

Abstract
The role played by cinnamic acid derivatives in treating cancer, bacterial infections, diabetes and neurological disorders, among many, has been reported. Cinnamic acid is obtained from cinnamon bark. Its structure is composed of a benzene ring, an alkene double bond and an acrylic acid functional group making it possible to modify the aforementioned functionalities with a variety of compounds resulting in bioactive agents with enhanced efficacy. The nature of the substituents incorporated into cinnamic acid has been found to play a huge role in either enhancing or decreasing the biological efficacy of the synthesized cinnamic acid derivatives. Some of the derivatives have been reported to be more effective when compared to the standard drugs used to treat chronic or infectious diseases in vitro, thus making them very promising therapeutic agents. Compound 20 displayed potent anti-TB activity, compound 27 exhibited significant antibacterial activity on S. aureus strain of bacteria and compounds with potent antimalarial activity are 35a, 35g, 35i, 36i, and 36b. Furthermore, compounds 43d, 44o, 55g–55p, 59e, 59g displayed potent anticancer activity and compounds 86f–h were active against both hAChE and hBuChE. This review will expound on the recent advances on cinnamic acid derivatives and their biological efficacy.
Keywords: cinnamic acid, antimicrobial, anticancer, antimalarial, Alzheimer’s disease
1. Introduction
Cinnamic acid, a natural aromatic carboxylic acid [1] (Figure 1), is a key chemical found in plants such as Cinnamomum cassia (Chinese cinnamon) and Panax ginseng, fruits, whole grains, vegetables and honey [1]. The presence of an acrylic acid group substituted on the phenyl ring gives cinnamic either a cis or a trans configuration with the latter being the most common of the two [2,3]. Studies have reported that cinnamic acid exhibit antioxidant, antimicrobial [4], anticancer [5], neuroprotective, anti-inflammatory and antidiabetic properties. [6]. Cinnamic acid terminates radical chain reactions by donating electrons that react with radicals forming stable products [7]. It is also used as a fragrant ingredient in toiletries, flavorings cosmetics and detergents [2]. Cinnamic acid can be prepared by enzymatic deamination of phenylalanine [8].
Figure 1.
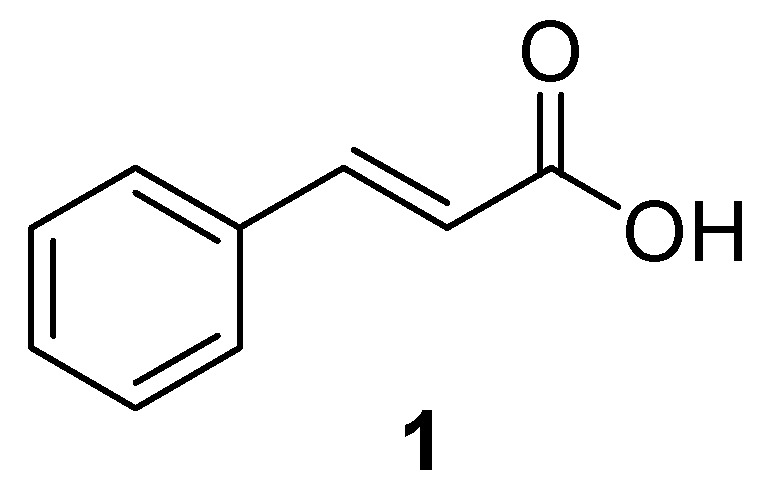
Chemical structure of cinnamic acid (1).
Besides the cinnamic acid derivatives that occur naturally in plants [2], the presence of a benzene ring and the acrylic acid group makes it possible to modify it resulting in synthetic cinnamic acid derivatives [9]. Apart from them being intermediates in producing other compounds like stilbenes and styrenes, cinnamic acid derivatives exhibit antibacterial, antifungal, anti-inflammatory [10], neuroprotective [11], anticancer [12] and antidiabetic activities [13]. Cinnamic acid derivatives that have been reported by researchers include ferulic acid, curcumin [11], caffeic acid, p-hydroxycinnamic acid [14] coumaric and chlorogenic acids [15], etc. These cinnamic acid analogues are differentiated by the presence of hydroxyl groups on the phenyl ring that are either free or methoxylated [15].
The biological activities of different cinnamic acid derivatives have been attributed to the nature and position of the substituent groups. Drug resistance and the absence of curative therapies with low adverse side effect profiles to control cancer, microbial growth, neurological disorders etc. has led to research on the development of therapeutic compounds based on cinnamic acid [16]. Due to the biological efficacy of derivatives of cinnamic acid, this review will report different recently synthesized derivatives of cinnamic acid and their biological activity in vitro and in vivo.
2. Cinnamic Acid Derivatives with Antimicrobial Activity
The World Health Organization (WHO) recognizes infectious diseases caused by bacteria, viruses and fungi as a global health threat, especially in developing and poor countries. About 3.5 million people die from infectious diseases annually [17]. In 2018, the WHO reported 37.9 million people to be living with Human Immunodeficiency Virus (HIV) worldwide with 770,000 dying from AIDS-related illnesses [18]. The 2019 Global tuberculosis (TB) reported an estimated 10.0 million TB infections and 1.2 million deaths in 2018. Of the TB cases, 8.6% were people living with HIV [19]. The appearance of drug-resistant microbial strains has led to difficulty in treating diseases such as TB and malaria. Cinnamic acid derivatives are known for their antimicrobial activity and researchers are searching for antimicrobial agents which are more effective than the currently used standard drugs [20].
Deng et al. extracted a 4-hydroxycinnamic acid derivative, methyl 2-{(E)-2-[4-(formyloxy)phenyl]ethenyl}-4-methyl-3-oxopentanoate (2) (Figure 2) from a plant-based endophytic fungus Pyronema sp. The biological activity of the compound was evaluated in vitro on a non-tuberculous mycobacterium, Mycobacterium marinum, responsible for skin infections. The compound exhibited a good inhibitory effect with an IC50 of 64 µM [21].
Figure 2.
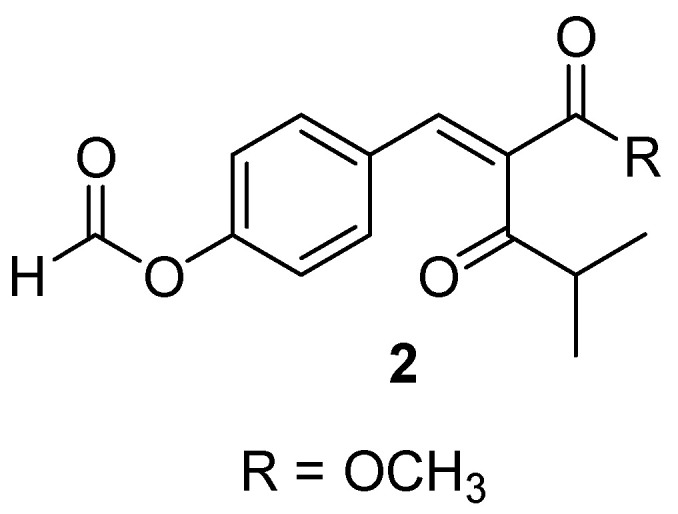
Chemical structure of 4-hydroxycinnamic acid derivative (2) [21].
Korosec et al. tested antifungal activity of cinnamic acid derivatives, most of them were synthesized [22]. The antifungal activity was based on targeting CYP53A15, a unique fungal enzyme known to play a role in demethyling lanosterol, subsequently promoting fungal growth [23]. Derivatives (3–9) (Figure 3) inhibited CYP53A15 enzymatic activity against the three fungi (Cochliobolus lunatus, Aspergillus niger and Pleurotus ostreatus) thereby revealing the potential antifungal activity of cinnamic acid derivatives [22].
Figure 3.

Chemical structures of cinnamic acid derivatives with antifungal activity (3–9) [22].
Based on the Table 1, compounds 3 and 4 were the best inhibitors of C. lunatus growth. The percentage fungal growth increased from compounds 3 to 9 revealing the weak antifungal activity of some of the compounds. Compound 4 was antifungal activity was potent on A. niger inhibitor and compound 6 was not effective in inhibiting P. ostreatus growth. Compound 5 exhibited broad antifungal activity in all the three fungal species. The presence of an electron-withdrawing group on the phenyl ring enhanced the antifungal activity of the compound. The type of phenyl ring substituent and its position also played an important role [23]. 2-nitro or 4-cyano substitution inhibited fungal growth to the optimum [20]. Compound 5 also exhibited the best inhibition on CYP53A15 activity making it a good candidate for the further development of antifungal drugs [22].
Table 1.
Comparing percentage fungal growth of the three fungi when treated with 0.5 mmol/L of cinnamic acid derivatives (3–9) [22].
| Compounds | %Fungal Growth in 0.5 mmol/L of Inhibitor | ||
|---|---|---|---|
| C. lunatus | A. niger | P. ostreatus | |
| 3 | 22 | 85 | 38 |
| 4 | 25 | 66 | 43 |
| 5 | 30 | 28 | 29 |
| 6 | 34 | 109 | 00 |
| 7 | 42 | 84 | 62 |
| 8 | 58 | 60 | 67 |
| 9 | 63 | 50 | 53 |
| control | 90 | 109 | 55 |
Atmaram Upare et al. synthesized cinnamic acid derivatives (10–26) (Figure 4) in which the carboxyl group was replaced with bioisostere 1,2,4-oxadiazole followed by substituting the phenyl ring of the cinnamic acid resulting in a series of novel styryl oxadiazoles. The compounds were tested in vitro at day 8 for their anti-tubercular activity against H37Ra strain of Mycobacterium tuberculosis (Table 2). Compound 10 was 20 times less active when compared to cinnamic acid. Compounds having electron-donating substituents on the para position of the phenyl ring (11–13) were inactive with IC50 > 30 µg/mL [24].
Figure 4.

Chemical structures of the synthesized novel styryl oxadiazoles [24].
Table 2.
In vitro anti-tubercular activity of compounds 10–26 against M.tb H37Ra on day 8 [24].
| Compound | IC50 (µg/mL) | Cpd. | IC50 (µg/mL) |
|---|---|---|---|
| 1 | 0.06 | 18 | ˃30 |
| 10 | 1.42 | 19 | 11.01 |
| 11 | ˃30 | 20 | 0.045 |
| 12 | ˃30 | 21 | 0.56 |
| 13 | ˃30 | 22 | ˃30 |
| 14 | 4.53 | 23 | 0.56 |
| 15 | 9.91 | 24 | ˃30 |
| 16 | 2.35 | 25 | ˃30 |
| 17 | 0.36 | 26 | 1.56 |
For the compounds with halogen substitutes on the phenyl ring, the position, number of substituents and the type of halogen affected the biological activity of the compounds. Para-substituted chloro-compound 14 (IC50 = 4.54 µg/mL) was more active when compared to ortho-substituted compound 15 (IC50 = 9.91 µg/mL), suggesting that the presence of chlorine on the para position enhanced the antibacterial activity of the compound [25]. Compound 17 with a 4-fluoro phenyl ring substitution was the most active of all the compounds containing fluorine with IC50 = 0.36 µg/mL indicating that fluoro substitution favored anti-tuberculosis activity of the synthesized compound. However, compound 24 displayed high IC50 value greater than 30 µg/mL suggesting the nature of the fluorine substituent was also important [24].
The introduction of an electron-withdrawing group improved their antibacterial activity (i.e., compounds 20 and 21). Outstandingly, compound 20 with a carboxylic acid at the para position exhibited potent anti-TB activity (IC50 = 0.045 µg/mL) when compared to cinnamic acid. Thus, generally, the presence of an electron-withdrawing group on the para position of the phenyl ring favored significant anti-TB activity [25]. Compounds with electron-withdrawing groups exhibited improved activity (compound 23 with IC50 = 0.56 µg/mL was more active than compound 16 with IC50 = 2.35 µg/mL) while compounds with electron-donating substituents on the phenyl ring did not display a significant anti-TB activity [24].
Malheiro et al. synthesized cinnamic acid derivatives by modifying the carboxyl group, the alkene and the phenyl ring (27–32) (Figure 5) and studied their effects, together with selected phytochemicals, on controlling planktonic Escherichia coli, Staphylococcus aureus and Enterococcus hirae. The phytochemicals and derivatives were also compared with the commonly used biocides such as sodium hypochlorite, triclosan, hydrogen peroxide, etc. Compound 27 appeared to be the most potent on all the three bacteria species, with better efficacy than sodium hypochlorite (Table 3) [16].
Figure 5.

Chemical structures of compounds, 27–32 [16].
Table 3.
Minimum inhibitory concentration (MIC) of representative biocides and derivatives against E. coli, S. aureus and E. hirae [16].
| Compounds | E. coli (mM) | S. aureus (mM) | E. hirae (mM) |
|---|---|---|---|
| Sodium hypochlorite | 5 | 8 | 5 |
| Triclosan | 0.003 | 0.003 | 0.02 |
| 27 | 3 | 3–5 | 5–8 |
| 28 | 20–25 | ˃25 | ˃25 |
| 29 | 15–20 | 15–20 | 20–25 |
| 30 | 15–20 | 15–25 | 18–25 |
| 31 | ˃13 | ˃13 | ˃13 |
| 32 | ˃25 | ˃25 | ˃25 |
Generally, compounds 28–30 inhibition of bacterial growth on all the three species was not significant when compared to the control (Table 3). However, the compound 27 antibacterial effect was significant on the S. aureus strain of bacteria studied when compared to the control, sodium hypochlorite. The results of the compounds showed that modifications on the alkene group, phenyl ring and carboxylic acid functional groups affect the biological activity of the compounds [16].
Antimicrobial activities of chlorogenic acid (compound 33) (Figure 6) include antibacterial activity against multi-drug resistant bacteria, antifungal and anti-viral activities against HSV-1 and HSV-2, adenovirus, Ebola and HIV. Antifungal effects of compound 33 are known as it is effective against Candida albicans through impacting the cell membrane. It is effective against Escherichia coli, Staphylococus aureus, Staphylococcus epidermidis and Klebsiella pneumonia, among other bacteria. It has broad applications in food due to its insensitivity against probiotic bacteria [26].
Figure 6.

