosis and GFR decline, e.g., by acting on residual podocyte injury. Thus, it is feasible to test novel drugs in an experimental setting of T2DM with residual progressive GFR loss despite antidiabetic therapy and a combination of RAS/SGLT2 inhibitors.
Most animal experiments in this domain mimic the early phase of diabetic nephropathy characterized by a stable total GFR, mesangial sclerosis, and mild albuminuria equivalent to CKD stage G1A1. In contrast, most clinical trials involve patients with diabetes receiving standard-of-care therapy for the progressive loss of GFR and macroproteinuria.31 Surgical nephron reduction, e.g., by uninephrectomy, is an established experimental tool to accelerate CKD progression by pushing single nephron hyperfiltration, a key pathomechanism of CKD progression, and feeding a sodium-deficient diet may augment this pathomechanism, as previously described.32–35 This enabled us to implement 4 weeks of therapy with MRE, which, on the basis of the recent results of several human randomized controlled trials (RCTs), qualifies to be the new standard of care for any RCT in T2DM in the future.4–6 Given the short MRE treatment period, we did not expect an attenuation of GFR decline because this required a minimum of 2–3 years of treatment in the Empagliflozin Cardiovascular Outcome Event Trial in Type 2 Diabetes Mellitus Patients (EMPA-REG OUTCOME)4; however, also consistent with the human trial, MRE immediately reduced macroproteinuria in our model. This antiproteinuric effect was associated with attenuation of podocyte loss in all nephrons and in superficial nephrons, even back to podocyte numbers of young untouched db/db mice (T−2). This can only be possible via de novo podocyte formation endorsed by one or several of the MRE drugs. For example, RAS inhibition can attenuate (diabetic) glomerulosclerosis by enhancing podocyte regeneration,36–40 a process that can also explain the observed increase in the densities of podocytes and their filtration slits along the glomerular filtration barrier.
Any putative nephroprotective effect of novel classes of drugs may significantly overlap with background therapy. For example, the direct mechanisms of action of MRE therapy, as well as their indirect consequences, could have completely altered the relative significance of the pathways targeted by novel drug candidates. Indeed, several drugs that have proven effective in rodent studies later showed no or only minor add-on effects when tested in a human RCT.41–49 For example, despite the renoprotective effects of BIO monotherapy,23 it was completely unclear whether BIO could elicit nephroprotective effects beyond the effects of the MRE combination. Our finding that BIO add-on therapy significantly attenuated our primary and clinically relevant end point, GFR decline, beyond MRE therapy is remarkable, especially given the short treatment period. Whether this effect on GFR relates to BIO’s capacity to further improve the residual structural damage of the filtration barrier and of glomerulosclerosis, or to additional effects on renal hemodynamics, vascular integrity or metabolic function remains in part uncertain. On the basis of previous reports, as well as our in vitro and in vivo data, BIO seems to endorse terminal podocyte differentiation and de novo podocyte production from local podocyte progenitors, mechanisms all supporting the structural integrity of the glomerular filtration barrier and attenuating glomerulosclerosis.22,50–57
Finally, our unprecedented safety analysis did not indicate any safety concerns for add-on BIO therapy inside and outside the kidney.
Several limitations remain. Our model does not mimic sex, age, and comorbidities of patients included in RCTs on diabetic kidney disease. Although our model mimics progressive GFR loss, this still occurs in the phase of an elevated total GFR, i.e., renal hyperfiltration, which still does not mimic advanced CKD. Therefore, in contrast to advanced CKD in human T2DM, interstitial inflammation and fibrosis remain negligible in our model. Our short treatment interval failed to replicate the preservation of GFR with MRE seen after 1–2 years of treatment in human RCTs, thus our model is sensitive only to immediate treatment effects. The low-salt diet, especially in the context of ramipril and empagliflozin therapy, may endorse hypovolemia and hypotension in db/db mice, whereas hypertension is prevalent in patients with diabetic kidney disease. On the other hand, reducing salt intake is part of the standard management of patients with hypertension and proteinuric kidney diseases, and our experimental setup modeled this scenario and not one of uncontrolled diabetes or hypertension. In patients with glucosuria-related osmotic diuresis, free access to fluid is essential, which was also granted to our mice. Nevertheless, volume issues may have contributed to the 30% mortality seen in our model and to the high variability of GFR in db/db 1K mice treated with MRE. In addition, quantification of podocyte numbers on single two-dimensional tissue sections was suboptimal, although we tried to optimize it by implementing the recently reported technical advances in counting podocytes and correction factors that apply depending on the thickness of the sections.28 Finally, podocyte loss into the urine was difficult to demonstrate because db/db mice excrete much-diluted urine, so all our attempts to demonstrate urinary podocytes failed, probably due to assay sensitivity. Nevertheless, our careful imaging analysis revealed podocyte detachment and denudated areas of the GBM in untreated db/db mice.
