

Significance Statement
Signaling to integrins is complex and depends on ligands and their binding sites. Signaling-competent integrin ligands that protect podocyte function remain unknown. This study demonstrates that the coagulation protease-activated protein C (aPC) binds via its RGD sequence to podocyte integrin-β3. Disruption of the aPC–integrin-β3 interaction results in excess RhoA activation and podocyte dysfunction. These findings identify the RGD-mediated aPC–integrin-β3 interaction as a rheostat of RhoA signaling, which is disrupted in diabetic nephropathy. Protease-activated receptor 1 (PAR1) antagonism could ameliorate excess RhoA signaling in the absence of aPC–integrin-β3 interaction. These data identify a new function of podocyte integrin-β3 and provide a mechanistic rationale for PAR antagonism as a therapeutic approach for diabetic nephropathy.
Keywords: diabetic nephropathy, integrin αvβ3, RhoA signaling, activated protein C, coagulation proteases
Visual Abstract

Abstract
Background
Diabetic nephropathy (dNP), now the leading cause of ESKD, lacks efficient therapies. Coagulation protease–dependent signaling modulates dNP, in part via the G protein–coupled, protease-activated receptors (PARs). Specifically, the cytoprotective protease-activated protein C (aPC) protects from dNP, but the mechanisms are not clear.
Methods
A combination of in vitro approaches and mouse models evaluated the role of aPC-integrin interaction and related signaling in dNP.
Results
The zymogen protein C and aPC bind to podocyte integrin-β3, a subunit of integrin-αvβ3. Deficiency of this integrin impairs thrombin-mediated generation of aPC on podocytes. The interaction of aPC with integrin-αvβ3 induces transient binding of integrin-β3 with Gα13 and controls PAR-dependent RhoA signaling in podocytes. Binding of aPC to integrin-β3 via its RGD sequence is required for the temporal restriction of RhoA signaling in podocytes. In podocytes lacking integrin-β3, aPC induces sustained RhoA activation, mimicking the effect of thrombin. In vivo, overexpression of wild-type aPC suppresses pathologic renal RhoA activation and protects against dNP. Disrupting the aPC–integrin-β3 interaction by specifically deleting podocyte integrin-β3 or by abolishing aPC’s integrin-binding RGD sequence enhances RhoA signaling in mice with high aPC levels and abolishes aPC’s nephroprotective effect. Pharmacologic inhibition of PAR1, the pivotal thrombin receptor, restricts RhoA activation and nephroprotects RGE-aPChigh and wild-type mice.
Conclusions aPC–integrin-αvβ3 acts as a rheostat, controlling PAR1-dependent RhoA activation in podocytes in diabetic nephropathy. These results identify integrin-αvβ3 as an essential coreceptor for aPC that is required for nephroprotective aPC-PAR signaling in dNP.
Diabetic nephropathy (dNP) is now the leading cause of ESKD in industrialized countries.1 Early disease stages of dNP are in principle reversible,2 yet specific medical approaches exploiting this therapeutic window are scarce. Recent findings have established that coagulation proteases and their receptors modulate dNP. Deciphering the underlying mechanism may offer new therapeutic strategies for this condition.
Beyond controlling hemostasis, coagulation proteases modulate cellular homeostasis via receptor-dependent mechanisms in a highly context-specific fashion.3–5 In particular, the cytoprotective effects of the anticoagulant serine protease-activated protein C (aPC) seem appealing, because these effects and the anticoagulant effects of aPC depend largely on disjunct molecular structures and can hence be specifically exploited.4,6 Accordingly, aPC variants with reduced anticoagulant efficacy are being investigated in clinical stroke trials, and small compounds called parmodulins that mimic biased aPC signaling via the pivotal G protein–coupled aPC receptor, protease-activated receptor 1 (PAR1), have been developed.6–8
Activation of the zymogen protein C (PC) by the thrombomodulin-thrombin complex is enhanced by the coreceptor endothelial PC receptor (EPCR) on endothelial cells. In addition, EPCR promotes biased signaling of aPC via PAR1 on endothelial cells.9–13 However, aPC also conveys cytoprotective effects in extravascular compartments—e.g., the skin, brain, and kidneys, targeting keratinocytes, neurons, and renal epithelial cells (podocytes, tubular cells), respectively14–16—in a manner partially independent of EPCR.15,17–19 aPC signaling on podocytes depends on species-specific PAR heterodimers (PAR2/PAR3 on human podocytes and PAR1/PAR3 on mouse podocytes), but appears to be independent of EPCR.15 It remains unknown whether receptors other than PARs are required for aPC-mediated signaling in podocytes.
In addition to PARs and EPCR, various other receptors interacting with aPC have been identified.20,21 aPC binds β-integrins via its RGD sequence, regulating the adhesion of neutrophils and macrophages.19,22 A potential role of an integrin-aPC interaction on podocytes appears plausible given aPC’s nephroprotective effects15,23,24 and the pivotal functions of integrins25,26 in podocytes. The functions of podocyte integrins appear to be twofold, because both integrin absence/integrin defects and excess integrin activation promote renal pathologies.27,28 Because podocytes express integrin-β1 and integrin-β3,25 we hypothesized that aPC interacts via its RGD sequence with podocyte integrin-β1 and/or integrin-β3 to modulate podocyte function.
Methods
Reagents
The following antibodies were used in this study: antibodies against integrin-β1 (sc-59827) and Gα13 (sc-410) from Santa Cruz Biotechnology; antibodies against mouse nephrin (AF3159) from R&D Systems; antibodies against integrin-β3 (13166), RhoA (2117), Rac1 (4651), and anti-rabbit horseradish peroxidase (HRP)–conjugated secondary antibody from Cell Signaling Technology; antibodies against active-conformation integrin-β3 AP5 (EBW107) from Kerafast, Inc.; antibodies against PC (ab36407) and WT-1 (ab89901) from Abcam; and an antibody against human PC (PA5-28321; Thermo Fisher Scientific). The mAb HAPC1573 has been described previously.8
The following inhibitors, agonists, and antagonists were used in this study: RGDS, Cyclo-RGDfv, human thrombin, and SCH79797 (Sigma). Other reagents included DMEM, trypsin-EDTA, penicillin, streptomycin, FCS, FBS, and HEPES from Thermo Fisher Scientific (Dreieich, Germany); BCA reagent from Perbio Science (Bonn, Germany); VECTASHIELD mounting medium with 4′,6-diamidino-2-phenylindole (DAPI), antigen-unmasking solution (Tris-based), and an M.O.M. kit from Vector Laboratories; RhoA and Rac1 pull-down assays from Cytoskeleton, Inc.; polyvinylidene difluoride membranes and Immobilon Western Chemiluminescent HRP Substrate from Merck Millipore; streptozotocin (STZ) from Enzo Life Sciences (Lörrach, Germany); Accu-Chek test strips, an Accu-Check glucometer, and protease inhibitor cocktail from Roche Diagnostics (Mannheim, Germany); albumin fraction V, Gill II hematoxylin, acrylamide, agarose, Periodic acid–Schiff (PAS) reagent, and ROTIHistokitt synthetic mounting medium from Carl Roth (Karlsruhe, Germany); and aqueous mounting medium from Zytomed (Berlin, Germany). Wild-type aPC and RGE-aPC were expressed and purified as described previously.22,29
Animals
APChigh and EPCRδ/δ mice have been previously described.10,30 Integrin-β3LoxP/LoxP mice (provided by Katherine Weilbaecher) were crossed with PodCre mice to generate mice with podocyte-specific deletion of integrin-β3.31,32 All mice were backcrossed onto the C57BL/6J background for at least eight generations and were routinely maintained on the C57BL/6J background. Only littermates were used as controls in this study, and the mice were randomly assigned to the experimental groups. The presence of targeted genes and transgenes was routinely determined by PCR analyses of tail DNA. Wild-type C57BL/6, db/dm, and db/db (C57BL/KSJRj-db) mice were obtained from Janvier (Le Genest-Saint-Isle, France). In some mice, we intraperitoneally injected aPC (1 mg/kg body wt, every alternate day) or an equal volume of PBS starting at 18 weeks of age until 1 day before the analyses.24,33 Mice were analyzed at age 24 weeks. The animal experiments were conducted following standards and procedures approved by the local Animal Care and Use Committee (Landesverwaltungsamt Halle, Germany).
Generation of RGE-APChigh Mutant Mice
To generate mice expressing a human hyperactivatable PC (PROC) mutant with an additional mutation in the RGD sequence (RGE-APChigh mice), the same construct previously used to generate APChigh mice was used. A point mutation (D222E) was generated by site-directed mutagenesis using the QuikChange mutagenesis kit (Agilent, Santa Clara, CA; Supplemental Figure 1A). Correct mutagenesis was confirmed by the presence of a newly generated BanII site resulting in a 521-bp and a 203-bp fragment upon BanII digestion of a PCR amplimer (using the primers P1, 5′-CAA GCC GGT TTA CTC TGA CCC-3′, and P2, 5′-CCT CTA TGC ACT CCC GCT CCA GGC-3′). Before pronucleus injection, correct mutagenesis was additionally confirmed by sequencing. Transgenic mice were generated in the transgenic core facility at the medical faculty of Heidelberg University. The transgenic RGE-APChigh mice were identified by PCR using the abovementioned primers, P1 and P2, followed by BanII digestion of the amplimer (Supplemental Figure 1B). Transgenic RGE-APChigh mice appeared grossly normal, reproduced normally, and did not show any signs of spontaneous bleeding.
Based on expression analyses using semiquantitative RT-PCR, the transgene is expressed predominantly in the liver but also in the brain and at very low levels in the kidney (Supplemental Figure 1C), which is consistent with the previously reported expression pattern of a hyperactivatable D167F/D172K PC mutant using the same promoter.10
The levels of APC in mice were determined using a capture assay with the antibody HAPC1555, which is highly specific for activated human PC and does not detect mouse (activated) PC.34 The mean plasma level of aPC was 5.3 ng/ml in RGE-APChigh mice, quite comparable to the values determined in parallel in APChigh mice (5.8 ng/ml; Supplemental Figure 1D). The total blood volume loss after a standardized tail injury was markedly greater in RGE-APChigh mice (102±11 µl) than in wild-type littermates (4.5±1.2 µl), but did not differ significantly from that observed in APChigh mice (98±31 µl; Supplemental Figure 1E), establishing that the mutant hyperactivatable RGE-PC mutant has an anticoagulant effect comparable to that of hyperactivatable RGD-PC.
STZ Model and In Vivo Interventions in Mice
Insulinopenia and persistent hyperglycemia were induced in experimental mice using low-dose STZ as described previously.10 Blood glucose levels were determined at least weekly in mice to ensure persistent hyperglycemia. Mice displaying blood glucose levels >500 mg/dl received individual injections of 1–2 U insulin glargine. Blood glucose levels shown within the manuscript reflect the last blood glucose measurement before euthanizing the mice. The STZ model results in mild albuminuria and thus reflects early stages of dNP, which are in principle reversible,2 making this an attractive model with which to study therapeutic approaches in early dNP. Subgroups of mice received various interventions. We intraperitoneally injected the mice in the different subgroups with the PAR1 antagonist SCH79797 (15 µg/kg), Cyclo-RGDfv (0.5 or 2 mg/kg body wt, dissolved in PBS), or PBS once daily starting 10 weeks after the last STZ injection until 1 day before the analyses (week 24).35–39
Determination of Albuminuria
Mouse urine albumin and creatinine were measured as previously described.10,40,41
Histology and PAS Staining
Tissues were processed as previously described.10,15 Sections (5 µm thick) were used for histologic analyses. At least 50 different superficial glomeruli per mouse were randomly chosen for analysis. Fractional mesangial area (FMA) was calculated as the percentage of PAS-positive area in the glomerular tuft area.33 All histologic analyses were conducted by a blinded investigator.