Chemical structure of chlorogenic acid (33) [26].
Wang et al. evaluated anti-Tobacco mosaic virus (TMV) activities of synthesized cinnamic acid derivatives containing dithioacetal moieties. Only 34a and 34b (Figure 7) displayed curative activities of 82.6% and 83.5%, respectively which were significant when compared to ribavirin (70.3%) and xiangcaoliusuobingmi (71.7%), used as the control [27].
Figure 7.

Cinnamic acid derivatives with dithioacetal moieties (34a and 34b) [27].
Compounds 34a and 34b antiviral activities were comparable to the activity of ningnanmycin (85.2%) used as the control. Treatment of TMV particles with 34b caused serious damage and fracture. Molecular docking results showed that 34b bonded excellently with amino acid residues GLN34, THR37, ARG90, and ARG46 of TMV coat protein via hydrogen bonds [27].
Despite the several reports on cinnamic acid derivatives, the compounds with potent antimicrobial activities are 3–5 which were effective against fungal strains such as C. lunatus, A. niger and P. ostreatus when compared to cinnamic acid [22]. Furthermore, compounds with high antibacterial activity are 20 which exhibited potent anti-TB activity when compared to cinnamic acid [24] and compound 27 which was effective on E. coli, S. aureus and E. hirae when compared to sodium hypochlorite [16].
3. Cinnamic Acid Derivatives with Anticancer Activity
In 2018, WHO reported 18.1 million new cancer cases and 9.6 million deaths making cancer one of the leading causes of death [28]. The high number of people who die from cancer has led to researchers making unremitted efforts to develop safe and efficient antitumor agents [29]. The effectiveness of most of the currently used chemotherapeutic agents is severely limited by drug resistance [30]. Most drugs fail during invasion and metastasis of cancers making patients succumb to the disease [30]. Cinnamic acid derivatives have anticancer effects and are effective against a wide range of cancers such as breast [5] colon, lung, etc. Antiproliferative activity of cinnamic derivatives against tumors has been reported [31]. Cinnamic acid derivatives, such as cinnamaldehyde, use apoptosis, among other ways, to destroy cancerous cells [32].
Perkoic et al. synthesized harmicine and cinnamic acid hybrids (35–37) (Figure 8). In vitro cytotoxicity of the hybrids on a human liver hepatocellular carcinoma cell line (HepG2) depended on the harmicine type and substituent in the cinnamic acid derivative. Compounds 36d, 36e and 36f were the most effective compounds against HepG2 cell line with IC50 values of 3.11, 2.19 and 0.74 µM, respectively, when compared to harmine with IC50 greater than 250 µM [33]. The increased cytotoxicity of compounds 36d, 36e and 36f is attributed to them being O-harmicines while the others are N-harmicines and N,O-bis-harmicines. However, no specific structure–activity relationship was reported [33].
Figure 8.

Structures of cinnamic acid-harmine hybrids (35–37) [33].
Pellerito et al. synthesized ferulate derivative, tributyltin(IV) ferulate. In vitro studies on colon cancer cells, HCT116, Caco-2 and HT-29 showed that the ferulic acid did not alter the viability of any of the three cell lines even up to 1 µM concentration while tributyltin(IV) ferulate showed remarkable cell viability reduction at 400 nM (−78.9% for HCT116, −64.1% for Caco-2 and −57.5% for HT-29). Tributyltin(IV) ferulate induced autophagic cell death and arrested the cell cycle thereby reducing colon cancer cell viability. Thus, apart from it showing remarkable cell viability against the tested cell lines, it also showed a very significant effect at very low doses making the novel ferulic acid derivative a promising colon cancer therapy candidate [34].
Matlou et al. synthesized zinc and indium tetra cinnamic acid phthalocyanine (Pc) complexes (38–40) (Figure 9) which were covalently linked through an amide bond to amino-functionalized magnetic nanoparticles (AMNPs) [35]. Cinnamic acid was connected to the nanoparticles due to its antitumor activity [36]. Compounds 38a–c were compared with compounds 39a–c for photodynamic therapy and in vitro cytotoxicity against human breast adenocarcinoma cell lines. Compounds 38a, 39a, 38b and 39b triplet quantum yield improved after linking to AMNPs but decreased for 39c (0.65) and 38c (0.73) due to photoisomerization on the alkene double bond of cinnamic acid [37]. The triplet lifetimes for complex 40 was shortened from 258 µs to 224 µs due to triplet energy lowering and non-radiative decays increasing after linking AMNPs to the complex. Singlet oxygen quantum yields also increased in the presence of AMNPs for 38a and 39a and decreased for 40.
Figure 9.

Chemical structures of zinc and indium tetra cinnamic acid phthalocyanine (Pc) complexes covalently linked to amino-functionalized magnetic nanoparticles (AMNPs) [35].
All the six compounds showed very low in vitro cytocidal effects in the dark with compound 38c showing the least cell viability at 78% at 80 µg/mL. This indicated that the used drug have low dark cytotoxicity. Photodynamic activity (PDT) of compounds 38a–c on MCF-7 cells increased as the drug concentration increased from 5–80 µg/mL and the cell death was evaluated based on the cell confluence of viable cells. PDT activity decreased for compounds 39a–c since phthalocyanine photoactivity decreased upon linking to AMNPs due to phthalocyanine complex aggregation. Compound 38b outperformed 38a and 38c due to spin-orbit coupling and efficient transfer of energy to ground state molecular oxygen, which gives it the significant cytocidal activity against the tumorigenic cells [35].
Lima et al. synthesized cinnamic acid derivatives having 1,2,3-triazolic moiety (41a–m and 42a–m) (Figure 10) and evaluated their antimetastatic activity. At 100 µmol/L, The compounds 41a, 41b, 41f, 41h–j, 41l, 41m, 42a–e, 42g–I and 42k–m significantly suppressed B16-F10 cell migration when compared to the cells treated with 0.4% DMSO. Compound 42a was the most effective with cell migration suppression of 86% when compared to the control. B16-F10 cell proliferation was also significantly impaired by compound 42a. There was a decrease in B16-F10 cell invasion and adhesion upon treatment of compound 42a for 24 h which confirmed the antimetastatic activity of cinnamic acid derivatives against colon carcinoma and human lung adenocarcinoma cells [38].
Figure 10.

Chemical structures of compounds 41a–m and 42a–m [38].
B16-F10 cell colonies were reduced when treated with different concentrations of compound 42a at 12.5 μmol/L (25.5%), 25 μmol/L (20.6%) and 50 μmol/L (93.1%) when compared to the control. Since cell proliferation, migration, invasion, adhesion and colonization are the central steps in the in vivo metastasis process [39], the positive results exhibited by compound 42a suggest its efficacy in reducing B16-F10 cells metastatic potential. Vero and fibroplast (NIH3T3) cells appeared somewhat sensitive to compound 42a at 100 µmol/L and no hemolysis was significant at different concentrations. Molecular binding prediction indicated that the compound 42a inhibitory ability is due to its interaction with MMP-9 and MMP-2, enzymes involved directly in melanoma progression [40] The heteroatoms in compound 42a have the capability to form hydrogen bonds with proteins thereby inhibiting the two enzymes [10].
Wang et al. designed and synthesized novel oleanolic acid (OA)-cinnamic acid ester derivatives (43a–d and 44a–o) and glycyrrhetinic acid (GA)-cinnamic acid ester derivatives (45a–p) (Figure 11) and assessed their in vitro cytotoxicity against MCF-7 (breast cancer), HeLa (cervical cancer) and L-O2 (a normal hepatic cell) [5].
Figure 11.

Chemical structures of the novel novel oleanolic acid (OA)-cinnamic acid ester derivatives (43a–d and 44a–o) and glycyrrhetinic acid (GA)-cinnamic acid ester derivatives (45a–p) [5].
Based on the Table 4 below with selected compounds, compound 45c exhibited the most significant cytotoxicity on L-O2 cells. The strong inhibitory activity (1.79 µM) against MCF-7 cells shown by compound 44e suggests that linking 4-methylcinnamic acid to the C-OH position of OA-2-chlorobenzyl ester has the ability to significantly improve the inhibition of MCF-7 cells. Compound 44o was the most potent compound against HeLa cells with IC50 of 1.35 µM, close to that of the control, gefitinib (1.28 µM) [5].
Table 4.
IC50 values of oleanolic acid (OA), glycyrrhetinic acid (GA) and their derivatives towards L-O2, MCF-7 and HeLa cells [5].
| Compounds | IC50 (µM) | Compounds | IC50 (µM) | ||||
|---|---|---|---|---|---|---|---|
| L-O2 | MCF-7 | HeLa | L-O2 | MCF-7 | HeLa | ||
| OA | 32.9 | ˃100 | ˃100 | 44m | ˃100 | 38.89 | 56.29 |
| GA | ˃100 | 64.16 | 98.99 | 44o | ˃100 | ˃100 | 1.35 |
| 43d | ˃100 | 32.49 | 1.55 | 45c | 10.19 | ˃100 | ˃100 |
| 43a | ˃100 | ˃100 | ˃100 | 45e | ˃100 | 36.84 | ˃100 |
| 43e | ˃100 | 1.79 | ˃100 | 45f | ˃100 | ˃100 | ˃100 |
| 44h | ˃100 | ˃100 | ˃100 | 45i | ˃100 | 46.53 | ˃100 |
Compounds 44a, 44h and 44f could not inhibit any of the three cell lines. The structure–activity relationships showed no clear trend when the substituent on the benzyl of GA was either electron-donating or electron-withdrawing. However, the electron-donating group (CH3) on the ortho position of the benzyl group on OA increased the inhibitory effect of the compounds on HeLa cells. Compounds 43d and 44o were further evaluated due to their cytotoxic effect against HeLa cells and they were found to induce apoptosis and increased reactive oxygen species levels in a dose-dependent manner. Both compound 43d and 44o induced autophagy in HeLa cells [5].
Endo et al. examined autophagy-inducing activity of cinnamic acid derivatives in Brazilian green propolis: p-coumaric acid (46), caffeic acid (47), chlorogenic acid (33), drupanin (48), 3,4-caffeoylquinic acid (3,4-CQA) (49), artepillin C (ArtC) (50), baccharin (51) and caffeic acid phenethyl ester (CAPE) (52) (Figure 12) by subjecting them to castration-resistant prostate cancer (CRPC) CWR22R1 cells. Compounds 33, 46, 47, and 48 were less cytotoxic but compounds 49–52 significantly decreased prostate cancer [41].
Figure 12.

Chemical structures of the cinnamic acid derivatives in propolis components [41].
CWR22Rv1 cells viability after 24 h treatment revealed compound 52 to be the most potent, even killing 90% of the cells at 30 µM dosage. Compounds 50 and 51 significantly increased LC3-II levels, showing high autophagy-inducing activity and compound 50 was more potent than 51. Further examinations showed that compound 46 induced autophagy through mechanisms different from lysosomal enzymes inhibition [41]. Choudhari et al. also reported the in vitro potency of Indian stingless bee propolis as anticancer agents [42]. Treatment of A549 cells with compound 52 (50 µM) significantly decreased, in a dose-dependent manner, the expression of claudin-2, a protein highly expressed in most cancer cells [43]. Compound 52 up-regulated lκB levels and inhibited lκB phosphorylation with no effect on Akt, thus decreasing the transcriptional activity of claudin-2 in A549 cells. Compound 52 enhanced the sensitivity of the cells to anticancer agents such as doxorubicin by increasing the paracellular permeability to anticancer agents. Shimizu and Suzuki reported the ability of compound 52 to reduce the inflammation stage in macrophages, supporting the results of Endo and his colleagues [44]. Positive results have been reported for bioavailability and pharmacokinetics of compound 52 in rodents but they still remain unclear in humans [45].
Cotreatment of CWR22Rv1 cells with compound 50 and wortmannin or U0126, which are autophagy inhibitors, greatly decreased the viable cells and significantly increased lactose dehydrogenase release when compared to treating with compound 50 alone indicating the enhancement of the cytotoxic potency of compound 50 by autophagy inhibition. The authors also reported the involvement of necroptosis in the cell death when treated with compound 50 and autophagy inhibitor. Thus based on their findings, compound 50 induces autophagy and apoptosis in CWR22Rv1 cells and most importantly, autophagy inhibition further increases apoptosis and induces necrosis [41].
Ge et al. designed and synthesized novel cinnamic acid-progenone hybrids (53a–o) (Figure 13) and evaluated their biological activity as potential antiproliferative agents against A549, H157, HepG2, MCF-7, and HL-60 cell lines (Table 5) [31].
Figure 13.