In conclusion, our attempt to improve drug testing for progressive CKD in type 2 diabetes included a model of progressive GFR loss, group size calculation, usual comedication, randomization to an intervention not established before disease, prespecified primary and secondary end points identical to those used in clinical trials, sophisticated morphologic assessment to gain mechanistic insights, and detailed safety analysis. In this setting, our drug candidate, BIO, showed significant renoprotective effects beyond metformin and dual RAS/SGLT2 inhibition. As the putative mechanism of action was augmenting podocyte numbers and morphology, drugs targeting these mechanisms might be a valuable add-on medication strategy in patients with residual CKD progression with T2DM despite dual RAS/SGLT2 inhibition.
DISCLOSURES
All authors have nothing to disclose.
Funding
M. Motrapu was supported by national overseas scholarship from the Government of India, Ministry of Social Justice and Empowerment. L. Anguiano and M.K. Świderska were supported by European Renal Association – European Dialysis and Transplant Association Long-Term Research Fellowships (LTF RLTF 855-2016 and 1586-2018). L. Anguiano also received a stipend for women postdoctoral reserachers from the Bavarian Gender Equality Grant. I. Mesas was supported by the Medical Faculty of Ludwig-Maximilians-Universität München (FöFoLe program). Y. Lei was supported by the PhD fellowship program of the Chinese Scholarship Council. P. Romagnani received funding from the European Research Council under the European Union’s Horizon 2020 research and innovation programme (grant agreement number 648274). H.-J. Anders was supported by the Deutsche Forschungsgemeinschaft (grant number AN372/24-1).
Supplementary Material
Acknowledgments
The expert technical support of Jana Mandelbaum and Dan Draganovici is gratefully acknowledged.
Dr. Hans-Joachim Anders and Dr. Lidia Anguiano designed the study; Dr. Manga Motrapu, Dr. Monika Katarzyna Świderska, Dr. Irene Mesas, Dr. Julian Marschner, Dr. Yutian Lei, Dr. Laura Martinez Valenzuela, Dr. Jia Fu, Dr. Kyung Lee, Dr. Maria Lucia Angelotti, Dr. Giulia Antonelli, and Dr. Lidia Anguiano carried out experiments; Dr. Manga Motrapu, Dr. Lidia Anguiano, Dr. Maria Lucia Angelotti, Dr. Paola Romagnani, and Dr. Hans-Joachim Anders analyzed the data; Dr. Lidia Anguiano, Dr. Jia Fu, Dr. Kyung Lee, Dr. Giulia Antonelli, and Dr. Maria Lucia Angelotti made the figures; Dr. Hans-Joachim Anders, Dr. Lidia Anguiano, Dr. Manga Motrapu, Dr. Paola Romagnani, Dr. Maria Lucia Angelotti, and Dr. Kyung Lee drafted and revised the paper. All authors further revised and approved the final version of the manuscript.
Dr. Hans-Joachim Anders reports personal fees from Previpharma, Secarna, Inositec, Janssen, Boehringer, and GSK, outside the submitted work.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
Supplemental Material
This article contains the following supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2019070703/-/DCSupplemental.
Supplemental Figure 1. Podocyte density analysis.
Supplemental Figure 2. Survival analysis.
Supplemental Figure 3. Podocyte detachment analysis.
Supplemental Figure 4. Healthy glomerulus with intact filtration barrier; injured glomerulus with damaged filtration barrier; and injured glomerulus with damaged filtration barrier.
Supplemental Figure 5. Single-cell expression of Gsk3β in glomerular cells in control and diabetic mice.
Supplemental Figure 6. Open capillaries analysis between db/db + MRE and db/db + MRE + BIO groups.
Supplemental Figure 7. Effect of BIO on glomeruli in vitro.