Immunohistochemistry
Animals were perfused with ice-cold PBS and then with 4% buffered paraformaldehyde (PFA). Tissues were fixed in 4% buffered PFA for 2 days, embedded in paraffin, and processed for sectioning. Double immunofluorescence of active integrin-β3 (AP5) and nephrin was performed on a subset of mouse renal tissue sections as previously described.26,42 WT-1 immunohistochemistry was performed on mouse renal tissue sections to estimate podocyte loss. In both cases, sections were fixed in ice-cold acetone for 1 minute after antigen retrieval in Tris-based antigen-unmasking solution, incubated in PBS (containing 0.025% Tween 20) for 5 minutes, and blocked in M.O.M. blocking solution (as applicable) according to the manufacturer’s instructions for 1 hour. The slides were then incubated with primary antibodies against active integrin-β3 (AP5; 1:100) and nephrin (1:250) or WT-1 (1:200) for 48 hours at 4°C. The slides were rinsed three times in PBS (containing 0.025% Tween 20). The sections were then incubated with corresponding fluorescently labeled secondary antibodies (AP5 and nephrin) or anti-rabbit HRP-conjugated secondary antibody (WT-1) for 120 minutes before again being rinsed three times in PBS (containing 0.025% Tween 20). The slides were covered with VECTASHIELD mounting medium containing the nuclear stain DAPI or hematoxylin counterstaining, and the specimens were analyzed on a Leica SP5 confocal microscope. WT-1–positive cells were counted to estimate podocyte loss in each experimental group. For immunofluorescence, DAPI-stained and fluorescently labeled images were acquired separately. The exposure settings and laser gain were kept the same for each condition. A total of 30 fields were acquired per condition, with a single focal plane per field. The images were analyzed with ImageJ/Fiji. For quantification, each individual glomerulus was selected using the drawing tool (freeform). The images were separated into their individual channels, and a threshold was determined to select only the active integrin-β3 fluorescence that overlapped with nephrin. The area, integrated intensity, and mean gray value were measured for each selected region of the glomerulus. This was repeated for a selected region within the glomeruli with no active integrin staining and therefore with no fluorescence. The fluorescence quantified for this region was considered to be the background. The mean fluorescence intensity for active integrin-β3 was calculated by subtracting the background fluorescence from the integrated density of active integrin-β3 fluorescence. These steps were repeated for at least 30 glomeruli per kidney section from three mice per group.
Transmission Electron Microscopy
Ultrastructural images of the glomerular filtration barrier were obtained by transmission electron microscopy as previously described.40 The thickness of the glomerular basement membrane (GBM) was analyzed using ImageJ software. For each image, the basement membrane thickness was determined at 15 adjacent and evenly distributed locations. Photomicrographs of the GBM were analyzed for the density of tight slit pores between the podocyte foot processes using randomly chosen electron micrographs of ten glomeruli per kidney for each mouse. Tight slit pores were identified by the obliteration of spaces between adjacent foot processes.24,43 The numbers of tight slit pores were counted and divided by the GBM length (µm) to determine the density of tight slit pores. A total of 500 foot processes from each group were evaluated for the analysis of slit pore density. All histologic analyses were conducted by a blinded investigator.
Cell Culture
Conditionally immortalized mouse podocytes and human podocytes were obtained from Prof. Jochen Reiser and cultured as described elsewhere.44 The cell lines tested negative for mycoplasma. Experiments were performed after 14 days of differentiation. For immunoblotting analysis, cells were serum starved for 8 hours before adding glucose for further 24 hours. Lysates were prepared at the desired time points after aPC or thrombin treatment. Murine trophoblast stem cells were cultured and differentiated as described previously.45
For in vitro AP5 immunocytochemical analyses, confluent differentiated podocytes were serum starved for 30 minutes and then stimulated with glucose (25 mM, 48 hours before the experiment; control, 5 mM glucose; osmotic control, 25 mM mannitol). Subsets of cells were treated with 20 nM aPC (for the last 3 hours).24 For cell migration assay (scratch assay), confluent differentiated podocytes were stimulated with puromycin aminonucleoside (PAN; 30 µg/ml, 30 minutes before the experiment; control, PBS) or with glucose (25 mM, 24 hours before the experiment; control, 5 mM glucose; osmotic control, 25 mM mannitol). Subsets of cells were pretreated (3 hours, then freshly added every 12 hours) with aPC alone (20 nM), aPC preincubated with the antibody HAPC1573 (1:1 ratio for 10 minutes under gentle agitation; HAPC1573 specifically inhibits aPC’s anticoagulant function), or RGE-aPC (20 nM).22,24 A standardized scratch injury was generated using a sterile pipette tip and images of the scratch injury were obtained after 24 hours, as previously described.36 The number of podocytes that migrated into the scratch were counted.
In Vitro aPC Generation Assay
Cell-dependent PC activation was determined as described previously with modifications.10,46 Here, 250,000 differentiated mouse podocytes (control and integrin-β3 knockdown [integrin-β3KD]) or mouse trophoblast cells were resuspended in 20 mM HEPES-hydrochloride (pH 7.5) containing 0.1% BSA, 0.15 M sodium chloride (NaCl), 5 mM calcium chloride, and 20 mM benzamidine and mixed at 37°C with a 25 µl volume of the same buffer containing 1 µg of PC and 10 nM human bovine thrombin to initiate activation. The reactions were stopped at each defined time interval by taking a 10-µl aliquot of the cell suspension and mixing it with 60 U/µl hirudin. aPC concentrations were determined based on the amidolytic activity toward 1 mM Spectrozyme PCa substrate in 0.15 M NaCl, 5 mM calcium chloride, and 20 mM HEPES-hydrochloride, pH 7.5. Substrate cleavage was measured in a BioTek microplate reader at 405 nm.
RhoA and Rac1 Pull-Down Assay
RhoA and Rac1 activation assays using Rhotekin-RBD and PAK-PBD affinity beads, respectively, were performed according to the manufacturer’s instructions (Cytoskeleton, Inc.). Conditionally immortalized human podocyte cultures were exposed to aPC (20 nM), thrombin (10 nM), or specific antagonists (RGDS, 50 µg/ml) for the desired period. The culture medium was aspirated, and the cells were rinsed twice with ice-cold PBS before ice-cold cell lysis buffer (50 mM Tris pH 7.5, 10 mM magnesium chloride, 0.5 M NaCl, and 2% Igepal) was added. The cells were harvested using a cell scraper, and the cell lysates were immediately clarified by centrifugation at 10,000 × g at 4°C for 1 minute. The protein concentration in each sample was determined, and equal concentrations of protein (200 µg) were incubated for 1 hour at 4°C with Rhotekin-RBD beads. The lysates were pelleted by centrifugation at 4000 × g, and the supernatant was aspirated. The pellet with Rhotekin-RBD beads was resuspended in 20 µl of 2× Laemmli buffer and boiled for 2 minutes. The active RhoA-GTP or Rac1-GTP and total RhoA or Rac1 in each sample were quantified by Western blot analysis using anti-RhoA or anti-Rac1 monoclonal antibodies, respectively (Cell Signaling Technology).
Immunocytochemistry (Active Integrin-β3 Staining)
Human podocytes were plated on collagen-coated glass coverslips and exposed to high glucose (25 mM) without or with additional aPC (20 nM) on the day of differentiation. The coverslips were washed with ice-cold 1× PBS and fixed in 3.7% PFA at room temperature for 15 minutes. The coverslips were washed (3× PBS, 0.3% Triton X-100, and 0.25% Tween 20 for 10 minutes per wash) and blocked (5% donkey serum in 1× PBS, 0.3% Triton X-100, and 0.25% Tween 20) for 1 hour. The sections were then incubated (overnight at 4°C) with the primary antibody AP5, which specifically recognizes the active conformation of integrin-β3. After washing (3× PBS for 10 minutes per wash), the sections were incubated with secondary anti-mouse tetramethylrhodamine B isothiocyanate–conjugated antibodies and FITC-conjugated phalloidin for 1 hour in the dark at 37°C. After washing with 1× PBS (3× for 10 minutes per wash in the dark), the coverslips were mounted on glass slides and prepared for imaging using an LSM 700 Imager.M2 (Carl Zeiss AG, Jena, Germany). The exposure settings and laser gain were kept the same for each condition. A total of 50 fields with z-stacks were acquired per condition per coverslip. The images were analyzed with ImageJ/Fiji. For quantification, the images were separated into their individual channels, and a threshold was determined to select only the active integrin-β3 fluorescence in the periphery of the cells per z-stack. The nuclei (reflecting the number of cells) were counted using the Point Picker plugin. The average integrated density was determined for every field.
In Situ Proximity Ligation Assay
The Duolink in situ proximity ligation assay (PLA) kit was used according to the manufacturer’s instructions (Olink Biosciences, Sigma Aldrich). Briefly, paraffin mouse kidney sections were dewaxed and rehydrated followed by antigen retrieval in Tris-based antigen-unmasking solution and then incubated in PBS (containing 0.025% Tween 20) for 30 minutes. Blocking was performed in M.O.M. blocking solution, according to the manufacturer’s instructions, for 30 minutes followed by PLA blocking solution for another 30 minutes. Then slides were incubated with three primary antibodies (overnight at 4°C) raised in different species and recognizing the target antigens of interest: rabbit anti-integrin-β3 (1:100), mouse anti-PC (1:100), and goat anti-nephrin (1:250). After washing (2× with buffers A and B provided in the PLA kit, each 10 minutes), sections were incubated with species-specific secondary antibodies (against integrin-β3 and PC) with a unique short DNA strand attached (PLA probes). Antibody-attached oligonucleotides were linked by enzymatic ligation, amplified via rolling circle amplification using a polymerase, and amplified DNA was detected by fluorescent-labeled complementary oligonucleotide probes. Additionally, slides were incubated with fluorescently labeled secondary antibody against nephrin for 120 minutes before being rinsed and mounted with VECTASHIELD mounting medium containing DAPI for nuclear staining.
Production of Lentiviral Particles
HEK293T cells (CRL-11268; ATCC) were grown at 37°C in a humidified atmosphere with 5% carbon dioxide in an incubator (Thermo Fisher Scientific). The culture medium contained DMEM (Gibco), 10% FBS, and penicillin-streptomycin (50 μg/ml and 50 μg/ml). HEK293T cells were seeded at a density of 11×106 cells per 15-cm2 dish 24 hours before transfection. Lentiviruses were produced by transfecting HEK293T cells with short hairpin RNA (targeting integrin-β3; the plasmids were obtained from Dharmacon) together with two helper plasmids (the packaging plasmid psPAX2 and the VSV-G–expressing plasmid pMD2.g; both from Addgene). The transfections were carried out using the calcium-phosphate method (2 M calcium chloride and 2× HEPES buffered saline, pH 7.4) with the plasmids in a 3:2:1 ratio (short hairpin RNA plasmid, psPAX2:pMDg.2). The virus-containing medium was harvested 48 hours and 72 hours after transfection and subsequently precleaned with a 0.45-μm filter (Millipore) as described.47 The virus-containing medium was mixed with 50% PEG 6000, 4 M NaCl, and 1× PBS (without calcium and magnesium) at volume percentages of 26%, 11%, and 12%, respectively, and incubated on a shaker at 4°C for 4 hours. After incubation, the solution was centrifuged at 1600 × g at 4°C for 60 minutes. After centrifugation, the supernatant was carefully removed without disturbing the viral pellet. Cell culture medium was added to the pellet for resuspension, and the 50 ml Falcon tubes were kept at 4°C in a shaker for recovery overnight. The viral particles were resuspended in 1/20th of the original volume. The protocol for concentrating the viral particles was adapted from that of Kutner et al.48 The cells were treated with two rounds of the enriched lentivirus, and knockdown efficiency was ascertained after 4 days by immunoblotting.
Microscale Thermophoresis Binding Assay
Protein-protein interactions between human integrin-αVβ3 and human aPC or human PC were determined by means of a microscale thermophoresis binding assay (NanoTemper Technologies, Munich, Germany). Integrin-αVβ3 was labeled with the fluorophore NT-647(Monolith NT Protein Labeling Kit; NanoTemper Technologies, Munich, Germany).49 A 14-fold titration series of aPC or PC (1 μM–0.122 μM) diluted 1:1 with PBS containing 0.05% Tween 20 was performed. The concentration of NT-647–labeled integrin-αVβ3 was kept constant (15 nM). The binding partners were incubated for 30 minutes in the dark to enable binding. The reaction was then aspirated into glass capillaries that were sealed with wax, and the thermophoretic movement of the labeled proteins was monitored with a laser (on for 30 seconds and off for 5 seconds) at a laser power of 80%. Fluorescence was measured before laser heating (FInitial) and after 30 seconds of laser-on time (FHot). The Monolith NT.115 device and the NT Analysis software version 1.427 (NanoTemper Technologies) were used for analysis. The normalized fluorescence FNorm=FHot/FInitial reflected the concentration ratio of the labeled molecules. FNorm was plotted directly and multiplied by a factor of 10, yielding a relative change in fluorescence per mill. Thus, the fraction of bound ligand molecules can be derived from the measured change in normalized FNorm. Kd was calculated from three independent thermophoresis measurements with NanoTemper Software (NanoTemper Technologies).
Statistical Analyses
The data are summarized as the mean±SEM. The Kolmogorov–Smirnov test or D’Agostino–Pearson normality test was used to determine whether the data followed a Gaussian distribution. Statistical analyses were performed with t test or ANOVA as appropriate. StatistiXL software (www.statistixl.com) and Prism 5 (www.graphpad.com) software were used for the statistical analyses. Statistical significance was accepted at values of P<0.05.