Chemical structures of designed and synthesized novel cinnamic acid-progenone hybrids (53a–o) [31].
Table 5.
In vitro antiproliferative activity of compounds 53a–o as IC50 (µM) [31].
| Cpd. | MCF-7 | A549 | H157 | HepG2 | HL-60 | Cpd. | MCF-7 | A549 | H157 | HepG2 | HL-60 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 53a | ˃50 | ˃50 | ˃50 | ˃50 | ˃50 | 53i | ˃50 | ˃50 | ˃50 | ˃50 | ˃50 |
| 53b | ˃50 | ˃50 | 3.72 | 5.46 | ˃50 | 53j | ˃50 | ˃50 | ˃50 | ˃50 | 10.19 |
| 53c | 4.16 | ˃50 | 24.50 | 6.55 | ˃50 | 53k | ˃50 | ˃50 | ˃50 | ˃50 | ˃50 |
| 53d | 8.85 | 30.98 | 21.33 | 22.59 | 15.68 | 53l | ˃50 | ˃50 | ˃50 | 5.5 | ˃50 |
| 53e | ˃50 | ˃50 | 24.54 | 3.81 | ˃50 | 53m | 6.55 | 36.20 | 30.67 | ˃50 | 11.26 |
| 53f | 4.91 | 3.22 | 4.28 | 3.45 | 6.87 | 53n | ˃50 | ˃50 | ˃50 | ˃50 | ˃50 |
| 53g | 6.46 | 5.16 | 3.77 | 8.86 | 3.51 | 53o | ˃50 | ˃50 | ˃50 | 3.10 | ˃50 |
| 53h | 6.11 | ˃50 | 17.98 | 5.22 | 25.64 | Dox | 1.51 | 0.31 | 0.48 | 1.82 | 0.91 |
Compounds 53f and 53g were the most potent compounds against all the five cancer cell lines (Table 5). Based on the IC50 values of compounds 53b–53g, 3-OH or 4-OH increased the potency of the compounds while methylation of the hydroxyl or changing its position decreased the compound activity especially towards A549 and HL-60 cancer cell lines. Antiproliferative activity also decreased upon introducing another methoxyl group (53h) or dialkylation of hydroxyl (53i–53l) indicating the role played by 3-OH and 4-OH on the phenyl ring on the compound’s activity which was observed in 53m being more active than 53n and 53o [31].
At 50 µM, all the compounds exhibited inhibition less than 50% on two non-tumor cell lines, BEAS2B and MCF-10A, thus the hybrids were considered to be selectively toxic towards the cancer cell lines compared to the non-tumor cells. Cell cycle results showed 53f arrested the cell progression in G1 phase and enhanced apoptosis in A549 cells, the regulation of Bax/Bcl-2 expression by 53f contributing to the latter [31].
After reviewing several cinnamamides and their structure–activity relationships in anticancer activity, Gaikwad et al. deduced that the compounds were more active when the ortho group remained unsubstituted but electron-donating groups were tolerated (Scheme 1). Electron-withdrawing groups such as NO2, CN and CF3 at the para position were important for potency and selectivity with tert-Bu and OCH3 being exceptions since they are electron-donating groups contributed to the activity of the compounds. The presence of electron-donating groups such as OCH3, vinyl and NH-R at the meta position was preferred for anticancer activity. For the R2 substitution, heterocyclic and aromatic amines significantly elevated anticancer activity while aliphatic amines showed relatively low potency [25].
Scheme 1.

General synthetic route for cinnamamide. Reagents and conditions: POCl3, TEA, DCM, 0 °C [25].
Most anticancer agents used to treat hepatocellular carcinoma suffer from drug resistance [46]. In order to overcome drug resistance, Ling et al. developed β-carboline and N-hydroxycinnamamide compounds (55a–p) (Figure 14) [30]. All the compounds, 55a–p, inhibited histone deacetylase 1 (HDAC1), a key HDAC subtype shown in cancer cell differentiation and proliferation [30]. Compound 55p displayed the best potency with IC50 of 1.3 Nm when compared to suberoylanilide hydroxamic acid (SAHA) (IC50 = 142 nM), an approved HDAC inhibitor. The presence of an electron-donating group such as the R group such as methoxyl (55g–j) or more than one methoxyl groups (5m–p) enhanced inhibitory potency compared to compounds with a hydrogen (55a,b) or methyl (55c,d) [30].
Figure 14.

Chemical structures of compounds 55a–p [30].
Antiproliferative activities of the compounds against human colon cancer cells (HCT116), human hepatocellular carcinoma (HCC) cells (HepG2 and SMMC-7721), and human lung cancer cells (H1299) showed that 55e, 55g–I and 55o,p (IC50 = 0.41–4.29 µM) showed better antiproliferative potency when compared to SAHA (IC50 = 4.79–7.02 µM) (Table 6). Compound 55p displayed the best antiproliferative effect against drug-sensitive Bel7402 and drug-resistant Bel7402/5-FU with IC50 values of 0.85 and 2.09 µM, respectively. Compound 55p induced better Bel7402/5-FU cell apoptosis than SAHA together with autophagic flux activity in Bel7402/5-FU cells by down-regulating p62 and LC3-1 and up-regulating LC3-ΙΙ proteins, making it a potential candidate in developing anticancer agents to treat multi-drug resistant human cancer [30].
Table 6.
IC50 values of compounds 55g,h, 55k,l, and 55o,p against drug-sensitive HCC Bel7402 and drug-resistant HCC Bel7402/5-FU cells [30].
| Compounds | IC50(µM) | |
|---|---|---|
| Bel7402 | Bel7402/5-FU | |
| SAHA | 4.72 | 9.83 |
| 5-FU | 15.6 | 61.7 |
| 55g | 1.03 | 2.17 |
| 55h | 2.15 | 2.90 |
| 55k | 3.24 | 4.38 |
| 55l | 2.71 | 3.06 |
| 55o | 1.97 | 3.66 |
| 55p | 0.85 | 2.09 |
Substituted cinnamic acyl sulfonamide derivatives (56a–58e) (Scheme 2) were synthesized and screened for their antiproliferative activity on MCF-7 human breast cancer cells. Based on Table 7, compound 56a displayed the best activity (IC50 = 0.17 µg/mL) when compared to the control, colchicine [47].
Scheme 2.

General synthesis of cinnamic acid sulfonamide derivatives (56a–58e). Reagents and conditions: (i) K2CO3, acetone, reflux, 29 h; (ii) piperidine, pyridine, 80–90 °C, 24 h; (iii) substituted benzene-sulfonamides, Dichloromethane (DCM), 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI), 4-Dimethylaminopyridine (DMAP), overnight [47].
Table 7.
Antiproliferative activities and anti-tubulin activities of 56a–58e against MCF-7 cells [47].
| Cpd. | n | R | MCF-7 IC50 (µg/mL) | ITP IC50 (µM) | Cpd. | n | R | MCF-7 IC50 (µg/mL) | ITP IC50 (µM) |
|---|---|---|---|---|---|---|---|---|---|
| 56a | 1 | H | 0.17 | 0.88 | 57d | 2 | Cl | 51.31 | 87.92 |
| 56b | 1 | CH3 | 48.95 | 40.85 | 57e | 2 | Br | ˃100 | ˃100 |
| 56c | 1 | F | 74.05 | 76.90 | 58a | 3 | H | 53.87 | 49.01 |
| 56d | 1 | Cl | 47.27 | 94.72 | 58b | 3 | CH3 | ˃100 | ˃100 |
| 56e | 1 | Br | 89.01 | 51.59 | 58c | 3 | F | ˃100 | ˃100 |
| 57a | 2 | H | 2.16 | 5.47 | 58d | 3 | Cl | ˃100 | ˃100 |
| 57b | 2 | CH3 | ˃100 | 93.33 | 58e | 3 | Br | ˃100 | ˃100 |
| 57c | 2 | F | 52.13 | 27.46 | Colchicine | 0.33 | 1.36 |
Compound 56a had no substituent on B phenyl ring but had a benzdioxan group on the A ring meaning the presence of a substituent on the B phenyl ring decreased the antiproliferative activity of the compound. Tubulin polymerization assay showed that compound 56a was the most potent compound at inhibiting tubulin polymerization in vitro. Molecular docking revealed that compound 56a formed one hydrogen bond and Van der Waals forces with tubulin [47].
Toolabi et al. synthesized 6-Cinnamoyl-4-arylaminothienopyrimidines and compounds 59e and 59g (Figure 15) were very effective against A549 and HeLa cell lines [48]. Compound 59e exhibited in vitro antiproliferative activity against both A549 and HeLa cell lines with IC50 values of 0.04 and 0.004 µM, respectively. Compound 59g had an IC50 of 0.033 µM against HeLa cells. Both compounds were better than the reference drug, erlotinib with IC50 values of 3.80 and 7.48 µM against A549 and HeLa cells, respectively. Compound 59e was further found to inhibit EGFR and ERH1/2 phosphorylation in HeLa cell lines [48].
Figure 15.

Structure of compounds 59e and 59g [48].
Yang et al. synthesized a number of cinnamic acid derivatives and found only compound 60 (Figure 16) which exhibited 25.78% cell viability when tested against leukemia (HL-60) cells at 20 µM for 48h [49]. The structure–cytotoxic activity relationship revealed that the presence of a trimethoxyl group on the phenyl ring, an ethylenediamine linker, inserting aryl substitutes at the C-α position and the presence of α,β-unsaturated ketone carbonyl substituent are all beneficial for the cytotoxicity of the compound. Compound 60 showed better uptake in HL-60 cells than in normal cells, with the mitochondria being the main site of accumulation. At 10 and 15 µM, compound 60 mainly induced apoptosis while at 20 µM it was elicited necrosis of the HL-60 cells [49].
Figure 16.

Chemical structure of compound 60 [49].
The potent derivatives with anticancer activities which need further studies are 36d, 36e, 36f, 43e, 56a, 43d, 44o, 55e and 55g–p. Cinnamic acid derivatives with potent anticancer activity against liver cancer are compounds 36d, 36e and 36f [33]. Compound 43e and 56a were potent on MCF-7 cell lines in vitro [5,47] and compounds 43d, 44o, 55e and 55g were active against cervical cancer cell lines when compared to other derivatives which have been reported [5,48]. Compounds 55g–55p were effective on human hepatocellular carcinoma cell line drug resistant cancer cell lines [30].
4. Cinnamic Acid Derivatives with Antiparasitic Activity
Parasitic diseases such as Psoroptic acariasis, malaria and leishmaniasis remain a major threat to public health especially to millions of people living in poor tropical countries [50]. In 2018, an estimated 405,000 people died of malaria, a parasitic infection calling for more measures to reduce the number of fatalities [51]. Resistance to broad spectrum anthelmintics in livestock helminths is a global threat to livestock such as sheep and goats [50]. The effects caused by these parasitic diseases on humans and livestock has led to the development of cinnamic acid derivatives with antiparasitic activity.
Perkovic et al. synthesized hybrids of harmine and cinnamic acid (35a–37i) (Figure 8) for antiplasmodial evaluation against the erythrocytic stage of Plasmodium falciparum (chloroquine-sensitive Pf3D7 and chloroquine-resistant PfDd2 strains) and hepatic stage of P. berghei as well as cytotoxicity against HepG2 cell line. From Table 8, compounds 36c, 36g, 36h and 37b (IC50 = 0.13–0.16 µM) were the most active against Pf3D7 strain with 37b exhibiting the most potency against Pf3D7 and PfDd2. Results on harmicines activity against P. berghei showed that they were more effective with (IC50 0.4–2.62 µM) when compared to the reference drug, primaquine (IC50 = 8.4 µM). Conjugating harmine with cinnamic acid derivatives via triazole linker produced hybrids with excellent antiplasmodial activity when compared to the parent compounds at the erythrocytic and hepatic stages of Plasmodium infection [33].
Table 8.
IC50 (µM) values for compounds 35–37 after in vitro screening against erythrocytic stage of P. falciparum (Pf3D7 and PfDd2 strains) and hepatic stages of P. berghei [33].
| Cpd | Pf3D7 | PfDd2 | P. berghei | Cpd | Pf3D7 | PfDd2 | P. berghei |
|---|---|---|---|---|---|---|---|
| 35a | 1.26 | 23.76 | 2.62 | 36g | 0.15 | 0.69 | - |
| 35b | 0.61 | 16.61 | - | 36h | 0.13 | 0.46 | - |
| 35c | 0.50 | 1.57 | - | 36i | 0.33 | 3.06 | 0.66 |
| 35d | 0.89 | 2.70 | - | 37a | 0.56 | 1.59 | - |
| 35e | 0.87 | 2.21 | - | 37b | 0.13 | 0.16 | 0.4 |
| 35f | 0.65 | 2.55 | - | 37c | 0.44 | 0.65 | - |
| 35g | 0.44 | 1.87 | 1.12 | 37d | 2.50 | 3.23 | - |
| 35h | 0.56 | 2.26 | 1.10 | 37e | 0.62 | 8.00 | - |
| 35i | 0.77 | 2.82 | - | 37f | 1.07 | 17.94 | - |
| 36a | 0.26 | 1.09 | - | 37g | 0.25 | 3.28 | - |
| 36b | 0.43 | 0.69 | - | 37i | 1.34 | 4.02 | - |
| 36c | 0.16 | 0.47 | - | CQ | 0.003 | 0.20 | - |
| 36d | 0.20 | 1.19 | - | Harmine | 8.25 | ˃27.7 | - |
| 36e | 0.20 | 0.84 | - | Primaquinee | - | - | 8.4 |
| 36f | 0.34 | 1.01 | - |
Structure–activity relationship showed that the general activity pattern was 36 ˃ 37 ˃ 35 except for the p-OCH3 substituted series which had 37b as the most potent compound. For series 35 and 37, the bulkiness of R2 enhanced their antiplasmodial activity in vitro. Replacing hydrogen with fluorine on the m- and p- positions of the cinnamic acid ring resulted in compounds with enhanced antiplasmodial activity for series 35 and 36 (for example 35e ˃ 35a, 35d ˃ 35a) but not for series 37 [33].
Rodrigues et al. synthesized cinnamic acid derivatives (61–63) (Figure 17) and tested their leishmanicidal activity against promastigote and amastigote forms of Leishmania braziensis. At 10 µM, all the derivatives with an isobenzofuranone moiety (compounds 61a–d) were toxic to promastigotes with survival less than 75%. Compounds 62a–d were not toxic to promastigotes of L. braziliensis. Compounds with a triazolic moiety (63a–d) revealed that only compound 63a and 63c were toxic with 80% and 75% survival. Compound 63b was the most active and its activity was similar to the positive control, amphotericin B (3.125 µg/mL) (Table 9) [3].
Figure 17.

