

Significance Statement
Cystic fibrosis (CF) is a multi-system disease caused by mutations in the gene encoding the cystic fibrosis transmembrane conductance regulator CFTR, an epithelial chloride channel. Patients with CF do not respond with increased urinary HCO3- excretion after stimulation with secretin and often present with metabolic alkalosis. Based on studies with several knockout (KO) mouse models, patients with CF, and cell studies, this paper defines the role of CFTR in renal HCO3- excretion. Secretin-induced renal HCO3- excretion occurs in the collecting duct and is pendrin- and CFTR-dependent. The study explains metabolic alkalosis in CF and suggests the feasibility of developing a test of urinary HCO3- excretion in CF patients who have two copies of the F508del mutation to assess CFTR function and response to drug treatment.
Keywords: cystic fibrosis, ion transport, kidney tubule, renal tubular acidosis
Visual Abstract

Abstract
Background
Patients with cystic fibrosis (CF) do not respond with increased urinary HCO3− excretion after stimulation with secretin and often present with metabolic alkalosis.
Methods
By combining RT-PCR, immunohistochemistry, isolated tubule perfusion, in vitro cell studies, and in vivo studies in different mouse models, we elucidated the mechanism of secretin-induced urinary HCO3− excretion. For CF patients and CF mice, we developed a HCO3- drinking test to assess the role of the cystic fibrosis transmembrane conductance regulator (CFTR) in urinary HCO3-excretion and applied it in the patients before and after treatment with the novel CFTR modulator drug, lumacaftor-ivacaftor.
Results
β-Intercalated cells express basolateral secretin receptors and apical CFTR and pendrin. In vivo application of secretin induced a marked urinary alkalization, an effect absent in mice lacking pendrin or CFTR. In perfused cortical collecting ducts, secretin stimulated pendrin-dependent Cl−/HCO3− exchange. In collecting ducts in CFTR knockout mice, baseline pendrin activity was significantly lower and not responsive to secretin. Notably, patients with CF (F508del/F508del) and CF mice showed a greatly attenuated or absent urinary HCO3−-excreting ability. In patients, treatment with the CFTR modulator drug lumacaftor-ivacaftor increased the renal ability to excrete HCO3−.
Conclusions
These results define the mechanism of secretin-induced urinary HCO3− excretion, explain metabolic alkalosis in patients with CF, and suggest feasibility of an in vivo human CF urine test to validate drug efficacy.
In 1902, Bayliss and Starling1 discovered the first hormone secretin and therewith established the hormone concept of organ function regulation. Today, the action of secretin is well understood in gastrointestinal physiology, where it acts as the main activator of exocrine pancreatic bicarbonate (HCO3−) secretion.2 Secretin has also been reported to elicit other physiologic effects, e.g., in the cardiovascular system.3–5 Its key physiologic role in the activation of pancreatic HCO3−-rich fluid secretion was used to study exocrine pancreas insufficiency in patients with cystic fibrosis (CF).6 This has contributed substantially to defining a major gastrointestinal phenotype in CF. At that time, researchers also collected urine samples in secretin-treated children with CF and healthy controls. The results were only published in abstract format but conveyed the remarkable finding that secretin elicits clear increases in urinary HCO3− excretion in normal test persons, an effect that was completely absent in patients with CF.7 Other human studies8 described the phenomenon of secretin-stimulated urinary HCO3− excretion. A mechanistic understanding of this renal effect is pending. Other renal effects of secretin have been reported and either show diuretic8,9 or antidiuretic actions.10,11 Intriguingly, substantial secretin receptor (SCTR) mRNA expression is present in rat connecting tubules (CNTs) and cortical collecting ducts (CCDs).12 Moreover, cell type–specific RNA-sequencing data from rat CNTs/CCDs show that SCTR mRNA expression is prominent in the β-intercalated cells (β-ICs), somewhat less in the α-intercalated cells, and nondetectable in the principal cells.13
In the exocrine pancreatic duct, the mechanism of secretin-activated secretion is well studied. The SCTR localizes to the basolateral membrane of this secretory epithelium. SCTR activation leads via stimulation of the cAMP/protein kinase A signaling pathway to activation of apical CFTR channels and the chloride ion (Cl−)/HCO3− exchanger SLC26A6, and hence a marked ductal secretion of sodium bicarbonate (NaHCO3).2,14,15
This project aimed to understand acute renal HCO3− excretion and to define the molecular mechanism of secretin-stimulated renal HCO3− excretion. By analogy with the established function of secretin in the exocrine pancreatic duct, we hypothesized that the SCTR localizes to the basolateral membrane in β-ICs and triggers acute activation of urinary HCO3− secretion by activation of apical pendrin (SLC26A4) and CFTR. We provide comprehensive evidence validating this hypothesis and show that secretin-stimulated renal HCO3− secretion by the β-ICs is absent in CF mice. Subsequently, we took a translational approach to verifying a greatly reduced ability to excrete an acute oral load of HCO3− in the urine of patients with CF. We applied this functional test to patients with CF before and after treatment with novel CFTR activator drugs and showed a sizable increase of urinary HCO3− excretion after treatment. We propose these results offer a fundamental new explanation of a basic physiologic function that offers direct clinical application to monitor treatment success in patients with CF.
Methods
Animals
The experiments in genetically modified mice were performed in littermates bred from heterozygotes. Mice of both genders were used at an approximate age of 2–4 months. Tubule-perfusion experiments used mice aged 4–6 weeks were used. The pendrin knockout (KO) mouse has an insertion of a neo cassette replacing exon 8 in the Scl26a4 gene, leading to loss of pendrin expression.16 It was purchased from The Jackson Laboratory (Bar Harbor, ME), JAX stock number 018424. The global CFTR (CFTRG) KO mouse (CFTR S489X-; FABP-hCFTR) was purchased from The Jackson Laboratory, JAX stock number 002364. This is a transgenic CFTRG KO mouse strain with expression of human CFTR under the activity of the rat fatty acid binding protein 2 (Fabp2), intestinal promoter, which corrects the otherwise lethal intestinal phenotype of CFTR KO mice.17
The genetic background was as follows: the renal tubule-specific CFTR (CFTRTS) KO mouse was produced in the laboratory of Dr. Kunzelmann and Dr. Schreiber in Regensburg/Germany. CFTRTS KO mice were generated by crossing the C57BL6/J CFTR fl10 mice18 with mice expressing renal tubule– specific Cre-recombinase driven by the Pax8 locus.19,20 To confirm exon 10 deletion of CFTR in the kidney, total RNA was isolated and RT-PCR was performed using the forward primer 5′-CCACAGGCATAATCATGGAA and the reverse primer 5′-TGTGACTCCACCTTCTCCAA. A 476-bp product was detected for wild-type (WT) CFTR, and a 293-bp product was detected for Δexon10 CFTR (Supplemental Figure 1).
In the experiments investigating plasma secretin, male C57Bl6 mice aged 10 weeks (Janvier Labs, Le Genest-Saint-Isle, France) were used to assess acute HCO3− load. The mice were acclimatized to the animal facility of the Department of Biomedicine, Aarhus University for 2 weeks before experiments.
All mice had ad libitum access to normal drinking water and standard rodent chow and were kept in a 12-hour light/dark cycle. Supplemental Table 1 provides weight, age, and genetic background for the mice used in the in vivo secretin experiments.
Enzymatic Digestion, Sorting of Renal Tubules, and End-Point RT-PCR
Mice were anesthetized with ketamine/xylazine and perfused with 20 ml PBS through the left ventricle. Kidneys were harvested, decapsulated, and cut into 8 to 10 transverse slices. The kidney slices were enzymatically digested for 10 minutes in incubation solution (140 mM sodium chloride [NaCl], 0.4 mM dipotassium phosphate [K2HPO4], 1.6 mM monopotassium phosphate [KH2PO4], 1 mM magnesium sulfate [MgSO4], 10 mM sodium [Na]-acetate, 1 mM α-ketoglutarate, 1.3 mM calcium [Ca]-gluconate, 5 mM glycine, 25 mg/L DNAseI [Roche], 1 mg/ml collagenase II [Pan Biotech, Aidenbach, Germany]) for 10 minutes at 37°C while shaken at 850 rpm in a thermomixer (Eppendorf). Supernatant was aspirated, 1 ml fresh incubation solution was added, and incubation was continued in 5-minute intervals, with removal of tubule-containing supernatant in between. Tubules were washed twice in incubation solution with 0.5 mg/ml albumin (sorting solution) and put on ice. Tubules were sorted to yield proximal tubule (PT), thick ascending limb (TAL), distal convoluted tubule (DCT), CNT/CCD, and medullary collecting duct (MCD) samples according to their appearance in phase-contrast microscopy, as published previously.21 PTs were long and highly convoluted with a smooth appearance; TALs were long, thin, and straight with a smooth appearance; DCTs and CNT/CCDs were identified together by the branching points where separate CNTs merge and by a composite appearance, resulting from the presence of intercalated cells. The transition between DCT and CNT/CCD was apparent as an abrupt change in diameter and transition from slightly convoluted to straight tubule, and DCTs were cut from the CNTs/CCDs at this transition. MCDs were identified as long, straight segments with the composite appearance caused by intercalated cells, connected to CNT/CCD branching points at the proximal part and with a progressive narrowing toward the distal part. A total of 25 PT, 75 TAL, 50 DCT, 50 CNT/CCD, and 50 MCD segments were gathered per sample. In unpublished experiments by M. Bleich and N. Himmerkus (University of Kiel, Germany), tubules gathered in this ratio produced comparable actin band densities in Western blots. mRNA was isolated from tubule samples with RNeasy Micro Kits (Qiagen), cDNA was generated with Superscript III (Invitrogen, Darmstadt, Germany) and Superase (Invitrogen). End-point RT-PCR was performed with HOT FIREPol DNA polymerase (Solis Biodyne, Tartu, Estonia) and primers against SCTR (forward, CTCCAACGCTTCCATCTGGT; reverse, GAACTTCCAGCTGCACCTCA) and CFTR (forward, GGGACGAGCCAAAAGCATTG; reverse, CTCCAGAAAAAGCATCGCCG). PCR products were run on 2% agarose gels for 20 minutes at 150 V on ice; bands were sequenced to verify amplification of the correct targets.
Pendrin mRNA Knockdown in Fischer Rat Thyroid Cells
Fischer rat thyroid (FRT) epithelial cells, stably coexpressing human CFTR and F508del (FRT-F508del) were used (generously supplied by L. J. Galietta from the Telethon Institute of Genetics and Medicine, Pozzuoli, Italy). Cells were cultured in F-12 Coon media (Sigma-Aldrich) with 10% FBS, 600 µg/ml zeocin (Invitrogen), 800 µg/ml G418 (Sigma-Aldrich), and penicillin-streptomycin (Gibco) at 37°C in a humidified incubator in 5% (v/v) carbon dioxide (CO2). Cells were transfected with small interfering RNA (siRNA) for Slc26a4 (5′-GGAAAUUGAGAUUCAAGUGtt, Silencer, Ambion; Life Technologies, Darmstadt, Germany) using Lipofectamine 3000 (Thermo Fisher Scientific) and investigated after 72 hours.
Western Blotting
Protein was isolated from cells using a sample buffer containing 25 mM Tris-hydrochloride, 150 mM NaCl, 100 mM dithiothreitol, 5.5% nonidet P-40, 5% glycerol, 1 mM EDTA, and 1% protease inhibitor mixture (Roche, Mannheim, Germany). Proteins were separated by 7% SDS-PAGE and transferred to a polyvinylidene difluoride membrane (GE Healthcare Europe GmbH, Munich, Germany) using a wet transfer unit (Bio-Rad). Membranes were incubated with primary rabbit anti-pendrin IgG (generous gift from Carsten Wagner, University of Zurich22) and rabbit anti–β-actin (1:10,000, A2066; Sigma-Aldrich). Proteins were visualized using horseradish peroxidase–conjugated secondary antibody and enhanced chemiluminescence detection, and analyzed using ImageJ.