Supplemental Table 1. Number of animals included for the analysis at endpoint.
Supplemental Table 2. Number of glomeruli quantified per section, for both superficial and juxtamedullary nephrons, in histological analysis.
Supplemental Table 3. Primers sequences.
Supplemental Table 4. Summary of statistical analysis performed for each figure and table.
Supplemental Table 5. Analysis of podocyte number and podocyte density at week 16 using the method by Venkatareddy, et al.1
Supplemental Table 6. BIO treatment in db/db mice from week 12 to 16.
Supplemental Table 7. Safety evaluation of BIO therapy in heart, pancreas, sciatic nerve, liver, plasma, skeletal muscle, and adipose tissue.
References
- 1.Cho NH, Shaw JE, Karuranga S, Huang Y, da Rocha Fernandes JD, Ohlrogge AW, et al.: IDF Diabetes Atlas: Global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res Clin Pract 138: 271–281, 2018. [DOI] [PubMed] [Google Scholar]
- 2.Ogurtsova K, da Rocha Fernandes JD, Huang Y, Linnenkamp U, Guariguata L, Cho NH, et al.: IDF Diabetes Atlas: Global estimates for the prevalence of diabetes for 2015 and 2040. Diabetes Res Clin Pract 128: 40–50, 2017. [DOI] [PubMed] [Google Scholar]
- 3.Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, et al.: EMPA-REG OUTCOME Investigators : Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med 373: 2117–2128, 2015. [DOI] [PubMed] [Google Scholar]
- 4.Wanner C, Inzucchi SE, Lachin JM, Fitchett D, von Eynatten M, Mattheus M, et al.: EMPA-REG OUTCOME Investigators : Empagliflozin and progression of kidney disease in type 2 diabetes. N Engl J Med 375: 323–334, 2016. [DOI] [PubMed] [Google Scholar]
- 5.Neal B, Perkovic V, Mahaffey KW, de Zeeuw D, Fulcher G, Erondu N, et al.: CANVAS Program Collaborative Group : Canagliflozin and cardiovascular and renal events in type 2 diabetes. N Engl J Med 377: 644–657, 2017. [DOI] [PubMed] [Google Scholar]
- 6.Perkovic V, Jardine MJ, Neal B, Bompoint S, Heerspink HJL, et al. DMet al. : Canagliflozin and renal outcomes in type 2 diabetes and nephropathy. N Engl J Med 380: 2295–2306, 2019 [DOI] [PubMed] [Google Scholar]
- 7.Anders H-J, Davis JM, Thurau K: Nephron protection in diabetic kidney disease. N Engl J Med 375: 2096–2098, 2016. [DOI] [PubMed] [Google Scholar]
- 8.Breyer MD, Böttinger E, Brosius FC 3rd, Coffman TM, Harris RC, Heilig CW, et al.: AMDCC : Mouse models of diabetic nephropathy. J Am Soc Nephrol 16: 27–45, 2005. [DOI] [PubMed] [Google Scholar]
- 9.Ninichuk V, Clauss S, Kulkarni O, Schmid H, Segerer S, Radomska E, et al.: Late onset of Ccl2 blockade with the Spiegelmer mNOX-E36-3’PEG prevents glomerulosclerosis and improves glomerular filtration rate in db/db mice. Am J Pathol 172: 628–637, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sayyed SG, Ryu M, Kulkarni OP, Schmid H, Lichtnekert J, Grüner S, et al.: An orally active chemokine receptor CCR2 antagonist prevents glomerulosclerosis and renal failure in type 2 diabetes. Kidney Int 80: 68–78, 2011. [DOI] [PubMed] [Google Scholar]
- 11.Menne J, Eulberg D, Beyer D, Baumann M, Saudek F, Valkusz Z, et al.: Emapticap Study Group : C-C motif-ligand 2 inhibition with emapticap pegol (NOX-E36) in type 2 diabetic patients with albuminuria. Nephrol Dial Transplant 32: 307–315, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de Zeeuw D, Bekker P, Henkel E, Hasslacher C, Gouni-Berthold I, Mehling H, et al.: CCX140-B Diabetic Nephropathy Study Group : The effect of CCR2 inhibitor CCX140-B on residual albuminuria in patients with type 2 diabetes and nephropathy: A randomised trial. Lancet Diabetes Endocrinol 3: 687–696, 2015. [DOI] [PubMed] [Google Scholar]