Chemical structures of compounds 61–63 (a–d) [3].
Table 9.
% toxicity of the compounds on Leishmania intracellular amastigotes after 48 h. All compounds were 10 µM [3].
| Compounds | % Toxicity | Cpd. | % Toxicity |
|---|---|---|---|
| 61a | 43.87 | 62c | 59.10 |
| 61b | 80.47 | 62d | 80.12 |
| 61c | 59.53 | 63a | 45.10 |
| 61d | 50.14 | 63b | 67.84 |
| 62a | 19.91 | 63c | 69.24 |
| 62b | 86.32 | 63d | 19.67 |
When treated against amastigote forms, compounds 61b, 62b and 62d were the most active with 80.47%, 86.32% and 80.12% toxicity, respectively, which was lower than amphotericin B (98.25%). Compounds 61b and 62b were further assayed because of their non-toxic nature to macrophages and their IC50 values were 7.58 µM and 3.05 µM, respectively, when tested in vitro on the promastigote forms [3].
These findings suggest that the presence of an electron-withdrawing group on the phenyl ring of cinnamic acid enhanced the compound’s anti-leishmanicidal activity. Halogenation of the phenyl ring of the triazole moiety plays an important role on the anti-leishmanicidal activity of the compounds. The results showed that the combination of two different bioactive chemical groups can yield effective novel anti-leishmanicidal drugs [3].
Mancilla-Montelongo et al. evaluated the in vitro anthelmintic (AH) activity of cinnamic acid (1) and its six derivatives (p-coumaric acid (46), caffeic acid (47), trans-ferulic acid (64), trans-sinapic acid (65), 3,4-dimethoxycinnamic acid (66) and chlorogenic acid (51) (Figure 18) against the larvae and eggs of a gastrointestinal nematode, Haemonchus contortus. Only ferulic acid and chlorogenic acid showed notable AH activity ˃74% and ˃39% egg inhibition, respectively, at 300 µg/mL. Although compounds 1, 46 and 47 showed no egg hatching inhibition even at 400 µg/mL, they damaged the larvae structurally, leading to their mortality [15].
Figure 18.

Chemical structures of cinnamic acid with in vitro anthelmintic (AH) activity [15].
The authors also made mixtures (M) of the analogues. M1 was a combination of all the seven analogues. M2 was a combination of compounds 64 and 33 which blocked larval hatching. M3 was a combination of compounds 1, 46, 64, 65 and 33. M4 was a combination of compounds 1, 46 and 65 and the combination resulted in the larval damage. M5, which is a combination of compounds 1 and 46, exhibited significant ovicidal activity. M1 exhibited an effective concentration 50% (EC50) of 1628.10 µg/mL, M4 (1143.75 µg/mL) and M3 (923.37 µg/mL). M2 and M5 showed no significant EC50 value revealing an antagonistic effect. The presence of two free hydroxyl groups in compound 47 did not produce any significant effect of the compound active against H. contortus eggs suggesting that the activity of the compounds is based on their ability to inhibit hatching-related processes and their ability to form a complex film by binding to the proteins [15].
Chen et al. synthesized a series of cinnamic acid derivatives (67–79) and evaluated their efficacy against Psoroptes cuniculi, a non-burrowing ear mite causing Psoroptic acariasis (Figure 19). The synthesized cinnamic acid derivatives included thiol esters, esters, ketones, amides and analogues with a heteroaromatic ring. The structure–activity relationship of the derivatives was studied. Compounds exhibiting higher activity were further studied for median lethal concentrations (LC50) and median lethal times (LT50) in comparison with the reference drug, ivermectin [52].
Figure 19.

Chemical structures of compounds 67–79 [52].
Based on Table 10, compounds 67–75 and 79 had a mean mortality of 84% and above with only compounds 77 and 78 exhibiting mean mortality below 40% at 0.5 mg/mL. These compounds were potent when compared to ivermectin, an acaricidal agent due to factors such as lower molecular weight and simplified structure, affordability and non-toxicity to mammals and higher activity [52,53]. All the compounds displayed LC50 values higher than ivermectin.
Table 10.
Mean mortality percentages of Psoroptes cuniculi when incubated with 0.5 mg/mL and 0.25 mg/mL of different compounds [52].
| Cpd. | Mean Mortality % | LC50 [mM] (24 h) | LT50 (h) | |
|---|---|---|---|---|
| 0.5 mg/mL | 0.25 mg/mL | |||
| 67 | 100.0 | 78.3 | 1.07 | 13.3 |
| 68 | 100.0 | 98.3 | 0.56 | 12.0 |
| 69 | 90.0 | 76.0 | 0.87 | 13.9 |
| 70 | 84.0 | 68.0 | 0.74 | 15.2 |
| 71 | 100.0 | 100.0 | 0.45 | 9.8 |
| 72 | 100.0 | 80.0 | 0.82 | 15.6 |
| 73 | 100.0 | 84.0 | 0.64 | 6.8 |
| 74 | 100.0 | 100.0 | 0.36 | 2.6 |
| 75 | 98.0 | 66.0 | ||
| 76 | 65.0 | 16.7 | ||
| 77 | 38.3 | - | ||
| 78 | 26.0 | - | ||
| 79 | 94.0 | 44.0 | ||
| Ivermectin | 75.0 | 68.3 | 0.28 | 8.9 |
The cinnamic acid esters (67–70), the type of alkyl group, the length of the chain and the degree of branching greatly affected their biological activity. The compounds with potent biological activity were compound 68, 71, 73 and 74. Steric hindrance close to the ester group decreased the activity of the compounds. The conversion of a carbonyl on the ester group to a methylene group led to a notable decrease in activity as seen when comparing 68 with 78. Activity decreased when ester (68) was changed to thiol ester (79). Replacing the phenyl group (68) with 2-pyridinyl (71) or 2-furanyl (74) increased the activity [52]. Based on the different reports on the antiparasitic activity of cinnamic derivatives, the potent cinnamic acid derivatives with antimalarial activity are 36c, 36g, 36h and 37b [33].
5. Cinnamic Acid Derivatives with Neurological Activity
The brain, an organ of soft nervous tissue is contained in the skull of vertebrates. It functions as the coordinating centre of sensation, intellectual and nervous activity [54]. When the brain is damaged, it can affect many different things, including memory, sensation, and even personality. A number of brain diseases and neurological disorders have been found which include brain tumor, traumatic brain injury and Alzheimer’s disease, and these need to be treated for the well-functioning of the brain [55]. The treatment of Alzheimer’s disease is of prime importance since the disease is characterized by memory loss, cognitive impairment and can be fatal. Treatment methods include restoring acetylcholine levels [56] and clearing the neurotoxic amyloid-beta protein [1]. Research has proven that cinnamic acid derivatives have neuroprotective ability and scientists keep on looking for neuroprotective agents based on cinnamic acid with better biological activity [6].
Lan et al. reacted N-benzyl pyridinium with cinnamic acids of different substitutions to obtain cinnamic acid derivatives (80a–n) (Scheme 3) that inhibit cholinesterase, suggesting potential efficacy in treating Alzheimer’s disease. Compounds 80a–n were highly selective towards acetylcholinesterace from electric eel (AChE) and butylcholinesterace from equine serum (BuChE). However, they were more selective towards AChE when compared to BuChE. Compound 80l was effective at inhibiting AChE with IC50 of 12.1 nM while compound 80n had the highest inhibitory activity against BuChE (IC50 = 1.9 nM). The low cinnamic acid inhibitory activity of IC50 ˃ 100 nM revealed the requirement of the N-benzyl pyridium moiety for enhanced activity [11].
Scheme 3.

Synthetic route for compounds 80a–n. Reagents and conditions: (i) DMAP/EDCl, DCM, rt, 12 h; (ii) CH3CN, reflux, 1–3 h [11].
The methoxy groups present at positions 3 and 4 of the N-benzyl pyridium moiety increased the inhibitory activity of the compound which was significant in compounds 80i–n when compared to 80b–g. Activity decreased upon the introduction of fluorine, methyl and bromine on the meta, para position (for example, compound 80a was more potent than 80g for AChE). Compounds with different substituents (80i–n) had better activity against BuChE which was observed in compound 80h and compounds with a Br substitution showed higher potency (80f, 80g, 80l and 80m) (Table 11). The compounds’ ability to inhibit BuChE was affected more by the substituent size when compared to the electronic properties. Compound 80l had a dual inhibitory potency and further studies showed that it exhibited good neuroprotective activity against Aβ1-42 toxicity in PC12 cells and it can penetrate into the brain, which was observed in PAMPA-BBB assay in vitro making compound 80l a leading novel compound for the treatment of Alzheimer’s disease [11].
Table 11.
Inhibition concentration (IC50) of ChEs activity in nM [11].
| Compound | eeAChE | eqBuChE | Cpd. | eeAChE | eqBuChE |
|---|---|---|---|---|---|
| 80a | 54.1 | 6.3 | 80i | 51.4 | 2.7 |
| 80b | 102.6 | 8.1 | 80j | 90.7 | 3.8 |
| 80c | 1263.8 | 25.0 | 80k | 29.6 | 3.1 |
| 80d | 93.5 | 7.4 | 80l | 12.1 | 2.6 |
| 80e | 90.8 | 10.0 | 80m | 92.6 | 2.5 |
| 80f | 153.6 | 2.5 | 80n | 50.3 | 1.9 |
| 80g | 1450.5 | 2.1 | Cinnamic acid | ˃100,000 | ˃100 |
| 80h | 135.2 | 4.5 | Donepezil | 40.2 | 4.5 |
Ghafary et al. designed cinnamic acid-tryptamide hybrids (81a–l) (Scheme 4) and evaluated their inhibitory activity against acetylcholinesterace (AChE) and butylcholinesterace (BChE) (Table 12) [56]. The presence of either an electron-donating or an electron-withdrawing group on position 4 of the unsubstituted tryptamine derivatives (81a–f) reduced the AChE inhibitory activity and the same effect was observed in compounds containing any substituent on the 4-position of the pendant benzyl group in the 5-methoxytryptamine series (81g–l), except for 81i. Replacing fluorine with different electron-withdrawing groups greatly improved BChE inhibition meaning inhibition of BChE potentially depended more on hydrophobic or hydrogen interactions between the chloro or nitro groups with the BChE active site than on the electron-withdrawing effect [56].
Scheme 4.

Synthetic route for compounds 81a–l. Reagents and conditions: (i) K2CO3/DMF/80 °C, (ii) pyrrolidine/pyridine/EtOH/reflux, (iii) EDCl/HOBt/CH3CN/rt [56].
Table 12.
Cholinesterace inhibitory activity IC50 (µM) of compounds 81a–l [56].
| Cpd | R1 | R2 | AChE | BChE | Cpd | R1 | R2 | AChE | BChE |
|---|---|---|---|---|---|---|---|---|---|
| 81a | H | H | 14.93 | 2.12 | 81h | F | OCH3 | 29.67 | 5.89 |
| 81b | F | H | 32.17 | 9.36 | 81i | Cl | OCH3 | 13.42 | 2.86 |
| 81c | Cl | H | 24.7 | 0.89 | 81j | NO2 | OCH3 | 31.45 | 4.28 |
| 81d | NO2 | H | 34.49 | 0.55 | 81k | CH3 | OCH3 | 31.85 | 12.25 |
| 81e | CH3 | H | 39.1 | 7.73 | 81l | OCH3 | OCH3 | 25.11 | 7.69 |
| 81f | OCH3 | H | 34.95 | 2.51 | Donepezil | 0.027 | 7.79 | ||
| 81g | H | OCH3 | 14.18 | 6.63 |
For compounds 81g–l, the presence of an electron-withdrawing group (81i; 81j and 81h) enhanced the anti-BChE activity with 81g–j and 81l being more potent than the standard drug, donepezil. Compound 81d displayed excellent neuroprotective ability on PC12 neuron cells at 10 µM with cell viability of 62.71. Molecular docking studies showed that 81d inhibited BChE via a mixed-type mode of inhibition [56].
In a recent study, Medvedeva et al. reported the ability of 3-methoxy-4-hydroxycinnamic acid (64), 3,4- dimethoxycinnamic acid (66) and 3-methoxy-4-acetamidoxycinnamic acid (82) (Figure 20) [57] to prevent alpha-synuclein amyloid transformation, thus aiding the treatment of Parkinson’s disease [58]. The IC50 values of the compounds were 251 µM for 64, 50 µM for 82 and 13 µM for 2. The compounds were also non-toxic towards human neuroblastoma cell line making them very promising for treating synucleinopathies [57].
Figure 20.

Chen et al. evaluated the ability of new tacrine-cinnamic acid hybrids in inhibiting cholinesterase as potential therapeutics for the treatment of Alzheimer’s disease. Only 83, 84 and 85 (Figure 21) were chosen to validate their inhibitory activities on human cholinesterases (AChE and BuChE). The IC50 values of the three compounds on human AChE and BuChE, respectively, were 10.2 nM and 6.3 nM for 83, 16.5 nM and 5.7 nM for 84 and 15.7 nM and 8.0 nM for 85. Binding patterns of compounds 83 and 84 with cholinesterases showed π-π stacking interactions, hydrogen bonds and van der Waals forces. At 25 µM, the three compounds inhibited Aβ1–42 aggregation with an inhibitory rate between 31.82% and 34.57% when compared to resveratrol (30.36%), a reference compound [59].
Figure 21.

Chemical structures of novel tacrine-cinnamic acid hybrids [59].
Behavioural studies revealed that all the three compounds, 83–85 showed enhanced cognitive function in the Institute of Cancer Research (ICR) mice in vivo. Administration of the three compounds caused no remarkable morphological liver changes making them safe and promising compounds for the treatment of Alzheimer’s disease [59].
Lan et al. reported on ferulic acid derivatives (86a–o) (Figure 22) as multi-target inhibitors against Alzheimer’s disease. All the ferulic acid derivatives inhibited eeAChE and eqBuChE with IC50 ranging from 0.055 µM to 42.26 µM (Table 13). When evaluated on human ChE (hChE), compounds 86f–h were potent against both hAChE and hBuChE with inhibitory activity higher than the reference compound, donepezil (Table 13) [60].
Figure 22.