Immunohistochemistry
Mouse kidneys were fixed by perfusion with 4% paraformaldehyde and postfixed in 0.5 mol/L sucrose and 4% paraformaldehyde solution. Cryosections of 5 µm were incubated in 0.1% SDS for 5 minutes, washed with PBS, and blocked with 5% BSA and 0.04% Triton X-100 in PBS for 30 minutes. Sections were incubated with primary antibodies against CFTR (rabbit IgG, ACL-006; Alomone Labs), SCTR (rabbit IgG; epitope, AA 170–220; bs-0089R; Bioss, Woburn, MA), and pendrin E-20 (goat IgG; Santa Cruz Biotechnology) in 0.5% BSA and 0.04% Triton X-100 overnight at 4°C, and with anti-rabbit Alexa Fluor 488 IgG or anti-goat Alexa Fluor 546 (Invitrogen) for 1 hour at 37°C. Sections were counterstained with HOE33342 (Sigma-Aldrich). Immunofluorescence was detected using an Axio Observer microscope equipped with ApoTome.2 and ZEN 2.6 (Zeiss, Oberkochen, Germany).
FRT cells grown on glass coverslips were fixed with 4% paraformaldehyde. After washing, the cells were permeabilized with 0.3% (v/v, PBS) Triton X-100 and blocked with 5% (w/v, PBS) BSA for 1 hour at 37°C. The cells were incubated overnight with primary antibodies (1:100) against rabbit anti-CFTR (rabbit IgG, ACL-006) and anti-pendrin (rabbit IgG, NBP1-60106; Novus Biologicals). Binding of the primary antibody was visualized by incubation with appropriate secondary antibodies conjugated with Alexa Fluor 488 or Alexa Fluor 546 (1:300, Molecular Probes; Invitrogen). Nuclei were stained with HOE33342 (0.1 g/ml PBS; AppliChem, Darmstadt, Germany). Glass coverslips were mounted on glass slides with fluorescent mounting medium (DakoCytomation, Hamburg, Germany) and examined with an Axio Observer microscope equipped with ApoTome.2 and ZEN 2.6.
In Vivo Urine pH Measurements
Anesthesia was introduced with an intraperitoneal (i.p.) injection of 10 µl/g body wt of a ketamine-xylazine mix (10 mg/ml ketamine and 1 mg/ml xylazine). An intravenous access was obtained by cannulation of one tail vein and the mice were kept under anesthesia by a continuous rate infusion corresponding to half of the induction dose per hour (on average, 125 µl 0.9% saline per hour) via a syringe pump (Model 33 Twin Syringe Pump; Harvard Apparatus). Mice were placed on a water-heated plate (37°C). Bladder catheterization was introduced and urine was collected every 5 minutes. Urinary pH (pHu) was measured continuously throughout the experiment by placement of a micro pH electrode (pH-200; Unisense, Aarhus, Denmark) and a reference electrode placed into the outflow of the catheter . Following a 30 minute control period, mice were i.p. injected with 100 µl saline containing either secretin (400 pg/g body wt) or just saline.
Urine HCO3− Concentration Measurements
The urine was collected and pooled in 30-minute intervals, allowing measurement of mean HCO3− concentration ([HCO3−]) in the time periods 0–30, 30–60, and 60–90 minutes. In a few experiments with too low urine output, urine was pooled to allow analysis of [HCO3−] before and after secretin application (0–30 minutes and 30–90 minutes). Measurements were performed by a Corning 960 analyzer23 calibrated to small volume samples in the pendrin mice series and by a custom-built infrared CO2 sensor–based system in the rest of the experimental series.24 In urine samples from human participants, the same principle was applied, but the system was redesigned using a GM70 Carbon Dioxide Meter (Vaisala, Helsinki, Finland) and 5 ml sample sizes.
Tubule Perfusion of the CCD
Mice were euthanized by cervical dislocation. The kidneys were removed, placed, and sliced in ice-cold (4°C) control solution (see below). The CCDs were isolated from the cortex with ultrafine forceps. The dissected CCDs were transferred to a tissue chamber and perfused with a concentric pipette system25 with control solution at 37°C. CCDs were mechanically stabilized near the bath bottom with an additional holding pipette. The perfusion chamber was mounted on an inverted fluorescence microscope (Axiovert 100 TV; Zeiss). The setup comprises an inverted microscope with a 63× C-Apochromat 1.2NA water objective, a monochromator (Polychrome IV; Till Photonics, Planegg, Germany) and a charge-coupled device camera (MicroMax, 5 MHz; Princeton Instruments, Trenton, NJ). Image acquisition and data analysis were performed with Metamorph/Metafluor (Universal Imaging, West Chester, PA).
Intracellular pH Measurements
Intracellular pH (pHi) was measured with the ratiometric fluorescent dye 2′,7′-bis-(2-carboxyethyl)-5-(and-6)-carboxyfluorescein, acetoxymethyl ester (BCECF-AM; Invitrogen). CCDs were incubated with 2 µM luminal BCECF-AM in control solution for 10 minutes at 37°C during continuous luminal and bath perfusion. The pHi was measured every 2 seconds as the emission ratio at 490 nm/436 nm excitation. The excitation duration was 10 milliseconds at 436 nm and 25 milliseconds at 490 nm, and a binning factor of four was used for image storage. A 500 nm beam splitter and a 520–560 nm band pass filter were used to collect the emitted light. Luminal BCECF-AM loading in CCDs led to selective dye loading into the intercalated cells.26 The experiments were carried out after a stable, sound fluorescence signal was achieved and the fluorescence of the entire tubule was recorded and used for data analysis of single intercalated cells. Tubules were perfused with a Na+-free solution to bypass any possible effects of the Na+-dependent Cl−/HCO3− exchanger SLC4A827 on the Cl− removal maneuver. The luminal fluid was then accordingly switched to a Cl−- and Na+-free solution following the described Cl− removal maneuver. To assess pendrin activity, we performed a maneuver previously described.28–30 In short, luminal Cl− was removed, which reverses the direction of pendrin-driven HCO3−/Cl− exchange, leading to cellular HCO3− uptake. This increases intracellular [HCO3−] and subsequently pH. The initial alkalization rate reflects pendrin activity. Experimental solutions were as follows: The control contained 118mM NaCl, 1 mM MgSO4, 1.3 mM Ca-gluconate, 5 mM d-glucose, 0.4 mM KH2PO4, 1.6 mM K2HPO4, 25 mM NaHCO3, and 5 mM HEPES. The Na+-free solution contained 125 mM N-methyl-D-glucamine (NMDG), 105 mM hydrochloride, 1 mM MgSO4, 1.3 mM Ca-gluconate, 5 mM d-glucose, 0.4 mM H2PO4, 1.6 mM K2HPO4, 24 mM choline-HCO3, and 5 mM HEPES. Cl−- and Na+-free solution contained 117 mM NMDG, 89 mM d-gluconic acid, 1 mM MgSO4, 4 mM Ca-gluconate, 5 mM d-glucose, 0.4 mM KH2PO4, 1.6 mM K2HPO4, 24 mM choline-HCO3, and 5 mM HEPES. All solutions were titrated to pH 7.40 and gassed at 37°C with 95% oxygen (O2)/5% CO2.
Calibration of the BCECF-AM signal was performed using nigericin in the presence of high potassium ion concentration.31 The calibration solution contained 95 mM potassium chloride, 15 mM NaCl, 0.4 mM NaH2PO4, 1.6 mM Na2HPO4, 5 mM glucose, 1 mM magnesium chloride (MgCl2), 1.3 mM Ca-gluconate, 25 mM HEPES, and 20 mM NMDG, supplemented with 2 µM nigericin. The calibration solutions were titrated to pH 6.5, 7.0, and 7.5 at 37°C.
pHi measurements in FRT cells were performed in great similarity to that described above. Cells were incubated in ringer solution containing 145 mM NaCl, 0.4 mM KH2PO4, 1.6 mM K2HPO4, 5 mM glucose, 1 mM MgCl2, 1.3 mM Ca-gluconate, and 5 mM probenecid, supplemented with 1 µM BCECF-AM and 0.01% pluronic (Life Technologies) for 60 minutes at room temperature. For pHi measurements, cells were mounted in a cell chamber and perfused with HCO3−/CO2 buffered solution containing 120 mM NaCl, 0.4 mM KH2PO4, 1.6 mM K2HPO4, 5 mM glucose, 1 mM MgSO4, 1.5 mM Ca-gluconate, and 25 mM NaHCO3 gassed with 95% O2/5% CO2. Pendrin activity was measured by the initial slope of pHi increase after applying Cl−-free HCO3−/CO2-containing solution containing 115 mM Na-gluconate, 0.4 mM KH2PO4, 1.6 mM K2HPO4, 5 mM glucose, 1 mM MgSO4, 8 mM Ca-gluconate, and 30 mM NaHCO3 gassed with 95% O2/5% CO2. For pHi calibration, cells were superfused with a buffer of pH 6.5, 7.5, and 8.5 containing 105 mM potassium chloride, 1 mM MgCl2, 30 mM HEPES, supplemented with 5 µM nigericin. Very similar optical parameters and the same analyzing software was used as described above with a high-speed polychromator system and a CoolSnap HQ camera (Visitron Systems, Puchheim, Germany).
Measurement of Plasma Secretin
Base Load through Gavage
Mice were given either a HCO3−-containing or a control gavage. Both gavages contained distilled water and 2% (mass) glucose to initiate a meal-typical metabolic response. The HCO3− gavage contained 4 mmol NaHCO3/kg body wt. The control gavage contained only glucose. The volume given by either gavage was 20 µl/g body wt.
Plasma Analysis
At 15 minutes after gavage, the mice were anesthetized with ketamine/xylazine. By puncturing the retro-orbital vein plexus, a small blood sample was collected 30 minutes after gavage using a heparinized glass capillary and plasma HCO3− was immediately measured using an ABL80 blood gas analyzer (Radiometer, Ballerup, Denmark). Immediately thereafter, the mice were euthanized and blood was taken from the heart, the plasma was separated by centrifugation (1000× g for 3 minutes) and snap frozen in liquid nitrogen and stored at −80°C for further use. Secretin plasma levels were measured with an ELISA (catalog number EKM2692; Nordic BioSite, Täby, Sweden) according to the supplier’s instructions.
The Development of the CF Urine Test in Mice
Base Load through Gavage
To test the role of CFTR and pendrin in urinary HCO3− excretion, CFTR and pendrin WT and KO mice were given either a gavage containing 2.24 mmol NaHCO3/kg body wt or a control gavage. Both gavages contained distilled water and 2% (mass) glucose. Each mouse received 20 µl/g body wt. Afterward, mice were placed in metabolic cages with parafilm covered collection funnels. Urine samples were collected immediately with a micropipette and frozen for pH and HCO3− measurements as described above.
Plasma Analysis
In a subset of experiments mice were anesthetized 50 minutes after the gavage load. A blood sample was collected from the retro-orbital vein plexus exactly 1 hour after gavage and plasma [HCO3−] and pH were immediately measured by using an ABL80 gas analyzer.
The CF Urine Test in Humans
From the CF Centre of Aarhus University Hospital, nine adult patients with CF who were homozygous for the ΔF508 mutation, aged 20–45 years old and of both sexes, were included in the study. All participants provided written informed consent before enrolling. The control group consisted of 11 aged-matched, healthy adult volunteers of both sexes. The protocol for the urine test was as follows: a baseline urine sample was obtained at time 0, afterward each participant drank 200 ml of tap water containing NaHCO3 corresponding to a dose of 0.94 mmol/kg body wt. For the following 3 hours, participants voided and drank 200 ml tap water hourly. For each voiding sample, the total volume was measured and 14 ml of urine was stored in 15 ml tubes at −20°C until measurement of pH and [HCO3−]. All samples were analyzed on the same day that the experiment was conducted. A subset of patients was studied at the next control visit at the CF clinic after 4 weeks of treatment with lumacaftor-ivacaftor. A subset of healthy controls and one patient with CF also completed an additional drinking test with only tap water. For all tests, participants fasted overnight and during the experiment, but were allowed to drink water until the start of the experiment. Experiments were initiated between 07:30 am and 11:30 am in the morning. To validate that the decreased urinary [HCO3−] observed in patients with CF was not due to malabsorption of NaHCO3, a venous blood gas analysis was obtained in a subset of patients and healthy controls. See Supplemental Figure 2 for a schematic outline of the protocol.