Results
Integrin-αvβ3 Binds To PC and aPC and Enhances PC Activation on Podocytes
We previously proposed that EPCR is not required for the cytoprotective effect of aPC in podocytes.15 To address the role of EPCR in vivo, we induced persistent hyperglycemia in mice expressing low levels of EPCR (below 10% compared with wild-type mice, EPCRδ/δ mice).30 Albuminuria and glomerular damage, as reflected by the FMA, were comparable in diabetic EPCRδ/δ and diabetic wild-type mice (Figure 1). This finding contrasts earlier results in mice with markedly impaired ability to activate PC (TMPro/Pro mice), which are also characterized by a limited capability to restrict thrombin activity and enhanced PAR1-signaling.10,50 These data support the conclusion that EPCR is not required for aPC’s cytoprotective effects in the context of dNP and raise the question whether other coreceptors are required for PC activation and aPC signaling via PARs on podocytes.
Figure 1.
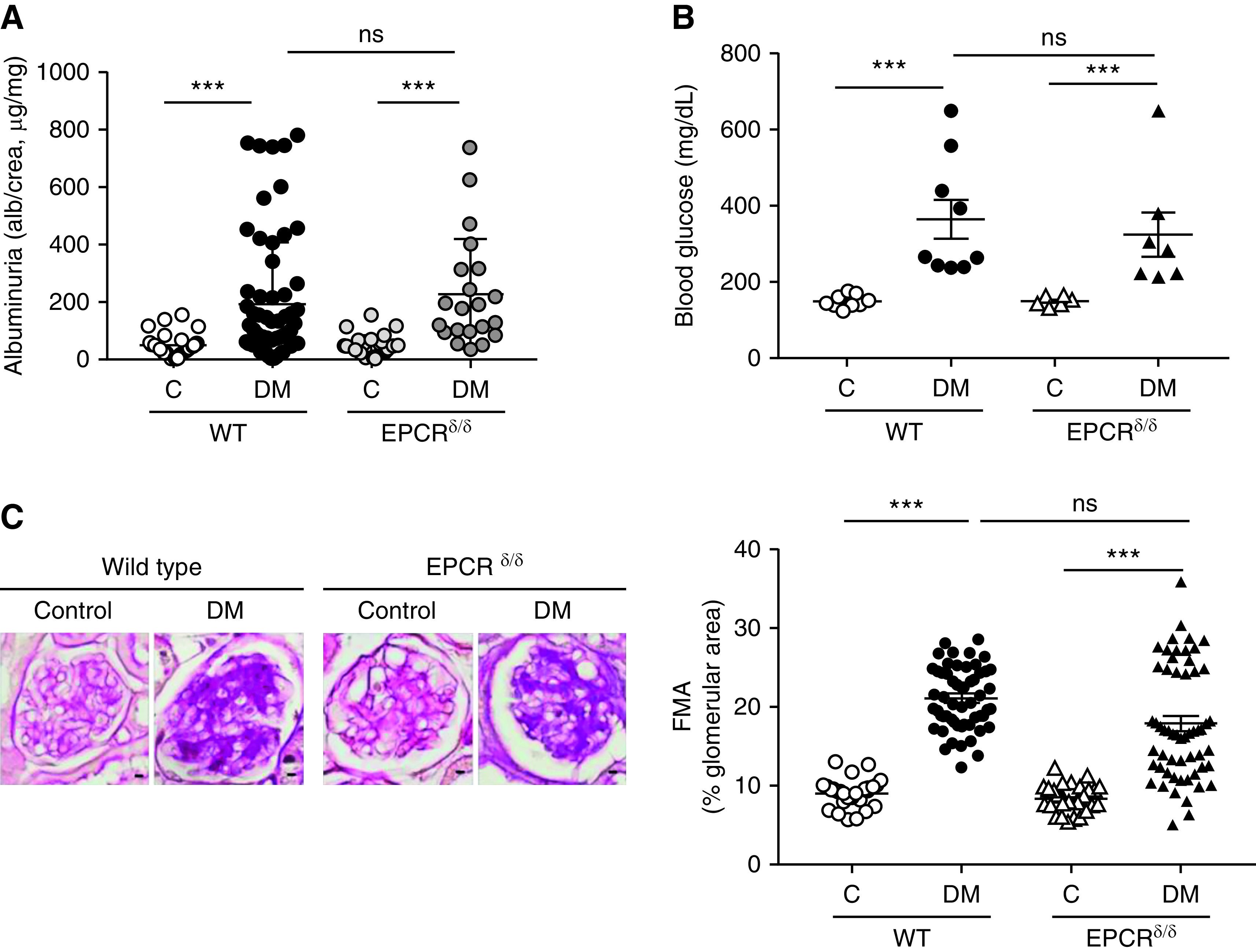
EPCR deficiency has no effect on dNP. (A and B) Dot plot summarizing (A) urine albumin levels (albumin-creatinine ratio) or (B) blood glucose levels in control and diabetic wild-type (WT) and EPCR-deficient (EPCRδ/δ) mice. Albuminuria and blood glucose levels are not different between control or diabetic EPCRδ/δ mice as compared with control or diabetic wild-type mice, respectively. (C) Representative images of glomeruli (left; PAS staining of paraffin-fixed sections; scale bar, 5 μm) and dot plot summarizing data for the FMA (right). Data shown as dot plots represent mean±SEM of at least six mice per group. ***P<0.005. (A and B) ANOVA with Tukey-adjusted post hoc comparison; diabetic mice (DM) were compared with corresponding (WT or EPCRδ/δ) nondiabetic control mice and diabetic WT mice were compared with diabetic EPCRδ/δ mice. C, control; DM, mice with persistent hyperglycemia after STZ injection.
Given the relevance of β1 and β3 integrins on podocytes25 and the known binding of aPC to integrins,19,22 we hypothesized that aPC’s cytoprotective effect requires aPC binding to podocyte integrins. Indeed, immunoprecipitation studies revealed that human aPC readily binds to integrin-β3 on human podocytes in a concentration-dependent manner, whereas very weak binding to integrin-β1 was observed (Figure 2A). The presence of PC and aPC in the urine of humans and mice and the binding of aPC to integrin-β3 on podocytes, as determined by PLA, indicate that PC/aPC can cross the glomerular filtration barrier and bind to podocyte integrin-β3 (Figure 2, B and C, Supplemental Figure 2). To determine the specific interaction of PC and aPC with the functionally important podocyte integrin heterodimer αvβ3, we analyzed protein-protein interactions in a cellfree system using microscale thermophoresis, which demonstrated a strong interaction of both PC and aPC with integrin-αvβ3. Intriguingly, the zymogen PC bound to integrin-αvβ3 with slightly higher affinity (Kd=12.4±3.4 nM) than aPC (Kd=61.5±10.3 nM; Figure 2D).
Figure 2.
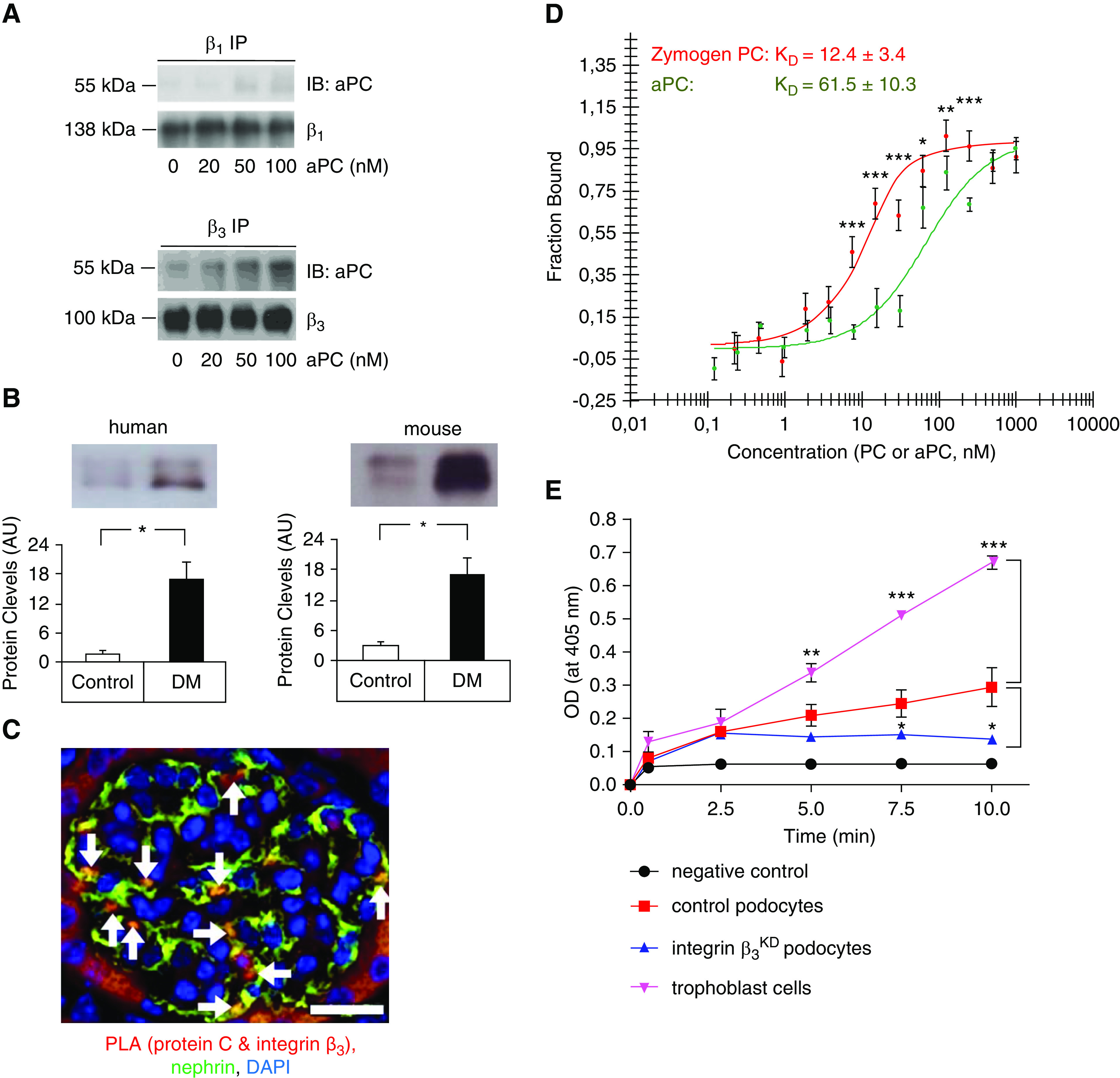
The PC–integrin-αvβ3 interaction enhances aPC generation in podocytes. (A) Representative immunoblot images of aPC (55 kDa, top) after immunoprecipitation of integrin-β1 (β1 IP, left, 138 kDa) or integrin-β3 (β3 IP, right, 100 kDa) in human podocytes. Human aPC binds in particular to integrin-β3 on human podocytes; the antibody used to detect aPC recognizes both the zymogen and the activated form. The lower gel shows the loading control (integrin-β1 or integrin-β3). The concentration of aPC is shown at the bottom in nanomolar, and the incubation time was 10 minutes. (B and C) Experimental evidence that PC/aPC is glomerularily filtered and interacts with β3 integrin on podocytes. (B) PC is detectable in the urine of nondiabetic humans and mice (control) and markedly increases in patients with diabetes (DM) and diabetic mice (db/db mice, DM). Representative immunoblot images and bar graph summarizing results of at least five humans or mice per group. *P<0.05, t test comparing DM versus control. (C) Representative image, showing colocalization (yellow, white arrows) of the PC–integrin-β3 complex (red, detected by PLA) with nephrin (green, podocyte marker, immunofluorescence staining) in mouse kidney sections. Scale bar, 20 μm. (D) Binding of the serine protease aPC (green) or the zymogen PC (red) to integrin-αvβ3 as analyzed by microscale thermophoresis. The data are presented as the mean±SEM for three independent repeat experiments. *P<0.05, **P<0.01, ***P<0.005 comparing PC versus aPC; t test with Bonferroni correction. (E) Cell-based in vitro aPC generation assay. Thrombin (10 nM)-mediated PC activation was determined in the presence of trophoblast cells (positive control, purple), control mouse podocytes (red), or integrin-β3–deficient (integrin-β3KD, blue) podocytes. aPC generation in the absence of cells served as a negative control (black). The data are presented as the mean±SEM for three independent repeat experiments. Comparative results were obtained with an independent integrin-β3KD cell line. *P<0.05, **P<0.01, ***P<0.005 versus control podocytes; one-way ANOVA with Bonferroni-adjusted post hoc comparison of trophoblast cells or integrin-β3KD podocytes with control podocytes.