Chemical structures of compounds 86a–o [60].
Table 13.
IC50 values of compounds 86f–h on human AChE (hAChE) and BuChE (hBuChE) [60].
| Cpd. | hAChE (nM) | HBuChE (µM) |
|---|---|---|
| 86f | 16.5 | 0.76 |
| 86g | 19.7 | 0.59 |
| 86h | 13.7 | 0.66 |
| Donepezil | 30.2 | 8.7 |
The presence of the methoxyl and hydroxyl groups favored the inhibition of Aβ aggregation. Kinetic studies using compound 86g showed competition for the binding site between the compounds and acetylcholine [60]. The interactions involved π-π stacking, hydrogen bond and cation-π [61]. At concentrations between 6 and 50 µM, compound 86g exhibited neuroprotective effects. Compounds 86f and 86g crossed the BBB thereby reaching the biological targets in the CNS [60].
Zhang et al. synthesized many compounds and found compound 87 (Figure 23) to be the most potent on both the nerve and endothelium cells. Structure–activity relationship analysis showed that the compounds with three methoxyl groups were the most potent indicating the role played by the methoxyl group in optimizing the compound’s activity [9].
Figure 23.

Chemical structure of compound 87 [9].
Compound 87 exhibited high neuroprotective ability against H2O2-induced damage with EC50 values of 3.26 µM and 2.41 µM in HBMEC-2 and SH-SY5Y cells, respectively. Compound 87 showed potential in inhibiting the apoptosis of HBMEC-2 and SH-SY5Y cells induced by H2O2, and also inhibited neuronal apoptosis and promoted angiogenesis [9].
Adisakwattana et al. evaluated the in vitro inhibitory effects of cinnamic acid derivatives (1, 27, 46, 47, 64, 66, 88–95) (Figure 24) against Protein Tyrosine Phosphatase 1B (PTP1B). Results showed that replacing the carboxyl group of cinnamic acid (1) (16.0% inhibition) with alcohol, aldehyde and methyl ester groups (89, 27 and 92) (12.5–18.0% inhibition) did not increase the PTP1B inhibition percentage. Compounds 46 and 94 exhibited the highest inhibition of 39.4% and 35.9%, respectively, showing the role played by the hydroxyl group on the ortho- or para-positions. Introducing two moieties on cinnamic acid resulted in compounds with better inhibition (21.5–24.4%) when compared to cinnamic acid except for compound 64 [13].
Figure 24.

Structures of cinnamic acid derivatives effective against Protein Tyrosine Phosphatase 1B (PTP1B) [13].
The most potent compounds with effective neurological activity are compounds 86f–h. They were active against both hAChE and hBuChE when compared to donepezil in vitro [60]. There is a pressing need for these compounds to be evaluated in vivo.
6. Cinnamic Derivatives with Antidiabetic Activity
Diabetes mellitus is characterized as a metabolic disorder that results from a defect in insulin action, secretion or both [62]. The International Diabetes Federation estimated the global prevalence of diabetes mellitus to increase to 591 million by 2035 from 382 million reported in 2013. The inability to regulate blood glucose also causes many complications such as angiopathy, retinopathy and neuropathy [63]. The inhibitory effect of cinnamic acid derivatives on Protein Tyrosine Phosphatase 1B (PTP1B) has been one of the several ways to regulate blood glucose [13].
Amalan et al. also reported the ability of compound 46 in lowering blood glucose and improving insulin levels [64]. Compounds 94 and 89 (Figure 24) inhibited p-NPP hydrolysis catalysed by PTP1B with IC50 values of 137.74 and 168.40 nM. Kinetic studies proved compounds 89 and 94 to be non-competitive inhibitors with the ability of being developed into highly selective PTP1B inhibitors [64], thus cinnamic acid derivatives can be further explored as PTP1B inhibitors for the treatment of diabetes [13]. Hypochlorous acid (HOCl) and H2O2 scavenging assay results showed compound 95 has the antioxidant capability to capture free radicals and reactive species such as O2·- and HOCl/OCl- [64] which causes damage to the organs in the body [65].
Adisakwattana et al. evaluated compounds 1, 46–47, 64, 66, 88–91 and 93–95 (Figure 24) for their inhibitory activity against intestinal α-glucosidase (maltase and sucrose). The most effective compounds (64, 90 and 47) exhibited a non-competitive inhibition, better on sucrase when compared to maltase (Table 14), although these compounds were less potent than acarbose with IC50 values of 2.2 and 4.8 µM for maltase and sucrase, respectively. Combining compounds 64, 90 and 47 with acarbose produced additive inhibition resulting in reduced postprandial hyperglycemia and hyperinsulinemia in type 2 diabetes [66].
Table 14.
IC50 values for maltase and sucrase inhibition by compounds 47, 64 and 90 [67].
| Compounds | Maltase | Sucrase |
|---|---|---|
| 47 | 0.74 | 0.49 |
| 64 | 0.79 | 0.45 |
| 90 | 0.76 | 0.45 |
Wang et al. isolated a cinnamic acid glycoside derivative, dissectumoside (96), from the roots of H. dissectum (Figure 25). When evaluated for its in vitro triglycerides accumulation activity in 3T3-L1 adipocytes, compound 96 increased activity from 7.8% at 1 µM through 32.1% at 10 µM to 40.1% at 30 µM. The authors reported that compound 96 provided antidiabetic effect [67] as previously reported [68] due to its ability to effectively transport glucose from the medium into adipocytes and triglycerides [67].
Figure 25.
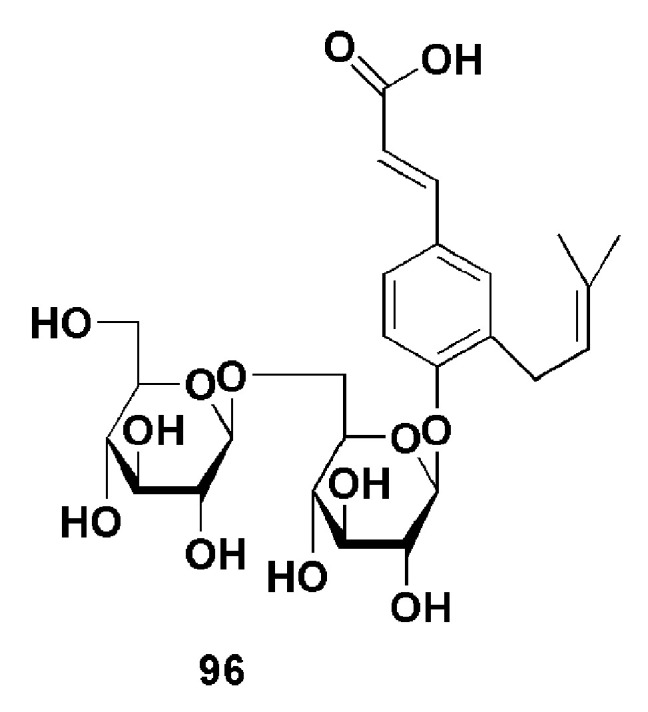
Chemical structure of dissectumoside, 96 [67].
Xu et al. evaluated the activity of coumarin derivatives (97a–i and 98a–i) (Figure 26) as α-glucosidase inhibitors with both the 97 series and the 98 series derivatives having the same substituents. Compounds 98a–i were less potent with 98b having the highest inhibition of 54.745 at 100 µM and IC50 of 94.52 µM, the rest had inhibition less than 50% and IC50 ˃100 µM [69].
Figure 26.

Chemical structures of coumarin derivatives (97a–i and 98a–i) [69].
Since compounds 97a–i were better inhibitors (Table 15) than 98a–i, it showed that conjugating cinnamic acid with 4-hydroxycoumarin was better than with 7-hydroxycoumarin. Compound 97b exhibited the most effective inhibition activity of 89.92% and IC50 of 12.98 µM suggesting that the presence of a methyl group, which is an electron-donating group, favored the withdrawing group (97e–i). The two most potent compounds, 97a and 97b, showed that their α-glucosidase inhibition was reversible. Compound 97b fitted well into the active site of α-glucosidase forming a hydrogen bond with the LYS293 amino acid sequences [69].
Table 15.
Inhibitory Activity of Compounds 97a–i on α-glucosidase [69].
| Compounds | 97a | 97b | 97c | 97d | 97e | 97f | 97g | 97h | 97i |
|---|---|---|---|---|---|---|---|---|---|
| R1 | H | CH3 | OCH3 | F | Cl | Br | CF3 | OH | OH |
| R2 | H | H | H | H | H | H | H | H | OCH3 |
| % inhibition | 83.51 | 89.92 | 73.15 | 68.50 | 33.10 | 60.99 | 41.87 | 33.19 | 19.51 |
| IC50 (µM) | 19.64 | 12.98 | 36.88 | 41.73 | ˃100 | 69.30 | ˃100 | ˃100 | ˃100 |
7. Cinnamic Acid Derivatives with Antioxidant and Anti-Inflammatory Activity and in Cosmetics
Biomolecular oxidations of reactive oxygen and nitrogen species create oxidative stress. Free radicals produce damaged membranes, produce DNA nicks and inflict other oxidative injuries. Accumulation of free radicals activates proto oncogenes causing carcinogenesis [70]. There is a need to scavenge free radicals in the body to minimize the damage inflicted by free radicals. Cinnamic acid derivatives are known to have antioxidant properties [4]. Three mechanisms are reported in phenolic compounds during free radical scavenging. The first one involves a hydrogen atom being abstracted from the antioxidant by the radical. The second step involves the formation of a radical cation ArOH+• through the transfer of an electron to the free radical from the antioxidant and the deprotonation yielding ArO• radical and ROH. In the last step, the phenolic compound is deprotonated and there is an electron transfer from the phenoxide anion to RO• to yield phenoxyl radicals [71].
Muhammad et al. isolated a new bioactive compound (99) (Figure 27) from the plant, Viola betonicifolia. At the concentration range of 2–50 µg/mL, compound 99 inhibited DPPH (diphenyl-2-picryl hydrazyl) free radicals with IC50 of 124 µM which was higher than for common antioxidants α-tocopherol (IC50 of 96 µM) and vitamic C (IC50 of 90 µM). As an anti-glycating agent, compound 99 showed moderate activity with IC50 of 355 µM when compared to the IC50 of rutin, a standard drug which was 294 µM. Thus, due to its free radical scavenging ability, compound 99 has the potential to delay and prevent diabetic complications [72].
Figure 27.
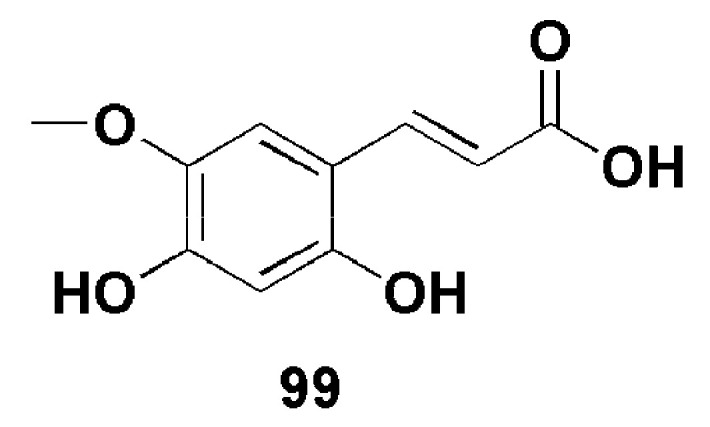
Structure of compound 99 [72].
Kortenska et al. reported the antioxidant activity of compounds 46, 64, 65, 100–103 (Figure 28) in pure triacylglycerols of sunflower oil (TGSO) at 80 °C. Compound 101 showed the highest antioxidant activity at 0.1 mM [73]. The reaction of a phenol antioxidant with lipid radicals produces phenoxyl radicals which are stabilized by unpaired electrons delocalized around the aromatic ring [61]. Replacing hydrogen atoms in the o-position with prenyl groups increased the electron density of the OH-moiety through inductive effects thereby enhancing its reactivity towards lipid radicals [74]. This resulted in compounds 101 and 102 being more active than compound 46 [73].
Figure 28.

Chemical structures of compounds 46, 64, 65, 100–103 [73].
Transformation of the phenol in compounds, 100 and 102 led to the loss of antioxidant activity as there was no hydrogen donating group left. The antioxidant efficiency was found to decrease in the order: 65 ˃ 101 ˃ 103 ˃ 64 ˃ 46 ≈ 100 ≈ 102. Inhibition degree (measure of antioxidant strength), that is the number of times the antioxidant shortened the oxidation chain length, followed the sequence α-tocopherol ˃ 101 ˃ 103 ˃ 65 ˃ 64 ˃ 46 ≈ 100 ≈ 102. The antioxidant activity, calculated as the ability of an inhibitor to terminate the oxidation chain and also the decreased oxidation rate during induction period, followed the sequence: α-tocopherol ˃ 101 ˃ 103 ˃ 65 ˃ 64 ˃ 46 ≈ 100 ≈ 102 [73].
Nazir et al. synthesized cinnamic acid derivatives (104a–c) (Scheme 5) with tyrosinase inhibition activity for hyperpigmentation treatment. Compound 104c (IC50 = 5.7 µM) was the most potent compound (Table 16) and kinetic studies revealed that compound 104c inhibited tyrosinase [75].
Scheme 5.