Statistical Analyses
GraphPad Prism 8.2.1 (GraphPad Software) was used for statistical analysis. All data sets were evaluated for normality by plotting QQ plots for each data set. Data sets not characterized as normally distributed were log-transformed before statistical analysis to improve normal distribution. In all cases this led to a good approximation of a normal distribution. Differences were tested with unpaired t test, paired t test, one-way ANOVA, or two-way ANOVA, with Bonferroni multiple comparison test where required. All tests were two sided and performed at a significance level of 5%. Each figure legend specifies which tests were used. All values are presented as mean values±SEM.
Study Approval
Handling and experiments of mice were approved by the Danish animal welfare regulations (Dyreforsøgstilsynet, approval number 2016-15-0201-01129). The CFTRTS KO mouse was generated and bred according to the guidelines for the welfare of experimental animals issued by the Federal Government of Germany.
The HCO3− drinking test and the use of blood samples were approved by our ethical research committee (approval number 1-10-72-237-18, Region Midtjylland). Written informed consent was given by all study participants.
Results
Expression and Immunolocalization of the SCTR and CFTR in Mouse CCDs
RNA-sequencing data show selective, marked expression of SCTR mRNA in rat CCD α- and β-ICs.13 Using RT-PCR, we found expression of SCTR in all samples of the mouse renal collecting system (DCT, CNT/CCD, and MCD). CFTR expression was found in all PT samples and approximately 50% of the DCT and the CNT/CCD samples (Supplemental Table 2, Supplemental Figure 3). Together, these data confirm the specific expression of the SCTR in the CCD and ascertain close similarity of rat and mouse tissue. Our data also confirm that CFTR is expressed in the CCD, which was reported to be primarily expressed in β-ICs.32
We next performed immunohistochemistry to localize SCTR and CFTR in native mouse kidney. Basolateral SCTR staining was found in pendrin-positive cells and also in other cellular regions of CCD cells (Figure 1A). CFTR staining decorated the luminal membrane of pendrin-positive cells (Figure 1, B and C). No CFTR staining was found in the cortex of CFTR KO mice (Supplemental Figure 4). Control staining without primary antibodies is shown in Supplemental Figure 5. These results show basolateral SCTR localization in β-ICs and CFTR localization in the same membrane domain as pendrin.
Figure 1.
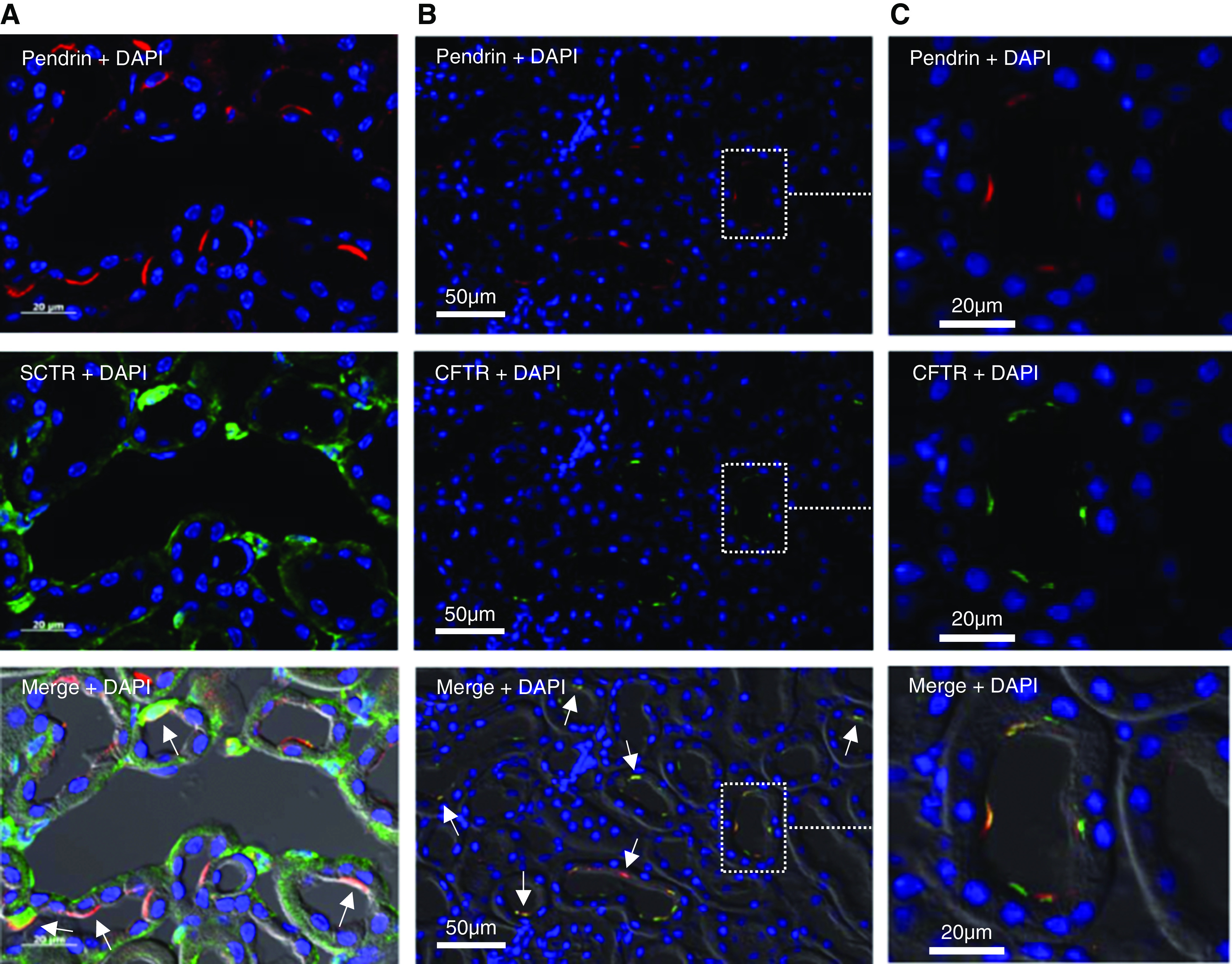
The secretin receptor and CFTR is present in pendrin-positive cells in the cortex of WT mice. (A) Immunohistochemical localization of pendrin (red, upper panel), the SCTR (green, middle panel), and double staining (lower panel) in the cortex of WT mice. (B) Immunohistochemical localization of pendrin (red, upper panel), CFTR (green, middle panel), and double staining (lower panel) in the cortex of WT mice. (C) Higher magnification of the indicated area in (B) showing apical colocalization of CFTR and pendrin in the apical membrane. DAPI, 4′,6-diamidino-2-phenylindole.
Secretin Induces an Acute Urinary Alkalization
pHu in anesthetized pendrin WT mice was monitored for a 30-minute baseline period before i.p. application of an equal saline volume (100 µl) containing either 400 pg/g body wt secretin or saline. A photographic image of the setup is shown in Figure 2A. In Figure 2B, two original pHu traces are shown, one with injection of 400 pg/g body wt secretin and one with control. Within a few minutes after application of secretin, an acute and robust urinary alkalization occurred. Maximal urine alkalization was observed between 20 and 30 minutes after secretin application. Subsequently, we studied the dose dependency of this secretin effect. An increase of pHu was observed with a dose of 4 pg/g body wt. Higher doses of 40, 200, and 400 pg/g body wt secretin (i.p.) caused increasing pHu alkalizations. At the highest used dose of 400 pg/g body wt, the peak pHu increase amounted to approximately 0.38 pH units (Figure 2C). In all ensuing experiments, the dose of 400 pg/g body wt was used. These results show that secretin causes an acute and dose-dependent urinary alkalization.
Figure 2.
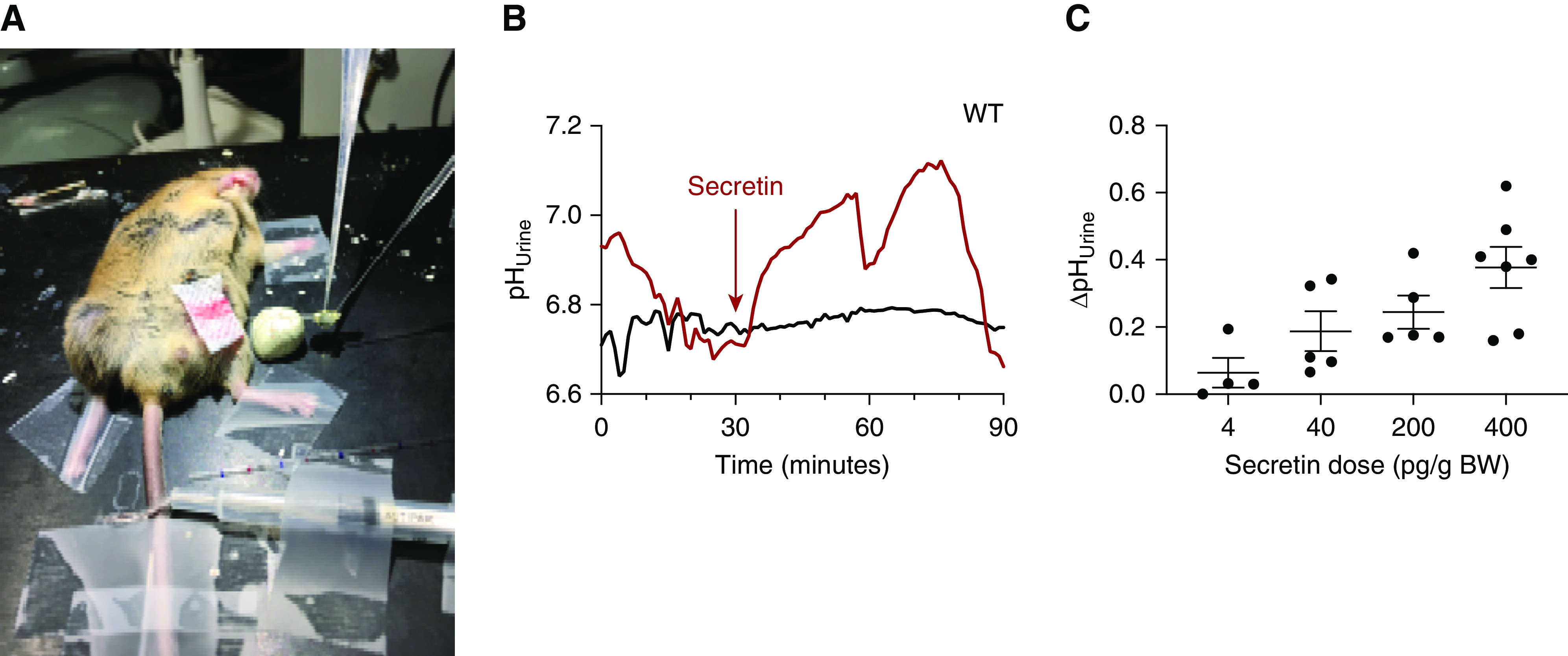
Secretin stimulates acute urine alkalization in mice. (A) Photographic image of the in vivo experimental setup. (B) Two original pHu traces are shown, indicating a prompt urine alkalization after application of 400 pg/g body wt (i.p.) secretin (red) and no effect in the control injected mouse (black). (C) Dose response relationship of the secretin effect on pHu, (n=4–7).
The Secretin-Induced Urine Alkalization Is Absent in Pendrin and CFTR KO Mice
We anticipated that pendrin and CFTR KO mice do not respond to secretin with an acute urine alkalization. We studied the effect of secretin on pHu, urine [HCO3−], and urine HCO3− excretion in six different mouse groups: pendrin, CFTRG, and CFTRTS WT and KO mice. Figure 3, A–C, shows that secretin-treated WT animals responded with a significant, transient urinary alkalization compared with vehicle-treated animals. Secretin-treated WT mice had significantly higher urine [HCO3−] compared with vehicle-treated mice (Figure 3, D–F). Urine HCO3− excretion rate was increased in secretin-treated WT mice as compared with those treated with vehicle (Supplemental Figure 6). No significant secretin-induced increases in pHu, urine [HCO3−], or urinary HCO3− excretion were observed in any KO mice models compared with vehicle-treated ones (Figure 3, A–F, Supplemental Figure 6). Both CFTR KO mice models had significantly lower baseline urine [HCO3−] compared with WT mice (Figure 3, H and I). In pendrin KO mice, secretin induced a urinary acidification starting after some 20 minutes postinjection (Figure 3A). A urinary acidification was absent in WT mice. A concurrent activation of hydrogen ion secretion would cause an underestimation of the activated HCO3− excretion observed in WTs. These results illustrate the absolute pendrin and CFTR dependence of secretin-induced renal HCO3− excretion and that loss of CFTR causes lower resting urine [HCO3−]. An in-depth description of the results from the three different mouse models can be found in the Supplemental Results.