The higher affinity of PC than aPC for integrin-αvβ3 raised the question of whether PC binding to integrin-αvβ3 promotes aPC generation. To answer this question, we assessed thrombin-mediated PC activation by control or integrin-β3–deficient (integrin-β3KD; Supplemental Figure 3 Figure 3Supplemental Figure 3) podocytes using trophoblast cells as positive controls (Figure 2E).51,52 PC activation was reduced to approximately 50% of that by control podocytes in two independent podocyte integrin-β3KD cell lines (Figure 2E). These results support a model in which integrin-β3 enhances aPC generation, potentially enhancing cytoprotective aPC-PAR signaling on podocytes while suppressing local thrombin generation and cell-disruptive thrombin-PAR signaling.15,53
Activated PC Induces a Temporal Interaction of Integrin-β3 with Gα13
Ligand binding to integrins may modify their conformation. Therefore, we determined the levels of active integrin-β3 on human podocytes using the conformation-specific mAb AP5, which detects an active conformation of integrin-β3.54 Interestingly, compared with control medium (5 mM glucose), exposure of podocytes to high glucose medium (25 mM), but not increased mannitol concentrations (25 mM), reduced the active integrin-β3 conformation. Concomitant exposure to aPC abolished the glucose-dependent reduction in active integrin-β3, whereas aPC (control medium, 5 mM glucose) had no effect (Figure 3A). Hence, aPC maintains an active integrin-β3 conformation congruent with signaling of aPC via integrin-β3. Integrins modulate the cytoskeleton in part via RhoA signaling.55 In agreement with the cytoskeletal changes observed, high glucose induced RhoA activation, which was not observed in the presence of aPC (Figure 3B).
Figure 3.
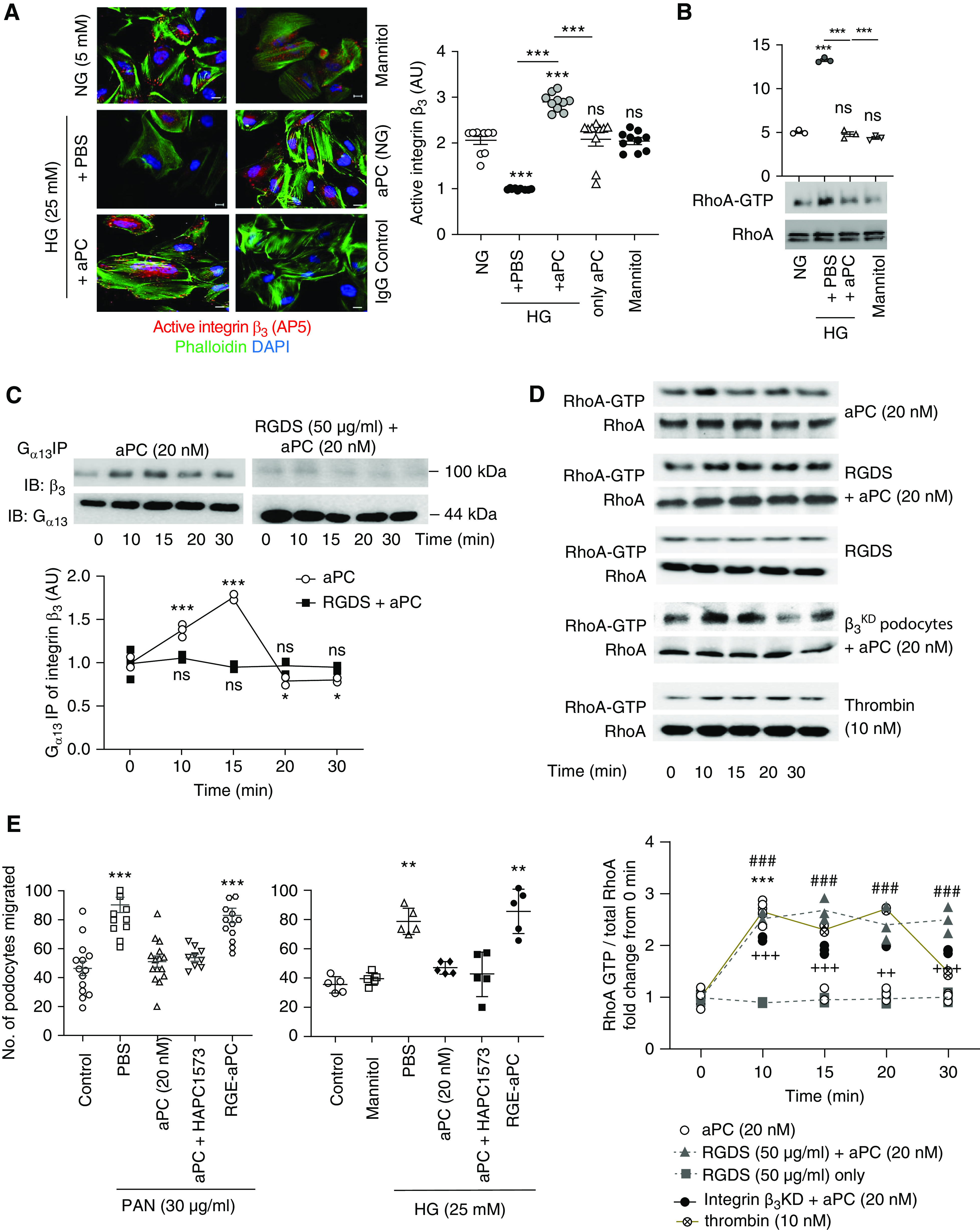
aPC–integrin-αvβ3 temporally regulates RhoA activation in podocytes. (A) Representative immunofluorescence images (left) of active integrin-β3, as determined by the conformation-specific antibody AP5, in human podocytes without (normal glucose concentration, 5 mM glucose, NG) or with high glucose (HG, 25 mM) stimulation in the absence (PBS) or presence of aPC (20 nM). Mannitol is used as an osmotic control. Cells stained with nonspecific IgG served as staining controls. The dot plot at the right summarizes the results. All groups were compared with control and HG+PBS to HG+aPC. Scale bar, 20 μm. (B) Representative immunoblot images showing levels of RhoA-GTP (21 kDa) and total RhoA (21 kDa) obtained from the RhoA pull-down assay and dot blot summarizing the data. Compared with control (5 mM glucose, NG) and the osmotic control mannitol (25 mM), high glucose concentrations (25 mM, HG, 3 hours) induce RhoA activation in human podocytes, which is prevented by concomitant exposure to aPC (aPC, 20 nM). (C) Representative integrin-β3 immunoblot images (top; IB: β3) of Gα13 immunoprecipitate showing time-dependent interaction of Gα13 with integrin-β3 upon stimulation of human podocytes with aPC. Gα13 (44 kDa) immunoblots were used as loading controls (top; IB: Gα13). Preincubation of cells with RGDS abolished the aPC-induced time-dependent interaction of Gα13 with integrin-β3. The line graph summarizes the results from three repeat experiments (bottom), with each dot representing an individual measurement. All groups were compared with control. (D) Representative immunoblot images (top) showing the levels of RhoA-GTP (21 kDa) and total RhoA (21 kDa) obtained from the RhoA pull-down assay and line graphs summarizing the kinetic data (bottom). RhoA activation was transient (peaking at 10 minutes) in human podocytes stimulated with aPC only (aPC), whereas sustained RhoA activation over 30 minutes was observed in podocytes stimulated with aPC and RGDS (aPC+RGDS), in integrin-β3–deficient podocytes stimulated with aPC (after lentiviral short hairpin RNA–mediated knockdown of integrin-β3, aPC-β3KD), or in thrombin-stimulated podocytes. RGDS alone had no effect (RGDS). All groups were compared with control. (E) Dot plot summarizing how PAN or high glucose (HG, 25 mM) induced podocyte migration (as determined by scratch assay) after treatment with PBS (PBS, control), aPC, aPC preincubated with the antibody HAPC1573 (a mouse mAb that blocks aPC’s anticoagulant effect), or RGE-aPC. In addition, mannitol as an osmotic control is shown for the glucose stimulation experiment. All groups were compared with control. The data are shown as dots of at least three (A–D) or five (E) independent repeat experiments in the dot plot, including mean and SEM in (A, B, and E), or as dots representing individual data points from three independent repeat experiments in the line graphs in (C and D). *P<0.05, **P<0.01, ***P<0.005 (A, B, and E). ***P<0.005 (aPC versus time point 0 minute), ### P<0.005 (RGDS + aPC versus time point 0 minute), ++P<0.01, +++P<0.005 (integrin β3KD + aPC versus time point 0 minute) in D. (A, B, D, and E) one-way ANOVA with Tukey-adjusted post hoc comparison of treated cells with untreated cells (time point, 0 minute).
Binding of the RGD-containing coagulation factor fibrin/fibrinogen to integrin-β3 induces interactions of the integrin-β3 cytoplasmic tail with components of canonical G protein-coupled receptor (GPCR) signaling, e.g., Gα13 and Gα12.42,56 To determine whether binding of aPC to integrin-β3 on podocytes induces G-protein recruitment, we analyzed the integrin-β3 interaction with Gα13 after stimulation with aPC. aPC transiently induced recruitment of Gα13 to integrin-β3, which peaked after 10–15 minutes (Figure 3C), and this effect was completely abolished by the antagonistic peptide RGDS. Fibrin/fibrinogen outside-in signaling via integrin-β3 recruits Gα13 in platelets, which negatively regulates thrombin–PAR–Src-dependent RhoA activation.42,56 Likewise, aPC inhibited Src activity, as deduced from an increased ratio of inactivating Y527 phosphorylation to activating Y416 phosphorylation in podocytes (Supplemental Figure 4A). The aPC-induced inhibition of Src activity was abolished or even reversed in the presence of an αvβ3 antagonist (Supplemental Figure 4A). These data suggest that aPC modulates Gα13 signaling upon binding to integrin-β3.
Integrin-β3–aPC Interaction Is Required for Transient RhoA Activation in Podocytes
Because outside-in signaling of integrin-β3 via Gα13 negatively regulates thrombin-PAR–dependent RhoA activation in platelets,42,56 we next exposed podocytes to aPC and determined RhoA activity. aPC transiently activated RhoA, with a peak after 10 minutes (Figure 3D). In contrast to the aPC-dependent transient RhoA activation, aPC induced a sustained suppression of Rac1 in podocytes (Supplemental Figure 4B). Blocking the aPC-integrin interaction with the RGDS peptide caused sustained aPC-dependent RhoA activation, whereas the RGDS peptide alone had no effect. Similarly, aPC triggered sustained RhoA activation in integrin-β3KD podocytes (Figure 3D). Intriguingly, stimulation of podocytes with thrombin (10 nM) also resulted in sustained RhoA activation, which was not altered in the presence of RGDS peptide (Figure 3D, Supplemental Figure 4C). These data indicate that both aPC and thrombin induce RhoA activation but that aPC specifically restricts RhoA activation by binding via its RGD sequence to integrin-β3.
To ascertain the mechanistic relevance of aPC–integrin-β3 binding on podocytes, we used the well established PAN-induced podocyte injury model. Treatment with PAN induces loss of actin fibers and promotes pathologic podocyte migration, which is an in vitro correlate of podocyte foot process effacement in vivo.36 Wild-type aPC, but not an integrin binding–deficient aPC mutant (RGE-aPC), inhibited PAN-induced podocyte migration, demonstrating the relevance of aPC’s RGD sequence for its cytoprotective effect in podocytes (Figure 3E). The effect of aPC was independent of its anticoagulation property, because preincubation with the mAb HAPC1573, which specifically blocks aPC’s anticoagulant activity, did not interfere with the aPC-mediated inhibition of podocyte migration (Figure 3E).15 Similar results were obtained when using high glucose (25 mM) instead of PAN as a stimulus, demonstrating the relevance of these observations for hyperglycemia-induced podocyte dysfunction (Figure 3E). Taken together, these results show that aPC–integrin-β3 interaction is required for suppressing of podocyte injury– or hyperglycemia-induced RhoA activation.
Podocyte-Specific Genetic Ablation of Integrin-β3 Abrogates the Cytoprotective Effect of aPC
We and others have previously shown that both genetically induced as well as injections of aPC ameliorate experimental dNP.10,23,24,33 To evaluate the relevance of aPC–integrin-β3 signaling for aPC-mediated nephroprotection in vivo, we first used the conformation-specific antibody AP5 to determine integrin-β3 regulation by aPC in vivo. In models, FSGS integrin activation, as reflected by AP5 staining, is induced via RGD-independent ligand binding to integrin-β3.36,57 In contrast, AP5 staining is reduced in models of dNP.54 Our above results (Figure 3) suggest that aPC, which is reduced in diabetes mellitus,3 induces transient integrin-β3 activation via its RGD sequence. Taken together, these observations suggest a concept of bimodal integrin activation through various binding sites. Congruently, we observed less integrin-β3 activation (as demonstrated by AP5 levels) in podocytes in 24-week-old db/db mice (model of type 2 diabetes mellitus) compared with age-matched nondiabetic db/m mice (Figure 4A, Supplemental Figure 5). In vivo treatment of db/db mice with aPC following an established protocol known to be nephroprotective (a dose of 1 mg/kg body wt for 6 weeks)24 restored AP5 staining to levels observed in db/m mice (Figure 4A). Similar results were obtained in an STZ-induced model of type 1 diabetes mellitus (DM, a murine model reflecting early and theoretically reversible dNP).2 Following sustained hyperglycemia for 16 weeks, colocalization of AP5 with nephrin was reduced, but treatment of mice with aPC restored AP5 staining (Figure 4A, Supplemental Figure 5). The maintained presence of podocyte-active integrin-β3 supports a model in which aPC conveys its cytoprotective effect by interacting with and signaling through integrin-β3 via its RGD sequence.