Synthesis of compounds (104a–c) [75].
Table 16.
Substituents, mushroom tyrosinase inhibitory activity, IC50 (µM) and kinetics of compounds 104a–c [75].
| Cpd | R | IC50 (µM) | Kf (µM) |
|---|---|---|---|
| 104a | H | 39.4 | - |
| 104b | 2-OH | 172.8 | - |
| 104c | 4-OH | 5.7 | 11 |
| Kojic acid | 16.7 | n.d. |
n.d., not determined. Kf is the kinetic reaction constant.
Treatment of B16F10 melanoma cells with compound 104c (30 µg/mL) showed cell viability of higher than 99%. Compound 104c proved a better inhibitor than kojic acid since 125 µg/mL of kojic acid was needed to achieve the same inhibition effected by 30 µg/mL of compound 104c. Thus compound 104c is a promising melanogenic regulator [75].
Gießel et al. synthesized cinnamic acid derivatives (105–112) (Figure 29) for use in flavors and fragrances. When evaluated for their biological activity, the compounds were weak inhibitors of both eeAChE and eqBuAChE with the highest inhibition being 45.5%. Cytotoxic studies against several human tumour cell lines and non-malignant mouse fibroblasts showed the compounds to be non-cytotoxic since they all had EC50 values of more than 30 µM, thus can be safely used as cosmetics and flavoring. The compounds had a typical vanillin smell suggesting their potential use in fragrances and flavor [12].
Figure 29.

Chemical structures of compounds 105–112 [12].
Taofiq et al. evaluated the anti-inflammatory and anti-tyrosinase activities of p-coumaric acid (compound 46). Its anti-inflammatory activity was measured to be EC50 of 152 µg/mL and anti-tyrosinase activity of EC50 ˃ 2.0 mg/mL. [76]. The ability of compound 46 to inhibit tyrosinase was credited to its ability to reduce microphthalmia-associated transcription factor (MITF) [77] and tyrosinase mRNA expression as high as 73% and 82%, respectively. The results showed that compound 46 has the potential to be used together with other bioactive ingredients for protection against hyperpigmentation [76]. Gunia-Krzyzak et al. reported compound 46-containing cream that decreased erythema and pigmentation when applied daily on the foreskin arm [78].
Lee et al. reported the ability of hydroxycinnamic acids (cinnamic acid (1), p-coumaric acid (46), caffeic acid (47) and chlorogenic acid (33) on reducing furan and trapping α-dicarbonyl compounds, which are possible human carcinogens. [79]. Addition of water-soluble antioxidants such as caffeic acid, chlorogenic acid and ferulic acid has been found to reduce levels of furan derivatives [80]. When compound 47 was added to canned-coffee model systems (CCMS) containing glyoxal (GO), furan was reduced from 59.76 µg/L to 48.31 µg/L while compound 51 reduced it to 41.38 µg/L.
Compound 47 reduced furan in methyglyoxal (MGO) from 45.79 µg/L to 35.41 µg/L while compound 33 reduced it to 32.65 µg/L. The results showed the capability of the compounds to suppress furan formation in CCMS. Compounds 47 and 33 showed better GO and MGO trapping efficiency when compared to compounds 1 and 46. This was attributed to the presence of two hydroxyl groups on the aromatic ring trapping the dicarbonyls thereby causing the reduction of furan concentration in CCMS [79].
Garcia-Jimenez et al. reported the catalysis and inhibition of tyrosinase when subjected to different cinnamic acid derivatives (1, 46, 47, 88, 91 and 93–95) (Figure 30). Results of the compounds in the presence of 4-tert-butylcatechol (TBC) showed that compounds 1, 91, 93, 94 and 95 are competitive inhibitors whereas compounds 46, 47 and 88 are substrates of tyrosinase. The inhibition constants were affected by the ability of the substituent group to increase the charge on the benzene ring [81].
Figure 30.

Chemical structures of compounds 1, 46, 47, 88, 91 and 93–95 [81].
The methoxy group, OCH3 is an electron donor and it increased the charge on the benzene ring making the inhibition constant to be in the order 95 ˃ 93 ˃ 91 ˃ 1. Tyrosinase could not hydroxylate compound 94 but was able to hydroxylate compound 88 because compound 94 has a great distance for ligand hydroxylation while compound 88 has a distance feasible for hydroxylation [81].
Ribeiro et al. evaluated the inhibition of cyclooxygenase enzymes (COX-1 and COX-2) by synthesized cinnamic acid derivatives (113a–h, 114a–h, 115a–c, 116a–c and 117) (Scheme 6). Table 17 showed the most important structure–activity relationship for COX-1 inhibition with the presence of the di-substituted aromatic rings having one hydroxyl group and one methoxyl group or two hydroxyl groups, an amide group and the absence of an α,β-double bond [82].
Scheme 6.