- 13.Ninichuk V, Kulkarni O, Clauss S, Anders H-J: Tubular atrophy, interstitial fibrosis, and inflammation in type 2 diabetic db/db mice. An accelerated model of advanced diabetic nephropathy. Eur J Med Res 12: 351–355, 2007. [PubMed] [Google Scholar]
- 14.Lin JS, Susztak K: Podocytes: The weakest link in diabetic kidney disease? Curr Diab Rep 16: 45, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tharaux P-L, Huber TB: How is proteinuric diabetic nephropathy caused by disturbed proteostasis and autophagy in podocytes? Diabetes 65: 539–541, 2016. [DOI] [PubMed] [Google Scholar]
- 16.Gödel M, Hartleben B, Herbach N, Liu S, Zschiedrich S, Lu S, et al.: Role of mTOR in podocyte function and diabetic nephropathy in humans and mice. J Clin Invest 121: 2197–2209, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sweetwyne MT, Gruenwald A, Niranjan T, Nishinakamura R, Strobl LJ, Susztak K: Notch1 and Notch2 in podocytes play differential roles during diabetic nephropathy development. Diabetes 64: 4099–4111, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lasagni L, Lazzeri E, Shankland SJ, Anders H-J, Romagnani P: Podocyte mitosis - a catastrophe. Curr Mol Med 13: 13–23, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hara M, Oohara K, Dai D-F, Liapis H: Mitotic catastrophe causes podocyte loss in the urine of human diabetics. Am J Pathol 189: 248–257, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guo D, Shen Y, Li W, Li Q, Zhao Y, Pan C, et al.: 6-Bromoindirubin-3′-Oxime (6BIO) suppresses the mTOR pathway, promotes autophagy, and exerts anti-aging effects in rodent liver. Front Pharmacol 10: 320, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lasagni L, Angelotti ML, Ronconi E, Lombardi D, Nardi S, Peired A, et al.: Podocyte regeneration driven by renal progenitors determines glomerular disease remission and can Be pharmacologically enhanced. Stem Cell Reports 5: 248–263, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Paeng J, Chang JH, Lee SH, Nam BY, Kang HY, Kim S, et al.: Enhanced glycogen synthase kinase-3β activity mediates podocyte apoptosis under diabetic conditions. Apoptosis 19: 1678–1690, 2014. [DOI] [PubMed] [Google Scholar]
- 23.Wan J, Li P, Liu DW, Chen Y, Mo HZ, Liu BG, et al.: GSK-3β inhibitor attenuates urinary albumin excretion in type 2 diabetic db/db mice, and delays epithelial-to-mesenchymal transition in mouse kidneys and podocytes. Mol Med Rep 14: 1771–1784, 2016. [DOI] [PubMed] [Google Scholar]
- 24.Vallon V, Huang DY, Deng A, Richter K, Blantz RC, Thomson S: Salt-sensitivity of proximal reabsorption alters macula densa salt and explains the paradoxical effect of dietary salt on glomerular filtration rate in diabetes mellitus. J Am Soc Nephrol 13: 1865–1871, 2002. [DOI] [PubMed] [Google Scholar]
- 25.Darisipudi MN, Kulkarni OP, Sayyed SG, Ryu M, Migliorini A, Sagrinati C, et al.: Dual blockade of the homeostatic chemokine CXCL12 and the proinflammatory chemokine CCL2 has additive protective effects on diabetic kidney disease. Am J Pathol 179: 116–124, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lei Y, Devarapu SK, Motrapu M, Cohen CD, Lindenmeyer MT, Moll S, et al.: Interleukin-1β inhibition for chronic kidney disease in obese mice with type 2 diabetes. Front Immunol 10: 1223, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schreiber A, Shulhevich Y, Geraci S, Hesser J, Stsepankou D, Neudecker S, et al.: Transcutaneous measurement of renal function in conscious mice. Am J Physiol Renal Physiol 303: F783–F788, 2012. [DOI] [PubMed] [Google Scholar]