Figure 3.
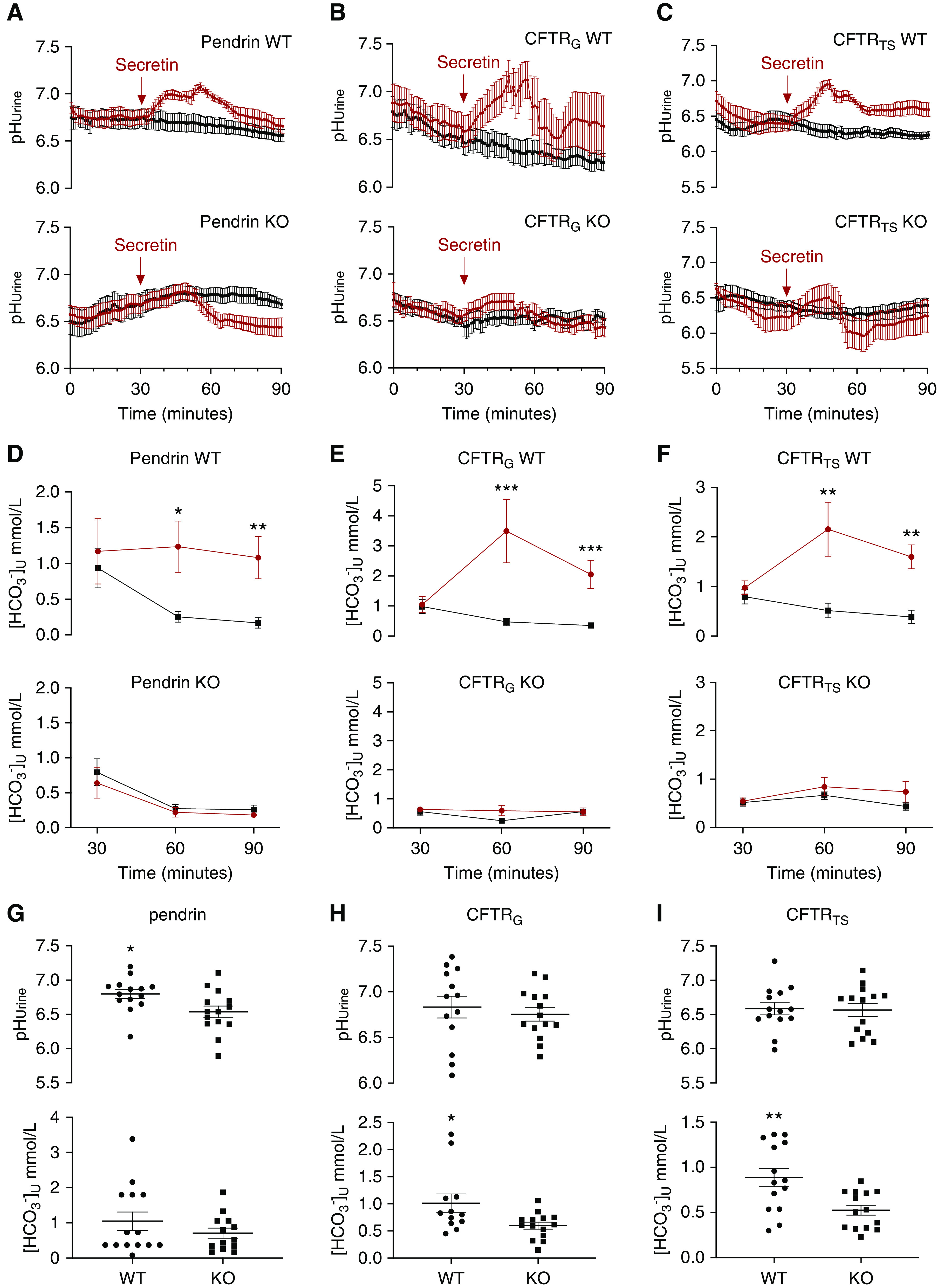
The secretin-stimulated urine alkalization is absent in mice lacking pendrin and dramatically reduced in global and tubule-specific CFTR KO mice. (A–C) Summary pHu curves of control (black) and secretin-injected mice (400 pg/g body wt, i.p.; red) in (A) pendrin WT and KO mice, (B) global CFTR (CFTRG) WT and KO mice, and (C) CFTRTS WT and KO mice. n=6–7 in all experimental series. Peak difference between vehicle- and secretin-treated mice was tested with t test. (D–F) Urine [HCO3−] is significantly higher in WT mice after stimulation with secretin as compared with controls. This effect is absent in KO mice. Urine [HCO3−] in control mice (black) and secretin-treated mice (red) in (D) pendrin WT and KO mice, (E) CFTRG WT and KO mice, and (F) CFTRTS WT and KO mice. n=6–7 in all experimental series. Differences between vehicle- and secretin-treated mice were tested by two-way ANOVA with Bonferroni multiple comparison test. (G–I) Baseline urine [HCO3−] is higher in CFTR WT mice. Baseline pHU and urine [HCO3−] in (G) pendrin WT and KO mice, (H) CFTRG WT and KO mice, and (I) CFTRTS WT and KO mice. n=12–14 in all experimental series. Difference in pHU and urine [HCO3−] between genotypes was tested with t tests. *P<0.05, **P<0.01, ***P<0.001.
Secretin Stimulates HCO3− Secretion in β-ICs of the Isolated Perfused CCD
The above evidence indicates that the action of secretin occurs via binding to the basolateral SCTR in β-ICs and activates CCD HCO3− secretion via this pathway. To study this directly, we used the isolated perfused CCD and pHi measurements of β-ICs. These cells can be identified after luminal loading with BCECF-AM and rapidly removing luminal chloride in the presence of luminal HCO3− (see Figure 4A and Methods section for details). Fast removal of luminal Cl− causes the reversal of the apical anion exchanger pendrin. In β-ICs, this leads to a pendrin-dependent33 cellular uptake of HCO3− and a marked intracellular alkalization (Figure 4, B and C).34 We therefore take the initial alkalization rate after luminal Cl− removal as the activity measure of the pendrin transport rate. First, we performed time control experiments, where the functional activity of pendrin was tested twice as shown in Figure 4B. In these time control experiments, the second luminal zero Cl− removal maneuver showed a slightly increased functional activity of pendrin (n=5 tubules, 5 mice, 25 cells; Figure 4, B and D). Thereafter, using the same protocol, we tested the effect of basolateral secretin addition (10 nM for 10 minutes). As shown in Figure 4C, secretin markedly increased the initial intracellular alkalization rate, an effect that was significantly larger when compared with the time controls (n=5 tubules, 5 mice, 28 cells). In Figure 4D, the summary of all experiments is shown. Interestingly, in CCDs isolated form CFTRG KO mice, basal pendrin function was significantly lower (n=10 tubules, 5 mice, 51–53 cells; Figure 4F) and could not be activated by secretin (n=5 tubules, 5 mice, 23–28 cells; Figure 4E). These results provide proof of principle that secretin directly stimulates β-IC–mediated HCO3− secretion. In the absence of CFTR, pendrin function is reduced and cannot be activated with secretin.
Figure 4.
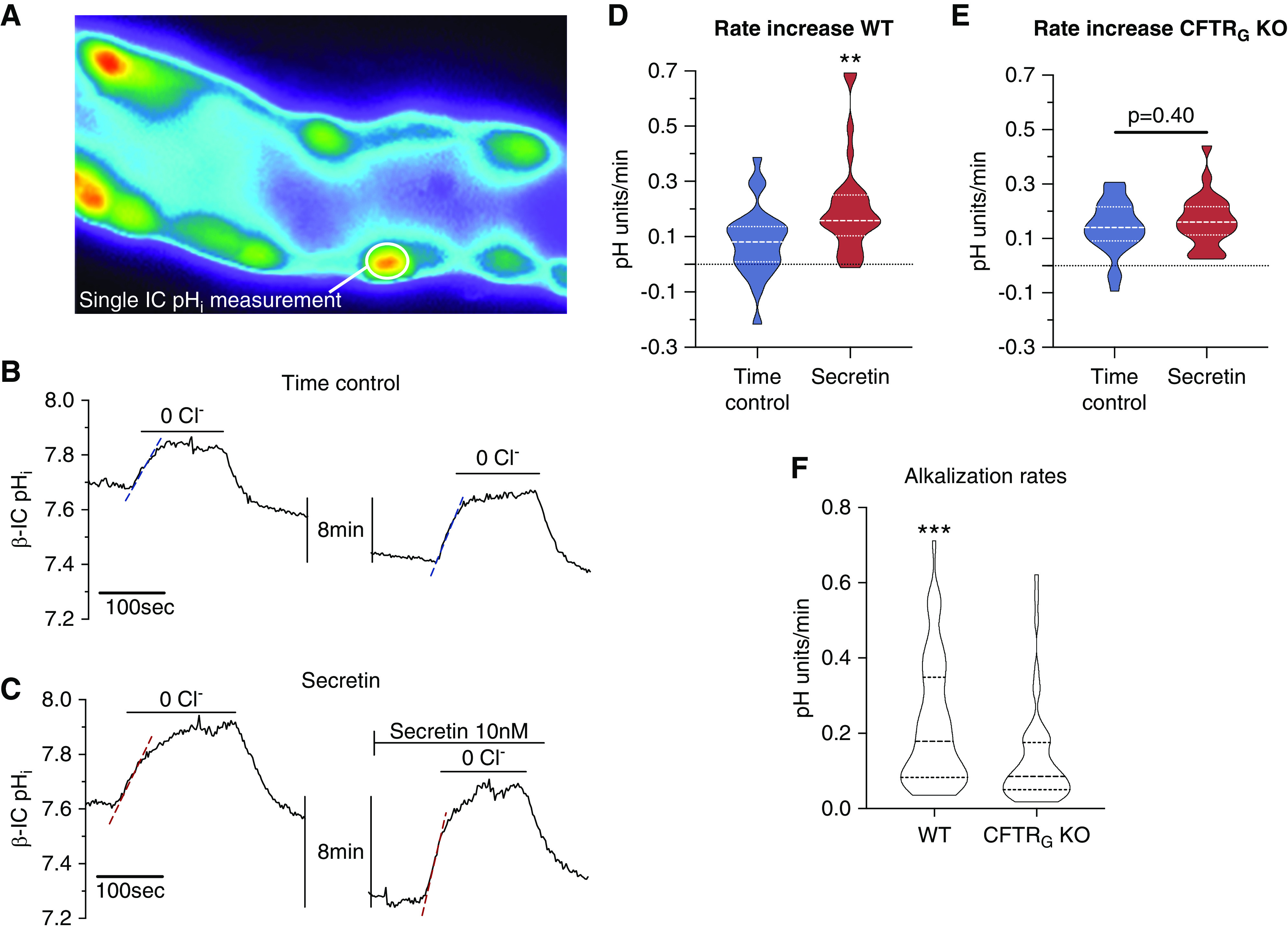
Secretin stimulates pendrin-mediated HCO3− transport in isolated mouse CCD. (A) Fluorescence image of an isolated perfused CCD loaded with luminal BCECF-AM. Multiple intercalated cells protruding into the lumen can be identified. Most of the intercalated cells in the CCD show functional pendrin positivity (prompt intracellular alkalization after luminal chloride removal, data not shown). (B) A time control experiment showing an original pHi recording of a single pendrin-positive cell after a first removal of luminal chloride followed by a second removal of luminal chloride 10 minutes later. (C) A secretin stimulation experiment showing an original pHi recording of a single pendrin-positive cell. Luminal chloride was removed twice, first without stimulation and second after stimulation with 10 nM basolateral secretin for 10 minutes. Note the markedly faster alkalization effect in this experiment after secretin stimulation. (D and E) Summary of the entire experimental series. Secretin stimulated a significantly faster intracellular alkalization rate as compared with time controls, indicating activation of pendrin-mediated HCO3− secretin (n=5 tubules, n=23–28 cells, t test). (F) Note the significantly lower baseline functional pendrin activity in CFTRG KO CCDs (n=10 tubules, n=51–53 cells, t test). **P<0.01, ***P<0.001.