Synthesis of compounds 113a–h, 114a–h, 115a–c, 116a–c and 117. Reagents and conditions: (i) Dimethylformamide (DMF), Triethylamine (TEA), Benzotriazol-1-yloxy)tris (dimethylamino) phosphonium hexafluorophosphate(BOP), Dichloromethane (DCM), room temperature (rt) [82].
Table 17.
IC50 and inhibition % of PGE2 production via COX-1 and COX-2, in human whole blood [82].
| Cpd. | COX-1 | COX-2 | Cpd. | COX-1 | COX-2 | ||||
|---|---|---|---|---|---|---|---|---|---|
| IC50 (µM) |
Inhibition (%) |
IC50 (µM) |
Inhibition (%) |
IC50 (µM) |
Inhibition (%) |
IC50 (µM) |
Inhibition (%) |
||
| 113a | N.A. | N.A. | N.A. | N.A. | 114g | 9 | 85 | 21 | 79 |
| 113b | N.A. | N.A. | N.A. | N.A. | 114h | N.A. | N.A. | N.A. | N.A. |
| 113c | ˃100 | 41 | N.A. | N.A. | 115b | 97 | 57 | N.A. | N.A. |
| 113e | ˃100 | 39 | 3.0 | 93 | 116a | 11 | 82 | 62 | 90 |
| 114a | 24 | 82 | 12.7 | 99.0 | 116b | ˃100 | 24 | 76 | 57 |
| 114b | 14 | 81 | 20 | 98.3 | 116c | 15 | 84 | 13.4 | 98.0 |
| 114c | 87 | 60 | 30 | 74 | 117 | 4.3 | 79 | 1.09 | 85 |
| 114d | 45 | 82 | 19 | 91 | Ind. | - | 91 | - | - |
| 114e | 24 | 65 | 2.4 | 99.4 | Cel. | - | - | - | 71 |
| 114f | N.A. | N.A. | N.A. | N.A. | |||||
N.A.—Non active up to highest tested concentration of 100 µM. All the compounds were tested at 100 µM except for compound 117 tested at 12.5 µM for COX-1 and 3.1 µM for COX-2. The two reference drugs were tested at 1 µM. Where Ind. and Cel. represent Indomethacin and Celecoxib, respectively.
These aforementioned features gave compounds 114e (IC50 = 24 µM and 65% inhibition), 114g (IC50 = 9 µM and 85% inhibition) and 116c (IC50 = 15 µM and 84% inhibition). Compounds 116a and 117 were also good COX-1 inhibitors meaning a tri-substituted aromatic ring with one hydroxyl group and two methoxyl groups and no α,β-double bond made compound 116a effective. The best COX-1 inhibitor was compound 117 (IC50 = 4.3 µM) meaning the 3,5-di-tert-butyl-4-hydroxyl aromatic substitution, the presence of tertiary diethylamide group and an α,β-double bond contributed to its biological activity [82].
Compounds 113e, 114a, 114e, 116c, and 117 appeared to be the best COX-2 inhibitors with IC50 values of 3.0, 12.7, 2.4, 13.4, and 1.09 µM, respectively. The structure–activity relationship of the compounds revealed the presence 3,5-di-tert-butyl-4-hydroxyl aromatic substitution, tertiary diethylamide group and an α,β-double bond (compound 132) and a disubstituted aromatic ring with two hydroxyl groups, an amide group and an α,β-double bond influenced the biological activity of these compounds. Since compounds 113e, 114e, 116d and 117 were the most potent compounds against COX-2, they are suggested as potential non-steroidal anti-inflammatory drugs [82].
Ruan et al. reported the potency of a resveratrol-based cinnamic acid derivative, 118 (Figure 31). Compound 118 inhibited LPS-mediated expression of inducible nitric oxide synthase (iNOS) and COX-2 in RAW264.7 cells when analysed by Western blot [83].
Figure 31.
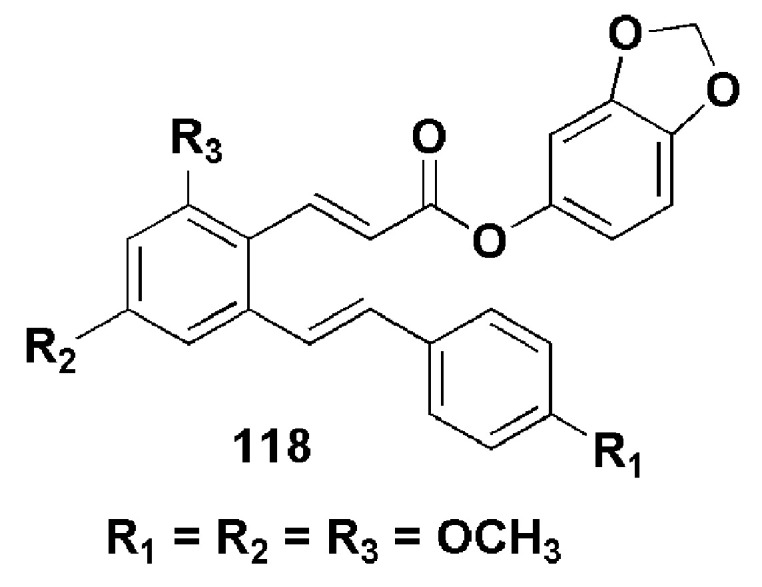
Chemical structure of compound 118 [83].
Inflammation-related diseases are characterized by the expressions of iNOS and COX-2. Compound 188 suppressed the production of NO in LPS-induced RAW264.7 cells in vitro. Furthermore, it inhibited LPS-induced protein expression and immunofluorescence study showed that the compound slightly lowered the activation p65 in the nuclei. The anti-inflammatory role of the compound is partially attributed to its inhibitory effect on the NF-κB signaling pathway LPS-induced RAW 264.7 cells. [83].
8. Conclusions
Research reports have shown that derivatives of cinnamic acid are active in vitro when compared to some of the currently used drugs such as the anticancer, antimicrobial drugs etc. which were used as control. The antimicrobial, anticancer and anti-oxidant activity of the derivatives was influenced by the nature and position of the substituent groups on the phenyl ring. The length of the linker also influenced the biological activity of the derivatives.
Furthermore, the toxicological studies performed on some of the derivatives revealed the non-toxic effects of the compounds suggesting their suitability for applications in vivo. However, the major draw-back pertaining to the synthesized derivatives is that most of them lack in vivo data. Thus, adding to the in vitro analyses that have been carried on most of the potent derivatives, there is a need to proceed to in vivo studies. More research on the development of cinnamic derivatives has the potential to result in potent compounds that will successfully reach clinical trials.
Acknowledgments
The financial assistance of the Medical Research Council and National Research Foundation, South Africa towards this research are hereby acknowledged. The views and opinions expressed in this manuscript are those of the authors and not of MRC or NRF.
Abbreviations
| Cpd. | Compound |
| IC50Half | maximal inhibitory concentration |
| EC50 | Half maximal effective concentration |
| HSV | Herpes simplex virus |
| HIV | Human immunodeficiency virus |
| DMSO | Dimethyl sulfoxide |
| TEA | Triethylamine |
| DCM | Dichloromethane |
| EDCl | 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide |
| DMAP | 4-Dimethylaminopyridine |
| DMF | D imethylformamide |
| HOBt | Hydroxybenzotriazole |
| CNS | Central Nervous System |
| BBB | Blood-Brain Barrier |
| AKT | Protein kinase B |
| ERK | extracellular signal-regulated kinase |
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- 1.Chandra S., Roy A., Jana M., Pahan K. Cinnamic Acid Activates PPARα to Stimulate Lysosomal Biogenesis and Lower Amyloid Plaque Pathology in an Alzheimer’s Disease Mouse Model. Neurobiol. Dis. 2019;124:379–395. doi: 10.1016/j.nbd.2018.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yilmaz S., Sova M., Ergün S. Antimicrobial Activity of Trans-Cinnamic Acid and Commonly Used Antibiotics against Important Fish Pathogens and Nonpathogenic Isolates. J. Appl. Microbiol. 2018;125:1714–1727. doi: 10.1111/jam.14097. [DOI] [PubMed] [Google Scholar]
- 3.Rodrigues M.P., Tomaz D.C., Ângelo de Souza L., Onofre T.S., Aquiles de Menezes W., Almeida-Silva J., Suarez-Fontes A.M., Rogéria de Almeida M., Manoel da Silva A., Bressan G.C. Synthesis of Cinnamic Acid Derivatives and Leishmanicidal Activity against Leishmania Braziliensis. Eur. J. Med. Chem. 2019;183:1–14. doi: 10.1016/j.ejmech.2019.111688. [DOI] [PubMed] [Google Scholar]
- 4.Abd El-Raouf O.M., El-Sayed M., Manie M.F. Cinnamic Acid and Cinnamaldehyde Ameliorate Cisplatin-Induced Splenotoxicity in Rats. J. Biochem. Mol. Toxicol. 2015;29:1–6. doi: 10.1002/jbt.21715. [DOI] [PubMed] [Google Scholar]
- 5.Wang S.R., Yang W., Fan Y., Dehaen W., Li Y., Li H., Wang W., Zheng Q., Huai Q. Design and Synthesis of the Novel Oleanolic Acid-Cinnamic Acid Ester Derivatives and Glycyrrhetinic Acid-Cinnamic Acid Ester Derivatives with Cytotoxic Properties. Bioorg. Chem. 2019;88:1–15. doi: 10.1016/j.bioorg.2019.102951. [DOI] [PubMed] [Google Scholar]
- 6.Guo S., Zhen Y., Zhu Z., Zhou G., Zheng X. Cinnamic Acid Rescues Behavioral Deficits in a Mouse Model of Traumatic Brain Injury by Targeting MiR-455-3p/HDAC2. Life Sci. 2019;235:1–9. doi: 10.1016/j.lfs.2019.116819. [DOI] [PubMed] [Google Scholar]
- 7.Ugazio E., Carlotti M.E., Sapino S., Trotta M., Vione D., Minero C. Photodegradation of Cinnamic Acid in Different Media. J. Dispers. Sci. Technol. 2008;29:641–652. doi: 10.1080/01932690701758491. [DOI] [Google Scholar]
- 8.Jin J.B., Cai B., Zhou J.M. Salicylic acid. In: Li J., Li C., Smith S., editors. Hormone Metabolism and Signallingin Plants. Elsevier; Amsterdam, The Netherlands: 2017. pp. 273–289. [DOI] [Google Scholar]
- 9.Zhang W.X., Wang H., Cui H.R., Guo W.B., Zhou F., Cai D.S., Xu B., Jia X.H., Huang X.M., Yang Y.Q. Design, Synthesis and Biological Evaluation of Cinnamic Acid Derivatives with Synergetic Neuroprotection and Angiogenesis Effect. Eur. J. Med. Chem. 2019;183:1–16. doi: 10.1016/j.ejmech.2019.04.033. [DOI] [PubMed] [Google Scholar]
- 10.De Lima G.D.A., Rodrigues M.P., de Mendes T.A.O., Moreira G.A., Siqueira R.P., da Silva A.M., Vaz B.G., Fietto J.L.R., Bressan G.C., Machado-Neves M. Synthesis and Antimetastatic Activity Evaluation of Cinnamic Acid Derivatives Containing 1,2,3-Triazolic Portions. Toxicol. In Vitro. 2018;53:1–9. doi: 10.1016/j.tiv.2018.07.015. [DOI] [PubMed] [Google Scholar]
- 11.Lan J.S., Hou J.W., Liu Y., Ding Y., Zhang Y., Li L., Zhang T. Design, Synthesis and Evaluation of Novel Cinnamic Acid Derivatives Bearing N-Benzyl Pyridinium Moiety as Multifunctional Cholinesterase Inhibitors for Alzheimer’s Disease. J. Enzyme Inhib. Med. Chem. 2017;32:776–788. doi: 10.1080/14756366.2016.1256883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gießel J.M., Loesche A., Hoenke S., Csuk R. In Search of New Cinnamic Acid Derived Flavours and Fragrances. Results Chem. 2019;1:1–8. doi: 10.1016/j.rechem.2019.100010. [DOI] [Google Scholar]
- 13.Adisakwattana S., Pongsuwan J., Wungcharoen C., Yibchok-Anun S. In Vitro Effects of Cinnamic Acid Derivatives on Protein Tyrosine Phosphatase 1B. J. Enzyme Inhib. Med. Chem. 2013;28:1067–1072. doi: 10.3109/14756366.2012.715286. [DOI] [PubMed] [Google Scholar]
- 14.Hu S., Yang X., Xue J., Chen X., Bai X.H., Yu Z.H. Graphene/Dodecanol Floating Solidification Microextraction for the Preconcentration of Trace Levels of Cinnamic Acid Derivatives in Traditional Chinese Medicines. J. Sep. Sci. 2017;40:2959–2966. doi: 10.1002/jssc.201700169. [DOI] [PubMed] [Google Scholar]
- 15.Mancilla-Montelongo G., Castañeda-Ramírez G.S., de JesúsTorres-Acosta J.F., Sandoval-Castro C.A., Borges-Argáez R. Evaluation of Cinnamic Acid and Six Analogues against Eggs and Larvae of Haemonchus Contortus. Vet. Parasitol. 2019;270:25–30. doi: 10.1016/j.vetpar.2019.05.009. [DOI] [PubMed] [Google Scholar]
- 16.Malheiro J.F., Maillard J.Y., Borges F., Simões M. Evaluation of Cinnamaldehyde and Cinnamic Acid Derivatives in Microbial Growth Control. Int. Biodeterior. Biodegrad. 2019;141:71–78. doi: 10.1016/j.ibiod.2018.06.003. [DOI] [Google Scholar]
- 17.World Health Organisation . Global Report for Research on Infectious Diseases of Poverty. WHO; Geneva, Switzerland: 2012. pp. 1–184. [Google Scholar]
- 18.UNAIDS . Global HIV and AIDS Statistics 2019 Fact Sheet. Volume 1. UNAIDS; Geneva, Switzerland: 2019. pp. 1–6. [Google Scholar]
- 19.World Health Organisation . Global Tuberculosis Report. WHO; Geneva, Switzerland: 2019. pp. 1–297. [Google Scholar]
- 20.Guzman J.D. Natural Cinnamic Acids, Synthetic Derivatives and Hybrids with Antimicrobial Activity. Molecules. 2014;19:19292–19349. doi: 10.3390/molecules191219292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Deng Z., Li C., Luo D., Teng P., Guo Z., Tu X., Zou K., Gong D. A New Cinnamic Acid Derivative from Plant-Derived Endophytic Fungus Pyronema Sp. Nat. Prod. Res. 2017;31:2413–2419. doi: 10.1080/14786419.2017.1311890. [DOI] [PubMed] [Google Scholar]
- 22.Korošec B., Sova M., Turk S., Kraševec N., Novak M., Lah L., Stojan J., Podobnik B., Berne S., Zupanec N., et al. Antifungal Activity of Cinnamic Acid Derivatives Involves Inhibition of Benzoate 4-Hydroxylase (CYP53) J. Appl. Microbiol. 2014;116:955–966. doi: 10.1111/jam.12417. [DOI] [PubMed] [Google Scholar]
- 23.Podobnik B., Stojan J., Lah L., Krasevec N., Seliskar M., Rizner T.L., Rozman D., Komel R. CYP53A15 of Cochliobolus lunatus, a target for natural antifungal compounds. J. Med. Chem. 2008;51:3480–3486. doi: 10.1021/jm800030e. [DOI] [PubMed] [Google Scholar]
- 24.Atmaram Upare A., Gadekar P.K., Sivaramakrishnan H., Naik N., Khedkar V.M., Sarkar D., Choudhari A., Mohana Roopan S. Design, Synthesis and Biological Evaluation of (E)-5-Styryl-1,2,4-Oxadiazoles as Anti-Tubercular Agents. Bioorg. Chem. 2019;86:507–512. doi: 10.1016/j.bioorg.2019.01.054. [DOI] [PubMed] [Google Scholar]
- 25.Gaikwad N., Nanduri S., Madhavi Y.V. Cinnamamide: An Insight into the Pharmacological Advances and Structure–Activity Relationships. Eur. J. Med. Chem. 2019;181:1–24. doi: 10.1016/j.ejmech.2019.07.064. [DOI] [PubMed] [Google Scholar]
- 26.Naveed M., Hejazi V., Abbas M., Kamboh A.A., Khan G.J., Shumzaid M., Ahmad F., Babazadeh D., FangFang X., Modarresi-Ghazani F. Chlorogenic Acid (CGA): A Pharmacological Review and Call for Further Research. Biomed. Pharmacother. 2018;97:67–74. doi: 10.1016/j.biopha.2017.10.064. [DOI] [PubMed] [Google Scholar]
- 27.Wang Y., He F., Wu S., Luo Y., Wu R., Hu D., Song B. Design, Synthesis, Anti-TMV Activity, and Preliminary Mechanism of Cinnamic Acid Derivatives Containing Dithioacetal Moiety. Pestic. Biochem. Physiol. 2020:1–7. doi: 10.1016/j.pestbp.2020.01.002. [DOI] [PubMed] [Google Scholar]
- 28.World Health Organisation Latest Global Cancer Data. [(accessed on 12 September 2018)]; Available online: https://www.who.int/cancer/PRGlobocanFinal.pdf.
- 29.Zhao Z., Song H., Xie J., Liu T., Zhao X., Chen X., He X., Wu S., Zhang Y., Zheng X. Research Progress in the Biological Activities of 3,4,5-Trimethoxycinnamic Acid (TMCA) Derivatives. Eur. J. Med. Chem. 2019;173:213–227. doi: 10.1016/j.ejmech.2019.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ling Y., Gao W.J., Ling C., Liu J., Meng C., Qian J., Liu S., Gan H., Wu H., Tao J. β-Carboline and N-Hydroxycinnamamide Hybrids as Anticancer Agents for Drug-Resistant Hepatocellular Carcinoma. Eur. J. Med. Chem. 2019;168:515–526. doi: 10.1016/j.ejmech.2019.02.054. [DOI] [PubMed] [Google Scholar]
- 31.Ge Y.X., Wang Y.H., Zhang J., Yu Z.P., Mu X., Song J.L., Wang Y.Y., Yang F., Meng N., Jiang C.S. New Cinnamic Acid-Pregenolone Hybrids as Potential Antiproliferative Agents: Design, Synthesis and Biological Evaluation. Steroids. 2019;152:1–9. doi: 10.1016/j.steroids.2019.108499. [DOI] [PubMed] [Google Scholar]
- 32.Sadeghi S., Davoodvandi A., Pourhanifeh M.H., Sharifi N., ArefNezhad R., Sahebnasagh R., Moghadam S.A., Sahebkar A., Mirzaei H. Anti-Cancer Effects of Cinnamon: Insights into Its Apoptosis Effects. Eur. J. Med. Chem. 2019;178:131–140. doi: 10.1016/j.ejmech.2019.05.067. [DOI] [PubMed] [Google Scholar]