- 28.Venkatareddy M, Wang S, Yang Y, Patel S, Wickman L, Nishizono R, et al.: Estimating podocyte number and density using a single histologic section. J Am Soc Nephrol 25: 1118–1129, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fu J, Akat KM, Sun Z, Zhang W, Schlondorff D, Liu Z, et al.: Single-cell RNA profiling of glomerular cells shows dynamic changes in experimental diabetic kidney disease. J Am Soc Nephrol 30: 533–545, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.The Human Protein Atlas : Tissue expression of GSK3B - Summary - The Human Protein Atlas. Available at: https://www.proteinatlas.org/ENSG00000082701-GSK3B/tissue. Accessed June 26, 2019
- 31.Anguiano Gómez L, Lei Y, Kumar Devarapu S, Anders H-J: The diabetes pandemic suggests unmet needs for ‘CKD with diabetes’ in addition to ‘diabetic nephropathy’-implications for pre-clinical research and drug testing. Nephrol Dial Transplant 33: 1292–1304, 2018. [DOI] [PubMed] [Google Scholar]
- 32.Vallon V, Wead LM, Blantz RC: Renal hemodynamics and plasma and kidney angiotensin II in established diabetes mellitus in rats: Effect of sodium and salt restriction. J Am Soc Nephrol 5: 1761–1767, 1995. [DOI] [PubMed] [Google Scholar]
- 33.Vallon V, Kirschenmann D, Wead LM, Lortie MJ, Satriano J, Blantz RC, et al.: Effect of chronic salt loading on kidney function in early and established diabetes mellitus in rats. J Lab Clin Med 130: 76–82, 1997. [DOI] [PubMed] [Google Scholar]
- 34.Ekinci EI, Clarke S, Thomas MC, Moran JL, Cheong K, MacIsaac RJ, et al.: Dietary salt intake and mortality in patients with type 2 diabetes. Diabetes Care 34: 703–709, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thomas MC, Moran J, Forsblom C, Harjutsalo V, Thorn L, Ahola A, et al.: FinnDiane Study Group : The association between dietary sodium intake, ESRD, and all-cause mortality in patients with type 1 diabetes. Diabetes Care 34: 861–866, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lichtnekert J, Kaverina NV, Eng DG, Gross KW, Kutz JN, Pippin JW, et al.: Renin-angiotensin-aldosterone system inhibition increases podocyte derivation from cells of renin lineage. J Am Soc Nephrol 27: 3611–3627, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang J, Yanez D, Floege A, Lichtnekert J, Krofft RD, Liu ZH, et al.: ACE-inhibition increases podocyte number in experimental glomerular disease independent of proliferation. J Renin Angiotensin Aldosterone Syst 16: 234–248, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gagliardini E, Corna D, Zoja C, Sangalli F, Carrara F, Rossi M, et al.: Unlike each drug alone, lisinopril if combined with avosentan promotes regression of renal lesions in experimental diabetes. Am J Physiol Renal Physiol 297: F1448–F1456, 2009. [DOI] [PubMed] [Google Scholar]
- 39.Macconi D, Sangalli F, Bonomelli M, Conti S, Condorelli L, Gagliardini E, et al.: Podocyte repopulation contributes to regression of glomerular injury induced by ACE inhibition. Am J Pathol 174: 797–807, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pichaiwong W, Hudkins KL, Wietecha T, Nguyen TQ, Tachaudomdach C, Li W, et al.: Reversibility of structural and functional damage in a model of advanced diabetic nephropathy. J Am Soc Nephrol 24: 1088–1102, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mann JF, Schmieder RE, McQueen M, Dyal L, Schumacher H, Pogue J, et al.: ONTARGET investigators : Renal outcomes with telmisartan, ramipril, or both, in people at high vascular risk (the ONTARGET study): A multicentre, randomised, double-blind, controlled trial. Lancet 372: 547–553, 2008. [DOI] [PubMed] [Google Scholar]
- 42.Barnett AH, Bain SC, Bouter P, Karlberg B, Madsbad S, Jervell J, et al.: Diabetics Exposed to Telmisartan and Enalapril Study Group : Angiotensin-receptor blockade versus converting-enzyme inhibition in type 2 diabetes and nephropathy. N Engl J Med 351: 1952–1961, 2004. [DOI] [PubMed] [Google Scholar]
- 43.Haller H, Ito S, Izzo JL Jr., Januszewicz A, Katayama S, Menne J, et al.: ROADMAP Trial Investigators : Olmesartan for the delay or prevention of microalbuminuria in type 2 diabetes. N Engl J Med 364: 907–917, 2011. [DOI] [PubMed] [Google Scholar]