cAMP-Dependent Activation of Pendrin Requires Functional CFTR in the FRT Cell Model
To study the role of CFTR on pendrin function and expression, we applied the FRT cell model.35 Heterologous expression of WT CFTR led to a marked upregulation of pendrin mRNA, protein expression, and apical staining, which were inhibitable by siRNA for pendrin (Figure 5, A–D, Supplemental Figure 7). In contrast, expression of mutated ΔF508 CFTR did not trigger an increase of pendrin mRNA and protein expression or staining. Functional testing of the apical Cl−/HCO3− activity showed relatively low intracellular alkalization rates in parental FRT cells with or without stimulation of IBMX/forskolin. In cells that expressed WT CFTR, the cytosolic elevation of cAMP with IBMX/forskolin triggered a marked activation of apical membrane Cl−/HCO3− activity, likely reporting augmented pendrin function (Figure 5, E and F). This increased functional activation could not be observed when FRT cells expressed ΔF508 CFTR (Figure 5C). Blocking CFTR with inh172 prevented the functional upregulation of Cl−/HCO3− activity in cells expressing WT CFTR (Figure 5C). Furthermore, siRNA knockdown of pendrin markedly reduced the IBMX/forskolin induced Cl−/HCO3− activity (Figure 5G), indicating the majority of the Cl−/HCO3− exchange in this cell model is pendrin dependent. These results indicate that WT CFTR facilitates functional and molecular expression of pendrin in the FRT cell model, aligning well with our data in the isolated perfused CCD that showed reduced pendrin function when CFTR is absent.
Figure 5.
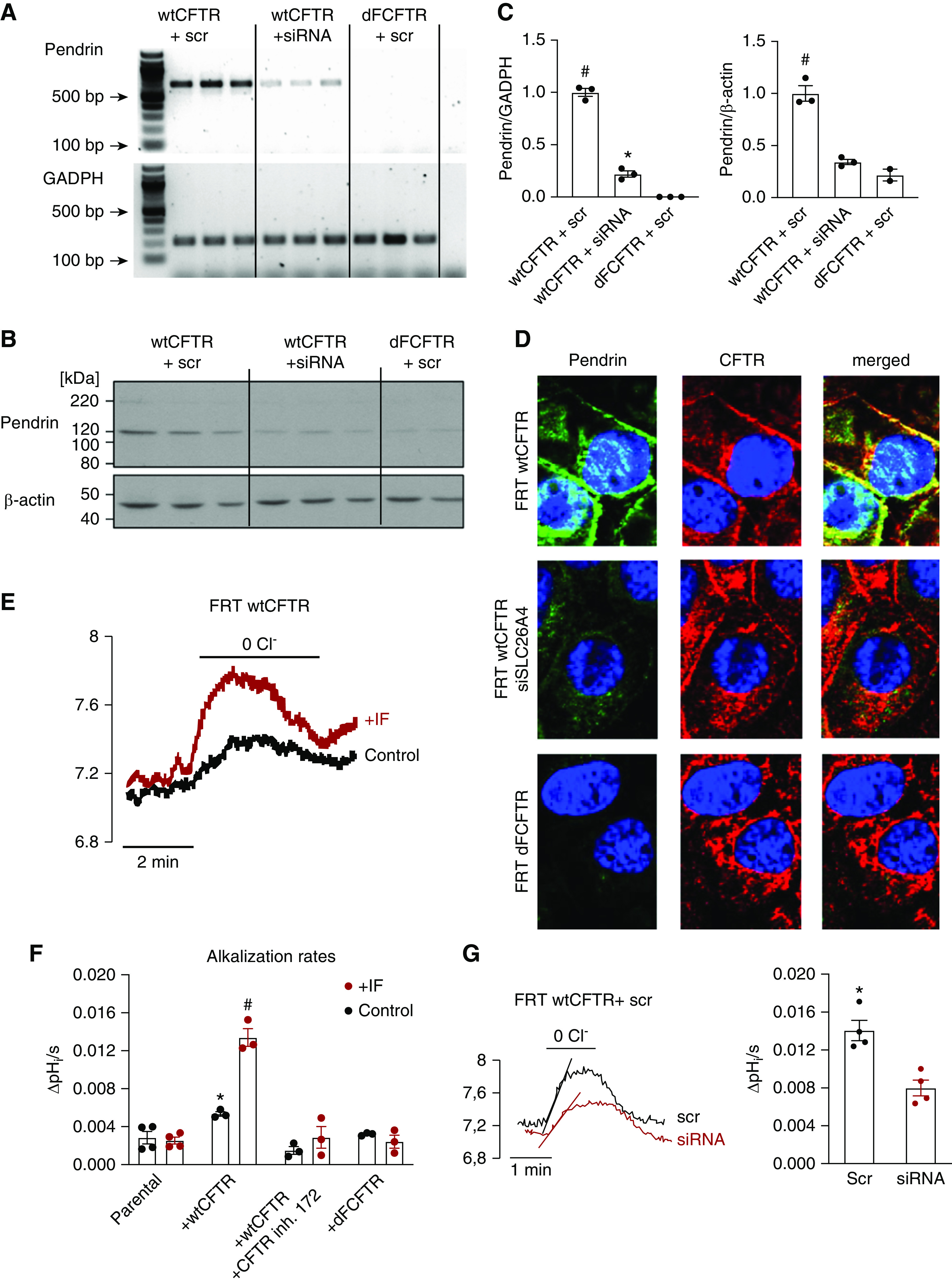
cAMP-dependent activation of pendrin requires functional CFTR in the FRT cell model. (A) mRNA expression of pendrin and glyceraldehyde 3-phosphate dehydrogenase (GADPH) in FRT cells transfected with WT CFTR+scr, WT CFTR+siRNA, or dFCFTR+scr. (B) Protein abundance of pendrin and β-actin in FRT cells transfected with WT CFTR+scr, WT CFTR+siRNA, or dFCFTR+scr. (C) Pendrin mRNA and protein abundance is greatly increased in FRT cells transfected with WT CFTR, can be downregulated with siRNA for pendrin, and is almost absent in FRT cells transfected with dFCFTR. #P<0.001 versus WT CFTR+siRNA and dFCFTR+scr. *P<0.01 versus dFCFTR+scr. One-way ANOVA with Bonferroni multiple comparisons test. (D) Pendrin colocalizes with CFTR at the apical surface of WT CFTR-transfected FRT cells (upper panels), can be downregulated with siRNA for pendrin (middle panels), and is absent in dFCFTR-transfected FRT cells (lower panels). (E) An original experiment showing marked functional upregulation of apical Cl−/HCO3− function after IF stimulation in WT CFTR transfected FRT cells. (F) Effect of IF stimulation on apical Cl−/HCO3− exchange activity in parental-, WT CFTR-, dFCFTR-transfected FRT cells and with preincubation with CFTR inhibitor 172. Note the absence of IF-induced Cl−/HCO3− in cells with either no expression of CFTR, expression of dFCFTR, and in FRT WT CFTR cells treated with CFTR inh. 172 (20 µM, 3 minutes preincubation). #P<0.0001, IF-treated FRT WT CFTR cells were significantly upregulated compared with control and all other IF-treated groups. *P=0.007, significant difference between control FRT WT CFTR cells with and without CFTR inhibitor 172. One-way ANOVA with Bonferroni multiple comparisons test (n=3–4 experiments; on average each experiment included measurement of 150 single cells). (G) An original experiment and summary data showing significantly downregulation of IF-stimulated Cl−/HCO3−-exchange in WT CFTR transfected FRT cells by siRNA knockdown of pendrin. *P<0.01, t test. dFCFTR, ΔF508 CFTR; IF, 100 µM IBMX and 2 µM forskolin; scr, scrambled siRNA; siRNA, siRNA for pendrin; ΔpHi/s, the intracelluar pH increase per second.
Plasma Secretin Is Elevated in Mice Challenged with an Acute Oral HCO3− Load
In mice receiving a gavage load of 4 mmol NaHCO3/kg body wt, we measured a significantly higher plasma [HCO3−] when compared with mice receiving a control gavage (26.7±0.42 versus 23.4±0.49 mM, P<0.001, n=8–9; Figure 6B). Interestingly, significantly elevated plasma secretin levels were measured in the HCO3−-loaded mice as compared with the controls (14.8±1.1 versus 11.0±0.9 ng/ml, P=0.02, n=8–9; Figure 6C). Plotting all measured data pairs of the plasma secretin concentration as a function of plasma [HCO3−] indicated a correlation that was significantly different from zero (Supplemental Figure 8). These results show that an acute plasma alkalosis stimulates an increase in plasma secretin levels.
Figure 6.
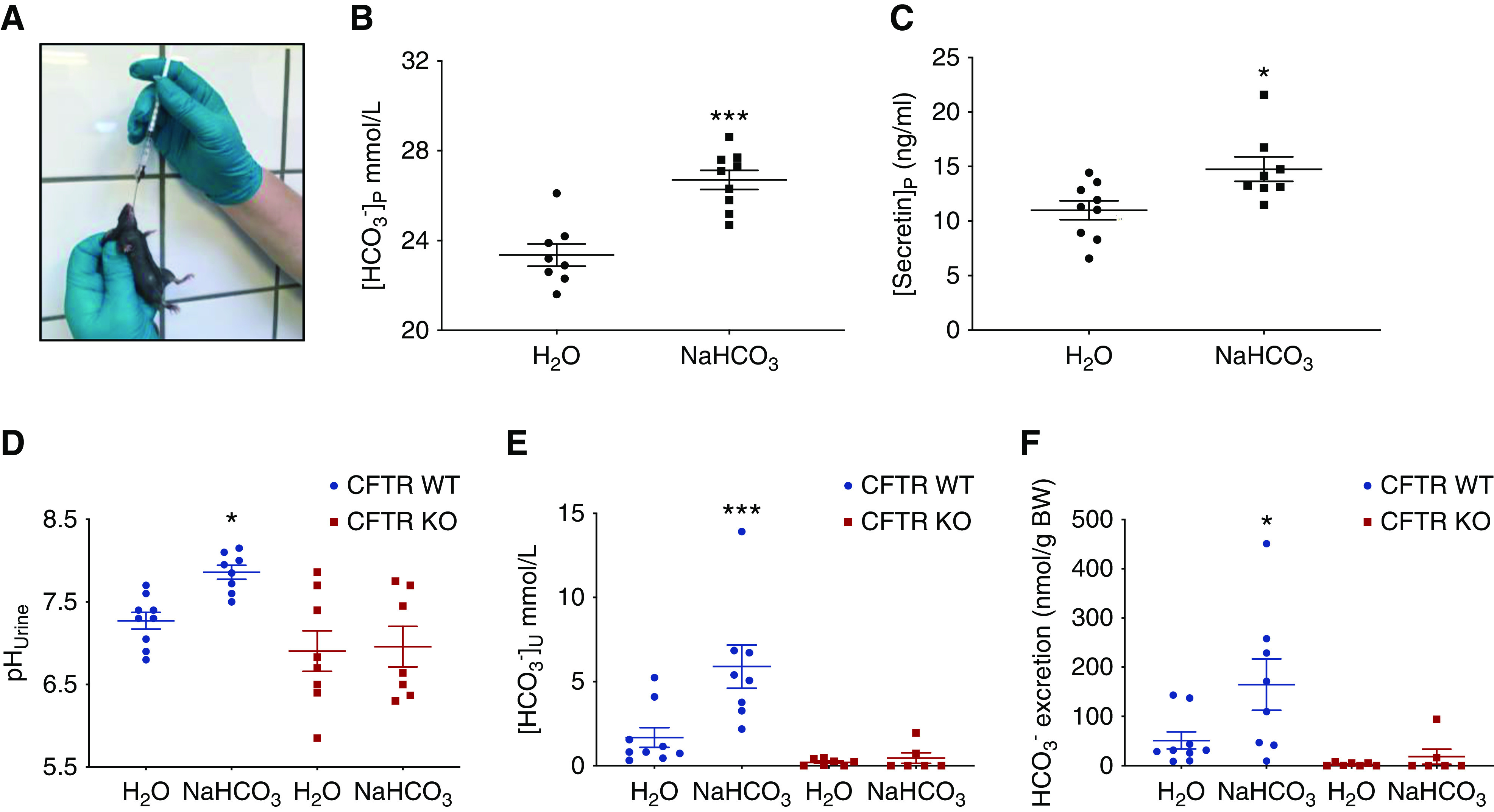
An acute metabolic alkalosis increases plasma secretin. CFTR KO mice are unable to increase their urine HCO3− excretion after an acute oral challenge. (A) Original image illustrating the oral fluid loading procedure. (B) Plasma [HCO3−] in mice loaded with either a control or a HCO3−-containing gastric gavage after 30 minutes. Note the significant elevation of plasma [HCO3−] ([HCO3−]p) in the base loaded mice, t test (n=8–9). (C) Plasma secretin concentration in mice loaded with either a control or a HCO3−-containing gastric gavage. Note the significant elevation in the plasma concentration of secretin ([secretin]p) in the base loaded mice, t test (n=8–9). (D–F) pHu, [HCO3−], and total HCO3− excretion in urine collected over a 3-hour period from awake mice subjected either to a control gavage or a gavage containing 2.24 mmol NaHCO3/kg body wt. Note the absent HCO3− excretion in CFTR KO mice. One-way ANOVA (n=6–9). *P<0.05, ***P<0.001.