- 33.Perković I., Raić-Malić S., Fontinha D., Prudêncio M., Pessanha de Carvalho L., Held J., Tandarić T., Vianello R., Zorc B., Rajić Z. Harmicines—Harmine and Cinnamic Acid Hybrids as Novel Antiplasmodial Hits. Eur. J. Med. Chem. 2020;187:1–16. doi: 10.1016/j.ejmech.2019.111927. [DOI] [PubMed] [Google Scholar]
- 34.Pellerito C., Emanuele S., Ferrante F., Celesia A., Giuliano M., Fiore T. Tributyltin(IV) Ferulate, a Novel Synthetic Ferulic Acid-Derivative, Induces Autophagic Cell Death in Colon Cancer Cells: From Chemical Synthesis to Biochemical Effects. J. Inorg. Biochem. 2020;205:1–14. doi: 10.1016/j.jinorgbio.2020.110999. [DOI] [PubMed] [Google Scholar]
- 35.Matlou G.G., Oluwole D.O., Nyokong T. Evaluation of the Photosensitizing Properties of Zinc and Indium Tetra Cinnamic Acid Phthalocyanines Linked to Magnetic Nanoparticles on Human Breast Adenocarcinoma Cells. J. Lumin. 2019;205:385–392. doi: 10.1016/j.jlumin.2018.09.054. [DOI] [Google Scholar]
- 36.De P., Baltas M., Bedos-Belval F. Cinnamic Acid Derivatives as Anticancer Agents-A Review. Curr. Med. Chem. 2011;18:1672–1703. doi: 10.2174/092986711795471347. [DOI] [PubMed] [Google Scholar]
- 37.Kaupp G. Encyclopedia of Physical OrganicChemistry. 1st ed. John Wiley & Sons, Inc.; Hoboken, NJ, USA: 2017. Organic Solid-State Reactions; pp. 1–79. [Google Scholar]
- 38.Tsai C.M., Yen G.C., Sun F.M., Yang S.F., Weng C.J. Assessment of the Anti-Invasion Potential and Mechanism of Select Cinnamic Acid Derivatives on Human Lung Adenocarcinoma Cells. Mol. Pharm. 2013;10:1890–1900. doi: 10.1021/mp3006648. [DOI] [PubMed] [Google Scholar]
- 39.Pijuan J., Barceló C., Moreno D.F., Maiques O., Sisó P. In Vitro Cell Migration, Invasion, and Adhesion Assays: From Cell Imaging to Data Analysis. Front. Cell Dev. Biol. 2019;7:1–16. doi: 10.3389/fcell.2019.00107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brewster R., Pincus P.A., Safran S.A. Hybrid Lipids as a Biological Surface-Active Component. Biophys. J. 2009;97:1087–1094. doi: 10.1016/j.bpj.2009.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Endo S., Hoshi M., Matsunaga T., Inoue T., Ichihara K., Ikari A. Autophagy Inhibition Enhances Anticancer Efficacy of Artepillin C, a Cinnamic Acid Derivative in Brazilian Green Propolis. Biochem. Biophys. Res. Commun. 2018;497:437–443. doi: 10.1016/j.bbrc.2018.02.105. [DOI] [PubMed] [Google Scholar]
- 42.Choudhari M.K., Haghniaz R., Rajwade J.M., Paknikar K.M. Anticancer Activity of Indian Stingless Bee Propolis: An In Vitro Study. Evid.-Based Complement. Altern. Med. 2013;2013:928280. doi: 10.1155/2013/928280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kinugasa T., Huo Q.U.N., Higashi D., Shibaguchi H. Selective Up-Regulation of Claudin-1 and Claudin-2 in Colorectal Cancer. Anticancer Res. 2007;27:3729–3734. doi: 10.1016/S0016-5085(08)62087-9. [DOI] [PubMed] [Google Scholar]
- 44.Shimizu Y., Suzuki T. Brazilian Propolis Extract Reduces Intestinal Barrier Defects and Inflammation in a Colitic Mouse Model. Nutr. Res. 2019;69:30–41. doi: 10.1016/j.nutres.2019.07.003. [DOI] [PubMed] [Google Scholar]
- 45.Sonoki H., Tanimae A., Furuta T., Endo S., Matsunaga T., Ichihara K., Ikari A. Caffeic Acid Phenethyl Ester Down-Regulates Claudin-2 Expression at the Transcriptional and Post-Translational Levels and Enhances Chemosensitivity to Doxorubicin in Lung Adenocarcinoma A549 Cells. J. Nutr. Biochem. 2018;56:205–214. doi: 10.1016/j.jnutbio.2018.02.016. [DOI] [PubMed] [Google Scholar]
- 46.Marin J.J., Herraez E., Lozano E., Macias R.I., Briz O. Models for Understanding Resistance to Chemotherapy in Liver Cancer. Cancers. 2019;11:1677. doi: 10.3390/cancers11111677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Luo Y., Zhou Y., Song Y., Chen G., Wang Y.X., Tian Y., Fan W.W., Yang Y.S., Cheng T., Zhu H.L. Optimization of Substituted Cinnamic Acyl Sulfonamide Derivatives as Tubulin Polymerization Inhibitors with Anticancer Activity. Bioorg. Med. Chem. Lett. 2018;28:3634–3638. doi: 10.1016/j.bmcl.2018.10.037. [DOI] [PubMed] [Google Scholar]
- 48.Toolabi M., Moghimi S., Bakhshaiesh T.O., Salarinejad S., Aghcheli A., Hasanvand Z., Nazeri E., Khalaj A., Esmaeili R., Foroumadi A. 6-Cinnamoyl-4-Arylaminothienopyrimidines as Highly Potent Cytotoxic Agents: Design, Synthesis and Structure-Activity Relationship Studies. Eur. J. Med. Chem. 2020;185:1–13. doi: 10.1016/j.ejmech.2019.111786. [DOI] [PubMed] [Google Scholar]
- 49.Yang K., Li Y., Tang Q., Zheng L., He D. Synthesis, Mitochondrial Localization of Fluorescent Derivatives of Cinnamamide as Anticancer Agents. Eur. J. Med. Chem. 2019;170:45–54. doi: 10.1016/j.ejmech.2019.03.001. [DOI] [PubMed] [Google Scholar]
- 50.Nabih M. Antiparasitic Activity of Medicinal Plant Extracts. Suez Canal University Faculty of Science Zoology Department; Ismailia, Egypt: 2019. pp. 1–36. [DOI] [Google Scholar]
- 51.World Health Organization . World Malaria Report 2019. WHO; Geneva, Switzerland: 2019. [Google Scholar]
- 52.Chen D.D., Zhang B.Y., Liu X.X., Li X.Q., Yang X.J., Zhou L. Bioactivity and Structure–Activity Relationship of Cinnamic Acid Derivatives and Its Heteroaromatic Ring Analogues as Potential High-Efficient Acaricides against Psoroptes Cuniculi. Bioorg. Med. Chem. Lett. 2018;28:1149–1153. doi: 10.1016/j.bmcl.2017.08.051. [DOI] [PubMed] [Google Scholar]
- 53.Shang X.F., Dai L.X., Liu Y.Q., Zhao Z.M., Li J.C., Yang G.Z., Yang C.J. Acaricidal Activity and Enzyme Inhibitory Activity of Active Compounds of Essential Oils against Psoroptes Cuniculi. Vet. Parasitol. 2019;267:54–59. doi: 10.1016/j.vetpar.2019.01.013. [DOI] [PubMed] [Google Scholar]
- 54.Benton A.L. Foundations of Physiological Psychology. 6th ed. Pearson A and B; London, UK: 2005. Chapter 3. Structure of the Nervous SystemNeurology; pp. 1–34. [DOI] [Google Scholar]
- 55.Loscher W., Potschka H. Drug resistance in brain diseases and the role of drug efflux transporters. Nat. Rev. Neurosci. 2005;6:591–602. doi: 10.1038/nrn1728. [DOI] [PubMed] [Google Scholar]
- 56.Ghafary S., Najafi Z., Mohammadi-Khanaposhtani M., Nadri H., Edraki N., Ayashi N., Larijani B., Amini M., Mahdavi M. Novel Cinnamic Acid–Tryptamine Hybrids as Potent Butyrylcholinesterase Inhibitors: Synthesis, Biological Evaluation, and Docking Study. Arch. Pharm. (Weinh.) 2018;351:1–10. doi: 10.1002/ardp.201800115. [DOI] [PubMed] [Google Scholar]
- 57.Medvedeva M., Barinova K., Melnikova A., Semenyuk P., Kolmogorov V., Gorelkin P., Erofeev A., Muronetz V. Naturally Occurring Cinnamic Acid Derivatives Prevent Amyloid Transformation of Alpha-Synuclein. Biochimie. 2020;170:128–139. doi: 10.1016/j.biochi.2020.01.004. [DOI] [PubMed] [Google Scholar]
- 58.Semenyuk P., Kurochkina L., Barinova K., Muronetz V. Alpha-Synuclein Amyloid Aggregation Is Inhibited by Sulfated Aromatic Polymers and Pyridinium Polycation. Polymers. 2020;12:517. doi: 10.3390/polym12030517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen Y., Zhu J., Mo J., Yang H., Jiang X., Lin H., Gu K., Pei Y., Wu L., Tan R. Synthesis and Bioevaluation of New Tacrine-Cinnamic Acid Hybrids as Cholinesterase Inhibitors against Alzheimer’s Disease. J. Enzyme Inhib. Med. Chem. 2018;33:290–302. doi: 10.1080/14756366.2017.1412314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lan J.S., Zeng R.F., Jiang X.Y., Hou J.W., Liu Y., Hu Z.H., Li H.X., Li Y., Xie S.S., Ding Y. Design, Synthesis and Evaluation of Novel Ferulic Acid Derivatives as Multi-Target-Directed Ligands for the Treatment of Alzheimer’s Disease. Bioorg. Chem. 2020;94:1–12. doi: 10.1016/j.bioorg.2019.103413. [DOI] [PubMed] [Google Scholar]
- 61.Novak I., Klasinc L., McGlynn S.P. Photoelectron Spectra and Biological Activity of Cinnamic Acid Derivatives Revisited. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2018;189:129–132. doi: 10.1016/j.saa.2017.08.030. [DOI] [PubMed] [Google Scholar]
- 62.Shairibha1 S.M.R., Rajadurai M., Kumar A.N. Effect of p -Coumaric Acid on Biochemical Parameters in Streptozotocin-Induced Diabetic Rats. JAIR. 2014;3:237–242. [Google Scholar]
- 63.Amalan V., Vijayakumar N., Indumathi D. Antidiabetic and Antihyperlipidemic Activity of p -Coumaric Acid in Diabetic Rats, Role of Pancreatic GLUT 2: In Vivo Approach. Biomed. Pharmacother. 2016;84:230–236. doi: 10.1016/j.biopha.2016.09.039. [DOI] [PubMed] [Google Scholar]
- 64.Amalan V., Vijayakumar N., Ramakrishnan A. p-Coumaric Acid Regulates Blood Glucose and Antioxidant Levels in Streptozotocin Induced Diabetic Rats. J. Chem. Pharm. Res. 2015;7:831–839. [Google Scholar]
- 65.Spagnol C.M., Assis R.P., Brunetti I.L., Isaac V.L.B., Salgado H.R.N., Corrêa M.A. In Vitro Methods to Determine the Antioxidant Activity of Caffeic Acid. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2019;219:358–366. doi: 10.1016/j.saa.2019.04.025. [DOI] [PubMed] [Google Scholar]
- 66.Adisakwattana S., Chantarasinlapin P., Thammarat H., Yibchok-Anun S. A Series of Cinnamic Acid Derivatives and Their Inhibitory Activity on Intestinal α-Glucosidase. J. Enzyme Inhib. Med. Chem. 2009;24:1194–1200. doi: 10.1080/14756360902779326. [DOI] [PubMed] [Google Scholar]
- 67.Wang Z.G., Mi J., Wang X.R., Huo Y.Y., Peng Y.J., Zhang H.M., Gao Y., Zhang H.L. A New Cinnamic Acid Glycoside from Roots of Heracleum Dissectum. Nat. Prod. Res. 2018;32:133–140. doi: 10.1080/14786419.2017.1340285. [DOI] [PubMed] [Google Scholar]
- 68.Mi J., Peng Y., Zhang H., Wang X., Huo Y., Wang Z. CHEMISTRY A New Benzofuran Derivative Glycoside and a New Coumarin Glycoside from Roots of Heracleum Dissectum Ledeb. Med. Chem. Res. 2018:470–475. doi: 10.1007/s00044-017-2073-9. [DOI] [Google Scholar]
- 69.Xu X.T., Deng X.Y., Chen J., Liang Q.M., Zhang K., Li D.L., Wu P.P., Zheng X., Zhou R.P., Jiang Z.Y. Synthesis and Biological Evaluation of Coumarin Derivatives as α-Glucosidase Inhibitors. Eur. J. Med. Chem. 2020;189:1–8. doi: 10.1016/j.ejmech.2019.112013. [DOI] [PubMed] [Google Scholar]
- 70.Kumar R., Mahey S., Arora R., Mahajan J., Kumar V., Arora S. Insights into Biological Properties of Less Explored Bark of Industrially Important Acacia Catechu Willd. Ind. Crops Prod. 2019;138:1–9. doi: 10.1016/j.indcrop.2019.111486. [DOI] [Google Scholar]
- 71.Mazzone G., Russo N., Toscano M. Antioxidant Properties Comparative Study of Natural Hydroxycinnamic Acids and Structurally Modified Derivatives: Computational Insights. Comput. Theor. Chem. 2016;1077:39–47. doi: 10.1016/j.comptc.2015.10.011. [DOI] [Google Scholar]
- 72.Muhammad N., Saeed M., Adhikari A., Khan K.M., Khan H. Isolation of a New Bioactive Cinnamic Acid Derivative from the Whole Plant of Viola Betonicifolia. J. Enzyme Inhib. Med. Chem. 2013;28:997–1001. doi: 10.3109/14756366.2012.702344. [DOI] [PubMed] [Google Scholar]
- 73.Kortenska V.D., Velikova M.P., Yanishlieva N.V., Totzeva I.R., Bankova V.S., Marcucci M.C. Kinetics of Lipid Oxidation in the Presence of Cinnamic Acid Derivatives. Eur. J. Lipid Sci. Technol. 2002;104:19–28. doi: 10.1002/1438-9312(200201)104:1<19::AID-EJLT19>3.0.CO;2-Q. [DOI] [Google Scholar]
- 74.Amić A., Marković Z., Klein E., Dimitrić Marković J.M., Milenković D. Theoretical Study of the Thermodynamics of the Mechanisms Underlying Antiradical Activity of Cinnamic Acid Derivatives. Food Chem. 2018;246:481–489. doi: 10.1016/j.foodchem.2017.11.100. [DOI] [PubMed] [Google Scholar]
- 75.Nazir Y., Saeed A., Rafiq M., Afzal S., Ali A., Latif M., Zuegg J., Hussein W.M., Fercher C., Barnard R.T. Hydroxyl Substituted Benzoic Acid/Cinnamic Acid Derivatives: Tyrosinase Inhibitory Kinetics, Anti-Melanogenic Activity and Molecular Docking Studies. Bioorg. Med. Chem. Lett. 2020;30:1–5. doi: 10.1016/j.bmcl.2019.126722. [DOI] [PubMed] [Google Scholar]
- 76.Taofiq O., Heleno S.A., Calhelha R.C., Fernandes I.P., Alves M.J., Barros L., González-Paramás A.M., Ferreira I.C.F.R., Barreiro M.F. Phenolic Acids, Cinnamic Acid, and Ergosterol as Cosmeceutical Ingredients: Stabilization by Microencapsulation to Ensure Sustained Bioactivity. Microchem. J. 2019;147:469–477. doi: 10.1016/j.microc.2019.03.059. [DOI] [Google Scholar]
- 77.Lee S., Chen Y., Lin C., Chen K. Hair Dyes Resorcinol and Lawsone Reduce Production of Melanin in Melanoma Cells by Tyrosinase Activity Inhibition and Decreasing Tyrosinase and Microphthalmia-Associated Transcription Factor (MITF) Expression. IJMS. 2015;16:1495–1508. doi: 10.3390/ijms16011495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gunia-Krzyżak A., Słoczyńska K., Popiół J., Koczurkiewicz P., Marona H., Pękala E. Cinnamic Acid Derivatives in Cosmetics: Current Use and Future Prospects. Int. J. Cosmet. Sci. 2018;40:356–366. doi: 10.1111/ics.12471. [DOI] [PubMed] [Google Scholar]
- 79.Lee S.M., Zheng L.W., Jung Y., Hwang G.S., Kim Y.S. Effects of Hydroxycinnamic Acids on the Reduction of Furan and α-Dicarbonyl Compounds. Food Chem. 2020;312:1–5. doi: 10.1016/j.foodchem.2019.126085. [DOI] [PubMed] [Google Scholar]
- 80.Sawant R.R., Torchilin V.P. Multifunctional Nanocarriers and Intracellular Drug Delivery. Curr. Opin. Solid State Mater. Sci. 2012;16:269–275. doi: 10.1016/j.cossms.2012.09.001. [DOI] [Google Scholar]
- 81.Garcia-Jimenez A., García-Molina F., Teruel-Puche J.A., Saura-Sanmartin A., Garcia-Ruiz P.A., Ortiz-Lopez A., Rodríguez-López J.N., Garcia-Canovas F., Munoz-Munoz J. Catalysis and Inhibition of Tyrosinase in the Presence of Cinnamic Acid and Some of Its Derivatives. Int. J. Biol. Macromol. 2018;119:548–554. doi: 10.1016/j.ijbiomac.2018.07.173. [DOI] [PubMed] [Google Scholar]
- 82.Ribeiro D., Poença C., Varela C., Janela J., Tavares da Silva E.J., Fernandes E., Roleira F.M.F. New Phenolic Cinnamic Acid Derivatives as Selective COX-2 Inhibitors. Design, Synthesis, Biological Activity and Structure-Activity Relationships. Bioorg. Chem. 2019;91:1–10. doi: 10.1016/j.bioorg.2019.103179. [DOI] [PubMed] [Google Scholar]
- 83.Ruan B.F., Ge W.W., Cheng H.J., Xu H.J., Li Q.S., Liu X.H. Resveratrol-Based Cinnamic Ester Hybrids: Synthesis, Characterization, and Anti-Inflammatory Activity. J. Enzyme Inhib. Med. Chem. 2017;32:1282–1290. doi: 10.1080/14756366.2017.1381090. [DOI] [PMC free article] [PubMed] [Google Scholar]