Figure 4.
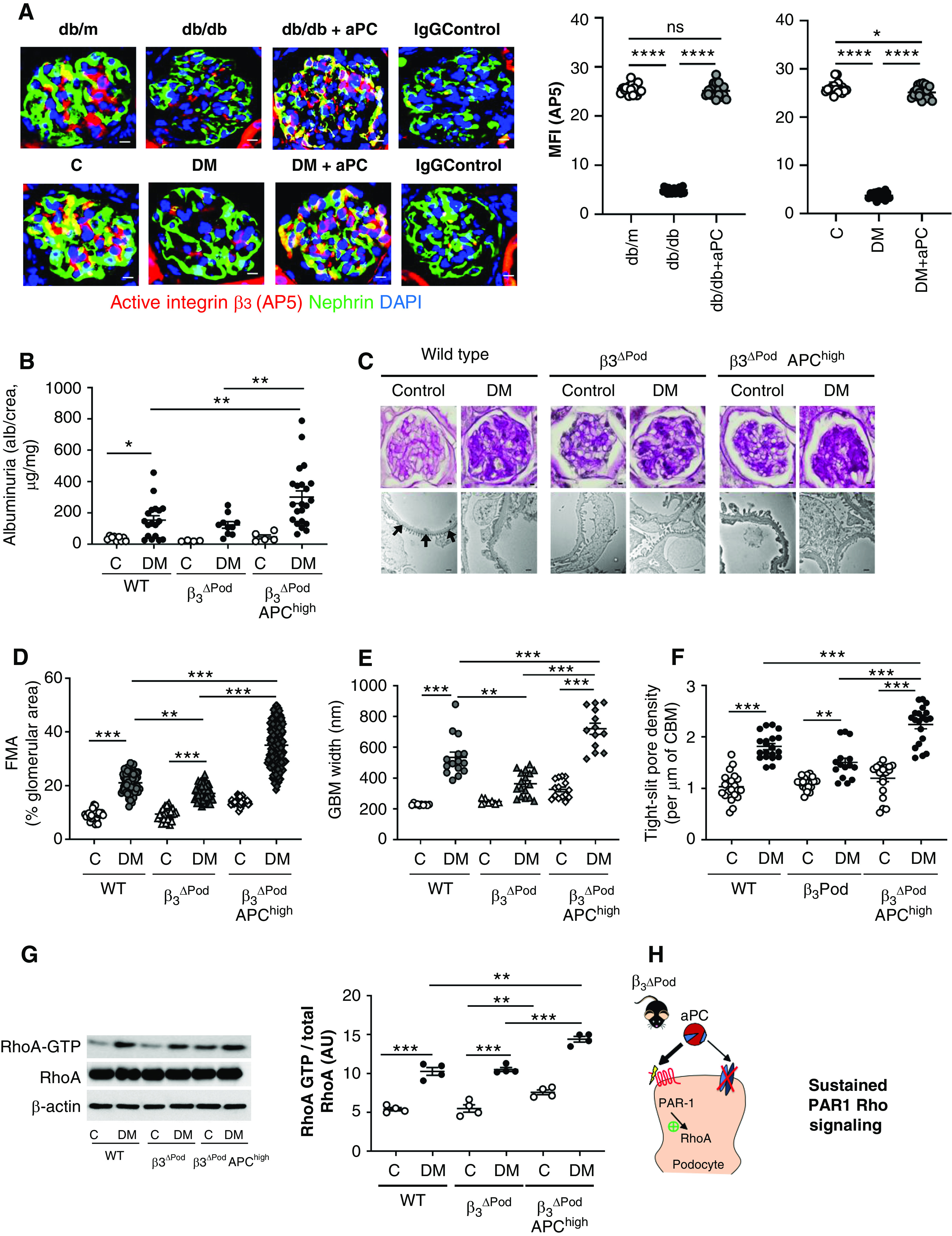
Podocyte-specific deletion of integrin-β3 abrogates the cytoprotective effect of aPC in dNP. (A) Representative immunofluorescence images (left) of glomeruli of nondiabetic mice (db/m or control [C]), diabetic mice (db/db or STZ-induced diabetes [DM]), or diabetic mice treated with aPC (db/db+aPC or DM+aPC). The conformation-specific antibody AP5 was used to detect active integrin-β3 (red). Podocytes were identified by nephrin staining (green); yellow reflects the colocalization of AP5 and nephrin. The nuclei were stained with DAPI (blue). Dot plots summarizing the results are shown at the right. Diabetic mice without or with aPC treatment were compared with nondiabetic control mice (db/m or C) and among each other (db/db versus db/db+aPC and DM versus DM+aPC). Scale bar, 10 μm. (B) Dot plot summarizing urine albumin levels (the albumin-creatinine ratio) in control (C) and diabetic (DM) wild-type (WT) mice, β3ΔPod mice and β3ΔPod mice crossed with APChigh mice (β3ΔPod APChigh). (C) Representative images of glomeruli (top; PAS staining of paraffin-fixed sections; scale bar, 5 μm) and the glomerular filtration barrier (bottom, transmission electron microscopy; scale bar, 0.2 μm) and dot plots summarizing (D) the data for the FMA, (E) the width of the GBM (representative of arrows in the far-left image of [C] only), and (F) tight slit pore density, reflecting foot process effacement. (G) Representative immunoblots (left) of RhoA-GTP (21 kDa), total RhoA (21 kDa), and β-actin (42 kDa) from renal tissue lysates from experimental mice and dot plots summarizing the data from the experimental groups (right). (H) Schematic representation of the working model: aPC cannot interact with integrin-αvβ3 in β3ΔPod mice, resulting in unopposed PAR1-RhoA signaling, aggravating podocyte dysfunction and hence promoting dNP in mice with increased aPC levels. The data shown in dot plots represent the mean±SEM of at least ten mice (A), five mice (B and D–F), or four mice (G) per group. *P<0.05, **P<0.01, ***P<0.005. (A and C–E) ANOVA with Tukey-adjusted post hoc comparison; for each genotype diabetic mice were compared do nondiabetic mice, and diabetic mutant mice were compared with diabetic wild-type mice. C, nondiabetic control mice; DM, mice with persistent hyperglycemia after STZ injection; MFI, mean fluorescent intensity.
To scrutinize the in vivo relevance of the aPC–integrin-β3 interaction, we first generated mice with podocyte-specific deletion of integrin-β3 by crossing integrin-β3LoxP/LoxP mice with PodCre mice (β3ΔPod mice). These mice were then crossed with mice expressing a hyperactivatable human PC mutant, resulting in high plasma levels of aPC (APChigh mice).10 Stable hyperglycemia, resembling diabetes mellitus, was induced in these mice using STZ (DM; Supplemental Figure 6A). Compared with diabetic wild-type mice, diabetic β3ΔPod mice were partially protected against dNP based on improved morphologic indices of dNP (FMA, GBM width; Figure 4, C–E). Strikingly, podocyte-specific deletion of integrin-β3 in APChigh mice (β3ΔPod-APChigh mice) not only abolished aPC’s protective effect but actually exacerbated albuminuria and morphologic features of dNP (FMA, podocyte loss, GBM width, and tight slit pore density; Figure 4, B–F, Supplemental Figure 6). Furthermore, integrin-β3 deficiency in podocytes of diabetic APChigh mice (β3ΔPod-APChigh mice) resulted in increased RhoA activity compared with that of diabetic wild-type or diabetic β3ΔPod mice (Figure 4G). These results are congruent with the above in vitro data, demonstrating that the aPC–integrin-β3 interaction via the RGD sequence is required to limit RhoA activation. Increased aPC levels and concomitantly reduced podocyte integrin-β3 expression in β3ΔPod-APChigh mice results in unbalanced PAR1 activation by aPC (Figure 4H). This triggers uncontrolled RhoA signaling and podocyte dysfunction, mimicking the effect of thrombin through PAR1 signaling.
The partial improvement of dNP in mice with isolated integrin-β3 deficiency in podocytes (the reduced FMA and GBM width in β3ΔPod mice) is consistent with a pathologic role of activated integrin-β3 in podocytes,26,36,58 whereas the aggravation of albuminuria, histologic indices, and RhoA activation in mice with combined podocyte integrin-β3 deficiency and increased aPC plasma levels (β3ΔPod-APChigh mice) elucidates a previously unknown protective function of integrin-β3 in dNP.
Integrin-αvβ3 Antagonism Dose-Dependently Modulates dNP
The partial protection from dNP in β3ΔPodmice and the aggravation of dNP in β3ΔPod-APChigh mice indicate a dual function of podocyte integrin-β3 that is reminiscent of the dose-dependent effects of integrin inhibitors on tumor angiogenesis.37 To address whether integrin inhibition has dose-dependent effects in the context of dNP, we tested two different concentrations of the integrin-αvβ3 inhibitor Cyclo-RGDfv (0.5 mg/kg and 2.0 mg/kg body wt). Interventions with Cyclo-RGDfv were initiated in mice with stable hyperglycemia for 10 weeks and hence after establishment of albuminuria (Figure 5A, Supplemental Figure 7).
Figure 5.
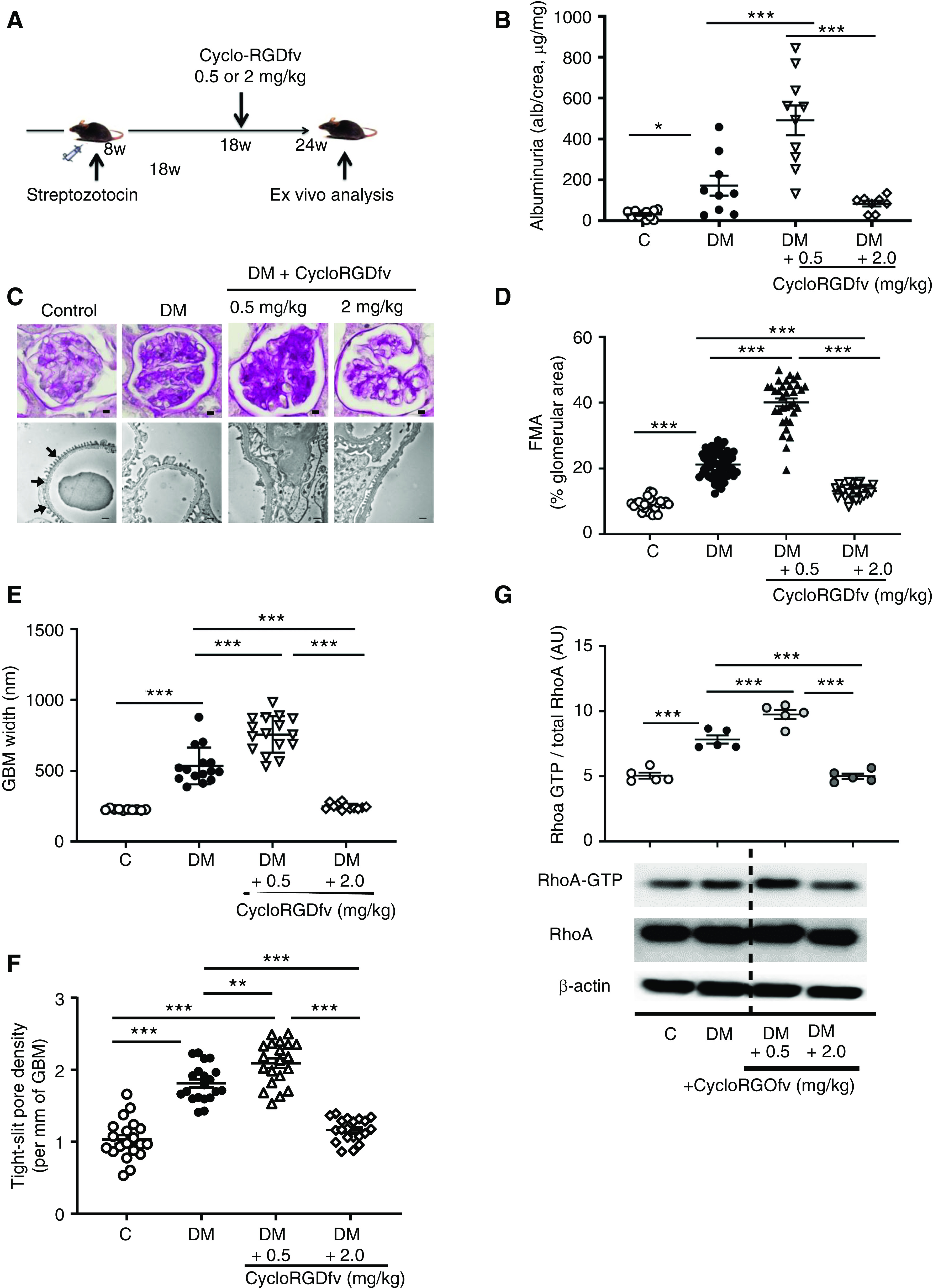
Integrin-αvβ3 antagonism dose-dependently modulates dNP. (A) Scheme showing the experimental approach and dosing of the integrin-αvβ3 antagonist Cyclo-RGDfv in wild-type mice with STZ-induced persistent hyperglycemia (DM). (B) Dot plot summarizing urine albumin levels (albumin-creatinine ratio) in control (C) and diabetic (DM) mice; diabetic control mice received PBS. (C) Representative images of glomeruli (top; PAS staining of paraffin-fixed sections; scale bar, 5 μm) and the glomerular filtration barrier (bottom, transmission electron microscopy; scale bar, 0.2 μm) and dot plots summarizing the data for (D) the FMA, (E) the width of the GBM (representative of arrows in the far-left image of [C] only), and (F) tight slit pore density, reflecting foot process effacement. (G) Representative immunoblots of RhoA-GTP (21 kDa), total RhoA (21 kDa), and β-actin (42 kDa) from renal tissue lysates from experimental mice and a dot plot summarizing the data. The data shown in the dot plots represent the mean±SEM of at least eight mice (B–F) or five mice (G) per group; each dot represents data from one mouse. *P<0.05, **P<0.01, ***P<0.001. (B and D–F) ANOVA with Tukey-adjusted post hoc comparison; diabetic mice were compared do nondiabetic mice, and diabetic mice treated with Cyclo-RGDfv were compared with diabetic control mice. C, nondiabetic control mice; DM, mice with persistent hyperglycemia after STZ injection.