CFTR KO Mice Are Unable To Increase Urine HCO3− Excretion after an Oral Challenge
We subsequently challenged awake CFTR WT and KO mice with an oral load of 2.24 mmol NaHCO3/kg body wt and measured their ability to increase urinary HCO3− excretion in the following 3 hours. Figure 6, D–F, shows the remarkable and complete absence to increase pHu, urine [HCO3−], and HCO3− excretion in CFTR KO mice. In additional experiments, mixed venous blood samples taken from anesthetized mice 1 hour after the same oral NaHCO3 challenge showed that CFTR KOs displayed a significant increase of blood pH and blood [HCO3−], where WT animals did not (Supplemental Figure 9, A and B). Similar experiments were performed in pendrin KOs that confirm their complete inability to acutely increase urine pH (Supplemental Figure 9, C–E). These results indicate that intact CFTR and pendrin function is necessary to permit the acute HCO3− excretion response in mice and likely reflect that both KO models have dysfunctional β-ICs.
The CF Urine Test: Human Patients with CF Have a Markedly Reduced Ability To Excrete a Defined Oral HCO3− Load and Show a Significant Improvement after 4 weeks Treatment with Lumacoftor-Ivacaftor
Intrigued by the above mouse data we questioned whether humans that suffer from CF may have a reduced ability to excrete HCO3− with their urine. An HCO3− drinking test was developed. A schematic outline is shown in Supplemental Figure 3. Nine adult patients with CF who were homozygous for the ΔF508 mutation, aged 20–45 years old and of both sexes, were included in the study. The urine HCO3− excretion in fasting participants was measured before and for 3 hours (hourly) after the oral challenge. The control group consisted of similarly aged, healthy adult volunteers of both sexes. We found that patients with CF have a markedly lower baseline urine [HCO3−] before the start of the drinking test (Figure 7B). The oral NaHCO3 drinking test led to a large increase in urine [HCO3−] in control subjects and this increase was greatly reduced in the patients with CF, reaching only about 40% of the response in healthy volunteers (Figure 7D). In addition, the cumulated HCO3− excretion was significantly lower in patients with CF, reaching only about 60% of the response in healthy volunteers (Figure 7E). To investigate whether the decreased urinary HCO3− excretion could be due to a compromised intestinal HCO3− absorption, venous acid-base status was analyzed before and 1 hour after the NaHCO3 challenge in a subset of healthy controls and patients with CF. Both patients with CF and healthy controls had a similar and significantly increased plasma [HCO3−] 1 hour after the challenge, indicating the observed differences are unlikely to be caused my malabsorption of HCO3− (Supplemental Figure 10). A subgroup of patients with CF was started on CFTR-modulating therapy with lumacaftor-ivacaftor treatment and, on their first control visit 4 weeks later, the same oral NaHCO3 drinking test was repeated. In Figure 8, D–F, the results of three patients with CF before and after lumacaftor-ivacaftor treatment are shown. In all patients, the challenged urinary HCO3− excretion after treatment was increased, although to a variable degree. After treatment, the peak urine [HCO3−] measured after 2 hours nearly doubled. These data suggest that lumacaftor-ivacaftor treatment improved the kidneys’ ability to acutely activate urinary HCO3− excretion after an oral challenge.
Figure 7.
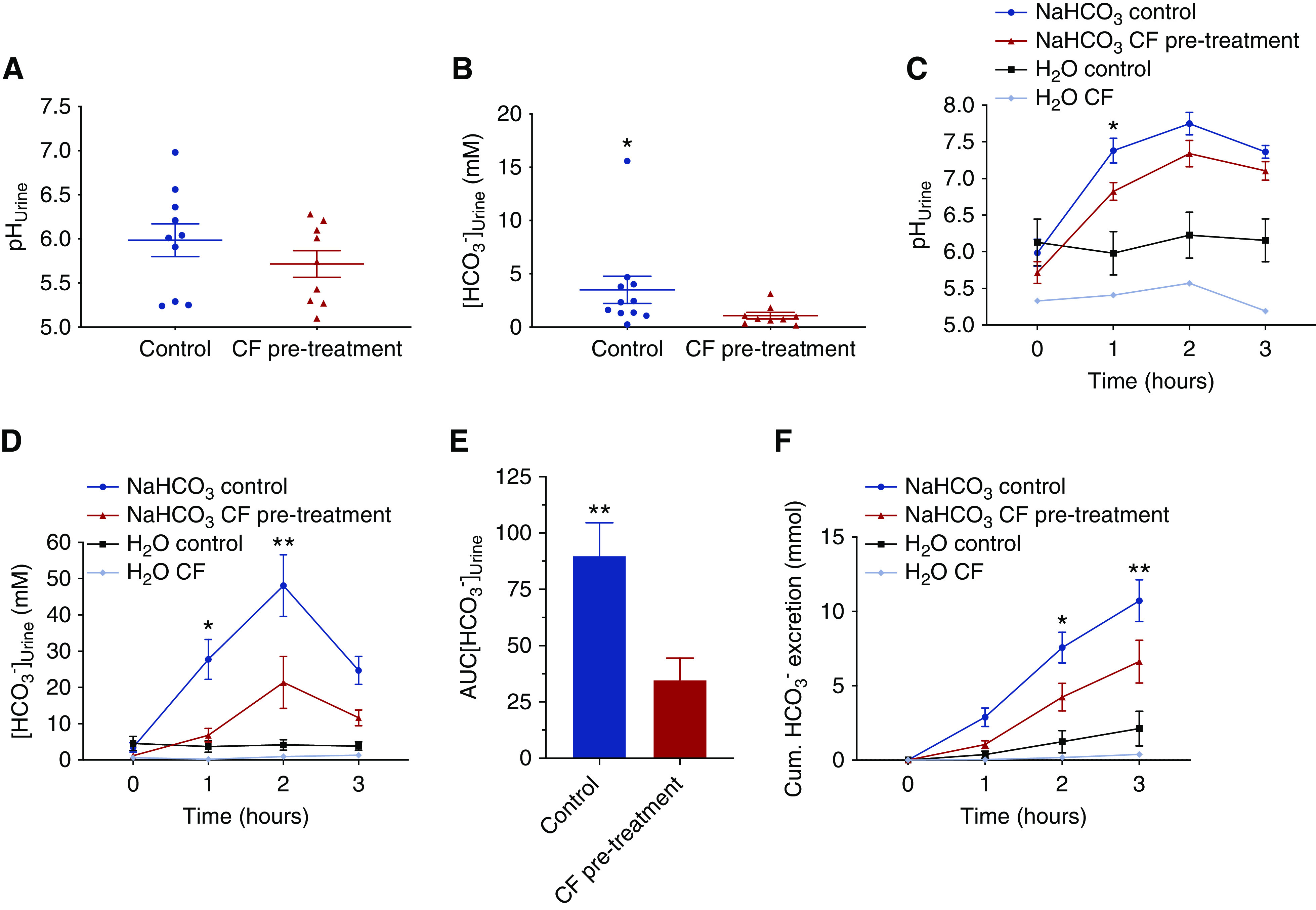
Patients with CF (ΔF508/ΔF508) have reduced urine [HCO3−] and a markedly reduced ability to excrete an oral HCO3− load. (A and B) Baseline urine pH and [HCO3−] in adult patients with CF (n=9) and healthy controls (n=10–11). Note the decreased urine [HCO3−] in patients with CF, t test. *P<0.05. (C, D, and F) Urine pH, urine [HCO3−], and cumulated urinary HCO3− excretion as response to a NaHCO3 drinking test (0.94 mmol NaHCO3/kg body wt) in fasting patients with CF (red, n=9), healthy controls (blue, n=11), and a water drinking test in one fasting patient with CF (bright blue) and fasting healthy controls (black, n=5). Note the greatly reduced urinary [HCO3−] and HCO3− excretion in patients with CF as compared with healthy controls. *P<0.05, **P<0.01 difference between healthy controls and patients with CF subjected to the oral NaHCO3 challenge, two-way ANOVA with Bonferroni multiple comparisons test. (E) Area under the curve (AUC) of urinary [HCO3−] in controls (n=11) and patients with CF (n=9). **P<0.01, t test.
Figure 8.
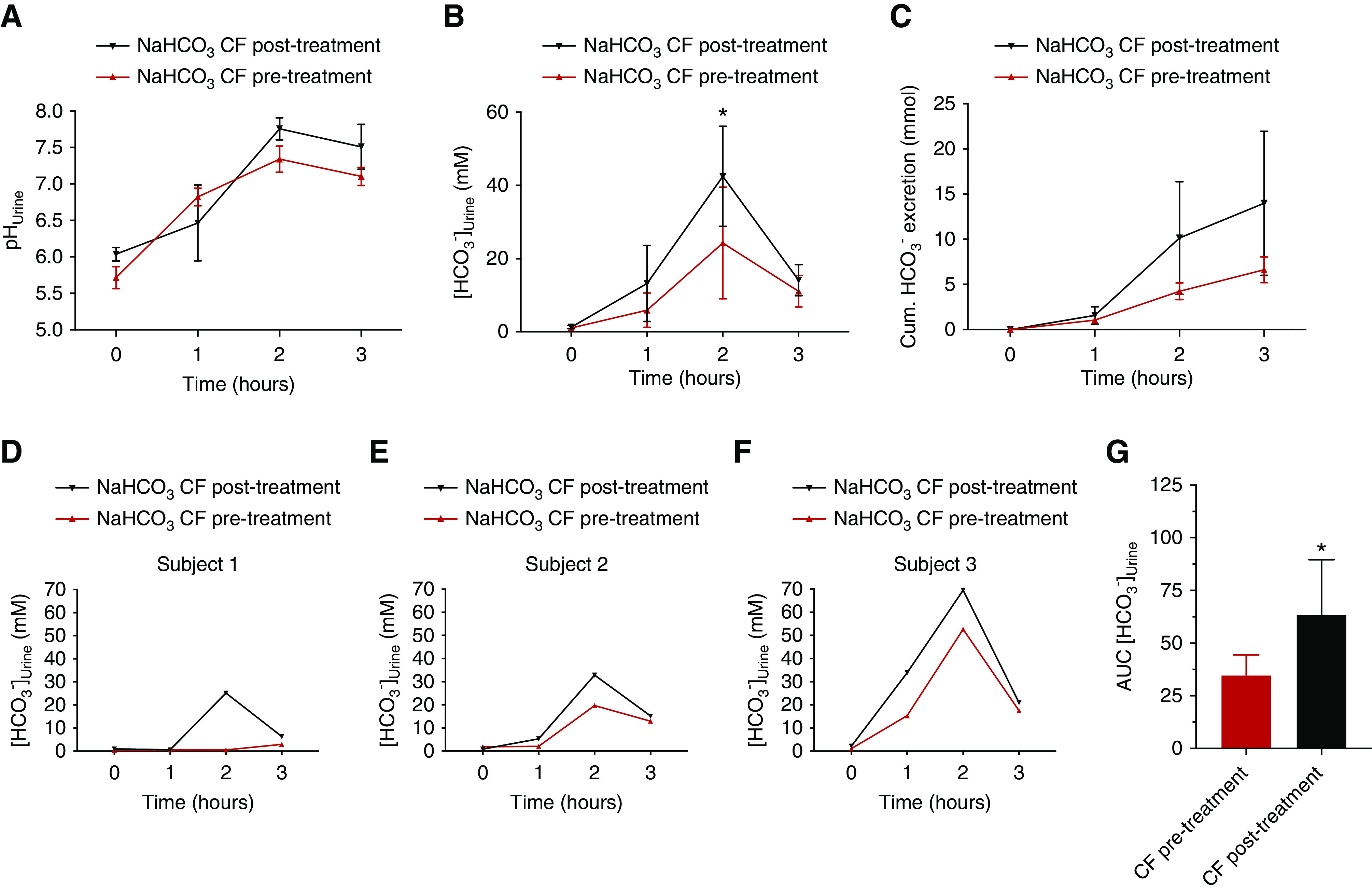
Treatment with lumacaftor-ivacaftor improves the HCO3− excretion deficit. (A–C) pHu, urine [HCO3−], and accumulated urinary HCO3− excretion as response to a NaHCO3 drinking test in patients with CF before (red, n=3) and after 4 weeks of lumacaftor-ivacaftor treatment (black, n=3), *P<0.05, paired t test. (D–F) Individual traces of the three lumacaftor-ivacaftor–treated participants before and after treatment. Note the approximately 100% increase in urine [HCO3−] in each participant. (G) Area under the curve (AUC) of urinary [HCO3−] in patients with CF before treatment (red, n=3) and after 4 weeks of lumacaftor-ivacaftor treatment (black, n=3). *P<0.05, difference between patients with CF before and after treatment, paired t test.