- 44.Fried LF, Emanuele N, Zhang JH, Brophy M, Conner TA, Duckworth W, et al.: VA NEPHRON-D Investigators : Combined angiotensin inhibition for the treatment of diabetic nephropathy. N Engl J Med 369: 1892–1903, 2013. [DOI] [PubMed] [Google Scholar]
- 45.Parving H-H, Brenner BM, McMurray JJ, de Zeeuw D, Haffner SM, Solomon SD, et al.: ALTITUDE Investigators : Cardiorenal end points in a trial of aliskiren for type 2 diabetes. N Engl J Med 367: 2204–2213, 2012. [DOI] [PubMed] [Google Scholar]
- 46.de Zeeuw D, Akizawa T, Audhya P, Bakris GL, Chin M, Christ-Schmidt H, et al.: BEACON Trial Investigators : Bardoxolone methyl in type 2 diabetes and stage 4 chronic kidney disease. N Engl J Med 369: 2492–2503, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pergola PE, Raskin P, Toto RD, Meyer CJ, Huff JW, Grossman EB, et al.: BEAM Study Investigators : Bardoxolone methyl and kidney function in CKD with type 2 diabetes. N Engl J Med 365: 327–336, 2011. [DOI] [PubMed] [Google Scholar]
- 48.Mann JFE, Green D, Jamerson K, Ruilope LM, Kuranoff SJ, Littke T, et al.: ASCEND Study Group : Avosentan for overt diabetic nephropathy. J Am Soc Nephrol 21: 527–535, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Heerspink HJL, Parving HH, Andress DL, Bakris G, Correa-Rotter R, Hou FF, et al.: SONAR Committees and Investigators : Atrasentan and renal events in patients with type 2 diabetes and chronic kidney disease (SONAR): A double-blind, randomised, placebo-controlled trial. Lancet 393: 1937–1947, 2019. [DOI] [PubMed] [Google Scholar]
- 50.Zhou S, Wang P, Qiao Y, Ge Y, Wang Y, Quan S, et al.: Genetic and pharmacologic targeting of glycogen synthase kinase 3β reinforces the Nrf2 antioxidant defense against podocytopathy. J Am Soc Nephrol 27: 2289–2308, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang Z, Bao H, Ge Y, Zhuang S, Peng A, Gong R: Pharmacological targeting of GSK3β confers protection against podocytopathy and proteinuria by desensitizing mitochondrial permeability transition. Br J Pharmacol 172: 895–909, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xu W, Ge Y, Liu Z, Gong R: Glycogen synthase kinase 3β orchestrates microtubule remodeling in compensatory glomerular adaptation to podocyte depletion. J Biol Chem 290: 1348–1363, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xu W, Ge Y, Liu Z, Gong R: Glycogen synthase kinase 3β dictates podocyte motility and focal adhesion turnover by modulating paxillin activity: Implications for the protective effect of low-dose lithium in podocytopathy. Am J Pathol 184: 2742–2756, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Naves MA, Requião-Moura LR, Soares MF, Silva-Júnior JA, Mastroianni-Kirsztajn G, Teixeira VP: Podocyte Wnt/ß-catenin pathway is activated by integrin-linked kinase in clinical and experimental focal segmental glomerulosclerosis. J Nephrol 25: 401–409, 2012. [DOI] [PubMed] [Google Scholar]
- 55.Lin C-L, Wang JY, Huang YT, Kuo YH, Surendran K, Wang FS: Wnt/beta-catenin signaling modulates survival of high glucose-stressed mesangial cells. J Am Soc Nephrol 17: 2812–2820, 2006. [DOI] [PubMed] [Google Scholar]
- 56.Unnersjö-Jess D, Scott L, Blom H, Brismar H: Super-resolution stimulated emission depletion imaging of slit diaphragm proteins in optically cleared kidney tissue. Kidney Int 89: 243–247, 2016. [DOI] [PubMed] [Google Scholar]
- 57.Siegerist F, Ribback S, Dombrowski F, Amann K, Zimmermann U, Endlich K, et al.: Structured illumination microscopy and automatized image processing as a rapid diagnostic tool for podocyte effacement. Sci Rep 7: 11473, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.