Compared with vehicle-treated diabetic wild-type (DM) mice, diabetic mice treated with low-dose Cyclo-RGDfv (0.5 mg/kg) exhibited enhanced albuminuria, extracellular matrix accumulation (FMA), GBM widths, tight slit pore density, podocyte loss (Figure 5, B–F, Supplemental Figure 7, B and C), and RhoA activation (Figure 5G). These data are consistent with cytoprotective aPC signaling via podocyte integrin-β326,36,58 and suggest that low doses of an integrin-αvβ3 inhibitor abolish the nephroprotective effect of endogenous aPC in the context of dNP.
We next used a higher dose of Cyclo-RGDfv (2 mg/kg) with the same experimental design (Figure 5A). In contrast to low-dose Cyclo-RGDfv–mediated inhibition, high-dose Cyclo-RGDfv–mediated integrin-αvβ3 inhibition partially reversed albuminuria, dNP morphologic features (FMA, GBM width, tight slit pore density, and podocyte loss; Figure 5, B–F, Supplemental Figure 7, B and C), and RhoA activation (Figure 5G).
These data illustrate that integrin-αvβ3 inhibition is a double-edged sword in dNP. The aggravation of dNP upon low-dose integrin-αvβ3 inhibition is consistent with the proposed cytoprotective signaling of aPC via integrin-αvβ3 in stressed podocytes.
Targeted Disruption of the aPC–Integrin Interaction In Vivo Promotes Excess PAR1 and RhoA Signaling and Exacerbates dNP
To directly demonstrate the relevance of the proposed nephroprotective aPC–integrin-β3 interaction via aPC’s RGD sequence in vivo, we generated transgenic mice expressing a human hyperactivatable aPC mutant lacking the endogenous RGD integrin-binding site (RGE-APChigh mice; Supplemental Figure 1). These model mice complemented the previously generated model mice expressing a hyperactivatable human aPC mutant (APChigh mice, containing the endogenous RGD sequence), enabling us to directly determine the role of aPC’s RGD sequence in vivo. Blood loss upon standardized tail injury and aPC plasma levels were comparable in APChigh mice and RGE-APChigh mice (Supplemental Figure 1, D and E), confirming similar expression and anticoagulant function.
Persistent hyperglycemia was induced in wild-type, APChigh, and RGE-APChigh mice (Figure 6A, Supplemental Figure 8A). Compared with wild-type mice, APChigh mice were protected against dNP, as reflected by reduced albuminuria and markedly reduced morphologic markers of dNP (FMA, GBM width, tight slit pore density, and podocyte numbers; Figure 6, B–F, Supplemental Figure 8, B and C), in agreement with the findings of previous studies.10 In contrast, disruption of aPC-integrin binding in RGE-APChigh mice exacerbated albuminuria, FMA, GBM width, tight slit pore density, and podocyte loss (Figure 6, B–F, Supplemental Figure 8, B and C), demonstrating a crucial role of aPC’s RGD site in its cytoprotective effect in dNP. These functional and morphologic changes were associated with corresponding changes in RhoA activation (Figure 6G). Thus, compared with that in diabetic wild-type mice, RhoA activation remained low in diabetic APChigh mice but was markedly enhanced in RGE-APChigh mice.
Figure 6.
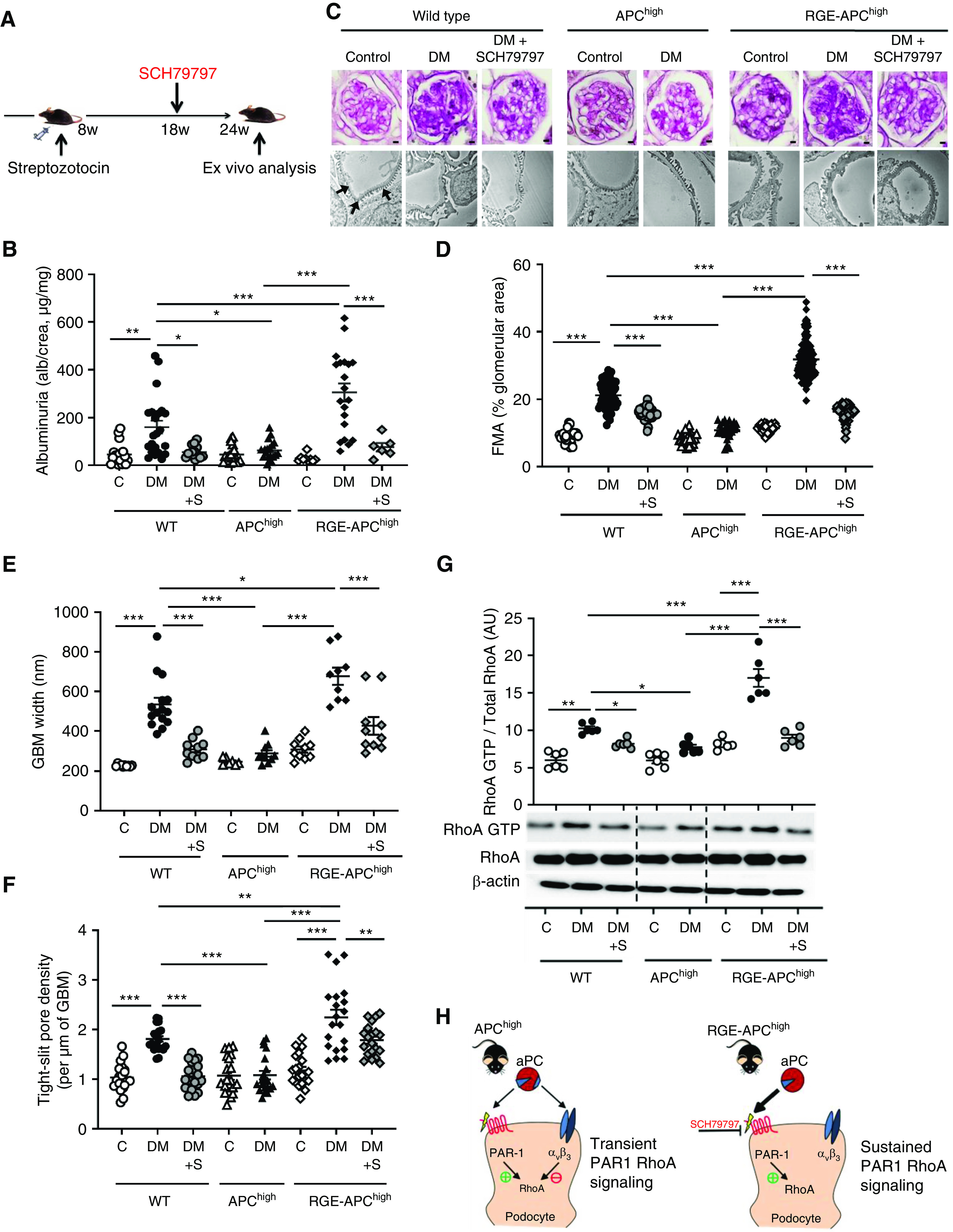
aPC protects against dNP via its RGD sequence. (A) Experimental design. SCH79797 was used as a PAR1 antagonist in a subgroup of mice to counteract excess PAR1 signaling. (B) Dot plot summarizing urine albumin levels (albumin-creatinine ratio) in control (C) and diabetic (DM) mice. Some diabetic mice received additional SCH79797 treatment (DM+S). (C) Representative images of glomeruli (top; PAS staining of paraffin-fixed sections; scale bar, 5 μm) and the glomerular filtration barrier (bottom; transmission electron microscopy; scale bar, 0.2 μm) and dot plots summarizing the data for (D) the FMA, (E) the width of the GBM (representative of arrows in the far-left image only), and (F) tight-slit pore density, reflecting foot process effacement. (G) Representative immunoblots (bottom) of RhoA-GTP (21 kDa), total RhoA (21 kDa), and β-actin (42 kDa) from renal tissue lysates from experimental mice and a dot plot summarizing the data (top). (H) Schematic representation of the working model: in APChigh mice, aPC activates PAR1 via its proteolytic activity, thus promoting RhoA activation, but at the same time binds to integrin-β3, restricting RhoA activity and resulting in transient RhoA signaling. In contrast, in RGE-APChigh mice, aPC cannot bind to integrin-β3, resulting in sustained RhoA signaling (bottom). SCH79797, a PAR1 antagonist, counteracts the enhanced PAR1 and RhoA signaling in RGE-APChigh mice. The data shown in dot plots represent the mean±SEM of at least six mice per group. *P<0.05, **P<0.01, ***P<0.001. (B and D–F) ANOVA with Tukey-adjusted post hoc comparison; for each genotype diabetic mice were compared do nondiabetic mice, diabetic mutant mice were compared with diabetic wild-type mice, and diabetic mice treated with SCH79797 were compared with untreated diabetic mice of the same genotype. C, nondiabetic control mice; DM, mice with persistent hyperglycemia after STZ injection; DM+S, diabetic mice treated with the PAR1 antagonist SCH79797.
Given the sustained RhoA activation upon thrombin stimulation of podocytes in vitro (Figure 3D), we next hypothesized that loss of nephroprotection and enhanced RhoA activation in RGE-APChigh mice reflects unopposed PAR1 signaling. To evaluate this hypothesis, we inhibited PAR1 using a selective nonpeptide PAR1 antagonist (SCH79797).35 Treatment with SCH79797 initiated 10 weeks after induction of hyperglycemia (Figure 6A) markedly reduced RhoA activation, reversed albuminuria, and attenuated morphologic features of dNP (FMA, GBM width, tight slit pore density, and podocyte loss) in diabetic RGE-APChigh mice (Figure 6, B–G, Supplemental Figure 8, B and C). Thus, pharmacologic mitigation of PAR1 signaling reverses the markedly aggravated renal phenotype in diabetic mice expressing RGE-APChigh.
Endogenous aPC ameliorates dNP, as indicated by the aggravated renal phenotype in diabetic mice with markedly impaired PC activation ability and exaggerated thrombin generation (TMPro/Pro mice).10 Considering the impairment of aPC generation in diabetic mice and patients with diabetes,10,59 we hypothesized that PAR1 inhibition may reduce RhoA activation and ameliorate dNP in wild-type mice. Indeed, PAR1 antagonism in diabetic wild-type mice reversed albuminuria, ameliorated morphologic features of dNP (FMA, GBM width, tight slit pore density, and podocyte loss), and reduced RhoA activation (Figure 6, B–G, Supplemental Figure 8, B and C). Thus, the aPC-integrin interaction is essential for aPC’s nephroprotective effect, and disruption of the aPC-integrin interaction or reduced aPC generation (as observed in diabetes mellitus) promotes PAR1-dependent excess RhoA signaling and thereby dNP (Figure 6H). Importantly, pharmacologic inhibition of aberrant PAR signaling counteracts excess RhoA signaling in the absence of the aPC–integrin-β3 interaction.