Discussion
The mechanism underlying secretin-induced urinary HCO3− excretion and postprandial urinary alkalization remains enigmatic. Here we show that secretin elicits a marked urinary alkalization, increasing urinary [HCO3−] and renal HCO3− excretion rates in anesthetized mice. This effect is present in multiple mouse models, corroborating data from older studies in healthy humans,8 thus proving mice to be a reliable animal model to study the molecular mechanism of the secretin effect in the kidney. Importantly, we identified that the secretin effect is completely absent in mice lacking the apical Cl−/HCO3− exchanger pendrin, which in the kidney is exclusively expressed in the apical membrane of β-ICs of the CCD and mediates HCO3− secretion and Cl− absorption.13,36–39 Moreover, we have shown that the secretin effect on pHu is dramatically diminished in mice with either global or renal tubule–specific KO of CFTR. The effect on urinary [HCO3−] and on renal HCO3− excretion is completely abolished in both CF animal models, closely corroborating early data in children with CF that were unable to increase renal HCO3− excretion after application of secretin.7 In the isolated perfused mouse CCD, the function of β-ICs was studied directly and secretin was shown to activate base secretion rates. Pendrin function was lower in CCDs lacking CFTR and could not be stimulated by secretin. In the same mouse strain, renal pendrin protein expression was previously shown to be reduced in mice subjected to 3 days of NaHCO3-enriched drinking water.40 The SCTR is a Gαs-coupled 7TM receptor that elicits its effect primarily via an increase of cAMP.11,41 In the FRT cell culture model, we studied the effect of increasing cellular cAMP concentrations with IMBX/forskolin. Parental FRT cells were unresponsive to IBMX/forskolin. When WT CFTR was expressed in FRT cells, a marked expression of pendrin mRNA, protein, and apical staining was detected, which was paralleled by a marked IBMX/forskolin-stimulated upregulation of pendrin function. This functional and molecular upregulation of pendrin was strongly inhibited by mutated CFTR, siRNA for pendrin, and CFTR inhibitor 172. Others have shown functional interaction between CFTR and the STAS domain of SLC26A6 and DRA,42 and our study strongly implies this molecular interaction might be at play for SLC26A4 as well. Altogether, this study provides a comprehensive explanation of the molecular mechanism of secretin-induced renal HCO3− excretion. Secretin leads to SCTR activation in the basolateral membrane of β-ICs of the collecting duct and activation of pendrin and CFTR-dependent HCO3− secretion. This mechanism closely mirrors the molecular mechanism of secretin-induced NaHCO3 secretion in the pancreatic duct, both being defective in patients with CF.2 Eventually, epithelial HCO3− secretion via a pendrin- and CFTR-dependent mechanism was also shown in respiratory epithelia and finds further support by our study.43,44
Our results open the important question of the integrative physiologic role of this secretin-stimulated action in the kidney. We pursued this question by proposing that an acute blood alkalization may cause an increase of plasma secretin that drives renal HCO3− excretion and thus permits homeostatic regulation of plasma HCO3−. Indeed, after an acute metabolic alkalosis, we found a significant increase in plasma secretin. Based on these results we suggest the following physiologic concept: a primary increase of plasma HCO3− triggers an increase of plasma secretin that drives renal HCO3− excretion and thus permits homeostatic regulation of plasma HCO3−. Secretin may originate from the hypothalamus because secretin-containing neurons are present in this neuroendocrine brain region.45 A secondary increase of secretin occurs after a meal, where activation of gastric acid secretion is mirrored by a transient blood alkalization, the so-called alkaline tide.46 During this digestive period, secretin is released from duodenal S cells following the intraluminal acidification of the upper small intestine caused by emptying of the stomach. In parallel, this meal-induced blood alkalization could also trigger secretin release from other sources, as suggested above, to drive renal HCO3− excretion. These considerations provide a plausible hypothesis of the urine component of the alkaline tide. The role of secretin would be to provide an adequate, acute signal to keep plasma HCO3− within tolerable ranges by facilitating renal HCO3− excretion. Our results also imply that secretin serves a so-far-undiscovered role as a “homeostatic HCO3− hormone” that, analogously to other hormones, can regulate ion (here HCO3−) and water homeostasis by controlling the transport processes in the collecting duct. A schematic illustration of the proposed mechanism is shown in Figure 9.
Figure 9.

Secretin-release is stimulated by alkalosis and triggers HCO3- secretion in the CCD. Schematic illustration of the proposed mechanism of secretin-induced urinary HCO3− excretion. Ingestion of a meal will cause a transient alkalosis. Ingestion of a meal itself and/or metabolic alkalosis stimulates secretin release. This will increase plasma concentration of secretin ([secretin]p). An increased plasma concentration of secretin will activate HCO3− secretion by β-ICs in the CCD. This will increase urine pH and [HCO3−] excretion and thereby help compensate metabolic alkalosis. This image was created with BioRender.com.
A major finding of this study is the inability of secretin-induced renal HCO3− excretion in mice that either lack CFTR globally or specifically in the renal tubular system. These results closely match those from human patients with CF that have been shown to be unresponsive in increasing their renal HCO3− excretion after secretin application.7 Patients with CF often present with metabolic alkalosis and a renal cause of this, namely a reduced ability to excrete HCO3−, has been suggested.40,47 Metabolic alkalosis has also been described in patients with pendred syndrome.48,49 It is also worth mentioning that patients with CF have been described with increased levels of plasma secretin.50 The cause of this is not established but one may suggest that metabolic alkalosis could trigger an increase of plasma secretin levels. Taken together, our results provide a mechanistic understanding that the loss of CFTR in the kidney leads to reduced urinary HCO3− excretion because β-IC function is compromised and this, in turn, explains the metabolic alkalosis in patients with CF. We also found that mice and humans require functional CFTR to activate the kidneys’ ability to acutely excrete a defined oral HCO3− load. Patients with CF who are homozygous for the most common mutation (ΔF508) have a greatly attenuated ability to acutely excrete HCO3− by their kidneys. Compared with the findings in the mouse model, we still see some residual function. We suggest that this is explained by the ΔF508 mutation, which still produces CFTR protein, but with significantly reduced function and apical membrane abundance.51 Eventually, we found that treatment with the novel CFTR modulator ivacaftor-lumacaftor increased the ability of patients with CF to acutely increase their urine HCO3− excretion. With these results, we propose that the acute urine HCO3− excretion ability, similar to the sweat test, qualifies as a CF test to quantify functional CFTR losses and monitor drug effectiveness as a possible precision treatment measure for each patient with CF.
Disclosures
P. Berg, K. Kunzelmann and J. Leipziger have a patent entitled “Cystic Fibrosis Urine Test” pending, filed by Aarhus University and University of Regensburg at the European Patent Office (TECH-2017-631-075, patent number 19173359.1-1112). All remaining authors have nothing to disclose.
Funding
This work was supported by Det Frie Forskningsråd (Danish Council for Independent Research) grants DFF 6110-00131 and DFG 387509280-SFB 1350.
Supplementary Material
Acknowledgments
We would like to thank Prof. Hans Beat Hadorn, Munich for his valuable hints and fruitful discussion. We also thank Sebastian Frische for provision of the custom-built small volume HCO3− measuring device. FRT epithelial cells, stably coexpressing human F508del (FRT-F508del) were a generous gift from L. J. Galietta (Telethon Institute of Genetics and Medicine, Pozzuoli, Italy). The pendrin antibody used for Western blotting was a generous gift from Carsten Wagner, University of Zurich. The floxed CFTR mouse was a kind gift from Mitchel Drumm and Dr. Craig Hodges, Case Western Reserve University, Cleveland, Ohio. Expert technical assistance from Karen Skjødt Sørensen, Yordanos Yehdego, and Ane Sørensen is greatly appreciated.
Dr. Peder Berg, Prof. Karl Kunzelmann, and Prof. Jens Leipziger developed the project idea; Dr. Jesper Frank Andersen, Dr. Peder Berg, Dr. Ines Cabrita, Dr. Casper K. Larsen, Prof. Jens Leipziger, Prof. Rainer Schreiber, Dr. Mads V. Sorensen, Dr. Samuel L. Svendsen performed and analyzed the experiments; Dr. Peder Berg, Dr. Søren Jensen-Fangel, Dr. Majbritt Jeppesen, and Prof. Jens Leipziger developed the CF urine test; Prof. Jens Leipziger drafted the manuscript. All authors critically revised the manuscript for important intellectual content and approved the final version.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
See related editorial, “No Zoom Required: Meeting at the β-Intercalated Cells,” on pages 1655–1657.
Supplemental Material
This article contains the following supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2020010053/-/DCSupplemental.
Supplemental Figure 1. Renal tubule-specific knockout of CFTR in mouse kidney.
Supplemental Figure 2. Schematic outline of the protocol for the HCO3−drinking test in humans.
Supplemental Figure 3. Representative gel images from RT-PCR results probing for the SCTR and CFTR along the mouse renal tubular system.
Supplemental Figure 4. Immunohistochemical localization of CFTR and pendrin in CFTR KO mice.
Supplemental Figure 5. Control staining without primary antibodies.
Supplemental Figure 6. Urinary HCO3− excretion rates from in vivo mouse experiments.
Supplemental Figure 7. mRNA expression of pendrin and CFTR in parental, WT CFTR- and deltaF508-transfected FRT cells.
Supplemental Figure 8. Correlation of plasma secretin concentration plotted as a function of plasma HCO3− concentration.
Supplemental Figure 9. Mixed venous blood gas parameters in CFTR- and pendrin KO/WT mice 1 hour after control or NaHCO3 gavage and urinary pH in pendrin WT/KO mice.
Supplemental Figure 10. Venous blood gas parameters before and 1 hour after a NaHCO3 challenge in healthy controls and CF patients prior CFTR modulator treatment.
Supplemental Table 1. Mice characteristics for the mice used in in vivo secretin experiments.
Supplemental Table 2. RT-PCR results probing for presence of CFTR and secretin receptor mRNA along mouse renal tubular system.