Discussion
The roles of integrins in podocyte function are well established. Likewise, accumulating evidence supports a role of coagulation proteases in regulating podocyte function. Within this study, we established a new mechanism through which integrins and coagulation proteases coordinately control podocyte function. Integrin-β3 and the serine protease aPC coordinately regulate transient RhoA activation in podocytes. The transient activation of RhoA depends on aPC binding to integrin-β3 via its RGD sequence. In the absence of aPC’s RGD sequence or of podocyte integrin-β3, aPC induces sustained RhoA signaling and podocyte dysfunction, mimicking the effect of thrombin. In support of the development of aberrant PAR1 signaling upon disruption of the aPC–integrin-β3 interaction, PAR1 inhibition counteracted the enhanced RhoA activation and maintained renal function in mice expressing an aPC variant lacking the RGE sequence. These data support a pathophysiologic role of increased RhoA and PAR1 signaling in dNP60–62 and identify aPC as a rheostat that controls RhoA activation by interacting with integrin-β3.
The roles of integrins in podocytes are well established.27 However, integrin activation is a complex process that can be mediated by RGD and non-RGD binding sites. Thus, suPAR regulates integrin activation and glomerular disease in mice through a non-RGD binding site.58 By contrast, this study demonstrates that PC/aPC crosses the glomerular filtration barrier and identifies the serine protease aPC as a physiologic integrin-β3 ligand at the RGD binding site that conveys cyto- and nephroprotective effects. Inducible costimulator ligand was recently identified as yet another RGD-binding integrin ligand that fine tunes integrin function in podocytes.63 Together, these studies demonstrate the need to fine tune integrin function and to maintain RhoA signaling in a physiologic balance in the context of glomerular disease.
This study identifies aPC as a RGD-dependent integrin-β3 ligand on podocytes, which promotes nephroprotection at two levels. First, as indicated by the decreased thrombin-dependent PC activation in integrin-β3 knockdown cells, binding of PC to podocyte integrin-β3 enhances thrombin-mediated aPC generation, inhibiting potentially harmful thrombin generation.53 Second, the binding of aPC to integrin-β3 restricts excessive and harmful RhoA signaling.60 The aPC-mediated regulation of RhoA activation is linked with integrin-dependent signaling through Gα13, as aPC induced a transient integrin-β3–Gα13 interaction via its RGD sequence.
The aPC-integrin–dependent inhibition of RhoA signaling via G-protein signaling has similarities with the pathway described by Gong et al.42 Fibrinogen/fibrin binding to integrin-β3 on platelets induces a functional interaction of the cytoplasmic integrin domain with Gα13 to restrict RhoA activation, counteracting agonist (thrombin)-induced GPCR signaling.42 Although the data of Gong et al.42 suggest that two different ligands control GPCR-Gα13 (thrombin) and integrin-Gα13 (fibrinogen/fibrin) signaling, our data indicate that a single protein, aPC, induces both G protein–dependent RhoA activation through PARs and RhoA inhibition through noncanonical integrin-Gα13 signaling (Figure 6H, left). Depletion of integrin-β3 or mutation of aPC’s RGD sequence abolishes aPC’s rheostat function and results in unopposed PAR1-mediated RhoA activation in mice that can be ameliorated by pharmacologic inhibition of PAR1 (Figure 6H, right). These results thus provide new insights into the mechanisms of cytoprotective signaling mediated by podocyte integrins.
Consistent with the existence of cytoprotective signaling through integrin-β3 in podocytes, inhibition of integrin-αvβ3 in dNP is a double-edged sword, as indicated by the dose-dependent effects of integrin-αvβ3 inhibition in experimental dNP models. This finding is consistent with the dose-dependent pro- and antiangiogenic effects of integrin inhibition.37 Enhancement of angiogenesis by low-dose integrin inhibitors has been attributed to altered αvβ3 and vascular endothelial growth factor receptor-2 trafficking.37 Integrins control endosomal and lysosomal trafficking of several distinct classes of transmembrane receptors, including receptors known to regulate podocyte function (e.g., uPAR, EGF receptor, and GPCRs).26 Whether integrin inhibitors likewise modulate PAR trafficking on podocytes remains unknown. Another potential explanation for the dose-dependent effect of αvβ3 inhibition in dNP is the expression of integrins by different renal cell types (e.g., podocytes versus endothelial cells). Alternatively, different concentrations of integrin-αvβ3 inhibitors may differentially regulate integrin inside-out versus outside-in signaling.
Intriguingly, podocyte integrin-β3 appears to substitute for known functions of EPCR on endothelial cells, i.e., PC activation and aPC signaling. EPCR, the pivotal coreceptor for aPC signaling in endothelial cells, is not required for aPC signaling in podocytes.15 Signaling of aPC independent of EPCR has been demonstrated to occur in several cell types and different contexts,15,17–19 but the coreceptors required in the absence of EPCR remain largely unknown. The current data and observations by Cao et al.19 show that RGD-binding integrins can functionally substitute for EPCR in propagating aPC-dependent cytoprotective effects. Importantly, this study demonstrates that integrin-β3 enhances local PC activation as well as conveys aPC signaling in podocytes. This identifies a new function of the PC/aPC–integrin-β3 interaction in addition to the previously shown functions related to neutrophil and macrophage adhesion.19,22 The existence of two distinct coreceptors of aPC-PAR signaling on podocytes and endothelial cells, integrin-β3 and EPCR, respectively, provides new insights into the cell specificity of aPC signaling and provides a plausible explanation for the disjunct aPC-mediated effects on RhoA and Rac1 signaling in podocytes and endothelial cells, respectively (Figure 3D, Supplemental 4B ).64,65
The study identifies additional questions. For example, expression of integrin-β3 is not restricted to podocytes and it remains to be evaluated whether binding of aPC to integrin-β3 modulates the function of cells other than podocytes within the kidney. However, the results obtained in mice with targeted deletion of integrin-β3 specifically in podocytes establish a salutary function for aPC–integrin-β3 signaling in dNP in vivo. Furthermore, species-specific differences in PAR signaling in podocytes (e.g., signaling of aPC via PAR3/PAR2 in humans but via PAR3/PAR1 in mouse podocytes) preclude the direct translation of these preclinical results from murine models into humans.15,53 However, in vitro work using human podocytes suggests that modulation of PAR signaling is a promising approach for the treatment of human glomerular disease (e.g.,15 and this study), which warrants further analyses.
Disclosures
J. Reiser is cofounder of Trisaq, a biotechnology company that develops new drugs for kidney diseases. All remaining authors have nothing to disclose.
Funding
This work was supported by DFG grants TH 1789/1-1 (to M. Thati); WA 3663/2-1 (to H. Wang); IS-67/8-1, IS-67/11-1, SFB854/B26, RTG2408/P7, and RTG2408/P9 (to B. Isermann); RTG2408/P5 and KO 5736/1-1 (to S. Kohli); MO 1082/7-1 (to M.J. Moeller); project number 236360313 – SFB 1118 (to B. Isermann); and project number 259130777 – SFB 1177 and project C2 SCHA 1082/6-1 (to L. Schaefer). This work was also supported by the BMBF STOP-FSGS consortium grant 01GM1518A (to M.J. Moeller); National Heart, Lung, and Blood Institute grant HL 101917 (to A.R. Rezaie); the Stiftung Pathobiochemie und Molekulare Diagnostik (to M. Thati); Center of Thrombosis and Hemostasis Mainz funded by BMBF grant 01EO1503 (to W. Ruf); Boehringer Ingelheim (to M. Thati and W. Ruf); and the Alexander von Humboldt-Stiftung (to W. Ruf). The work was also supported by a Deutscher Akademischer Austauschdienst scholarship (to M.M. Al-Dabet).
Supplementary Material
Acknowledgments
We thank Dr. Katherine Weilbaecher, Washington University School of Medicine, for providing β3LoxP/LoxP mice and Elliot Rosen for providing EPCRδ/δ mice. We also thank Ibrahim Sögüt, Kuheli Banerjee, Anubhuti Gupta, Satish Ranjan, Kathrin Deneser, Julia Judin, Juliane Friedrich, René Rudat, and Rumiya Makarova for their excellent technical support.
Dr. Sanschita Ghosh, Dr. Madhusudhan Thati, and Dr. Hongjie Wang designed and conducted in vitro work, mouse experiments, and ex vivo analysis; Dr. Moh’d Mohanad Al-Dabet, Dr. Wei Dong, Dr. Ahmed Elwakiel, Dr. Ihsan Gadi, Dr. Dheerendra Gupta, Dr. Sumra Nazir, Dr. Rajiv Rana, and Dr. Silke Zimmermann assisted with animal experiments and ex vivo analysis; Dr. Shrey Kohli, Dr. Shruthi Krishnan, and Dr. Akash Mathew conducted in vitro experiments; Dr. Ronald Biemann and Dr. Stoyan Stoyanov assisted with image acquisition and analyses; Dr. Jinyang Zeng-Brouwers and Dr. Liliana Schaefer conducted the microscale thermophoresis binding assay; Dr. Charles T. Esmon, Dr. Marcus J. Moeller, Dr. Jochen Reiser, Dr. Alireza R. Rezaie, and Dr. Wolfram Ruf provided reagents and critically reviewed the manuscript; and Dr. Berend Isermann and Dr. Madhusudhan Thati conceptually designed the study and prepared the manuscript.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
Data Sharing Statement
All data associated with this study are available in the main text or the Supplemental Materials.
Supplemental Material
This article contains the following supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2019111163/-/DCSupplemental.
Supplemental Figure 1. Protein C/aPC is glomerularily filtered.
Supplemental Figure 2. Characterizing knockdown of integrin-β3 in podocytes.
Supplemental Figure 3. aPC-induced signaling in podocytes.
Supplemental Figure 4. Characteristics of experimental mice, corresponding to Figure 4A.
Supplemental Figure 5. Characteristics of experimental mice, corresponding to Figure 4, B–G.
Supplemental Figure 6. Characterization of experimental mice, corresponding to Figure 5.
Supplemental Figure 7. Generation and characterization of RGE-APChigh mice.
Supplemental Figure 8. Characterization of experimental mice, corresponding to Figure 6.
References
- 1.Fouli GE, Gnudi L: The future: experimental therapies for renal disease in diabetes. Nephron 143: 3–7, 2019. [DOI] [PubMed] [Google Scholar]
- 2.Gaede P, Tarnow L, Vedel P, Parving HH, Pedersen O: Remission to normoalbuminuria during multifactorial treatment preserves kidney function in patients with type 2 diabetes and microalbuminuria. Nephrol Dial Transplant 19: 2784–2788, 2004. [DOI] [PubMed] [Google Scholar]
- 3.Bock F, Shahzad K, Vergnolle N, Isermann B: Activated protein C based therapeutic strategies in chronic diseases. Thromb Haemost 111: 610–617, 2014. [DOI] [PubMed] [Google Scholar]
- 4.Isermann B: Homeostatic effects of coagulation protease-dependent signaling and protease activated receptors. J Thromb Haemost 15: 1273–1284, 2017. [DOI] [PubMed] [Google Scholar]
- 5.Arakaki AKS, Pan WA, Trejo J: GPCRs in cancer: protease-activated receptors, endocytic adaptors and signaling. Int J Mol Sci 19: 1886, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Griffin JH, Zlokovic BV, Mosnier LO: Activated protein C: biased for translation. Blood 125: 2898–2907, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aisiku O, Peters CG, De Ceunynck K, Ghosh CC, Dilks JR, Fustolo-Gunnink SF, et al.: Parmodulins inhibit thrombus formation without inducing endothelial injury caused by vorapaxar. Blood 125: 1976–1985, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nazir S, Gadi I, Al-Dabet MM, Elwakiel A, Kohli S, Ghosh S, et al.: Cytoprotective activated protein C averts Nlrp3 inflammasome-induced ischemia-reperfusion injury via mTORC1 inhibition. Blood 130: 2664–2677, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Riewald M, Petrovan RJ, Donner A, Mueller BM, Ruf W: Activation of endothelial cell protease activated receptor 1 by the protein C pathway. Science 296: 1880–1882, 2002. [DOI] [PubMed] [Google Scholar]
- 10.Isermann B, Vinnikov IA, Madhusudhan T, Herzog S, Kashif M, Blautzik J, et al.: Activated protein C protects against diabetic nephropathy by inhibiting endothelial and podocyte apoptosis. Nat Med 13: 1349–1358, 2007. [DOI] [PubMed] [Google Scholar]
- 11.Mosnier LO, Zlokovic BV, Griffin JH: The cytoprotective protein C pathway. Blood 109: 3161–3172, 2007. [DOI] [PubMed] [Google Scholar]
- 12.Rezaie AR: The occupancy of endothelial protein C receptor by its ligand modulates the par-1 dependent signaling specificity of coagulation proteases. IUBMB Life 63: 390–396, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mosnier LO, Sinha RK, Burnier L, Bouwens EA, Griffin JH: Biased agonism of protease-activated receptor 1 by activated protein C caused by noncanonical cleavage at Arg46. Blood 120: 5237–5246, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guo H, Liu D, Gelbard H, Cheng T, Insalaco R, Fernández JA, et al.: Activated protein C prevents neuronal apoptosis via protease activated receptors 1 and 3. Neuron 41: 563–572, 2004. [DOI] [PubMed] [Google Scholar]
- 15.Madhusudhan T, Wang H, Straub BK, Gröne E, Zhou Q, Shahzad K, et al.: Cytoprotective signaling by activated protein C requires protease-activated receptor-3 in podocytes. Blood 119: 874–883, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McKelvey K, Jackson CJ, Xue M: Activated protein C: a regulator of human skin epidermal keratinocyte function. World J Biol Chem 5: 169–179, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang XV, Banerjee Y, Fernández JA, Deguchi H, Xu X, Mosnier LO, et al.: Activated protein C ligation of ApoER2 (LRP8) causes Dab1-dependent signaling in U937 cells. Proc Natl Acad Sci U S A 106: 274–279, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.O’Brien LA, Richardson MA, Mehrbod SF, Berg DT, Gerlitz B, Gupta A, et al.: Activated protein C decreases tumor necrosis factor related apoptosis-inducing ligand by an EPCR- independent mechanism involving Egr-1/Erk-1/2 activation. Arterioscler Thromb Vasc Biol 27: 2634–2641, 2007. [DOI] [PubMed] [Google Scholar]
- 19.Cao C, Gao Y, Li Y, Antalis TM, Castellino FJ, Zhang L: The efficacy of activated protein C in murine endotoxemia is dependent on integrin CD11b. J Clin Invest 120: 1971–1980, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weiler H: Multiple receptor-mediated functions of activated protein C. Hamostaseologie 31: 185–195, 2011. [DOI] [PubMed] [Google Scholar]
- 21.Shahzad K, Kohli S, Al-Dabet MM, Isermann B: Cell biology of activated protein C. Curr Opin Hematol 26: 41–50, 2019. [DOI] [PubMed] [Google Scholar]
- 22.Elphick GF, Sarangi PP, Hyun YM, Hollenbaugh JA, Ayala A, Biffl WL, et al.: Recombinant human activated protein C inhibits integrin-mediated neutrophil migration. Blood 113: 4078–4085, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gil-Bernabe P, D’Alessandro-Gabazza CN, Toda M, Boveda Ruiz D, Miyake Y, Suzuki T, et al.: Exogenous activated protein C inhibits the progression of diabetic nephropathy. J Thromb Haemost 10: 337–346, 2012. [DOI] [PubMed] [Google Scholar]
- 24.Madhusudhan T, Wang H, Ghosh S, Dong W, Kumar V, Al-Dabet MM, et al.: Signal integration at the PI3K-p85-XBP1 hub endows coagulation protease activated protein C with insulin-like function. Blood 130: 1445–1455, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Greka A, Mundel P: Cell biology and pathology of podocytes. Annu Rev Physiol 74: 299–323, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hayek SS, Koh KH, Grams ME, Wei C, Ko YA, Li J, et al.: A tripartite complex of suPAR, APOL1 risk variants and αvβ3 integrin on podocytes mediates chronic kidney disease. Nat Med 23: 945–953, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kreidberg JA, Symons JM: Integrins in kidney development, function, and disease. Am J Physiol Renal Physiol 279: F233–F242, 2000. [DOI] [PubMed] [Google Scholar]
- 28.Pozzi A, Jarad G, Moeckel GW, Coffa S, Zhang X, Gewin L, et al.: Beta1 integrin expression by podocytes is required to maintain glomerular structural integrity. Dev Biol 316: 288–301, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dong W, Wang H, Shahzad K, Bock F, Al-Dabet MM, Ranjan S, et al.: Activated protein C ameliorates renal ischemia-reperfusion injury by restricting Y-box binding protein-1 ubiquitination. J Am Soc Nephrol 26: 2789–2799, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Castellino FJ, Liang Z, Volkir SP, Haalboom E, Martin JA, Sandoval-Cooper MJ, et al.: Mice with a severe deficiency of the endothelial protein C receptor gene develop, survive, and reproduce normally, and do not present with enhanced arterial thrombosis after challenge. Thromb Haemost 88: 462–472, 2002. [PubMed] [Google Scholar]
- 31.Morgan EA, Schneider JG, Baroni TE, Uluçkan O, Heller E, Hurchla MA, et al.: Dissection of platelet and myeloid cell defects by conditional targeting of the beta3-integrin subunit. FASEB J 24: 1117–1127, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moeller MJ, Sanden SK, Soofi A, Wiggins RC, Holzman LB: Podocyte-specific expression of cre recombinase in transgenic mice. Genesis 35: 39–42, 2003. [DOI] [PubMed] [Google Scholar]
- 33.Bock F, Shahzad K, Wang H, Stoyanov S, Wolter J, Dong W, et al.: Activated protein C ameliorates diabetic nephropathy by epigenetically inhibiting the redox enzyme p66Shc. Proc Natl Acad Sci U S A 110: 648–653, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liaw PC, Esmon CT, Kahnamoui K, Schmidt S, Kahnamoui S, Ferrell G, et al.: Patients with severe sepsis vary markedly in their ability to generate activated protein C. Blood 104: 3958–3964, 2004. [DOI] [PubMed] [Google Scholar]
- 35.Strande JL, Hsu A, Su J, Fu X, Gross GJ, Baker JE: SCH 79797, a selective PAR1 antagonist, limits myocardial ischemia/reperfusion injury in rat hearts. Basic Res Cardiol 102: 350–358, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wei C, Möller CC, Altintas MM, Li J, Schwarz K, Zacchigna S, et al.: Modification of kidney barrier function by the urokinase receptor. Nat Med 14: 55–63, 2008. [DOI] [PubMed] [Google Scholar]
- 37.Reynolds AR, Hart IR, Watson AR, Welti JC, Silva RG, Robinson SD, et al.: Stimulation of tumor growth and angiogenesis by low concentrations of RGD-mimetic integrin inhibitors. Nat Med 15: 392–400, 2009. [DOI] [PubMed] [Google Scholar]
- 38.Yan J, Manaenko A, Chen S, Klebe D, Ma Q, Caner B, et al.: Role of SCH79797 in maintaining vascular integrity in rat model of subarachnoid hemorrhage. Stroke 44: 1410–1417, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sonin DL, Wakatsuki T, Routhu KV, Harmann LM, Petersen M, Meyer J, et al.: Protease-activated receptor 1 inhibition by SCH79797 attenuates left ventricular remodeling and profibrotic activities of cardiac fibroblasts. J Cardiovasc Pharmacol Ther 18: 460–475, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marquardt A, Al-Dabet MM, Ghosh S, Kohli S, Manoharan J, ElWakiel A, et al.: Farnesoid X receptor agonism protects against diabetic tubulopathy: potential add-on therapy for diabetic nephropathy. J Am Soc Nephrol 28: 3182–3189, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shahzad K, Bock F, Al-Dabet MM, Gadi I, Kohli S, Nazir S, et al.: Caspase-1, but not caspase-3, promotes diabetic nephropathy. J Am Soc Nephrol 27: 2270–2275, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gong H, Shen B, Flevaris P, Chow C, Lam SC, Voyno-Yasenetskaya TA, et al.: G protein subunit Galpha13 binds to integrin alphaIIbbeta3 and mediates integrin “outside-in” signaling. Science 327: 340–343, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hasegawa K, Wakino S, Simic P, Sakamaki Y, Minakuchi H, Fujimura K, et al.: Renal tubular Sirt1 attenuates diabetic albuminuria by epigenetically suppressing Claudin-1 overexpression in podocytes. Nat Med 19: 1496–1504, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Madhusudhan T, Wang H, Dong W, Ghosh S, Bock F, Thangapandi VR, et al.: Defective podocyte insulin signalling through p85-XBP1 promotes ATF6-dependent maladaptive ER-stress response in diabetic nephropathy. Nat Commun 6: 6496, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kohli S, Ranjan S, Hoffmann J, Kashif M, Daniel EA, Al-Dabet MM, et al.: Maternal extracellular vesicles and platelets promote preeclampsia via inflammasome activation in trophoblasts. Blood 128: 2153–2164, 2016. [DOI] [PubMed] [Google Scholar]
- 46.Galvin JB, Kurosawa S, Moore K, Esmon CT, Esmon NL: Reconstitution of rabbit thrombomodulin into phospholipid vesicles. J Biol Chem 262: 2199–2205, 1987. [PubMed] [Google Scholar]
- 47.Jiang W, Hua R, Wei M, Li C, Qiu Z, Yang X, et al.: An optimized method for high-titer lentivirus preparations without ultracentrifugation. Sci Rep 5: 13875, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kutner RH, Zhang XY, Reiser J: Production, concentration and titration of pseudotyped HIV-1-based lentiviral vectors. Nat Protoc 4: 495–505, 2009. [DOI] [PubMed] [Google Scholar]
- 49.Wienken CJ, Baaske P, Rothbauer U, Braun D, Duhr S: Protein-binding assays in biological liquids using microscale thermophoresis. Nat Commun 1: 100, 2010. [DOI] [PubMed] [Google Scholar]
- 50.Yokota N, Zarpellon A, Chakrabarty S, Bogdanov VY, Gruber A, Castellino FJ, et al.: Contributions of thrombin targets to tissue factor-dependent metastasis in hyperthrombotic mice. J Thromb Haemost 12: 71–81, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Isermann B, Sood R, Pawlinski R, Zogg M, Kalloway S, Degen JL, et al.: The thrombomodulin-protein C system is essential for the maintenance of pregnancy. Nat Med 9: 331–337, 2003. [DOI] [PubMed] [Google Scholar]
- 52.Faioni EM, Fontana G, Razzari C, Avagliano L, Bulfamante G, Calvi E, et al.: Activation of protein C in human trophoblasts in culture and downregulation of trophoblast endothelial protein C receptor by TNF-α. Reprod Sci 22: 1042–1048, 2015. [DOI] [PubMed] [Google Scholar]
- 53.Sharma R, Waller AP, Agrawal S, Wolfgang KJ, Luu H, Shahzad K, et al.: Thrombin-induced podocyte injury is protease-activated receptor dependent. J Am Soc Nephrol 28: 2618–2630, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yoo TH, Pedigo CE, Guzman J, Correa-Medina M, Wei C, Villarreal R, et al.: Sphingomyelinase-like phosphodiesterase 3b expression levels determine podocyte injury phenotypes in glomerular disease. J Am Soc Nephrol 26: 133–147, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Huveneers S, Danen EH: Adhesion signaling - crosstalk between integrins, Src and Rho. J Cell Sci 122: 1059–1069, 2009. [DOI] [PubMed] [Google Scholar]
- 56.Shen B, Estevez B, Xu Z, Kreutz B, Karginov A, Bai Y, et al.: The interaction of Gα13 with integrin β1 mediates cell migration by dynamic regulation of RhoA. Mol Biol Cell 26: 3658–3670, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Madsen CD, Ferraris GM, Andolfo A, Cunningham O, Sidenius N: uPAR-induced cell adhesion and migration: vitronectin provides the key. J Cell Biol 177: 927–939, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wei C, El Hindi S, Li J, Fornoni A, Goes N, Sageshima J, et al.: Circulating urokinase receptor as a cause of focal segmental glomerulosclerosis. Nat Med 17: 952–960, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Matsumoto K, Yano Y, Gabazza EC, Araki R, Bruno NE, Suematsu M, et al.: Inverse correlation between activated protein C generation and carotid atherosclerosis in Type 2 diabetic patients. Diabet Med 24: 1322–1328, 2007. [DOI] [PubMed] [Google Scholar]
- 60.Kolavennu V, Zeng L, Peng H, Wang Y, Danesh FR: Targeting of RhoA/ROCK signaling ameliorates progression of diabetic nephropathy independent of glucose control. Diabetes 57: 714–723, 2008. [DOI] [PubMed] [Google Scholar]
- 61.Waasdorp M, Duitman J, Florquin S, Spek CA: Protease-activated receptor-1 deficiency protects against streptozotocin-induced diabetic nephropathy in mice. Sci Rep 6: 33030, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Waasdorp M, Duitman J, Florquin S, Spek CA: Vorapaxar treatment reduces mesangial expansion in streptozotocin-induced diabetic nephropathy in mice. Oncotarget 9: 21655–21662, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wei C, Li J, Adair BD, Zhu K, Cai J, Merchant M, et al.: uPAR isoform 2 forms a dimer and induces severe kidney disease in mice. J Clin Invest 129: 1946–1959, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rezaie AR: Protease-activated receptor signalling by coagulation proteases in endothelial cells. Thromb Haemost 112: 876–882, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Russo A, Soh UJ, Paing MM, Arora P, Trejo J: Caveolae are required for protease-selective signaling by protease-activated receptor-1. Proc Natl Acad Sci U S A 106: 6393–6397, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.