References
- 1.Bayliss W, Starling E: The mechanism of pancreatic secretion. J Physiol 28: 325–353, 1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee M, Ohana E, Park H, Yang D, Muallem S: Molecular mechanism of pancreatic and salivary gland fluid and HCO3 secretion. Physiol Rev 92: 39–74, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gunnes P, Waldum H, Rasmussen K, Ostensen H, Burhol P: Cardiovascular effects of secretin infusion in man. Scand J Clin Lab Invest 43: 637–642, 1983. [PubMed] [Google Scholar]
- 4.Marchand G, Ott C, Lang F, Greger R, Knox F: Effect of secretin on renal blood flow, interstitial pressure, and sodium excretion. Am J Physiol 232: F147–F151, 1977. [DOI] [PubMed] [Google Scholar]
- 5.Bell D, McDermott B: Secretin and vasoactive intestinal peptide are potent stimulants of cellular contraction and accumulation of cyclic AMP in rat ventricular cardiomyocytes. J Cardiovasc Pharmacol 23: 959–969, 1994. [DOI] [PubMed] [Google Scholar]
- 6.Kopelman H, Durie P, Gaskin K, Weizman Z, Forstner G: Pancreatic fluid secretion and protein hyperconcentration in cystic fibrosis. N Engl J Med 312: 329–334, 1985. [DOI] [PubMed] [Google Scholar]
- 7.Bretscher D, Hagmann R, Hadorn B, Howald B, Lüthi C, Oetliker O: Response of renal handling of sodium (Na) and bicarbonate (HCO3) to secretin in normals and patients with cystic fibrosis. Pediatr Res 8: 899, 1974 [Google Scholar]
- 8.Viteri A, Poppell J, Lasater J, Dyck W: Renal response to secretin. J Appl Physiol 38: 661–664, 1975. [DOI] [PubMed] [Google Scholar]
- 9.Barbezat G, Isenberg J, Grossman M: Diuretic action of secretin in dog. Proc Soc Exp Biol Med 139: 211–215, 1972. [DOI] [PubMed] [Google Scholar]
- 10.Chu J, Chung S, Lam A, Tam S, Chung S, Chow B: Phenotypes developed in secretin receptor-null mice indicated a role for secretin in regulating renal water reabsorption. Mol Cell Biol 27: 2499–2511, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Procino G, Milano S, Carmosino M, Barbieri C, Nicoletti M, Li J, et al.: Combination of secretin and fluvastatin ameliorates the polyuria associated with X-linked nephrogenic diabetes insipidus in mice. Kidney Int 86: 127–138, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee J, Chou C, Knepper M: Deep sequencing in microdissected renal tubules identifies nephron segment-specific transcriptomes. J Am Soc Nephrol 26: 2669–2677, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen L, Lee J, Chou C, Nair A, Battistone M, Păunescu T, et al.: Transcriptomes of major renal collecting duct cell types in mouse identified by single-cell RNA-seq. Proc Natl Acad Sci U S A 114: E9989–E9998, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gray M, Greenwell J, Argent B: Secretin-regulated chloride channel on the apical plasma membrane of pancreatic duct cells. J Membr Biol 105: 131–142, 1988. [DOI] [PubMed] [Google Scholar]
- 15.Wang Y, Soyombo A, Shcheynikov N, Zeng W, Dorwart M, Marino C, et al.: Slc26a6 regulates CFTR activity in vivo to determine pancreatic duct HCO3- secretion: Relevance to cystic fibrosis. EMBO J 25: 5049–5057, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Everett L, Belyantseva I, Noben-Trauth K, Cantos R, Chen A, Thakkar S, et al.: Targeted disruption of mouse Pds provides insight about the inner-ear defects encountered in Pendred syndrome. Hum Mol Genet 10: 153–161, 2001. [DOI] [PubMed] [Google Scholar]
- 17.Zhou L, Dey C, Wert S, DuVall M, Frizzell R, Whitsett J: Correction of lethal intestinal defect in a mouse model of cystic fibrosis by human CFTR. Science 266: 1705–1708, 1994. [DOI] [PubMed] [Google Scholar]
- 18.Hodges C, Cotton C, Palmert M, Drumm M: Generation of a conditional null allele for Cftr in mice. Genesis 46: 546–552, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kos C: Cre/loxP system for generating tissue-specific knockout mouse models. Nutr Rev 62: 243–246, 2004. [DOI] [PubMed] [Google Scholar]
- 20.Bouchard M, Souabni A, Busslinger M: Tissue-specific expression of cre recombinase from the Pax8 locus. Genesis 38: 105–109, 2004. [DOI] [PubMed] [Google Scholar]
- 21.Larsen C, Jensen I, Sorensen M, de Bruijn P, Bleich M, Praetorius H, et al.: Hyperaldosteronism after decreased renal K+ excretion in KCNMB2 knock-out mice. Am J Physiol Renal Physiol 310: F1035–F1046, 2016 [DOI] [PubMed] [Google Scholar]
- 22.Wagner C, Finberg K, Stehberger P, Lifton R, Giebisch G, Aronson P, et al.: Regulation of the expression of the Cl-/anion exchanger pendrin in mouse kidney by acid-base status. Kidney Int 62: 2109–2117, 2002. [DOI] [PubMed] [Google Scholar]
- 23.Savory D, Pryce J: Quality control of measurements of pH, carbon dioxide tension, and total carbon dioxide in plasma. Clin Chem 24: 1618–1619, 1978. [PubMed] [Google Scholar]
- 24.Trepiccione F, Iena F, Catalini L, Carpi F, Koed M, Frische S: Measurement of total CO2 in microliter samples of urine and other biological fluids using infrared detection of CO2. Pflugers Arch 469: 1267–1275, 2017. [DOI] [PubMed] [Google Scholar]
- 25.Greger R, Hampel W: A modified system for in vitro perfusion of isolated renal tubules. Pflugers Arch 389: 175–176, 1981. [DOI] [PubMed] [Google Scholar]
- 26.Weiner I, Hamm L: Use of fluorescent dye BCECF to measure intracellular pH in cortical collecting tubule. Am J Physiol 256: F957–F964, 1989. [DOI] [PubMed] [Google Scholar]
- 27.Leviel F, Hübner C, Houillier P, Morla L, El Moghrabi S, Brideau G, et al.: The Na+-dependent chloride-bicarbonate exchanger SLC4A8 mediates an electroneutral Na+ reabsorption process in the renal cortical collecting ducts of mice. J Clin Invest 120: 1627–1635, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weiner I, Weill A, New A: Distribution of Cl-/HCO3- exchange and intercalated cells in rabbit cortical collecting duct. Am J Physiol 267: F952–F964, 1994. [DOI] [PubMed] [Google Scholar]
- 29.Lynch I, Greenlee M, Gumz M, Rudin A, Xia S, Wingo C: Heterogeneity of H-K-ATPase-mediated acid secretion along the mouse collecting duct. Am J Physiol Renal Physiol 298: F408–F415, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tokonami N, Morla L, Centeno G, Mordasini D, Ramakrishnan S, Nikolaeva S, et al.: α-Ketoglutarate regulates acid-base balance through an intrarenal paracrine mechanism. J Clin Invest 123: 3166–3171, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thomas J, Buchsbaum R, Zimniak A, Racker E: Intracellular pH measurements in Ehrlich ascites tumor cells utilizing spectroscopic probes generated in situ. Biochemistry 18: 2210–2218, 1979. [DOI] [PubMed] [Google Scholar]
- 32.Todd-Turla K, Rusvai E, Náray-Fejes-Tóth A, Fejes-Tóth G: CFTR expression in cortical collecting duct cells. Am J Physiol 270: F237–F244, 1996. [DOI] [PubMed] [Google Scholar]
- 33.Amlal H, Petrovic S, Xu J, Wang Z, Sun X, Barone S, et al.: Deletion of the anion exchanger Slc26a4 (pendrin) decreases apical Cl(-)/HCO3(-) exchanger activity and impairs bicarbonate secretion in kidney collecting duct. Am J Physiol Cell Physiol 299: C33–C41, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roos A, Boron W: Intracellular pH. Physiol Rev 61: 296–434, 1981. [DOI] [PubMed] [Google Scholar]
- 35.Sheppard D, Carson M, Ostedgaard L, Denning G, Welsh M: Expression of cystic fibrosis transmembrane conductance regulator in a model epithelium. Am J Physiol 266: L405–L413, 1994. [DOI] [PubMed] [Google Scholar]
- 36.Wall S, Lazo-Fernandez Y: The role of pendrin in renal physiology. Annu Rev Physiol 77: 363–378, 2015. [DOI] [PubMed] [Google Scholar]
- 37.Royaux I, Wall S, Karniski L, Everett L, Suzuki K, Knepper M, et al.: Pendrin, encoded by the Pendred syndrome gene, resides in the apical region of renal intercalated cells and mediates bicarbonate secretion. Proc Natl Acad Sci U S A 98: 4221–4226, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Soleimani M, Greeley T, Petrovic S, Wang Z, Amlal H, Kopp P, et al.: Pendrin: an apical Cl-/OH-/HCO3- exchanger in the kidney cortex. Am J Physiol Renal Physiol 280: F356–F364, 2001. [DOI] [PubMed] [Google Scholar]
- 39.Wall S, Kim Y, Stanley L, Glapion D, Everett L, Green E, et al.: NaCl restriction upregulates renal Slc26a4 through subcellular redistribution: Role in Cl- conservation. Hypertension 44: 982–987, 2004. [DOI] [PubMed] [Google Scholar]
- 40.Varasteh Kia M, Barone S, McDonough A, Zahedi K, Xu J, Soleimani M: Downregulation of the Cl-/HCO3-exchanger pendrin in kidneys of mice with cystic fibrosis: Role in the pathogenesis of metabolic alkalosis. Cell Physiol Biochem 45: 1551–1565, 2018. [DOI] [PubMed] [Google Scholar]
- 41.Ulrich C 2nd, Holtmann M, Miller L: Secretin and vasoactive intestinal peptide receptors: Members of a unique family of G protein-coupled receptors. Gastroenterology 114: 382–397, 1998. [DOI] [PubMed] [Google Scholar]
- 42.Ko S, Zeng W, Dorwart M, Luo X, Kim K, Millen L, et al.: Gating of CFTR by the STAS domain of SLC26 transporters. Nat Cell Biol 6: 343–350, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Garnett J, Hickman E, Burrows R, Hegyi P, Tiszlavicz L, Cuthbert A, et al.: Novel role for pendrin in orchestrating bicarbonate secretion in cystic fibrosis transmembrane conductance regulator (CFTR)-expressing airway serous cells. J Biol Chem 286: 41069–41082, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim D, Huang J, Billet A, Abu-Arish A, Goepp J, Matthes E, et al.: Pendrin mediates bicarbonate secretion and enhances cystic fibrosis transmembrane conductance regulator function in airway surface epithelia. Am J Respir Cell Mol Biol 60: 705–716, 2019. [DOI] [PubMed] [Google Scholar]
- 45.Chu J, Lee L, Lai C, Vaudry H, Chan Y, Yung W, et al.: Secretin as a neurohypophysial factor regulating body water homeostasis. Proc Natl Acad Sci U S A 106: 15961–15966, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Niv Y, Fraser G: The alkaline tide phenomenon. J Clin Gastroenterol 35: 5–8, 2002. [DOI] [PubMed] [Google Scholar]
- 47.Al-Ghimlas F, Faughnan M, Tullis E: Metabolic alkalosis in adults with stable cystic fibrosis. Open Respir Med J 6: 59–62, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kandasamy N, Fugazzola L, Evans M, Chatterjee K, Karet F: Life-threatening metabolic alkalosis in Pendred syndrome. Eur J Endocrinol 165: 167–170, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pela I, Bigozzi M, Bianchi B: Profound hypokalemia and hypochloremic metabolic alkalosis during thiazide therapy in a child with Pendred syndrome. Clin Nephrol 69: 450–453, 2008. [DOI] [PubMed] [Google Scholar]
- 50.Windstetter D, Schaefer F, Schärer K, Reiter K, Eife R, Harms H, et al.: Renal function and renotropic effects of secretin in cystic fibrosis. Eur J Med Res 2: 431–436, 1997. [PubMed] [Google Scholar]
- 51.O’Sullivan B, Freedman S: Cystic fibrosis. Lancet 373: 1891–1904, 2009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.